

Background: Macrophage and dendritic cell inflammatory responses to malaria remain undefined despite their crucial roles in controlling infections, pathogenesis, and protective immunity development.
Results: Dendritic cells but not macrophages produce inflammatory cytokines to parasites.
Conclusion: Rapid acidification prevents endosomal receptor-mediated recognition of parasites and cytokine responses by macrophages.
Significance: Blocking endosomal acidification might enhance the efficacy of whole parasite-based vaccine.
Keywords: cytokine induction, inflammation, innate immunity, macrophage, malaria, phagocytosis, plasmodium, vacuolar acidification
Abstract
Inflammatory cytokines produced at the early stages of malaria infection contribute to shaping protective immunity and pathophysiology. To gain mechanistic insight into these processes, it is important to understand the cellular origin of cytokines because both cytokine input and cytokine-producing cells play key roles. Here, we determined cytokine responses by monocytes, macrophages, and dendritic cells (DCs) to purified Plasmodium falciparum and Plasmodium berghei ANKA, and by spleen macrophages and DCs from Plasmodium yoelii 17NXL-infected and P. berghei ANKA-infected mice. The results demonstrate that monocytes and macrophages do not produce inflammatory cytokines to malaria parasites and that DCs are the primary source early in infection, and DC subsets differentially produce cytokines. Importantly, blocking of phagosomal acidification by inhibiting vacuolar-type H+-ATPase enabled macrophages to elicit cytokine responses. Because cytokine responses to malaria parasites are mediated primarily through endosomal Toll-like receptors, our data indicate that the inability of macrophages to produce cytokines is due to the phagosomal acidification that disrupts endosomal ligand-receptor engagement. Macrophages efficiently produced cytokines to LPS upon simultaneously internalizing parasites and to heat-killed Escherichia coli, demonstrating that phagosomal acidification affects endosomal receptor-mediated, but not cell surface receptor-mediated, recognition of Toll-like receptor agonists. Enabling monocytes/macrophages to elicit immune responses to parasites by blocking endosomal acidification can be a novel strategy for the effective development of protective immunity to malaria. The results have important implications for enhancing the efficacy of a whole parasite-based malaria vaccine and for designing strategies for the development of protective immunity to pathogens that induce immune responses primarily through endosomal receptors.
Introduction
Malaria, a complex disease caused by Plasmodium parasites is a huge public health problem in endemic countries (1, 2). In both human and mouse malaria, excessive production of inflammatory cytokines, including TNF-α, IL-6, IL-12, and IFN-γ, at the early stages of infection is a key contributor to pathogenesis (3–6). However, appropriate production of inflammatory cytokines is essential for the resolution of infection with positive outcomes as they play critical roles in parasite growth control by up-regulating phagocytic clearance by monocytes and macrophages. Early inflammatory cytokine responses are also crucial for modulating Th1 and Th2 responses and the development of protective adaptive immunity. Furthermore, the interaction of cytokine-producing cells with T cells results in the activation of the latter for subsequent modulation of Th1/Th2 responses. Therefore, knowledge on the cellular sources of early cytokine responses is crucial in gaining insight into mechanisms that underlie the development of malaria immunity and pathology. Despite this importance, the cells that elicit cytokine responses at the early stages of malaria infection have not been clearly defined.
Macrophages and dendritic cells (DCs)3 are two major sentinel cell types of the innate immune system that are critical for the control of infections and the development of adaptive immunity. DCs are primarily involved in recognizing pathogens, initiating cytokine responses, and processing and presenting antigens (7, 8). Macrophages, however, not only function as effector cells to efficiently eliminate pathogens through phagocytosis, but also produce cytokines and present antigens (9, 10). In the case of malaria infection, in which parasites harbor red blood cells, monocytes, macrophages, and DCs can readily access parasites for initiating innate immune responses and acquiring the capacity to direct the adaptive immune responses. However, the development of protective immunity to malaria is inefficient and requires repeated infections over a considerable period of time (11, 12). The cellular and molecular mechanisms that underlie this phenomenon remain poorly understood. Although some studies have reported that monocytes and macrophages produce inflammatory cytokines in response to malaria parasites (13, 14), others showed that macrophages do not produce cytokines, and the cells become nonfunctional (15–17). Thus, it has been unclear whether or to what extent monocytes and macrophages produce cytokines to malaria parasites. If indeed macrophages become nonfunctional upon phagocytosis of parasites, the underlying mechanisms have not been clearly understood. Previously, it was suggested that macrophages become nonfunctional due to damages caused by the oxidative burst induced by phagocytosis of parasites (17). However, this suggestion is inconsistent with the property of macrophages because these cells are known to induce low levels of oxidative burst upon phagocytosis and digest pathogens by phagosomal acidification (18); a marked oxidative burst is the characteristic of neutrophils. Given that information on the functional capacity of the innate immune system in malaria immunity development is limited, and early cytokine responses determine the effectiveness of adaptive immunity, it is important to understand inflammatory cytokine responses to malaria by macrophages and DCs.
Here, in malaria-infected mice, we show that DCs are the main source of inflammatory cytokines at the early stages of infection and that spleen DC subsets differentially elicit cytokine responses. Importantly, we also show that human monocytes and mouse monocytes and macrophages do not produce cytokines to parasites and that phagosomal acidification is the primary reason for the inability of macrophages to produce cytokines. Thus, the development of adaptive immunity to malaria is mainly dependent on DCs, which are fewer in number compared with monocytes and macrophages. This might be a reason as to why the development of protective immunity to malaria is inefficient and requires several repeated infections (11, 12).
Experimental Procedures
Mice and Malaria Infection
C57BL/6 wild type mice were bred and maintained in a pathogen-free environment. Mice were infected with either nonlethal Plasmodium yoelii 17XNL (Py17XNL) or lethal Plasmodium berghei ANKA (PbANKA) strains by intraperitoneal injection of blood containing about 0.5 million IRBCs from an infected donor mice in 100 μl of saline (19). Experiments in mice were performed in accordance with the recommendations in Care and Use of Laboratory Animals from the National Institutes of Health. The Institutional Animal Care and Use Committee of the Pennsylvania State University College of Medicine has approved the protocols.
P. falciparum Culturing
The human malaria parasite, P. falciparum (3D7 strain), was cultured using O-positive human red blood cells in RPMI 1640 medium containing 10% human O-positive plasma and 50 μg/ml gentamycin under 90% nitrogen, 5% oxygen, and 5% carbon dioxide as described previously (20). Human blood and plasma were obtained from the Blood Bank of Hershey Medical Center. The cultures were synchronized using 5% sorbitol (21) and tested for mycoplasma every 10–15 days by using Myco Sensor PCR assay kit from Stratagene (La Jolla, CA). Cultures containing parasites at the late trophozoite and early schizont stages were harvested by centrifuging at 250 × g for 5 min. The cell pellets were resuspended in RPMI 1640 medium, overlaid on 65% Percoll, and centrifuged at 1200 × g for 15 min. The cell layer, containing predominantly IRBCs on the top of Percoll cushions, was collected, washed twice with RPMI 1640 medium, and used for cell stimulation.
For the isolation of merozoites (22), P. falciparum cultures having 20–25% IRBCs and 0.1–0.2% hematocrit were incubated at 37 °C without supplementing fresh red blood cells. After schizonts burst, the cultures were centrifuged at 250 × g for 5 min. The supernatants were collected and centrifuged further at 2500 × g for 15 min. The pellets from this step were suspended in RPMI 1640 medium and overlaid on top of stepwise 30, 45, and 65% Percoll cushions. After centrifuging at 2500 × g for 15 min, the dark layer on the top of 30% Percoll containing merozoites and parasite food vacuoles was collected. The food vacuoles were removed by passing through magnetic LS columns. The purity of the prepared merozoites was >95%.
Isolation of Mouse Parasites
For the isolation of IRBCs (19), blood collected from PbANKA-infected mice was diluted with 2 volumes of PBS, pH 7.4, centrifuged on Isolymph (CTL Scientific Supply, Deer Park, NY) cushions at 1200 × g at room temperature for 15 min, and the buffy coat removed. The cell pellets were resuspended in 2 volumes of PBS and centrifuged on 70% Percoll (Sigma) at 1200 × g at room temperature for 15 min. The IRBCs on the top of Percoll cushions were collected and washed with PBS.
Heat-killed Escherichia coli
E. coli cells (DH10B strain) were cultured in LB medium in the presence of 30 μg/ml streptomycin sulfate overnight at 37 °C, harvested by centrifugation at 3200 × g, and washed twice with PBS, pH 7.4. The bacterial pellet was suspended in PBS, heated at 80 °C for 20 min, and centrifuged. The pellet was suspended in PBS and used for cell stimulation.
Preparation of Mouse Bone Marrow Cell-derived Macrophages and DCs
Bone marrow cell-derived macrophages (BMDMs) were prepared by culturing the mouse bone marrow cells in DMEM containing 10% FBS, 1 mm sodium pyruvate, 50 μm 2-mercaptoethanol, nonessential amino acids, 10 units/ml penicillin/streptomycin (complete DMEM), and 30% L929 cell culture conditioned medium (19, 22). After 7 days, BMDMs were harvested by scraping, suspended in complete DMEM, and used for stimulation experiments. GM-DCs were prepared by culturing mouse bone marrow cells for 7–8 days in complete DMEM containing 10% conditioned DMEM from GM-CSF-producing cells (23). FL-DCs were prepared by culturing mouse bone marrow cells in complete DMEM supplemented with 15% Fms-like tyrosine kinase 3 (FLT3) ligand-containing conditioned medium, which was obtained by culturing B16 cells expressing retrovirus-encoded FLT3 ligand (24).
Isolation of Mouse Monocytes and Macrophages
Blood from naive mice was collected into tubes containing 100 μl of 1 mm heparin per mouse, diluted with 2 volumes of PBS, and centrifuged on Isolymph (CTL Scientific Supply, Deer Park, NY) at 1200 × g at room temperature for 15 min. PBMCs present on the top of the Isolymph cushions were collected. For the isolation of liver macrophages (25), mouse livers were flushed with 10 ml of PBS and digested with 1 mg/ml collagenase D (Roche Applied Science, Mannheim, Germany) at 37 °C. After 30 min, tissues were homogenized and centrifuged at 30 × g at 4 °C for 5 min. The supernatants were collected and centrifuged at 300 × g at 4 °C for 10 min. The cell pellets were resuspended in 5 ml of 40% Percoll, loaded onto 70% Percoll cushions, and centrifuged at 1200 × g at room temperature for 15 min. The cell layers in the interface were collected and washed with PBS. For the isolation of splenic macrophages, spleens were digested with 1 mg/ml collagenase D, homogenized, and single cell suspensions prepared as reported previously (22). Monocytes from PBMCs and macrophages from liver and spleen cell preparations were isolated by magnetic separation using anti-human/mouse CD11b microbeads (Miltenyi Biotec, Auburn, CA). In all cases, monocytes and macrophages that adhered to culture plates were used for induction of cytokine responses.
Isolation of Human Monocytes
PBMCs from the buffy coat of healthy human blood were isolated by centrifugation on ISOLYMPH cushions (22). Monocytes from the PBMCs were isolated by magnetic separation using anti-human/mouse CD11b microbeads. The cells were plated into 96-well plastic plates and incubated for 2–3 h; floating cells were removed, and the adhered cells were used for stimulation with P. falciparum IRBCs.
Cell Stimulation and Cytokine Analysis by ELISA
The human and mouse blood monocytes, mouse spleen and liver macrophages, GM-DCs and FL-DCs (in each case 1 × 105 cells/well), and BMDM (0.4 × 105 cells/well) in 96-well plates were cultured in 200 μl of complete DMEM (for mouse cells) or RMPI 1640 medium (for human cells) per well. The cells were stimulated with either IRBCs or merozoites for 48 h in the case of monocytes and macrophages and for 24 h in the case of DCs. Cells stimulated with either 2 μg/ml CpG ODN-1826 (Coley Pharmaceutical, Kanata, Ontario, Canada) or 100 ng/ml LPS (catalogue no. L9764, Sigma) were analyzed as controls. To test the effects of phagosomal acidification and oxidative burst on cytokine production, BMDMs (0.4 × 105 cell/well in 200 μl of complete DMEM) were stimulated with IRBCs, LPS plus IRBCs, LPS plus silica particles (0.3 μm size, Sigma), LPS plus synthetic hemozoin prepared as described previously (26) or with heat-killed E. coli in the absence or presence of the indicated amounts of bafilomycin A1 (LC Laboratories, Woburn, MA) and/or theophylline (Sigma). The cytokines secreted into culture media were analyzed by using ELISA kits from R & D Systems (Minneapolis, MN). For the analysis of intracellular IL-12 in BMDMs stimulated with IRBCs in the presence of bafilomycin A1 and/or theophylline, the cells were cultured in ultra-low attachment 24-well plates (0.5 × 106 cells/well in 0.5 ml) and stimulated with IRBCs. After 14 h of culturing, Golgi-Plug was added, and cells were harvested at 20 h for the analysis of intracellular IL-12 by flow cytometry.
Analysis of Macrophages and DCs from Infected Mice for Cytokine Production
Spleens from mice infected with either Py17XNL or PbANKA parasites were collected at different days postinfection and single cell suspensions prepared as described above. Cells (1 × 107/well) were cultured in 24-well plates in 1 ml of complete DMEM in the presence of GolgiPlug for 6 h and harvested, and the cytokine-producing cells were analyzed by flow cytometry (see below).
Flow Cytometry
Prior to staining cells with specific antibodies, the surface Fc receptors were blocked with the anti-CD16/32 antibody (clone 93, e-Bioscience). Spleen DCs and macrophages from parasite-infected mice were first stained with antibodies against surface marker proteins and then stained for intracellular proteins with FITC-anti-IFN-β, eFluor 450-anti-TNF-α, and PerCP-Cy5.5-anti-IL-12p40 antibodies (see below). BMDMs stimulated with IRBCs in the presence and absence of bafilomycin A1 and/or theophylline were surface-stained with PE-conjugated anti-mouse CD11b antibody followed by intracellular staining with PerCP-Cy5.5-anti-IL-12p40 antibody. The stained cells were analyzed by using the LSRII (BD Biosciences) flow cytometer, and the results were analyzed using FlowJo software (Treestar, Ashland, OR).
The anti-mouse antibodies used for cell staining were as follows: FITC-, PE-, and APC-anti-CD11c (clone 418N), PE- and PerCP-Cy5.5-anti-CD11b (M1/70), PE- and APC-anti-I-A/I-E (M5/114.15.2), PE-Cy7-anti-Siglec H (eBio440c), APC-eFluor 780-anti-F4/80 (BM8), eFluor 450-anti-TNF-α (MP6-XT22), and PerCP-Cy5.5-anti-IL-12p40 (C17.8) antibodies were from e-Biosciences, San Diego; Alexa Fluor 700-anti-CD4 (RM4-5) and brilliant violet 605-anti-CD8α (53–6.7) antibodies were from BD Biosciences; FITC-anti-IFN-β antibody (RMMB-1) was from PBL Assay Science, Piscataway, NJ; BV605-anti-CD169 (3D6.112) and PE-Cy7-anti-CD68 (FA-11) antibodies were from BioLegend, San Diego; and APC-anti-MARCO (579511) antibody was from R & D Systems, Minneapolis, MN.
Statistical Analysis
Statistical significance for the differences in the levels of cytokines produced from the control and experimental groups were determined by one-way analysis of variance followed by the Newman-Keuls test or t test using GraphPad Prism version 6.01. The results are presented as the means ± S.D. p values <0.05 were considered statistically significant.
Results
Unlike DCs, Macrophages Do Not Produce Inflammatory Cytokines in Response to Malaria Parasites
IRBCs and merozoites are the major immunostimulatory forms of P. falciparum that induce the production of inflammatory cytokines, such as TNF-α, IL-6, and IL-12 by DCs, and the soluble components of parasites released during the schizont rupture induce only negligible inflammatory responses (22). Here, to determine the extent of de novo inflammatory cytokine production by macrophages, we analyzed TNF-α, IL-6, and IL-12 responses by BMDMs to the purified P. falciparum and PbANKA parasites. Both unprimed and IFN-γ-primed BMDMs did not produce cytokines, although the cells efficiently produced cytokines in response to control CpG ODN (Fig. 1, A and B). The unprimed and IFN-γ-primed mouse blood monocytes and spleen and liver macrophages were also unable to produce TNF-α and IL-12 in response to parasites (Fig. 1, C and D). Furthermore, human peripheral blood monocytes did not produce cytokines to P. falciparum (Fig. 1E). Together these results demonstrated that monocytes and macrophages do not produce typical inflammatory cytokines such as TNF-α, IL-6, and IL-12 in response to malaria parasites. In contrast, consistent with the results of previous studies (22, 27–29), GM-DCs and FL-DCs, the DCs derived by the differentiation of bone marrow cells, by using granulocyte/macrophage-colony stimulating factor and Fms-like tyrosine kinase 3 ligand, respectively, efficiently produced TNF-α, IL-6, and IL-12 in response to P. falciparum IRBCs and merozoites (Fig. 2). We have previously shown that human peripheral blood DCs robustly produce TNF-α and IL-12 to P. falciparum (30).
FIGURE 1.
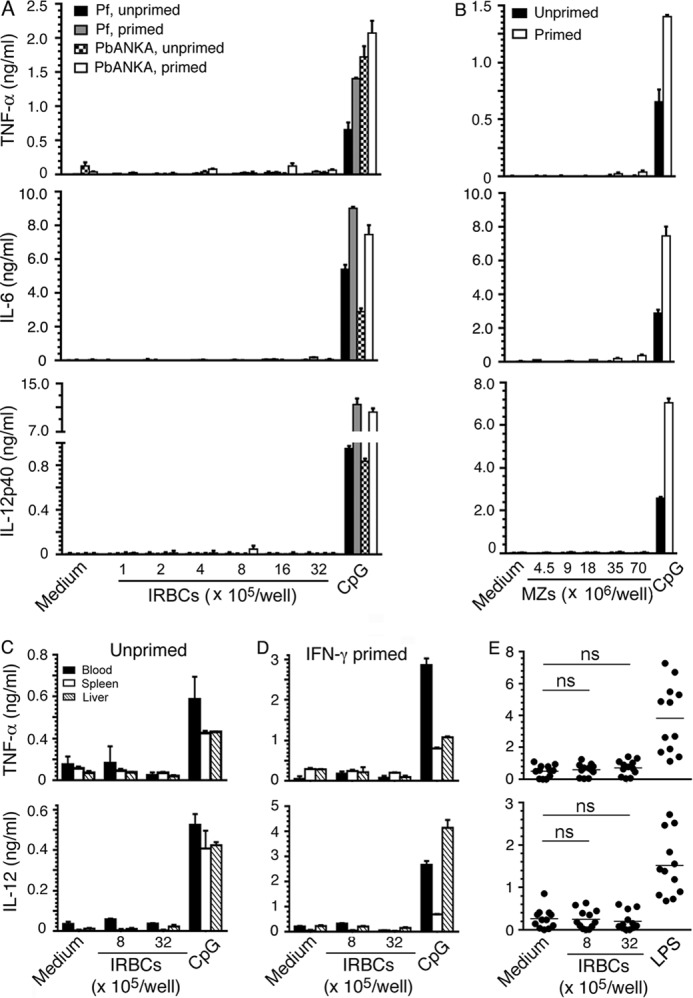
Monocytes and macrophages do not produce inflammatory cytokines in response to malaria parasites. A, BMDMs (0.4 × 105/well in 200 μl of DMEM), either unprimed or primed with 20 units of IFN-γ, were incubated with P. falciparum (Pf) or PbANKA IRBCs for 48 h. B, BMDMs, either unprimed or primed with 20 units of IFN-γ, were incubated with P. falciparum merozoites (MZs) for 48 h. C and D, mouse blood monocytes, spleen macrophages, and liver macrophages (in each case 1 × 105/well in 200 μl of DMEM), either unprimed or primed with 20 units of IFN-γ, were incubated with P. falciparum IRBCs for 48 h. E, CD11b+ adherent cells from human PBMCs were incubated with P. falciparum IRBCs. Cells stimulated with 2 μg/ml CpG ODN or 100 ng/ml LPS were used as controls. After 48 h, TNF-α, IL-6, and IL-12 secreted into the medium were analyzed by ELISA. Shown are data (mean values ± S.D.) from a representative of five independent experiments (A–D) or for each individual (E). ns, statistically not significant. Note: the x axis labeling in the left margin refers to each column in all five rows.
FIGURE 2.
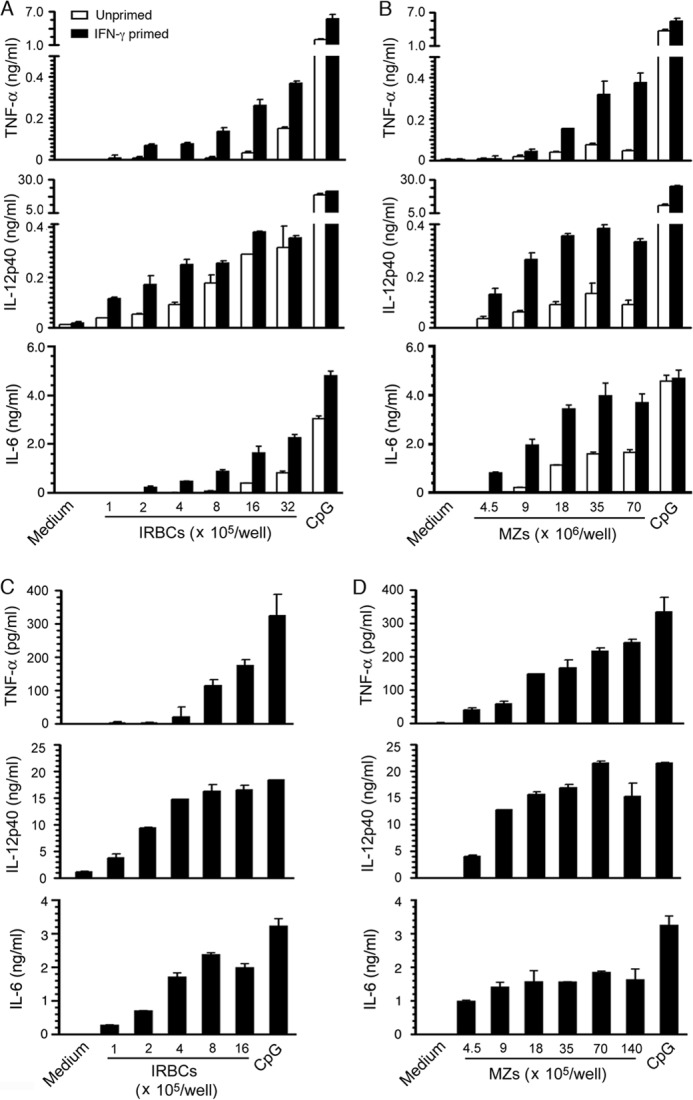
DCs produce inflammatory cytokines in response to malaria parasites. A–D, GM-DCs (A and B) and FL-DCs (C and D) in 96-well plates (1 × 105/well in 200 μl of DMEM) were stimulated with P. falciparum IRBCs or merozoites (MZs) for 24 h. DCs stimulated with 2 μg/ml CpG ODN were analyzed as a control. TNF-α, IL-6, and IL-12 secreted into the culture medium were measured by ELISA. The mean values ± S.D. from four independent experiments each performed in duplicate are plotted.
DCs but Not Macrophages Produce Cytokines at the Early Stages of Malaria Infection
To determine the physiological relevance of the results observed in the above in vitro studies, we analyzed macrophages and DCs from mice infected with either Py17XNL or PbANKA. Measurable levels of cytokines by flow cytometry were evident 3 days postinfection, and only DCs, but not monocytes/macrophages, produced cytokines. To ensure that the measured cytokines were those produced in direct response to parasites, we analyzed DCs and macrophages mice from at day 3 postinfection. This is the earliest time point when measurable cytokine proteins were evident. CD8α+ and CD8α− conventional DCs as well as CD11cintSiglecH+ plasmacytoid DCs isolated from both Py17XNL- and PbANKA-infected mice produced TNF-α (Fig. 3 and supplemental Fig. S1). Interestingly, however, only CD8α− DCs additionally produced IL-12 and IFN-β. Analysis of CD8α− DCs for the presence of CD4 showed that both CD4+CD8α− and CD4−CD8α− DCs expressed all three cytokines, TNF-α, IL-12, and IFN-β. Although the production of IFN-β by CD8α− DCs agrees with the recent findings (31), the expression of IL-12 by CD8α− DCs, but not by CD8α+ DCs, in response to malaria has not been previously shown. This differential production of inflammatory cytokines by different DC subsets to malaria parasites is likely related to their distinctive functions.
FIGURE 3.

Spleen DC subsets in malaria parasite-infected mice differentially produce inflammatory cytokines. Spleen cells from mice at 3 days postinfection with either Py17NXL or PbANKA parasites were cultured in the presence of Golgi-Plug for 6 h. The cells were then surface-stained with antibodies against the indicated marker proteins followed by intracellular staining with anti-cytokine antibodies and analyzed by flow cytometry. A, gating strategies for different DC subsets are shown. pDCs, plasmacytoid DCs. B, percent cytokine-producing cells (mean values ± S.D.) of a representative of four independent experiments, each performed using a group of three mice; cells from each mouse were separately analyzed. The contour plots of cytokine-expressing DCs are shown in supplemental Fig. S1. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant.
Mouse spleen macrophages are classified into four subsets (32), namely marginal zone macrophages (F4/80−CD11b−MARCO+), marginal zone metallophilic macrophages (F4/80−CD11b−CD169+), tingible body macrophages (F4/80−CD11b−CD68+), and red pulp macrophages (F4/80hiCD11blo/−CD169loMHCIIlo). In contrast to DCs, there was no TNF-α, IL-12, or IFN-β production by three major subsets of macrophages, marginal zone, marginal zone metallophilic, and tingible body macrophages from both Py17XNL- and PbANKA-infected mice (Fig. 4 and supplemental Fig. S2). However, the red pulp macrophages produced TNF-α but not IL-12 and IFN-β. Given their cytokine-producing ability, it is possible that red pulp macrophages are somewhat related to DCs. To test this possibility, we analyzed spleen macrophage subsets for CD11c expression (Fig. 5, A and B). The expression of CD11c and MHCII at high levels is considered to be the characteristic of DCs (33); macrophages express low levels of MHCII. Only red pulp macrophages, but not other macrophage subsets, showed high levels of CD11c (Fig. 5B). Considering high level expression of CD11c and low expression of MHCII, red pulp macrophages appear to have some characteristics of DCs and thus produce TNF-α. Because these cells represent only a minor population among spleen macrophages (Fig. 5C), it appears that DCs are the major source of inflammatory cytokines at the early stages of malaria infection.
FIGURE 4.

Majority of spleen macrophages in malaria parasite-infected mice does not produce inflammatory cytokines. The spleen cells from mice at 3 days postinfection with either Py17NXL or PbANKA were cultured and treated with the Golgi-Plug. The cells were then surface-stained with antibodies against the indicated marker proteins followed by intracellular staining with anti-cytokine antibodies and analyzed by flow cytometry. A, shown is the gating strategy for macrophage subsets: MM, marginal zone macrophages; MMM, marginal zone metallophilic macrophages; TBM, tingible body macrophages; RPM, red pulp macrophages. B, percent cytokine-producing macrophages from each subset; the mean values ± S.D. of a representative of four independent experiments, each performed using a group of three mice; cells from each mouse were separately analyzed. The contour plots of cytokine-expressing macrophages are shown in supplemental Fig. S2. ***, p < 0.001; ns, not significant.
FIGURE 5.

Analysis of CD11c in the spleen macrophage subsets from parasite-infected mice. A, spleen macrophages, gated as shown in Fig. 3, were further gated for the expression of CD11c. The data shown are a representative of two independent experiments each performed using a group of three mice; cells from each mouse were separately analyzed. B, percentage (mean values ± S.D.) of CD11chi cells in each macrophage subset. Arrowheads indicate values below detection limit. Only red pulp macrophages (RPM) expressed high levels of CD11c. C, percentage (mean values ± S.D.) of each macrophage subset relative to the total spleen macrophages. MM, marginal zone macrophages; MMM, marginal zone metallophilic macrophages; TBM, tingible body macrophages.
Phagosomal Acidification Prevents Cytokine Responses by Macrophages
Because monocytes and macrophages produced little or no cytokines in response to malaria parasites, we next sought to determine the underlying mechanisms. Previous studies have reported that, upon internalization of matured IRBCs, monocytes and macrophages were unable to digest ingested parasites or produce cytokines because oxidative burst induced upon phagocytosis causes functional impairment (15–17). To test this notion, we analyzed the effect of theophylline, an inhibitor of oxidative burst (34, 35), in producing cytokines in response to IRBCs. Only negligible levels of TNF-α production were apparent (Fig. 6A), suggesting that the oxidative burst is not the main reason for the inability of macrophages to produce cytokines to malaria parasites. The data agree with the fact that, unlike neutrophils that produce a rapid and potent oxidative burst upon ingestion of microbes or foreign particles, macrophages induce relatively low levels of oxidative burst, and these cells kill pathogens by rapidly acidifying phagosomes (18, 36, 37).
FIGURE 6.

Inhibition of phagosomal acidification but not oxidative burst enables macrophages to produce inflammatory cytokines in response to malaria parasites. BMDMs were incubated with P. falciparum IRBCs in the presence of theophylline (A), bafilomycin A1 (B), or mixture of 5 nm of bafilomycin and 10 or 100 μm theophylline (C). Th, theophylline; BA1, bafilomycin A1. TNF-α released into culture medium was analyzed by ELISA. Values for treatment with theophylline and/or bafilomycin A1 in medium (control), all values in A, and some values in B and C are below the detection limit. D, BMDMs in 24-well plates were incubated with IRBCs in the absence and presence of bafilomycin A1 (5 nm), theophylline (100 μm), and a mixture of bafilomycin A1 (5 nm) and theophylline (100 μm). The cells were stained with anti-mouse CD11b followed by intracellular staining with anti-mouse IL-12 antibodies and analyzed by flow cytometry. The results of a representative of three independent experiments each performed in triplicate are shown. E, plots of mean values ± S.D. of the experiment shown in D. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Several studies have shown that macrophages, neutrophils, and epithelial cells induce a strong acidification of phagosomes within 20–30 min of the initiation of internalizing microbes or inert particles/microbeads (18, 36, 38, 39). Thus, the pH value of phagosomes drops from neutral to ∼5 within 2–3 min after phagosome formation through a rapid and massive recruitment of vacuolar-type proton pump-ATPase (V-type H+-ATPase) to phagosomal membranes. Given this information, we predicted that rapid acidification of phagosomes is the reason for the lack of cytokine responses to malaria parasites by macrophages. We tested this hypothesis by inhibiting the phagosomal acidification with bafilomycin A1, a specific inhibitor of V-type H+-ATPase. Macrophages exposed to IRBCs in the presence of bafilomycin A1 produced substantial levels of TNF-α (Fig. 6B), suggesting that the phagosomal acidification and consequently endosomes becoming highly acidic upon fusion with phagosomes prevent parasite ligand-endosomal receptor interactions and cytokine responses. Furthermore, when macrophages were treated with bafilomycin A1 plus theophylline, the production of TNF-α was significantly increased compared with treatment with bafilomycin A1 alone (Fig. 6C). Because the inhibition of oxidative burst without blocking phagosomal acidification does not lead to cytokine responses, rapid acidification of phagosomes is the primary reason for the inability of macrophages to produce cytokines in response to malaria parasites. However, oxidative burst also contributes, but only after phagosomal acidification is regulated (see Fig. 6C). Furthermore, macrophages treated with either bafilomycin A1 alone or a mixture of bafilomycin A1 and theophylline secreted very low levels of IL-12. We previously showed that the transcription and production of TNF-α by macrophages in response to malaria glycosylphosphatidylinositols were significantly faster than those of IL-12 (40). Thus, we predicted that the low level of IL-12 secretion was at least partially due to the relatively delayed kinetics in its expression and secretion. Additionally, it is possible that the expressed IL-12 remained intracellular as a result of cells becoming progressively damaged upon phagocytosis. Therefore, we assessed the intracellular IL-12 in macrophages exposed to IRBCs in the presence of bafilomycin A1 and/or theophylline. Treatment with theophylline alone resulted in a low level of IL-12 expression (Fig. 6, D and E). However, a 10-fold increase of IL-12 was observed when treated with bafilomycin A1, and treatment with both bafilomycin A1 and theophylline further moderately enhanced the level of IL-12. Together, the above data demonstrated that the acidification of phagosomes is primarily responsible for the inability of macrophages to produce cytokines to malaria parasites.
Phagosomal Acidification Does Not Prevent Plasma Membrane Receptor-mediated Cytokine Responses by Macrophages
Because cytokine response to malaria parasites is primarily dependent on TLR9 and TLR7 recognition (19, 22, 27, 28, 41), we predicted that the phagosomal acidification in parasite-phagocytosed macrophages has no bearing on plasma membrane receptor-ligand interactions. To test this prediction, we assessed the TNF-α response by macrophages stimulated with LPS alone and LPS in the presence of IRBCs. In both cases, similar levels of TNF-α production were observed (Fig. 7A). We also tested TNF-α response to LPS by macrophages in the presence and absence of synthetic hemozoin or silica particles. Phagocytosis of either hemozoin or silica particles, which can induce phagosomal acidification (42), had no effect on the LPS-induced TNF-α production by macrophages. Together, these data suggested that phagosomal acidification in response to phagocytosed IRBCs or inert particles does not affect the plasma membrane receptor-mediated signaling. To confirm this inference, we analyzed cytokine production by macrophages to heat-killed E. coli, which can activate cells through LPS interacting with TLR4 on the cell surface, CpG motifs of its DNA interacting with TLR9 in endosomes, and likely also through other signaling mechanisms (43). Unlike malaria parasites, E. coli efficiently induced TNF-α production by macrophages (Fig. 7B), and the inhibition of phagosomal acidification by bafilomycin A1 resulted in increased production of TNF-α. Treatment with theophylline resulted in a small increase in TNF-α. Furthermore, treatment with theophylline plus bafilomycin A1 caused a moderate increase in TNF-α as compared to treatment with bafilomycin A1 alone. Together these data suggested that although phagosomal acidification drastically affects cytokine production mediated by the endosomal receptors, it does not markedly affect the cytokine responses mediated through the cell surface receptor such as TLR4.
FIGURE 7.
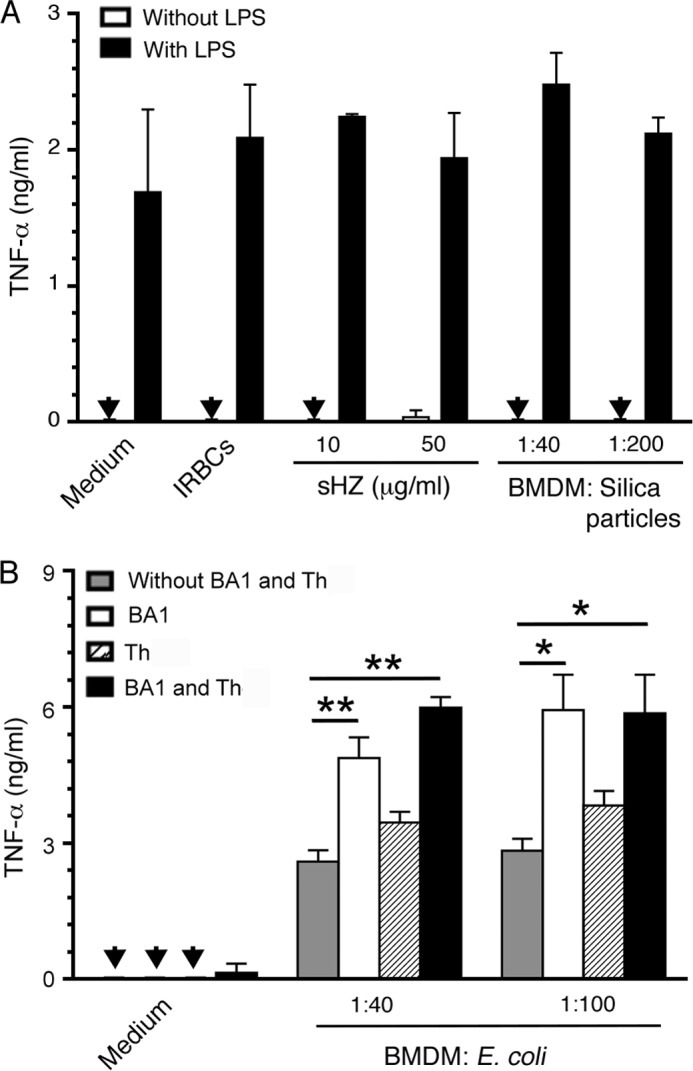
Phagosomal acidification does not significantly affect the cytokine production to LPS and to heat-killed E. coli. A, BMDMs were incubated with P. falciparum IRBCs, synthetic hemozoin (sHz), or silica particles in the absence or presence of 100 ng/ml LPS. B, BMDMs were incubated with heat-killed E. coli at the ratios of 1:40 or 1:100 in the absence and presence of bafilomycin A1 (BA1, 5 nm), theophylline (Th, 100 μm), and a mixture of bafilomycin A1 (5 nm) and theophylline (100 μm). After 48 h, the TNF-α released into the culture medium was measured by ELISA. The results (mean values ± S.D.) shown are representative of three independent experiments each performed in duplicate. Arrowheads indicate values below the detection limit.
Discussion
The results of this study clearly show that monocytes and macrophages do not produce inflammatory cytokines in response to malaria parasite-infected erythrocytes and merozoites, the physiological forms of parasites recognized by the innate immune system. The results also show that DCs efficiently produce inflammatory cytokines to malaria parasites and that these cells are the primary source of cytokines at the early stage of malaria infection. Additionally, our results demonstrate that mouse DC subsets differentially produce cytokines to malaria parasites. Notably, whereas both CD8α+ conventional DCs and plasmacytoid DCs produced TNF-α but not IL-12 and IFN-β, CD4+CD8α− and CD4−CD8α− DCs produced all three cytokines. CD8α− DCs have been shown to acquire increased capacity to inhibit Th1 development in a type I IFN signaling-dependent manner (31), although these cells initially promote Th1 development (44). This DC subtype expresses type I IFN receptor and amplifies their type I IFN-producing capacity via a positive feedback signaling, thereby contributing to a feed-forward signaling loop for dampening the Th1 development (31). Interestingly, in Plasmodium chabaudi malaria infection, both CD8α+ DCs and CD8α− DCs promote Th1 by inducing IFN-γ production in CD4 T cells at the early stages of infection (44). However, only CD8α− DCs induce CD4 T cells to produce IL-4 and IL-10, promoting Th1 to Th2 switching at the acute phase of infection (44). Thus, the differential cytokine responses by DC subsets observed here appear to be related to the modulation of Th1 and Th2 development during the course of malaria infection.
Our observation that CD8α− DCs but not CD8α+ DCs produce IL-12 at the early stage of malaria infection contrasts with the prevailing notion that CD8α+ DCs are the predominant IL-12-producing cells (8, 45) and that the IL-12-producing CD8α+ DCs but not CD8α− DCs guide Th1 responses (8). It appears that in malaria infection, the IL-12-producing CD8α− DCs, which also produce type I IFN, play a dual role during the course of malaria infection as follows: Th1 development at the early stage and switching of Th1 to Th2 as the infection progresses (44). This conclusion is supported by our previous findings that in P. yoelii 17XNL infection, DCs robustly produce IL-12 at 5 days postinfection and that the IL-12 production ceases by 10 days postinfection (19). Furthermore, S-27609, a TLR7 imidazoquinoline agonist has been shown to induce IL-12 production only by CD8α− DCs; CD8α+ DCs lack TLR7 expression and hence do not respond to imidazoquinolines (46, 47). In the light of this information, it appears that TLR7-dependent recognition of mouse parasite RNA contributes significantly to the production of IL-12 by CD8α− DCs (see Fig. 3) (41, 48). In any case, type I IFN- and IL-12-producing CD8α− DCs promote Th1 development at the early stage of infection (19). However, these cells appear to program themselves to gain the capacity to gradually reduce Th1 development while increasing their ability to promote Th2 development as the infection progresses (19).
Although this study shows that the mouse DC subsets in both lethal P. berghei ANKA and nonlethal P. yoelii infections produced similar inflammatory cytokine responses (see Fig. 3), these results cannot be generalized to human malaria infection because human DC subsets differ from those of mice with respect to the expression of various pathogen recognition receptors and hence in their cytokine outputs. This is evident from our earlier observations that although human plasmacytoid DCs efficiently produced IFN-α and TNF-α to malaria parasite lysate, the conventional DC subset failed to induce these cytokines (22). Therefore, detailed studies on human malaria infection are necessary to understand the pattern of cytokine responses by human DC subsets.
More importantly, our study demonstrates that rapid acidification of phagosomes upon internalization of malaria parasites is the primary reason for macrophages not producing cytokines (see Fig. 6). It is well known that upon ingestion of pathogens, a rapid and strong induction of oxidative burst by the massive recruitment of NADPH oxidase complex to phagosomes is the characteristic of neutrophils (18). Macrophages, however, recruit relatively low levels of NADPH oxidase to phagosomes, and these cells depend mainly on acidifying the phagosomes to digest the pathogens by rapidly recruiting markedly high levels of V-type H+-ATPase to the phagosomal membrane. The subsequent fusion of phagosomes with endosomes results in a quick drop in endosomal pH. Consequently, the ligand-receptor interactions cannot occur in endosomes, preventing downstream cytokine responses, if the pathogens are recognized exclusively through endosomal pathogen recognition receptors. In contrast, if pathogens are recognized either solely through plasma membrane receptors or through both plasma membrane and endosomal membrane receptors, it is likely that macrophages still produce significant levels of cytokines via plasma membrane receptor-initiated signaling. This is because neither plasma membrane receptor-mediated recognition nor signaling thereof occurs in an acidic environment. Thus, macrophages are unable to elicit cytokine responses to malaria parasites, which are primarily recognized by endosomal receptors (19, 22, 27, 28, 41). In contrast, TLR4-mediated, LPS-induced cytokine responses were not affected by the endosomal acidification induced upon simultaneous internalization of parasites and inert particles (see Fig. 7A). Macrophages could also efficiently produce cytokines in response to phagocytosis of E. coli (see Fig. 7B), presumably through the recognition of LPS by TLR4 and possibly by recognition of other ligands via corresponding plasma membrane receptors. Thus, our data clearly demonstrate that the cytokine production by macrophages during infections depends on the intrinsic molecular signatures of pathogens. If pathogens contain only the endosomal receptor-specific ligands such as TLR7, TLR8, and TLR9, little or no cytokine responses are produced by macrophages upon phagocytosis. However, if pathogens are recognized via plasma membrane receptors or both endosomal and plasma membrane receptors, as in the case of bacteria, macrophages most likely produce considerable levels of cytokines even though phagosomal acidification abolishes the endosomal receptor-dependent cytokine responses.
Our conclusion that macrophages do not produce cytokines in response to malaria parasites due to endosomal acidification disrupting receptor-ligand interaction agrees with the previous findings by us and others that parasite-induced cytokine responses are mediated mainly through endosomal receptors, TLR9 and TLR7; no observable TLR2- and TLR4-dependent cytokine responses were evident (19, 22, 27, 28, 41). Although some studies have shown that parasite factors such as glycosylphosphatidylinositols (40) and microparticles produced by IRBCs (49) enriched by purification can induce cytokine responses by macrophages, these factors may not present in parasites at the threshold levels required for inducing cytokine responses as evident from the lack of TLR2- and TLR4-dependent cytokine responses even at acute stages of infection (19, 41). However, it is possible that, in infected people, macrophages produce low levels of secondary cytokines at later stages of infection in response to cytokines such as TNF-α that have already been produced against parasites. It is also possible that people in endemic areas are co-infected with other pathogens for which both macrophages and DCs could produce cytokines. These processes may account for some levels of cytokine responses by monocytes observed in malaria-infected patients at the acute or chronic phases of infection (13, 50).
In contrast to macrophages, DCs recruit low levels of V-type H+-ATPase to the phagosomal membrane upon phagocytosis, limiting phagosomal acidification and maintaining a neutral or slightly above neutral pH for a long period of time (18). Thus, DCs are activated through the interaction of their endosomal receptors with malaria parasites and produce cytokines, thereby having the ability to induce adaptive immunity. In contrast, although monocytes and macrophages together outnumber DCs, the former cells becoming functionally impaired upon the uptake of parasites substantially limits the capacity of the innate immune system to effectively induce adaptive immunity to malaria. This may be partly responsible for the slow and inefficient development of protective immunity to malaria, requiring repeated infections. Thus, we predict that allowing macrophages to be functionally active at the early stages of malaria infection by blocking phagosomal acidification with suitable pharmacological inhibitors leads to an efficient development of protective immunity. Given that vaccination using whole parasites is thought to be more effective than the subunit antigen-based approach, and the former strategy is being actively considered (51–53), enabling monocytes/macrophages to remain functionally responsive after parasite uptake may enhance the vaccine efficacy. Thus, the results presented here have implication for enhancing protective immunity not only to malaria but also to other pathogens that are mainly recognized by endosomal receptors, and hence cytokine responses are not induced by macrophages due to phagosomal acidification.
Author Contributions
D. C. G. and X. W. designed research, analyzed data, and wrote the paper; X. W. and N. M. G. performed research. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Glenn Dranoff (Dana-Farber Cancer Institute) for providing the FLT3 ligand-expressing B16 cell line; Dr. Christopher Norbury (Microbiology and Immunology, Hershey Medical Center) for GM-CSF producing X63 melanoma cells; and Dr. Sanjai Kumar (Laboratory of Emerging Pathogens, Food and Drug Administration) for P. yoelii XNL parasites.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 AI41139 from NIAID. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Figs. S1 and S2.
- DC
- dendritic cell
- BMDM
- bone marrow cell-derived macrophage
- IRBC
- infected RBC
- Py17XNL
- P. yoelii 17XNL strain
- PbANKA
- P. berghei ANKA strain
- CpG ODN
- CpG oligodeoxynucleotide
- GM-DC
- DC obtained by the differentiation of mouse bone marrow cells with GM-CSF
- FL-DC
- DC obtained by the differentiation of mouse bone marrow cells with Fms-like tyrosine kinase 3 (FLT3) ligand
- PBMC
- peripheral blood mononuclear cell
- V-type H+-ATPase
- vacuolar-type proton pump-ATPase
- PE
- phycoerythrin.
References
- 1. Murray C. J., Rosenfeld L. C., Lim S. S., Andrews K. G., Foreman K. J., Haring D., Fullman N., Naghavi M., Lozano R., Lopez A. D. (2012) Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet 379, 413–431 [DOI] [PubMed] [Google Scholar]
- 2. Snow R. W., Guerra C. A., Noor A. M., Myint H. Y., Hay S. I. (2005) The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature 434, 214–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schofield L., Grau G. E. (2005) Immunological processes in malaria pathogenesis. Nat. Rev. Immunol. 5, 722–735 [DOI] [PubMed] [Google Scholar]
- 4. Stevenson M. M., Riley E. M. (2004) Innate immunity to malaria. Nat. Rev. Immunol. 4, 169–180 [DOI] [PubMed] [Google Scholar]
- 5. Urban B. C., Ing R., Stevenson M. M. (2005) Early interactions between blood-stage Plasmodium parasites and the immune system. Curr. Top. Microbiol. Immunol. 297, 25–70 [DOI] [PubMed] [Google Scholar]
- 6. Sharma S., DeOliveira R. B., Kalantari P., Parroche P., Goutagny N., Jiang Z., Chan J., Bartholomeu D. C., Lauw F., Hall J. P., Barber G. N., Gazzinelli R. T., Fitzgerald K. A., Golenbock D. T. (2011) Innate immune recognition of an AT-rich stem-loop DNA motif in the Plasmodium falciparum genome. Immunity 35, 194–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Banchereau J., Steinman R. M. (1998) Dendritic cells and the control of immunity. Nature 392, 245–252 [DOI] [PubMed] [Google Scholar]
- 8. Shortman K., Heath W. R. (2010) The CD8+ dendritic cell subset. Immunol. Rev. 234, 18–31 [DOI] [PubMed] [Google Scholar]
- 9. Murray P. J., Wynn T. A. (2011) Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 11, 723–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. den Haan J. M., Kraal G. (2012) Innate immune functions of macrophage subpopulations in the spleen. J. Innate Immun. 4, 437–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Langhorne J., Ndungu F. M., Sponaas A. M., Marsh K. (2008) Immunity to malaria: more questions than answers. Nat. Immunol. 9, 725–732 [DOI] [PubMed] [Google Scholar]
- 12. Struik S. S., Riley E. M. (2004) Does malaria suffer from lack of memory? Immunol. Rev. 201, 268–290 [DOI] [PubMed] [Google Scholar]
- 13. Chua C. L., Brown G., Hamilton J. A., Rogerson S., Boeuf P. (2013) Monocytes and macrophages in malaria: protection or pathology? Trends Parasitol. 29, 26–34 [DOI] [PubMed] [Google Scholar]
- 14. Malaguarnera L., Musumeci S. (2002) The immune response to Plasmodium falciparum malaria. Lancet Infect. Dis. 2, 472–478 [DOI] [PubMed] [Google Scholar]
- 15. Erdman L. K., Cosio G., Helmers A. J., Gowda D. C., Grinstein S., Kain K. C. (2009) CD36 and TLR interactions in inflammation and phagocytosis: implications for malaria. J. Immunol. 183, 6452–6459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schwarzer E., Kuhn H., Valente E., Arese P. (2003) Malaria-parasitized erythrocytes and hemozoin nonenzymatically generate large amounts of hydroxy fatty acids that inhibit monocyte functions. Blood 101, 722–728 [DOI] [PubMed] [Google Scholar]
- 17. Schwarzer E., Turrini F., Ulliers D., Giribaldi G., Ginsburg H., Arese P. (1992) Impairment of macrophage functions after ingestion of Plasmodium falciparum-infected erythrocytes or isolated malarial pigment. J. Exp. Med. 176, 1033–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Savina A., Amigorena S. (2007) Phagocytosis and antigen presentation in dendritic cells. Immunol. Rev. 219, 143–156 [DOI] [PubMed] [Google Scholar]
- 19. Gowda N. M., Wu X., Gowda D. C. (2012) TLR9 and MyD88 are crucial for the development of protective immunity to malaria. J. Immunol. 188, 5073–5085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Trager W., Jensen J. B. (1976) Human malaria parasites in continuous culture. Science 193, 673–675 [DOI] [PubMed] [Google Scholar]
- 21. Lambros C., Vanderberg J. P. (1979) Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 65, 418–420 [PubMed] [Google Scholar]
- 22. Wu X., Gowda N. M., Kumar S., Gowda D. C. (2010) Protein-DNA complex is the exclusive malaria parasite component that activates dendritic cells and triggers innate immune responses. J. Immunol. 184, 4338–4348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Inaba K., Inaba M., Romani N., Aya H., Deguchi M., Ikehara S., Muramatsu S., Steinman R. M. (1992) Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 176, 1693–1702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brasel K., De Smedt T., Smith J. L., Maliszewski C. R. (2000) Generation of murine dendritic cells from flt3-ligand-supplemented bone marrow cultures. Blood 96, 3029–3039 [PubMed] [Google Scholar]
- 25. Stout-Delgado H. W., Getachew Y., Miller B. C., Thiele D. L. (2007) Intrahepatic lymphocyte expression of dipeptidyl peptidase I-processed granzyme B and perforin induces hepatocyte expression of serine proteinase inhibitor 6 (Serpinb9/SPI-6). J. Immunol. 179, 6561–6567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Egan T. J., Chen J. Y., de Villiers K. A., Mabotha T. E., Naidoo K. J., Ncokazi K. K., Langford S. J., McNaughton D., Pandiancherri S., Wood B. R. (2006) Haemozoin (β-haematin) biomineralization occurs by self-assembly near the lipid/water interface. FEBS Lett. 580, 5105–5110 [DOI] [PubMed] [Google Scholar]
- 27. Parroche P., Lauw F. N., Goutagny N., Latz E., Monks B. G., Visintin A., Halmen K. A., Lamphier M., Olivier M., Bartholomeu D. C., Gazzinelli R. T., Golenbock D. T. (2007) Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proc. Natl. Acad. Sci. U.S.A. 104, 1919–1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pichyangkul S., Yongvanitchit K., Kum-arb U., Hemmi H., Akira S., Krieg A. M., Heppner D. G., Stewart V. A., Hasegawa H., Looareesuwan S., Shanks G. D., Miller R. S. (2004) Malaria blood stage parasites activate human plasmacytoid dendritic cells and murine dendritic cells through a Toll-like receptor 9-dependent pathway. J. Immunol. 172, 4926–4933 [DOI] [PubMed] [Google Scholar]
- 29. Seixas E., Cross C., Quin S., Langhorne J. (2001) Direct activation of dendritic cells by the malaria parasite, Plasmodium chabaudi chabaudi. Eur. J. Immunol. 31, 2970–2978 [DOI] [PubMed] [Google Scholar]
- 30. Gowda N. M., Wu X., Kumar S., Febbraio M., Gowda D. C. (2013) CD36 contributes to malaria parasite-induced pro-inflammatory cytokine production and NK and T cell activation by dendritic cells. PLoS ONE 8, e77604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Haque A., Best S. E., Montes de Oca M., James K. R., Ammerdorffer A., Edwards C. L., de Labastida Rivera F., Amante F. H., Bunn P. T., Sheel M., Sebina I., Koyama M., Varelias A., Hertzog P. J., Kalinke U., et al. (2014) Type I IFN signaling in CD8–DCs impairs Th1-dependent malaria immunity. J. Clin. Invest. 124, 2483–2496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hashimoto D., Miller J., Merad M. (2011) Dendritic cell and macrophage heterogeneity in vivo. Immunity 35, 323–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Geissmann F., Gordon S., Hume D. A., Mowat A. M., Randolph G. J. (2010) Unravelling mononuclear phagocyte heterogeneity. Nat. Rev. Immunol. 10, 453–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dent G., Giembycz M. A., Rabe K. F., Wolf B., Barnes P. J., Magnussen H. (1994) Theophylline suppresses human alveolar macrophage respiratory burst through phosphodiesterase inhibition. Am. J. Respir. Cell Mol. Biol. 10, 565–572 [DOI] [PubMed] [Google Scholar]
- 35. Marwick J. A., Wallis G., Meja K., Kuster B., Bouwmeester T., Chakravarty P., Fletcher D., Whittaker P. A., Barnes P. J., Ito K., Adcock I. M., Kirkham P. A. (2008) Oxidative stress modulates theophylline effects on steroid responsiveness. Biochem. Biophys. Res. Commun. 377, 797–802 [DOI] [PubMed] [Google Scholar]
- 36. Russell D. G., Vanderven B. C., Glennie S., Mwandumba H., Heyderman R. S. (2009) The macrophage marches on its phagosome: dynamic assays of phagosome function. Nat. Rev. Immunol. 9, 594–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Blander J. M., Medzhitov R. (2006) On regulation of phagosome maturation and antigen presentation. Nat. Immunol. 7, 1029–1035 [DOI] [PubMed] [Google Scholar]
- 38. Blanchette C. D., Woo Y. H., Thomas C., Shen N., Sulchek T. A., Hiddessen A. L. (2009) Decoupling internalization, acidification and phagosomal-endosomal/lysosomal fusion during phagocytosis of InlA coated beads in epithelial cells. PLoS ONE 4, e6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lukacs G. L., Rotstein O. D., Grinstein S. (1991) Determinants of the phagosomal pH in macrophages. In situ assessment of vacuolar H+-ATPase activity, counterion conductance, and H+ “leak”. J. Biol. Chem. 266, 24540–24548 [PubMed] [Google Scholar]
- 40. Zhu J., Wu X., Goel S., Gowda N. M., Kumar S., Krishnegowda G., Mishra G., Weinberg R., Li G., Gaestel M., Muta T., Gowda D. C. (2009) MAPK-activated protein kinase 2 differentially regulates Plasmodium falciparum glycosylphosphatidylinositol-induced production of tumor necrosis factor-α and interleukin-12 in macrophages. J. Biol. Chem. 284, 15750–15761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Baccarella A., Huang B. W., Fontana M. F., Kim C. C. (2014) Loss of Toll-like receptor 7 alters cytokine production and protects against experimental cerebral malaria. Malar. J. 13, 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Steinberg B. E., Huynh K. K., Grinstein S. (2007) Phagosomal acidification: measurement, manipulation and functional consequences. Biochem. Soc. Trans. 35, 1083–1087 [DOI] [PubMed] [Google Scholar]
- 43. Elson G., Dunn-Siegrist I., Daubeuf B., Pugin J. (2007) Contribution of Toll-like receptors to the innate immune response to Gram-negative and Gram-positive bacteria. Blood 109, 1574–1583 [DOI] [PubMed] [Google Scholar]
- 44. Sponaas A. M., Cadman E. T., Voisine C., Harrison V., Boonstra A., O'Garra A., Langhorne J. (2006) Malaria infection changes the ability of splenic dendritic cell populations to stimulate antigen-specific T cells. J. Exp. Med. 203, 1427–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hochrein H., Shortman K., Vremec D., Scott B., Hertzog P., O'Keeffe M. (2001) Differential production of IL-12, IFN-α, and IFN-γ by mouse dendritic cell subsets. J. Immunol. 166, 5448–5455 [DOI] [PubMed] [Google Scholar]
- 46. Doxsee C. L., Riter T. R., Reiter M. J., Gibson S. J., Vasilakos J. P., Kedl R. M. (2003) The immune response modifier and Toll-like receptor 7 agonist S-27609 selectively induces IL-12 and TNF-α production in CD11c+CD11b+CD8− dendritic cells. J. Immunol. 171, 1156–1163 [DOI] [PubMed] [Google Scholar]
- 47. Edwards A. D., Diebold S. S., Slack E. M., Tomizawa H., Hemmi H., Kaisho T., Akira S., Reis e Sousa C. (2003) Toll-like receptor expression in murine DC subsets: lack of TLR7 expression by CD8α+ DC correlates with unresponsiveness to imidazoquinolines. Eur. J. Immunol. 33, 827–833 [DOI] [PubMed] [Google Scholar]
- 48. Wu J., Tian L., Yu X., Pattaradilokrat S., Li J., Wang M., Yu W., Qi Y., Zeituni A. E., Nair S. C., Crampton S. P., Orandle M. S., Bolland S. M., Qi C. F., Long C. A., et al. (2014) Strain-specific innate immune signaling pathways determine malaria parasitemia dynamics and host mortality. Proc. Natl. Acad. Sci. U.S.A. 111, E511–E520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Couper K. N., Barnes T., Hafalla J. C., Combes V., Ryffel B., Secher T., Grau G. E., Riley E. M., de Souza J. B. (2010) Parasite-derived plasma microparticles contribute significantly to malaria infection-induced inflammation through potent macrophage stimulation. PLoS Pathog. 6, e1000744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Antonelli L. R., Leoratti F. M., Costa P. A., Rocha B. C., Diniz S. Q., Tada M. S., Pereira D. B., Teixeira-Carvalho A., Golenbock D. T., Gonçalves R., Gazzinelli R. T. (2014) The CD14+CD16+ inflammatory monocyte subset displays increased mitochondrial activity and effector function during acute Plasmodium vivax malaria. PLoS Pathog. 10, e1004393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Good M. F. (2011) A whole parasite vaccine to control the blood stages of Plasmodium: the case for lateral thinking. Trends Parasitol. 27, 335–340 [DOI] [PubMed] [Google Scholar]
- 52. Seder R. A., Chang L. J., Enama M. E., Zephir K. L., Sarwar U. N., Gordon I. J., Holman L. A., James E. R., Billingsley P. F., Gunasekera A., Richman A., Chakravarty S., Manoj A., Velmurugan S., Li M., et al. (2013) Protection against malaria by intravenous immunization with a nonreplicating sporozoite vaccine. Science 341, 1359–1365 [DOI] [PubMed] [Google Scholar]
- 53. Targett G. A., Moorthy V. S., Brown G. V. (2013) Malaria vaccine research and development: the role of the WHO MALVAC committee. Malar. J. 12, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.