

Background: Phenylalanine 508 (Phe-508) deletion in the first nucleotide-binding domain (NBD1) of the cystic fibrosis transmembrane conductance regulator (CFTR) causes cystic fibrosis.
Results: The Phe-508 deletion reduces the ability of NBD1 to homodimerize.
Conclusion: Deletion of Phe-508 likely also inhibits the NBD1/NBD2 interaction required for CFTR production and function.
Significance: The NBD1/NBD2 domain interface is an important therapeutic target.
Keywords: ABC transporter, cystic fibrosis transmembrane conductance regulator (CFTR), nuclear magnetic resonance (NMR), protein processing, protein/protein interaction, F508del, NBD dimerization, dynamics, nucleotide-binding domain (NBD)
Abstract
Deletion of Phe-508 (F508del) in the first nucleotide-binding domain (NBD1) of the cystic fibrosis transmembrane conductance regulator (CFTR) results in destabilization of the domain, intramolecular interactions involving the domain, and the entire channel. The destabilization caused by F508del manifests itself in defective channel processing and channel gating defects. Here, we present NMR studies of the effect of F508del and the I539T stabilizing mutation on NBD1 dynamics, with a view to understanding these changes in stability. Qualitatively, F508del NMR spectra exhibit significantly more peak broadening than WT spectra due to the enhanced intermediate time scale (millisecond to microsecond) motions in the mutant. Unexpectedly, studies of fast (nanosecond to picosecond) motions revealed that F508del NBD1 tumbles more rapidly in solution than WT NBD1. Whereas F508del tumbles at a rate nearly consistent with the monomeric state, the WT protein tumbles significantly more slowly. Paramagnetic relaxation enhancement experiments confirm that NBD1 homodimerizes in solution in the expected head-to-tail orientation. NMR spectra of WT NBD1 reveal significant concentration-dependent chemical shift perturbations consistent with NBD1 dimerization. Chemical shift analysis suggests that the more rapid tumbling of F508del is the result of an impaired ability to dimerize. Based on previously published crystal structures and NMR spectra of various NBD1 mutants, we propose that deletion of Phe-508 affects Q-loop conformational sampling in a manner that inhibits dimerization. These results provide a potential mechanism for inhibition of channel opening by F508del and support the dimer interface as a target for cystic fibrosis therapeutics.
Introduction
Cystic fibrosis is an inherited disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR)2 chloride channel (1), which is expressed on the apical surface of epithelial cells. The dominant disease-causing mutation is deletion of Phe-508 (F508del), which results in greatly reduced folding and trafficking efficiency and rapid degradation of the few CFTR molecules that do reach the cell membrane (2, 3). F508del also reduces the probability of channel opening (4, 5). One effect of these deficiencies is to reduce the amount of chloride entering the lumen of the lungs via lung epithelial cells, disrupting the salt balance in the airway fluid, thickening the fluid, and preventing proper clearance of bacteria.
CFTR consists of two modules each containing a membrane-spanning domain (MSD1 and MSD2) and a cytoplasmic nucleotide-binding domain (NBD1 and NBD2). The C terminus of NBD1 is joined to the N terminus of MSD2 by a long intrinsically disordered segment termed the regulatory (R) region. The cytoplasmic extensions of the MSDs form the intracellular domains and loops (ICLs), which couple the NBDs to the MSDs. The first 30 residues of the R region are alternatively considered to be part of NBD1 and are termed the regulatory extension (RE). NBD1 also contains a 30-residue intrinsically disordered regulatory insertion (RI) (6–10). Channel activation is thought to involve phosphorylation of sites in the R region, RE, and RI and concomitant reduction of dimer inhibiting interactions between these regulatory regions and the NBDs, binding of two ATP molecules at the NBD dimer interface, and NBD dimerization (11–17).
The mechanism by which F508del causes the observed processing, gating, and stability defects has been the subject of intense study. Crystal structures of mouse and human, WT, and mutant NBD1 indicate that deletion of Phe-508, which is part of a loop within NBD1, does not cause a major structural perturbation in NBD1 (6–10). Rather, F508del has been shown to decrease the stability of NBD1 and inhibit NBD1/ICL interactions (18–25). F508del lowers the thermal stability of the isolated NBD1 domain in a variety of sequence backgrounds (18, 19, 26). Evidence that NBD1 destabilization is problematic for proper processing was provided by NBD1-thermostabilizing mutations distant from the F508del site, including G550E, R553Q, R555K, and deletion of the RI. These NBD1-stabilizing mutations substantially improve processing efficiency of F508del CFTR without correcting structural perturbations near the F508del location (20, 27–29). Furthermore, the degree to which mutations stabilize or destabilize the isolated NBD1 domain is roughly correlated with full-length CFTR-processing efficiency (24, 25), establishing F508del destabilization of NBD1 as a key mechanism for the deleterious effects of the mutation.
Nonetheless, even with suppressor mutations that stabilize F508del NBD1 beyond the stability of the WT domain, the full-length F508del CFTR is not processed as efficiently as the WT, implying that F508del has additional defects. NBD1 crystal structures and CFTR models indicate that the F508del site is at the interface between NBD1 and ICL4 and that F508del creates a void at this interface. The interface defect can be corrected by mutations at this site, which presumably restore the strength of the interaction (24, 25, 30). Simultaneous introduction of NBD1-thermostabilizing and NBD1/ICL4 interface-correcting mutations synergistically improve CFTR F508del processing to near WT levels (24, 25), indicating that both defects contribute to F508del misprocessing.
Here, we report on NMR studies of human CFTR NBD1 ΔRIΔRE, an NBD1 construct ending at residue 646 and missing residues 405–436, which correspond to the RI. We refer to this construct as “WT” NBD1 ΔRIΔRE, and it is the background for further mutations. Completion of >82% of assignments for WT, F508del, and I539T NBD1 ΔRIΔRE allowed extensive characterization of NBD1. Overall, F508del NMR spectra exhibit significantly more peak broadening than WT spectra, likely due to enhanced intermediate time scale motions in the mutant, which is consistent with the greater instability of F508del NBD1. However, our data indicate that F508del may also impair the ability of NBD1 to dimerize, an ability that is likely required for proper folding of NBD2 (31) and channel gating. Unexpectedly, studies of fast (nanosecond to picosecond) motions revealed that isolated F508del NBD1 ΔRIΔRE tumbles more rapidly in solution than WT or I539T NBD1 ΔRIΔRE. F508del NBD1 ΔRIΔRE tumbles at a rate close to that expected for monomeric NBD1, whereas the WT protein tumbles more slowly consistent with some degree of homodimerization. The concentration dependence of chemical shift perturbations observed in NMR spectra of WT NBD1 ΔRIΔRE and paramagnetic relaxation enhancement data provide further evidence for NBD1 dimerization. Chemical shift analysis suggests that the more rapid tumbling of F508del is the result of impaired dimerization competency. Considering previously published crystal structures and NMR spectra of various NBD1 ΔRIΔRE mutants, we propose that deletion of Phe-508, which directly interacts with the Q-loop, promotes a Q-loop conformation that is unfavorable for homodimerization and, by extension, heterodimerization with NBD2.
Experimental Procedures
Protein Expression and Purification
WT, F508del, and I539T variants of human NBD1 ΔRIΔRE (387–646, Δ405–436) were expressed and purified as described previously (32). A single cysteine mutant of NBD1, E402C NBD1 ΔRIΔRE, was generated on a Cys-less NBD1 (C491V, C524T, C590V, and C592V) background by site-directed mutagenesis and confirmed by DNA sequencing. Mutations were chosen for the Cys-less background by predicting the change in free energy of unfolding (ΔΔG) for particular amino acid substitutions using Rosetta3.4 (33). Protein quantification was performed using the Bradford assay calibrated using a BSA standard. Samples were filter-sterilized to prevent bacterial growth and resulting ATP hydrolysis and acidification of the samples. The tempo-maleimide spin label was conjugated to E402C, by first reducing the cysteine with 5 mm of freshly added DTT followed by extensive buffer exchange into a buffer with no DTT and subsequent incubation with a 5-fold excess of tempo-maleimide. The spin label tempo-maleimide was chosen because it forms a stable thioether linkage with cysteine side chains that is not disrupted by addition of the reductant tris(2-carboxyethyl) phosphine (TCEP) and because it does not form mixed disulfides. Following the labeling reaction, the sample was purified by gel filtration to remove excess tempo-maleimide. Tempo-maleimide-labeled protein was used soon after preparation, because we have found that hydrolysis of the succinimide ring occurs over time increasing the range of motion available for the tempo moiety. Mass spectrometry was performed on all samples to confirm that their masses matched the expected values.
NMR Spectroscopy
All NMR experiments were carried out in the previously described NMR buffer (32) at pH 7.5 unless otherwise stated. Experiments were run at 20 °C on Varian Innova 500 MHz spectrometers equipped with pulsed field gradient triple resonance probes or a Varian Innova 600 MHz spectrometer equipped with a pulsed field gradient triple resonance cryoprobe. Data were processed with NMRPipe (34) and analyzed in NMRView (35) and Sparky (36). Assignments were made using TROSY versions of HNCO, HN(CA)CO, HNCA, HN(CO)CA, HN(CA)CB, HN(COCA)CB, and three-dimensional NOESY experiments (37–41). Assignment samples were 15N- and 13C-labeled and partially (70%) or fully deuterated at all nonlabile protons. TALOS+ was used to derive secondary structure from NBD1 chemical shifts (42). Paramagnetic relaxation enhancement experiments were measured using standard HSQC experiments on a sample containing 525 μm tempo-maleimide-labeled natural abundance E402C NBD1 ΔRIΔRE and 175 μm 15N-labeled WT NBD1 ΔRIΔRE in the absence of reductant. To demonstrate the specificity of the PRE effect, TCEP was then added to the sample at a concentration of 7 mm and incubated for 100 h to reduce the spin label and eliminate the PRE effect. HSQC spectra were then recorded for use as the control experiment.
Backbone dynamics of NBD1 ΔRIΔRE were assessed using T1, T1ρ, and heteronuclear NOE measurements (43–45), measured at 600 MHz using 15N-labeled samples that were 70% deuterated at all nonlabile protons. T1 relaxation times were acquired in an interleaved fashion with delays of 10, 171, 352, 564, 827, 1129, and 1512 ms. T1ρ times were recorded in an interleaved fashion using delay values of 2, 4, 9, 15, 21, 30, and 40 ms and a 1.9 kHz spin-lock field. Following spectral processing using NMRPipe software, peaks intensities were fit using the software FuDA (developed by D. F. Hansen and S. Kristensen and available on line). Exponential decays were fit to the equation I(Trelax) = I0·exp(−RTrelax), where Trelax is the delay time and I0 is the initial peak intensity. T2 values were calculated from the measured T1 and T1ρ values using the equation T2 = (sin2θ·T1·T1ρ)/(T1 − T1ρ·cos2θ) with θ = tan−1(υSL/Δυ), where υSL is the strength of the spin lock, and Δυ is the offset from the carrier frequency. {1H}15N heteronuclear NOE values were calculated as the ratio of peak intensities obtained with 5 s of proton presaturation and a 7-s interscan delay over those obtained with no presaturation and a 12-s interscan delay. The overall rotational correlation time for each protein was calculated from T1 and T2 using the program DASHA (46). The expected τc was calculated using HydroNMR (47).
We analyzed the relaxation data, taking into account the observation that NBD1 likely undergoes exchange between monomeric and dimeric (or oligomeric) forms. Assuming that exchange occurs rapidly, the apparent relaxation rate corresponds to the population-weighted average according to Equation 1,
 |
where pn and (R)n are the relative population and spin relaxation rate of the nth exchanging state, respectively, and there are N oligomeric states in total. Relaxation rates can be cast as linear combinations of the spectral density function, J(ω), evaluated at various frequencies, ω (48). For instance, the longitudinal relaxation rate, R1, is given by Equation 2,
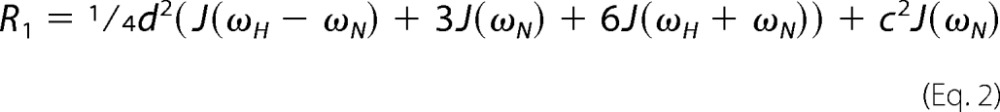 |
where d = (μ0hγNγH/(8π2))〈1/rNH3〉; c = (ωN/30.5)(CSApar −CSAprp); ωH and ωN are the 1H and 15N Larmor frequencies; μ0 is the permeability of free space; γH and γN are the 1H and 15N gyromagnetic ratios; h is Planck's constant; rNH is the length of the 1H–15N bond, and CSApar and CSAprp are the parallel and perpendicular components of the assumed axially symmetric 15N chemical shift tensor. Thus, with averaging, the apparent longitudinal relaxation rate is a linear combination of population-weighted average values of the spectral density function shown in Equation 3,
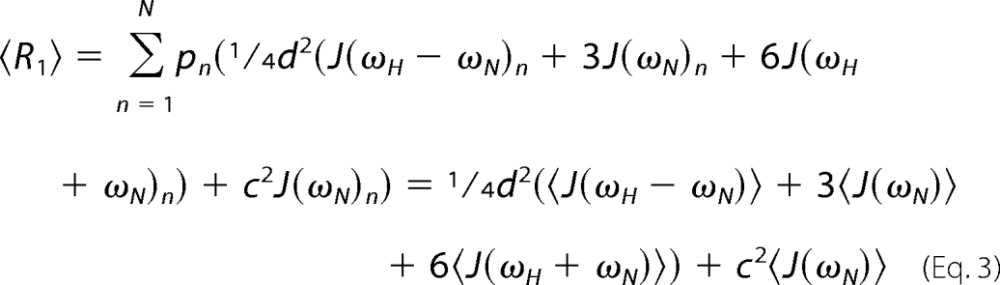 |
where J(ω)n is the spectral density function at a frequency ω for the nth oligomeric state. Similarly, the apparent transverse relaxation rate is given by Equation 4,
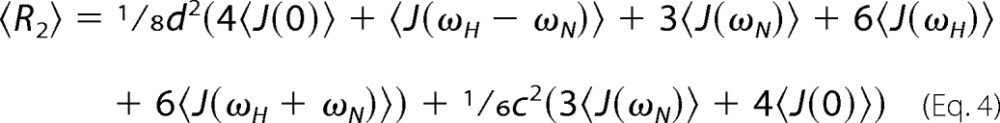 |
In the case of the NOE, a steady state is reached when the condition of Equation 5 is met.
where NzSS and Nzeq are 15N magnetization with and without 1H saturation; Izeq is the magnitude of 1H magnetization at equilibrium, and σ is the cross-relaxation rate (49). Thus with rapid averaging among oligomeric states Equation 6 applies.
and the apparent steady-state NOE is given by Equation 7,
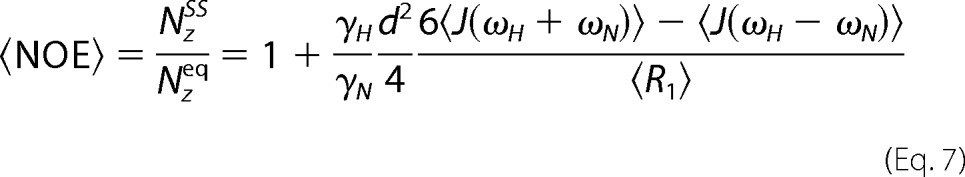 |
Equations 2, 3, and 7 can be rearranged to isolate values of J(ω) using the reduced spectral density mapping approximation to yield Equations 8–10,
 |
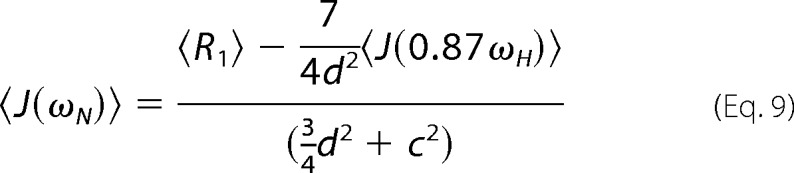 |
 |
The spectral density function can be related to the motions of NH bond vectors according to the Lipari-Szabo formalism shown in Equation 11,
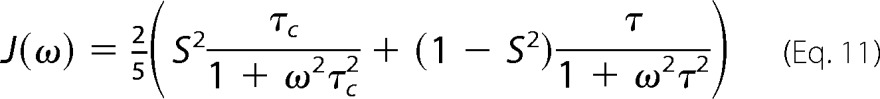 |
where τc is the rotational correlation time for molecular tumbling in solution; 1/τ = 1/τc + 1/τe, and τe is the effective correlation time for internal motions (50). S2 is an order parameter describing the magnitude of internal motions, and can take values between 1, for an NH bond-vector that is perfectly rigid in the molecular frame, and 0 for one that is isotropically averaging. Assuming that the order parameter, S2 is the same in all oligomeric states, the exchange-averaged value of J(0) is shown in Equation 12,
 |
where τe≪τc, so we get Equation 13
In the case of J(ωN) and J(0.87ωH), we get Equations 14 and 15,
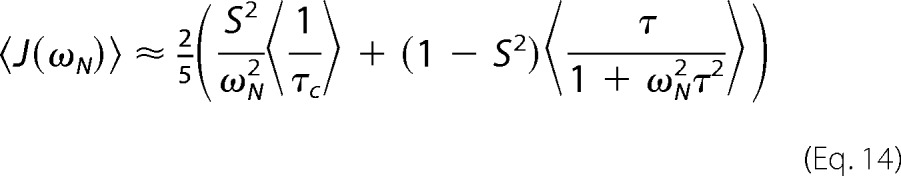 |
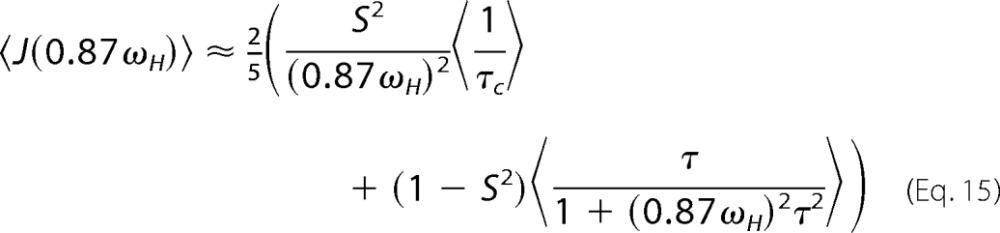 |
To calculate the molecular tumbling time from relaxation measurements, it is typically assumed that internal motions are in the narrowing, τ ≈ 0, limit (43). The ratio of T1 and T2 relaxation times is then a function of τc only. Values of τc were extracted for each protein based on T1/T2 ratios using DASHA software (46). The dependence of T1/T2 ratios on exchange-averaged tumbling times is clearly evident when ωτc ≫ 1 and ωN−2 ≫ {ωH−2, (ωH − ωN)−2, (ωH + ωN)−2}, as is the case here. The ratio then yields Equation 16,
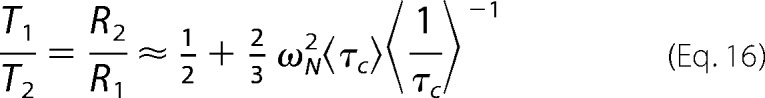 |
By assuming that 〈τc〉〈1/τc〉−1 ≈ 〈τc〉2, the average molecular tumbling time, 〈τc〉, can be obtained on a per residue basis. This approximation is particularly applicable to the exchange processes studied here, which we believe are dominated by monomer/dimer equilibria. For these systems, it can be shown that 〈τc〉 = λ〈1/τc〉−1, where λ = 1 + fD(1 − fD)(r + (1/r) − 2); fD is the fraction of protein in the dimeric state, and r is the dimer/monomer ratio of correlation times (∼2). For highly asymmetric populations, λ is very close to 1, and the approximation is excellent. For equal populations fD = 0.5; λ is maximal at λ = 1.125 for r = 2, which still represents fairly minimal error.
Results
Assignments
As a prerequisite for NMR solution studies, we assigned human NBD1 ΔRIΔRE chemical shifts. Removal of the RI (residues 405–436) dramatically improves expression and solubility of human NBD1 (10), resulting in NBD1 NMR spectra amenable to detailed analysis (Fig. 1a). All of the work described in this study utilized NBD1 with the RI (and RE) removed. NBD1 ΔRIΔRE with the I539T-stabilizing mutation (23) was assigned first, with 91% completion of backbone 15N, 13C′, and 1HN resonances. Full assignment experiments were also recorded for WT and, together with the I539T assignments, enabled us to complete assignment of 89% of backbone 15N, 13C′, and 1HN resonances for WT. These assignments and a partial set of assignment experiments were then used as a basis for the F508del assignment, which achieved 82% completion, as reported previously (32).
FIGURE 1.
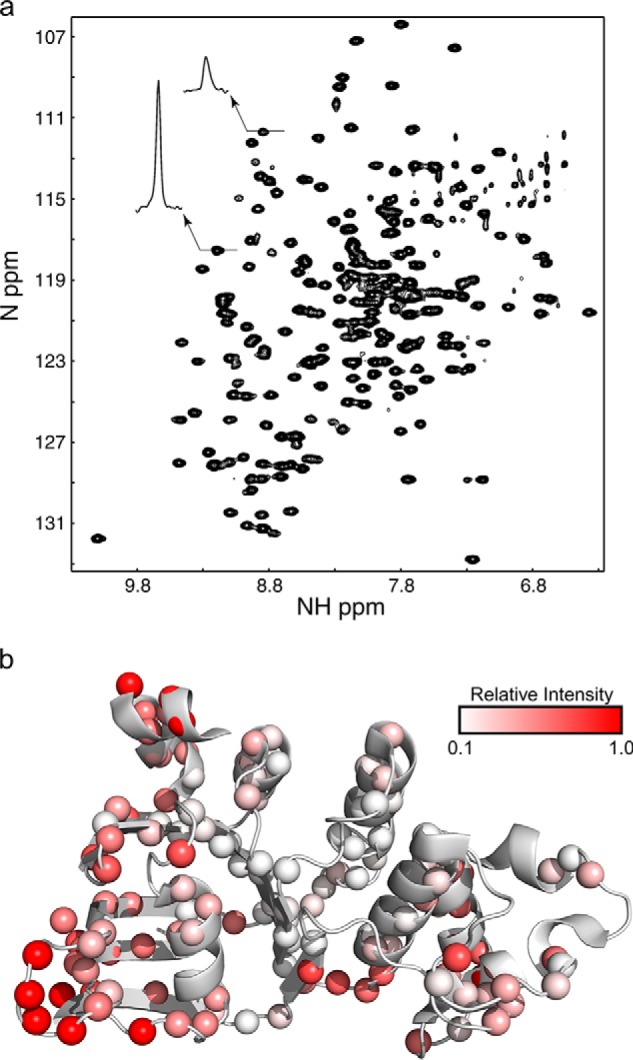
a, 1H-15N HSQC spectrum of 180 μm human CFTR NBD1 ΔRIΔRE recorded at 20 °C. Note the variation in peak intensity that results from chemical exchange as illustrated by the two peak cross-sections. b, ribbon diagram of NBD1 showing relative peak intensities observed in the 1H-15N HSQC spectrum of partially deuterated human NBD1 ΔRIΔRE recorded at 600 μm and at 20 °C in the presence of 5 mm ATP at a field strength of 600 MHz. Data points are shown as spheres colored according to their relative intensity as shown in the legend.
Comparison of WT, I539T, and F508del Spectra
Human NBD1 ΔRIΔRE spectra, while significantly better than full-length mouse NBD1 spectra with the RI (13), still exhibit inhomogeneous peak intensities (Fig. 1). Inhomogeneity in peak intensity can result from differences in flexibility across the protein with regions that can rapidly access multiple conformations on a nanosecond to picosecond time scale (for example flexible loops) exhibiting greater peak intensities. Conversely, loss of peak intensity frequently results when atoms in the protein exchange between different environments on a slower microsecond to millisecond time scale, a process known as chemical exchange. Chemical exchange can result from ligand binding and exchange between oligomeric states or conformational exchange. For WT NBD1 ΔRIΔRE, weaker peaks are observed for many residues in the ATP-binding core and the α-subdomain. The weak peaks in the interior of the ATP-binding core are consistent with NBD1 microsecond to millisecond conformational lability extending through these regions and are most likely not due to exchange of ATP. This NBD1 construct binds tightly to ATP and ATP exchange appears to be minimal and likely does not make a significant contribution to the variable peak intensities. Corroborating the high ATP affinity, we were unable to obtain NMR spectra of this NBD1 construct in the absence of ATP, despite two attempts using either extensive dialysis (three times 1000-fold) or buffer exchange in a protein concentrator (2000-fold). This is consistent with the sub-micromolar ATP affinity reported in the literature for the NBD1 ΔRIΔRE construct (18).
Comparison of WT, I539T, and F508del demonstrates overlapping unassigned regions clustered on or near the α-subdomain, including most of the Q-loop, portions of helix 5 (H5), and the adjacent ABC signature sequence and residues immediately following the Walker B motif (Fig. 2). Although there is a caveat that transferring WT assignments to F508del could make it difficult to assign residues not assigned in the WT, this does suggest the presence of chemical exchange processes that are shared by all three variants in these areas. Many of the unassigned residues are situated at the NBD1 homodimer interface (10), including Gln-493, Gly-550, Ile-546, Thr-547, Leu-548, Gly-576, and Tyr-577 (Fig. 2d), hinting at exchange between monomer and dimer or conformational exchange that is linked to dimerization. Note that besides these unassigned residues, several other residues at the interface are weak, reinforcing the possibility of dimerization-related exchange broadening. The bulk of the ATP-binding core (Fig. 2, gray) and the β-subdomain (green) are assigned in all three variants.
FIGURE 2.
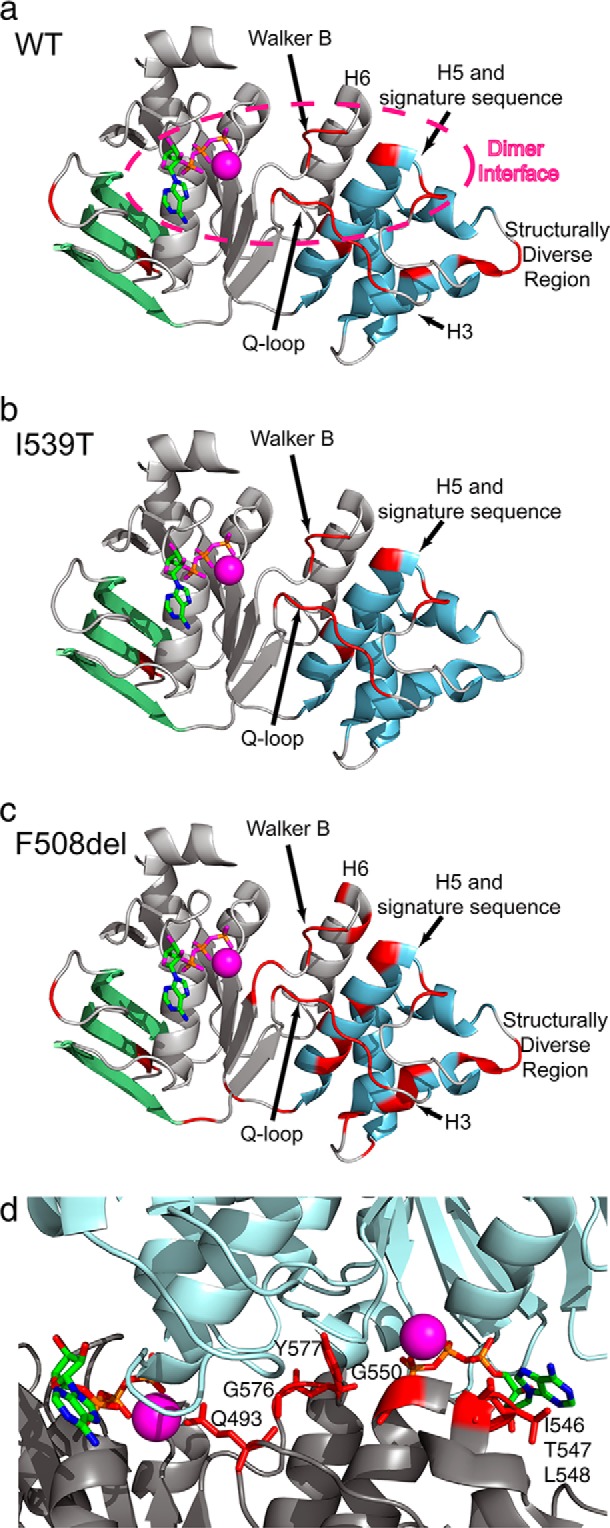
a–c, ribbon diagrams of WT, I539T, and F508del NBD1 ΔRIΔRE. The β-subdomain, ATP-binding core, and α-helical subdomain are labeled in green, gray, and blue, respectively. The stick representation of ATP is shown bound to the Walker A motif and colored by element type (green, carbon; blue, nitrogen; pink, oxygen; orange, phosphorus). Magnesium is shown as a magenta sphere. Unassigned regions are shown in red. All ribbon diagrams in this work use the 2PZE coordinates unless stated otherwise. d, WT unassigned residues shown in the context of the homodimer interface. Ribbon diagrams of the two subunits of NBD1 in the 2PZE structure are shown in gray and light blue, respectively. ATP is colored by element type (green, carbon; blue, nitrogen; red, oxygen; orange, phosphorus), and magnesium is colored magenta. Side chains of unassigned residues are shown and colored red. Note that there are additional assigned residues (data not shown) in this interface with weak overlapped peak intensities for which we were unable to obtain precise chemical shifts in HSQC experiments.
Comparison of WT, I539T, and F508del spectra demonstrate that the two mutations do not cause any major structural changes. To confirm that NBD1 ΔRIΔRE has the same structure in solution as observed in the crystal structures, we used TALOS+ (42) to predict NBD1 secondary structure. We used the set of I539T chemical shift assignments for our prediction, because it was the most complete. As seen in Fig. 3, chemical shift-derived secondary structure closely matches secondary structure observed in the crystal structures. The most notable exceptions are for H5 and H9, which do not have chemical shifts consistent with α-helical structure. In the case of H5, this can be attributed to missing assignments. H9 is observed in both helical and random coil conformations in the crystal structures, and our previously published results indicate that H9 exists in equilibrium between helical and random coil conformations (32). We also recorded NOESY spectra for WT NBD1 ΔRIΔRE and looked for NOE distance restraints between residues that are far apart in the primary sequence but close in space based on the crystal structure. More than 60 NOEs fitting these criteria were identified in a nonexhaustive analysis (Table 1). Thus, as expected, both the chemical shift and NOE data confirm that NBD1 has the same structure in solution as that observed in the crystal structures.
FIGURE 3.

Plot of TALOS+ chemical shift derived secondary structure for I539T NBD1 ΔRIΔRE as a function of residue number. Values of 1 on the graph indicate β-strand, and values of −1 indicate α-helix. The colored rectangles at the top of the graph indicate the position of β-strands (blue) and α-helices (pink) in the NBD1 ΔRIΔRE crystal structure (2PZE).
TABLE 1.
Partial list of observed long range amide proton NOEs for WT CFTR NBD1 ΔRIΔRE and corresponding distance in PDB 2PZE
| Proton 1 | Proton 2 | Distance | Proton 1 | Proton 2 | Distance |
|---|---|---|---|---|---|
| Å | Å | ||||
| 619 HN | 458 HN | 4.9 | 397 HN | 444 HN | 2.7 |
| 619 HN | 626 HN | 5.3 | 397 HN | 481 HN | 4.8 |
| 483 HN | 393 HN | 2.8 | 397 HN | 443 HN | 4.7 |
| 483 HN | 394 HN | 4.9 | 397 HN | 445 HN | 5.0 |
| 399 HN | 441 HN | 2.8 | 441 HN | 399 HN | 2.8 |
| 399 HN | 440 HN | 4.1 | 522 HN | 526 HN | 4.9 |
| 541 HN | 500 HN | 2.7 | 553 HN | 549 HN | 4.4 |
| 572 HN | 491 HN | 3.4 | 489 HN | 567 HN | 4.9 |
| 393 HN | 483 HN | 2.8 | 489 HN | 568 HN | 3.6 |
| 393 HN | 484 HN | 5.0 | 452 HN | 599 HN | 5.3 |
| 395 HN | 481 HN | 2.9 | 567 HN | 488 HN | 4.9 |
| 443 HN | 397 HN | 4.7 | 567 HN | 487 HN | 3.1 |
| 394 HN | 447 HN | 5.0 | 481 HN | 395 HN | 2.9 |
| 440 HN | 401 HN | 3.5 | 446 HN | 395 HN | 5.1 |
| 440 HN | 399 HN | 4.1 | 628 HN | 616 HN | 2.8 |
| 599 HN | 451 HN | 3.4 | 549 HN | 553 HN | 4.4 |
| 599 HN | 452 HN | 5.3 | 624 HN | 442 HN | 5.0 |
| 442 HN | 624 HN | 5.0 | 451 HN | 599 HN | 3.4 |
| 398 HN | 478 HN | 3.8 | 625 HN | 620 HN | 3.5 |
| 445 HN | 396 HN | 5.0 | 500 HN | 542 HN | 5.0 |
| 502 HN | 539 HN | 3.9 | 500 HN | 541 HN | 2.7 |
| 603 HN | 573 HN | 2.8 | 616 HN | 628 HN | 2.8 |
| 396 HN | 444 HN | 5.0 | 442 HN | 399 HN | 5.0 |
| 396 HN | 445 HN | 5.0 | 442 HN | 623 HN | 4.8 |
| 448 HN | 393 HN | 5.1 | 488 HN | 567 HN | 4.9 |
| 453 HN | 614 HN | 3.1 | 489 HN | 570 HN | 3.6 |
| 453 HN | 615 HN | 4.6 | 568 HN | 487 HN | 4.7 |
| 614 HN | 453 HN | 3.1 | 583 HN | 579 HN | 4.6 |
| 487 HN | 567 HN | 3.1 | 589 HN | 593 HN | 5.2 |
| 517 HN | 513 HN | 4.0 | 604 HN | 573 HN | 5.4 |
| 444 HN | 396 HN | 5.0 | 621 HN | 462 HN | 4.9 |
| 444 HN | 397 HN | 2.7 | 501 HN | 541 HN | 4.3 |
| 620 HN | 625 HN | 3.5 | 505 HN | 501 HN | 4.3 |
The similarity between WT, I539T, and F508del spectra strongly supports the conclusion that all share the same fold. Nonetheless, comparison of WT and F508del spectra (Fig. 4a) reveal significant chemical shift perturbations and changes in peak intensities. The chemical shift perturbations are primarily clustered around the mutation, with the Phe-508 loop, H3 and H4, and the C terminus of H5 exhibiting the largest differences (Fig. 4b). Smaller chemical shift perturbations are observed throughout the ATP-binding core, but differences are minimal in the β-subdomain, which includes the loop where the RI is normally found. Smaller chemical shift perturbations in the 20 Hz range are observed as far away as H6. These distant perturbations may be the result of subtle structural or dynamic changes propagated through the structure upon Phe-508 deletion (51), or they could result indirectly from changes in dimerization or ATP binding affinity caused by deletion of Phe-508. Qualitatively, F508del spectra have far more peak broadening, which manifests as much weaker peak intensities. The increase in peak broadening is due to increased chemical exchange in the intermediate (millisecond to microsecond) time scale regime, which could be attributed to increased exchange between oligomeric states or increased conformational exchange within monomeric NBD1.
FIGURE 4.
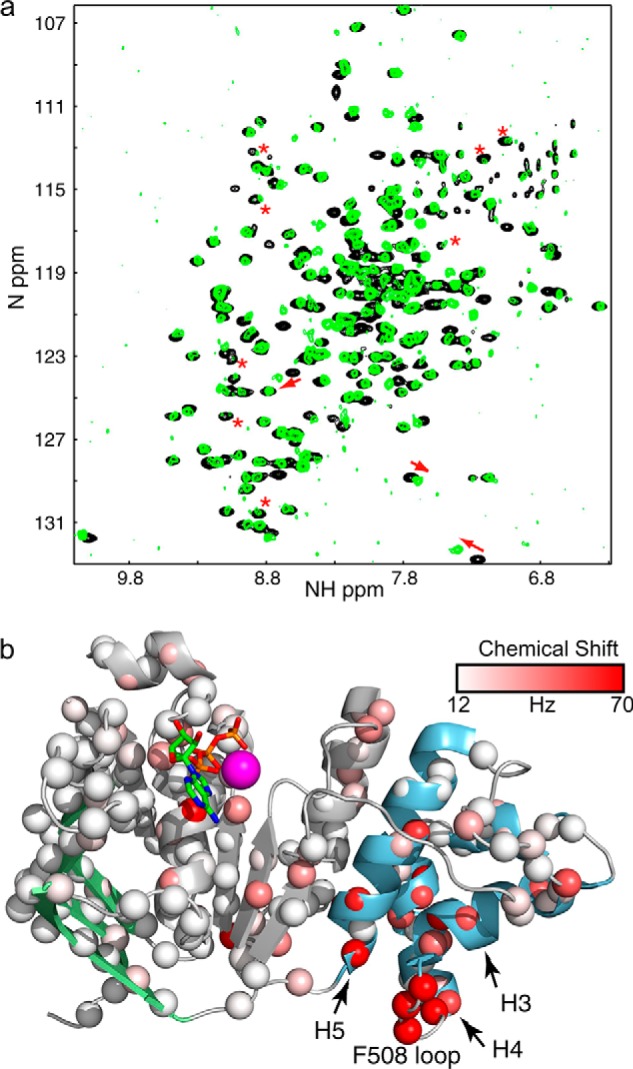
a, overlay of 1H-15N HSQC spectrum of WT (black) and F508del NBD1 ΔRIΔRE (green). The largest chemical shift perturbations are highlighted by red arrows. Changes in peak intensity are highlighted by red asterisks. b, ribbon diagram of NBD1 showing the size and location of chemical shift perturbations between WT and F508del NBD1 ΔRIΔRE. Data points are shown as spheres colored according to the size of the chemical shift perturbation as shown in the legend. ATP is shown and color-coded by element type. The magenta sphere is magnesium.
NBD1 15N Relaxation Studies
Because changes in NBD1 thermostability underlie F508del defects, we probed the changes in dynamics resulting either from deletion of Phe-508 or the I539T stabilizing mutation. We measured T1, T2, and heteronuclear NOE values, which are responsive to fast time scale (nanosecond to picosecond) motions, for WT, I539T, and F508del NBD1 ΔRIΔRE. Based on T1 and T2 measurements, we calculated the overall rotational correlation time (τc) for each protein sample as a prerequisite for analyzing the internal bond motions, to determine whether the NBD1 variants are tumbling at a similar rate, reflective of being a similar size (Table 2). Because we did not expect to see a significant concentration effect on the τc values, no attempt was made to match the protein concentration in the samples. To calculate τc values, we first removed from the analysis residues with rapid internal motion not reflective of the overall tumbling rate, i.e. those residues with heteronuclear NOEs less than 0.6. As can be seen in Table 2, τc varied significantly between the three variants. F508del has the lowest τc, whereas WT had the highest τc despite being at the lowest concentration. These τc values are an average value across the NMR observable portion of the sample. Highly aggregated protein and visible precipitates, which are too large to observe by NMR and are more prevalent in F508del samples, do not contribute to the observed τc values. The F508del τc is close to the expected value for monomeric NBD1 ΔRIΔRE, which we calculated using HydroNMR (47), suggesting that F508del is close to being monomeric under these conditions. In contrast, the WT and I539T protein appear to be in exchange between the monomeric form and dimeric or higher order oligomeric forms resulting in higher τc values. Given the more extensive broadening in F508del spectra, the lower τc was unexpected. The lower τc suggests that the broadening of NMR-observable F508del NBD1 ΔRIΔRE is due to increased intermediate time scale dynamics in the mutant and not due to oligomerization or aggregation within NMR-observable species. Thus, F508del peak broadening is consistent with F508del NBD1 more readily making excursions from the ground state to one or more higher energy states and with the published lower stability of F508del NBD1.
TABLE 2.
Measured or predicted τc values for CFTR NBD1 ΔRIΔRE
Predicted values were obtained using HydroNMR(47) and the 2PZE structure. Measured values were calculated from observed T1, T2, and heteronuclear NOE values after exclusion of residues with NOE values below 0.6.
| Variant | Sample concentration | τc |
|---|---|---|
| mm | ns | |
| Predicted for monomer | 16–20 | |
| Predicted for dimer | 32–38 | |
| WT | 0.6 | 29.4 ± 2.5 |
| F508del | 0.9 | 19.9 ± 1.1 |
| I539T | 1.5 | 27.3 ± 1.5 |
The τc differences also complicate the quantitative interpretation of T1 and T2 in terms of fast time scale dynamics because it is likely that the WT and I539T samples, in particular, contain a mixture of monomers, dimers, and possibly higher order oligomers. As mentioned previously, all three variants also show signs of chemical exchange, which decreases the apparent T2 values in a residue-specific manner. We accounted for this by excluding residues with T1/T2 ratios more than one standard deviation above the mean from our analysis. Because of these limitations, we did not use model-free analysis to obtain S2 order parameter values representative of overall amplitudes of fast time scale dynamics. Instead, we fit spectral density functions to our data to derive a measure of the motion at particular frequencies (Fig. 5). As outlined under “Experimental Procedures,” the analysis yields population-weighted average values of the spectral density function, assuming that exchange among oligomeric forms is rapid. Strikingly, similar profiles are obtained for all three variants, indicating that fast time scale motions are similar. In the case of 〈J(0)〉, smaller values correspond to internal motions with larger amplitudes. Overall, the magnitudes of J(0) for the different variants scale with the apparent correlation times, 〈τc〉, as expected, according to Equation 13. Conversely, for 〈J(0.87ωH)〉, the values for the three variants are nearly superimposable, implying that they are independent of global tumbling and that the second term in Equation 15 dominates. In this case, larger values of 〈J(0.87ωH)〉 correspond to greater mobility and slower time scale internal motions. Interestingly, values of 〈J(0)〉 and 〈J(0.87ωH)〉 are anti-correlated indicating that they are both reporting on the same dynamics. For 〈J(ωN)〉, values decrease with increasing 〈τc〉, yet are roughly anti-correlated with 〈J(0)〉. This suggests that both terms in Equation 14 contribute to the data, consistent with internal motions on the time scale of hundreds of picoseconds.
FIGURE 5.

Spectral density function J(0. 87ωH) ribbon diagram plots for WT (a), F508del (b), and I539T NBD1 ΔRIΔRE (c) are shown. Data points for specific residues are plotted as spheres that are colored according to the legend, from white to orange to red reflecting the range from least to most mobile. d, spectral density functions at three different frequencies as a function of NBD1 residue number for WT (green), F508del (red), and I539T (blue).
Regions with the highest mobility include H9, the S1-S2 loop, and the structurally diverse region ((SDR) amino acids 526–547) (Fig. 5). The high degree of mobility for H9 and the SDR is consistent with the high structural variability observed in the crystal structures for these regions (6–10). H9 flexibility is also consistent with our previous observation that H9 exchanges between helix and coil conformations (32). The mobility of the S1-S2 loop indicates that a high degree of flexibility persists in this region from which the intrinsically disordered RI has been excised. Additional flexibility is observed for the Phe-508-containing loop, the H5-S7 loop at the α-subdomain/ATP-binding core interface, and the S8-H7 loop, where we were able to obtain data. Given that the fast time scale motions involve small local bond motions rather than larger scale conformational rearrangements, these results are consistent with the high degree of similarity observed when comparing the WT and F508del crystal structures and strongly support the conclusion that F508del does not grossly alter the structure of NBD1.
Although the analysis of fast time scale motions matched expectations developed on the basis of the crystal structures, the smaller correlation time of F508del NBD1 ΔRIΔRE was unexpected, because F508del NBD1 has an increased propensity to aggregate that is well documented in the literature (18, 19). However, the observed τc values indicate that F508del is closer to monomeric than WT, for the NMR observable fraction that excludes highly aggregated NBD1.
NBD1 Homodimerization
NBD dimerization is of central interest to CFTR studies because NBD heterodimerization is thought to be important for proper channel folding and required for channel opening. Because of NBD2 insolubility, we could not examine heterodimerization directly and therefore we decided to explore the homodimerization properties of WT NBD1 as a mimic of heterodimerization. We looked at the effects of increasing NBD1 ΔRIΔRE concentrations on chemical shifts and peak intensities at equilibrium and following rapid dilution. In equilibrium studies, NBD1 ΔRIΔRE peak intensities were less than commensurate with increasing protein concentrations, hinting that NBD1 homo-oligomerizes in a concentration-dependent fashion, leading to a decrease in normalized peak intensity. Increasing protein concentrations also increase the solution viscosity, which confounded quantitative analysis of the decrease in normalized peak intensities. Chemical shifts, however, are not directly responsive to changes in viscosity.
Therefore, we turned to the subtle but reproducible concentration-dependent chemical shift perturbations that were observed in these spectra for our quantitative analysis (Fig. 6a). Moving from 0.78 to 0.08 mm NBD1 ΔRIΔRE, amide chemical shift perturbations were observed for Cys-491 (35 Hz), Gly-509 (∼30 Hz), Gly-486 (27 Hz), Gly-551 (16 Hz), Ser-557 (12 Hz), Leu-571 (11 Hz), and Ser-549 (10 Hz), as well as a perturbation for the Trp-496 indole NϵH (27 Hz). Note that the relatively small size of the perturbation at Cys-491 and the concentration dependence, an indicator of fast exchange, rules out disulfide bond formation as an explanation for the chemical shift perturbation. At first glance, the mapping of chemical shift perturbations onto the NBD1 structure (Fig. 7a) does not suggest dimerization at the presumed physiological dimer interface, which is expected to be similar to the NBD1 homodimer interface observed in many of the crystal structures. In particular, Gly-551 and Ser-549 are at this interface; however Cys-491, Ser-557, Leu-571, Trp-496, and Gly-509 are outside of this region but are likely linked to the dimerization interface, as shown below. A critical point to note is that very little chemical shift data could be obtained for residues directly in the interface (Figs. 2d and 7a). This is partly because many key residues within the interface are not assigned, including Gln-493, Tyr-577, Gly-550, Ile-546, Thr-547, Leu-548, and Gly-576. Residues in the interface for which we could not extract chemical shift information due to broadening and/or spectral overlap include Thr-460, Leu-578, Asp-529, and Thr-582. In addition, we do not expect many large chemical shift perturbations near the Walker A motif, as ATP is expected to remain tightly bound at this site independent of dimerization. Collectively, this may explain why the majority of the larger shift perturbations do not map to the dimer interface. Notably, for I539T NBD1 ΔRIΔRE, similar shift perturbations were confirmed for the indole of Trp-496 and the amides of Cys-491, Leu-571, and Glu-403 (Fig. 6e). For both WT and I539T, these chemical shift perturbations are modulated by concentration causing the peaks to “move” along a straight vector across the concentration series.
FIGURE 6.

Concentration dependence of 1H-15N HSQC spectra of NBD1 ΔRIΔRE. a, overlay of spectra of WT NBD1 ΔRIΔRE recorded at 0.78 mm (black), 0.39 mm (red), 0.20 mm (green), and 0.083 mm (blue). Arrows indicate the direction in which the peaks move following dilution. b, overlay of spectra of Y577K NBD1 ΔRIΔRE recorded at 1.7 mm (black) and 0.1 mm (red). The location of peaks that are severely broadened is shown by the dashed circles. c, overlay of spectra of Y577E NBD1 ΔRIΔRE recorded at 1.9 mm (black) and 0.1 mm (red). d, overlay of spectra of WT NBD1 ΔRIΔRE (black) at 0.78 mm and F508del NBD1 ΔRIΔRE (red) at 1.1 mm. e, overlay of spectra of I539T NBD1 ΔRIΔRE recorded at 0.8 mm (black) and 0.2 mm (red). f, overlay of spectra of R487S NBD1 ΔRIΔRE recorded at 1.3 mm (black) and 0.16 mm (red). g, overlay of spectra of F508del NBD1 ΔRIΔRE recorded at 2.1 mm (black), 1.2 mm (red), 0.53 mm (green), 0.26 mm (blue), and 0.13 mm (yellow).
FIGURE 7.
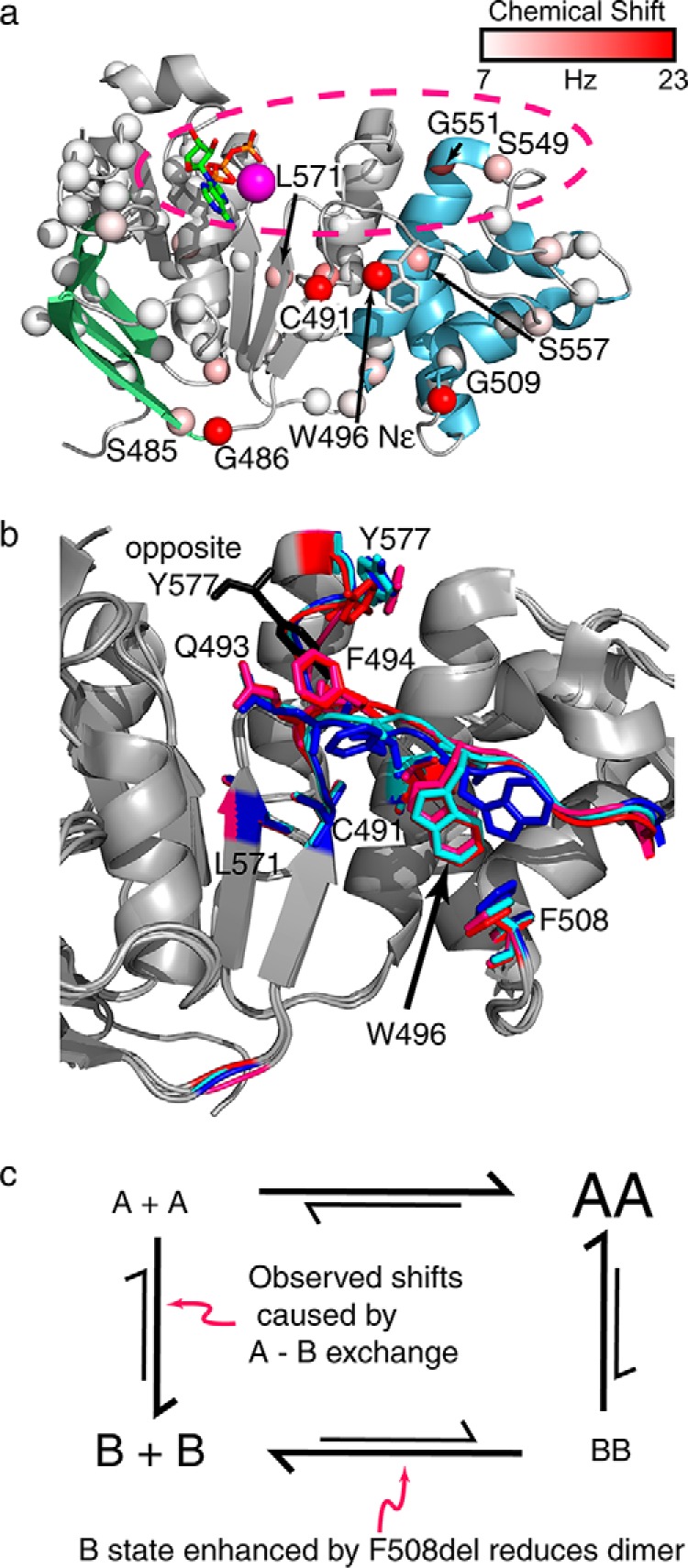
a, mapping of the concentration-dependent chemical shift perturbations for NBD1 on a ribbon diagram of NBD1 using spheres colored according to the legend. The approximate location of the physiological dimer interface is shown by the pink dashed oval. Note the very limited number of data points (spheres) in the dimer interface. b, overlay of the Q-loop and dimerization surface of both 2PZE crystal structure subunits (blue and cyan; NBD1 is dimerized with an interface at the expected physiological interface in the 2PZE structure) and both 2PZG subunits (pink and red; physiological dimer interface is not formed in 2PZG). The position of the Tyr-577 from the opposite subunit in the 2PZE structure is indicated in black. Note the differences at Tyr-577 and Phe-494 that result from dimer formation, as well as the variable orientations of the Trp-496 side chain. Structural alignments were performed using residues 440–600, encompassing both the ATP-binding core and the α-helical subdomain. c, proposed model explaining the concentration-dependent chemical shift perturbations. Both A and B forms are sampled by WT NBD1, in a manner that depends on dimerization. The A form dimerizes more readily and is favored by dimers. The B form is favored by F508del NBD1 and disfavors dimerization.
As shown in Fig. 7a, some residues for which shift perturbations were observed (Ser-485, Gly-486, and Gly-509) are quite far from the dimerization interface (see below for a discussion of the shift perturbations at Gly-486 (27 Hz) and Ser-485 (10 Hz)). To test whether the observed chemical shift perturbations are due to dimerization at the expected dimer interface, we mutated Tyr-577, which is in the center of this interface, and looked for modulation of the chemical shifts. Peaks corresponding to the amide groups of Gly-486, Cys-491, Gly-509, and Gly-551 and the Trp-496 indole NϵH, which have the largest concentration-dependent chemical shift perturbations in the WT, become invisible at high concentrations and nearly so at low concentrations for the Y577K NBD1 mutant (Fig. 6b). Although the effect of Y577K on the amount of dimer is unclear from these experiments, this mutation in the dimer interface has a pronounced effect (i.e. extensive broadening) on Gly-486, Cys-491, Trp-496, and Gly-509 confirming that it is reasonable to interpret the concentration-dependent chemical shift perturbations at these residues in the WT as a response to dimerization at the expected interface. Interestingly, a Y577E mutant did not result in significant broadening of Cys-491and Trp-496 resonances relative to WT or to significant concentration-dependent chemical shift perturbations (Fig. 6c). However, concentration-dependent changes in broadening of Gly-486, Ser-485, and Gly-551 are observed, confirming that these residues are also linked to the dimer interface. Visual inspection indicates changes in linewidth and relative peak intensity throughout the Y577E spectra at 0.1 and 1.9 mm, strongly hinting that significant dimerization is occurring in this mutant. Combined with observations seen in Y577K, these data indicate that chemical shift perturbations at Ser-485, Gly-486, Cys-491, Trp-496, and Gly-509 are linked to changes at the presumed heterodimer interface. However, the differences in the responses indicate that the link is not direct, as might be expected because they are not in the interface (Fig. 7a). Rather, the chemical shift perturbations are likely due to a conformational exchange process that is coupled to dimerization and the dimerization interface.
The relatively large chemical shift perturbations at Gly-486 and Ser-485 caused us to examine whether our observed chemical shift perturbations might be due to dimerization at a secondary interface, specifically the nonphysiological dimer interface observed in the 2PZG NBD1 crystal structure. In this structure, the positively charged Arg-487 forms intermolecular contacts with the negatively charged Asp-565 and Asp-567 of the adjacent NBD1 subunit. To determine whether we were observing intermolecular interactions between Arg-487 and Asp-565 and Asp-567, which form the di-acidic motif, we introduced the R487S mutation to remove the positive charge and disrupt this interface. R487S NBD1 ΔRIΔRE still exhibits concentration-dependent linear chemical shift perturbations for Cys-491 and Trp-496 NϵH (Fig. 6f). We could not confirm that the Ser-485 and Gly-486 chemical shifts still are perturbed, as these residues were not assigned for this mutant form of the protein. This implies at a minimum that the Cys-491 and Trp-496 chemical shift perturbations are due to dimerization at the presumed heterodimer interface and not the secondary interface observed in 2PZG. The observation that all of the chemical shift perturbations for WT NBD1 ΔRIΔRE can be fit to a single dimerization constant (see below) and the fact that mutation of Tyr-577 in the heterodimer interface affects the same set of residues indicates that all of the concentration-dependent chemical shift perturbations observed for WT are linked to dimerization at the heterodimer interface.
For WT NBD1 ΔRIΔRE, the progressive concentration-dependent peak position changes for residues Ser-485, Gly-486, Cys-491, Trp-496, Gly-509, and Leu-571 indicate an equilibrium between two states that are in fast exchange, with the changing peak positions reflective of the shifting populations of these states. To look for insight into the nature of the conformational exchange we analyzed CFTR NBD1 crystal structures. The Phe-494 and Tyr-577 side chain orientations, which are at the dimer interface, are variable in the crystal structures, and the differences appear to be related to the presence (2PZE, blue) or absence (2PZG, magenta) of dimerization (Fig. 7b). Stacking interactions between the Tyr-577 of one subunit and Tyr-577 and Gln-493 of the opposite subunit are lost if dimerization at the ATP-containing dimer interface is not present. In the 2PZG structure, in which this physiological dimer interface is not formed, Tyr-577 and Phe-494 change orientation and move into the void that is filled by Tyr-577 from the opposite subunit when the dimer is formed. As Phe-494 is part of the Q-loop, these changes propagate to the Q-loop backbone conformational ensemble. We thus attribute the chemical shift perturbations at Cys-491, Ser-557, Leu-571, and Trp-496 to reorientation of Tyr-577 and Phe-494 upon dimerization and the resulting Q-loop conformational changes. Notably, most of the largest perturbations occur on a contiguous segment that includes the loop and β-strand preceding the Q-loop and the Q-loop and the α-helix and loop following the Q-loop, which we collectively term the Q-loop segment (amino acids 485–509). The data suggest a model in which the entire Q-loop segment is in equilibrium between two different conformations (A and B in Fig. 7c). Dimerization affects the position of residues Tyr-577 and Phe-494 and indirectly affects the population of the two conformations. Mutations at the dimer interface (Y577K and Y577E) also affect the Q-loop segment conformational sampling. In all three cases the same set of residues is affected, but differences in the time scale or kinetics of the sampling of the A and B conformations lead to either linear chemical shift perturbations, broadening, or both. Implicit in this model is the fact that, for the WT, one of the Q-loop conformations is more compatible with dimerization. Dimerization pushes the conformational equilibrium toward this conformation. As an extension of this hypothesis, mutations that push the conformation of the Q-loop into the B conformation disfavor dimerization.
Although the chemical shift perturbations are not directly modulated by dimerization, within a single variant they still report on the amount of dimer. Therefore, we were able to use chemical shift perturbations from spectra recorded at multiple WT concentrations and fit a dimer dissociation constant of 280 ± 200 μm (Fig. 8). The full set of 19 peaks could be globally fit to the same Kd value. The large error reflects the relatively small chemical shift perturbations and the limited number of concentrations that were recorded. The dimer dissociation constant is likely modulated by ATP affinity and concentration, given the prominent place of ATP at the interface. Within the context of full-length CFTR, the inherent NBD dimer dissociation constant is likely modulated by covalent attachment to other CFTR regions. The weak affinity is consistent with the difficulty that the cystic fibrosis research community has had in observing NBD homo- or heterodimerization.
FIGURE 8.

Amide proton chemical shifts of WT NBD1 ΔRIΔRE as a function of concentration for 19 different resonances. The circles represent data, and the line represents the fitted curve, which yielded a dimerization constant of 280 ± 200 μm.
Confirmation of NBD1 Homodimers by Paramagnetic Relaxation Enhancement (PRE)
To confirm that NBD1 is homodimerizing in the expected head-to-tail orientation, we covalently linked a paramagnetic spin label to a single cysteine within NBD1. Paramagnetic spin labels have an unpaired electron that causes broadening (and associated signal loss) of nearby NMR resonances via a process known as PRE. The single cysteine residue, E402C, was introduced on a Cys-less NBD1 ΔRIΔRE (C491V, C524T, C590V, and C592V). This enabled conjugation of a single molecule of tempo-maleimide to each NBD1 molecule. Position 402 was chosen because it is at the periphery of the expected dimer interface and thus not likely to disrupt dimerization, but it is still close enough to the interface for the tempo-maleimide to broaden NMR signals on an opposing protamer if a dimer is formed. E402C NBD1 ΔRIΔRE could be expressed and purified in the amounts required for NMR. Of note, we were not able to express or purify NBD1 using the previously published mutations C491S, C524S, C590V, and C592V (12), probably because these mutations destabilize NBD1.
To ensure that E402C NBD1 ΔRIΔRE is folded similarly to WT NBD1 ΔRIΔRE, spectra of the mutant were recorded and compared with the WT (Fig. 9a). The five mutations lead to numerous chemical shift perturbations. However, many of the resonances of the mutant overlap precisely with their WT counterparts, providing very strong evidence that this construct assumes the same fold as the WT. To determine whether free tempo-maleimide interacts with NBD1, we added 2 mm free tempo-maleimide into fully tempo-maleimide-labeled E402C NBD1 ΔRIΔRE. Because the only cysteine is already conjugated with tempo-maleimide, any further loss of NMR signals must be interpreted as an intermolecular interaction between NBD1 and free tempo-maleimide. We quantified the ratio of these signals and plotted it on NBD1. Following addition of free tempo-maleimide, we noted loss of signal intensity near H7, S9, and S10 (red spheres in Fig. 9b), suggesting that tempo-maleimide on its own interacts with the hydrophobic patch previously identified as a binding site for the small molecule CFFT-001 (32).
FIGURE 9.

PRE data for NBD1 homodimer formation. a, overlay of 1H-15N HSQC spectra of WT (black) and E402C (red) NBD1 ΔRIΔRE. b, ribbon diagram of NBD1 showing the degree of signal loss following addition of tempo-maleimide in trans. Data points are shown as spheres colored according to degree of signal loss as shown in the legend. ATP is shown as a stick figure and color-coded by element type. The region of broadening is circled in orange. c, overlay of spectra of 15N WT NBD1 ΔRIΔRE recorded in the presence of a 3-fold excess of isotopically natural abundance tempo-maleimide-labeled E402C NBD1 ΔRIΔRE recorded before (red) and after (black) reduction of the spin label with TCEP. d, ribbon diagram of an NBD1 dimer with 15N WT NBD1 ΔRIΔRE (teal) bound to E402C WT NBD1 ΔRIΔRE (gray, not observable in this experiment). Data points are shown as spheres colored on a gradient from white to red, with the most severely spin label-broadened residues shown in red as indicated in the legend. The region of broadening also observed in the control experiment is circled in orange. The dimer induced broadening is circled in black. The green sphere represents the amide nitrogen of Cys-402. The proposed inward motion of the Cys-402 containing loop is illustrated with a green arrow. The two ATP molecules at the dimer interface are shown and colored by element type.
To detect dimers, a sample containing 525 μm tempo-maleimide-labeled, isotopically natural abundance E402C NBD1 ΔRIΔRE and 175 μm 15N-labeled WT NBD1 ΔRIΔRE was prepared. Spectra were recorded in the absence of reductant (spin label broadens NMR signals in this case) and following reduction of the spin label with TCEP (broadened signals restored). The spectra provide clear evidence for loss of NMR signal due to an intermolecular interaction between the (unobservable) tempo-maleimide-labeled NBD1 and the (observable) 15N-labeled WT NBD1 ΔRIΔRE (Fig. 9c). We measured the ratio of signal intensities before and after addition of TCEP and mapped them onto the NBD1 structure (Fig. 9d). Besides broadening on the SDR and Asp-565, we observed two foci of significant broadening. The first region (Fig. 9d, circled in orange) includes residues close to H7, S9, and S10 as observed in the control experiment with free tempo-maleimide. Broadening in this area is likely the result of an interaction with tempo-maleimide that does not require dimer formation. The second region is at the N terminus of H5 and H6 close to the dimer interface (Fig. 9d, circled in black). No broadening was observed at the N terminus of H5 and H6 when free tempo-maleimide was added, leading us to conclude that the severe broadening at this position is due to NBD1 homodimer formation. The position of the broadening is consistent with a head-to-tail homodimer, but it requires an inward movement of the Cys-402 loop as indicated by the green arrow allowing tempo-maleimide to bind close to the N terminus of H5 and H6. This would position the Cys-402 loop (residual RI) adjacent to ATP and may influence the ATP conformation, as was previously observed for the full RI in mouse NBD1 crystal structures (6). Moderate broadening on the SDR may be the result of dynamic conformational sampling of this region by the Cys-402 loop and tempo-maleimide. Note that no broadening is expected or observed near Cys-402, because the tempo-maleimide-labeled E402C NBD1 ΔRIΔRE is not 15N-labeled and thus not visible in these spectra. In summary, the PRE data confirm that NBD1 forms homodimers and supports a head-to-tail assembly.
Effect of F508del on Dimerization
We next examined concentration-dependent chemical shift perturbations for F508del NBD1 ΔRIΔRE (Fig. 6g). The perturbations are much smaller than observed for the WT. For example, Cys-491 moves by 11 Hz going from a concentration of 0.13 to 2.1 mm, and the Trp-496 indole NϵH is perturbed by 4 Hz. Although the effects are smaller, these residues are still responsive to dimerization. Comparison of WT and F508del spectra demonstrates that at high concentration the F508del peaks are closer to the position of the monomer peaks in the WT spectra. This indicates that F508del favors the Q-loop conformation that is less compatible with dimerization (Fig. 6d). Chemical shift perturbations consistent with this interpretation are seen for Trp-496, Glu-403, Cys-491, and Leu-571. The small size of the F508del chemical shift perturbations precluded fitting the data to a dimerization constant. Taken together with the τc values, our data indicate that F508del pushes the Q-loop into a conformation that hampers dimerization and thus inhibits NBD1 homodimerization.
Analysis of the crystal structures provides a plausible mechanism for how F508del affects the Q-loop conformation. Trp-496, in the middle of the Q-loop, has a direct side chain interaction with Phe-508 in many structures (Fig. 7b), which is necessarily disrupted in the mutant. Interestingly, even within a single crystal structure Trp-496 can occupy multiple positions, consistent with the notion that the Q-loop can occupy multiple conformations. Together with our data, this suggests that deletion of Phe-508 directly affects the distribution of Trp-96 side chain conformations, a change that we suspect is propagated through the Q-loop to the dimer interface. Evidence supporting the connection between dimerization and the Phe-508 position comes from the large concentration-dependent chemical shift change of the adjacent residue Gly-509 (∼30 Hz) seen in WT spectra (Fig. 6a) and the concentration-dependent broadening of this signal in the Y577K spectra (Fig. 6b). Thus, the dimer interface and the Phe-508 loop appear to be tightly linked. Collectively, the data indicate that F508del reduces the ability of NBD1 ΔRIΔRE to homodimerize, probably by disrupting the Phe-508/Trp-496 side chain interaction that stabilizes the Q-loop.
Discussion
NBD heterodimerization is thought to be integral to CFTR folding and processing (31) as well as channel gating (14–17). In the absence of NBD2, the open probability of the channel decreases by almost 10-fold (52), supporting the hypothesis that NBD1/NBD2 interaction drives channel opening. Despite the dramatic difference in open probability in the absence of NBD2, direct monitoring of NBD heterodimerization has been limited by the difficulty in detecting any interaction between the isolated NBDs. Deletion of the RI may enhance NBD1 dimerization, because the RI may perturb the configuration of ATP in the dimer interface, as observed in a mouse CFTR NBD1 crystal structure (6). We speculate that this facilitated our observation of NBD1 homodimers. Phosphorylation of the RI is proposed to relieve RI inhibition of NBD dimerization (13). Our PRE data suggest that even the residual RI (402C containing loop) may influence the ATP conformation.
Data on NBD dimerization in other ABC transporters is similarly scarce. Transporter associated with antigen processing (TAP) is composed of two half-channels (TAP1 and TAP2). Heterodimerization of the two TAP NBDs has not been observed, but the affinity of mutant TAP1 NBD homodimers is between 20 and 170 μm (53). Unlike TAP, CFTR is a full channel and NBD1 and NBD2 are covalently linked; thus, one would expect the affinity of NBD dimers to be weaker than that observed for TAP, which is what we observed. Thus, NBD potentiation of channel opening likely involves a modest energetic contribution.
Although F508del is known to impair both folding and channel opening, the effect of F508del on NBD heterodimerization is not well studied, due to the difficulty in directly measuring dimerization and producing isolated NBD2. The notion that F508del inhibits NBD heterodimerization has been suggested in previously published studies of CFTR processing (31) and channel activity (54, 55) as well as a study of a related ABC transporter (56). We demonstrate that in solution F508del inhibits NBD1 ΔRIΔRE oligomerization and provides chemical shift evidence of concentration-dependent dimerization of WT NBD1 ΔRIΔRE. We also show that F508del chemical shifts are consistent with a Q-loop segment conformation that inhibits homodimerization. We propose that in addition to its effects on NBD1 stability and NBD1/ICL4 interactions, F508del inhibits NBD1/NBD2 heterodimerization, and this contributes to the poor folding and gating properties of the mutant. Notably, F508del CFTR can still transition to the open conformation, suggesting that the energetic effect of the mutation on NBD dimerization is modest.
In addition to a significant effect on NBD1 stability, F508del is known to disrupt folding through impairment of interdomain interactions. As mentioned in the Introduction, much evidence points to the importance of the Phe-508 side chain in disrupting the NBD1/ICL4 interaction. However, other evidence suggests that F508del also disrupts the NBD1/NBD2 interaction, including the increased proteolytic susceptibility of NBD2 (31) in the context of F508del. The simplest explanation for this result is that NBD1 stabilizes NBD2 or at least helps it to fold and that deletion of Phe-508 destabilizes the NBD1/NBD2 interaction. F508del is known to disrupt processing of CFTR that is missing NBD2, indicating that the F508del defect is certainly not limited to the disruption of the NBD interface (52). CFTR missing NBD2 can also be exported from the endoplasmic reticulum (52), demonstrating that NBD2 is not required for the folding process per se. However, if NBD2 is present and the NBD1/NBD2 interface is required for NBD2 folding and stability, then disruption of this interaction would lead to misfolded NBD2 and targeting of CFTR for degradation. Supporting this contention, at least two mutations in NBD2 (N1303K, which interacts with the Q-loop of NBD2, and p.Ile1234_Arg1239del) are known to reduce the amount of mature CFTR (52, 57). Interestingly, the combined suppressor mutations I539T, G550E, R553M, and R555K have a bigger positive effect on F508del CFTR when NBD2 is present (58), suggesting the importance of the NBD interaction and hinting that these NBD1-stabilizing mutations may also improve the ability of F508del NBD1 to dimerize with NBD2. Structural models have led to the prediction that the suppressor mutation G550E enhances dimerization through an electrostatic interaction with basic surfaces on NBD2 (59). The side chain of Phe-508 has been shown to be important for the interdomain interactions required for channel opening and open time residency (55), hinting that the Phe-508 side chain plays a role in NBD dimerization. The gating deficiency of F508del CFTR could also be explained by impairment of NBD interactions. In addition to the established deficiencies in stability and NBD1/ICL4 interaction, the evidence outlined above and data contained in this study indicate that F508del also impairs NBD1/NBD2 interaction.
The mechanism by which F508del might impair NBD dimerization is not immediately obvious, because Phe-508 is relatively distant from the dimer interface. In addition to being distant from the dimer interface, previously published crystal structures demonstrate that Phe-508 does not grossly perturb the ground state structure of NBD1 (8, 9). This conclusion is strongly supported by our NMR structural data, as well as studies of fast time scale dynamics in NBD1 ΔRIΔRE indicating that overall patterns of flexibility are shared by the ground states of WT, F508del, and I539T NBD1. We propose that F508del disrupts NBD dimerization by promoting a higher energy Q-loop conformation that inhibits dimerization. Data presented here indicate that residues in the Q-loop segment of WT NBD1 exist in equilibrium between two states. Furthermore, mutations in the dimer interface (Y577K and Y577E) clearly alter the Q-loop segment equilibrium, with changes observed as far away from the dimer interface as Gly-509, which is adjacent to the Phe-508 position.
The structural differences between the two Q-loop states observed by our NMR studies are still unclear. In a related molecular dynamics study (60), the entire Q-loop was observed to dissociate from the main portion of F508del NBD1, a result that the authors attribute to the loss of the stabilizing role of Phe-508 on Trp-496 and Met-498. It is not clear that such a drastic transformation is observed in our studies. Nevertheless, the molecular dynamics results hint at the poor stability of the ground state Q-loop segment conformation, particularly in the absence of Phe-508. The populations of the two Q-loop segment states that are observed in our study are modulated in a concentration-dependent fashion, thus indicating that one of these states is more compatible with the monomeric state, whereas the other state is more compatible with a higher order oligomer that is most likely dimer. Analysis of the 2PZE and 2PZG crystal structures of NBD1 also suggests conformational heterogeneity. The homodimer state in particular has poor electron density for some Q-loop side chains (Phe-494 through Trp-496) and different side chain positions for the two subunits. Conformational changes on the Q-loop appear to be linked to reorientation of Tyr-577 and Phe-494 in response to homodimerization. As our chemical shift data for the observed Q-loop resonances show, deletion of F508del, which interacts with Trp-496 in the Q-loop, pushes the conformation of the Q-loop toward the state that is less compatible with homodimerization. The relevance of the Phe-508/Trp-496 interaction for NBD1 folding has previously been noted (9), suggesting that the Q-loop segment conformational ensemble plays an important role in folding efficiency. The shift in the conformational ensemble of the Q-loop upon deletion of Phe-508 is also supported by the differential accessibility of Cys-491 in WT and F508del NBD1 (58). Because the mechanism of dimer inhibition involves an alternate Q-loop segment conformation and changes at the dimer interface, it seems likely that F508del will disrupt NBD1/NBD2 heterodimerization as well as the NBD1 homodimers observed here. Other published Q-loop segment suppressor mutations such as S492P and S495P (21, 29) are also likely to modulate NBD dimerization. Besides these Q-loop effects, other influences on NBD1/NBD2 heterodimerization include nucleotide binding, the RI and R region and their phosphorylation. A remaining challenge is to determine the relative importance of F508del disruption of heterodimerization.
Our results suggest that targeting the NBD1/NBD2 interface for stabilization may be one useful way to suppress the processing effects of Phe-508 deletion, as well as the gating defects. Even WT CFTR folds inefficiently, and the temperature-corrected channel has poor stability and gating properties once at the cell membrane, consistent with the idea that CFTR exists in a shallow energetic well and readily accesses unfolded states after assembly. Although stabilizing NBD1 is known to improve CFTR processing (19, 24, 25, 27, 28), it may not be an exclusive solution to the problem. Mutations that inhibit processing, either by destabilizing intra- or interdomain interactions or by disrupting membrane integration in the case of the MSDs, are spread throughout the ordered regions of CFTR (61), suggesting that multiple domains or domain/domain interactions in CFTR have marginal stability, which could be suitable targets for stabilizing CFTR and correcting processing defects. Both MSD1 and NBD2 are known to require stabilization by NBD1, stabilization that is lost upon deletion of Phe-508 (31, 52). Conversely, VX-809 stabilization of MSD1 is postulated to improve maturation by enhancing stabilizing interactions with MSD2 and NBD1 (62). Gain-of-function mutations, which presumably stabilize an open conformation, have been identified in both MSD1 and MSD2 (63, 64). It has also been suggested that low temperature rescue may be an NBD2-related effect (58), which does not occur in the absence of NBD2. Together with the presented data, this implies that enhancing NBD2 stability or stabilizing the NBD1/NBD2 interface may be an avenue for improving processing and channel gating through therapeutics.
Author Contributions
P. A. C. and J. D. F.-K. designed the research; P. A. C., P. J. F., and A. K. M. performed research. P. A. C., R. V., and R. P. H. contributed new reagents. P. A. C., P. J. F., A. K. M., and J. D. F.-K. analyzed the data. P. A. C., A. K. M., and J. D. F.-K. wrote the paper.
Acknowledgments
We thank Drs. Ranjith Muhandiram and Lewis Kay for assistance with NMR experiments and helpful comments. We thank Drs. Lewis Kay, Chi Wang, and John Hunt for critical reading of the manuscript. We also thank Drs. Jennifer Dawson and Algridas Velyvis for helpful discussions.
This work was supported by Cystic Fibrosis Foundation Therapeutics Grant FORMAN05XX0 (to J. D. F.-K.) and a Cystic Fibrosis Canada grant (to J. D. F.-K.). The authors declare that they have no conflicts of interest with the contents of this article.
This article was selected as a Paper of the Week.
WT, I539T and F508del NBD1 assignments have been deposited in the BMRB.
- CFTR
- cystic fibrosis transmembrane conductance regulator
- NBD
- nucleotide-binding domain
- R
- regulatory
- RI
- regulatory insertion
- MSD
- membrane spanning domain
- SDR
- structurally diverse region
- RE
- regulatory extension
- ICL
- intracellular loop
- TCEP
- tris(2-carboxyethyl) phosphine
- PRE
- paramagnetic relaxation enhancement
- TAP
- transporter associated with antigen processing.
References
- 1. Riordan J. R., Rommens J. M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., Chou J.-L., Drumm M. L., Iannuzzi M. C., Collins F. S., Tsui L.-C. (1989) Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 245, 1066–1073 [DOI] [PubMed] [Google Scholar]
- 2. Cheng S. H., Gregory R. J., Marshall J., Paul S., Souza D. W., White G. A., O'Riordan C. R., Smith A. E. (1990) Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63, 827–834 [DOI] [PubMed] [Google Scholar]
- 3. Lukacs G. L., Chang X. B., Bear C., Kartner N., Mohamed A., Riordan J. R., Grinstein S. (1993) The ΔF508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J. Biol. Chem. 268, 21592–21598 [PubMed] [Google Scholar]
- 4. Dalemans W., Barbry P., Champigny G., Jallat S., Dott K., Dreyer D., Crystal R. G., Pavirani A., Lecocq J. P., Lazdunski M. (1991) Altered chloride ion channel kinetics associated with the ΔF508 cystic fibrosis mutation. Nature 354, 526–528 [DOI] [PubMed] [Google Scholar]
- 5. Drumm M. L., Wilkinson D. J., Smit L. S., Worrell R. T., Strong T. V., Frizzell R. A., Dawson D. C., Collins F. S. (1991) Chloride conductance expressed by ΔF508 and other mutant CFTRs in Xenopus oocytes. Science 254, 1797–1799 [DOI] [PubMed] [Google Scholar]
- 6. Lewis H. A., Buchanan S. G., Burley S. K., Conners K., Dickey M., Dorwart M., Fowler R., Gao X., Guggino W. B., Hendrickson W. A., Hunt J. F., Kearins M. C., Lorimer D., Maloney P. C., Post K. W., et al. (2004) Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J. 23, 282–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lewis H. A., Wang C., Zhao X., Hamuro Y., Conners K., Kearins M. C., Lu F., Sauder J. M., Molnar K. S., Coales S. J., Maloney P. C., Guggino W. B., Wetmore D. R., Weber P. C., Hunt J. F. (2010) Structure and dynamics of NBD1 from CFTR characterized using crystallography and hydrogen/deuterium exchange mass spectrometry. J. Mol. Biol. 396, 406–430 [DOI] [PubMed] [Google Scholar]
- 8. Lewis H. A., Zhao X., Wang C., Sauder J. M., Rooney I., Noland B. W., Lorimer D., Kearins M. C., Conners K., Condon B., Maloney P. C., Guggino W. B., Hunt J. F., Emtage S. (2005) Impact of the ΔF508 mutation in first nucleotide-binding domain of human cystic fibrosis transmembrane conductance regulator on domain folding and structure. J. Biol. Chem. 280, 1346–1353 [DOI] [PubMed] [Google Scholar]
- 9. Thibodeau P. H., Brautigam C. A., Machius M., Thomas P. J. (2005) Side chain and backbone contributions of Phe508 to CFTR folding. Nat. Struct. Mol. Biol. 12, 10–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Atwell S., Brouillette C. G., Conners K., Emtage S., Gheyi T., Guggino W. B., Hendle J., Hunt J. F., Lewis H. A., Lu F., Protasevich I. I., Rodgers L. A., Romero R., Wasserman S. R., Weber P. C., et al. (2010) Structures of a minimal human CFTR first nucleotide-binding domain as a monomer, head-to-tail homodimer, and pathogenic mutant. Protein Eng. Des. Sel. 23, 375–384 [DOI] [PubMed] [Google Scholar]
- 11. Chappe V., Irvine T., Liao J., Evagelidis A., Hanrahan J. W. (2005) Phosphorylation of CFTR by PKA promotes binding of the regulatory domain. EMBO J. 24, 2730–2740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mense M., Vergani P., White D. M., Altberg G., Nairn A. C., Gadsby D. C. (2006) In vivo phosphorylation of CFTR promotes formation of a nucleotide-binding domain heterodimer. EMBO J. 25, 4728–4739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kanelis V., Hudson R. P., Thibodeau P. H., Thomas P. J., Forman-Kay J. D. (2010) NMR evidence for differential phosphorylation-dependent interactions in WT and ΔF508 CFTR. EMBO J. 29, 263–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vergani P., Lockless S. W., Nairn A. C., Gadsby D. C. (2005) CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature 433, 876–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Berger A. L., Ikuma M., Welsh M. J. (2005) Normal gating of CFTR requires ATP binding to both nucleotide-binding domains and hydrolysis at the second nucleotide-binding domain. Proc. Natl. Acad. Sci. U.S.A. 102, 455–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Aleksandrov L., Aleksandrov A. A., Chang X. B., Riordan J. R. (2002) The first nucleotide binding domain of cystic fibrosis transmembrane conductance regulator is a site of stable nucleotide interaction, whereas the second is a site of rapid turnover. J. Biol. Chem. 277, 15419–15425 [DOI] [PubMed] [Google Scholar]
- 17. Aleksandrov A. A., Cui L., Riordan J. R. (2009) Relationship between nucleotide binding and ion channel gating in cystic fibrosis transmembrane conductance regulator. J. Physiol. 587, 2875–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Protasevich I., Yang Z., Wang C., Atwell S., Zhao X., Emtage S., Wetmore D., Hunt J. F., Brouillette C. G. (2010) Thermal unfolding studies show the disease causing F508del mutation in CFTR thermodynamically destabilizes nucleotide-binding domain 1. Protein Sci. 19, 1917–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang C., Protasevich I., Yang Z., Seehausen D., Skalak T., Zhao X., Atwell S., Spencer Emtage J., Wetmore D. R., Brouillette C. G., Hunt J. F. (2010) Integrated biophysical studies implicate partial unfolding of NBD1 of CFTR in the molecular pathogenesis of F508del cystic fibrosis. Protein Sci. 19, 1932–1947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aleksandrov A. A., Kota P., Aleksandrov L. A., He L., Jensen T., Cui L., Gentzsch M., Dokholyan N. V., Riordan J. R. (2010) Regulatory insertion removal restores maturation, stability and function of ΔF508 CFTR. J. Mol. Biol. 401, 194–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Aleksandrov A. A., Kota P., Cui L., Jensen T., Alekseev A. E., Reyes S., He L., Gentzsch M., Aleksandrov L. A., Dokholyan N. V., Riordan J. R. (2012) Allosteric modulation balances thermodynamic stability and restores function of ΔF508 CFTR. J. Mol. Biol. 419, 41–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pissarra L. S., Farinha C. M., Xu Z., Schmidt A., Thibodeau P. H., Cai Z., Thomas P. J., Sheppard D. N., Amaral M. D. (2008) Solubilizing mutations used to crystallize one CFTR domain attenuate the trafficking and channel defects caused by the major cystic fibrosis mutation. Chem. Biol. 15, 62–69 [DOI] [PubMed] [Google Scholar]
- 23. DeCarvalho A. C., Gansheroff L. J., Teem J. L. (2002) Mutations in the nucleotide binding domain 1 signature motif region rescue processing and functional defects of cystic fibrosis transmembrane conductance regulator ΔF508. J. Biol. Chem. 277, 35896–35905 [DOI] [PubMed] [Google Scholar]
- 24. Rabeh W. M., Bossard F., Xu H., Okiyoneda T., Bagdany M., Mulvihill C. M., Du K., di Bernardo S., Liu Y., Konermann L., Roldan A., Lukacs G. L. (2012) Correction of both NBD1 energetics and domain interface is required to restore ΔF508 CFTR folding and function. Cell 148, 150–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mendoza J. L., Schmidt A., Li Q., Nuvaga E., Barrett T., Bridges R. J., Feranchak A. P., Brautigam C. A., Thomas P. J. (2012) Requirements for efficient correction of ΔF508 CFTR revealed by analyses of evolved sequences. Cell 148, 164–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Denning G. M., Anderson M. P., Amara J. F., Marshall J., Smith A. E., Welsh M. J. (1992) Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive (see comments). Nature 358, 761–764 [DOI] [PubMed] [Google Scholar]
- 27. Teem J. L., Berger H. A., Ostedgaard L. S., Rich D. P., Tsui L. C., Welsh M. J. (1993) Identification of revertants for the cystic fibrosis ΔF508 mutation using STE6-CFTR chimeras in yeast. Cell 73, 335–346 [DOI] [PubMed] [Google Scholar]
- 28. Teem J. L., Carson M. R., Welsh M. J. (1996) Mutation of R555 in CFTR-ΔF508 enhances function and partially corrects defective processing. Receptors Channels 4, 63–72 [PubMed] [Google Scholar]
- 29. He L., Aleksandrov A. A., An J., Cui L., Yang Z., Brouillette C. G., Riordan J. R. (2015) Restoration of NBD1 thermal stability is necessary and sufficient to correct F508 CFTR folding and assembly. J. Mol. Biol. 427, 106–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Loo T. W., Bartlett M. C., Clarke D. M. (2010) The V510D suppressor mutation stabilizes ΔF508-CFTR at the cell surface. Biochemistry 49, 6352–6357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Du K., Sharma M., Lukacs G. L. (2005) The ΔF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat. Struct. Mol. Biol. 12, 17–25 [DOI] [PubMed] [Google Scholar]
- 32. Hudson R. P., Chong P. A., Protasevich I. I., Vernon R., Noy E., Bihler H., An J. L., Kalid O., Sela-Culang I., Mense M., Senderowitz H., Brouillette C. G., Forman-Kay J. D. (2012) Conformational changes relevant to channel activity and folding within the first nucleotide binding domain of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 287, 28480–28494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Leaver-Fay A., Tyka M., Lewis S. M., Lange O. F., Thompson J., Jacak R., Kaufman K., Renfrew P. D., Smith C. A., Sheffler W., Davis I. W., Cooper S., Treuille A., Mandell D. J., Richter F., et al. (2011) ROSETTA3:an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 487, 545–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 35. Johnson B. A., Blevins R. A. (1994) NMR View: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4, 603–614 [DOI] [PubMed] [Google Scholar]
- 36. Goddard T. D., Kneller D. G. (2008) SPARKY 3. University of California, San Francisco [Google Scholar]
- 37. Yang D., Kay L. E. (1999) Improved 1HN-detected triple resonance TROSY-based experiments. J. Biomol. NMR 13, 3–10 [DOI] [PubMed] [Google Scholar]
- 38. Konrat R., Yang D., Kay L. E. (1999) A 4D TROSY-based pulse scheme for correlating 1HNi,15Ni,13Cα,13C'i-1 chemical shifts in high molecular weight, 15N,13C, 2H labeled proteins. J. Biomol. NMR 15, 309–313 [DOI] [PubMed] [Google Scholar]
- 39. Salzmann M., Pervushin K., Wider G., Senn H., Wüthrich K. (1999) [13C]-constant-time [15N,1H]-TROSY-HNCA for sequential assignments of large proteins. J. Biomol. NMR 14, 85–88 [DOI] [PubMed] [Google Scholar]
- 40. Zhang O., Kay L. E., Olivier J. P., Forman-Kay J. D. (1994) Backbone 1H and 15N resonance assignments of the N-terminal SH3 domain of drk in folded and unfolded states using enhanced-sensitivity pulsed field gradient NMR techniques. J. Biomol. NMR 4, 845–858 [DOI] [PubMed] [Google Scholar]
- 41. Zhang O., Forman-Kay J. D., Shortle D., Kay L. E. (1997) Triple-resonance NOESY-based experiments with improved spectral resolution: applications to structural characterization of unfolded, partially folded and folded proteins. J. Biomol. NMR 9, 181–200 [DOI] [PubMed] [Google Scholar]
- 42. Shen Y., Delaglio F., Cornilescu G., Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kay L. E., Torchia D. A., Bax A. (1989) Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry 28, 8972–8979 [DOI] [PubMed] [Google Scholar]
- 44. Farrow N. A., Muhandiram R., Singer A. U., Pascal S. M., Kay C. M., Gish G., Shoelson S. E., Pawson T., Forman-Kay J. D., Kay L. E. (1994) Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33, 5984–6003 [DOI] [PubMed] [Google Scholar]
- 45. Korzhnev D. M., Skrynnikov N. R., Millet O., Torchia D. A., Kay L. E. (2002) An NMR experiment for the accurate measurement of heteronuclear spin-lock relaxation rates. J. Am. Chem. Soc. 124, 10743–10753 [DOI] [PubMed] [Google Scholar]
- 46. Orekhov V. Y., Nolde D. E., Golovanov A. P., Korzhnev D. M., Arseniev A. S. (1995) Processing of heteronuclear NMR relaxation data with the new software DASHA. Appl. Magn. Reson. 9, 581–588 [Google Scholar]
- 47. García de la Torre J., Huertas M. L., Carrasco B. (2000) HYDRONMR: prediction of NMR relaxation of globular proteins from atomic-level structures and hydrodynamic calculations. J. Magn. Reson. 147, 138–146 [DOI] [PubMed] [Google Scholar]
- 48. Farrow N. A., Zhang O., Szabo A., Torchia D. A., Kay L. E. (1995) Spectral density function mapping using 15N relaxation data exclusively. J. Biomol. NMR 6, 153–162 [DOI] [PubMed] [Google Scholar]
- 49. Cavanagh J., Fairbrother W., Palmer A., Skelton N. (1996) Protein NMR Spectroscopy Principles and Practice, pp. 243–300, Academic Press, Inc., San Diego [Google Scholar]
- 50. Lipari G., Szabo A. (1982) Model-free approach to the interpretation of nuclear magnetic relaxation in macromolecules: 1. Theory and range of validity. J. Am. Chem. Soc. 104, 4546–4559 [Google Scholar]
- 51. Dawson J. E., Farber P. J., Forman-Kay J. D. (2013) Allosteric coupling between the intracellular coupling helix 4 and regulatory sites of the first nucleotide-binding domain of CFTR. PLoS One 8, e74347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cui L., Aleksandrov L., Chang X. B., Hou Y. X., He L., Hegedus T., Gentzsch M., Aleksandrov A., Balch W. E., Riordan J. R. (2007) Domain interdependence in the biosynthetic assembly of CFTR. J. Mol. Biol. 365, 981–994 [DOI] [PubMed] [Google Scholar]
- 53. Grossmann N., Vakkasoglu A. S., Hulpke S., Abele R., Gaudet R., Tampé R. (2014) Mechanistic determinants of the directionality and energetics of active export by a heterodimeric ABC transporter. Nat. Commun. 5, 5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jih K. Y., Li M., Hwang T. C., Bompadre S. G. (2011) The most common cystic fibrosis-associated mutation destabilizes the dimeric state of the nucleotide-binding domains of CFTR. J. Physiol. 589, 2719–2731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cui L., Aleksandrov L., Hou Y. X., Gentzsch M., Chen J. H., Riordan J. R., Aleksandrov A. A. (2006) The role of cystic fibrosis transmembrane conductance regulator phenylalanine 508 side chain in ion channel gating. J. Physiol. 572, 347–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Woodward O. M., Tukaye D. N., Cui J., Greenwell P., Constantoulakis L. M., Parker B. S., Rao A., Köttgen M., Maloney P. C., Guggino W. B. (2013) Gout-causing Q141K mutation in ABCG2 leads to instability of the nucleotide-binding domain and can be corrected with small molecules. Proc. Natl. Acad. Sci. U.S.A. 110, 5223–5228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Molinski S. V., Gonska T., Huan L. J., Baskin B., Janahi I. A., Ray P. N., Bear C. E. (2014) Genetic, cell biological, and clinical interrogation of the CFTR mutation c.3700 A→G (p.Ile1234Val) informs strategies for future medical intervention. Genet. Med. 16, 625–632 [DOI] [PubMed] [Google Scholar]
- 58. He L., Aleksandrov L. A., Cui L., Jensen T. J., Nesbitt K. L., Riordan J. R. (2010) Restoration of domain folding and interdomain assembly by second-site suppressors of the ΔF508 mutation in CFTR. FASEB J. 24, 3103–3112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Xu Z., Pissarra L. S., Farinha C. M., Liu J., Cai Z., Thibodeau P. H., Amaral M. D., Sheppard D. N. (2014) Revertant mutants modify, but do not rescue, the gating defect of the cystic fibrosis mutant G551D-CFTR. J. Physiol. 592, 1931–1947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wieczorek G., Zielenkiewicz P. (2008) ΔF508 mutation increases conformational flexibility of CFTR protein. J. Cyst. Fibros. 7, 295–300 [DOI] [PubMed] [Google Scholar]
- 61. Van Goor F., Yu H., Burton B., Hoffman B. J. (2014) Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J. Cyst. Fibros. 13, 29–36 [DOI] [PubMed] [Google Scholar]
- 62. Ren H. Y., Grove D. E., De La Rosa O., Houck S. A., Sopha P., Van Goor F., Hoffman B. J., Cyr D. M. (2013) VX-809 corrects folding defects in CFTR through action on membrane-spanning domain1 (MSD1). Mol. Biol. Cell 24, 3016–3024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wei S., Roessler B. C., Chauvet S., Guo J., Hartman J. L. 4th, Kirk K. L. (2014) Conserved allosteric hot spots in the transmembrane domains of cystic fibrosis transmembrane conductance regulator (CFTR) channels and multidrug resistance protein (MRP) pumps. J. Biol. Chem. 289, 19942–19957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang W., Okeyo G. O., Tao B., Hong J. S., Kirk K. L. (2011) Thermally unstable gating of the most common cystic fibrosis mutant channel (ΔF508): “rescue” by suppressor mutations in nucleotide binding domain 1 and by constitutive mutations in the cytosolic loops. J. Biol. Chem. 286, 41937–41948 [DOI] [PMC free article] [PubMed] [Google Scholar]