

Background: PCSK9 is a modulator of LDLR and the tetraspanin CD81.
Results: Although CD81 is targeted for degradation by PCSK9 in an LDLR-independent manner, it can associate with the LDLR.
Conclusion: CD81 and LDLR are two independent targets of PCSK9 that bind to each other.
Significance: Structure-based mutagenesis of PCSK9 reveals functional interactions of CD81 with LDLR and PCSK9.
Keywords: cell biology, cell surface protein, lipoprotein receptor, proprotein convertase subtilisin/kexin type 9 (PCSK9), protein sorting, protein-protein interaction
Abstract
Proprotein convertase subtilisin/kexin type 9 (PCSK9) is an important factor in plasma cholesterol regulation through modulation of low density lipoprotein receptor (LDLR) levels. Naturally occurring mutations can lead to hyper- or hypocholesterolemia in human. Recently, we reported that PCSK9 was also able to modulate CD81 in Huh7 cells. In the present study, several gain-of-function and loss-of-function mutants as well as engineered mutants of PCSK9 were compared for their ability to modulate the cell surface expression of LDLR and CD81. Although PCSK9 gain-of-function D374Y enhanced the degradation both receptors, D374H and D129N seemed to only reduce LDLR levels. In contrast, mutations in the C-terminal hinge-cysteine-histidine-rich domain segment primarily affected the PCSK9-induced CD81 degradation. Furthermore, when C-terminally fused to an ACE2 transmembrane anchor, the secretory N-terminal catalytic or hinge-cysteine-histidine-rich domain domains of PCSK9 were able to reduce CD81 and LDLR levels. These data confirm that PCSK9 reduces CD81 levels via an intracellular pathway as reported for LDLR. Using immunocytochemistry, a proximity ligation assay, and co-immunoprecipitation, we found that the cell surface level of PCSK9 was enhanced upon overexpression of CD81 and that both PCSK9 and LDLR interact with this tetraspanin protein. Interestingly, using CHO-A7 cells lacking LDLR expression, we revealed that LDLR was not required for the degradation of CD81 by PCSK9, but its presence strengthened the PCSK9 effect.
Introduction
Extensive studies on familial hypercholesterolemia (FH)2 and the accompanied risk for coronary heart disease led to a breakthrough in 2003, the discovery of PCSK9 (1) as a positive regulator of low density lipoprotein-cholesterol (LDL-C) in plasma (2). The underlying mechanism is related to the ability of PCSK9, which is expressed mostly in liver hepatocytes and in the small intestine (1, 3), to bind and escort the LDLR to endosomal/lysosomal degradation pathways (4, 5).
PCSK9 belongs to the mammalian proprotein convertase family of subtilisin-like serine proteinases (1, 6). The newly synthesized PCSK9, a 72-kDa protein, comprises a signal peptide (amino acids 1–30), a prodomain (amino acids 31–152), a catalytic domain (amino acids 153–404), a hinge region (HR) (amino acids 405–452), and a cysteine-histidine-rich domain (CHRD) (amino acids 453–692) (Fig. 1). In the endoplasmic reticulum (ER), the zymogen pro-PCSK9 (amino acids 31–692) autocatalytically cleaves itself into PCSK9 (amino acids 153–692) but remains non-covalently tightly bound to its inhibitory prodomain (amino acids 31–152). Mature functional PCSK9 (∼60 kDa) is thus secreted as a catalytically inactive prodomain-PCSK9 complex (1, 7). This property places PCSK9 in a class of its own, different from the other proprotein convertases, as it has no known substrate other than itself (6). PCSK9 is mostly synthesized in liver hepatocytes from which circulating PCSK9 originates (3, 8) and in the small intestine (1, 3).
FIGURE 1.

Schematic representation of PCSK9 constructs used in this study. PCSK9 comprises a signal peptide (SP), a prodomain (Pro), a catalytic domain, a HR, and a CHRD. The numbers and arrows denote amino acid positions at boundaries between domains. The N-terminal CAT construct and the HR-CHRD were individually subcloned for further study. Full-length PCSK9 and the truncated constructs (CAT and HR-CHRD) were fused with the TM-CT of LAMP1 or ACE2 to drive the proteins to the lysosomal compartment or to the cell surface, respectively.
PCSK9 has been extensively studied for its ability to bind cell surface receptor proteins such as the LDLR (4, 5), very low density lipoprotein (VLDL) receptor, and apolipoprotein E receptor 2 (8, 9) and sort them to endosomes/lysosomes for degradation (5, 6). In that context, the PCSK9-LDLR interaction has been shown to occur between the catalytic domain of PCSK9 (10) and the N-terminal part of epidermal growth factor-like repeat A (EGF-A) domain of the LDLR (10, 11), although the CHRD and the prodomain seem to possess some additional binding affinity for the β-barrel ligand-binding domain of the LDLR (12–15).
There are two pathways by which PCSK9 leads to the degradation of the LDLR. In the extracellular pathway, following secretion from cells, extracellular PCSK9 binds to the LDLR at the cell surface, triggering PCSK9-LDLR complex internalization and its traffic to acidic endosomes/lysosomes where degradation occurs. In the intracellular pathway, mature PCSK9 traffics from the ER to the Golgi apparatus where it binds to the LDLR and sorts the receptor directly to lysosomes for degradation (16). In liver, PCSK9 seems to enhance the degradation of the LDLR mostly by the extracellular pathway (1, 6).
Interestingly, several natural mutations in the human PCSK9 gene have been reported. They confer gain-of-function (GOF) or loss-of-function (LOF) activity on the LDLR. Therefore, individuals with GOF mutations will have reduced LDLR levels in hepatocytes and thus suffer from hypercholesterolemia. Various LOF and GOF mutants were found to be associated with hypocholesterolemia and hypercholesterolemia, respectively (17). For example, the LOF mutations G236S and N354I are due to failure to exit the ER or to undergo autocatalytic cleavage, respectively (18). In contrast, typical GOF mutations include D129N, D374H, and D374Y, which enhance the binding of PCSK9 to the LDLR (19, 20). The various strategic mutants used in this study are described in Table 1.
TABLE 1.
List of PCSK9 mutants used in this study
| No. | Mutant | Effect | Remarks |
|---|---|---|---|
| 1 | V4I | GOF | Found in individuals with high LDL-C levels (24). |
| 2 | E32K | GOF | E32K mutant bearers had higher plasma PCSK9 level and higher level of LDL-C. Transiently transfected HepG2 cells produced more E32K than the wild-type protein (25). |
| 3 | E54A | GOF | Found in individuals with high LDL-C levels (24). |
| 4 | R93C | LOF | Found in individuals with low LDL-C levels (24). |
| 5 | L108R | GOF | Found in French autosomal dominant hypercholesterolemia probands. Normal autocatalytic activity and secretion but strong effect on LDLR degradation (14). L108R involved in van der Waals contact of PCSK9 prodomain and LDLR β-propeller (15). |
| 6 | S127R | GOF | Poor autocatalytic activity (29) but strong affinity to LDLR in neutral and acidic pH (20). |
| 7 | D129G | GOF | Found associated with FH. Autocatalytic activity reduced by 80%, causing low level of mature PCSK9 (27). |
| 8 | D129N | GOF | Detected in FH patients. Normal maturation and secretion (19). |
| 9 | R194A | LOFa | Arg194 forms salt bridges with EGF-A (10). R194A showed reduced binding for LDLR (28). |
| 10 | R215H | GOF | Normal autocatalytic activity, but the mature form was resistant to furin cleavage (18). |
| 11 | F216L | GOF | Partially resistant to furin and PC5/6A cleavages (29). |
| 12 | R218S | GOF | Found related to FH patients (30). Resistant to furin and PC5/6A cleavages (29). |
| 13 | G236S | LOF | Failed to produce secreted forms (18). |
| 14 | D238A | LOFa | Asp238 forms a hydrogen bond with EGF-A (10). D238A showed reduced affinity for LDLR binding (28). |
| 15 | A239D | LOF | Found in individuals with low LDL-C levels (24). |
| 16 | N354I | LOF | Asn354 has hydrogen bonds with other residues that stabilize and keep proper folding for PCSK9. N354I lacked autocatalytic activity in vitro to produce secreted forms (18). |
| 17 | R357H | GOF | Found related to FH patients (30). |
| 18 | D374H | GOF | Found in Portuguese FH patients (31). |
| 19 | D374Y | GOF | Found in Norwegian patients with FH (32). Has partial resistance to furin (29) and higher affinity for LDLR by 25 times compared with wild type (33). Degrades LDLR 10-fold higher than wild type (23). |
| 20 | F379A | LOFa | Phe379 is in the center of PCSK9-EGF-A interface; F379A reduces the binding by 90% (10). |
| 21 | H391N | LOF | Found in individuals with low LDL-C levels (34). |
| 22 | N425S | GOF | Detected in FH patients with higher LDL-C levels (19, 35). |
| 23 | R434W | LOF | Poor secretion and low effect on LDLR degradation; resistant to cleavage by furin (36). |
| 24 | G452D | LOF | Found in individuals with low LDL-C levels (24). |
| 25 | S462P | LOF | Trapped in the ER after autocatalytic cleavage (37). |
| 26 | R469W | GOF | Found in individuals with high LDL-C levels (30, 34). |
| 27 | R496W | GOF | Detected in FH patients with higher LDL-C levels (19, 35). Normal maturation and secretion and similar effect on LDLR (19). |
| 28 | A514T | GOF | Found in individuals with high LDL-C levels (24). |
| 29 | A522T | LOF | Detected in hypocholesterolemia blood donors (38). |
| 30 | H553R | GOF | Found in individuals with high LDL-C (34). |
| 31 | Q554E | LOF | Found in individuals with low LDL-C (34). |
| 32 | P616L | LOF | Detected in hypocholesterolemia blood donors (38). |
| 33 | V624M | GOF | Found in individuals with high LDL-C levels (24). |
| 34 | S668R | LOF | Found in individuals with low LDL-C levels (24). |
a Based on in vitro data.
Recently, we identified CD81 as a new cell surface protein that could be targeted by PCSK9 (21). Because PCSK9 reduces the protein levels of the LDLR and hepatitis C virus (HCV) uses both LDLR and CD81 as entry factors, we showed that PCSK9 could prevent Huh7 cells from infection with the virus. Surprisingly, we also found that stable PCSK9 overexpression in HuH7 cells could also decrease CD81 levels. In this study, we investigated further how PCSK9, CD81, and LDLR interact with each other and probed the effect of the expression of various PCSK9 mutants on CD81 levels.
Experimental Procedures
cDNA Constructs
Human full-length PCSK9 and its truncated constructs catalytic domain (CAT), HR, and CHRD with or without the transmembrane-cytosolic tail (TM-CT) of lysosomal-associated membrane protein 1 (LAMP1) or angiotensin I-converting enzyme 2 (ACE2) proteins (hereby abbreviated as PCSK9-LAMP1, PCSK9-ACE2, CAT-ACE2, and HR-CHRD-ACE2) were cloned into pIRES2-eGFP (Clontech) and phCMV3 vectors as described (12). Plasmids coding for human PCSK9 mutants were produced from phCMV3-hPCSK9-ACE2 using the QuikChange Lightning kit (Agilent Technologies). The primer sequence information will be given if required. The integrity of all mutants was verified by sequencing.
Cell Culture and Transfection
Huh7 cells were cultured in Dulbecco's modified Eagle's medium (DMEM), and Chinese hamster ovary (CHO) and CHO-A7 cells were grown in DMEM/F-12 (Ham's) (Invitrogen) supplemented with 10% fetal bovine serum (FBS) (Wisent), ampicillin/tetracycline, and sodium pyruvate. Transfections were done using Lipofectamine 2000 (Invitrogen) and TransIT-2020 (Mirus) according to the manufacturers' protocols.
Western Blotting
Cells were lysed in radioimmune precipitation assay buffer (50 mm Tris (pH 7.5), 150 mm NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Nonidet P-40, 1 mm PMSF (Sigma-Aldrich), and Complete mixture protease inhibitor (Roche Applied Science)). The cell lysates were loaded and separated by 10% Tris-glycine SDS-PAGE or on 4–15% gradient gels (Bio-Rad). Proteins were transferred onto PVDF membranes (Bio-Rad). The membranes were blocked for 1 h and then incubated with in-house rabbit anti-human PCSK9 antibody (1:2500), goat anti-ACE2 antibody (clone C-18, 1:1000; Santa Cruz Biotechnology), or mouse anti-β-actin antibody (1:1000; Ambion) for 1 h. The membranes were then incubated with appropriate HRP-conjugated secondary antibodies (1:10,000; Jackson ImmunoResearch Laboratories) for 1 h. The membranes were incubated with enhanced chemiluminescent HRP substrate and exposed to x-ray films. Densities of protein bands were measured by the AlphaImager 3400 Transilluminator Gel Imaging Detection System (Alpha Innotech).
Immunofluorescence Assay (IFA)
After fixation, coverslips were permeabilized with 0.5% Triton X-100 in PBS for 5 min at room temperature. To visualize co-localization of proteins with lysosomes, cells were incubated with 100 nm LysoTracker Red DND-99 (Molecular Probes) for 2 h at 37 °C. Permeabilization was omitted if only surface proteins were to be detected. The coverslips were blocked in blocking buffer (10% FBS, 3% bovine serum albumin (BSA), and 0.1% sodium azide in PBS) for 30 min at room temperature prior to incubation with primary antibodies rabbit anti-PCSK9 (in-house; 1:150 dilution), mouse anti-CD81 (JS-81, BD Pharmingen; 1:50 dilution), goat anti-LDLR (R&D Systems; 1:50 dilution), mouse anti-LDLR (C7, Santa Cruz Biotechnology; 1:20 dilution), and mouse anti-CD9 (BD Pharmingen; 1:25 dilution). The coverslips were washed with PBS and incubated with appropriate secondary antibodies (Alexa Fluor 647 chicken anti-mouse, Alexa Fluor 568 donkey anti-rabbit, Alexa Fluor 647 chicken anti-rabbit, and Alexa Fluor 568 donkey anti-goat; 1:500 dilution) from Molecular Probes. Coverslips were stained with DAPI for 5 min and mounted on slides with Prolong Gold antifade mountant (Invitrogen). Imaging was performed on a confocal microscope system (Zeiss LSM780). Protein co-localization analysis was done by using ImageJ software and Just Another Colocalization Plugin (JACoP) (22).
In Situ Proximity Ligation Assay (PLA)
PLA experiments were done using Duolink In Situ reagents (Olink Bioscience). The blocking and primary antibody staining steps were conducted as described above. Coverslips were then stained with the corresponding mixture of PLA probes (anti-rabbit PLUS (catalog number DUO92002) and anti-mouse MINUS (catalog number DUO92004)) for 1 h at 37 °C. The samples were then incubated with the ligation ligase solution for 30 min at 37 °C to hybridize oligonucleotides tagged on probes. The coverslips were then incubated with the amplification polymerase solution for 100 min at 37 °C to amplify hybridized oligonucleotides and fluorescently label the amplification products. Finally, the coverslips were mounted on slides with Duolink In Situ Mounting Medium with DAPI. Imaging was done using a confocal microscope system (LSM780) as mentioned above. Duolink ImageTool software (Olink Bioscience) was used to analyze PLA signals.
Co-immunoprecipitation (CoIP)
Plasmids coding for PCSK9-V5, PCSK9-ACE2, LDLR, or proteins were co-transfected with CD81 plasmid with a 1:1 ratio into HEK293T cells on 100-mm dishes using Lipofectamine 3000 (Invitrogen) according to the manufacturer's protocol. Co-tranfections of PCSK9-V5 and LDLR or CD81 and ACE2 were done as positive and negative controls, respectively. Transfected cells were washed with ice-cold PBS and lysed with 1 ml of 1% Brij-97 lysis buffer (50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1 mm CaCl2, 1 mm MgCl2, 1% Brij-97 (Sigma-Aldrich), 1 mm PMSF, and Complete mixture protease inhibitor). After centrifugation to pellet cell debris, the lysates were precleared by incubation with 2 μg of mouse IgG1 (Santa Cruz Biotechnology) and 20 μl of Protein A/G PLUS-agarose beads (Santa Cruz Biotechnology) for 1 h. Next, the precleared lysates were incubated with 2 μg of mouse anti-CD81 (Clone 5A6, Santa Cruz Biotechnology) and 20 μl of Protein A/G PLUS-agarose beads or with 10 μl of anti-V5 affinity gel (Biotool) for 2 h. The incubation steps were carried out at room temperature with rotation. Afterward, the beads were washed with 1% Brij-97 lysis buffer four times and then transferred into new tubes to avoid carrying over any trace of proteins that may bind to the tube wall during incubation steps. Next, the proteins were eluted from the beads by adding 50 μl of elution buffer (0.2 m glycine and 2.5% acetic acid in 0.2% Brij-97 buffer). Finally, the eluants were neutralized by 10 m NaOH and subjected to Western blot analysis using rabbit anti-PCSK9, goat anti-LDLR (R&D Systems), mouse anti-CD81 (clone 5A6), goat anti-ACE2 (clone C-18), and rabbit anti-calnexin (Enzo Life Sciences).
Fluorescence-activated Cell Sorting (FACS) Analysis
Plasmids coding for PCSK9 constructs or mutants were co-transfected in Huh7 cells with peGFP-C1 plasmid (Clontech) with a 3:1 ratio. GFP plasmid was used for the gating of transfected cells. The cells were detached and suspended using Versene or trypsin (Life Technologies), washed with ice-cold PBS, incubated with the fixable viability dye eFluor® 780 (eBioscience) at 4 °C for 30 min, and then fixed with 4% paraformaldehyde in PBS for 10 min at room temperature. After fixation, cells were blocked and then incubated with allophycocyanin- or phycoerythrin-mouse anti-human CD81 (BD Pharmingen), allophycocyanin- or phycoerythrin-mouse anti-human LDLR(R&D Systems), allophycocyanin-mouse IgG1 κ isotype control (BD Pharmingen), or mouse IgG1-phycoerythrin isotype control (R&D Systems) for 1 h. To analyze total protein expressions, FACS buffer and FACS washing buffer were complemented with saponin (0.3%). Cells were resuspended in FACS buffer and analyzed on a BD LSRFortessa flow cytometer (BD Biosciences). The data were analyzed with CyflogicTM software version 1.2.1 (CyFlo Ltd., Finland). The geometric mean values were used to indicate protein expression levels. Geometric mean values of isotypes were considered as the background and were subtracted from the values from the samples.
Statistical Analysis
All data were calculated and statistically analyzed using GraphPad Prism 5.03 (GraphPad Prism Software, Inc.) as indicated in the legends (*, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001).
Results
PCSK9 Is Recruited to the Cell Surface by LDLR and CD81
Mature PCSK9 is known to be secreted from cells where it can bind its cell surface receptor ligands (LDLR, VLDL receptor, and apolipoprotein E receptor 2) in an autocrine and/or paracrine manner (11, 23). We first determined by confocal IFA whether PCSK9 could bind cell surface CD81. As a positive control, binding of PCSK9 to LDLR was analyzed in parallel. The results depicted in Fig. 2A show that, upon co-transfection of PCSK9 cDNA with cDNA of either LDLR or CD81, PCSK9 immunoreactivity was detected at the plasma membrane by IFA of non-permeabilized Huh7 cells. Notably, Huh7 cells overexpressing PCSK9 in a background of endogenous levels of CD81 and LDLR showed no detectable PCSK9 signal at the plasma membrane (Fig. 2, B and C) even though the cells were expressing PCSK9 at a significant level (Fig. 2, D and E). Quantitative analysis by Western blotting revealed that the expression of PCSK9 or its co-expression with LDLR or CD81 does not change the ratio of secreted versus intracellular PCSK9, which was estimated to be about 9-fold higher in the medium as compared with cell extracts (not shown). Note also that only transfected cells were positive for PCSK9 as verified by the co-transfection of PCSK9 cDNA with that of GFP (Fig. 2A). To eliminate any possibility of nonspecific binding of antibodies used in this experiment, we carried out the same experiments excluding secondary antibodies to CD81 or LDLR. The tests showed that the anti-PCSK9 antibody and corresponding secondary antibodies did not bind to CD81 (Fig. 2, F and G) or LDLR (Fig. 2, H and I). These results show that in the context of overexpression of LDLR (Fig. 2A, upper) or CD81 (Fig. 2A, lower) some PCSK9 is retained at the cell surface and co-localizes with these ligands.
FIGURE 2.

Recruitment of PCSK9 to the cell surface by LDLR and CD81. A–C, immunofluorescence analysis of PCSK9 localization. In addition to peGFP as an indicator of transfection, Huh7 cells were co-transfected with plasmids expressing PCSK9 and LDLR (A, upper), PCSK9 and CD81 (A, lower), PCSK9 (B), or empty vector (C). Cells were not permeabilized and were stained with rabbit anti-PCSK9 (red) and goat-anti LDLR (A, upper, green) or rabbit anti-PCSK9 and mouse anti-CD81 (B, lower, green) antibodies. D--I, verification of antibody specificity. Huh7 cells were co-transfected with peGFP and PCSK9 (D), CD81 (F), LDLR (H), or empty vector (E, G, and I). Transfected cells were treated with Triton X-100 (D and E) or left untreated (F–I) and stained with rabbit anti-PCSK9 (D and E), rabbit anti-PCSK9 and mouse anti-CD81 (F and G), or rabbit anti-PCSK9 and goat anti-LDLR (H and I) antibodies. Following primary antibody incubation, cells were stained with Alexa Fluor 568 chicken anti-rabbit IgG (D--I, red). The nuclei were stained with DAPI. The co-localization measurements in A were based on Manders' coefficients using ImageJ and JACoP.
Effects of PCSK9-ACE2 on LDLR and CD81 Cell Surface Expression in Huh7 Cells
In a previous study, we have shown that it takes at least ∼8–10 μg/ml pure PCSK9 added to HuH7 cells to achieve a ∼50% reduction in cellular CD81 (21). In addition, upon medium swap of PCSK9 isolated from media of HEK293 cells overexpressing wild-type (WT) PCSK9, we usually achieved less than ∼15% reduction of CD81. Therefore, in an effort to develop a simple assay to analyze the structure-function activity of PCSK9 on CD81 degradation, we used a chimera of PCSK9 fused to the TM-CT of ACE2. Indeed, we previously showed that fusion of the TM-CT of either ACE2 or LAMP1 to the C terminus of PCSK9 prevents its secretion and greatly enhances its ability to escort its ligands toward degradation compartments (21). Therefore, to develop a simple FACS screening assay to analyze the activities of PCSK9 and its mutants on CD81, we expressed the PCSK9-ACE2 chimera in Huh7 cells. As expected, PCSK9 immunoreactivity of PCSK9-ACE2 was detected both intracellularly and at the plasma membrane as opposed to WT PCSK9 that is secreted and hence only detected in the cells but not at the cell surface (Fig. 3A). We then confirmed by Western blotting that the autocatalytic zymogen processing of pro-PCSK9-ACE2 into PCSK9-ACE2 still occurs unabated in these cells (Fig. 3B). The activity of PCSK9-ACE2 on both the LDLR and CD81 was verified using flow cytometry, and as expected (21), overexpression of PCSK9-ACE2 resulted in significantly reduced cell surface levels and enhanced intracellular degradation of both ligands (Fig. 3C). Transfections with an empty vector or with a plasmid encoding the full-length ACE2 were used as controls, which had no effect on the levels of either LDLR or CD81.
FIGURE 3.

Effects of PCSK9 constructs on endogenous LDLR and CD81 expression in Huh7 cells. A, immunofluorescence analysis of PCSK9 construct localization. Huh7 cells were co-transfected with plasmids expressing eGFP and wild-type PCSK9 (left panel) or eGFP and PCSK9 fused to the TM-CT of ACE2 (right panel). Anti-PCSK9 staining (red) was compared in non-permeabilized ((−) Triton) and permeabilized ((+) Triton) Huh7 cells. B, Western blot analysis of the expression and maturation of overexpressed PCSK9 constructs in Huh7 cells using rabbit anti-PCSK9. Cells were transfected with plasmids encoding the indicated proteins. C, effects of PCSK9-ACE2 on total and surface expressions of LDLR and CD81 in Huh7 cells. The cells were transfected with plasmids encoding PCKS9-ACE2, ACE2, or an empty vector and stained with anti-LDLR or anti-CD81 antibodies prior to analysis by FACS. Saponin was added when the detection of intracellular protein was required (total LDLR and total CD81). The experiments were repeated at least three times. Statistics analysis was based on one-way analysis of variance (ANOVA) Dunnett's test. *, p ≤ 0.05; ***, p ≤ 0.001. The error bars represent S.E.
Effects of Mutations in the Signal Peptide and Prodomain of PCSK9-ACE2 on the Levels of Surface LDLR and CD81
Based on findings that PCSK9-ACE2 could efficiently lower both LDLR and CD81, we investigated the effects of transiently overexpressed specific PCSK9-ACE2 mutants on these proteins at the cell surface. Documented natural and engineered GOF and LOF mutations located on signal peptide, prodomain, catalytic domain, hinge region, and CHRD were selected for this study (Table 1) (10, 14, 15, 18–20, 23–25, 27–38). All PCSK9 mutants were fused with the TM-CT of ACE2 because PCSK9-ACE2 had much stronger effects than free PCSK9, making it easier to detect changes in protein levels by FACS. In addition to the measurement of the levels of surface LDLR and CD81 by FACS analysis, we also evaluated the zymogen processing of all PCSK9-ACE2 mutants by Western blotting. The maturation level is reflected by the ratio of mature PCSK9 to pro-PCSK9. Maturation of PCSK9-ACE2 mutants and surface LDLR and CD81 levels were compared with that of WT PCSK9-ACE2 and as control in the presence of ACE2 alone. From the signal peptide and prodomain region, eight single point mutations were inserted into WT PCSK9 (Fig. 4, A and B). Although most mutations did not overtly change the activity of PCSK9-ACE2 on LDLR or CD81, only R93C, known to result in LOF (24), significantly reduced this activity on LDLR but not CD81, and L108R did the reverse. In contrast, the GOF D129G completely lost its ability to enhance the degradation of CD81 compared with WT PCSK9-ACE2. Clearly D129G and less so D129N differentiate the effect of PCSK9-ACE2 on LDLR versus CD81.
FIGURE 4.
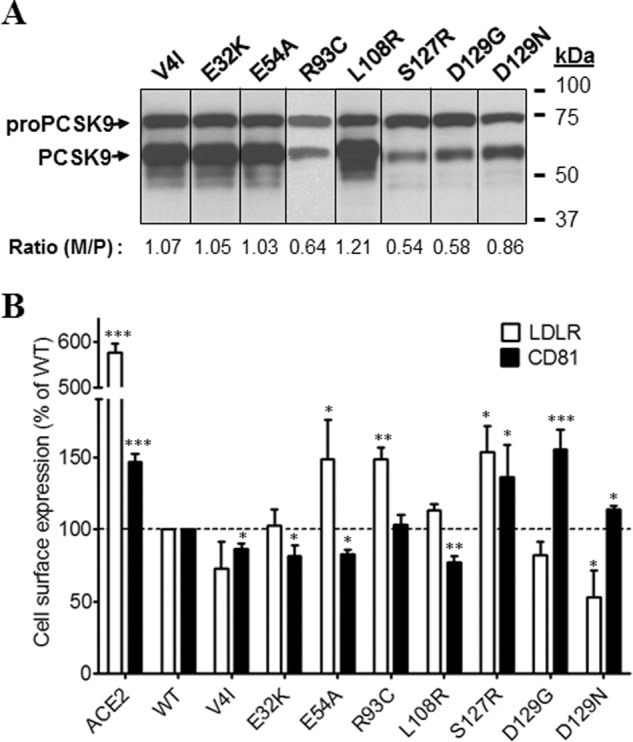
Effects of PCSK9 mutations in the signal peptide or prodomain on endogenous LDLR and CD81 expression. Huh7 cells were transfected with plasmids coding for PCSK9-ACE2 mutants in the signal peptide or prodomain. A, Western blot analysis of PCSK9-ACE2 mutant expression and maturation using rabbit anti-PCSK9 antibody. B, FACS analysis of surface expression of the LDLR and CD81 in Huh7 cells transfected with PCSK9-ACE2 mutants. All experiments were repeated at least three times. Statistics analysis was based on one-way ANOVA Dunnett's test. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001. The error bars represent S.E. M/P, mature PCSK9 to pro-PCSK9.
Effects of Mutations in the Catalytic Domain of PCSK9-ACE2 on the Expression of Surface LDLR and CD81
Thirteen mutations in the catalytic domain were investigated (Fig. 5, A and B). The mutant N354I that expressed only pro-PCSK9, thus exhibiting lack of zymogen processing, and the LOF mutants G236S and A239D (18) had no activity on either LDLR or CD81 (Fig. 5, A and B). It is well known that Asp374 is critical for the binding of PCSK9 to the EGF-A domain of the LDLR (10). Analysis of various mutants of this residue revealed that most other amino acids enhance the activity of PCSK9 on the LDLR (10, 33). Thus, as expected, both D374Y and D374H enhanced the activity of PCSK9-ACE2 toward the LDLR (Fig. 5B). Unexpectedly and different from D374Y, the mutant D374H lost its enhanced activity on CD81, emphasizing the difference in the critical residues in these receptors that interact with PCSK9 Asp374.
FIGURE 5.
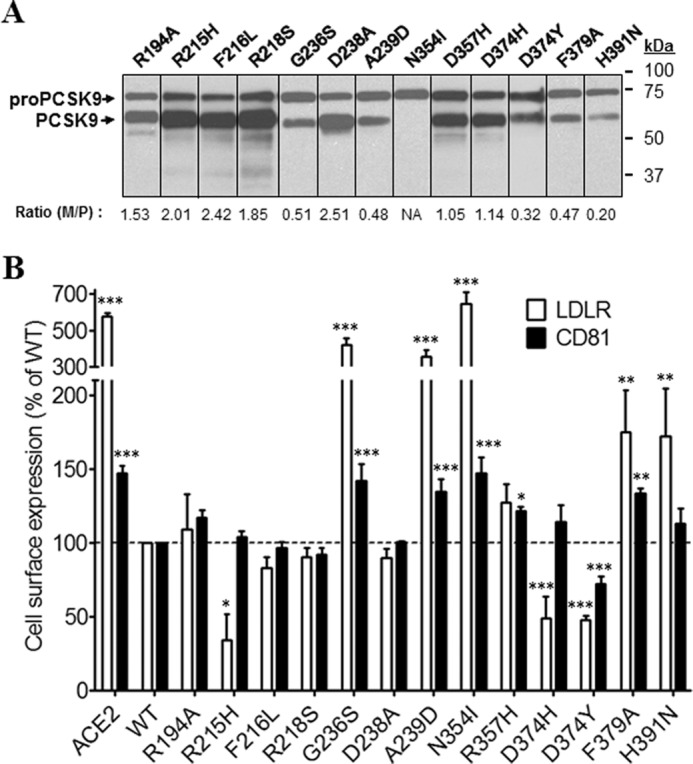
Effects of PCSK9 mutations in the CAT on endogenous LDLR and CD81 expression. Huh7 cells were transfected with plasmids coding for PCSK9-ACE2 mutants in the CAT. A, Western blot analysis of PCSK9 expression and maturation using rabbit anti-PCSK9 antibody. B, FACS analysis of surface expression of the LDLR and CD81 in Huh7 cells transfected with PCSK9-ACE2 mutants. All experiments were repeated at least three times. Statistics analysis was based on one-way ANOVA Dunnett's test. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001. The error bars represent S.E. M/P, mature PCSK9 to pro-PCSK9; NA, not applicable.
Effects of Mutations in the Hinge Region and CHRD of PCSK9-ACE2 on the Expressions of Surface LDLR and CD81
Among investigated mutations in the HR-CHRD region, A522T was half as well processed as WT and exhibited a clear LOF on both receptors (Fig. 6, A and B). We also noted that most HR-CHRD mutations investigated did not significantly modify the activity of PCSK9 toward the LDLR compared with WT. However, they generally enhanced by 10–30% the activity of PCSK9 toward CD81, suggesting that the modulation of CD81 by PCSK9 is sensitive to the HR-CHRD.
FIGURE 6.
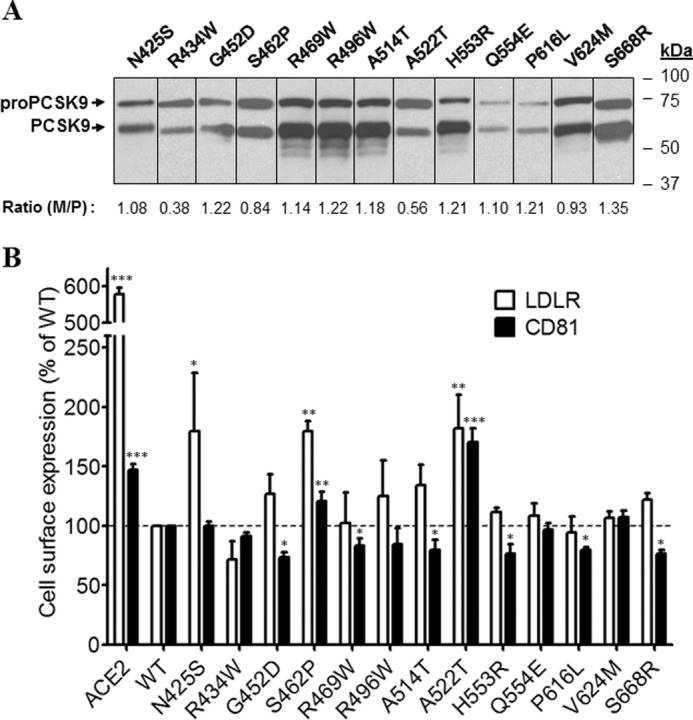
Effects of PCSK9 mutants in HR-CHRD on endogenous LDLR and CD81 expression. Huh7 cells were transfected with plasmids coding for PCSK9-ACE2 mutants in the HR-CHRD. A, Western blot analysis of PCSK9 expression and maturation using rabbit anti-PCSK9 antibody. B, FACS analysis of surface expression of the LDLR and CD81 in Huh7 cells transfected with PCSK9 mutants. All experiments were repeated at least three times. Statistics analysis was based on one-way ANOVA Dunnett's test. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001. The error bars represent S.E. M/P, mature PCSK9 to pro-PCSK9.
PCSK9 Catalytic and C-terminal Domains Could Down-regulate LDLR and CD81
Next, we tried to define the PCSK9 domain(s) responsible for modulation of CD81 levels. PCSK9 CAT and C-terminal (HR-CHRD) domains fused or not to the TM-CT of ACE2 were investigated. The expression of the domains was confirmed by Western blotting using rabbit anti-PCSK9 antibody (Fig. 7A). The HR-CHRDs appeared as single bands, but CATs appeared as two distinct bands that correspond to immature and mature forms as observed in full-length PCSK9. In the absence of the TM-CT of ACE2, CAT and HR-CHRD of PCSK9 had no effect on either surface or total expression of LDLR and CD81 (Fig. 7B). However, as for the full-length protein, the ACE2-fused truncated PCSK9 constructs showed stronger effects than the non-fused forms. The levels of surface and total LDLR and CD81 were decreased significantly by CAT-ACE2 and HR-CHRD-ACE2 as compared with control (Fig. 7B). Notably, HR-CHRD-ACE2 reduced surface CD81 expression to 0.56, but only a marginal decrease was observed for total CD81. Altogether, the results suggest that both domains of PCSK9 have the potential to regulate LDLR and CD81 when fused to TM-CT of ACE2.
FIGURE 7.
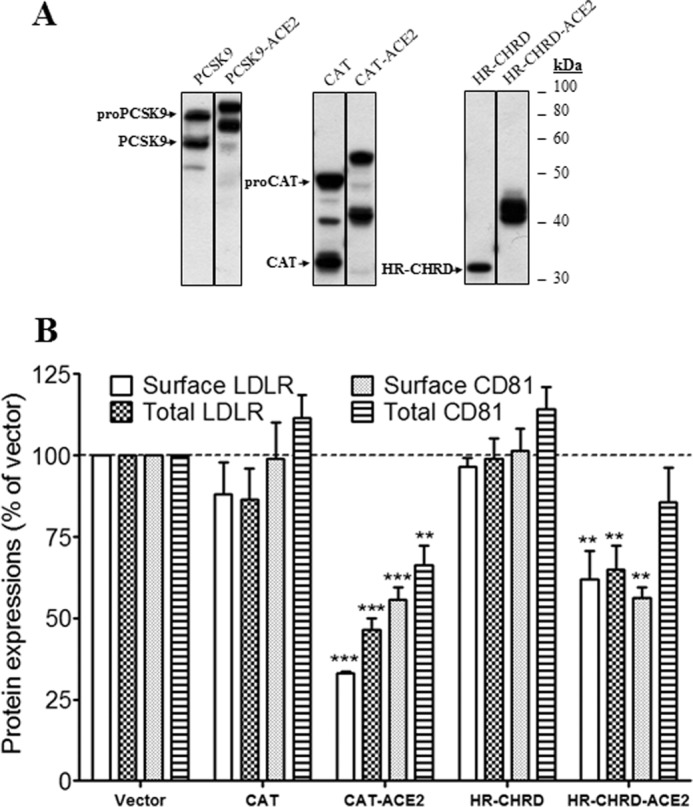
Effects of CAT and HR-CHRD on the endogenous LDLR and CD81 expression. Huh7 cells were transfected with plasmids coding for PCSK9 constructs (full length, CAT, and HR-CHRD) with or without the TM-CT of ACE2. A, Western blot analysis of PCSK9 expression and maturation using rabbit anti-PCSK9 antibody. B, FACS analysis of surface expression of the LDLR and CD81 in Huh7 cells transfected with PCSK9 constructs. All experiments were repeated at least three times. Statistic analysis was based on one-way ANOVA Dunnett's test. **, p ≤ 0.01; ***, p ≤ 0.001. The error bars represent S.E.
Effects of Intracellular PCSK9 on LDLR and CD81 Expressions
PCSK9 is known to modulate LDLR from the cell surface as well as through an intracellular pathway (9, 12). To evaluate the ability of PCSK9 to modulate CD81 through the intracellular pathways, we used a PCSK9 fused to the TM-CT domain of LAMP1 as reported previously (9, 39). Indeed, this chimeric protein cannot reach the plasma membrane and is mostly directed to lysosomes where degradation occurs (9). Because maturation of PCSK9-LAMP1 is normal in Huh7 cells (Fig. 8A), we compared the expression of PCSK9-LAMP1 by IFA in permeabilized and non-permeabilized transiently transfected cells. Results showed that PCSK9-LAMP1 was intracellularly localized but was not detected at the cell surface (Fig. 8B). To evaluate the lysosomal localization of PCSK9-LAMP1, we stained the cells expressing this chimera with LysoTracker Red. PCSK9-LAMP1 co-localized with lysosomes but was also found in other compartments of the cell (Fig. 8C). This is not unexpected as PCSK9-LAMP1 traverses the various cellular compartments including the ER and Golgi and cycles back to endosomes from the plasma membrane (40). We next analyzed the ability of the chimeric protein to degrade LDLR and CD81 (Fig. 8D). Similarly to PCSK9-ACE2, the chimera PCSK9-LAMP1 significantly reduced expression of both CD81 and LDLR. Although the interaction between PCSK9-LAMP1 and its targets occurs within cells, a significant depletion of LDLR and CD81 can be observed at the plasma membrane. This indicates that depletion of the intracellular pool affects the expression of CD81 and LDLR at the cell surface (Fig. 8D) or that the PCSK9-LAMP1 can transiently cycle to the cell surface as was reported for LAMP1 itself (40).
FIGURE 8.

Effects of intracellular PCSK9 on total and surface endogenous LDLR and CD81 expression. A, Huh7 cells were transfected with plasmids expressing wild-type PCSK9 or PCSK9 with the TM-CT of LAMP1 protein (PCSK9-LAMP1) that targets the protein to the lysosomes. Total cellular proteins were separated by 10% SDS-PAGE and then analyzed by rabbit anti-PCSK9 antibody. B, localization of PCSK9-LAMP1 in the cells was analyzed by immunofluorescence. Huh7 cells were co-transfected with eGFP- and PCSK9-LAMP1-coding plasmids before staining with rabbit anti-PCSK9 antibody. To detect intracellular PCSK9, cells were treated with Triton X-100. C, co-localization of PCSK9-LAMP1 with a lysosome marker. Huh7 cells were transiently transfected with PCSK9-LAMP1. Before fixation and staining with rabbit anti-PCSK9 (green), cells were incubated with LysoTracker (red). Imaging was done by a confocal microscope. The co-localization measurement was based on Manders' coefficients using ImageJ and JACoP. D, effects of PCSK9-LAMP1 on surface and total expressions of LDLR and CD81 in Huh7 cells by FACS. The cells were transfected with PCSK9-LAMP1, LAMP1, or empty vector as a control and then subjected to FACS using anti-LDLR or anti-CD81 antibody. Intracellular detection was obtained through saponin treatment (total LDLR and total CD81). The experiments were repeated at least three times. Statistics analysis was based on one-way ANOVA Dunnett's test. *, p ≤ 0.05; ***, p ≤ 0.001. The error bars represent S.E.
In Situ Interactions among PCSK9 and LDLR and CD81
The interaction between PCSK9 and the LDLR has been extensively characterized (41). Here, we demonstrated that PCSK9 is recruited at the cell surface by CD81 (Fig. 2A, lower) and that full-length PCSK9 as well as the CAT and HR-CHRD subdomains can modulate CD81 and LDLR (Figs. 3 and 7, respectively). To demonstrate the interaction between PCSK9 and CD81, we performed in situ PLA. This new approach allows the identification of single interactions in their natural environment (42–44) and has been used successfully in recent studies on PCSK9 (45) and tetraspanin proteins (46). In our experiment, the PCSK9-LDLR interaction was used as a positive control, and transfection with an empty vector served as a negative control. As expected, the result depicted in Fig. 9 clearly shows that in Huh7 cells endogenous LDLR can interact in situ with PCSK9-ACE2 (average of 82 dots per cell). In addition, the results showed that CD81 makes several interactions (>280 dots per cell) with PCSK9. Interestingly, we observed more steady-state CD81-PCSK9 interactions than LDLR-PCSK9. This might be due to a more efficient degradation of the LDLR by PCSK9, resulting in lower steady-state levels of the LDLR-PCSK9 complex than the CD81-PCSK9 complex. However, we cannot rule out that the LDLR antibody used is not less avid to LDLR than is the anti-CD81 toward its antigen.
FIGURE 9.

Interaction among PCSK9 and endogenous membrane-anchored proteins LDLR and CD81 by in situ PLA. Huh7 cells were co-transfected with eGFP- and PCSK9-ACE2-encoding plasmids or eGFP and empty phCMV3 vector as a control and grown on coverslips. The cells were permeabilized, and the PLA procedure was performed according to the manufacturer's instructions using the rabbit anti-PCSK9 and mouse anti-LDLR or rabbit anti-PCSK9 and mouse anti-CD81 antibodies. Duolink ImageTool software was used to analyze PLA signals (red dots). Histograms (right) show quantification of specific and nonspecific signal. Statistics analysis was based on unpaired t test. ***, p ≤ 0.001. The error bars represent S.E.
PCSK9 Is Co-immunoprecipitated with CD81
The PLA results (Fig. 9) show the in situ interaction between CD81 and PCSK9. To make sure that the two proteins have physical binding, we carried out a coIP assay. By co-expression of CD81 and PCSK9 or PCSK9-ACE2 in HEK293T cells and using anti-CD81 antibody to pull down CD81, we detected the coexistences of PCSK9 (Fig. 10A) or PCSK9-ACE2 (Fig. 10B) with CD81 in the coIP eluants. To confirm that CD81-PCSK9-ACE2 binding does not come from the TM-CT of ACE2, we tried to pull down full-length ACE2 protein with CD81. However, no CD81-ACE2 binding was detected by coIP (Fig. 10B). PCSK9-LDLR interaction was also confirmed by coIP of PCSK9 with LDLR using anti-V5 (Fig. 10C). These results suggest that PCSK9 can bind not only to LDLR but also to CD81.
FIGURE 10.
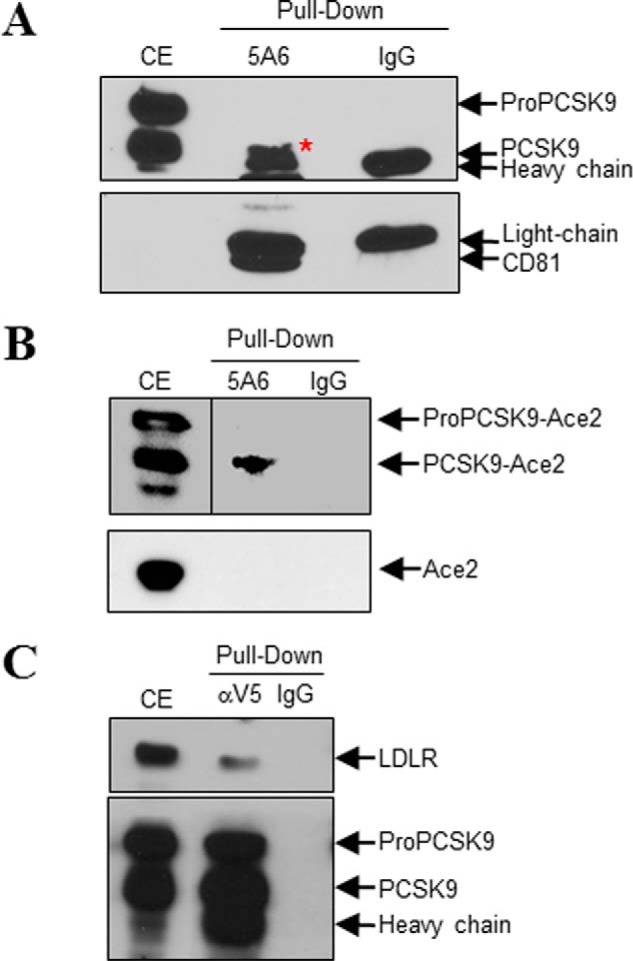
CoIP of CD81 with PCSK9. HEK293T cells were co-transfected with CD81- and PCSK9-V5- (A), PCSK9-ACE2- (B, upper), CD81- and ACE2- (B, lower), or PCSK9-V5- and LDLR (C)-encoding plasmids. Cell lysates were pulled down using anti-CD81 (clone 5A6) (A and B) or anti-V5 (C) antibody; mouse IgG1 was used as the mock antibody. A and B, coIP of CD81 with PCSK9 (A) or PCSK9-ACE2 but not ACE2 (B) using anti-CD81. C, coIP of PCSK9-V5 with LDLR using anti-V5. The red asterisk pinpoints PCSK9, which partially overlaps with the mouse 5A6 heavy chain. CE, cells extract.
Degradation of CD81 by PCSK9 Is Independent of LDLR
Because PCSK9-ACE2 can modulate the endogenous LDLR and CD81 levels and PCSK9 was recruited to the cell surface when either protein was overexpressed, we raised the question of whether PCSK9 recruitment to the cell surface in the presence of CD81 required LDLR expression. To address this question, we compared the plasma membrane recruitment of PCSK9 by CD81 in CHO and CHO-A7 cells that lack endogenous LDLR (39, 47). Indeed, LDLR modulation by PCSK9 was previously shown in normal CHO cells (39). CHO-A7 cells are thus suitable for the characterization of CD81 regulation by PCSK9 in the absence of LDLR. As in Huh7 cells (Fig. 2A, lower), upon CD81 transfection in CHO cells, PCSK9 was also clearly recruited at the plasma membrane (Fig. 11A). Interestingly, PCSK9 recruitment was also observed in the LDLR-deficient CHO-A7 cells overexpressing CD81. This result indicates that the LDLR is not required for PCSK9-CD81 interaction (Figs. 9 and 10). Although LDLR is not required for the latter interaction, it may contribute to the internalization and trafficking of the CD81-PCSK9 to endosomes/lysosomes. Therefore, we investigated the involvement of LDLR in the degradation of CD81 in CHO-A7 cells. First, we observed that PCSK9-ACE2 could normally decrease co-expressed LDLR levels in these cells (Fig. 11B). Similarly to what has been discussed in Huh7 cells, PCSK9-ACE2 could also efficiently decrease CD81 expression on the cell surface of both parental and LDLR-deficient CHO cells (Fig. 11C), indicating that PCSK9-induced CD81 degradation could occur in the absence of LDLR. We then analyzed the effect of LDLR overexpression on CD81 degradation by PCSK9. Results showed that in normal CHO cells the addition of LDLR did not change the effect of PCSK9-ACE2 on CD81. However, in CHO-A7 cells, overexpression of LDLR improved PCSK9 activity on CD81 (Fig. 11C), suggesting that PCSK9 activity on CD81 benefits from LDLR expression.
FIGURE 11.

Recruitment of PCSK9 by CD81 and its function in modulation of LDLR and CD81 in LDLR non-expressing CHO cells. A, parental CHO cells and CHO cells lacking LDLR expression (CHO-A7) were co-transfected with a set of plasmids expressing eGFP, PCSK9, and CD81 or eGFP, PCSK9, and empty phCMV3 vector as a negative control. Cells were stained with rabbit anti-PCSK9 (red) and mouse-anti CD81 (green) antibodies. The nuclei were stained with DAPI. Imaging was done by a confocal microscope. B, effect of PCSK9 on LDLR in CHO-A7 cells. In addition to eGFP- and LDLR-encoding plasmids, CHO and CHO-A7 cells were co-transfected with ACE2 (red), PCSK9-ACE2 (blue), or empty vector (gray). Cells were stained with allophycocyanin-conjugated mouse anti-LDLR and subjected to FACS analysis. The histograms represent triplicate experiments. C, in addition to eGFP-encoding plasmid as the indicator of transfection, CHO and CHO-A7 cells were co-transfected with CD81 and plasmids coding for the indicated proteins. Cells were stained with phycoerythrin-conjugated mouse anti-CD81 and subjected to FACS analysis. Expressions of CD81 in transfected CHO cells were compared with empty vector-transfected cells. The experiments were repeated at least three times. Statistics analysis was based on one-way ANOVA Tukey test. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ns, not significant. The error bars represent S.E.
CD81 Interacts with LDLR
We next investigated in situ the putative LDLR-CD81 interaction. This was inspired by the facts that (i) CD81 and LDLR interact with PCSK9, (ii) the levels of both are down-regulated by PCSK9, and (iii) LDLR expression seems to facilitate CD81 modulation by PCSK9 (Fig. 11C). Therefore, we analyzed the co-localization signal by IFA and observed that overexpressed CD81 and LDLR co-localized with a co-localization Manders' coefficient of 0.503 (Fig. 12A). To ensure that CD81-LDLR co-localization was relevant and to rule out false results arising from nonspecific binding of antibodies, we carried out an antibody specificity test. We showed that the primary and secondary antibodies used to detect CD81 (Fig. 12B) do not bind to LDLR or its corresponding primary antibody (Fig. 12, F and G). We also confirmed that the primary and secondary antibodies for LDLR (Fig. 12E) have no affinity for CD81 and anti-CD81 primary antibody (Fig. 12, C and D).
FIGURE 12.

CD81-LDLR co-localization in Huh7 cells. A, Huh7 cells were co-transfected with eGFP- and LDLR-encoding plasmids. Cells were stained with goat anti-LDLR (red) and mouse-anti CD81 (green) antibodies. B–G, verification of antibody specificity. Huh7 cells were co-transfected with eGFP and CD81 (B and C), eGFP and LDLR (E and F), or eGFP and empty phCMV3 vector (D and G). The cells were treated with Triton X-100 and stained with mouse anti-CD81 (B) or goat anti-LDLR (E) or co-stained with mouse anti-CD81 and goat anti-LDLR (C, D, F, and G) antibodies. Following primary antibody incubation, the cells were stained with Alexa Fluor chicken anti-mouse (B, F, and G) or donkey anti-goat (C–E) IgG. The nuclei were stained with DAPI. Imaging was done by a confocal microscope. The co-localization measurement in A was based on Manders' coefficients using ImageJ and JACoP.
In addition to co-localization of LDLR and CD81 observed by IFA, we carried out PLA to determine the in situ interactions of these proteins (Fig. 13). This approach revealed that overexpressed CD81 could interact with endogenous LDLR and that overexpressed LDLR could interact with endogenous CD81. The extent of interaction increased significantly when both proteins were overexpressed. To verify the LDLR-CD81 interaction specificity, we assessed the interaction between LDLR and CD9. We chose CD9 as a control for CD81 because as members of the tetraspanin family both are present in tetraspanin-enriched microdomains, and their interaction has been proposed previously (48, 49). As expected, LDLR-CD9 interaction could not be detected by PLA, demonstrating the specificity of the assay. To ensure that CD81 has a physical interaction with LDLR, we co-expressed in HEK293 cells CD81 and LDLR (Fig. 14). The data show that CD81 co-immunoprecipitates with both forms of the LDLR, the ∼100-kDa non-O-glycosylated ER form and the ∼150-kDa mature fully glycosylated LDLR. These results suggest that CD81 and LDLR meet very early along the secretory pathway, likely in the ER, but also at the Golgi and/or cell surface.
FIGURE 13.

Interaction between LDLR and CD81 by in situ PLA. Huh7 cells were co-transfected with eGFP-expressing plasmid and with plasmids coding for the indicated proteins. The cells were permeabilized and co-stained with goat anti-LDLR and mouse anti-CD81 or goat anti-LDLR and mouse anti-CD9 antibodies. The PLA was done as described under “Experimental Procedures.” Duolink ImageTool software was used to analyze PLA signals (red dots). Statistics analysis was based on unpaired t test. The nuclei were stained with DAPI. ***, p ≤ 0.001. The error bars represent S.E.
FIGURE 14.
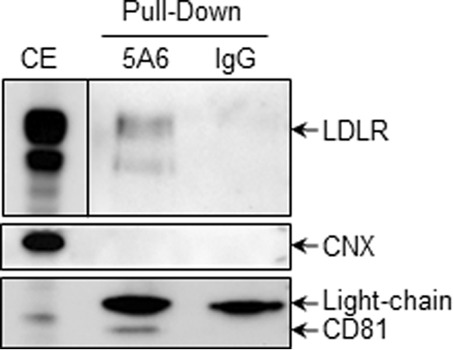
CoIP of CD81 with LDLR. HEK293T cells were co-transfected with CD81- and LDLR-encoding plasmids. Cell lysates were pulled down using anti-CD81 (clone 5A6) or mouse IgG1 as the mock antibody. The membrane-bound protein calnexin (CNX) was used as an internal control.
Because CD81 and LDLR are both membrane proteins, it was possible that anti-CD81 pulled down not only CD81 but also membrane debris containing CD81 and/or LDLR. However, the absence of membrane-bound calnexin in the eluants (Fig. 14) confirmed that there were no membrane-associated contaminating proteins pulled down by anti-CD81.
Discussion
PCSK9 is a modulator of LDLR levels through an early interaction in ER (12) and later post-ER compartments (endosomes or Golgi apparatus) or at the cell surface. These interactions ultimately lead to LDLR degradation in endosomes/lysosomes (12). PCSK9 was reported to bind the EGF-A domain of the LDLR (11, 23). Among extracellular domains of LDLR, the EGF-A domain is central for the binding and internalization of cell surface PCSK9-LDLR complex, but the cytoplasmic domain of LDLR is not essential in this process (39, 50). Similarly, our results showed that PCSK9 co-localizes at the cell surface not only with the LDLR but also with CD81 (Fig. 2A) where it interacts with these proteins (Figs. 9 and 10). To evaluate the effect of point mutations of PCSK9 on both LDLR and CD81, we developed a simple FACS assay using PCSK9 fused at its C terminus with the TM-CT domain of ACE2, resulting in an enhanced concentration of PCSK9 at the plasma membrane. In all our FACS analyses, we overexpressed PCSK9 and its mutants and analyzed their effect on the endogenous levels of LDLR or CD81.
An extensive analysis demonstrated that the LDLR and CD81 are similarly affected by PCSK9-ACE2 and its mutants except for some specific mutations. The most notable GOF mutations are D129N and D374H, which greatly enhanced LDLR degradation but not that of CD81 compared with WT. In contrast, the LOF mutations E54A, R93C, H391N, and N425S either completely or partially lost activity on LDLR but exhibited the same or enhanced activity on CD81. These data suggest that the PCSK9-sensitive determinants in CD81 and LDLR are similar but not identical. However, we could not find any significant alignment of the EGF-A domain of LDLR (interacting with the catalytic subunit of PCSK9) with any segment of the CD81 including its EC1 or EC2 ectodomains. Therefore, the binding domain of CD81 to PCSK9 remains obscure.
PCSK9 catalytic domain is known for its affinity for the LDLR, whereas the CHRD participates in the PCSK9 trafficking to the cell surface. Previously, it has been shown that PCSK9 truncated forms (CAT and CHRD) can both interact with the LDLR but are unable to modulate its levels (12, 51, 52). Here, the same observation was made in Huh7 cells. Indeed, overexpression of CAT and HR-CHRD did not change the total and surface levels of LDLR and CD81. Interestingly, ACE2-tagged CAT or HR-CHRD could efficiently decrease both surface and total levels of LDLR and CD81 (Fig. 7B). Normally, these two truncated constructs could interact with LDLR, although the CHRD had lower affinity than CAT (12). Therefore, we suspect that addition of the ACE2 TM-CT to the CAT or HR-CHRD stabilizes the complex formation and helps in its sorting to lysosomes for degradation (23). Moreover, by retaining them in cells, the fusion increases their local protein concentration.
We previously reported on the enhanced degradation of LDLR and CD81 in Huh7 cells expressing wild-type PCSK9, PCSK9-ACE2, or PCSK9-LAMP1 (21). The fusion of the TM-CT of ACE2 to PCSK9 allows its targeting to the cell surface, whereas the fusion to the TM-CT of LAMP1 primarily targets the protein to the lysosomal compartment. In this study, we sought to gain insight into the propensity of both surface and intracellular PCSK9 to modulate LDLR and CD81 levels. In agreement with our previous findings, we found that PCSK9-ACE2 and PCSK9-LAMP1 decreased total and surface expression of LDLR and CD81 (Figs. 3C and 8D). It is known that PCSK9 can interact with the LDLR at the cell surface or intracellularly and trigger its degradation in endosomes/lysosomes (12). PCSK9-ACE2 and/or PCSK9-LAMP1 is likely to act on CD81 in a similar intracellular pathway as WT PCSK9 on LDLR. However, further investigations are required to better address this question.
Interestingly, we found that CD81 and LDLR co-localized (Fig. 12A) and interacted with each other (Figs. 13 and 14). Because PCSK9 and CD81 can interact with the LDLR in situ (Figs. 9 and 13), they might form a trimeric complex in Huh7 cells. However, an extensive future study is required to determine the stoichiometry of the complex and the interacting domains of each protein. Nevertheless, our data indicated that the presence of the LDLR is not mandatory for PCSK9-CD81 interaction or for PCSK9-dependent CD81 degradation (Fig. 11A). Finally, in HepG2 cells (devoid of CD81), the PCSK9-induced degradation of the LDLR occurs with high efficiency (53), suggesting that CD81 is not necessary for PCSK9 to enhance the degradation of the LDLR.
It is noteworthy that the LDLR and CD81 are both important receptors for HCV entry (54–56). The actual model of HCV entry proposes that the initial attachment of the infectious particle occurs through the LDLR. The virus is then transferred to a series of entry factors that include SR-B1, CD81, CLDN1, and occludin (57). CD81-CLDN1 interaction has been observed and plays an important role in HCV entry (58, 59). Here, we have described a novel LDLR-CD81 interaction that may contribute to HCV entry. However, we can only speculate whether this mechanism/complex may modulate the likeliness of the CD81/LDLR-mediated viral invasion through interaction of this complex with the HCV envelope glycoprotein E2.
PCSK9-ACE2 interacts with and modulates the levels of both LDLR and CD81. This may result in a competition between the two proteins for their modulator, PCSK9. Indeed, it has been shown that PCSK9-modulated LDLR degradation could be prevented by a surface protein competitor (namely annexin A2), which could bind to the CHRD of PCSK9 (26, 60). To address this question, we investigated the effect of PCSK9-ACE2 on LDLR in normal CHO cells and in LDLR-deficient CHO-A7 cells. The results showed that LDLR levels were reduced by PCSK9-ACE2 in the two cell lines (Fig. 11B) similar to what was observed in Huh7 cells (Fig. 3C). Additionally, we showed that PCSK9-ACE2 can down-regulate CD81 expression levels in CHO and CHO-A7 cells. This indicates that the presence of LDLR is not necessary for PCSK9-mediated CD81 modulation (Fig. 11C). It is notable that the overexpression of LDLR in parental CHO cells did not change the effect of PCSK9-ACE2 on CD81 levels, suggesting that LDLR is not limiting and that it does not compete with CD81 for PCSK9. Moreover, the addition of LDLR in CHO-A7 cells enhanced CD81 degradation by PCSK9-ACE2, suggesting that LDLR favors PCSK9 modulation of CD81. Altogether, our results in CHO and CHO-A7 cells showed that LDLR was not required for CD81 regulation by PCSK9, albeit its presence may enhance PCSK9-dependent CD81 degradation. It is thus possible that expression of CD81 in HepG2 cells (lacking endogenous CD81) may enhance the ability of PCSK9 to enhance the degradation of endogenous LDLR.
In summary, this study confirms CD81 as a receptor targeted for degradation by PCSK9 by an intracellular pathway that may require different conformational determinants in PCSK9 than for the LDLR. Furthermore, our data revealed formation a novel complex between PCSK9 and CD81 and between CD81 and LDLR, suggesting the existence of a complex array of partners modulated by PCSK9.
Author Contributions
Q.-T. L. performed all experiments, analyzed the data, and wrote the paper. M. B. supervised Q.-T. L. and analyzed the data. N. G. S. provided reagents, analyzed the data, and made a substantial contribution to the manuscript writing. P. L. directed the whole process, analyzed the data, and wrote the manuscript.
Acknowledgments
We thank Jessy Tremblay for technical assistance in flow cytometry and confocal microscopy. We thank Ahmed Fahmy for helpful discussions.
This work was supported by Canadian Institutes of Health Research Operating Grant 93792 (to P. L.) and by Leducq Foundation Grant 13 CVD-03 (to N. G. S.). The authors declare that they have no conflicts of interest with the contents of this article.
- FH
- familial hypercholesterolemia
- PCSK9
- proprotein convertase subtilisin/kexin type 9
- LDLR
- low density lipoprotein receptor
- GOF
- gain-of-function
- LOF
- lost-of-function
- LDL-C
- low density lipoprotein cholesterol
- HR
- hinge region
- CHRD
- cysteine-histidine-rich domain
- ER
- endoplasmic reticulum
- EGF-A
- epidermal growth factor-like repeat A
- HCV
- hepatitis C virus
- CAT
- catalytic domain
- TM-CT
- transmembrane-cytosolic tail
- LAMP1
- lysosomal-associated membrane protein 1
- ACE2
- angiotensin I-converting enzyme 2
- eGFP
- enhanced green fluorescent protein
- IFA
- immunofluorescence assay
- PLA
- proximity ligation assay
- coIP
- co-immunoprecipitation
- ANOVA
- analysis of variance
- JACoP
- Just Another Colocalization Plugin.
References
- 1. Seidah N. G., Benjannet S., Wickham L., Marcinkiewicz J., Jasmin S. B., Stifani S., Basak A., Prat A., Chretien M. (2003) The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc. Natl. Acad. Sci. U.S.A. 100, 928–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abifadel M., Varret M., Rabès J. P., Allard D., Ouguerram K., Devillers M., Cruaud C., Benjannet S., Wickham L., Erlich D., Derré A., Villéger L., Farnier M., Beucler I., Bruckert E., Chambaz J., Chanu B., Lecerf J. M., Luc G., Moulin P., Weissenbach J., Prat A., Krempf M., Junien C., Seidah N. G., Boileau C. (2003) Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 34, 154–156 [DOI] [PubMed] [Google Scholar]
- 3. Zaid A., Roubtsova A., Essalmani R., Marcinkiewicz J., Chamberland A., Hamelin J., Tremblay M., Jacques H., Jin W., Davignon J., Seidah N. G., Prat A. (2008) Proprotein convertase subtilisin/kexin type 9 (PCSK9): hepatocyte-specific low-density lipoprotein receptor degradation and critical role in mouse liver regeneration. Hepatology 48, 646–654 [DOI] [PubMed] [Google Scholar]
- 4. Maxwell K. N., Breslow J. L. (2004) Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc. Natl. Acad. Sci. U.S.A. 101, 7100–7105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Benjannet S., Rhainds D., Essalmani R., Mayne J., Wickham L., Jin W., Asselin M. C., Hamelin J., Varret M., Allard D., Trillard M., Abifadel M., Tebon A., Attie A. D., Rader D. J., Boileau C., Brissette L., Chrétien M., Prat A., Seidah N. G. (2004) NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J. Biol. Chem. 279, 48865–48875 [DOI] [PubMed] [Google Scholar]
- 6. Seidah N. G., Awan Z., Chrétien M., Mbikay M. (2014) PCSK9: a key modulator of cardiovascular health. Circ. Res. 114, 1022–1036 [DOI] [PubMed] [Google Scholar]
- 7. Seidah N. G., Prat A. (2012) The biology and therapeutic targeting of the proprotein convertases. Nat. Rev. Drug Discov. 11, 367–383 [DOI] [PubMed] [Google Scholar]
- 8. Roubtsova A., Munkonda M. N., Awan Z., Marcinkiewicz J., Chamberland A., Lazure C., Cianflone K., Seidah N. G., Prat A. (2011) Circulating proprotein convertase subtilisin/kexin 9 (PCSK9) regulates VLDLR protein and triglyceride accumulation in visceral adipose tissue. Arterioscler. Thromb. Vasc. Biol. 31, 785–791 [DOI] [PubMed] [Google Scholar]
- 9. Poirier S., Mayer G., Benjannet S., Bergeron E., Marcinkiewicz J., Nassoury N., Mayer H., Nimpf J., Prat A., Seidah N. G. (2008) The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. J. Biol. Chem. 283, 2363–2372 [DOI] [PubMed] [Google Scholar]
- 10. Kwon H. J., Lagace T. A., McNutt M. C., Horton J. D., Deisenhofer J. (2008) Molecular basis for LDL receptor recognition by PCSK9. Proc. Natl. Acad. Sci. U.S.A. 105, 1820–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang D. W., Lagace T. A., Garuti R., Zhao Z., McDonald M., Horton J. D., Cohen J. C., Hobbs H. H. (2007) Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J. Biol. Chem. 282, 18602–18612 [DOI] [PubMed] [Google Scholar]
- 12. Nassoury N., Blasiole D. A., Tebon Oler A., Benjannet S., Hamelin J., Poupon V., McPherson P. S., Attie A. D., Prat A., Seidah N. G. (2007) The cellular trafficking of the secretory proprotein convertase PCSK9 and its dependence on the LDLR. Traffic 8, 718–732 [DOI] [PubMed] [Google Scholar]
- 13. Yamamoto T., Lu C., Ryan R. O. (2011) A two-step binding model of PCSK9 interaction with the low density lipoprotein receptor. J. Biol. Chem. 286, 5464–5470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Abifadel M., Guerin M., Benjannet S., Rabès J. P., Le Goff W., Julia Z., Hamelin J., Carreau V., Varret M., Bruckert E., Tosolini L., Meilhac O., Couvert P., Bonnefont-Rousselot D., Chapman J., Carrié A., Michel J. B., Prat A., Seidah N. G., Boileau C. (2012) Identification and characterization of new gain-of-function mutations in the PCSK9 gene responsible for autosomal dominant hypercholesterolemia. Atherosclerosis 223, 394–400 [DOI] [PubMed] [Google Scholar]
- 15. Lo Surdo P., Bottomley M. J., Calzetta A., Settembre E. C., Cirillo A., Pandit S., Ni Y. G., Hubbard B., Sitlani A., Carfí A. (2011) Mechanistic implications for LDL receptor degradation from the PCSK9/LDLR structure at neutral pH. EMBO Rep. 12, 1300–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Poirier S., Mayer G., Poupon V., McPherson P. S., Desjardins R., Ly K., Asselin M. C., Day R., Duclos F. J., Witmer M., Parker R., Prat A., Seidah N. G. (2009) Dissection of the endogenous cellular pathways of PCSK9-induced low density lipoprotein receptor degradation: evidence for an intracellular route. J. Biol. Chem. 284, 28856–28864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Abifadel M., Rabès J. P., Devillers M., Munnich A., Erlich D., Junien C., Varret M., Boileau C. (2009) Mutations and polymorphisms in the proprotein convertase subtilisin kexin 9 (PCSK9) gene in cholesterol metabolism and disease. Hum. Mutat. 30, 520–529 [DOI] [PubMed] [Google Scholar]
- 18. Cameron J., Holla O. L., Laerdahl J. K., Kulseth M. A., Ranheim T., Rognes T., Berge K. E., Leren T. P. (2008) Characterization of novel mutations in the catalytic domain of the PCSK9 gene. J. Intern. Med. 263, 420–431 [DOI] [PubMed] [Google Scholar]
- 19. Fasano T., Sun X. M., Patel D. D., Soutar A. K. (2009) Degradation of LDLR protein mediated by 'gain of function' PCSK9 mutants in normal and ARH cells. Atherosclerosis 203, 166–171 [DOI] [PubMed] [Google Scholar]
- 20. Fisher T. S., Lo Surdo P., Pandit S., Mattu M., Santoro J. C., Wisniewski D., Cummings R. T., Calzetta A., Cubbon R. M., Fischer P. A., Tarachandani A., De Francesco R., Wright S. D., Sparrow C. P., Carfi A., Sitlani A. (2007) Effects of pH and low density lipoprotein (LDL) on PCSK9-dependent LDL receptor regulation. J. Biol. Chem. 282, 20502–20512 [DOI] [PubMed] [Google Scholar]
- 21. Labonté P., Begley S., Guévin C., Asselin M. C., Nassoury N., Mayer G., Prat A., Seidah N. G. (2009) PCSK9 impedes hepatitis C virus infection in vitro and modulates liver CD81 expression. Hepatology 50, 17–24 [DOI] [PubMed] [Google Scholar]
- 22. Bolte S., Cordelières F. P. (2006) A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224, 213–232 [DOI] [PubMed] [Google Scholar]
- 23. Lagace T. A., Curtis D. E., Garuti R., McNutt M. C., Park S. W., Prather H. B., Anderson N. N., Ho Y. K., Hammer R. E., Horton J. D. (2006) Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J. Clin. Investig. 116, 2995–3005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miyake Y., Kimura R., Kokubo Y., Okayama A., Tomoike H., Yamamura T., Miyata T. (2008) Genetic variants in PCSK9 in the Japanese population: rare genetic variants in PCSK9 might collectively contribute to plasma LDL cholesterol levels in the general population. Atherosclerosis 196, 29–36 [DOI] [PubMed] [Google Scholar]
- 25. Noguchi T., Katsuda S., Kawashiri M. A., Tada H., Nohara A., Inazu A., Yamagishi M., Kobayashi J., Mabuchi H. (2010) The E32K variant of PCSK9 exacerbates the phenotype of familial hypercholesterolaemia by increasing PCSK9 function and concentration in the circulation. Atherosclerosis 210, 166–172 [DOI] [PubMed] [Google Scholar]
- 26. Mayer G., Poirier S., Seidah N. G. (2008) Annexin A2 is a C-terminal PCSK9-binding protein that regulates endogenous low density lipoprotein receptor levels. J. Biol. Chem. 283, 31791–31801 [DOI] [PubMed] [Google Scholar]
- 27. Homer V. M., Marais A. D., Charlton F., Laurie A. D., Hurndell N., Scott R., Mangili F., Sullivan D. R., Barter P. J., Rye K. A., George P. M., Lambert G. (2008) Identification and characterization of two non-secreted PCSK9 mutants associated with familial hypercholesterolemia in cohorts from New Zealand and South Africa. Atherosclerosis 196, 659–666 [DOI] [PubMed] [Google Scholar]
- 28. Bottomley M. J., Cirillo A., Orsatti L., Ruggeri L., Fisher T. S., Santoro J. C., Cummings R. T., Cubbon R. M., Lo Surdo P., Calzetta A., Noto A., Baysarowich J., Mattu M., Talamo F., De Francesco R., Sparrow C. P., Sitlani A., Carfí A. (2009) Structural and biochemical characterization of the wild type PCSK9-EGF(AB) complex and natural familial hypercholesterolemia mutants. J. Biol. Chem. 284, 1313–1323 [DOI] [PubMed] [Google Scholar]
- 29. Benjannet S., Rhainds D., Hamelin J., Nassoury N., Seidah N. G. (2006) The proprotein convertase (PC) PCSK9 is inactivated by furin and/or PC5/6A: functional consequences of natural mutations and post-translational modifications. J. Biol. Chem. 281, 30561–30572 [DOI] [PubMed] [Google Scholar]
- 30. Allard D., Amsellem S., Abifadel M., Trillard M., Devillers M., Luc G., Krempf M., Reznik Y., Girardet J. P., Fredenrich A., Junien C., Varret M., Boileau C., Benlian P., Rabès J. P. (2005) Novel mutations of the PCSK9 gene cause variable phenotype of autosomal dominant hypercholesterolemia. Hum. Mutat. 26, 497. [DOI] [PubMed] [Google Scholar]
- 31. Bourbon M., Alves A. C., Medeiros A. M., Silva S., Soutar A. K., and Investigators of Portuguese FH Study (2008) Familial hypercholesterolaemia in Portugal. Atherosclerosis 196, 633–642 [DOI] [PubMed] [Google Scholar]
- 32. Leren T. P. (2004) Mutations in the PCSK9 gene in Norwegian subjects with autosomal dominant hypercholesterolemia. Clin. Genet. 65, 419–422 [DOI] [PubMed] [Google Scholar]
- 33. Cunningham D., Danley D. E., Geoghegan K. F., Griffor M. C., Hawkins J. L., Subashi T. A., Varghese A. H., Ammirati M. J., Culp J. S., Hoth L. R., Mansour M. N., McGrath K. M., Seddon A. P., Shenolikar S., Stutzman-Engwall K. J., Warren L. C., Xia D., Qiu X. (2007) Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat. Struct. Mol. Biol. 14, 413–419 [DOI] [PubMed] [Google Scholar]
- 34. Kotowski I. K., Pertsemlidis A., Luke A., Cooper R. S., Vega G. L., Cohen J. C., Hobbs H. H. (2006) A spectrum of PCSK9 alleles contributes to plasma levels of low-density lipoprotein cholesterol. Am. J. Hum. Genet. 78, 410–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pisciotta L., Priore Oliva C., Cefalù A. B., Noto D., Bellocchio A., Fresa R., Cantafora A., Patel D., Averna M., Tarugi P., Calandra S., Bertolini S. (2006) Additive effect of mutations in LDLR and PCSK9 genes on the phenotype of familial hypercholesterolemia. Atherosclerosis 186, 433–440 [DOI] [PubMed] [Google Scholar]
- 36. Dubuc G., Tremblay M., Paré G., Jacques H., Hamelin J., Benjannet S., Boulet L., Genest J., Bernier L., Seidah N. G., Davignon J. (2010) A new method for measurement of total plasma PCSK9: clinical applications. J. Lipid Res. 51, 140–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cameron J., Holla Ø. L., Laerdahl J. K., Kulseth M. A., Berge K. E., Leren T. P. (2009) Mutation S462P in the PCSK9 gene reduces secretion of mutant PCSK9 without affecting the autocatalytic cleavage. Atherosclerosis 203, 161–165 [DOI] [PubMed] [Google Scholar]
- 38. Fasano T., Cefalù A. B., Di Leo E., Noto D., Pollaccia D., Bocchi L., Valenti V., Bonardi R., Guardamagna O., Averna M., Tarugi P. (2007) A novel loss of function mutation of PCSK9 gene in white subjects with low-plasma low-density lipoprotein cholesterol. Arterioscler. Thromb. Vasc. Biol. 27, 677–681 [DOI] [PubMed] [Google Scholar]
- 39. Canuel M., Sun X., Asselin M. C., Paramithiotis E., Prat A., Seidah N. G. (2013) Proprotein convertase subtilisin/kexin type 9 (PCSK9) can mediate degradation of the low density lipoprotein receptor-related protein 1 (LRP-1). PLoS One 8, e64145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lippincott-Schwartz J., Fambrough D. M. (1987) Cycling of the integral membrane glycoprotein, LEP100, between plasma membrane and lysosomes: kinetic and morphological analysis. Cell 49, 669–677 [DOI] [PubMed] [Google Scholar]
- 41. Urban D., Pöss J., Böhm M., Laufs U. (2013) Targeting the proprotein convertase subtilisin/kexin type 9 for the treatment of dyslipidemia and atherosclerosis. J. Am. Coll. Cardiol. 62, 1401–1408 [DOI] [PubMed] [Google Scholar]
- 42. Blokzijl A., Nong R., Darmanis S., Hertz E., Landegren U., Kamali-Moghaddam M. (2014) Protein biomarker validation via proximity ligation assays. Biochim. Biophys. Acta 1844, 933–939 [DOI] [PubMed] [Google Scholar]
- 43. Söderberg O., Leuchowius K. J., Gullberg M., Jarvius M., Weibrecht I., Larsson L. G., Landegren U. (2008) Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods 45, 227–232 [DOI] [PubMed] [Google Scholar]
- 44. Söderberg O., Gullberg M., Jarvius M., Ridderstråle K., Leuchowius K. J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L. G., Landegren U. (2006) Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000 [DOI] [PubMed] [Google Scholar]
- 45. Sun H., Samarghandi A., Zhang N., Yao Z., Xiong M., Teng B. B. (2012) Proprotein convertase subtilisin/kexin type 9 interacts with apolipoprotein B and prevents its intracellular degradation, irrespective of the low-density lipoprotein receptor. Arterioscler. Thromb. Vasc. Biol. 32, 1585–1595 [DOI] [PubMed] [Google Scholar]
- 46. Huang W., Febbraio M., Silverstein R. L. (2011) CD9 tetraspanin interacts with CD36 on the surface of macrophages: a possible regulatory influence on uptake of oxidized low density lipoprotein. PLoS One 6, e29092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Krieger M., Brown M. S., Goldstein J. L. (1981) Isolation of Chinese hamster cell mutants defective in the receptor-mediated endocytosis of low density lipoprotein. J. Mol. Biol. 150, 167–184 [DOI] [PubMed] [Google Scholar]
- 48. Hemler M. E. (2005) Tetraspanin functions and associated microdomains. Nat. Rev. Mol. Cell Biol. 6, 801–811 [DOI] [PubMed] [Google Scholar]
- 49. Horváth G., Serru V., Clay D., Billard M., Boucheix C., Rubinstein E. (1998) CD19 is linked to the integrin-associated tetraspans CD9, CD81, and CD82. J. Biol. Chem. 273, 30537–30543 [DOI] [PubMed] [Google Scholar]
- 50. Strøm T. B., Holla Ø. L., Tveten K., Cameron J., Berge K. E., Leren T. P. (2010) Disrupted recycling of the low density lipoprotein receptor by PCSK9 is not mediated by residues of the cytoplasmic domain. Mol. Genet. Metab. 101, 76–80 [DOI] [PubMed] [Google Scholar]
- 51. Saavedra Y. G., Day R., Seidah N. G. (2012) The M2 module of the Cys-His-rich domain (CHRD) of PCSK9 protein is needed for the extracellular low-density lipoprotein receptor (LDLR) degradation pathway. J. Biol. Chem. 287, 43492–43501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang D. W., Garuti R., Tang W. J., Cohen J. C., Hobbs H. H. (2008) Structural requirements for PCSK9-mediated degradation of the low-density lipoprotein receptor. Proc. Natl. Acad. Sci. U.S.A. 105, 13045–13050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Butkinaree C., Canuel M., Essalmani R., Poirier S., Benjannet S., Asselin M. C., Roubtsova A., Hamelin J., Marcinkiewicz J., Chamberland A., Guillemot J., Mayer G., Sisodia S. S., Jacob Y., Prat A., Seidah N. G. (2015) Amyloid precursor-like protein 2 and sortilin do not regulate the PCSK9 convertase-mediated low density lipoprotein receptor degradation but interact with each other. J. Biol. Chem. 290, 18609–18620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pileri P., Uematsu Y., Campagnoli S., Galli G., Falugi F., Petracca R., Weiner A. J., Houghton M., Rosa D., Grandi G., Abrignani S. (1998) Binding of hepatitis C virus to CD81. Science 282, 938–941 [DOI] [PubMed] [Google Scholar]
- 55. Akazawa D., Date T., Morikawa K., Murayama A., Miyamoto M., Kaga M., Barth H., Baumert T. F., Dubuisson J., Wakita T. (2007) CD81 expression is important for the permissiveness of Huh7 cell clones for heterogeneous hepatitis C virus infection. J. Virol. 81, 5036–5045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Albecka A., Belouzard S., Op de Beeck A., Descamps V., Goueslain L., Bertrand-Michel J., Tercé F., Duverlie G., Rouillé Y., Dubuisson J. (2012) Role of low-density lipoprotein receptor in the hepatitis C virus life cycle. Hepatology 55, 998–1007 [DOI] [PubMed] [Google Scholar]
- 57. Fénéant L., Levy S., Cocquerel L. (2014) CD81 and hepatitis C virus (HCV) infection. Viruses 6, 535–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Harris H. J., Davis C., Mullins J. G., Hu K., Goodall M., Farquhar M. J., Mee C. J., McCaffrey K., Young S., Drummer H., Balfe P., McKeating J. A. (2010) Claudin association with CD81 defines hepatitis C virus entry. J. Biol. Chem. 285, 21092–21102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Evans M. J., von Hahn T., Tscherne D. M., Syder A. J., Panis M., Wölk B., Hatziioannou T., McKeating J. A., Bieniasz P. D., Rice C. M. (2007) Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446, 801–805 [DOI] [PubMed] [Google Scholar]
- 60. Seidah N. G., Poirier S., Denis M., Parker R., Miao B., Mapelli C., Prat A., Wassef H., Davignon J., Hajjar K. A., Mayer G. (2012) Annexin A2 is a natural extrahepatic inhibitor of the PCSK9-induced LDL receptor degradation. PLoS One 7, e41865. [DOI] [PMC free article] [PubMed] [Google Scholar]