

Background: Copper amine oxidases catalyze amine oxidation using copper and a quinone cofactor.
Results: Halides bind axially to the copper center, preventing the reduced cofactor from adopting an on-copper conformation.
Conclusion: The cofactor undergoes large conformational changes during the catalytic reaction that enable transitions between different types of chemistry.
Significance: Molecular details of cofactor movement have been unveiled based on structural and kinetic evidence.
Keywords: conformational change, electron transfer, oxidase, quinone, radical, x-ray crystallography, catalytic intermediate, catalytic mechanism, copper amine oxidase, topa quinone
Abstract
The catalytic reaction of copper amine oxidase proceeds through a ping-pong mechanism comprising two half-reactions. In the initial half-reaction, the substrate amine reduces the Tyr-derived cofactor, topa quinone (TPQ), to an aminoresorcinol form (TPQamr) that is in equilibrium with a semiquinone radical (TPQsq) via an intramolecular electron transfer to the active-site copper. We have analyzed this reductive half-reaction in crystals of the copper amine oxidase from Arthrobacter globiformis. Anerobic soaking of the crystals with an amine substrate shifted the equilibrium toward TPQsq in an “on-copper” conformation, in which the 4-OH group ligated axially to the copper center, which was probably reduced to Cu(I). When the crystals were soaked with substrate in the presence of halide ions, which act as uncompetitive and noncompetitive inhibitors with respect to the amine substrate and dioxygen, respectively, the equilibrium in the crystals shifted toward the “off-copper” conformation of TPQamr. The halide ion was bound to the axial position of the copper center, thereby preventing TPQamr from adopting the on-copper conformation. Furthermore, transient kinetic analyses in the presence of viscogen (glycerol) revealed that only the rate constant in the step of TPQamr/TPQsq interconversion is markedly affected by the viscogen, which probably perturbs the conformational change. These findings unequivocally demonstrate that TPQ undergoes large conformational changes during the reductive half-reaction.
Introduction
Copper amine oxidases (CAOs2; EC 1.4.3.6) catalyze the oxidative deamination of various primary amines to produce the corresponding aldehydes and ammonia, coupled with the reduction of molecular oxygen to hydrogen peroxide (1–3). CAOs play distinct physiological roles in prokaryotes and eukaryotes. Prokaryotic CAOs mainly function to assimilate primary amines as carbon and nitrogen sources for growth. Eukaryotic CAOs have versatile functions, being involved in the detoxification of bio-active amines, such as histamine (4); cell adhesion (5); cell death (6); collagen cross-linking in animals (7); and germination, root growth, and healing of wounded cell walls in plants (8). It has been reported that human serum CAOs cause angiopathy in diabetes (9). Therefore, various CAO inhibitors have been developed as therapeutic drugs (10, 11).
The catalytic center common to the CAO family consists of a mononuclear copper ion and a redox-active organic cofactor, topa quinone (TPQ), which is generated through posttranslational and self-catalytic processes (12–15). As summarized in a recent review (3), x-ray crystal structures of CAOs have been determined for a number of enzymes from various sources, including bacteria (Escherichia coli (16) and Arthrobacter globiformis (AGAO) (17–19)), yeasts (Hansenula polymorpha (recently reclassified as Pichia angusta) (HPAO-1 and HPAO-2) (20, 21) and Pichia pastoris (lysyl oxidase) (22)), a fungus (Aspergillus nidulans (23)), a plant (Pisum savitum (24)), a mammal (Bos taurus (25)), and Homo sapiens (diamine oxidase and vascular adhesion protein 1, which is identical to semicarbazide-sensitive amine oxidase (26–28)). The active site structures of these enzymes, including the positions of TPQ and Cu(II), are highly conserved, suggesting that they have a common mechanism for single-turnover TPQ biogenesis and catalytic amine oxidation. The latter process has been shown to proceed through a ping-pong mechanism consisting of two half-reactions (Scheme 1) (1–3).
SCHEME 1.

Presumed catalytic mechanism of AGAO.
In the initial reductive half-reaction, the C5 carbonyl group of the oxidized cofactor (TPQox) undergoes nucleophilic attack by amine substrate to form the substrate Schiff base (TPQssb). Stereospecific proton abstraction by a conserved base (Asp-298 in AGAO) (29) converts TPQssb to the product Schiff base (TPQpsb). Concomitantly with the release of the corresponding aldehyde, TPQpsb is hydrolyzed to the reduced cofactor (an aminoresorcinol form, TPQamr) that is in equilibrium with a semiquinone radical form (TPQsq) via the single electron transfer reduction of Cu(II) to Cu(I). In these steps of the reductive half-reaction, the cofactor ring is not directly ligated to the copper atom (a configuration designated the “off-copper” conformation), whereas in the TPQsq·Cu(I) state, the cofactor ring is assumed to be directed toward the metal in the so-called “on-copper” conformation to facilitate electron transfer. This suggests that the equilibrium between TPQamr·Cu(II) and TPQsq·Cu(I) is accompanied by a substantial conformational reorganization of the TPQ ring. Indeed, the conformational flexibility of TPQ within the active site of CAOs has previously been suggested to be important in catalysis (24, 30, 31). In the subsequent oxidative half-reaction, the reduced cofactor is reoxidized by dioxygen to produce hydrogen peroxide and an iminoquinone intermediate (TPQimq), which is further hydrolyzed to form the oxidized cofactor, releasing ammonia in the following step (1–3). In the presence of excess substrate, it is assumed that TPQimq reacts directly with the substrate amine to form TPQssb (via a trans-imination reaction) rather than regenerating TPQox (Scheme 1).
Depending on the enzyme sources and reaction conditions, such as pH and temperature, various amounts of TPQsq·Cu(I) are known to be formed by adding an amine substrate under anaerobic conditions, which is assumed to induce the reductive half-reaction. However, the mechanistic role of the TPQsq·Cu(I) form in the subsequent O2 reduction remains unclear and controversial (32–39). Two reaction pathways for the O2 reduction have been proposed, depending on the enzyme sources. One is an inner sphere mechanism in which O2 is coordinated on Cu(I) and reduced by the transfer of two electrons from TPQsq·Cu(I) to ultimately produce a Cu(II)-bound peroxide species and TPQimq (Scheme 1). The necessary singlet to triplet spin transition is allowed by the coordination of O2 to Cu(I). AGAO and P. savitum CAO are CAOs that have been suggested to follow this mechanism (37, 38). The formation of the TPQsq·Cu(I) state at the beginning of the oxidative half-reaction is believed to be essential in this process. The other mechanism that has been proposed is an outer sphere process that is suggested to occur in bovine serum CAO (34) and HPAO-1 (36, 39). In both of these enzymes, O2 binds to a hydrophobic pocket close to the cofactor and is initially reduced by TPQamr via a single electron transfer that does not change the oxidation state of the Cu(II) center. The resulting superoxide anion then coordinates to Cu(II), inducing the second electron transfer. The TPQsq·Cu(I) complex is not formed in this catalytic cycle and is assumed to be an off-pathway product generated only by the anaerobic reduction of the CAO by the amine substrate.
We have previously determined x-ray crystal structures of the intermediates, including TPQssb and TPQpsb, formed during the reductive half-reaction of AGAO with 2-phenylethylamine (2-PEA) (29), tyramine (40), and ethylamine (41). In each case, the TPQ cofactor in these structures had an off-copper conformation. Here, we present new high resolution structures of TPQsq formed in the reductive half-reaction of AGAO with 2-PEA and histamine. The TPQsq formed with these substrates exists exclusively in the on-copper conformation with the 4-OH group ligating axially to the copper center, which is probably in the Cu(I) oxidation state. Moreover, we found that the off-copper TPQamr is formed when the crystals are soaked with substrate in the presence of halide ions that act as uncompetitive inhibitors with respect to the amine substrate and noncompetitive inhibitors with respect to dioxygen. Halide ions bind axially to the copper center, preventing TPQamr from coordinating to copper. Combined with the results of spectrophotometric, steady-state, and transient kinetics analyses, the results presented herein provide unequivocal evidence for the occurrence of a large conformational change in the TPQ cofactor during the reductive half-reaction of AGAO.
Experimental Procedures
Materials
Recombinant AGAO was purified as its inactive precursor and converted to the copper- and TPQ-containing active form as reported previously (12, 19). Protein and TPQsq concentrations were determined spectrophotometrically using molar extinction coefficients of ϵ280 = 93,200 m−1 cm−1 (12) and ϵ468 = 4500 m−1 cm−1, respectively (35). All amine substrates used for kinetic analyses and crystal soaking were neutralized with 1 m H2SO4.
Spectrophotometric Measurements
To achieve fully anaerobic conditions, the enzyme and substrate solutions were kept in a vacuum-type glove box (Iuchi, SGV-65V) filled with 99.999% (v/v) argon gas for at least 2 h, as described previously (18). The enzyme (final concentration, 100 μm monomer) was anaerobically mixed with 1 mm 2-PEA in 50 mm HEPES, pH 6.8, in the presence or absence of various concentrations of sodium, potassium, or ammonium salts of halide ions. For measurements of pH dependence, the enzyme (100 μm monomer) was reduced with substrate in 100 mm MES (pH 5.7, 5.8, 6.0, 6.3, 6.5, and 6.7), 100 mm HEPES (pH 7.0, 7.3, 7.5, 7.8, and 8.0), 100 mm TAPS (pH 8.5 and 9.0), or 100 mm CHES (pH 9.5 and 10.1) at a nearly constant ionic strength (I = 0.35 ± 0.03) after adjustment with 100 mm Na2SO4. The enzyme was mixed with its substrate under anaerobic conditions in a quartz cuvette with a gas-tight screw cap, and after 5 min, the absorption spectrum was measured at 25 °C with an Agilent 8453 photodiode array spectrophotometer. An apparent pKa value for the absorbance change at 468 nm was determined by fitting the data to Equation 1,
where y represents the absorbance at a particular pH, C1 and C2 are the pH-independent values of the absorbance, and Ka1 and Ka2 are the acid dissociation constants associated with the pH profile. Data fitting was performed by nonlinear regression using Kaleidagraph version 4.1 (Abelbeck Software).
Steady-state Kinetic Analysis
Steady-state kinetic analyses were conducted at 30 °C with 2-PEA, histamine, or ethylamine (hydrochloride) as the substrate using the colorimetric assay protocol described previously (12). Inhibition by halide ions was studied using assay solutions containing 2–40 μm 2-PEA and 0–50 mm NaF, NaCl, NaBr, or NaI. Inhibition constants (Ki) were determined on the basis of uncompetitive inhibition (42) with respect to the amine substrate using Equation 2,
where v0 represents the initial reaction rate and [S] and [I] are the concentrations of substrate and inhibitor, respectively. Data fitting was performed by multiple regression analysis using R (available from the R Project Web site). Inhibition by halide ions was also studied using assay solutions containing 8.8–1160 μm dissolved oxygen and a fixed concentration (40 μm) of 2-PEA in the presence of 0–25 mm NaCl. Various concentrations of dissolved oxygen were established by mixing 99.99% (v/v) O2 and 99.99% (v/v) N2 gas with a gas mixer (KOFLOC, PMG-1) and bubbling the resulting mixture through a needle into the reaction mixture contained in a tightly sealed cuvette with a silicon rubber cap. The mixed gas line was also branched into the cell of a Clark-type oxygen electrode (YSI Inc., model 5300) for determination of the dissolved oxygen concentration. The reaction was initiated by adding a small amount of the anaerobic enzyme solution with a microsyringe. Inhibition constants (Ki) were determined on the basis of noncompetitive inhibition (42) with respect to dissolved oxygen using Equation 3,
where v0 represents the initial reaction rate, and [S] and [I] are the concentrations of dissolved oxygen and inhibitor, respectively. Data fitting was performed by multiple regression analysis using R.
The effect of solvent viscosity was investigated by measuring the enzyme's activity toward 2-PEA at 4 °C in a solution of 0–30% (w/v) glycerol or sucrose as a viscogen; the solvent's viscosity was determined relative to a buffer-only solution with an Ostwald viscometer.
Stopped-flow Measurements
Transient kinetic analyses were done at 4 °C with an Applied Photophysics stopped-flow spectrophotometer (40, 41). Typically, equal volumes (about 30 μl each) of enzyme (200 μm monomer in 50 mm HEPES buffer, pH 6.8) and substrate (1 mm 2-PEA) solutions were mixed in a 20-μl mixing cell by triggering with an N2 gas piston; the mixing dead time was generally 2.3 ms at an N2 gas pressure of 500 kilopascals. To avoid spectral changes associated with the oxidative half-reaction, both enzyme and substrate solutions were maintained under fully anaerobic conditions as described above. The substrate solution was supplemented with 200 mm NaCl, 600 mm NaBr, or 600 mm Na2SO4, as appropriate. The effect of solvent viscosity on transient kinetics was also studied as described above by adding a viscogen (0–30% (w/v) glycerol or sucrose) to both the enzyme and substrate solutions. UV-visible absorption spectra were recorded every 2.5 ms at wavelengths of 250–800 nm. Spectral data were analyzed using Pro-Kineticist II (Applied Photophysics) to obtain the spectra of the reaction intermediates and to calculate the rate constants for each reaction step.
Single-crystal Microspectrophotometry
AGAO crystals prepared as described below were subjected to microspectrophotometry before x-ray diffraction as reported previously (29).
X-ray Crystallographic Analysis
AGAO was crystallized by microdialysis essentially according to the method described previously (29). Briefly, a 15 mg/ml protein solution was dialyzed in a 50-μl dialysis button at 16 °C against 1.05 m potassium-sodium tartrate in 25 mm HEPES buffer, pH 6.8. After 2 weeks of crystal growth, the dialysis buttons were transferred into a fresh reservoir solution supplemented with 45% (v/v) glycerol as a cryoprotectant, and the crystals were soaked at 16 °C for 24 h, followed by further soaking in the fully anaerobic reservoir solution containing 45% (v/v) glycerol for 24 h. The crystals were then incubated in a solution (pH 6.8) containing 4 mm 2-PEA, 10 mm histamine, or 50 mm ethylamine (hydrochloride) with or without 100 mm NaCl or 300 mm NaBr for about 1 h until their color faded. They were then mounted on thin nylon loops (φ, 0.5–0.7 mm) and frozen by flash cooling in liquid CF4. All procedures were done in the anaerobic box, and the frozen crystals were kept in liquid N2 until x-ray diffraction analysis.
Diffraction data sets were collected at 100 K with synchrotron X-radiation using a DIP6040 imaging plate (Bruker AXS, Billerica, MA) in the BL44XU station or using a Quantum 210 CCD detector (ADSC) in the BL38B1 station at SPring-8 (Hyogo, Japan). X-rays with a wavelength of 0.919 Å were used to detect anomalous peaks derived from bromine; a wavelength of 0.9 Å was used otherwise. The collected data sets were processed and scaled using HKL2000 (43) or MOSFLM (44) and SCALA (45), respectively. The starting model was obtained by molecular replacement with Phaser (46). The search model was based on the coordinates of the AGAO monomer (Protein Data Bank code 1IU7) after removing all water molecules and a metal ion. Refinements, electron density map calculations, and assignment of solvent molecules were initially done using Refmac 5 (47) and later with Phenix (48). Manual rebuilding was performed using Coot (49), and water molecules and other ligands, such as metal ions, were added step by step to the model during the refinement process. The models of the catalytic intermediates of TPQ and phenylacetaldehyde were built using the Monomer library sketcher from the CCP4 package (50), and then the dictionary files used with Refmac 5, Phenix, and Coot were generated using the PRODRG server (51). PyMOL version 1.5 (Schrödinger, LLC) was used for figure drawings. Anomalous maps for bromine atoms were generated using fft (52) from the CCP4 package (50) based on anomalous difference and phase data for the final model. Details and statistics pertaining to the data collection and refinement are summarized in Table 1.
TABLE 1.
Data collection and crystallographic refinement statistics
| AGAOPEA | AGAOHTA | AGAOETA/HCl | AGAOPEA/NaBr | AGAOPEA/NaCl | AGAONaBr | |
|---|---|---|---|---|---|---|
| Soaking conditions | 4 mm 2-PEA, anaerobic | 10 mm histamine, anaerobic | 50 mm ethylamine-HCl, anaerobic | 4 mm 2-PEA, 300 mm NaBr, anaerobic | 4 mm 2-PEA, 100 mm NaCl, anaerobic | 300 mm NaBr, aerobic |
| Protein Data Bank code | 3X3X | 3X3Y | 3X3Z | 3X41 | 3X40 | 3X42 |
| Data collection | ||||||
| Temperature (K) | 100 | 100 | 100 | 100 | 100 | 100 |
| Wavelength (Å) | 0.9 | 0.9 | 0.9 | 0.919 | 0.9 | 0.919 |
| Space group | C2 | C2 | C2 | C2 | C2 | C2 |
| Unit-cell dimensions | ||||||
| a, b, c (Å) | 191.66, 62.89, 158.01 | 193.47, 63.25, 157.91 | 192.55, 62.73, 157.65 | 191.62, 63.29, 157.85 | 191.81, 63.02, 158.11 | 192.68, 63.51, 158.10 |
| β (degrees) | 117.48 | 117.73 | 117.62 | 117.21 | 117.29 | 117.55 |
| Resolution limit (Å) | 38.5–1.57 (1.65–1.57) | 50.0–1.50 (1.53–1.50) | 38.5–1.51 (1.59–1.51) | 47.3–1.87 (1.97–1.87) | 26.5–1.85 (1.95–1.85) | 100–1.89 (1.96–1.89) |
| No. of observations | 1,511,400 | 1,700,730 | 1,862,064 | 503,626 | 520,004 | 935,217 |
| No. of unique reflections | 228,324 | 264,870 | 260,515 | 137,175 | 142,096 | 267,101 |
| I/σ (I)a | 5.3 (1.9) | 26.1 (19.1) | 17.3 (4.0) | 13.0 (2.6) | 10.1 (3.5) | 6.6 (1.7) |
| Redundancya | 6.6 (6.2) | 6.4 (6.0) | 7.1 (6.9) | 3.7 (3.5) | 3.7 (3.8) | 3.5 (3.2) |
| Overall completeness (%)a | 98.1 (96.0) | 98.1 (97.4) | 99.8 (99.8) | 98.7 (97.2) | 99.2 (98.8) | 99.3 (98.7) |
| Overall Rmerge (%)a,b | 9.3 (39.2) | 8.8 (42.5) | 9.2 (40.4) | 9.1 (35.1) | 11.2 (38.2) | 5.6 (34.7) |
| Wilson B factor (Å2) | 13.85 | 13.84 | 11.87 | 16.52 | 16.85 | 19.81 |
| No. of molecules/asymmetric unit | 2 | 2 | 2 | 2 | 2 | 2 |
| Refinement statistics | ||||||
| Resolution limit (Å) | 20.4–1.57 (1.59–1.57) | 36.5–1.50 (1.52–1.50) | 22.3–1.51 (1.53–1.51) | 37.1–1.87 (1.89–1.87) | 26.2–1.85 (1.87–1.85) | 24.7–1.89 (1.90–1.88) |
| Residues in the core φψ region (%) | 96.4 | 96.7 | 96.3 | 96.4 | 96.2 | 96.5 |
| No. of atoms/asymmetric unit | 11,032 | 11,413 | 11,677 | 11,201 | 11,118 | 11,191 |
| No. of solvent atoms | 1170 | 1236 | 1527 | 1134 | 1239 | 1198 |
| Average temperature factors | ||||||
| Protein | 18.5 | 17.5 | 17.5 | 21.5 | 24.0 | 23.2 |
| Ligand/ion | 29.1 | 35.0 | 33.2 | 38.9 | 37.3 | 48.2 |
| Solvent | 29.2 | 29.2 | 31.8 | 34.7 | 33.3 | 34.5 |
| Root mean square deviation from ideal values | ||||||
| Bond lengths (Å) | 0.011 | 0.012 | 0.011 | 0.014 | 0.013 | 0.009 |
| Bond angles (degrees) | 1.049 | 1.078 | 1.153 | 1.112 | 1.077 | 1.083 |
| Rwork (%)a,c | 21.5 (26.2) | 16.2 (22.4) | 16.1 (20.3) | 15.2 (20.5) | 21.2 (28.1) | 15.8 (22.6) |
| Rfree (%)a,d | 24.3 (30.5) | 17.7 (25.7) | 17.8 (21.9) | 18.0 (23.7) | 26.4 (34.1) | 19.0 (27.9) |
| Ramachandran plot statistics (%) | ||||||
| Residues in favored regions | 96.4 | 96.7 | 96.3 | 96.3 | 96.1 | 96.6 |
| Residues in allowed regions | 3.6 | 3.3 | 3.5 | 3.7 | 3.7 | 3.4 |
| Outliers | 0 | 0 | 0.2 | 0 | 0.2 | 0 |
a Values in parentheses refer to data for the highest resolution shells.
b Rmerge = ΣhΣi|Ih,1 − 〈Ih〉|/ΣhΣi Ih,1, where Ih,1 is the intensity value of the ith measurement of h, and 〈Ih〉 is the corresponding mean value of Ih for all i measurements.
c Rwork = Σ‖Fo| − |Fc‖/Σ|Fo|.
d Rfree is an R factor of the refinement evaluated for 5% of reflections that were excluded from the refinement.
Results
Effect of Halide Ions on the Absorption Spectrum of TPQsq
It is well established that the absorption spectrum of the bound cofactor TPQ changes rapidly during the reductive half-reaction of CAOs (1–3), as demonstrated by stopped-flow spectrophotometry of the reaction with AGAO (29, 40, 53). The final form of TPQ in the reductive half-reaction without turnover (measured under anaerobic conditions) is TPQsq, which exhibits characteristic absorption maxima at about 440 and 470 nm with a shoulder at about 350 nm. We incidentally noted that the TPQsq spectrum of AGAO was markedly depressed in the presence of NaCl, which was added to the reaction mixture to maintain a constant ionic strength. For example, in the presence of 10 mm NaCl, the intensities of the absorption bands at 352, 438, and 468 nm decreased to about 32% of those without NaCl (Fig. 1). When calculated using the molar extinction coefficient at 468 nm (ϵ468 = 4500 m−1 cm−1) reported for TPQsq (35), the presence of NaCl reduced the calculated TPQ content relative to total AGAO from 38 to 9.8%. This was attributed to a shift of the equilibrium between TPQamr and TPQsq (see Scheme 1) toward the former, which has no absorption bands above 300 nm (54). To identify the chemical species that caused this equilibrium shift, we investigated the effects of adding 10 mm NH4Cl, KCl, Na2SO4, or NaH2PO4 in the reductive half-reaction and found that comparable shifts were induced by NH4Cl and KCl but not Na2SO4 or NaH2PO4 (Fig. 1), clearly indicating that the Cl− ion was preventing the formation of TPQsq. Other halide ions (F−, Br−, and I−) behaved similarly (Fig. 2), and their effects were roughly concentration-dependent, with an order of effectiveness of Cl− ≈ Br− > F− > I−. A similar effect (bleaching of TPQsq absorption) by a high concentration of Cl− (0.45 m KCl) was reported in an early study on methylamine oxidase from Arthrobacter P1 (55), but the mechanism of bleaching was not further pursued.
FIGURE 1.
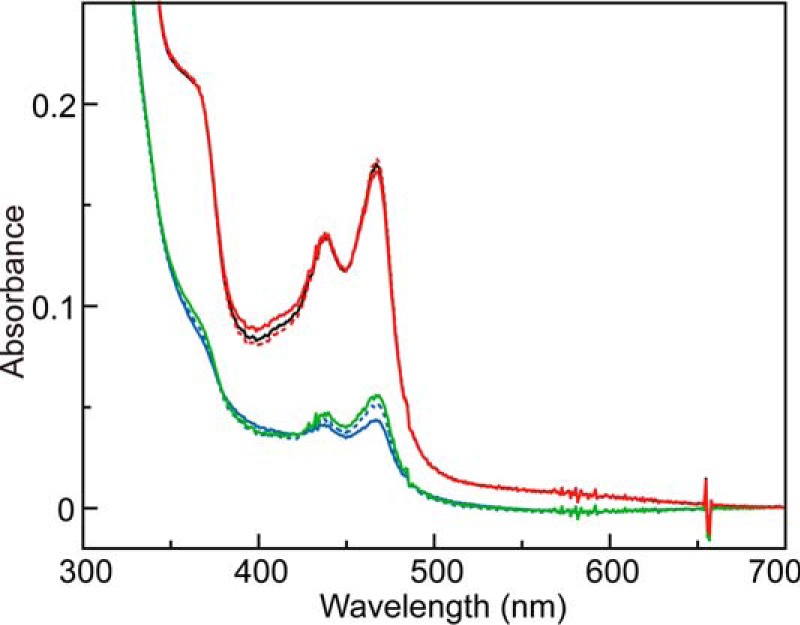
Effects of salts on TPQsq formation. UV-visual absorption spectra of TPQsq generated by anaerobic reaction with 2-PEA (1 mm) as the substrate were measured with 100 μm AGAO monomer in 100 mm HEPES, pH 6.8, in the absence (black solid line) and presence of various salts at a final concentration of 10 mm: NaCl (green solid line), NH4Cl (blue solid line), KCl (blue broken line), Na2SO4 (red broken line), or NaH2PO4 (red solid line).
FIGURE 2.
Effects of halide ion concentration on TPQsq formation. UV-visual absorption spectra of TPQsq (generated from 100 μm AGAO monomer and 1 mm 2-PEA) were measured in 100 mm HEPES, pH 6.8, containing 0 (black), 5 (blue), 10 (green), 30 (orange), and 50 mm (red) NaF (A), NaCl (B), NaBr (C), or NaI (D).
Effect of Halide Ions on Catalytic Activity
We have briefly reported the inhibition of AGAO activity by Cl− ion (56) in the past. In this work, more precise steady-state kinetic analyses were conducted by systematically varying the concentrations of the substrate amine (2-PEA) or dissolved oxygen in the presence of 0–50 mm solutions of different halide ions. Double reciprocal plots (1/v versus 1/s) revealed that the Cl− ion was an uncompetitive inhibitor with respect to the substrate amine and a noncompetitive inhibitor with respect to dissolved oxygen (Fig. 3); other halide ions (F−, Br−, and I−) exhibited similar inhibition patterns (data not shown). These results show that halide ions bind to a substrate·enzyme complex but not to the free enzyme and that they bind equally to the O2-bound and O2-unbound enzyme forms at a site distinct from the O2-binding site (41). The Ki values calculated for uncompetitive inhibition were 97.2 ± 6.5 mm for F−, 26.2 ± 1.4 mm for Cl−, 58.2 ± 2.8 mm for Br−, and 69.6 ± 9.4 mm for I−, whereas that for noncompetitive inhibition by Cl− was 32.8 ± 1.7 mm. The inhibition of catalytic activity by halide ions may be unique to AGAO; the Cl− ion reportedly has no inhibitory effect on the activities of human kidney diamine oxidase, P. pastoris amine oxidase, or P. savitum CAO (57).
FIGURE 3.

Steady-state kinetics analysis in the presence of NaCl. A, AGAO activity was measured at 30 °C in 50 mm HEPES (pH 6.8) in the presence of 0 (○), 10 (□), 30 (▵), or 50 (♢) mm NaCl with systematically varied 2-PEA concentrations. B, AGAO activity was measured in solutions containing 40 μm 2-PEA and 0 (○), 12.5 (□), 25 (▵), or 50 (♢) mm NaCl with systematically varied solvent O2 concentrations. Each point represents the mean ± S.E. (error bars) from two independent experiments.
Effects of Halide Ions on Transient Kinetics
To identify the substrate·enzyme complex to which the halides bind, we performed transient kinetic analyses of the reductive half-reaction in the presence of 100 mm NaCl, 300 mm NaBr, or 300 mm Na2SO4 (as a control for the ionic strength). As reported previously (29, 40, 53), rapid spectral changes associated with changes in the redox and chemical state of TPQ (TPQox → TPQssb → TPQpsb → TPQamr → TPQsq) were observed (Fig. 4). We noted that in the presence of 100 mm NaCl or 300 mm NaBr, the TPQsq absorption band appeared transiently within ∼117 ms and then gradually declined in intensity to about 30–40% of the value achieved in the absence of halide ions or in the presence of 300 mm Na2SO4 (Fig. 4, A and B). The final spectra (at 1023 ms) acquired in the presence of NaCl or NaBr (Fig. 4, C and D) mostly lacked the TPQsq-characteristic peaks, suggesting a shift of the equilibrium toward TPQamr, as discussed above (Fig. 2, B and C).
FIGURE 4.

Spectral changes during the reductive half-reaction with 2-PEA. UV-visible absorption spectra were recorded after mixing AGAO (100 μm monomer) with 2 mm 2-PEA (A), 2 mm 2-PEA plus 300 mm Na2SO4 (B), 2 mm 2-PEA plus 100 mm NaCl (C), or 2 mm 2-PEA plus 300 mm NaBr (D) in 50 mm HEPES buffer, pH 6.8, at 4 °C under anaerobic conditions. The spectra obtained at 0 (red), 2.30, 3.84, 6.4, 8.96, 14.1, 24.3, 44.8, 117, 209, 332, 600, and 1023 ms are shown using darker colors to represent later times. The arrows indicate the direction of the spectral changes. Deduced absorption spectra are shown for TPQox (red), TPQssb (yellow), TPQpsb (green), TPQamr (black), and TPQsq (blue) in E, F, G, and H, calculated from the measurements of A (no salt), B (300 mm Na2SO4), C (100 mm NaCl), and D (300 mm NaBr), respectively. The absorption spectra for TPQamr·X− (cyan) generated during the experiments using 100 mm NaCl (C), and 300 mm NaBr (D) are shown in G and H, although they are defined to be identical to that of TPQamr (black). The inset of A shows the time course of the absorbance at 468 nm in the spectral changes of A (no salt, red), B (300 mm Na2SO4, black), C (100 mm NaCl, blue), and D (300 mm NaBr, green).
Initially, the multiwavelength data of all spectral changes were fitted to the four-step mechanism connecting TPQox, TPQssb, TPQpsb, TPQamr, and TPQsq (Scheme 1) by global analysis as reported previously (29). The spectral changes in the presence of 300 mm Na2SO4 (Fig. 4B) were solved to provide rate constants that were essentially identical to those obtained without the salt (Fig. 4A) (Table 2), and the deduced UV-visible absorption spectra (Fig. 4, E and F) of TPQox, TPQssb, TPQpsb, TPQamr, and TPQsq were similar to those reported previously (29). However, the spectral changes observed in the presence of 100 mm NaCl or 300 mm NaBr could not be fitted to the four-step model. On the basis of the spectral changes and inhibition mechanism described above, it appeared that halide ions (X−) bound to the TPQamr state. We therefore proposed a branched model (with the branch connecting TPQox to TPQamr·X−; Scheme 1), in which the TPQamr·X− complex accumulates. The new model provided a reasonable solution to the data fitting of the spectral changes in the presence of NaCl or NaBr. As shown in Table 2, the rate constants of the steps between TPQox and TPQsq (k±1, k±2, k±3, and k±4) were comparable with those observed without halide ions, although the k−3 value was approximately halved. The rate constant of the branching step from TPQamr to TPQamr·X− (k+5) (Scheme 1) was estimated to be half that for TPQsq formation (k+4). The magnitude of these parameters well explains the slow accumulation of TPQamr·X−; TPQsq is formed initially but gradually converted to TPQamr·X− via TPQamr. The deduced absorption spectra of TPQox, TPQssb, TPQpsb, TPQamr, and TPQsq in the presence of NaCl (Fig. 4G) and NaBr (Fig. 4H) were essentially identical to those observed without halide ions (Fig. 4E) and in the presence of 300 mm Na2SO4 (Fig. 4F). Altogether, these results show that halide ions bind to the TPQamr form in the reductive half-reaction, thereby inhibiting the formation of TPQsq from TPQamr.
TABLE 2.
Kinetic constants for each step of the reductive half-reaction in the absence and presence of various salts and a viscogen at 4 °C
| No addition | +300 mm Na2SO4 | +100 mm NaCl | +300 mm NaBr | +30% (w/v) glycerol | |
|---|---|---|---|---|---|
| k+1 (s−1)a | 887 ± 0.2 | 880 ± 0.3 | 887 ± 0.6 | 880 ± 0.08 | 871 ± 0.03 |
| k−1 (s−1) | 531 ± 0.8 | 529 ± 0.05 | 530 ± 0.2 | 528 ± 2 | 560 ± 0.02 |
| k+2 (s−1) | 206 ± 0.04 | 207 ± 0.1 | 206 ± 0.1 | 207 ± 0.3 | 201 ± 0.006 |
| k−2 (s−1) | 34 ± 0.008 | 34 ± 0.02 | 34 ± 0.02 | 34 ± 0.07 | 34 ± 0.001 |
| k+3 (s−1) | 102 ± 0.06 | 102 ± 0.05 | 102 ± 0.06 | 103 ± 0.2 | 100 ± 0.003 |
| k−3 (s−1) | 127 ± 8 | 127 ± 5 | 63 ± 3 | 76 ± 7 | 134 ± 0.007 |
| k+4 (s−1) | 39 ± 9 | 39 ± 6 | 32 ± 6 | 23 ± 11 | 15 ± 0.001 |
| k−4 (s−1) | 17 ± 3 | 12 ± 2 | 15 ± 4 | 21 ± 6 | 6 ± 0.001 |
| k+5 (s−1)a | 15 ± 7 | 21 ± 12 | |||
| k−5 (s−1) | 5 ± 2 | 9 ± 3 |
a Determined as a first-order rate constant at constant and excess concentrations of amine substrate (2-PEA) and halide ions.
Effect of Solvent Viscosity on AGAO Activity
To further probe the equilibrium shift between TPQamr and TPQsq, we examined the dependence of catalytic activity on solvent viscosity, which can perturb diffusion-controlled steps, including substrate binding, product release, and conformational changes of the enzyme (58, 59). Glycerol (Mr = 92.1) and sucrose (Mr = 342.3) were used as viscogens. Steady-state kinetic analyses in viscogenic solutions, in which the relative solvent viscosity (η/η0, where η and η0 denote viscosities in the presence and absence of the viscogen, respectively) was increased by adding glycerol or sucrose, yielded values of kcat/Km for 2-PEA that were basically identical to that observed in non-viscogenic solution, whereas the kcat values decreased. The ratio of the viscogen-free kcat value to that in the presence of viscogen (kcat0/kcat, where kcat0 and kcat denote the rate constants in the absence and presence of the viscogen, respectively) was roughly proportional to the relative solvent viscosity (Fig. 5). This suggests that a diffusion-controlled step(s) is included in the overall reaction consisting of TPQimq, TPQssb, TPQpsb, TPQamr, TPQsq, and TPQimq under steady-state conditions (Scheme 1).
FIGURE 5.

Dependence of steady-state kinetic parameters on solvent viscosity. The ratios of kcat/Km (A) and kcat (B) in the presence and absence of viscogen at 4 °C were plotted against the relative solvent viscosity, η/η0, where zero denotes the value in the absence of viscogen. Black circles and squares, data points for glycerol and sucrose, respectively. A thin black line denotes the theoretical result for a completely diffusion-controlled process with a slope of 1.0. Error bars, S.D. (n = 2).
To evaluate the effect of solvent viscosity on each step of the reductive half-reaction, transient kinetic analyses were conducted (Fig. 6). Stopped-flow measurements in the absence and presence of 30% (w/v) glycerol generally produced similar spectral changes, but the formation of TPQsq was clearly slower in the viscogen's presence (Fig. 6, A and B, insets). Slow TPQsq formation was evident from the comparison of the traces of absorbance changes at 468 nm specific to TPQsq (Fig. 6C). Further, the rate constants of each step (k+1, k+2, k+3, k−1, k−2, and k−3) in the reductive half-reaction were determined by global analysis according to the four-step model starting from TPQox and ending at TPQsq (Scheme 1). Of these rate constants, k+1, k+2, k+3, k−1, k−2, and k−3 were independent of the glycerol concentration, but the values of k+4 and k−4 (which relate to the interconversion of TPQamr and TPQsq) decreased in proportion to the glycerol concentration (Table 2), showing that this step is diffusion-controlled. For detailed analysis, the ratios of k0/k were plotted against the relative solvent viscosity, η/η0 (Fig. 6D), where k0 and k are the rate constants (k+2, k−2, k+4, and k−4) in the absence and presence of viscogen (glycerol), respectively. The ratios of k+40/k+4 and k−40/k−4 increased significantly in proportion to the relative solvent viscosity, although the slope of the fitted line (0.50) indicated a partial effect; an entirely diffusion-controlled reaction would give a slope of 1.0 (60) (Fig. 6D). The ratios of the other rate constants were almost independent of the relative solvent viscosity (e.g. slope = 0.003 for k+20/k+2) (Fig. 6D). The reaction step connecting TPQamr and TPQsq involves neither substrate binding nor product release. Thus, these findings show that the interconversion of TPQamr and TPQsq is accompanied by a conformational change(s) of the enzyme. Reducing the viscogen's access to the region undergoing the conformational change could reduce its effective concentration, explaining the partial effect indicated by the k0/k slope of ∼0.5. It is therefore likely that the conformational change(s) occurs somewhere in the protein interior, such as the buried active site (most likely in the TPQ cofactor itself). Supporting this, we were unable to observe any effect of solvent viscosity on the rate constants k+4 and k−4 when using 30% (w/v) sucrose as the viscogen; sucrose's molecular size is 3.7-fold greater than that of glycerol, so it probably cannot penetrate into the active site cavity (Fig. 6D).
FIGURE 6.
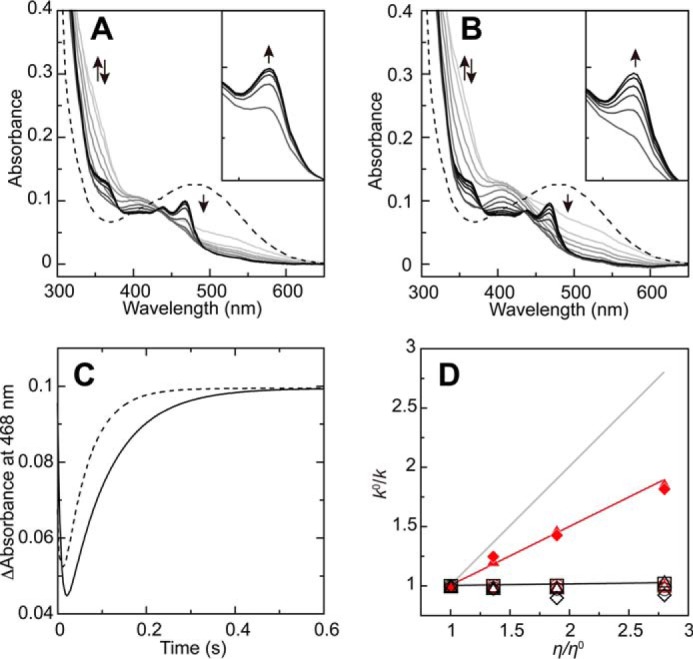
Effects of viscosity on the reductive half-reaction. UV-visible absorption spectra were recorded in 50 mm HEPES buffer, pH 6.8, at 4 °C under anaerobic conditions upon mixing AGAO (100 μm monomer) with 2 mm 2-PEA (A) or 2 mm 2-PEA (B) in the presence of 30% (w/v) glycerol. The spectra obtained at 0 (broken line), 2.30, 3.84, 6.4, 8.96, 14.1, 24.3, 44.8, 117, 209, 332, 600, and 1023 ms are shown with darker colors representing later times. The arrows indicate the direction of the spectral changes. The insets expand the 440–500 nm region for easier inspection of the spectral changes. C, absorbance at 468 nm was monitored during the spectral change of 2 mm 2-PEA (broken line) (A) or 2 mm 2-PEA (B) in the presence of 30% (w/v) glycerol (solid line) at 0–0.6 s. D, ratios of the rate constants (k+20/k+2 (red open circle), k−20/k−2 (red open triangle), k+40/k+4 (red triangle), and k−40/k−4 (red rhombus) determined in solutions of up to 30% (w/v) glycerol and k+20/k+2 (black open circle), k−20/k−2 (black open square), k+40/k+4 (black open triangle), and k−40/k−4 (black open rhombus) determined in solutions of up to 30% (w/v) sucrose) were plotted against the relative viscosity. S.D. values were less than 0.03% at all points (n = 2–4) and therefore are not shown with error bars. Thick lines (black and red) denote fitted linear lines for the k+40/k+4 values for sucrose and glycerol, respectively. The gray solid line denotes the theoretical result for a completely diffusion-controlled process with a slope of 1.0.
Reductive Half-reaction in Crystals
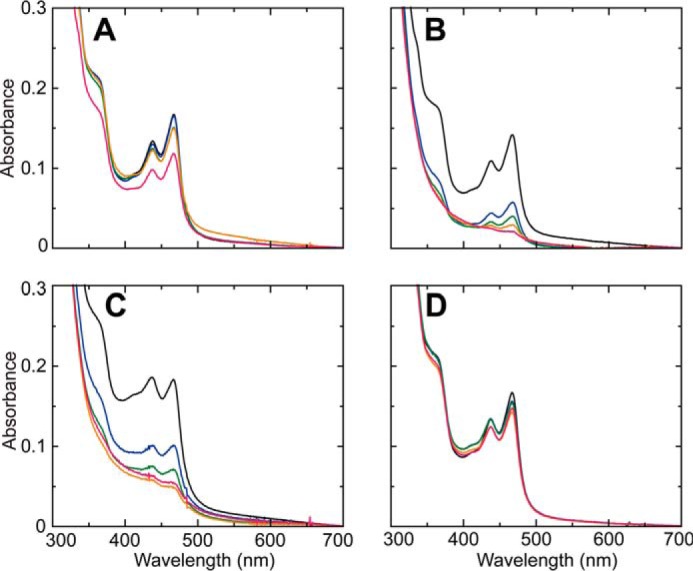
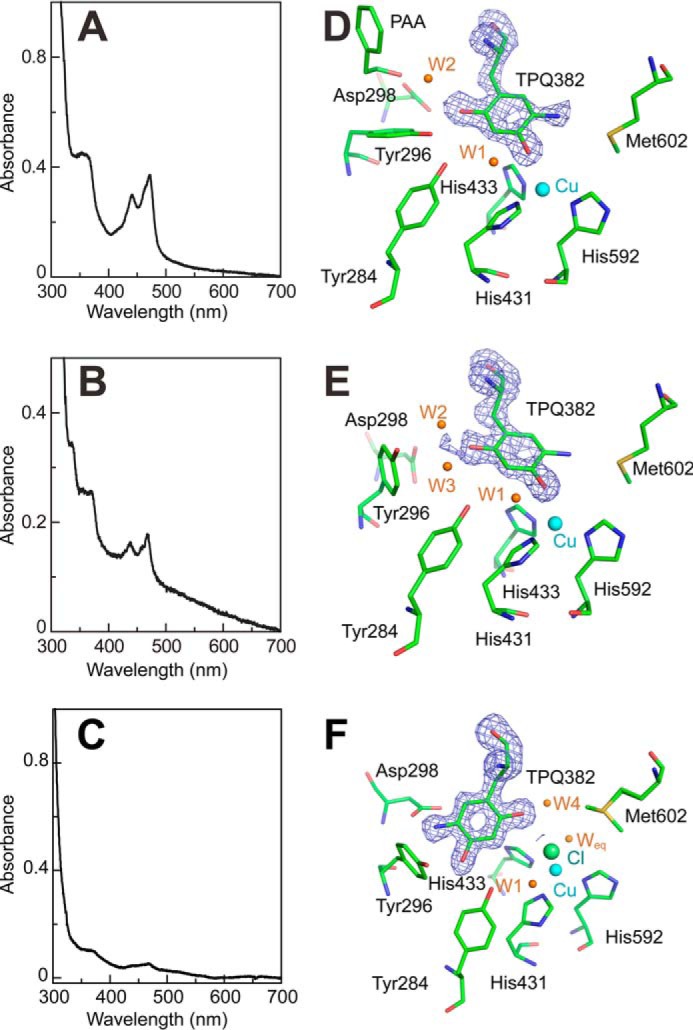
To obtain structural insights into the interconversion between TPQamr and TPQsq, the AGAO crystals were reacted anaerobically with substrates in the presence or absence of halide ions under six different conditions (Table 3), and the intermediates formed were freeze-trapped for structural determination. Before x-ray analysis, these crystals were subjected to single-crystal microspectrophotometry to identify the reaction intermediates present within them (29, 40, 41). The absorption spectra of the frozen crystals anaerobically soaked with 4 mm 2-PEA or 10 mm histamine (these crystals were designated AGAOPEA and AGAOHTA, respectively) (Fig. 7, A and B) were very similar to that of TPQsq for the enzyme in solution, showing specific absorption peaks around 365, 438, and 465 nm (Fig. 1). In contrast, the crystals anaerobically soaked with 50 mm ethylamine (hydrochloride) (AGAOETA/HCl) gave a rather peakless spectrum resembling that of TPQamr (Fig. 7C), although small TPQsq-like peaks remained. Furthermore, in the presence of 100 mm NaCl or 300 mm NaBr, the crystals prepared by anaerobic soaking with 4 mm 2-PEA (designated AGAOPEA/NaCl and AGAOPEA/NaBr, respectively) exhibited absorption spectra (Fig. 8, A and B) that were distinct from those of TPQsq and TPQamr but comparable with that of TPQpsb for the enzyme in solution (Fig. 4E). To identify the halide ion-binding site(s) in the unreacted enzyme, the crystal was aerobically soaked with 300 mm NaBr alone (AGAONaBr). This yielded a spectrum identical to that of TPQox because the reaction could not proceed (Fig. 8C). These crystal absorption spectra revealed that the reductive half-reaction occurred in all of the crystals except for AGAONaBr and that the accumulation of the TPQpsb, TPQamr, or TPQsq intermediates could be induced by soaking the crystals with appropriate amine substrates in the presence or absence of halides (Table 3).
TABLE 3.
Summary of crystal soaking conditions and active-site features determined by x-ray crystallographic analysis
| AGAOPEA | AGAOHTA | AGAOETA/HCla | AGAOPEA/NaCl | AGAOPEA/NaBr | AGAONaBr | |
|---|---|---|---|---|---|---|
| Soaking conditions | 2-PEA, anaerobic | Histamine, anaerobic | Ethylamine, anaerobic | 2-PEA, NaCl, anaerobic | 2-PEA, NaBr, anaerobic | NaBr, aerobic |
| Assigned TPQ species | TPQsq | TPQsq | TPQamr | TPQpsb | TPQpsb | TPQox |
| Conformation of TPQ | On copper | On copper | Off copper | Off copper | Off copper | Off copper |
| Copper coordination geometry | Tetrahedral | Tetrahedral | Square pyramidal | Square pyramidal | Square pyramidal | Square pyramidal |
| Presumed copper valence | Cu(I) | Cu(I) | Cu(II) | Cu(II) | Cu(II) | Cu(II) |
| Axial ligand of copper atom (distance, Å) | TPQsq C4-OH (2.8) | TPQsq C4-OH (2.7) | Cl− (2.5) | Cl− (2.3) | Br− (2.5) | Water (2.6) |
a Data for chain A.
FIGURE 7.
Catalytic intermediate structures and UV-visible absorption spectra of AGAO after reaction with 2-PEA, histamine, and ethylamine hydrochloride. AGAO crystals were anaerobically soaked with excess 2-PEA, histamine, or ethylamine hydrochloride, and the x-ray structures of the resulting crystals were determined. UV-visible absorption spectra for single crystals of AGAOPEA (A), AGAOHTA (B), and AGAOETA/HCl (C) were measured before x-ray exposure. The active-site structures of AGAOPEA (D), AGAOHTA (E), and AGAOETA/HCl (F) are shown superimposed on the Fo − Fc omit map (blue mesh) for residue 382 contoured at 3.5 σ. Active-site residues are represented by green stick models. Water molecules and copper centers are represented by brown and cyan spheres, respectively. All molecular drawings were generated using PyMOL.
FIGURE 8.
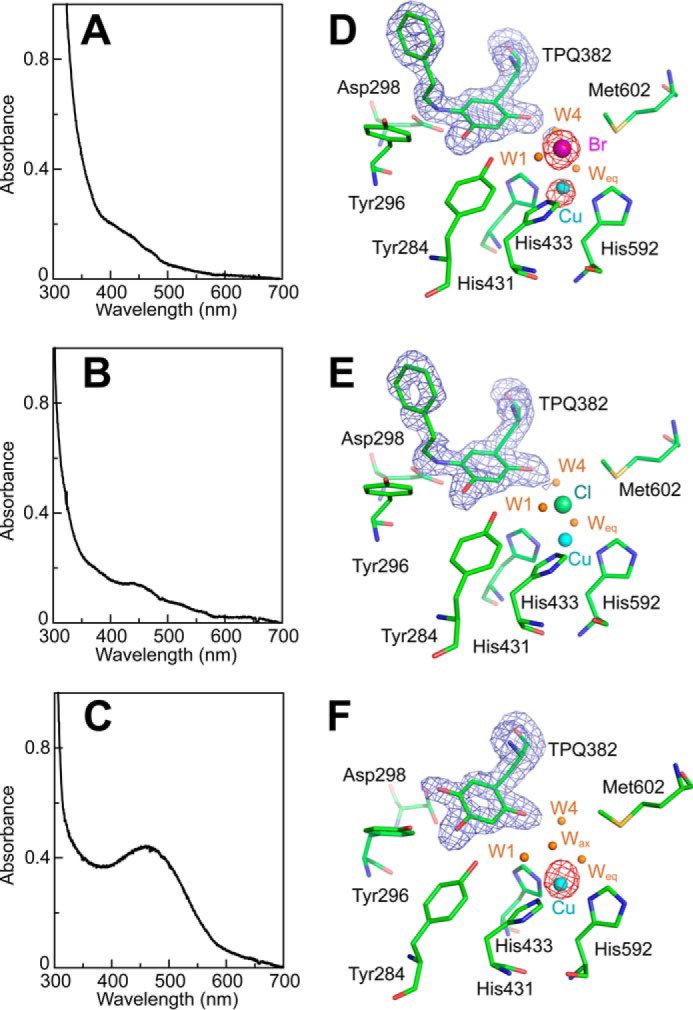
Active-site structures and UV-visible absorption spectra of AGAO soaked with halide salts and/or substrates. AGAO crystals were anaerobically soaked with excess 2-PEA in the presence of NaBr (A and D) or NaCl (B and E). C and F show results for AGAO crystals soaked only with NaBr under aerobic conditions. UV-visible absorption spectra for the single crystals of AGAOPEA/NaBr (A), AGAOPEA/NaCl (B), and AGAONaBr (C) were measured before x-ray exposure. The refined model of the active site structures of AGAOPEA/NaBr (D), AGAOPEA/NaCl (E), and AGAONaBr (F) are shown superimposed on the Fo − Fc omit map (blue mesh) for residue 382 contoured at 3.5 σ. Bromine-anomalous maps contoured at 8 σ are represented by red meshes in the active sites of AGAOPEA/NaBr (D) and AGAONaBr (F). The anomalous dispersion of the copper atom is pronounced although the wavelength of the used x-ray (0.919 Å) deviated from the peak wavelength of the copper atom (1.3808 Å), and an anomalous peak (about 18 σ) was detected on the copper site of the active center as well as for the bromine atom. Active-site residues are represented by green stick models. Water molecules and copper atoms are represented by brown and cyan spheres, respectively. All molecular drawings were generated using PyMOL.
The overall structures determined for AGAOPEA, AGAOHTA, AGAOETA/HCl, AGAOPEA/NaCl, AGAOPEA/NaBr, and AGAONaBr were comparable with that of resting AGAO (Protein Data Bank code 1IU7), with root mean square deviations for the main-chain atoms within ∼0.4–0.5 Å. In these crystal structures, we assigned the chemical structures of TPQ based on its absorption spectra (Table 3) and constructed the active-site structures using the assigned models of TPQ in different conformations, which were built to coincide with the Fo − Fc omit maps (Figs. 7 and 8). The two monomers (chains A and B) of the homodimer in the asymmetric unit of the crystals showed essentially identical active-site structures except for those in the AGAOETA/HCl crystal, in which active-site residues, including TPQ and water molecules, had slightly different conformations and electron densities between the two monomers.
The most notable finding from the crystal structures was that the TPQsq moieties in AGAOPEA and AGAOHTA had an on-copper conformation with ∼100% occupancy, with the 4-OH group of TPQsq projecting toward the copper center at a distance of 2.7–2.9 Å (Fig. 7, D and E) and the 5-NH2 group positioned opposite to the catalytic base (Asp-298) in close proximity to Met-602. This is the first x-ray structure of CAO with TPQsq being exclusively copper-ligating, although an on-copper TPQsq structure with ∼65–70% occupancy was recently reported for HPAO-1 crystals reduced with methylamine in a low oxygen environment at pH 8.5 (39). The copper center in AGAOPEA and AGAOHTA was tetrahedrally coordinated with the 4-OH group of TPQ at the “axial” position and the imidazole groups of three histidines (His-431, His-433, and His-592) at the “equatorial” positions. No water molecules were coordinated to the copper center. On the other hand, TPQamr in AGAOETA/HCl had an off-copper conformation with ∼100% occupancy, in which the 5-NH2 group of TPQamr was positioned close to Asp-298 and the 4-OH group was hydrogen-bonded to the Tyr-284 side chain rather than coordinating to the copper center (Fig. 7F). The off-copper conformation was the sole conformer in chain A. However, both the off-copper and on-copper conformers were present (at a relative abundance of about 6:4) in chain B. Probably, the minor on-copper conformation in chain B was partly responsible for the crystals' small TPQsq-like absorption peaks (Fig. 7C). The spectrophotometrically assigned TPQpsb moiety of AGAOPEA/NaCl and AGAOPEA/NaBr also had an off-copper conformation, in which the C5 position of TPQ was connected to additional electron density corresponding to the phenylethyl moiety of the product phenylacetaldehyde (PAA) via a covalent linkage in the form of an imine bond (Fig. 8, D and E).
The binding of several halide ions was detected in the AGAOETA/HCl, AGAOPEA/NaCl, AGAOPEA/NaBr, and AGAONaBr crystals (2 and 5 Cl− ions/dimer in AGAOETA/HCl and AGAOPEA/NaCl, respectively, and 8 Br− ions/dimer in AGAOPEA/NaBr and AGAONaBr), as judged by the anomalous peaks (over 7 σ) generated by the Br− ion and the electron densities (over 8 σ) of the Cl− ion, which were clearly greater than those of water molecules (less than 5 σ). The Cl− ions identified in AGAOETA/HCl derived from the substrate (ethylamine hydrochloride), which was present at a high concentration (50 mm). In the halide-bound complexes, a Br− or Cl− ion was found to occupy the axial coordination site of the active-site copper center in the AGAOETA/HCl, AGAOPEA/NaCl, and AGAOPEA/NaBr structures (Figs. 7F and 8 (D and E) and Table 3). These are the first x-ray crystal structures of CAO with an anionic inhibitor (57, 61, 62) bound to the active site copper center. In contrast, the axial position in AGAONaBr was occupied by a water molecule rather than Br− (Fig. 8F and Table 3); Br− ions instead bound to the protein surface in a seemingly nonspecific fashion. Similar behavior was observed in AGAOPEA/NaBr. Specific binding of a halide ion at the axial position of the active-site copper center only occurred in crystals that had been anaerobically soaked with substrate, strongly suggesting that the uncompetitive inhibition of the steady-state reaction by halide ions with respect to 2-PEA is due to their ability to bind to the copper center in the reaction intermediates, in which TPQ is reduced with substrates, rather than to the copper center in the free enzyme. It also suggests that the axial ligand-binding position of the copper center exhibits a stronger preference for halide ions in the substrate-reduced form of AGAO than in the resting form.
Comparison of the on-copper TPQsq structures of AGAOPEA and AGAOHTA with the off-copper TPQox structure in resting AGAO (Protein Data Bank code 1IU7) revealed that most active-site residues (except for Tyr-296 and Met-602), water molecules, and the copper center are retained in almost the same positions and conformations (Fig. 9). In the TPQsq structures, the side-chain phenol ring of Tyr-296 rotates ∼80° around the Cα–Cβ bond to participate in a hydrogen-bonding network involving two water molecules (W1 and W2); the 2-OH, 4-OH, and 5-NH2 groups of TPQsq; the 4-OH group of Tyr-284; and the Sδ atom of Met-602 (Fig. 9A). The on-copper conformation is probably stabilized by this hydrogen-bonding network, allowing the direct coordination of the 4-OH group of TPQ to the copper center, which would facilitate rapid, ligand-to-metal charge transfer-like electron transfer from TPQamr to Cu(II) to form the TPQsq·Cu(I) state.
FIGURE 9.

Interactions in the on-copper and off-copper conformations. The active sites of AGAOPEA (A) and the substrate-free and oxidative form of AGAO (Protein Data Bank code 1IU7) (B), in which the TPQ ring has on-copper and off-copper conformations, respectively, are drawn showing hydrogen bonds (dotted lines) and ligation to the copper centers (red lines). Estimated hydrogen bond lengths are shown in Å. The superposition of A and B is shown in C, in which the on-copper (A) and off-copper (B) structures are colored in purple and gray, respectively. All molecular drawings were generated using PyMOL.
In keeping with the single-crystal microspectrophotometric observations, a TPQpsb-like intermediate was identified in the AGAOPEA/NaBr and AGAOPEA/NaCl structures, which was probably formed by a condensation reaction between the amino group of TPQamr (in the off-copper form) and the aldehyde group of the reaction product PAA, which remains in the substrate-binding hydrophobic pocket (Fig. 8, D and E); PAA was indeed found to remain bound in the AGAOPEA structure (Fig. 7D), as reported previously for the 2-PEA-reduced E. coli CAO crystals (63). This assumption is also supported by the finding that the TPQpsb observed in AGAOPEA/NaBr and AGAOPEA/NaCl had a cis-configuration, whereas the TPQpsb formed during the reductive half-reaction of the D298A mutant of AGAO had a trans-configuration (29). The absence of the product aldehydes (imidazole-4-acetaldehyde and acetaldehyde) in the AGAOHTA and AGAOETA/HCl structures is probably due to their low affinities for the substrate-binding pocket of AGAO; for comparative purposes, the Km values for histamine (1.2 mm) and ethylamine (170 mm) are 220- and 30,000-fold higher, respectively, than that for 2-PEA (5.4 μm) (41). Overall, these findings indicate that in the AGAOPEA/NaBr and AGAOPEA/NaCl crystals, the binding of halide ions (Br− and Cl−) to the axial position of the copper center prevented TPQamr from adopting the on-copper conformation, causing it to back-react with PAA to form a TPQpsb configuration distinct from that formed during the reductive half-reaction.
Effect of pH on the Equilibrium between TPQamr and TPQsq
The equilibrium between TPQamr and TPQsq in AGAO has been reported to shift toward TPQsq under alkaline conditions (35), which suggests that an ionizable group(s) plays a role in triggering the conformational change of the cofactor. We therefore used spectrophotometry to investigate the effect of pH on the equilibrium between TPQamr and TPQsq. The reductive half-reaction was performed with 2-PEA as the substrate at pH values ranging from 5.7 to 10.1 with a constant ionic strength (Fig. 10A); AGAO is stable in this pH range. The TPQsq-specific absorption peaks at about 440 and 470 nm, and the shoulder at about 350 nm increased in intensity as the pH rose from 5.7 to 8.5 but did not change further above pH 8.5, indicating that the equilibrium shifted toward TPQsq above pH 8.5. By plotting the absorbance at 468 nm, to which TPQamr makes no contribution, against the pH (Fig. 10B) and fitting the data to Equation 1, two ionizable groups with apparent pKa values of 5.96 ± 0.05 (pKa1) and 7.74 ± 0.19 (pKa2) were found to be involved in the equilibrium shift from TPQamr to TPQsq. Judging from the magnitude of pH-independent absorbance values (C1 = 0.164, C2 = 0.053), deprotonation of the ionizable group with pKa = 5.96 contributes predominantly to the equilibrium shift.
FIGURE 10.

pH dependence of TPQsq formation. A, 100 μm AGAO monomer was anaerobically reduced with 1 mm 2-PEA at various pH values in the presence of 100 mm Na2SO4, and UV-visual absorption spectra of AGAO were measured at 25 °C after a 5-min preincubation. B, absorbance at 468 nm specific to TPQsq was plotted against pH. A solid line indicates the theoretical line obtained by data fitting. The spectra at pH 5.67, 6.02, 6.53, 6.99, 7.52, 8.03, 8.54, 9.04, 9.55, and 10.12 are shown with darker colors corresponding to higher pH values. Each point represents the mean ± S.E. (error bars) from 2–4 independent experiments.
Discussion
The results presented above demonstrate that the off-copper and on-copper conformations of TPQ are readily interconvertible during the reductive half-reaction with various amine substrates and in the presence of halide ions. In the steps prior to the formation of TPQamr (Scheme 1), the catalytic reaction proceeds with TPQ always maintained in the off-copper conformation irrespective of its chemical state (29, 40, 41). In the TPQssb and TPQpsb states, the distal part of the substrate amine is anchored to the substrate-binding pocket, preventing the TPQ ring from adopting the on-copper conformation (29). However, once TPQpsb is hydrolyzed, the reduced TPQ gains the conformational flexibility that enables facile interconversion between the off-copper and on-copper conformations.
In the on-copper conformation of TPQsq as observed in the AGAOPEA and AGAOHTA structures (Fig. 9, A and B), the active-site copper center is equatorially coordinated by three imidazole groups from His residues without the equatorial water ligand seen in the resting TPQox·Cu(II) state (17) and other intermediates formed in the reductive half-reaction (Figs. 7 and 8). A similar decrease in the number of equatorial ligands at the copper center was observed in extended x-ray absorption fine structure studies on various dithionite-treated CAOs, in which Cu(II) is reduced to Cu(I) (64). It is therefore suggested that the tetrahedrally coordinated copper centers observed in the AGAOPEA and AGAOHTA crystals are probably in the Cu(I) oxidation state, as was proposed in a recent paper on the structure of methylamine-reduced HPAO-1 (39). Based on the full occupancy of the modeled TPQsq in the electron density map, essentially all of their TPQ is assumed to be in the TPQsq state. Taken together, these results show that the on-copper conformation is a consequence of 1e− transfer from TPQamr to Cu(II) to form the TPQsq·Cu(I) state. In contrast, a water molecule (Weq) was identified as an equatorial ligand of the copper atom in the AGAOETA/HCl crystal, with observed electron densities of 3.7 and 2.3 σ in crystallographically distinguishable chains A and B, respectively. In addition, a Cl− ion was found to occupy an axial position in the copper complex (Fig. 7C), whose five-coordinate square pyramidal structure suggests that the copper is in the Cu(II) oxidation state, especially in chain A, for which TPQ is mostly in the off-copper TPQamr state based on its absorption spectrum and x-ray structure. We have so far observed neither the off-copper TPQsq·Cu(I) form nor the on-copper TPQamr·Cu(II) form, strongly suggesting that the 1e− transfer occurs exclusively in the on-copper conformation of TPQamr. This also leads to the suggestion that before the formation of the TPQsq·Cu(I) state, the TPQ quinone ring moves from the off-copper to the on-copper conformation.
The spectroscopic data from the transient kinetics experiments showed that the formation of TPQsq is dependent on solvent viscosity (Fig. 6D), further supporting the occurrence of a conformational change in TPQ during the last step of the reductive half-reaction. Because the rate constants of this step (k±4) were determined from the absorbance changes associated with TPQsq, they represent the rates of both the conformational change of TPQ and the subsequent electron transfer from TPQamr to Cu(II). The same presumption holds for the rate constants of the electron transfer (kET) determined previously by temperature-jump relaxation studies (33, 35, 65), which varied from 60–75 s−1 (Arthrobacter P1 methylamine oxidase) (65) to 20,000 s−1 (P. savitum CAO) (33). The latter extremely large rate constant was attributed to the intrinsic kET and interpreted to mean that the TPQ cofactor is in close proximity (∼3 Å) to the copper center (33) (cf. the distance of 2.6 Å between the TPQ 4-O atom and the copper center; Fig. 9). It is thus likely that the smaller rate constants determined previously (kET) and in this study (k+4) mainly reflect the rate constants for the conformational change of the TPQ cofactor, in agreement with the suggestion that conformationally “gated” or controlled electron transfer is plausible (33). An extended x-ray absorption fine structure study (64) also raised the possibility that variations in the redox potentials or the effective electron transfer distance between TPQamr and Cu(II) may control kET. The difference between the rate constants for AGAO determined here (Table 2; k+4 = 39 s−1 at 4 °C, pH 6.8) and previously (kET = 73 s−1 at 5 °C, pH 7.2) (37) is probably due to subtle differences in temperature and pH, both of which strongly affect the equilibrium between TPQamr·Cu(II) and TPQsq·Cu(I) (33, 35, 37, 39, 65) (Fig. 10); at higher pH values and temperatures, the equilibrium shifts toward TPQsq·Cu(I), which suggests a ΔH value of >0 for the conformational change of TPQ. Finally, it should be noted that the rate constants of the conformational change are appreciably larger than the kcat value (17 s−1) determined by steady-state kinetics under the same conditions (at 4 °C and pH 6.8), supporting the hypothesis that the conformational change of the cofactor can occur within the overall turnover reaction.
Based on the pH dependence of the equilibrium between TPQamr and TPQsq, it is suggested that deprotonation of two ionizable groups with pKa values of 5.96 ± 0.05 and 7.74 ± 0.19 (Fig. 10B) facilitates the equilibrium shift toward TPQsq. Among several ionizable groups in the active site (5-NH2 (estimated pKa, 5.88), 4-OH (9.59), and 2-OH (11.62) groups of TPQamr (54); the carboxyl group of Asp-298 (7.5 ± 0.20) (29); and the water axially coordinated to the copper atom (∼7.5) (66)), the 5-NH2 group of TPQamr is most likely assigned to the group with the lower pKa value (5.96) and mainly contributes to the equilibrium shift toward TPQsq, with the neutral form of TPQamr that is the major form at pH >7 being the direct precursor to TPQsq (Scheme 1). Furthermore, the neutral form of TPQamr is only weakly tethered in the active site, forming neither electrostatic interactions nor charge-assisted hydrogen bonds (67) with surrounding residues. It should thus be amenable to facile conformational change. The ionizable group with the higher pKa value (7.74), although contributing insignificantly to the equilibrium shift toward TPQsq, may be ascribed to the 4-OH group of TPQsq with a pKa value of 6.39 determined with a model compound (68), rather than the same group in TPQamr (pKa 9.59) (54); deprotonation of the 4-OH group of TPQsq is expected to stabilize the TPQsq form (see Scheme 1). If this is the case, deprotonation of the 4-OH group would occur after the conformational change of the TPQ ring and the electron transfer from TPQamr to Cu(II). A notable increase of the pKa value (from 6.39 to 7.74) may be conceivable to occur in the hydrophobic active site of CAOs, as observed for the carboxyl group of the catalytic base Asp-298 with a significantly high pKa value (7.5 ± 0.20) (29).
The TPQamr structures have been determined previously with the substrate-reduced forms of E. coli CAO and HPAO-1 (Protein Data Bank codes 1D6U and 4EV2, respectively), in which the axial positions of the copper atom are occupied by water and a dioxygen species, respectively. Comparison of these structures with the TPQamr of AGAO bound with a chloride ion (AGAOETA/HCl) has revealed that the TPQamr ring is tilted anticlockwise by about 20° (rigid body rotation around the Cβ–Cγ bond) in AGAO (Fig. 11A). This ∼20° tilting of the TPQ ring appears to result from the minuscule movement (by 0.4 Å) of the position of the 2-OH group of TPQamr, probably due to the repulsion from the axially coordinated chloride ion that has a larger van der Waals radius than water and a dioxygen species. Consequently, the 5-NH2 group approaches within hydrogen bond distance (2.8 Å) to the carboxyl group of Asp-298 (Fig. 11A), which is predominantly protonated at crystallization pH of 6.8, thereby lowering the nucleophilicity of the 5-NH2 group. The hydrogen bond may also stabilize the tilted conformation of TPQamr even after the halide ion is released (in the step of k−5 in Scheme 1). In addition, the aldehyde group of the product PAA that remains bound in AGAOPEA is located rather distant (3.2 Å) from the 5-NH2 group of TPQamr in the AGAOETA/HCl structure to undergo the nucleophilic attack (Fig. 11B). These structural consequences well explain the significantly decreased rate constant (k−3) for the back-formation of TPQpsb in the presence of halide ions (Table 2). Moreover, the geometry of the 5-NH2 group of TPQamr relative to the aldehyde carbon atom of PAA strongly suggests that the nucleophilic attack occurs from the si face of the carbonyl carbon, leading to the formation of TPQpsb in cis-configuration (cis-TPQpsb) (Fig. 11D), unlike the formation of TPQpsb in trans-configuration (trans-TPQpsb) from TPQssb in the forward reductive half-reaction of the D298A mutant (Fig. 11C) (29).
FIGURE 11.
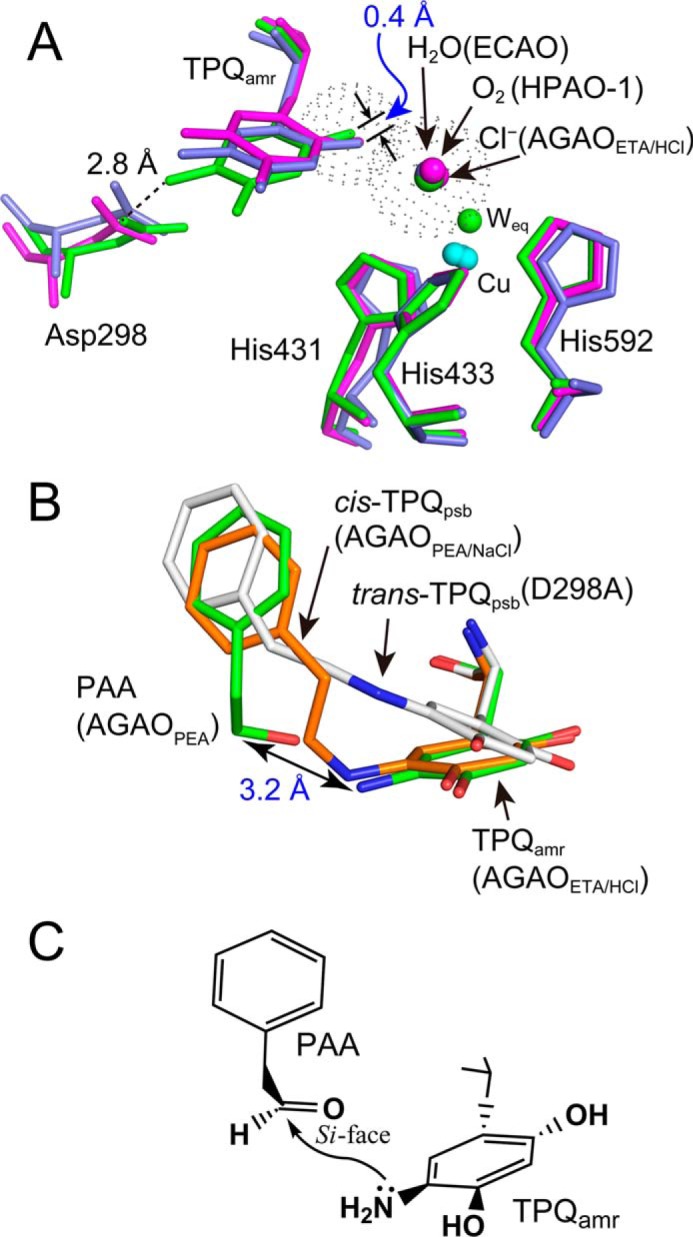
Effect of binding of chloride ion at the axial position of the copper center on the conformation of TPQamr. A, conformation of the TPQamr ring in the AGAOETA/HCl structure (green) is compared with those of the substrate-reduced E. coli CAO (ECAO) (purple) and HPAO-1 (magenta). Cyan spheres, copper atoms. Residue numbers are referred to those of AGAO. van der Waals surfaces of the chloride ion and the oxygen atom of the 2-OH group of TPQamr are represented with gray dots. B, comparison of cis-TPQpsb formed in AGAOPEA/NaCl (orange), cis-TPQpsb formed in the D298A mutant of AGAO (29) (gray), TPQamr formed in AGAOETA/HCl (green), and PAA formed in AGAOPEA (green). C, schematic drawing of the presumed mechanism of the formation of cis-TPQpsb in AGAOPEA/NaCl.
Monovalent anions, such as cyanide and azide, were reported to be inhibitors of various CAOs, showing competitive, noncompetitive, uncompetitive, or mixed type inhibition with respect to the amine substrate and dioxygen (57, 61, 62). For AGAO, cyanide is an uncompetitive inhibitor with respect to the amine substrate, and azide is a noncompetitive inhibitor with respect to both amine and dioxygen (57, 61). Although the inhibition patterns of halide ions (uncompetitive and noncompetitive with respect to amine substrate and dioxygen, respectively) (Fig. 3) for AGAO are similar to those of azide and cyanide, their effects on the equilibrium between TPQamr and TPQsq are very different. Halide ions inhibit TPQsq formation (Fig. 2) by axially coordinating to the copper center (Figs. 7F and 8 (D and E)), whereas azide converts TPQsq into a ligand-to-metal charge transfer complex (57), and cyanide facilitates TPQsq formation (61), both of which were suggested to bind at an equatorial position of the copper center (69, 70). It is assumed that azide is probably too large to bind axially to the copper atom and probably cyanide too. Although we cannot currently explain why halide ions, but not azide/cyanide, bind at the axial position of the copper center in the reduced form of TPQ and inhibit TPQsq formation, it is reasonable to assume that halide ion binding at the axial position of the copper atom would block the coordination of the 4-OH group of TPQamr to the copper center and prevent the subsequent electron transfer to Cu(II). The inability of halide ions to bind to the axial position of the copper center in the resting TPQox state (Fig. 8F) may be due to electrostatic repulsion from the delocalized negative charge through the 4-O− to 2-C=O group of TPQox; in the TPQamr state, both the 2-OH and 4-OH groups (whose estimated pKa values are 11.62 and 9.59, respectively) (54) are neutral and therefore would not electrostatically repel halide ions. The uncompetitive inhibition of halide ions with respect to the amine substrate is consistent with their binding only to the reaction intermediate, whereas noncompetitive inhibition with respect to dioxygen indicates halide binding at a site distinct from the O2-binding site. The exclusive binding of halide ions to Cu(II) with TPQamr in the off-copper conformation (which does not bind dioxygen) is challenging to reconcile with their activity as inhibitors of the oxidative half-reaction that are noncompetitive with respect to dioxygen. This issue can be resolved by supposing that dioxygen binds directly to Cu(I) with TPQsq in the on-copper conformation during the oxidative half-reaction and undergoes 1 e−-reduction by Cu(I) through the inner sphere mechanism proposed for AGAO (37, 38). Alternatively, halide ions may inhibit the oxidative half-reaction by binding to the O2-bound enzyme.
The off-copper to on-copper conformational change of TPQamr involves three motions of the TPQ ring: sliding (∼53° rotation around the Cα–Cβ bond), tilting up (∼20° rigid body rotation centered at the Cα carbon), and revolution (180° rotation around the Cβ–Cγ bond) (Fig. 12). In the off-copper conformation, the TPQ ring is sandwiched between the side chains of Asn-381 and Tyr-384/Val-282 in a narrow wedge-shaped space (29, 53). The initial step of the conformational change is a simultaneous combination of sliding and tilting up in order to avoid a steric clash between the TPQ ring and the side chain of His-433, which would come within ∼1 Å of the ring if it only slid. This combined sliding/tilting-up motion leads to the axial coordination of the 4-OH group of TPQamr to the copper atom, where there is sufficient space for the TPQ ring to rotate by 180° around the Cβ–Cγ bond. The final 180° rotation of the TPQ ring can only occur in the clockwise direction because anticlockwise rotation would lead to a clash between the 5-NH2 group and the His-433 side chain while permitting a minor movement of the Tyr-384 side chain (Fig. 12). It is unclear whether the electron transfer from TPQamr to Cu(II) occurs immediately upon formation of the on-copper conformation of the TPQ ring or after the 180° rotation to the final conformation stabilized by the hydrogen-bonding network (Fig. 9A).
FIGURE 12.
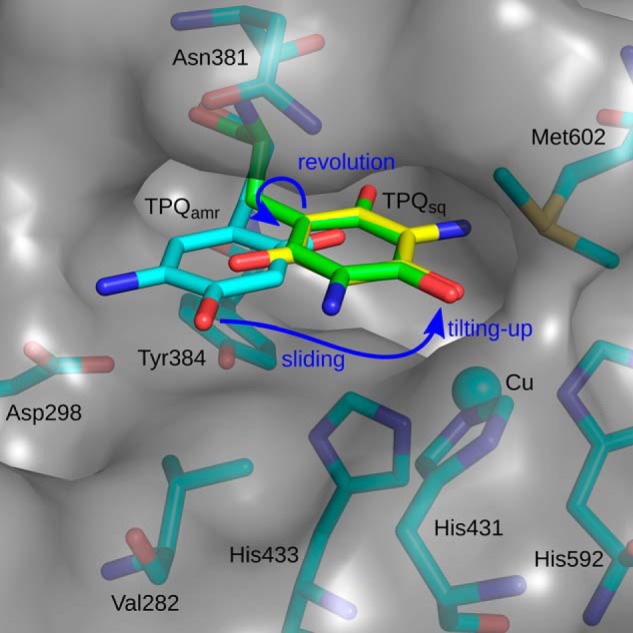
Possible route for the conformational change of TPQ in the active site of AGAO. Stick models of the active-site residues in AGAOETA/HCl (TPQamr, cyan) and AGAOPEA (TPQsq, yellow) are shown within the cavity with its surface drawn in half-transparent gray. The on-copper TPQamr conformer predicted after the first combined sliding/tilting up motion is colored green. The following 180° rotation of the TPQ ring provides a conformation identical with that of the on-copper TPQsq. The rotation direction and movement of the TPQ ring are shown with blue arrows. The figure was generated with PyMOL.
Finally, it is noteworthy that the on-copper TPQsq structure stabilizes the conformation of the side chain of Met-602 by hydrogen bond formation between the 5-NH2 group of TPQsq and the Sδ atom of Met-602 (Fig. 9). Met-602 is located at the end of the predicted O2 pathway from the O2-prebinding site to the copper center and has conformational flexibility with dual extreme conformers (19). Thus, it is tempting to speculate that the tethering of the Met-602 side chain could act as a gate to allow O2 to enter into the copper center in the initial phase of the oxidative half-reaction.
In conclusion, the results presented herein show that TPQ undergoes a large conformational change during the reductive half-reaction of AGAO, which efficiently mediates between the acid/base chemistry conducted in the off-copper conformation of TPQ by the conserved catalytic base (Asp-298 in AGAO) and the redox chemistry conducted in the on-copper conformation of TPQ at the metal center.
Author Contributions
T. M., A. H., S. N., T. N., H. Y., K. T., and T. O. participated in research design. T. M., A. H., S. N., M. K., Y. K., and T. O. conducted experiments. All authors performed data analysis and wrote or contributed to the writing of the manuscript.
Acknowledgments
This work was performed using synchrotron beamline stations BL44XU and BL38B1 at SPring-8 under the Cooperative Research Program of the Institute for Protein Research, Osaka University (Proposals 2007A6904, 2007B6904, 2008A6808, 2008B6808, 2009A6911, 2009B6911, 2013A6810, 2013B6810, 2014A6912, and 2014B6912) and with approval from the Japan Synchrotron Radiation Institute (Proposals 2009A1148, 2009B1106, 2010A1203, 2014A1144). The MX225HE CCD detector (Rayonix) at BL44XU was supported by Academia Sinica and the National Synchrotron Radiation Research Center (Taiwan).
This work was supported by Japan Society for the Promotion of Science Grants-in-Aid for Scientific Research 23770127 and 26440037 (to T. M.) and 18350085 and 15K07391 (to T. O.); Operational Program Education for Competitiveness-European Social Fund Project CZ.1.07/2.3.00/20.0165) (to K. T.); and by funding from the Network Joint Research Center for Materials and Devices. The authors declare that they have no conflicts of interest with the contents of this article.
- CAO
- copper amine oxidase
- AGAO and HPAO
- CAO from A. globiformis and H. polymorpha, respectively
- 2-PEA
- 2-phenylethylamine
- TPQ
- topa quinone
- TPQimq
- iminoquinone form of TPQ
- TPQpsb
- product Schiff base of TPQ
- TPQssb
- substrate Schiff base of TPQ
- TPQox
- oxidized form of TPQ
- TPQamr
- reduced (aminoresorcinol) form of TPQ
- TPQsq
- semiquinone radical form of TPQ
- TAPS
- 3-{[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]amino}-1-propanesulfonic acid
- CHES
- 2-(cyclohexylamino)ethanesulfonic acid
- PAA
- phenylacetaldehyde.
References
- 1. MacIntire W. S., Hartmann C. (1993) in Principles and Applications of Quinoproteins (Davidson V. L., ed) pp. 97–171, Marcel Dekker, New York [Google Scholar]
- 2. Floris G., Mondovi B. (eds) (2009) Copper Amine Oxidases: Structures, Catalytic Mechanisms and Role in Pathophysiology, CRC Press, Inc., Boca Raton, FL [Google Scholar]
- 3. Klema V. J., Wilmot C. M. (2012) The role of protein crystallography in defining the mechanisms of biogenesis and catalysis in copper amine oxidase. Int. J. Mol. Sci. 13, 5375–5405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Maintz L., Novak N. (2007) Histamine and histamine intolerance. Am. J. Clin. Nutr. 85, 1185–1196 [DOI] [PubMed] [Google Scholar]
- 5. Jalkanen S., Karikoski M., Mercier N., Koskinen K., Henttinen T., Elima K., Salmivirta K., Salmi M. (2007) The oxidase activity of vascular adhesion protein-1 (VAP-1) induces endothelial E- and P-selectins and leukocyte binding. Blood 110, 1864–1870 [DOI] [PubMed] [Google Scholar]
- 6. Hernandez M., Solé M., Boada M., Unzeta M. (2006) Soluble semicarbazide sensitive amine oxidase (SSAO) catalysis induces apoptosis in vascular smooth muscle cells. Biochim. Biophys. Acta 1763, 164–173 [DOI] [PubMed] [Google Scholar]
- 7. Lucero H. A., Kagan H. M. (2006) Lysyl oxidase: an oxidative enzyme and effector of cell function. Cell Mol. Life Sci. 63, 2304–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cona A., Rea G., Angelini R., Federico R., Tavladoraki P. (2006) Functions of amine oxidases in plant development and defense. Trends Plant Sci. 11, 80–88 [DOI] [PubMed] [Google Scholar]
- 9. Mészáros Z., Karádi I., Csányi A., Szombathy T., Romics L., Magyar K. (1999) Determination of human serum semicarbazide-sensitive amine oxidase activity: a possible clinical marker of atherosclerosis. Eur. J. Drug Metab. Pharmacokinet. 24, 299–302 [DOI] [PubMed] [Google Scholar]
- 10. Yu P. H., Davis B. A., Deng Y. (2001) 2-Bromoethylamine as a potent selective suicide inhibitor for semicarbazide-sensitive amine oxidase. Biochem. Pharmacol. 61, 741–748 [DOI] [PubMed] [Google Scholar]
- 11. Inoue T., Morita M., Tojo T., Nagashima A., Moritomo A., Miyake H. (2013) Novel 1H-imidazol-2-amine derivatives as potent and orally active vascular adhesion protein-1 (VAP-1) inhibitors for diabetic macular edema treatment. Bioorg. Med. Chem. 21, 3873–3881 [DOI] [PubMed] [Google Scholar]
- 12. Matsuzaki R., Fukui T., Sato H., Ozaki Y., Tanizawa K. (1994) Generation of the topa quinone cofactor in bacterial monoamine oxidase by cupric ion-dependent autooxidation of a specific tyrosyl residue. FEBS Lett. 351, 360–364 [DOI] [PubMed] [Google Scholar]
- 13. Klinman J. P., Mu D. (1994) Quinoenzymes in biology. Annu. Rev. Biochem. 63, 299–344 [DOI] [PubMed] [Google Scholar]
- 14. Choi Y. H., Matsuzaki R., Fukui T., Shimizu E., Yorifuji T., Sato H., Ozaki Y., Tanizawa K. (1995) Copper/topa quinone-containing histamine oxidase from Arthrobacter globiformis. Molecular cloning and sequencing, overproduction of precursor enzyme, and generation of topa quinone cofactor. J. Biol. Chem. 270, 4712–4720 [DOI] [PubMed] [Google Scholar]
- 15. Okajima T., Tanizawa K. (2009) Mechanism of TPQ biogenesis in prokaryotic copper amine oxidase in Copper Amine Oxidases: Structures, Catalytic Mechanisms and Role in Pathophysiology (Floris G., Mondovi B., eds) pp. 103–118, CRC Press, Inc., Boca Raton, FL [Google Scholar]
- 16. Parsons M. R., Convery M. A., Wilmot C. M., Yadav K. D., Blakeley V., Corner A. S., Phillips S. E., McPherson M. J., Knowles P. F. (1995) Crystal structure of a quinoenzyme: copper amine oxidase of Escherichia coli at 2 Å resolution. Structure 3, 1171–1184 [DOI] [PubMed] [Google Scholar]
- 17. Wilce M. C. J., Dooley D. M., Freeman H. C., Guss J. M., Matsunami H., McIntire W. S., Ruggiero C. E., Tanizawa K., Yamaguchi H. (1997) Crystal structures of the copper-containing amine oxidase from Arthrobacter globiformis in the holo and apo forms: implications for the biogenesis of topaquinone. Biochemistry 36, 16116–16133 [DOI] [PubMed] [Google Scholar]
- 18. Kishishita S., Okajima T., Kim M., Yamaguchi H., Hirota S., Suzuki S., Kuroda S., Tanizawa K., Mure M. (2003) Role of copper ion in bacterial copper amine oxidase: spectroscopic and crystallographic studies of metal-substituted enzymes. J. Am. Chem. Soc. 125, 1041–1055 [DOI] [PubMed] [Google Scholar]
- 19. Murakawa T., Hayashi H., Sunami T., Kurihara K., Tamada T., Kuroki R., Suzuki M., Tanizawa K., Okajima T. (2013) High-resolution crystal structure of copper amine oxidase from Arthrobacter globiformis: assignment of bound diatomic molecules as O2. Acta Crystallogr. D Biol. Crystallogr. 69, 2483–2494 [DOI] [PubMed] [Google Scholar]
- 20. Li R., Klinman J. P., Mathews F. S. (1998) Copper amine oxidase from Hansenula polymorpha: the crystal structure determined at 2.4 Å resolution reveals the active conformation. Structure 6, 293–307 [DOI] [PubMed] [Google Scholar]
- 21. Chang C. M., Klema V. J., Johnson B. J., Mure M., Klinman J. P., Wilmot C. M. (2010) Kinetic and structural analysis of substrate specificity in two copper amine oxidases from Hansenula polymorpha. Biochemistry 49, 2540–2550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Duff A. P., Cohen A. E., Ellis P. J., Kuchar J. A., Langley D. B., Shepard E. M., Dooley D. M., Freeman H. C., Guss J. M. (2003) The crystal structure of Pichia pastoris lysyl oxidase. Biochemistry 42, 15148–15157 [DOI] [PubMed] [Google Scholar]
- 23. McGrath A. P., Mithieux S. M., Collyer C. A., Bakhuis J. G., van den Berg M., Sein A., Heinz A., Schmelzer C., Weiss A. S., Guss J. M. (2011) Structure and activity of Aspergillus nidulans copper amine oxidase. Biochemistry 50, 5718–5730 [DOI] [PubMed] [Google Scholar]
- 24. Kumar V., Dooley D. M., Freeman H. C., Guss J. M., Harvey I., McGuirl M. A., Wilce M. C., Zubak V. M. (1996) Crystal structure of a eukaryotic (pea seedling) copper-containing amine oxidase at 2.2 Å resolution. Structure 4, 943–955 [DOI] [PubMed] [Google Scholar]
- 25. Lunelli M., Di Paolo M. L., Biadene M., Calderone V., Battistutta R., Scarpa M., Rigo A., Zanotti G. (2005) Crystal structure of amine oxidase from bovine serum. J. Mol. Biol. 346, 991–1004 [DOI] [PubMed] [Google Scholar]
- 26. McGrath A. P., Hilmer K. M., Collyer C. A., Shepard E. M., Elmore B. O., Brown D. E., Dooley D. M., Guss J. M. (2009) Structure and inhibition of human diamine oxidase. Biochemistry 48, 9810–9822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Airenne T. T., Nymalm Y., Kidron H., Smith D. J., Pihlavisto M., Salmi M., Jalkanen S., Johnson M. S., Salminen T. A. (2005) Crystal structure of the human vascular adhesion protein-1: unique structural features with functional implications. Protein Sci. 14, 1964–1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jakobsson E., Nilsson J., Ogg D., Kleywegt G. J. (2005) Structure of human semicarbazide-sensitive amine oxidase/vascular adhesion protein-1. Acta Crystallogr. D Biol. Crystallogr. 61, 1550–1562 [DOI] [PubMed] [Google Scholar]
- 29. Chiu Y. C., Okajima T., Murakawa T., Uchida M., Taki M., Hirota S., Kim M., Yamaguchi H., Kawano Y., Kamiya N., Kuroda S., Hayashi H., Yamamoto Y., Tanizawa K. (2006) Kinetic and structural studies on the catalytic role of the aspartic acid residue conserved in copper amine oxidase. Biochemistry 45, 4105–4120 [DOI] [PubMed] [Google Scholar]
- 30. Plastino J., Green E. L., Sanders-Loehr J., Klinman J. P. (1999) An unexpected role for the active site base in cofactor orientation and flexibility in the copper amine oxidase from Hansenula polymorpha. Biochemistry 38, 8204–8216 [DOI] [PubMed] [Google Scholar]
- 31. Murray J. M., Saysell C. G., Wilmot C. M., Tambyrajah W. S., Jaeger J., Knowles P. F., Phillips S. E., McPherson M. J. (1999) The active site base controls cofactor reactivity in Escherichia coli amine oxidase: x-ray crystallographic studies with mutational variants. Biochemistry 38, 8217–8227 [DOI] [PubMed] [Google Scholar]
- 32. Dooley D. M., McGuirl M. A., Brown D. E., Turowski P. N., McIntire W. S., Knowles P. F. (1991) A Cu(I)-semiquinone state in substrate-reduced amine oxidases. Nature 349, 262–264 [DOI] [PubMed] [Google Scholar]
- 33. Turowski P. N., McGuirl M. A., Dooley D. M. (1993) Intramolecular electron transfer rate between active-site copper and topa quinone in pea seedling amine oxidase. J. Biol. Chem. 268, 17680–17682 [PubMed] [Google Scholar]
- 34. Su Q., Klinman J. P. (1998) Probing the mechanism of proton coupled electron transfer to dioxygen: the oxidative half-reaction of bovine serum amine oxidase. Biochemistry 37, 12513–12525 [DOI] [PubMed] [Google Scholar]
- 35. Shepard E. M., Dooley D. M. (2006) Intramolecular electron transfer rate between active-site copper and TPQ in Arthrobacter globiformis amine oxidase. J. Biol. Inorg. Chem. 11, 1039–1048 [DOI] [PubMed] [Google Scholar]
- 36. Welford R. W., Lam A., Mirica L. M., Klinman J. P. (2007) Partial conversion of Hansenula polymorpha amine oxidase into a “plant” amine oxidase: implications for copper chemistry and mechanism. Biochemistry 46, 10817–10827 [DOI] [PubMed] [Google Scholar]
- 37. Shepard E. M., Okonski K. M., Dooley D. M. (2008) Kinetics and spectroscopic evidence that the Cu(I)-semiquinone intermediate reduces molecular oxygen in the oxidative half-reaction of Arthrobacter globiformis amine oxidase. Biochemistry 47, 13907–13920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mukherjee A., Smirnov V. V., Lanci M. P., Brown D. E., Shepard E. M., Dooley D. M., Roth J. P. (2008) Inner-sphere mechanism for molecular oxygen reduction catalyzed by copper amine oxidases. J. Am. Chem. Soc. 130, 9459–9473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Johnson B. J., Yukl E. T., Klema V. J., Klinman J. P., Wilmot C. M. (2013) Structural snapshots from the oxidative half-reaction of a copper amine oxidase: implications for O2 activation. J. Biol. Chem. 288, 28409–28417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Murakawa T., Okajima T., Kuroda S., Nakamoto T., Taki M., Yamamoto Y., Hayashi H., Tanizawa K. (2006) Quantum mechanical hydrogen tunneling in bacterial copper amine oxidase reaction. Biochem. Biophys. Res. Commun. 342, 414–423 [DOI] [PubMed] [Google Scholar]
- 41. Taki M., Murakawa T., Nakamoto T., Uchida M., Hayashi H., Tanizawa K., Yamamoto Y., Okajima T. (2008) Further insight into the mechanism of stereoselective proton abstraction by bacterial copper amine oxidase. Biochemistry 47, 7726–7733 [DOI] [PubMed] [Google Scholar]
- 42. Fersht A. (1998) in Structure and Mechanism in Protein Science, pp. 103–131, W. H. Freeman and Co., New York [Google Scholar]
- 43. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 44. Leslie A. G. W. (1992) Joint CCP4 and EESF-EACMB Newsletter on Protein Crystallography, SERC Daresbury Laboratory, Warrington, UK [Google Scholar]
- 45. Collaborative Computational Project, Number 4 (1994) The CCP 4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 46. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 48. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G. W., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schüttelkopf A. W., van Aalten D. M. F. (2004) PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 60, 1355–1363 [DOI] [PubMed] [Google Scholar]
- 52. Ten Eyck L. F. (1973) Crystallographic fast Fourier transforms. Acta Crystallogr. A Found. Adv. 29, 183–191 [Google Scholar]
- 53. Murakawa T., Hayashi H., Taki M., Yamamoto Y., Kawano Y., Tanizawa K., Okajima T. (2012) Structural insights into the substrate specificity of bacterial copper amine oxidase obtained by using irreversible inhibitors. J. Biochem. 151, 167–178 [DOI] [PubMed] [Google Scholar]
- 54. Mure M., Klinman J. P. (1993) Synthesis and spectroscopic characterization of model compounds for the active site cofactor in copper amine oxidase. J. Am. Chem. Soc. 115, 7117–7127 [Google Scholar]
- 55. Dooley D. M., McIntire W. S., McCuirl M. A., Cote C. E., Bates J. L. (1990) Characterization of the active site of Arthrobacter P1 methylamine oxidase: evidence for copper-quinone interactions. J. Am. Chem. Soc. 112, 2782–2789 [Google Scholar]
- 56. Shimizu E., Ohta K., Takayama S., Kitagaki Y., Tanizawa K., Yorifuji T. (1997) Purification and properties of phenylethylamine oxidase of Arthrobacter globiformis. Biosci. Biotechnol. Biochem. 61, 501–505 [Google Scholar]
- 57. Juda G. A., Shepard E. M., Elmore B. O., Dooley D. M. (2006) A comparative study of the binding and inhibition of four copper-containing amine oxidases by azide: implications for the role of copper during the oxidative half-reaction. Biochemistry 45, 8788–8800 [DOI] [PubMed] [Google Scholar]
- 58. Nakatani H., Dunford H. B. (1979) Meaning of diffusion-controlled association rate constants in enzymology. J. Phys. Chem. 83, 2662–2665 [Google Scholar]
- 59. Masson P., Lushchekina S., Schopfer L. M., Lockridge O. (2013) Effects of viscosity and osmotic stress on the reaction of human butyrylcholinesterase with cresyl saligenin phosphate, a toxicant related to aerotoxic syndrome: kinetic and molecular dynamics studies. Biochem. J. 454, 387–399 [DOI] [PubMed] [Google Scholar]
- 60. Hardy L. W., Kirsch J. F. (1984) Diffusion-limited component of reactions catalyzed by Bacillus cereus β-lactamase I. Biochemistry 23, 1275–1282 [DOI] [PubMed] [Google Scholar]
- 61. Shepard E. M., Juda G. A., Ling K. Q., Sayre L. M., Dooley D. M. (2004) Cyanide as a copper and quinone-directed inhibitor of amine oxidases from pea seedlings (Pisum sativum) and Arthrobacter globiformis: evidence for both copper coordination and cyanohydrin derivatization of the quinone cofactor. J. Biol. Inorg. Chem. 9, 256–268 [DOI] [PubMed] [Google Scholar]
- 62. Schwartz B., Olgin A. K., Klinman J. P. (2001) The role of copper in topa quinone biogenesis and catalysis, as probed by azide inhibition of a copper amine oxidase from yeast. Biochemistry 40, 2954–2963 [DOI] [PubMed] [Google Scholar]
- 63. Wilmot C. M., Hajdu J., McPherson M. J., Knowles P. F., Phillips S. E. (1999) Visualization of dioxygen bound to copper during enzyme catalysis. Science 286, 1724–1728 [DOI] [PubMed] [Google Scholar]
- 64. Dooley D. M., Scott R. A., Knowles P. F., Colangelo C. M., McGuirl M. A., Brown D. E. (1998) Structures of the Cu(I) and Cu(II) forms of amine oxidases from x-ray absorption spectroscopy. J. Am. Chem. Soc. 120, 2599–2605 [Google Scholar]
- 65. Dooley D. M., Brown D. E. (1996) Intramolecular electron transfer in the oxidation of amines by methylamine oxidase from Arthrobacter P1. J. Biol. Inorg. Chem. 1, 205–209 [Google Scholar]
- 66. Kyte J. (1995) Mechanism in Protein Chemistry, pp. 350–371, Garland Publishing, Inc., New York [Google Scholar]
- 67. Lopes Jesus A. J., Redinha J. S. (2011) Charge-assisted intramolecular hydrogen bonds in disubstituted cyclohexane derivatives. J. Phys. Chem. A 115, 14069–14077 [DOI] [PubMed] [Google Scholar]
- 68. Bisby R. H., Johnson S. A., Parker A. W., Tavender S. M. (1999) Time-resolved resonance Raman studies of radicals from 4-aminoresorcinol as models for the active site radical intermediate in copper amine oxidases. Laser Chem. 19, 201–208 [Google Scholar]
- 69. Dooley D. M., Cote C. E. (1985) Copper(II) coordination chemistry in bovine plasma amine oxidase: azide and thiocyanate binding. Inorg. Chem. 24, 3996–4000 [Google Scholar]
- 70. McGuirl M. A., Brown D. E., Dooley D. M. (1997) Cyanide as a copper-directed inhibitor of amine oxidases: implications for the mechanism of amine oxidation. J. Biol. Inorg. Chem. 2, 336–342 [Google Scholar]