

Background: NMDA receptor trafficking to synapses is essential for excitatory neurotransmission.
Results: We have identified novel residues within 14-3-3ζ that determine isoform-specific interaction with GluN2C.
Conclusion: 14-3-3 is a critical mediator of GluN2C-containing NMDA receptor trafficking.
Significance: This is the first study to uncover the isoform-specific role of 14-3-3 in targeting NMDA receptors to the membrane.
Keywords: 14-3-3 protein, ionotropic glutamate receptor, membrane trafficking, protein phosphorylation, protein-protein interaction, GluN2C, NMDA
Abstract
The 14-3-3 family of proteins is widely distributed in the CNS where they are major regulators of essential neuronal functions. There are seven known mammalian 14-3-3 isoforms (ζ,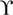 , τ, ϵ, η, β, and σ), which generally function as adaptor proteins. Previously, we have demonstrated that 14-3-3ϵ isoform dynamically regulates forward trafficking of GluN2C-containing NMDA receptors (NMDARs) in cerebellar granule neurons, that when expressed on the surface, promotes neuronal survival following NMDA-induced excitotoxicity. Here, we report 14-3-3 isoform-specific binding and functional regulation of GluN2C. In particular, we show that GluN2C C-terminal domain (CTD) binds to all 14-3-3 isoforms except 14-3-3σ, and binding is dependent on GluN2C serine 1096 phosphorylation. Co-expression of 14-3-3 (ζ and ϵ) and GluN1/GluN2C promotes the forward delivery of receptors to the cell surface. We further identify novel residues serine 145, tyrosine 178, and cysteine 189 on α-helices 6, 7, and 8, respectively, within ζ-isoform as part of the GluN2C binding motif and independent of the canonical peptide binding groove. Mutation of these conserved residues abolishes GluN2C binding and has no functional effect on GluN2C trafficking. Reciprocal mutation of alanine 145, histidine 180, and isoleucine 191 on 14-3-3σ isoform promotes GluN2C binding and surface expression. Moreover, inhibiting endogenous 14-3-3 using a high-affinity peptide inhibitor, difopein, greatly diminishes GluN2C surface expression. Together, these findings highlight the isoform-specific structural and functional differences within the 14-3-3 family of proteins, which determine GluN2C binding and its essential role in targeting the receptor to the cell surface to facilitate glutamatergic neurotransmission.
, τ, ϵ, η, β, and σ), which generally function as adaptor proteins. Previously, we have demonstrated that 14-3-3ϵ isoform dynamically regulates forward trafficking of GluN2C-containing NMDA receptors (NMDARs) in cerebellar granule neurons, that when expressed on the surface, promotes neuronal survival following NMDA-induced excitotoxicity. Here, we report 14-3-3 isoform-specific binding and functional regulation of GluN2C. In particular, we show that GluN2C C-terminal domain (CTD) binds to all 14-3-3 isoforms except 14-3-3σ, and binding is dependent on GluN2C serine 1096 phosphorylation. Co-expression of 14-3-3 (ζ and ϵ) and GluN1/GluN2C promotes the forward delivery of receptors to the cell surface. We further identify novel residues serine 145, tyrosine 178, and cysteine 189 on α-helices 6, 7, and 8, respectively, within ζ-isoform as part of the GluN2C binding motif and independent of the canonical peptide binding groove. Mutation of these conserved residues abolishes GluN2C binding and has no functional effect on GluN2C trafficking. Reciprocal mutation of alanine 145, histidine 180, and isoleucine 191 on 14-3-3σ isoform promotes GluN2C binding and surface expression. Moreover, inhibiting endogenous 14-3-3 using a high-affinity peptide inhibitor, difopein, greatly diminishes GluN2C surface expression. Together, these findings highlight the isoform-specific structural and functional differences within the 14-3-3 family of proteins, which determine GluN2C binding and its essential role in targeting the receptor to the cell surface to facilitate glutamatergic neurotransmission.
Introduction
The 14-3-3 family of proteins is highly conserved, ubiquitously expressed, and essential for many cellular functions (1, 2). It was originally identified as an abundant protein in brain tissue that showed a characteristic migration pattern following two-dimensional chromatography and electrophoresis, which gave rise to the peculiar terminology of 14-3-3. The mammalian 14-3-3 proteins contain seven highly homologous isoforms (ζ, , τ, ϵ, η, β, and σ) and form homo- and/or heterodimeric cup-shaped structures. Some isoforms primarily form homodimers such as the 14-3-3σ isoform, while some preferentially form heterodimers such as the 14-3-3ϵ isoform (3). Dimeric 14-3-3s interact with a variety of signaling proteins to regulate a wide range of cellular processes. Although all 14-3-3 isoforms are expressed in the brain, each isoform displays unique expression pattern. For example, the 14-3-3ϵ isoform is expressed throughout the brain with the most distinctive staining in the molecular and granular layers of the cerebellum, whereas the β, γ, η, and ζ isoforms are mainly expressed in the gray matter area of the brain including the hippocampus, thalamus, and cortex, with similar distribution pattern revealing subtle differences in location (4).
14-3-3 proteins are known to regulate the activity and subcellular localization of target proteins, and in particular control membrane receptor trafficking (5, 6). A number of studies have shown that 14-3-3 proteins can facilitate efficient cell surface expression of several membrane proteins including potassium channels, acetylcholine receptors, and NMDA receptors (NMDARs)2 (7–10). In any case, 14-3-3 proteins directly bind to these membrane proteins and regulate their surface expression in a phosphorylation-dependent manner. Although different 14-3-3 isoforms appear to have overlapping and redundant roles, some isoform-specific functions clearly exist. Emerging evidence suggests that the individual 14-3-3 isoforms can significantly differ in brain function (4, 11, 12). Additionally, it is well known that slight variations in structure and amino acid composition can result in major biological differences (13–15). Therefore, 14-3-3 isoform-specific structural differences and regulation of its host interacting proteins should be further clarified.
NMDARs are ligand-gated and voltage-dependent ionotropic glutamate receptors that are critical for synaptic plasticity and memory function (16, 17). NMDARs are tetramers composed of two obligatory GluN1 subunits and two regulatory subunits from the GluN2 (GluN2A-GluN2D) subunits and/or, less commonly, the GluN3 (GluN3A-GluN3B) subunits. The GluN1 subunit is alternatively spliced resulting in a total of eight splice variants. While GluN1 is widely distributed, each GluN2 subunit displays unique spatiotemporal expression patterns throughout the brain and receptors assembled with different GluN2 subunits exhibit distinct pharmacological and functional properties. GluN2A and GluN2B are the major GluN2 subunits expressed in the cortex and hippocampus. GluN2C was thought to have limited expression pattern, mainly restricted to the cerebellum, thalamus, and olfactory bulb (18). However, using knock-in mice expressing β-galactosidase as a reporter under the control of GluN2C promoter, it has been shown that GluN2C is also expressed in previously unidentified areas such as the retrosplenial cortex, striatum, and hippocampus (19). Whether GluN2C is expressed at synapses and what is the function of GluN2C in these newly identified areas remains unknown.
The trafficking of NMDARs to synapses plays an important role in regulating excitatory neurotransmission (20, 21). The properly assembled NMDARs, which contain both GluN1 and GluN2 subunits, are targeted onto plasma membrane via the biosynthetic secretory pathway. The most abundant GluN1 splice variant, GluN1-1, has been demonstrated to contain an ER-retention motif, which prevents surface expression of homomeric GluN1-1 (22). GluN2A, GluN2B, and GluN2C are also ER-retained and possess ER-retention signals in the transmembrane domain 3 and putative ER-retention motifs at the N terminus and/or C terminus (23, 24). It is generally believed that assembled protein complexes mask the ER-retention signal within individual subunits to retain unassembled subunits in the ER. Surprisingly, the assembled complex of NMDAR subunits does not override the ER-retention of the individual subunits and the ER export of the assembled NMDARs is dependent on a specific motif, suggesting that a novel mechanism must be employed. Compared with GluN2A and GluN2B, the trafficking of GluN2C is less well understood.
We have previously identified 14-3-3ϵ as a GluN2C binding partner, whose interaction is dependent on Ser-1096 phosphorylation on GluN2C (7). In the current study, we find that GluN2C interacts with all 14-3-3 proteins except the σ isoform. Co-expression of 14-3-3 isoforms (ϵ and ζ) that bind to GluN2C increases surface expression of GluN2C-containing NMDARs, but no increase is observed when 14-3-3σ is co-expressed. We also identify three critical residues in 14-3-3 that are required for the GluN2C binding and its effect on GluN2C trafficking. Interestingly, these three residues are not located in the ligand-binding groove of 14-3-3. Moreover, reciprocal mutations on these three residues in 14-3-3σ enable its binding to GluN2C. Thus, our findings reveal a novel regulatory region in 14-3-3 that modulates isoform-specific interaction with GluN2C.
Experimental Procedures
cDNA Constructs and Site-directed Mutagenesis
The human full-length 14-3-3 cDNA isoforms (ζ,, τ, ϵ, η, β, and σ) and enhanced cyan fluorescent protein (pECFP)-tagged difopein in pSCM137 vector were kindly provided by Haian Fu (Emory University). 14-3-3 constructs were amplified by PCR, and subcloned into the Gal4 activation domain-fusion vector pGAD10 and pCMV-Myc (BD Biosciences) using the In-Fusion® HD Cloning Kit (Clontech, CA). Full-length GluN2C in the mammalian expression vector pRK5 was epitope tagged with the 3xFLAG epitope (DYKDHDGDYKDHDIDYKDDDDK) between amino acids 36 and 37 using site-directed mutagenesis (Stratagene, La Jolla, CA). The GluN1–1a-IRES-DsRed plasmid was constructed by subcloning GluN1-1a into pIRES2-DsRed vector (BD Biosciences). All mutations were generated by site-directed mutagenesis. All cDNA constructs used were verified by DNA sequencing.
Yeast Two-hybrid Assay
Yeast two-hybrid assay was performed using the L40 yeast strain (MATa his3Δ200 trp1-901 leu2-3,112 ade2 LYS2::(lexAop)4-HIS3 URA3::(lexAop)8-lacZ GAL4) as described previously (25). Briefly, constructs in LexA-fusion vector pBHA (TRP1 plasmid) and the Gal4 activation domain fusion vector pGAD10 (LEU2 plasmid) were co-transformed into L40 yeast. After transformation, the yeast were plated in synthetic complete medium lacking leucine and tryptophan. Three independent yeast colonies were selected and assayed for expression of the reporter HIS3 gene in synthetic complete medium lacking leucine, tryptophan, and histidine.
Co-immunoprecipitation and Western Blot
HEK-293LTV cells were transiently transfected with GFP-GluN2C, GluN1–1a-IRES-DsRed, and Myc-14-3-3ε (positive control), Myc-14-3-3ζ WT, Myc-14-3-3ζ S145A/Y178H/C189I, Myc-14-3-3σ WT, Myc-14-3-3σ A147S/I191C, or empty Myc vector (negative control). 48 h post-transfection, cells were resuspended in cold PBS lysis buffer (pH 7.4, containing Pierce Protease and Phosphatase Inhibitor mini tablets, 1 mm EDTA), lysed by sonication and centrifuged for 20 min. The resulting pellet was solubilized using 0.5% SDS in PBS for 15 min at 37 °C. Five volumes of PBS containing 2% Triton-X 100 (TX-100) were added to the lysate resulting in a final concentration of 0.1% SDS. Insoluble material was removed by centrifugation for 15 min. The isolated membrane fraction was immunoprecipitated with anti-GFP antibodies overnight at 4 °C and then incubated with protein A magnetic beads (Dynabeads) for 1 h at 4 °C and washed with 0.05% SDS/1% TX-100 in PBS. Immunoprecipitates were resolved by 10% SDS-PAGE and immunoblotted with anti-Myc antibodies to detect 14-3-3 and anti-GFP antibodies to detect GluN2C (data not shown). The experiment was repeated three times and quantified using ImageQuant software. A ratio of immunoprecipitated 14-3-3 protein to input (2% of total lysate) was determined. The average normalized ratio is shown.
HeLa Cell Culture and Transfections
HeLa cells used for transient transfections were maintained in DMEM supplemented with 10% fetal calf serum (Invitrogen) and 2 mm glutamine and kept at 37 °C and 5% CO2. Cells were plated 30% confluent in 6-well dishes containing glass coverslips for 24 h. On the day of transfection, GFP-GluN2C, GluN1–1a-IRES-DsRed, and Myc-14-3-3 constructs were co-transfected into cells using the calcium phosphate method for 16 h. Then the cells were cultured in fresh medium for an additional 20 h for staining and subsequent immunocytochemistry experiments.
Primary Culture of Hippocampal Neurons and Transfection
Primary hippocampal cultures were prepared from embryonic day 18 (E18) Sprague-Dawley rats as previously described (28). Briefly, hippocampal tissue was dissociated with trypsin and plated on poly-d-lysine-coated coverslips at a density of 2 × 106 cells/ml. The cells were cultured in Neurobasal medium supplemented with B27 and l-glutamine (all from Invitrogen). The cultures were maintained at 37 °C in 5% CO2. Cells were transfected at 13 days in vitro (DIV) using Lipofectamine (Lipofectamine 2000; Invitrogen). Immunocytochemistry experiments were performed at DIV15.
Immunocytochemistry
Cells were stained using the following antibodies in PBS containing 3% normal goat serum (NGS) for 30 min: rabbit polyclonal anti-GFP (1:1000, Invitrogen), mouse monoclonal anti-FLAG M2 (1:1000, Sigma), mouse monoclonal anti-Myc (1:1000, Santa Cruz) and mouse monoclonal anti-synaptophysin (1:1000, Sigma). Images were collected using a Zeiss LSM 700 confocal microscope using 40× and 63× objectives, and series of 0.5 μm optical sections were captured through the z axis and used to create maximum projection images. For quantitative analysis, images from three dendrites per neuron (three to four neurons per experiment, three to four independent experiments from separate neuronal cultures) were collected and quantitated using ImageJ software. For surface labeling, cells were incubated with anti-GFP or anti-FLAG antibodies for 30 min to label the surface pool of receptors, washed with PBS, fixed in 4% paraformaldehyde in PBS for 15 min, and incubated with Alexa 647-conjugated secondary antibodies (Invitrogen) in PBS/3% NGS for 30 min. Cells were then permeabilized with 0.25% Triton-X 100 in PBS, incubated with anti-GFP or anti-FLAG antibodies to label the intracellular pool of receptors and anti-Myc to label Myc-14-3-3, and incubated with Alexa 488- and 405-conjugated secondary antibodies, respectively (Invitrogen). The fluorescence signal of GFP-tagged GluN2C is very low to nearly nonexistent (data not shown). Therefore, any non-immunolabeled GFP-GluN2C signal will have minimal effect on quantification of surface/intracellular GFP-GluN2C receptors. The cells were washed and mounted on glass slides with ProLong® Gold Antifade mounting medium (Molecular Probes). Surface fluorescence intensity was divided by intracellular fluorescence intensity to control for the GluN2C protein expression level. For surface receptor co-localization analysis with synaptophysin, cells were incubated with either polyclonal anti-GFP or anti-FLAG antibodies (depending on the cDNA expressed) for 30 min, washed with PBS, fixed in 4% paraformaldehyde in PBS for 15 min, and incubated with Alexa 647- (for labeling surface GluN2C). Cells were then permeabilized and labeled with anti-synaptophysin and Alexa 568-conjugated antibodies. The cells were washed and mounted as described above. Measurements from individual dendritic regions were first grouped and averaged; means from different neurons in each group were then averaged. Receptor puncta distribution was analyzed for colocalization with synaptophysin (defined as having overlapping or adjacent pixels).
Immunofluorescent Internalization Assay in HeLa Cells
Receptor endocytosis was analyzed using a fluorescence-based antibody uptake assay, as previously reported (26, 27) with minor changes. Transfected cells were incubated with anti-GFP antibody for 30 min on ice, washed three times with PBS, and returned to conditioned medium at 37 °C for 30 min to allow internalization. The subsequent steps were performed at room temperature. The cells were washed three times in PBS, fixed in 4% paraformaldehyde-4% sucrose in PBS for 15 min, blocked with 10% Normal Goat Serum (NGS from Vector Laboratories, Burlingame, CA) in PBS for 30 min and incubated with Alexa 647-conjugated anti-rabbit secondary antibodies (Invitrogen) for 30 min for labeling the surface population. The cells were then washed three times, permeabilized in 0.25% Triton X-100 in PBS for 5 min, blocked with 10% NGS in PBS for 1 h, and incubated for 30 min with mouse monoclonal anti-Myc antibody (1:1000, Santa Cruz Biotechnology) to label Myc-14-3-3-transfected cells. The cells were then incubated with Alexa 405-conjugated anti-mouse secondary antibody for labeling Myc-14-3-3 and Alexa 488-conjugated anti-rabbit secondary antibody to specifically label the internalized population of receptors. The cells were washed in PBS and mounted on glass slides with ProLong® Gold Antifade mounting medium (Molecular Probes) for imaging analysis.
Data Analysis and Statistics
Results from multiple repeats are expressed as average ± S.E. Data from immunohistochemistry experiments are the average of four cells/condition/experiment, quantified over four independent experiments. The statistical significance analysis for the difference between two groups was performed with Student's t test. The statistical significance analysis for the difference among multiple groups was performed with One-Way ANOVA. Following ANOVA, post hoc Bonferroni procedure was used to determine whether the data are statistically different from each other with *, p < 0.05 or **, p < 0.01.
Results
The C Terminus of GluN2C Interacts with 14-3-3 Isoforms
We assessed the 14-3-3 isoform-specific interactions with GluN2C-containing receptors using yeast two-hybrid direct protein-protein interaction assays. The last 175 amino acids within the C terminus of GluN2A, GluN2B, or GluN2C were fused to LexA DNA binding domain and co-transformed with the different 14-3-3 isoforms fused to Gal4 activation domain into yeast (Fig. 1A). By evaluating yeast cell growth, we observed that GluN2C interacted with 14-3-3ζ, ϵ, , τ, η, and β isoforms with similar degrees of binding strength and did not interact with 14-3-3σ (Fig. 1B). In contrast, we observed no specific binding when the C terminus of GluN1, GluN2A, or GluN2B was co-expressed with each 14-3-3 isoform, demonstrating that the interaction between 14-3-3 and NMDA receptors is specific to GluN2C (data not shown).
FIGURE 1.

GluN2C C terminus interacts with various 14-3-3 isoforms, and binding is regulated by Ser-1096.
A, schematic representation of yeast two-hybrid constructs used, including an alignment of GluN2A, GluN2B, and GluN2C. GluN2C Ser-1096 is indicated with an arrowhead and 14-3-3 binding motif is indicated with an arrow. The last 175 amino acids within the C terminus of GluN2A, GluN2B, or GluN2C were fused to LexA DNA binding domain. Each 14-3-3 isoform was fused to Gal4 activation domain and used for subsequent yeast two-hybrid binding assay. B, yeast were co-transformed with LexA-GluN2C WT C terminus and either Gal4 vector or Gal4-14-3-3, Gal4-14-3-3ζ, Gal4-14-3-3τ, Gal4- 14-3-3ϵ, Gal4-14-3-3η, Gal4-14-3-3β, or Gal4-14-3-3σ, and growth was evaluated on appropriate yeast selection medium. C, yeast were co-transformed with either LexA-GluN2C S1096A or LexA-GluN2C S1144A and either Gal4 vector or Gal4-14-3-3ϵ, τ, η, , ζ, or β, and growth was evaluated on appropriate yeast selection medium. B and C, results shown are 10-fold serial dilutions of yeast cells. All data shown are representative of at least three independent experiments.
GluN2C Ser-1096 Regulates GluN2C Binding to 14-3-3
14-3-3 interacts with its target proteins through binding to consensus phosphoserine-containing binding motifs. GluN2C contains two regions (1093-RHASLP-1098 and 1141-RLPSYP-1146) that are similar to previously identified 14-3-3 binding motifs, RSXSPXP and RXXXSPXP, where X can be any amino acid and SP indicates a phosphorylated serine (15, 29). Previously, we have identified that 14-3-3ϵ binding to GluN2C is regulated by Ser-1096 located within one of the two predicted 14-3-3 binding motifs on the C-terminal tail of GluN2C. To determine whether the other 14-3-3 isoforms bind to the same 14-3-3 binding motif in GluN2C, we examined the interaction of each 14-3-3 isoform with GluN2C containing either S1096A or S1144A phosphodeficient mutation. The S1096A mutation disrupted the binding to all GluN2C-interacting 14-3-3 isoforms (Fig. 1C), whereas it had no effect on binding to PSD-95 (positive control, data not shown). In contrast, GluN2C S1144A mutant had no effect on binding to all GluN2C-interacting 14-3-3 isoforms (Fig. 1C). Thus, GluN2C Ser-1096 plays a significant role in mediating binding to 14-3-3s.
GluN2C Interaction with 14-3-3ζ Is Mediated by Ser-145, Tyr-178, and Cys-189 on 14-3-3ζ
To identify the binding site contained within 14-3-3ζ that is required for the interaction with GluN2C, we generated truncated forms of wild-type (WT) 14-3-3ζ containing regions spanning α-helices 1–3 (a.a. 1–69), α-helices 4–6 (a.a. 70–161), or α-helices 7–9 (a.a. 162–253). We examined the interaction of GluN2C with WT 14-3-3ζ compared with each truncated 14-3-3ζ form using yeast two-hybrid binding assay. All 3 of the truncated forms of 14-3-3ζ did not interact with GluN2C (data not shown). These results suggest that an intact 14-3-3 structure is required for its interaction with GluN2C. Since this initial attempt to localize binding sites was unsuccessful, we employed a different experimental strategy. The observation that GluN2C exclusively binds to 14-3-3ϵ, ζ, , τ, η, and β isoforms but not with the σ isoform allowed us to identify putative amino acid residues within 14-3-3 that could mediate the isoform-specific interaction with GluN2C. Upon sequence examination, we found several identical amino acid residues at analogous parts of GluN2C-interacting 14-3-3 isoforms that are not conserved within 14-3-3σ. In previous studies, it has been shown that the 14-3-3ζ isoform is highly enriched in the hippocampus where they are up-regulated and exert neuroprotective effects following excitotoxic insults (30, 31). Interestingly, GluN2C has also been shown to be up-regulated in the hippocampus following excitotoxic insults (32, 33). Therefore, we chose to focus additional binding studies and functional analysis on the ζ isoform. To determine whether the 14-3-3 amino acid residues described above are required for isoform-specific GluN2C binding, we performed site-directed mutagenesis to replace the identified conserved amino acid residues of 14-3-3ζ by the corresponding amino acid residues of 14-3-3σ and examined the interaction of these 14-3-3ζ mutants with GluN2C using yeast two-hybrid binding assay. The interaction between 14-3-3ζ and GluN2C was reduced by single amino acid replacement S145A, Y178H, or C189I compared with the WT 14-3-3ζ, whereas the other mutations had no effect on reducing the interaction (Fig. 2A). Furthermore, the double mutants of 14-3-3ζ (S145A/Y178H, S145A/C189I, or Y178H/C189I) substantially reduced the binding to GluN2C and the triple mutations (S145A/Y178/C189I) completely abolished the GluN2C binding (Fig. 2B). We also assessed the interaction between GluN2C and 14-3-3 proteins using a co-immunoprecipitation approach. As shown in Fig. 2C and consistent with our yeast two-hybrid results, 14-3-3ϵ and ζ WT were effectively co-immunoprecipitated with GFP-GluN2C from HEK-293 lysate, whereas S145A/Y178/C189I mutations in 14-3-3ζ significantly reduced the interaction with GluN2C (1.000 ± 0.046 for ζ WT and 0.291 ± 0.024 for ζ S145A/Y178/C189I; *, p = 0.044). These results suggest that Ser-145, Tyr-178, and Cys-189 residues identical at analogous parts of GluN2C-interacting 14-3-3 isoforms are critical for mediating the isoform-specific interaction of 14-3-3ζ with GluN2C (Fig. 2D).
FIGURE 2.

GluN2C interaction with 14-3-3ζ is mediated by Ser-145, Tyr-178, and Cys-189 on 14-3-3ζ. A, yeast were co-transformed with LexA-GluN2C WT C terminus and either Gal4-14-3-3ζ WT or Gal4-14-3-3ζ amino acid mutants (R54S, H164N, A195T, I200M, D204H, S230A, D20E, V29A, K115R, E202D, E241Q, S145A, Y178H, or C189I, respectively). Growth was evaluated on appropriate yeast selection medium. Red box highlights single amino acid replacements that reduce GluN2C binding compared with the wild-type 14-3-3ζ. B, yeast were co-transformed with LexA-GluN2C WT C terminus and either Gal4-14-3-3ζ WT or Gal4-14-3-3ζ double and triple amino acid mutants (S145A/Y178H, S145A/C189I, Y178H/C189I, or S145A/Y178/C189I, respectively). Growth was evaluated on appropriate yeast selection medium. C, HEK-293LTV cells were transfected with GFP-GluN2C, GluN1–1a-IRES-DsRed, and 14-3-3ζ WT or 14-3-3ζ S145A/Y178/C189I. Receptors were immunoprecipitated from cell lysates with anti-GFP antibodies. Immunoprecipitates were resolved by SDS-PAGE and immunoblotted with anti-Myc antibodies. The data were quantified by measuring co-IP/input 14-3-3 band intensity ratios using ImageQuant software. Graph represents normalized means ± S.E., *, p < 0.05 (data normalized against 14-3-3ζ WT). All data shown are representative of at least three independent experiments (n = 3 separate cultures). Input = 2% total lysate. D, schematic diagram of GluN2C binding sites on 14-3-3 protein, including an alignment of all 7 mammalian 14-3-3 isoforms. α-helices (α1 to α9) are indicated as black boxes above the alignment. The critical amino acid residues that mediate binding to GluN2C (Ser-145, Tyr-178, and Cys-189 for 14-3-3ζ) within the positive interacting isoforms are indicated with asterisks in red. The corresponding amino acid residues within 14-3-3σ, which do not interact with GluN2C are indicated in green. A and B, results shown are 10-fold serial dilutions of yeast cells. All data shown are representative of at least three independent experiments.
Reciprocal Mutation of 14-3-3σ A147S and I191C is sufficient to promote GluN2C interaction with 14-3-3σ
To determine whether Ser-145, Tyr-178, and/or Cys-189 are sufficient for isoform-specific GluN2C interaction, we mutagenized each of the corresponding residues of 14-3-3σ including Ala-147, His-180, and Ile-191 to generate the reciprocal A147S, H180Y, and I191C single or double amino acid mutants. We hypothesized that the mutated form of 14-3-3σ, which contains the three residues critical for GluN2C interacting with all other 14-3-3 isoforms would promote binding with 14-3-3σ. Indeed, we found that A147S or I191C single amino acid mutation was sufficient to promote GluN2C binding, and double mutation A147S/H180Y and A147S/I191C further strengthened the interaction as we observed more cell growth by the yeast two-hybrid assay (Fig. 3A). In addition, 14-3-3σ A147S/I191C bound to GluN2C with similar binding strength as between 14-3-3ζ WT and GluN2C (lower panel, Fig. 3A). These results indicate that the Ser-145/Tyr-178/Cys-189 residues are sufficient to determine isoform binding specificity. (We note that the single H180Y mutation had no effect on GluN2C binding, although mutating this amino acid in the ζ isoform contributes to disruption of the GluN2C binding.) Again, we examined the interaction using a co-immunoprecipitation approach. As shown in Fig. 3B, 14-3-3σ WT bound very weakly to GluN2C (0.173 ± 0.017) and reciprocal mutation of the corresponding residues in 14-3-3σ (A147S/I191C) significantly promoted GluN2C interaction (1.000 ± 0.087, *, p = 0.038), supporting the results with the yeast two-hybrid experiments. To assess whether any obvious structural differences exist between 14-3-3σ and 14-3-3ζ to alternatively explain the isoform-specificity in complex formation with GluN2C, we compared the structural organization of the dimer interface by overlaying the previously resolved x-ray crystal structures (Fig. 3C). The view shown is in ribbon representation with the monomer structure highlighted and rotated 45° to allow visualization of the corresponding side-chains within the newly identified residues contained in 14-3-3ζ involved in the GluN2C binding. We determined that there are no potentially interacting residues in neighboring α-helices within 5 Å of the Ser-145/Tyr-178/Cys-189 residues. Structural alignment of these two isoforms reveals subtle differences and shifts in helical orientation. This reveals that differences in a few amino acids are sufficient to alter phosphopeptide client interaction and subsequent function. The Ser-145/Tyr-178/Cys-189 residues we identified are not contained within the peptide binding groove (a conserved region among all the different isoforms). Taken together, these data demonstrate a novel GluN2C binding motif contained within 14-3-3 comprised of Ser-145/Tyr-178/Cys-189 residues that are critical for 14-3-3 isoform-specific interaction.
FIGURE 3.

Reciprocal mutation of 14-3-3σ A147S is sufficient to promote GluN2C interaction with 14-3-3σ. A, yeast were co-transformed with LexA-GluN2C WT C terminus and either Gal4-14-3-3σ WT, Gal4-14-3-3ζ WT, or Gal4-14-3-3σ amino acid mutants (A147S, H180Y, I191C, A147S/H180Y, A147S/I191C, or H180Y/I191C, respectively), and growth was evaluated on appropriate yeast selection medium. Results shown are 10-fold serial dilutions of yeast cells. All data shown are representative of at least three independent experiments. B, HEK-293LTV cells were transfected with GFP-GluN2C, GluN1–1a-IRES-DsRed, and 14-3-3σ WT or 14-3-3σ A147S/I191C. Receptors were immunoprecipitated from cell lysates with anti-GFP antibodies. Immunoprecipitates were resolved by SDS-PAGE and immunoblotted with anti-Myc antibodies. The data were quantified by measuring co-IP/input14-3-3 band intensity ratios using ImageQuant software. Graph represents normalized means ± S.E., *, p < 0.05 (data normalized against 14-3-3σ A147S/I191C). All data shown are representative of at least three independent experiments (n = 3 separate cultures). Input = 2% total lysate. C, comparison of the structural organization of 14-3-3ζ and 14-3-3σ. The previously resolved x-ray crystal structures of dimeric 14-3-3ζ isoform (depicted in yellow) was superimposed onto dimeric 14-3-3σ isoform (blue). The view shown is in ribbon representation with the monomer structure highlighted by a box and rotated 45° to allow visualization of the corresponding amino acid side-chains within the newly identified residues (Ser-145, Tyr-178, and Cys-189) contained within 14-3-3ζ that mediate GluN2C interaction (indicated by arrows and highlighted in red). Previously identified phosphopeptide interacting residues include the Arginine-Arginine-Tyrosine triad (green), a hydrophobic patch (orange), and other essential residues for binding (purple).
14-3-3 Isoforms Differentially Regulate GluN2C Forward Trafficking in HeLa Cells
Previous studies have shown that 14-3-3ϵ mediates forward trafficking of GluN2C in cerebellar granule neurons (7). Although GluN2C is primarily expressed in the cerebellum, thalamus, and olfactory bulbs, it has been shown that GluN2C is endogenously expressed in other regions of the brain, such as in the hippocampus and cortex (19), where other 14-3-3 isoforms are also predominant. In addition, subcellular localization of ϵ, η, , ζ, and β isoforms in the rat forebrain has previously been determined using isoform-specific antibodies and subcellular fractionation techniques (35). The same study observed that although most of these isoforms exist in cytosolic fractions, ∼6% exist in the synaptic plasma membrane fraction. However, little is currently known about how the number and localization of GluN2C-containing NMDARs are regulated in non-cerebellar regions of the brain where they likely mediate essential excitatory neurotransmission. We next explored the possibility that specific 14-3-3 isoforms in complex with GluN2C may be critical in mediating effective receptor forward trafficking to the synaptic plasma membrane. To examine whether 14-3-3 regulates GluN2C trafficking in an isoform-specific manner, we co-expressed in HeLa cells, a heterologous expression system, GFP-GluN2C (which contains a GFP tag in the extracellular N-terminal domain of the receptor to enable labeling surface-expressed receptors), GluN1–1a-IRES-DsRed and Myc-tagged 14-3-3ϵ, 14-3-3ζ WT, 14-3-3σ WT, 14-3-3ζ S145A/Y178H/C189I mutant, or 14-3-3σ A147S/I191C mutant (Fig. 4A). We labeled the surface-expressed receptors with anti-GFP antibody, followed by membrane permeabilization and labeling of the intracellular pool of receptors using anti-GFP antibody. We found that the surface expression of GluN2C was substantially increased when co-expressed with both 14-3-3ϵ (1.87 ± 0.16 normalized surface/intracellular index; p < 0.001, ANOVA) and 14-3-3ζ (1.69 ± 0.12; *, p = 0.007) isoforms (Fig. 4B). Co-expression of 14-3-3σ WT isoform had no effect on GluN2C surface expression, which corroborates with our findings that GluN2C does not interact with the 14-3-3σ isoform. 14-3-3σ-transfected cells showed similar mean surface/intracellular ratio as compared with cells not transfected with 14-3-3 (0.97 ± 0.07; n.s. versus GluN2C/vector). Moreover, when we co-expressed ζ S145A/Y178H/C189I triple mutant construct, as previously shown to completely disrupt GluN2C binding, we found no effect on GluN2C surface expression (1.01 ± 0.04; n.s. versus GluN2C/vector). To determine if promoting GluN2C binding to σ isoform results in any functional effect, we co-expressed σ A147S/I191C double mutant construct (sufficient to promote GluN2C binding with similar binding affinity to ζ WT) and GluN2C. Given the unique role of 14-3-3σ as a critical mediator in cell cycle arrest and apoptosis (36, 37), we did not expect that promoting 14-3-3σ binding to GluN2C would necessarily affect receptor forward trafficking. However, we found that A147S/I191C mutations not only promoted 14-3-3σ binding to GluN2C, but also did result in increased GluN2C surface expression (1.78 ± 0.17; *, p = 0.002). We have also performed additional experiments to examine whether mutating the same corresponding residues in another 14-3-3 isoform, 14-3-3ϵ, would affect GluN2C trafficking. Cells co-expressing 14-3-3ϵ S148A/Y181H/C192I had significantly reduced surface/intracellular ratio of receptors (0.84 ± 0.28; *, p < 0.05; n = 12) compared with 14-3-3ϵ WT transfected cells (1.38 ± 0.25), similar to the effect observed with 14-3-3ζ WT versus 14-3-3ζ S145A/Y178H/C189I (data not shown). These results demonstrate that 14-3-3 isoform-specific binding to GluN2C functions to promote ER exit and subsequent trafficking to the cell surface in a mammalian heterologous expression system.
FIGURE 4.

14-3-3 isoforms differentially regulate GluN2C forward trafficking in HeLa cells. A, HeLa cells were co-transfected with GFP-GluN2C (containing a GFP protein tag in the extracellular N-terminal domain), GluN1–1a-IRES-DsRed (channel turned off) and Myc empty vector, Myc-14-3-3ϵ WT, Myc-14-3-3ζ WT, Myc-14-3-3ζ S145A/Y178H/C189I, Myc-14-3-3σ WT, or Myc-14-3-3σ A147S/I191C. Two days after transfection, cells were incubated with anti-GFP antibody for 30 min on ice. Cells were fixed and incubated with Alexa 647-conjugated anti-rabbit secondary antibody (depicted in red for optimal visual contrast effect) to visualize the surface receptors. Cells were then washed, permeabilized, and labeled with anti-GFP antibody and Alexa 488-conjugated anti-rabbit secondary antibody (green) to visualize intracellular pool of receptors and anti-Myc monoclonal antibody and Alexa 405-conjugated anti-mouse secondary antibody (gray) to visualize the 14-3-3-transfected cells. B, data were quantified by measuring ratios of surface GluN2C/intracellular GluN2C receptors using ImageJ software. Data represent means ± S.E. (n = 12 cells/group; **, p < 0.001; *, p < 0.05).
14-3-3 Isoforms Do Not Impair GluN2C Endocytosis in HeLa Cells
The increase in GluN2C surface receptors in cells expressing 14-3-3ϵ, 14-3-3ζ, or 14-3-3σ A147S/I191C isoforms could be due to other trafficking events, such as impaired endocytosis of GluN2C receptors. To rule this possibility out, we performed an immunofluorescence-based internalization assay of GluN2C containing an extracellular GFP tag to allow labeling of surface-expressed receptors as previously described (26, 27). We co-transfected HeLa cells with GFP-GluN2C, GluN1–1a-IRES-DsRed and Myc-14-3-3ζ WT, Myc-14-3-3ζ S145A/Y178H/C189I, Myc-14-3-3σ WT, Myc-14-3-3σ A147S/I191C, or empty Myc vector (Fig. 5A). The endocytosis assay demonstrates no significant difference in GluN2C internalization by co-expression of various 14-3-3 constructs (Fig. 5B), in particular when comparing 14-3-3ζ WT with 14-3-3ζ S145A/Y178H/C189I (p = 0.733, n = 15) and 14-3-3σ WT with 14-3-3σ A147S/I191C (p = 0.689, n = 15). These data suggest that the increased expression of GluN2C-containing NMDA receptors on the surface is not a result of impaired endocytosis and provides us with additional mechanistic evidence that 14-3-3 indeed regulates GluN2C surface expression by driving forward trafficking to plasma membrane.
FIGURE 5.
Co-expression of 14-3-3 isoforms does not affect GluN2C endocytosis in HeLa cells. A, an endocytosis assay was carried out using HeLa cells co-transfected with GFP-GluN2C, GluN1–1a-IRES-DsRed, and Myc-14-3-3ζ WT, Myc-14-3-3ζ S145A/Y178H/C189I, Myc-14-3-3σ WT, Myc-14-3-3σ A147S/I191C, or empty Myc vector. To visualize surface receptors, cells were incubated with anti-GFP antibody on ice, washed, and returned to conditioned medium for 30 min at 37 °C to allow receptor internalization. Cells were fixed and surface-expressed proteins were labeled with Alexa 647-conjugated secondary antibody (depicted in red). After permeabilization, internalized receptors were labeled with Alexa 488-conjugated secondary antibody (green) and Myc-14-3-3 was labeled with anti-Myc antibody and Alexa 405-conjugated anti-mouse secondary antibody (depicted in gray). B, data were quantified by measuring ratios of internalized/surface GluN2C receptors using ImageJ software. Data represent means ± S.E. (n = 15 cells/group; n.s. denotes no significant difference between groups).
Immunostaining of GluN2C-containing Receptors in Hippocampal Neurons Reveals Co-localization with Synaptophysin, a Presynaptic Marker
Currently, it is well accepted that GluN2A and GluN2B are the predominant NMDAR subunits at distinct developmental stages in hippocampal neurons (38). However, there is overwhelming evidence to suggest that GluN2C is not confined to cerebellar granule neurons as previously thought, but also exist widely throughout the adult brain, including but not limited to cortical, striatal, hippocampal, and thalamic neurons and non-neuronal cell types (19, 39–44). It is critical to establish the function and trafficking mechanisms of GluN2C in these brain regions and this will allow a more complete understanding of GluN2C regulation within pathological contexts. However, because there is currently no strong, specific anti-GluN2C antibody that is commercially available and does not cross-react with GluN2A/2B, many studies examining GluN2C are limited to expression of a tagged protein.
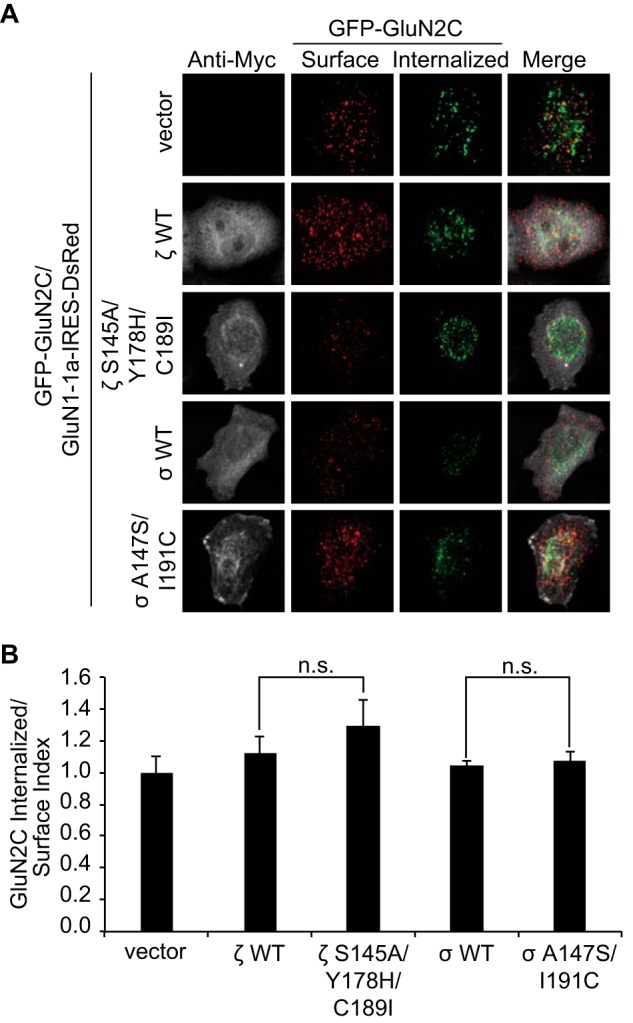
To determine if GluN2C can be trafficked to the synapse in mature hippocampal neurons, we transfected at DIV13 GluN2C construct, either GFP-tagged or 3xFLAG-tagged GluN2C to rule out any secondary effects due to the large size of GFP (3xFLAG data not shown as no significant differences were found between GFP-GluN2C and 3xFLAG-GluN2C) or GFP-tagged GluN2A used as a positive control (Fig. 6A). The distribution of surface-labeled GluN2 subunits was compared with that of the presynaptic vesicle marker, synaptophysin, a well-accepted synaptic marker (45, 46). At DIV15, surface receptors were labeled with anti-GFP. Receptor puncta distribution was analyzed for colocalization with synaptophysin (defined as having overlapping or adjacent pixels). Imaging analysis reveals 57% ± 4.6 of total GFP-GluN2C surface receptors (and 58% ± 3.7 for 3xFLAG-GluN2C surface receptors, data not shown) colocalize with synaptophysin, providing evidence that GluN2C can exist at hippocampal excitatory synapses. However, GluN2C synaptic colocalization is moderately, but significantly lower (*, p = 0.017) than the percentage for GFP-GluN2A (65% ± 4) surface receptors colocalized with synaptophysin (Fig. 6B). Despite these differences, our results suggest that transfected GluN2C-containing NMDARs can be trafficked to the synaptic cell surface of hippocampal neurons.
FIGURE 6.
Surface expressed GluN2C co-localizes with synaptophysin, a presynaptic marker. A, hippocampal neurons were transfected with GFP-GluN2C or GFP-GluN2A. To visualize the surface receptors, cells were incubated with anti-GFP antibody, fixed and incubated with Alexa 647-conjugated anti-rabbit secondary antibody (depicted in red for optimal visual contrast effect). Cells were then permeabilized and labeled with anti-synaptophysin antibody and Alexa 568-conjugated anti-mouse secondary antibody (depicted in green), a pre-synaptic vesicle protein marker. The right panels display representative zoomed in higher magnification images of 10 μm dendritic segments. Scale bar = 10 μm. B, co-localization of GFP puncta to synaptophysin puncta was measured. Value is given in percent co-localization (defined as the number of GFP puncta overlapping or adjacent to synaptophysin puncta, divided by the total number of pre-synaptic puncta). Data represent means ± S.E. (n = 12 cells per group; n = 36 dendritic segments per group; Student's t test, *, p < 0.05).
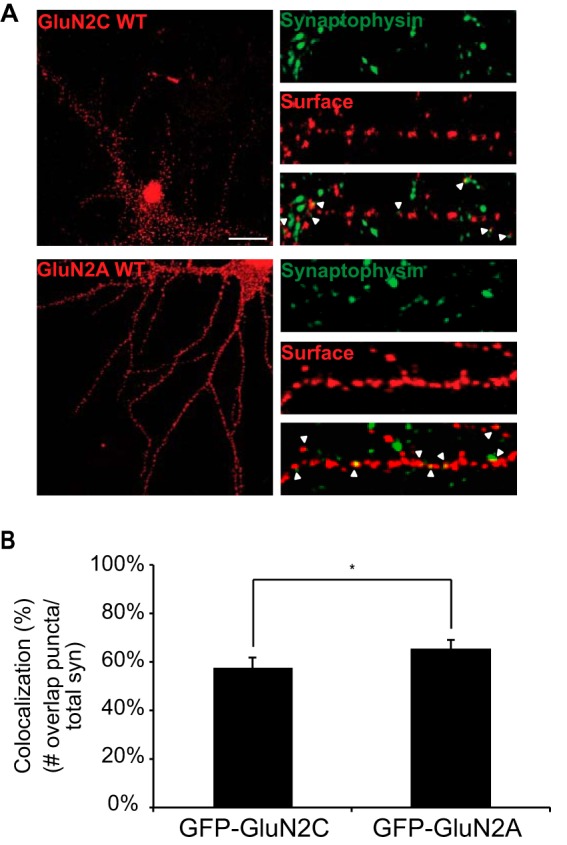
14-3-3 Interaction with GluN2C in Hippocampal Neurons Differentially Regulates Intracellular Trafficking
Dynamic trafficking of NMDARs to the neuronal plasma membrane is critical in maintaining chemical communication at excitatory synapses, as well as involved in numerous pathological outcomes such as overactivation following ischemia. We next tested the hypothesis that 14-3-3 binding to GluN2C can promote the forward delivery of ER-retained GluN2C to synaptic plasma membrane in hippocampal neurons, as was previously shown in HeLa cells. We co-expressed in mature DIV13 hippocampal neurons GFP-tagged GluN2C and Myc-tagged 14-3-3ϵ WT, 14-3-3ζ WT, 14-3-3ζ S145A/Y178H/C189I mutant, 14-3-3σ WT, 14-3-3σ A147S/I191C mutant, or Myc vector alone (Fig. 7A). Strikingly similar to the effect seen in HeLa cells, quantification reveals that the surface expression of GluN2C was significantly increased (Fig. 7B) when co-expressed with 14-3-3ϵ (1.77 ± 0.02 normalized surface/intracellular index; *, p = 0.022) and 14-3-3ζ isoforms (2.08 ± 0.01; **, p < 0.001). The 14-3-3σ WT and 14-3-3ζ S145A/Y178H/C189I mutant isoforms had no significant effect (1.11 ± 0.01 and 0.85 ± 0.01 respectively; n.s. versus GluN2C/vector) on GluN2C surface expression, whereas 14-3-3σ A147S/I191C mutation was sufficient to not only promote GluN2C binding but also functionally promote GluN2C surface expression. Taken together, these data demonstrate a 14-3-3 isoform-specific regulation of GluN2C trafficking in hippocampal neurons.
FIGURE 7.
14-3-3 interaction with GluN2C in hippocampal neurons differentially regulates intracellular trafficking. A, hippocampal neurons were co-transfected with GluN2C containing an extracellular GFP protein and Myc empty vector, Myc-14-3-3ϵ WT, Myc-14-3-3ζ WT, Myc-14-3-3ζ S145A/Y178H/C189I, Myc-14-3-3σ WT, or Myc-14-3-3σ A147S/I191C. Two days after transfection, cells were incubated with anti-GFP antibody for 30 min at room temperature. Cells were fixed and incubated with Alexa 647-conjugated anti-rabbit secondary antibody (depicted in red for optimal visual contrast effect) to visualize the surface receptors. Cells were then washed, permeabilized, and labeled with anti-GFP antibody and Alexa 488-conjugated anti-rabbit secondary antibody (green) to visualize intracellular pool of receptors and anti-Myc monoclonal antibody and Alexa 405-conjugated anti-mouse secondary antibody (gray) to visualize the 14-3-3-transfected cells. The right panels display representative zoomed in higher magnification images of 10 μm dendritic segments. Scale bars = 10 μm. B, data were quantified by measuring ratios of surface GluN2C/intracellular GluN2C receptors using ImageJ software. Data represent means ± S.E. (n = 9 cells per group; n = 27 dendritic segments per group; **, p < 0.001; *, p < 0.05).
Difopein Inhibition of 14-3-3 Binding to GluN2C Reduces GluN2C Surface Expression
To determine whether endogenous 14-3-3 isoforms are required for GluN2C trafficking, we used a well-characterized specific inhibitor of 14-3-3/ligand interactions, difopein (Dimeric Fourteen-three-three Peptide Inhibitor) (47), to disrupt interactions between endogenous 14-3-3 and GluN2C. We co-expressed 3xFLAG-tagged GluN2C and eCFP-vector or eCFP-difopein in DIV13 hippocampal neurons (Fig. 8A). As expected, quantitative analysis revealed that surface levels of GluN2C were dramatically reduced by nearly 70% in neurons expressing eCFP-difopein (0.33 ± 0.01 surface/intracellular index; **, p < 0.001) compared with that in the eCFP-vector-transfected neurons (Fig. 8B). These data are consistent with our overexpression study in hippocampal neurons (Fig. 7B), further demonstrating the critical role of endogenous 14-3-3 proteins in trafficking GluN2C receptors to the cell surface.
FIGURE 8.
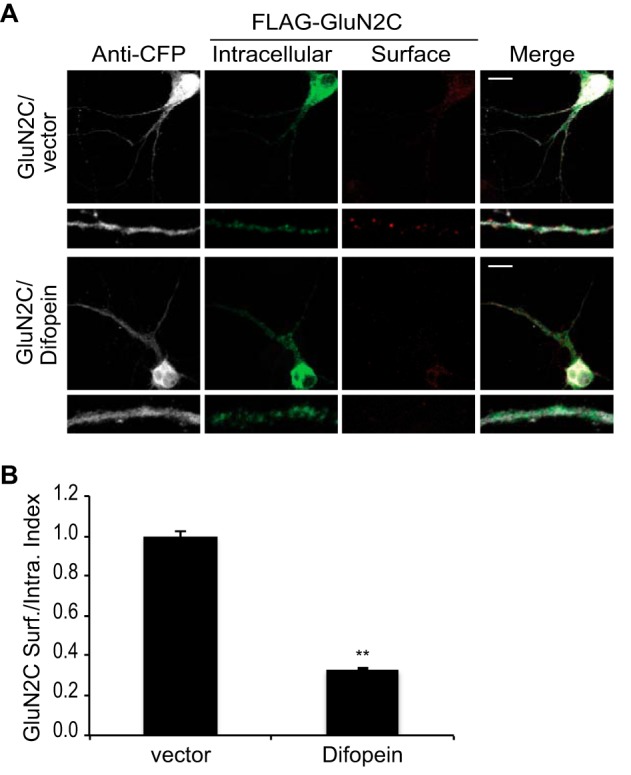
Difopein inhibition of 14-3-3 binding to GluN2C reduces GluN2C surface expression. A, hippocampal neurons were co-transfected with 3x FLAG-tagged GluN2C, eCFP empty vector, or eCFP-difopein as indicated. Two days after transfection, cells were incubated with monoclonal anti-FLAG antibody for 30 min at room temperature. Cells were fixed and incubated with Alexa 647-conjugated anti-mouse secondary antibody (depicted in red for optimal visual contrast effect) to visualize the surface receptors. Cells were then washed, permeabilized, and labeled with polyclonal anti-FLAG antibody and Alexa 488-conjugated anti-rabbit secondary antibody (green) to visualize intracellular pool of receptors and with anti-GFP antibody and Alexa 405-conjugated anti-rabbit secondary antibody (gray) to visualize the difopein-transfected cells. The bottom panels display representative zoomed in higher magnification images of 10 μm dendritic segments. Scale bars = 10 μm. B, data were quantified by measuring ratios of surface GluN2C/intracellular GluN2C using ImageJ software. Data represent means ± S.E. (n = 9 cells per group; n = 27 dendritic segments per group; **, p < 0.001).
Discussion
In this study, we have uncovered isoform-specific interactions of 14-3-3 proteins with GluN2C and its role in GluN2C trafficking. First, we show that GluN2C binds to all 14-3-3 proteins with the exception of the 14-3-3σ isoform. Second, we identify three critical residues in 14-3-3ζ that regulate its binding to GluN2C. These three residues are identical among all 14-3-3 proteins except the 14-3-3σ isoform. Additionally, 14-3-3σ carrying the reciprocal mutations of these three residues is able to bind to GluN2C, indicating that this newly identified regulatory region determines isoform-specific interactions with GluN2C. Third, we show that 14-3-3 binding promotes surface expression of GluN2C and the interaction between 14-3-3 and GluN2C is required for the increased GluN2C-containing NMDAR trafficking. Finally, using a well-characterized tool to functionally knock out all endogenous 14-3-3 proteins in neurons, we demonstrate that 14-3-3 binding plays a critical role in the regulation of GluN2C-containing NMDAR trafficking.
The 14-3-3 family of proteins are evolutionarily highly conserved, with seven isoforms identified in mammals, at least thirteen isoforms in plants, and two isoforms in yeast. Knocking out one of the yeast 14-3-3 genes has little effect on cell viability, whereas knocking out both genes is lethal to the yeast cell, indicating the functional redundancy of the 14-3-3 isoforms (48). In mammals, 14-3-3 proteins exist ubiquitously in almost all tissues with the highest expression in the brain. In addition, many organisms express multiple 14-3-3 isoforms. Although most 14-3-3 isoforms interact with their target proteins with similar affinity, some isoform-specific regulations have been observed. For example, two different 14-3-3 proteins are required to prevent activation of the protein kinase cdc2 that initiate mitosis in the nucleus after DNA damage (49). In one pathway, 14-3-3σ is required to sequester cdc2-cyclin B1 complexes in the cytoplasm. In a separate pathway, 14-3-3 isoforms other than 14-3-3σ binds to cdc25C (activator of cdc2-cyclin B1 complexes), which results in its retention in the cytoplasm, to ensure that mitosis does not occur in the presence of DNA damage. Interestingly, similar to GluN2C, cdc25C does not bind to 14-3-3σ although it interacts with most 14-3-3 isoforms in vitro (34, 49). However, the molecular basis for differential interaction between cdc25C and 14-3-3 proteins remains elusive.
Unlike other 14-3-3 isoforms, 14-3-3σ is primarily expressed in epithelial cells and forms homodimers almost exclusively. Structural studies have shown that 14-3-3σ contains unique interactions at the dimer interface that account for its strong propensity to form homodimers (36). It has been suggested that the C-terminal domain of 14-3-3σ comprises a second ligand binding site in addition to the phospho-dependent ligand binding, which is involved in 14-3-3σ-specific ligand interaction. Furthermore, a patch of three amino acid residues (Met-202, Asp-204, and His-206) in 14-3-3σ has been shown to mediate selectivity against cdc25C binding (36). Given that 14-3-3σ does not bind to GluN2C, potentially, the same three residues (Met-202, Asp-204, and His-206) could modulate isoform-specific interaction of 14-3-3 with GluN2C. However, we did not find any effect on GluN2C binding after mutating each of the corresponding amino acids (Ile-200, Glu-202, and Asp-204) individually in 14-3-3ζ (Fig. 2A). Although our data did not support a role of these previously identified residues in isoform-specific interaction of 14-3-3 with GluN2C, we could not rule out the possibility that all three residues need to be mutated simultaneously for the effect on disrupting interaction between 14-3-3ζ and GluN2C. Instead, we identify three novel amino acid residues (Ser-145, Tyr-178, and Cys-189) in the C-terminal domain of 14-3-3ζ that modulate isoform-specific interactions with GluN2C. We further demonstrate that the corresponding amino acid residues (Ala-147, His-180, and Ile-191) in 14-3-3σ are responsible for selectivity against GluN2C binding. Consistently, based on structural analysis, Ser-145 and Ala-147 have been suggested to be important for ligand specificity of 14-3-3ζ and 14-3-3σ, respectively (37). Taken together, our data reveal three critical residues that are important for isoform-specific interaction of 14-3-3 proteins with GluN2C.
14-3-3 proteins modulate many cellular processes including cell cycle control, apoptosis, metabolic regulation, and mitogenic signal transduction. In addition, 14-3-3 binding is important in regulating the trafficking of target proteins. There is evidence that 14-3-3 proteins preferentially associate with assembled proteins in the ER-Golgi intermediate compartment thus facilitating export of properly assembled multimers from the ER. For example, KCNK3 potassium channels are released from ER-retention through the phosphorylation-dependent binding of 14-3-3β (8). We have previously shown that 14-3-3ϵ specifically interacts with GluN2C and that phosphorylation of Ser-1096 on GluN2C regulates NMDAR binding to 14-3-3ϵ (7). We further demonstrate that 14-3-3ϵ preferentially associates with oligomerized GluN1/GluN2C, supporting a role in promoting the forward trafficking of correctly assembled GluN1/GluN2C-containing NMDARs. Since there are seven 14-3-3 isoforms highly expressed throughout the brain and emerging evidence to suggest that isoform-specific regulatory functions exist, we explored the key question of whether 14-3-3 isoforms differentially regulate trafficking of GluN2C-containing NMDARs. We now find that all 14-3-3 isoforms except 14-3-3σ bind to GluN2C and that Ser1096 is part of the 14-3-3 binding motif on GluN2C. Although in the current study we have only shown that 14-3-3ζ binding is required for the increased suface expression of GluN2C-containing NMDARs, the observation that 14-3-3σ mutant that binds to GluN2C also facilitates GluN2C trafficking to the plasma membrane strongly suggests that the interaction between GluN2C and 14-3-3 isoforms is critical for GluN2C-containing NMDAR trafficking.
NMDARs are essential for neuronal development and synaptic plasticity. Although they are expressed throughout the brain, their subunit composition varies considerably both spatially and temporally. The GluN2 subunit composition of NMDARs governs important receptor functions including channel properties, receptor trafficking, and synaptic expression. For example, GluN2C-containing NMDARs generate low conductance channel openings with a lower sensitivity to extracellular Mg2+ compared with GluN2A/GluN2B-containing receptors. GluN2C is highly enriched in the cerebellum, suggesting a unique role in cerebellar neurotransmission. GluN2C is also found in the other areas of the brain including cortex and hippocampus although at a low level. We have previously shown that expression of GluN2C protects neurons from excitotoxicity (7). Interestingly, GluN2C expression is up-regulated in the hippocampus following in vitro oxygen-glucose deprivation (an in vitro ischemia model), suggesting that it plays an important role during ischemia (32, 33). However, little information is available about GluN2C in the hippocampus. Here, we show that GluN2C colocalizes with synaptophysin in cultured hippocampal neurons, suggesting its localization at synapses. We also show that the physical interaction of 14-3-3 with GluN2C promotes surface expression of GluN2C in hippocampal neurons, consistent with our previous findings in cerebellar granule neurons. In conclusion, 14-3-3 binding to GluN2C is important in regulating the forward trafficking of the receptor to the cell surface to mediate glutamatergic neurotransmission. Understanding how these two proteins interact and particularly, the identification of the specific critical amino acid residues that mediate the interaction, could lead to the development of targeted peptide therapy to promote or disrupt this interaction under pathological conditions.
Author Contributions
C. C. and B.-S. C. designed the study. C. C. performed and analyzed all experiments. W.-H. W. and B.-S. C. supervised the project and helped with data analysis and interpretation. All authors reviewed the results, wrote the manuscript, and approved the final version of the manuscript.
Acknowledgments
We thank Haian Fu (Emory University) for the 14-3-3 and difopein constructs. We also thank Zhe Wei and Nick Mank for technical assistance.
This work was supported by National Institutes of Health NINDS Career Transition Award R00NS057266 (to B.-S. C.) and an NCI Career Transition Award 1K22CA122453 (to W.-H. W.). The authors declare that they have no conflicts of interest with the contents of this article.
- NMDAR
- NMDA receptor
- NMDA
- N-methyl-d-aspartate
- difopein
- dimeric 14-3-3 peptide inhibitor.
References
- 1. Fu H., Subramanian R. R., Masters S. C. (2000) 14-3-3 proteins: structure, function, and regulation. Annu. Rev. Pharmacol. Toxicol. 40, 617–647 [DOI] [PubMed] [Google Scholar]
- 2. Berg D., Holzmann C., Riess O. (2003) 14-3-3 proteins in the nervous system. Nat. Rev. Neurosci. 4, 752–762 [DOI] [PubMed] [Google Scholar]
- 3. Gardino A. K., Smerdon S. J., Yaffe M. B. (2006) Structural determinants of 14-3-3 binding specificities and regulation of subcellular localization of 14-3-3-ligand complexes: a comparison of the X-ray crystal structures of all human 14-3-3 isoforms. Semin. Cancer Biol. 16, 173–182 [DOI] [PubMed] [Google Scholar]
- 4. Baxter H. C., Liu W. G., Forster J. L., Aitken A., Fraser J. R. (2002) Immunolocalisation of 14-3-3 isoforms in normal and scrapie-infected murine brain. Neuroscience 109, 5–14 [DOI] [PubMed] [Google Scholar]
- 5. Shikano S., Coblitz B., Wu M., Li M. (2006) 14-3-3 proteins: regulation of endoplasmic reticulum localization and surface expression of membrane proteins. Trends Cell Biol. 16, 370–375 [DOI] [PubMed] [Google Scholar]
- 6. Smith A. J., Daut J., Schwappach B. (2011) Membrane proteins as 14-3-3 clients in functional regulation and intracellular transport. Physiology 26, 181–191 [DOI] [PubMed] [Google Scholar]
- 7. Chen B. S., Roche K. W. (2009) Growth factor-dependent trafficking of cerebellar NMDA receptors via protein kinase B/Akt phosphorylation of NR2C. Neuron 62, 471–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. O'Kelly I., Butler M. H., Zilberberg N., Goldstein S. A. (2002) Forward transport. 14-3-3 binding overcomes retention in endoplasmic reticulum by dibasic signals. Cell 111, 577–588 [DOI] [PubMed] [Google Scholar]
- 9. Yuan H., Michelsen K., Schwappach B. (2003) 14-3-3 dimers probe the assembly status of multimeric membrane proteins. Curr. Biol. 13, 638–646 [DOI] [PubMed] [Google Scholar]
- 10. Rosenberg M. M., Yang F., Giovanni M., Mohn J. L., Temburni M. K., Jacob M. H. (2008) Adenomatous polyposis coli plays a key role, in vivo, in coordinating assembly of the neuronal nicotinic postsynaptic complex. Mol. Cell Neurosci. 38, 138–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Umahara T., Uchihara T., Nakamura A., Iwamoto T. (2009) Isoform-dependent immunolocalization of 14-3-3 proteins in developing rat cerebellum. Brain Res. 1253, 15–26 [DOI] [PubMed] [Google Scholar]
- 12. Paul A. L., Denison F. C., Schultz E. R., Zupanska A. K., Ferl R. J. (2012) 14-3-3 phosphoprotein interaction networks - does isoform diversity present functional interaction specification? Front. Plant Sci. 3, 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Obsil T., Obsilova V. (2011) Structural basis of 14-3-3 protein functions. Semin. Cell Dev. Biol. 22, 663–672 [DOI] [PubMed] [Google Scholar]
- 14. Yang X., Lee W. H., Sobott F., Papagrigoriou E., Robinson C. V., Grossmann J. G., Sundström M., Doyle D. A., Elkins J. M. (2006) Structural basis for protein-protein interactions in the 14-3-3 protein family. Proc. Natl. Acad. Sci. U.S.A. 103, 17237–17242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yaffe M. B., Rittinger K., Volinia S., Caron P. R., Aitken A., Leffers H., Gamblin S. J., Smerdon S. J., Cantley L. C. (1997) The structural basis for 14-3-3:phosphopeptide binding specificity. Cell 91, 961–971 [DOI] [PubMed] [Google Scholar]
- 16. Cull-Candy S. G., Leszkiewicz D. N. (2004) Role of distinct NMDA receptor subtypes at central synapses. Sci. STKE 2004, re16. [DOI] [PubMed] [Google Scholar]
- 17. Paoletti P., Bellone C., Zhou Q. (2013) NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 14, 383–400 [DOI] [PubMed] [Google Scholar]
- 18. Wenzel A., Fritschy J. M., Mohler H., Benke D. (1997) NMDA receptor heterogeneity during postnatal development of the rat brain: differential expression of the NR2A, NR2B, and NR2C subunit proteins. J. Neurochem. 68, 469–478 [DOI] [PubMed] [Google Scholar]
- 19. Karavanova I., Vasudevan K., Cheng J., Buonanno A. (2007) Novel regional and developmental NMDA receptor expression patterns uncovered in NR2C subunit-β-galactosidase knock-in mice. Mol. Cell Neurosci. 34, 468–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wenthold R. J., Prybylowski K., Standley S., Sans N., Petralia R. S. (2003) Trafficking of NMDA receptors. Annu. Rev. Pharmacol. Toxicol. 43, 335–358 [DOI] [PubMed] [Google Scholar]
- 21. Lau C. G., Zukin R. S. (2007) NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat. Rev. Neurosci. 8, 413–426 [DOI] [PubMed] [Google Scholar]
- 22. Standley S., Roche K. W., McCallum J., Sans N., Wenthold R. J. (2000) PDZ domain suppression of an ER retention signal in NMDA receptor NR1 splice variants. Neuron 28, 887–898 [DOI] [PubMed] [Google Scholar]
- 23. Kaniakova M., Krausova B., Vyklicky V., Korinek M., Lichnerova K., Vyklicky L., Horak M. (2012) Key amino acid residues within the third membrane domains of NR1 and NR2 subunits contribute to the regulation of the surface delivery of N-methyl-D-aspartate receptors. J. Biol. Chem. 287, 26423–26434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lichnerova K., Kaniakova M., Skrenkova K., Vyklicky L., Horak M. (2014) Distinct regions within the GluN2C subunit regulate the surface delivery of NMDA receptors. Front. Cell Neurosci. 8, 375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wei Z., Behrman B., Wu W. H., Chen B. S. (2015) Subunit-Specific Regulation of N-Methyl-D-aspartate (NMDA) Receptor Trafficking by SAP102 Splice Variants. J. Biol. Chem. 290, 5105–5116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lavezzari G., McCallum J., Dewey C. M., Roche K. W. (2004) Subunit-specific regulation of NMDA receptor endocytosis. J. Neurosci. 24, 6383–6391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Suh Y. H., Pelkey K. A., Lavezzari G., Roche P. A., Huganir R. L., McBain C. J., Roche K. W. (2008) Corequirement of PICK1 binding and PKC phosphorylation for stable surface expression of the metabotropic glutamate receptor mGluR7. Neuron 58, 736–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen B. S., Gray J. A., Sanz-Clemente A., Wei Z., Thomas E. V., Nicoll R. A., Roche K. W. (2012) SAP102 mediates synaptic clearance of NMDA receptors. Cell Rep. 2, 1120–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Muslin A. J., Tanner J. W., Allen P. M., Shaw A. S. (1996) Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell 84, 889–897 [DOI] [PubMed] [Google Scholar]
- 30. Brennan G. P., Jimenez-Mateos E. M., McKiernan R. C., Engel T., Tzivion G., Henshall D. C. (2013) Transgenic overexpression of 14-3-3ζ protects hippocampus against endoplasmic reticulum stress and status epilepticus in vivo. PLoS ONE 8, e54491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schindler C. K., Heverin M., Henshall D. C. (2006) Isoform- and subcellular fraction-specific differences in hippocampal 14-3-3 levels following experimentally evoked seizures and in human temporal lobe epilepsy. J. Neurochem. 99, 561–569 [DOI] [PubMed] [Google Scholar]
- 32. Perez-Velazquez J. L., Zhang L. (1994) In vitro hypoxia induces expression of the NR2C subunit of the NMDA receptor in rat cortex and hippocampus. J. Neurochem. 63, 1171–1173 [DOI] [PubMed] [Google Scholar]
- 33. Small D. L., Poulter M. O., Buchan A. M., Morley P. (1997) Alteration in NMDA receptor subunit mRNA expression in vulnerable and resistant regions of in vitro ischemic rat hippocampal slices. Neurosci. Lett. 232, 87–90 [DOI] [PubMed] [Google Scholar]
- 34. Dalal S. N., Yaffe M. B., DeCaprio J. A. (2004) 14-3-3 family members act coordinately to regulate mitotic progression. Cell Cycle 3, 672–677 [PubMed] [Google Scholar]
- 35. Martin H., Rostas J., Patel Y., Aitken A. (1994) Subcellular localisation of 14-3-3 isoforms in rat brain using specific antibodies. J. Neurochem. 63, 2259–2265 [DOI] [PubMed] [Google Scholar]
- 36. Wilker E. W., Grant R. A., Artim S. C., Yaffe M. B. (2005) A structural basis for 14-3-3σ functional specificity. J. Biol. Chem. 280, 18891–18898 [DOI] [PubMed] [Google Scholar]
- 37. Benzinger A., Popowicz G. M., Joy J. K., Majumdar S., Holak T. A., Hermeking H. (2005) The crystal structure of the non-liganded 14-3-3σ protein: insights into determinants of isoform specific ligand binding and dimerization. Cell Res. 15, 219–227 [DOI] [PubMed] [Google Scholar]
- 38. Monyer H., Burnashev N., Laurie D. J., Sakmann B., Seeburg P. H. (1994) Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12, 529–540 [DOI] [PubMed] [Google Scholar]
- 39. Káradóttir R., Cavelier P., Bergersen L. H., Attwell D. (2005) NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature 438, 1162–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Binshtok A. M., Fleidervish I. A., Sprengel R., Gutnick M. J. (2006) NMDA receptors in layer 4 spiny stellate cells of the mouse barrel cortex contain the NR2C subunit. J. Neurosci. 26, 708–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kumar S. S., Huguenard J. R. (2003) Pathway-specific differences in subunit composition of synaptic NMDA receptors on pyramidal neurons in neocortex. J. Neurosci. 23, 10074–10083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pollard H., Khrestchatisky M., Moreau J., Ben Ari Y. (1993) Transient expression of the NR2C subunit of the NMDA receptor in developing rat brain. Neuroreport 4, 411–414 [DOI] [PubMed] [Google Scholar]
- 43. Salter M. G., Fern R. (2005) NMDA receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature 438, 1167–1171 [DOI] [PubMed] [Google Scholar]
- 44. Micu I., Jiang Q., Coderre E., Ridsdale A., Zhang L., Woulfe J., Yin X., Trapp B. D., McRory J. E., Rehak R., Zamponi G. W., Wang W., Stys P. K. (2006) NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature 439, 988–992 [DOI] [PubMed] [Google Scholar]
- 45. Roqué P. J., Guizzetti M., Giordano G., Costa L. G. (2011) Quantification of synaptic structure formation in cocultures of astrocytes and hippocampal neurons. Methods Mol. Biol. 758, 361–390 [DOI] [PubMed] [Google Scholar]
- 46. Prybylowski K., Chang K., Sans N., Kan L., Vicini S., Wenthold R. J. (2005) The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron 47, 845–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Masters S. C., Fu H. (2001) 14-3-3 proteins mediate an essential anti-apoptotic signal. J. Biol. Chem. 276, 45193–45200 [DOI] [PubMed] [Google Scholar]
- 48. van Heusden G. P., Griffiths D. J., Ford J. C., Chin A. W. T. F., Schrader P. A., Carr A. M., Steensma H. Y. (1995) The 14-3-3 proteins encoded by the BMH1 and BMH2 genes are essential in the yeast Saccharomyces cerevisiae and can be replaced by a plant homologue. Eur. J. Biochem. 229, 45–53 [PubMed] [Google Scholar]
- 49. Chan T. A., Hermeking H., Lengauer C., Kinzler K. W., Vogelstein B. (1999) 14-3-3σ is required to prevent mitotic catastrophe after DNA damage. Nature 401, 616–620 [DOI] [PubMed] [Google Scholar]