

ABSTRACT
Kaposi's sarcoma-associated herpesvirus (KSHV) is a gammaherpesvirus known to establish lifelong latency in the human host. We and others have previously shown that three KSHV homologs of cellular interferon regulatory factors (IRFs), known as viral IRFs (vIRFs), participate in evasion of the host interferon (IFN) response. We report that vIRF1 interacts with the cellular interferon-stimulated gene 15 (ISG15) E3 ligase, HERC5, in the context of Toll-like receptor 3 (TLR3) activation and IFN induction. The ISG15 protein is covalently conjugated to target proteins upon activation of the interferon response. Interaction between vIRF1 and HERC5 was confirmed by immunoprecipitation, and the region between amino acids 224 and 349 of vIRF1 was required for interaction with HERC5. We further report that expression of vIRF1 in the context of TLR3 activation results in decreased ISG15 conjugation of proteins. Specifically, TLR3-induced ISG15 conjugation and protein levels of cellular IRF3, a known ISG15 target, were decreased in the presence of vIRF1 compared to the control. vIRF1 itself was also identified as a target of ISG15 conjugation. KSHV-infected cells exhibited increased ISG15 conjugation upon reactivation from latency in coordination with increased IFN. Furthermore, knockdown of ISG15 in latently infected cells resulted in a higher level of KSHV reactivation and an increase in infectious virus. These data suggest that the KSHV vIRF1 protein affects ISG15 conjugation and interferon responses and may contribute to effective KSHV replication.
IMPORTANCE The KSHV vIRF1 protein can inhibit interferon activation in response to viral infection. We identified a cellular protein named HERC5, which is the major ligase for ISG15, as a vIRF1 binding partner. vIRF1 association with HERC5 altered ISG15 modification of cellular proteins, and knockdown of ISG15 augmented reactivation of KSHV from latency.
INTRODUCTION
Upon detection of viral infection, cells activate the quintessential antiviral immune response, which results in production of type I interferon (IFN), including interferon alpha (IFN-α) and IFN-β. Type I IFN induces an antiviral transcriptional program, producing proteins that cooperate to inhibit the spread of infection. One of the most abundantly produced transcripts upon induction of the type I IFN response is interferon-stimulated gene 15 (ISG15) (1, 2). ISG15 is a ubiquitin-like 17-kDa protein that is covalently conjugated to target proteins via a process called ISGylation (3–5). Similar to ubiquitin, ISG15 requires three enzymatic steps for conjugation onto target proteins. They include the E1 enzyme Ube1L, the conjugating E2 enzyme UbcH8, and the major E3 ligase HERC5 (6). Multiple large-scale screens have identified hundreds of potential cellular targets of ISGylation; however, in most cases, the specific function of ISG15 conjugation to cellular proteins remains undetermined (7).
Thus far, identified ISG15 target proteins function in a variety of cellular pathways, including glycolysis, cell motility, translation, and stress responses, with the most studied ISG15 targets lying within the type I IFN pathway. ISG15 targets in the IFN pathway include protein kinase R (PKR), retinoic acid-inducible gene 1 (RIG-I), signal transducer and activator of transcription 1 (STAT1), and cellular interferon regulatory factor 3 (IRF3) (7). The IRF3 transcription factor is activated upon detection of a pathogen and is in part responsible for transcription and production of type I IFNs. Additionally, IRF3 is ISGylated on lysine residues 193, 360, and 366 (8). When IRF3 is not ISGylated, IRF3 interacts more tightly with Pin1, resulting in increased polyubiquitination and IRF3 degradation (8). While the specific function of ISGylation on many other target proteins remains to be clarified, it is clear that ISG15 plays a crucial role in antiviral immunity. ISG15 conjugation has been found to inhibit the growth of influenza A and B virus, Ebola virus, Sindbis virus, HIV, and herpes simplex virus 1 (HSV-1), among others (9). In contrast, vesicular stomatitis virus (VSV) and lymphocytic choriomeningitis virus (LCMV) do not seem to be inhibited by ISG15 conjugation (10). To date, the only gammaherpesvirus that has been examined for a correlation between ISG15 expression and viral replication is murine gammaherpesvirus 68 (MHV-68). Infection of ISG15 knockout mice exhibited a 10-fold increase in MHV-68 viral titers, although no change in viral lethality was observed (11). The relationship between the human gammaherpesvirus Kaposi's sarcoma-associated herpesvirus (KSHV) and ISG15 has not been previously investigated.
KSHV is a double-stranded DNA virus and the etiological agent for Kaposi's sarcoma, an endothelial-cell-driven cancer (12). KSHV also contributes to two different lymphoproliferative disorders: primary effusion lymphoma (PEL) and multicentric Castleman's disease (MCD) (13, 14). Like all herpesviruses, KSHV establishes lifelong latent infection and, in order to do so, must evade host immune responses, including the type I IFN response. KSHV encodes four viral homologs of cellular interferon regulatory factors (vIRF1, -2, -3, and -4) with pleiotropic effects, including evasion of cell death, increased proliferation, and evasion of immune responses (15). Our previous work examined how KSHV vIRF1, -2, and -3 modulated Toll-like receptor 3 (TLR3)-mediated IFN production (16). We found that expression of vIRF1 and vIRF2 inhibited increases in IFN-β mediated by TLR3 (16), a pathogen-associated molecular pattern receptor that is upregulated in response to KSHV infection (17).
To further elucidate the mechanisms of KSHV and vIRF1 inhibition of type I IFN, we performed an unbiased analysis to discover novel cellular protein interactions with KSHV vIRF1 in the context of TLR3 activation and identified the cellular ISG15 E3 ligase, HERC5. Expression of vIRF1 resulted in a global decrease in ISGylation of target proteins. Additionally, ISG15 conjugation increased in latently infected cells following reactivation, and we found that knockdown of ISG15 allowed increased viral reactivation compared to a scrambled control. Collectively, these data suggest that modulation of the ISG15 pathway by KSHV vIRF1 may inhibit host immune responses and represent a mechanism through which KSHV evades IFN responses to establish and maintain lifelong infection of the human host.
MATERIALS AND METHODS
Plasmids.
pcDNA3 Ube1L, UbcH8, HA-HERC5, HIS6-HA-ISG15, and HIS6-3×FLAG-ISG15 (18, 19) were generous gifts of Robert Krug (University of Texas at Austin, Austin, TX). pcDNA3 HERC5 (without an epitope tag) was produced with a QuikChange XL II site-directed mutagenesis kit (Agilent). pcDNA3 Myc-vIRF1 was a generous gift from Jae Jung (University of Southern California, Los Angeles, CA). Myc-vIRF1 was cloned into pLenti CMV Puro DEST (w118-1; Addgene) via pENTR TOPO and the Gateway system (Invitrogen). 3×FLAG-vIRF1 was codon optimized and generated by Genescript and cloned into pcDNA3.1. Deletion mutants of vIRF1 were amplified from the codon-optimized construct and cloned with a pcDNA3.1 Directional TOPO Expression kit (Invitrogen). Mission short hairpin RNA (shRNA) constructs for ISG15 and HERC5 were purchased (Sigma).
Cell culture.
293 cells were cultured at 37°C with 5% CO2 in Dulbecco's modified Eagle medium (DMEM) (Cellgro) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Cellgro), 100 U/ml (each) of penicillin and streptomycin, and 2 mM l-glutamine. 293-TLR3 cells (InvivoGen) were maintained in 10 μg/ml Blasticidin S (InvivoGen). TREx BCBL1 inducible cells expressing either empty vector (EV) or replication and transcription activator (RTA) protein (20) were cultured in RPMI 1640 medium (Corning) containing tetracycline -free FBS (Clontech) and 20 μg/ml hygromycin B (Roche). BCBL1 PEL cells were cultured in RPMI 1640 medium (Corning) containing 0.05 mM β-mercaptoethanol. iSLK.219 cells harboring latent recombinant KSHV.219 (rKSHV.219) (21) were maintained in DMEM supplemented with 10% FBS (Cellgro), 1% penicillin and streptomycin, G418 (250 μg/ml), hygromycin (400 μg/ml), and puromycin (10 μg/ml).
293-TLR3 cells were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. The cells were transfected with twice as much HERC5 and ISG15 as Ube1L and UbcH8, as previously reported (22). To activate TLR3 signaling, cells were treated with high-molecular-weight poly(I·C) (Invivogen) at 20 μg/ml for 24 h. TREx BCBL1 cells were reactivated with 1 μg/ml doxycycline for the times indicated. BCBL1 PEL cells were reactivated with 1 mM sodium butyrate and 25 ng/ml 12-O-tetradecanoyl-phorbol 13-acetate (TPA) where indicated. iSLK.219 cells were reactivated with 0.1 μg/ml doxycycline.
Immunoprecipitation and immunoblotting.
For mass spectrometry, 10-cm2 dishes were seeded with 1.25 × 106 293-TLR3 cells, and after equilibration overnight, the cells were transfected for 36 h with 20 μg pcDNA3.1 3×FLAG-vIRF1, after which the cells were stimulated for 24 h with either poly(I·C) (Invivogen) or vehicle control. After stimulation, the cells were lysed, and FLAG affinity purification was performed according to the manufacturer's instructions (Sigma). The pulldown products were run on a 10% SDS gel and subjected to silver staining according to the manufacturer's suggestions (Invitrogen). Bands of interest were excised and submitted to the Taplin Mass Spectrometry Facility (Harvard University).
For immunoprecipitation assays, equal amounts of protein were precleared with the appropriate IgG antibody (normal mouse IgG; Santa Cruz), followed by overnight incubation with FLAG or hemagglutinin (HA) antibody-conjugated beads (EZview Red Anti-FLAG M2 Affinity Gel or EZveiw Red Anti-HA Affinity Gel; Sigma). The beads were washed 3 times with lysis buffer, and the bound proteins were released through competitive elution with 150 ng/μl 3× FLAG peptide (Sigma) or 100 μg/ml influenza virus HA peptide (Sigma) during a 1-h incubation. The supernatants were separated from the beads prior to addition of SDS loading dye and immunoblotting.
For immunoblotting, cells were washed once with PBS and lysed in 0.1% NP-40 with the addition of protease inhibitor complete tabs (Roche). The protein concentration was determined by Bradford assay (Bio-Rad), and equal amounts of proteins were separated using SDS-PAGE and transferred onto Hybond-ECL nitrocellulose membranes (GE Healthcare). The antibodies used included anti-Myc-horseradish peroxidase (HRP) (Invitrogen), anti-enterokinase cleavage site (xxxDDDDK)-HRP (Bethyl), anti-ISG15 (Cell Signaling), anti-IRF3 (Cell Signaling), anti-HA-HRP (Cell Signaling), anti-tubulin-HRP (Cell Signaling), anti-Ku70/80 (a generous gift from Dale Ramsden, University of North Carolina, Chapel Hill, NC), and anti-actin (Santa Cruz). The blots were exposed to appropriate secondary antibodies conjugated to horseradish peroxidase when necessary and visualized using SuperSignal West Pico (Pierce), Clarity (Bio-Rad), or Amersham Prime ECL Western blotting detection reagent (GE).
Lentiviral transduction.
pLenti Myc-vIRF1 and mission shRNA constructs for ISG15 and HERC5 were used to generate lentivirus-expressing vIRF1 or shRNAs via the ViraPower system (Invitrogen), according to the manufacturer's instructions. Briefly, 293T cells were transfected with 3 μg of shRNA construct and 9 μg of ViraPower packing mix. The transfection medium was replaced with antibiotic-free medium 4 h posttransfection; 48 h posttransfection, the medium was harvested and spun at 3,000 rpm for 15 min and stored in aliquots at −80°C. To transduce iSLK.219 cells, 2 ml of lentiviral supernatant was applied to the cells in conjunction with 10 μg/ml Polybrene (Sigma), and the cells were centrifuged at 2,500 rpm for 90 min at 32°C. Following transduction, 2 ml of complete medium was added to the cells. To transduce BCBL1 cells, 4 million cells were resuspended in 4 ml of lentiviral supernatant with 10 μg/ml Polybrene (Sigma) and centrifuged at 2,500 rpm for 90 min at 32°C. The lentiviral supernatant was removed, and the cells were resuspended in standard PEL medium for 24 h. Then, the cells were infected a second time in the same manner with 2 ml of lentiviral supernatant.
Quantitation of viral reactivation via real-time PCR or microscopy.
DNA was isolated with a DNeasy blood and tissue kit (Qiagen) according to the manufacturer's instructions. RNA was isolated using an RNeasy Plus minikit or an RNeasy Micro kit (Qiagen), and reverse transcription was performed using SuperScript III reverse transcriptase (Invitrogen) and oligo(dT) (Invitrogen). Real-time PCR was performed on an ABI 7300 using SYBR green PCR master mix (Bio-Rad). The PCR was carried out with 1 cycle of 50°C for 2 min and 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. All fold activations were normalized to β-actin. The primer sequences used were as follows: β-Actin Forward, 5′-TCATGAAGTGTGACGTGGACATC, and Reverse, 5′-CAGGAGGAGCAATGATCTTGATCT (23); Orf57 Forward, 5′-TGGACATTATGAAGGGCATCCT, and Reverse, 5′ –CGGGTTCGGACAATTGCT; IFN-α Forward, 5′-GTGAGGAAATACTTCCAAAGAATCAC, and Reverse, 5′-TCTCATGATTTCTGCTCTGACAA; and IFN-β Forward, 5′-CAGCAATTTTCAGTGTCAGAAGC, and Reverse, 5′-TCATCCTGTCCTTGAGGCAGT.
To determine levels of KSHV infection (green fluorescent protein [GFP]) or KSHV reactivation to the lytic state (red fluorescent protein [RFP]), cells were examined with a Nikon Eclipse Ti microscope using Nikon Elements software.
RESULTS
vIRF1 interacts with the ISG15 E3 ligase HERC5.
To identify vIRF1 functional roles in an unbiased manner, we sought to find novel cellular interacting proteins using a system in which vIRF1 was previously shown to inhibit IFN responses (16). To this end, 293-TLR3 stable expressing cells were transfected for 36 h with vIRF1-FLAG constructs and stimulated for 24 h with poly(I·C), a double-stranded RNA analogue and ligand for TLR3. After stimulation, FLAG affinity purification was performed, the products were separated via PAGE, and the gel was silver stained. Bands were excised and subjected to mass spectrometry. The results indicated that vIRF1 associated with CREBBP and p300, as previously reported (24, 25), in the presence or absence of poly(I·C) (Fig. 1A). However, only in the context of TLR3 activation with poly(I·C) did we find that vIRF1 coimmunoprecipitated with HERC5, the E3 ligase for ISG15.
FIG 1.
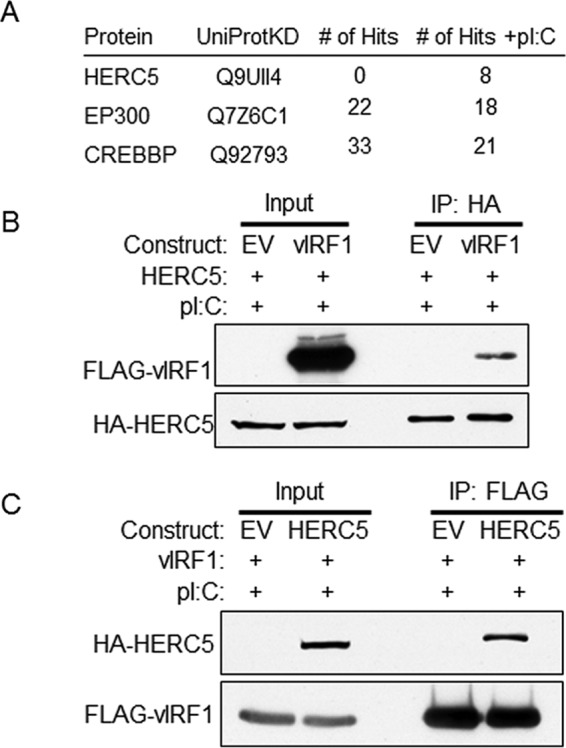
KSHV vIRF1 binds to cellular HERC5 in the context of TLR3 activation. (A) 293-TLR3 stable expressing cells were transfected for 36 h with vIRF1-FLAG and stimulated for 24 h with poly(I·C) (pI:C). After stimulation, FLAG affinity purification was performed, and the products were separated via PAGE and silver stained. Bands of interest were excised and subjected to mass spectrometry. Selected results for proteins interacting with KSHV vIRF1 with or without treatment with poly(I·C) are shown. (B) 293-TLR3 cells were transfected with HA-HERC5 and control EV or FLAG-vIRF1 plasmids. Twenty-four hours after transfection, the cells were treated with poly(I·C) and harvested 24 h after treatment. The cell lysates were immunoprecipitated (IP) with anti-HA antibody-conjugated beads; resolved by SDS-PAGE, along with input control lysates; and probed with antibodies as indicated. (C) 293-TLR3 cells were transfected with FLAG-vIRF1 and control EV or HA-HERC5 plasmid. Twenty-four hours after transfection, the cells were treated with poly(I·C) and harvested 24 h after treatment. The cell lysates were immunoprecipitated with anti-FLAG antibody-conjugated beads and were subjected to SDS-PAGE along with a fraction of the input lysates, and immunoblotting was performed with various antibodies as indicated. The data are representative of the results of more than four independent experiments.
To confirm the interaction between vIRF1 and HERC5, 293-TLR3 cells were transfected with equal concentrations of FLAG-vIRF1 or EV control and HA-HERC5 plasmids. HERC5 was immunoprecipitated with anti-HA antibody-conjugated beads, and HERC5-bound proteins were eluted with HA peptide. The eluate and input samples were resolved via SDS-PAGE and subjected to immunoblotting with an anti-FLAG antibody to detect vIRF1. This experiment showed that HERC5 and vIRF1 coimmunoprecipitate (Fig. 1B), and we confirmed this interaction with reverse immunoprecipitation, in which vIRF1 protein was captured using FLAG antibody-conjugated beads. Proteins were eluted with 3× FLAG peptide, and eluate and input samples were resolved via SDS-PAGE and subjected to immunoblotting with an anti-HA antibody. This immunoprecipitation also demonstrated an association between HERC5 and vIRF1 (Fig. 1C). These data indicate that KSHV vIRF1 associates with the ISG15 E3 ligase, HERC5.
HERC5 interacts with the C-terminal region of vIRF1.
As HERC5 was found to associate with vIRF1, we next sought to determine which portion of vIRF1 is required for the interaction between vIRF1 and HERC5. Two C-terminal deletion mutants of vIRF1, one containing only amino acids (aa) 1 to 349 and one containing only aa 1 to 224, were generated to compare to the full-length (FL) 449-aa vIRF1 (Fig. 2A). To determine the portion of vIRF1 required for binding to HERC5, 293-TLR3 cells were transfected with HA-HERC5 or control vector and either the FL or the aa 1 to 349 or aa 1 to 224 mutant FLAG-vIRF1 construct. The cell lysates were immunoprecipitated with FLAG antibody-conjugated beads and eluate, and input samples were subjected to SDS-PAGE and immunoblotted with anti-HA antibody to visualize HERC5. As shown in Fig. 2B, only FL and aa 1 to 349 vIRF1 were capable of binding to HERC5, while aa 1 to 224 vIRF1 did not demonstrate an association with HERC5. To confirm this result, the reverse immunoprecipitation was performed. 293-TLR3 cells were transfected with the FL or the aa 1 to 349 or aa 1 to 224 mutant FLAG-vIRF1 construct or control vector and HA-HERC5. The cell lysates were immunoprecipitated with anti-HA antibody, and eluates and input samples were resolved via SDS-PAGE. Immunoblotting with anti-FLAG antibodies was performed to detect vIRF1. As seen previously, only FL vIRF1 and the aa 1 to 349 vIRF1 mutant were capable of interacting with HERC5 (Fig. 2C). These data suggest that the region of vIRF1 between amino acids 224 and 349 interacts with HERC5.
FIG 2.
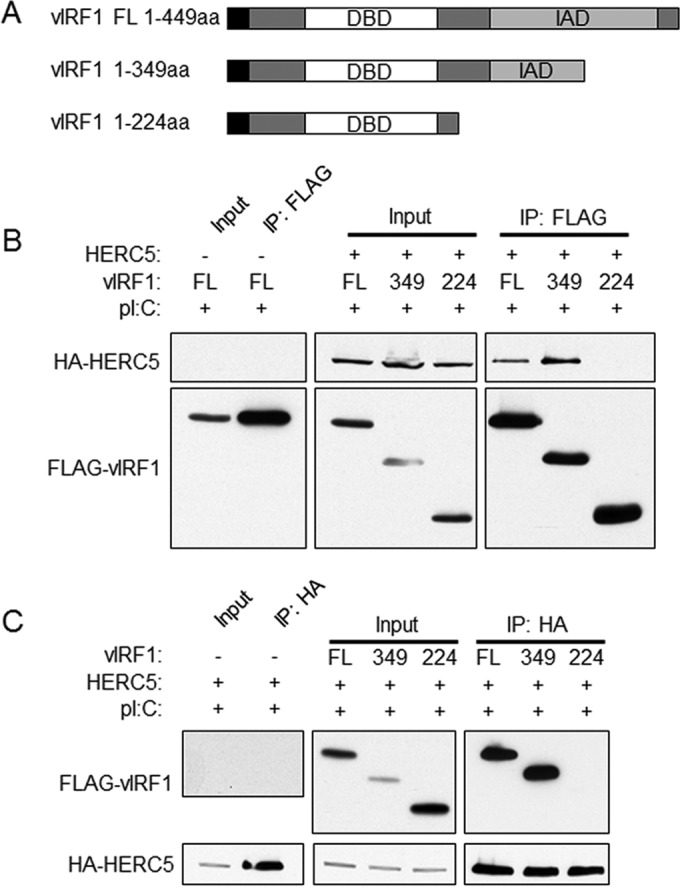
vIRF1 interacts with HERC5 between amino acids 224 and 349. (A) Two vIRF1 truncation mutants and full-length (FL) vIRF1. The black squares indicate the location of the FLAG tag. DBD, DNA-binding domain; IAD, IRF association domain. (B) 293-TLR3 cells were transfected with HA-HERC5 or control vector (−) and FL or aa 1 to 349 or aa 1 to 224 mutant FLAG-vIRF1 constructs. Twenty-four hours after transfection, the cells were treated with poly(I·C) and harvested 24 h after treatment. The cell lysates were immunoprecipitated with FLAG antibody-conjugated beads. Eluate and input samples were subjected to SDS-PAGE and immunoblotted as indicated. (C) 293-TLR3 cells were transfected with FL or aa 1 to 349 or aa 1 to 224 mutant FLAG-vIRF1 constructs or control vector (−) and HA-HERC5. Twenty-four hours after transfection, the cells were treated with poly(I·C) and harvested 24 h posttreatment. The cell lysates were immunoprecipitated with anti-HA antibody-conjugated beads prior to resolution with SDS-PAGE and immunoblotting with the indicated antibodies. The data are representative of the results of three independent experiments.
vIRF1 expression decreases total cellular ISG15 conjugation to proteins.
We demonstrated that vIRF1 associates with the ISG15 E3 ligase HERC5; however, the functional significance of this interaction remained unclear. To address this, the effect of vIRF1 expression on global ISG15 conjugation was examined. 293-TLR3 cells were transfected with constructs expressing ISG15 and its accompanying E1, E2, and E3 ligases and different ratios of control EV or vIRF1. Cells received 0 μg, 1 μg, or 4 μg of vIRF1 plasmid DNA, and EV plasmid was added to keep the total amount of transfected DNA at 4 μg for each sample. One day posttransfection, the cells received poly(I·C) to stimulate TLR3-mediated type I interferon production, ISG15 production, and ISG15 conjugation (2). Regardless of poly(I·C) treatment, cells that received 4 μg of vIRF1 construct demonstrated a decreased level of ISGylation of target proteins compared to cells that received 1 μg of vIRF1 plasmid DNA (Fig. 3A). Furthermore, cells that received only 1 μg of vIRF1 exhibited decreased ISG15 conjugation compared to cells that did not express vIRF1. These data indicate that expression of vIRF1 may serve to block the normal conjugation and function of ISG15.
FIG 3.
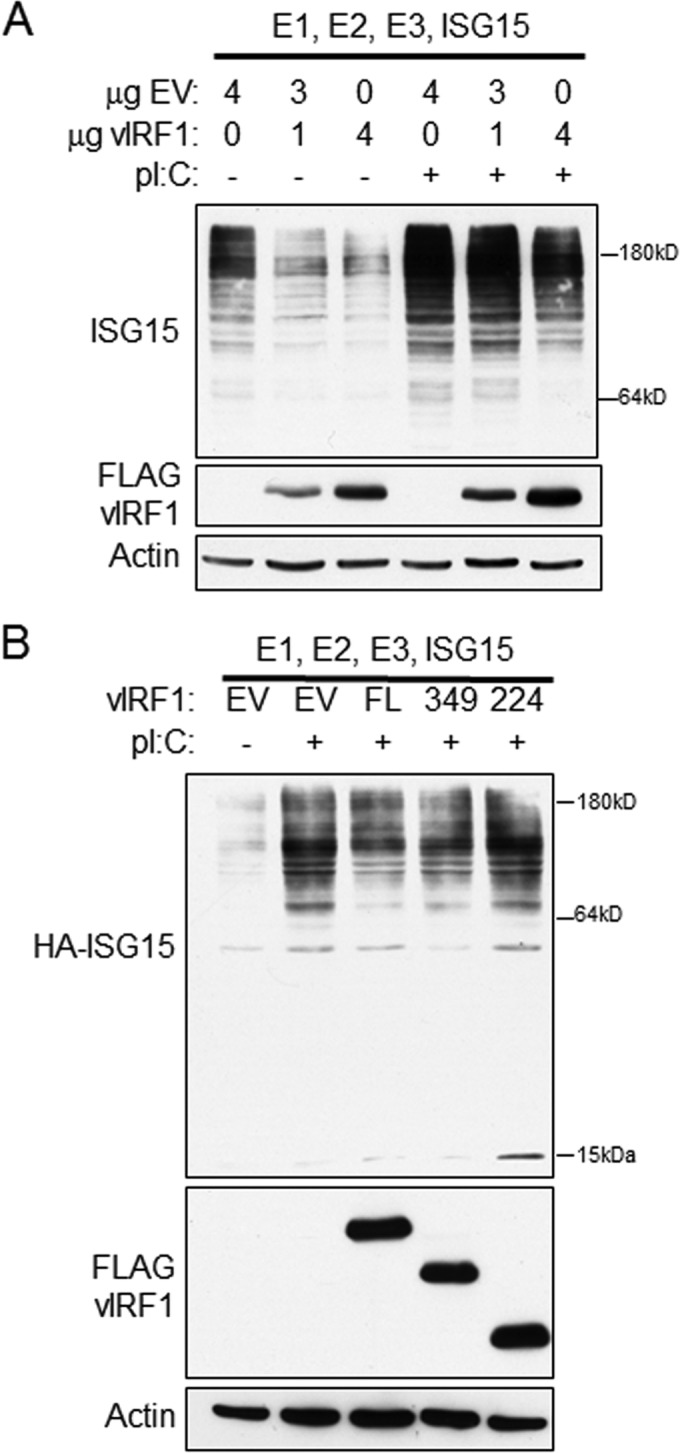
vIRF1 decreases conjugation of ISG15 to cellular proteins in the context of TLR3 activation. (A) 293-TLR3 cells were transfected with 0, 1, or 4 μg of FLAG-vIRF1 and control EV to a total of 4 μg DNA. The cells were also transfected with the E1, E2, and E3 ligases for ISG15, as well an ISG15 construct. Twenty-four hours after transfection, the cells were treated with poly(I·C) as indicated and harvested 24 h after treatment. The cell lysates were subjected to SDS-PAGE and immunoblotting and probed with antibodies as indicated. (B) 293-TLR3 cells were transfected with FL or aa 1 to 349 or aa 1 to 224 mutant FLAG-vIRF1 constructs and the E1, E2, and E3 ligases for ISG15 and HA-ISG15. Twenty-four hours after transfection, the cells were treated with poly(I·C) and harvested 24 h posttreatment. The cell lysates were subjected to SDS-PAGE, followed by immunoblotting with the antibodies indicated. The data are representative of the results of three independent experiments.
We hypothesized that vIRF1 interaction with HERC5 may be required for vIRF1-mediated alterations in the ISGylation of cellular proteins. To determine the importance of the vIRF1 and HERC5 association in altering ISG15 conjugation of target proteins, 293-TLR3 cells were transfected with constructs expressing ISG15 and its accompanying E1, E2, and E3 ligases and equal amounts of EV or FL, aa 1 to 349, or aa 1 to 224 mutant vIRF1 plasmid. The cells were treated with poly(I·C) to induce ISG15 conjugation, followed by cell lysis, SDS-PAGE, and immunoblotting with an anti-ISG15 antibody to assess ISGylation levels. As seen previously, expression of FL vIRF1 resulted in decreased levels of ISG15 compared to EV, and expression of the aa 1 to 349 mutant exhibited levels of ISG15 conjugation similar to that of FL vIRF1 (Fig. 3B). However, the aa 1 to 224 mutant vIRF1 sample showed the same degree of ISGylation as EV and more than that seen in the FL and aa 1 to 349 vIRF1 samples. These data suggest that vIRF1 expression alters the amount of ISG15 conjugation to target proteins and that the interaction between vIRF1 and HERC5 is necessary for this blockade.
vIRF1 expression decreases protein levels of IRF3 in the context of TLR3 activation.
Expression of vIRF1 resulted in decreased levels of ISG15 conjugation on cellular target proteins. Therefore, we examined the consequences of vIRF1 expression for specific targets of ISGylation. Many signaling molecules involved in the IFN response have been identified as ISG15 targets (7), and as vIRF1 is known to inhibit the IFN response (15), this area became the focus of our studies. Specifically, cellular IRF3, required for maximal transcription of IFN, associates with HERC5 and is ISGylated (8). IRF3 was also of interest, as our previous report indicated that vIRF1 expression led to decreased phosphorylation of IRF3 and was correlated with a significant reduction in interferon production (16). To determine the effect of vIRF1 expression on IRF3 protein levels in the context of TLR3 activation, 293-TLR3 cells were transfected with constructs expressing ISG15 and its accompanying E1, E2, and E3 ligases or EV control, and vIRF1 or EV control. After transfection, half of the samples were treated with poly(I·C) to activate the IFN response. The cells were then lysed and subjected to SDS-PAGE analysis and immunoblotted with antibodies that recognize endogenous IRF3 and ISG15. In cells that did not exogenously express the ISG15 pathway components, ISG15 conjugation of target proteins could not be detected (Fig. 4A), as reported previously (18, 19). As shown above, ISGylation of target proteins was increased following poly(I·C) stimulation compared to untreated cells, and expression of vIRF1 resulted in decreased ISG15 conjugation compared to EV. Importantly, in the context of poly(I·C) stimulation, IRF3 protein levels were reduced in the presence of vIRF1 compared to EV (Fig. 4A). It has previously been reported that ISGylation of IRF3 stabilizes the protein compared to an IRF3 mutant that cannot be ISGylated (8). The decrease in IRF3 protein levels correlated with decreased ISG15 conjugation of cellular genes, suggesting that one potential function of vIRF1 interaction with HERC5 may be to decrease ISGylation of IRF3 and thus decrease IRF3 protein stability and IRF3 transcription of IFN-stimulated genes.
FIG 4.
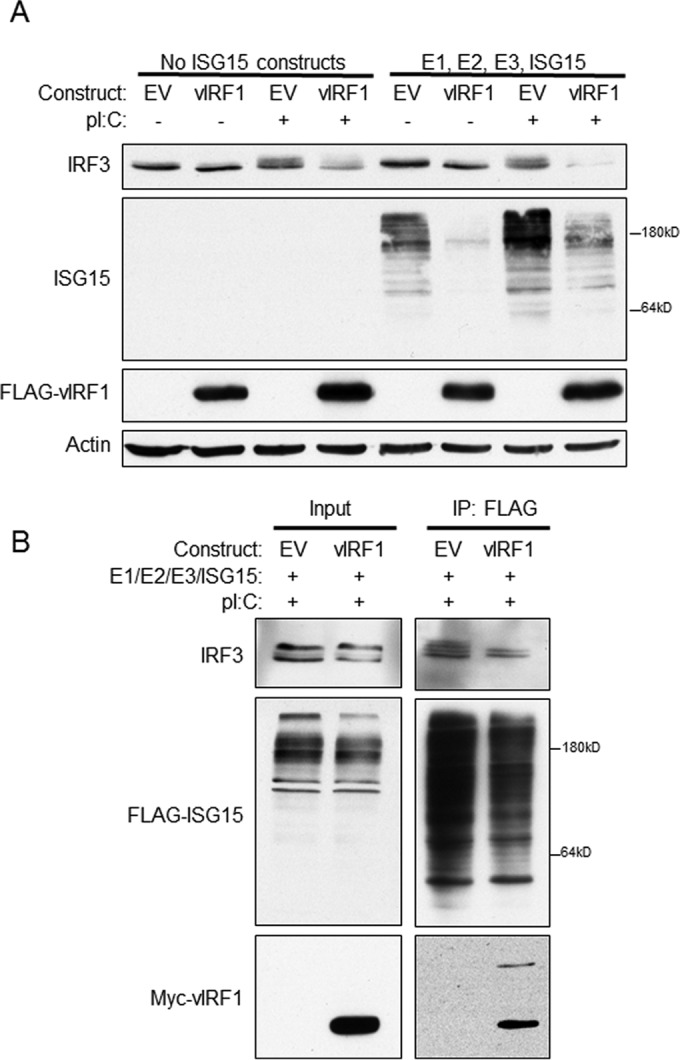
vIRF1 expression decreases protein levels of IRF3 following TLR3 activation. (A) 293-TLR3 cells were transfected with expression plasmids for E1, E2, and E3 ligases for ISG15 and ISG15 where indicated. The cells were also transfected with constructs of FLAG-vIRF1 or control EV. Twenty-four hours after transfection, the cells were treated with poly(I·C) as indicated and harvested 24 h after treatment. The cells were lysed, resolved via SDS-PAGE, and subjected to immunoblotting with the antibodies indicated. (B) 293-TLR3 cells were transfected with the E1, E2, and E3 ligases for ISG15 and FLAG-ISG15 and Myc-vIRF1 or control EV. Twenty-four hours after transfection, the cells were treated with poly(I·C) and harvested 24 h after treatment. The cells were lysed and immunoprecipitated with anti-FLAG antibody-conjugated beads prior to SDS-PAGE and immunoblotting with the indicated antibodies. The data are representative of the results of more than five independent experiments.
To confirm that IRF3 was ISGylated in our system, 293-TLR3 cells were transfected with constructs expressing FLAG-ISG15 and its accompanying E1, E2, and E3 ligases and EV control or Myc epitope-tagged vIRF1. The cells were then treated with poly(I·C) for 24 h. Cells were harvested, and the lysates were incubated with anti-FLAG antibody-conjugated beads to immunoprecipitate FLAG-ISG15 and ISG15-conjugated proteins. The resulting eluates and input samples were resolved via SDS-PAGE and immunoblotted with antibodies against IRF3 to compare the amount of ISGylated IRF3 in EV-expressing cells to that in vIRF1-expressing cells. More ISGylated IRF3 immunoprecipitated with the ISG15 antibody in EV-expressing cells than in vIRF1-expressing cells (Fig. 4B). This suggested that vIRF1 expression resulted in decreased ISGylation of IRF3 and, together with Fig. 4A, suggests that decreased ISG15 conjugation of IRF3 results in decreased IRF3 protein levels, which is consistent with previous reports (8).
KSHV vIRF1 is ISG15 conjugated.
Surprisingly, examination of vIRF1 after immunoprecipitation with an anti-ISG15 antibody indicated that vIRF1 was conjugated to ISG15. Furthermore, a higher-molecular-weight form of vIRF1 was visible after electrophoresis (Fig. 4B). This suggested that vIRF1 may itself be conjugated to ISG15. Other viral proteins are known to be ISGylated, including NS1A of influenza virus (26). To test the ISG15 conjugation of vIRF1, 293-TLR3 cells were transfected with the E1, E2, and E3 ligases for ISG15, FLAG-ISG15, and Myc-vIRF1 or control empty vector. The cell lysates were immunoprecipitated with anti-FLAG antibody-conjugated beads, bound proteins were eluted with 3× FLAG peptide, and the resultant eluates and input samples were subjected to SDS-PAGE prior to immunoblotting with anti-Myc antibodies to detect vIRF1. Our data indicated that vIRF1 is conjugated to ISG15 (Fig. 5A). Multiple forms of vIRF1 that appeared to be larger than canonical vIRF1 were visible in the immunoprecipitation eluates, suggesting that vIRF1 is ISGylated at multiple sites.
FIG 5.

KSHV vIRF1 is ISG15 conjugated. (A) 293-TLR3 cells were transfected with the E1, E2, and E3 ligases for ISG15, FLAG-ISG15, and Myc-vIRF1 or control empty vector (−). Twenty-four hours after transfection, the cells were treated with poly(I·C) and harvested 24 h after treatment. The cell lysates were immunoprecipitated with anti-FLAG antibody-conjugated beads, followed by elution and resolution of eluate and input lysates via SDS-PAGE and immunoblotting with the indicated antibodies. (B) 293-TLR3 cells were transfected with the E1, E2, and E3 ligases for ISG15, HA-ISG15, and FLAG-vIRF1 or control empty vector (−). Twenty-four hours after transfection, the cells were treated with poly(I·C) and harvested 24 h posttreatment. The cell lysates were immunoprecipitated with anti-FLAG antibody-conjugated beads, followed by elution and resolution of eluate and input lysates via SDS-PAGE and immunoblotting with the indicated antibodies. The data are representative of the results of three independent experiments.
To confirm this finding, reverse immunoprecipitation was also performed. After transfection with E1, E2, and E3 ligases for ISG15, FLAG-vIRF1, and HA-ISG15 or control empty vector, cells were lysed and incubated with anti-FLAG antibody-conjugated beads to isolate FLAG-vIRF1 and interacting partners. Eluates and input samples were resolved via SDS-PAGE and immunoblotted with anti-HA antibody to visualize ISG15. Again, more than one band of ISG15 was visible, suggesting covalent conjugation of ISG15 to vIRF1 (Fig. 5B). Together, these data indicate that vIRF1 is conjugated to ISG15 during TLR3 activation.
Reactivation of KSHV from latency results in induction of IFN and increased ISGylation.
The interaction between HERC5 and vIRF1 seems to inhibit the ISG15 pathway and may play a role in vIRF1 blockade of the IFN response. To assess normal ISG15 activity in the context of the whole virus, we employed a latent model of infection for reactivation of KSHV. Specifically, we utilized TREx BCBL1 PEL cells with an inducible empty vector (pc) or inducible replication and transcription activator protein (RTA), the major viral-lytic-switch protein of KSHV (20). The TREx cells were treated with doxycycline to activate RTA for various lengths of time, and cells were harvested and examined for expression of endogenous ISG15 by SDS-PAGE and immunoblotting. KSHV reactivation correlated with increased conjugation of ISG15, which peaked 24 h after reactivation, and the levels remained elevated 48 h after reactivation (Fig. 6A). Reactivation of TREx cells with inducible RTA expression also resulted in increased IFN-α and IFN-β message levels at 24 and 48 h compared to nonreactivated cells (Fig. 6B).
FIG 6.

Reactivation of KSHV increases ISG15 conjugation and IFN message. TREx BCBL1 cells expressing empty-vector control (pc) or RTA under a doxycycline (Dox)-inducible promoter were treated with doxycycline for the number of hours indicated. (A) Cells were harvested and lysed prior to separation with SDS-PAGE and immunoblotting with the antibodies indicated. (B) Doxycycline-treated TREx BCBL1 cells were harvested, RNA was extracted, and cDNA was generated prior to quantification of IFN-α and IFN-β message levels by real-time (RT) PCR. (C) BCBL1 PEL cells were reactivated with sodium butyrate treatment or vehicle control (−) for 24 or 48 h. The cells were harvested, and the lysates were resolved via SDS-PAGE, which was followed by immunoblotting with the indicated antibodies. (D) BCBL1 PEL cells reactivated with sodium butyrate for 0 (−), 24, 48, or 72 h were harvested. RNA was extracted, and cDNA was generated prior to quantification of IFN-α and IFN-β message levels by RT-PCR. The data are representative of the results of three independent experiments, and the values represent the means ± the standard deviations of the means from triplicate biological replicates.
To further examine the role of ISG15 during KSHV reactivation, normal BCBL1 PEL cells were reactivated with sodium butyrate for 24 or 48 h. The cells were harvested, and the lysates were separated via SDS-PAGE and probed for endogenous ISG15 expression. Similar to our observation in the inducible RTA system, an increase in ISG15 conjugation following KSHV reactivation was seen in the BCBL1 cells (Fig. 6C). As expected, reactivation of KSHV in BCBL1 cells resulted in increased IFN-α and IFN-β message levels (Fig. 6D). Together, these data suggest that the switch from latency to the lytic cycle activates the interferon pathway and induces ISG15 conjugation and that these processes are tightly correlated.
vIRF1 expression in PEL cells alters ISGylation and KSHV reactivation.
To specifically address the role of vIRF1 on ISG15 during reactivation of PEL cells, BCBL1 cells were transduced with a vIRF1-expressing lentivirus or EV control lentivirus. Two days after transduction, the cells were reactivated by addition of TPA and sodium butyrate for 48 h. The cells were harvested, and the lysates were separated via SDS-PAGE and probed for endogenous protein expression. Blotting confirmed the expression of vIRF1 and indicated that expression of vIRF1 resulted in a decrease in endogenous cellular IRF3 levels and ISG15 conjugation compared to the empty-vector control (Fig. 7A). To address the effect of vIRF1 expression on reactivation, reactivation was quantified via real-time PCR for message levels of a KSHV lytic gene, the Orf57 gene (27). At 48 h postreactivation, there was an 8-fold increase in KSHV lytic gene transcription in vIRF1-expressing cells compared to the empty-vector control (Fig. 7B). To further examine the effect of vIRF1 expression on the relative production of infectious virions, supernatants from reactivated BCBL1 cells expressing either the empty-vector control or vIRF1 were used to infect 293 cells. Infection of 293 cells was quantified via real-time PCR for Orf57 DNA levels compared to a standard curve. The number of viral genomes was increased in 293 cells infected with supernatants from vIRF1-expressing cells compared to empty-vector control cells (Fig. 7C). These data suggest that vIRF1 expression prior to reactivation results in a decrease in ISG15 conjugation, reduced expression of IRF3, and an increase in the production of infectious virions.
FIG 7.
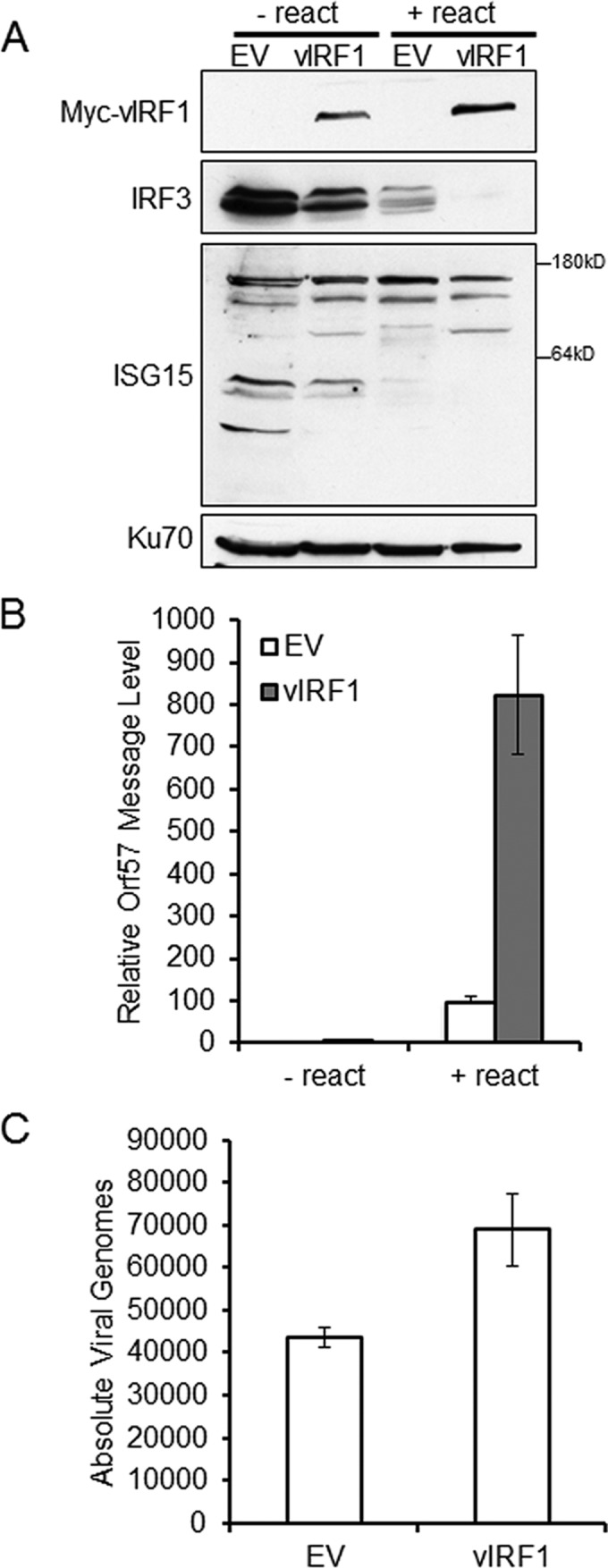
Expression of vIRF1 in PEL cells decreases ISG15 conjugation and increases KSHV reactivation. BCBL1 cells were infected with lentivirus containing EV control or Myc-vIRF1 for 48 h prior to reactivation with TPA and sodium butyrate. (A) Cells were harvested 48 h after reactivation, lysed, and resolved via SDS-PAGE, followed by immunoblotting with the indicated antibodies. − react, no reactivation; + react, with reactivation. (B) Lentivirus-transduced BCBL-1 cells were harvested 48 h after reactivation, RNA was extracted, and cDNA was generated prior to quantification of KSHV Orf57 message levels by real-time quantitative PCR (qPCR). (C) BCBL-1 cell supernatants were harvested 48 h after reactivation and used to infect naive 293 cells, and infected cells were harvested 48 h postinfection; DNA was isolated, and Orf57 DNA levels were compared to a standard curve to assess the absolute number of viral genomes by real-time qPCR. The data are representative of the results of two independent experiments, and the values represent the means ± the standard deviations of the means from triplicate biological replicates.
ISG15 knockdown prior to reactivation results in increased KSHV production.
As reactivation of latent KSHV was associated with increased ISG15 expression and ISG15 is involved in antiviral IFN responses (9), we hypothesized that ISG15 may affect KSHV virion production. To allow ease of assessment of latent and lytically infected cells, we utilized an iSLK.219 cell line—cells of epithelial origin that harbor the rKSHV.219 clone with GFP expression under a constitutively active promoter and RFP expression under the control of the lytic PAN promoter (21). First, iSLK.219 cells were infected with a scrambled (scr) or ISG15-targeting shRNA lentivirus. Efficient knockdown of ISG15 in iSLK.219 cells was detected by immunoblotting (Fig. 8A). Seventy-two hours after lentiviral infection, iSLK.219 cells were reactivated by addition of doxycycline and examined for reactivation by observing RFP expression using fluorescence microscopy. As expected, cells that were not reactivated exhibited no RFP expression (Fig. 8B). Cells that received the scrambled shRNA exhibited less RFP expression following reactivation than cells that were infected with the ISG15 shRNA lentivirus (Fig. 8B and C). Reactivation was quantified via real-time PCR for message levels of a KSHV lytic gene, the Orf57 gene (27). At 24 and 48 h postreactivation, there was a 7-fold and a 4-fold increase, respectively, in KSHV lytic gene transcription in ISG15 knockdown cells compared to a scrambled control (Fig. 8D).
FIG 8.

Knockdown of ISG15 increases KSHV replication upon reactivation. (A) iSLK.219 cells were infected with scrambled control (scr) or ISG15 shRNA lentivirus for 72 h. Cells were then lysed and subjected to SDS-PAGE and immunoblotting for the detection of ISG15 knockdown. (B) Seventy-two hours after lentiviral infection with scrambled control or ISG15 shRNA lentivirus, iSLK.219 cells were reactivated for 48 h with doxycycline. Lytic virus was detected by RFP expression via fluorescence microscopy. (C) RFP-expressing iSLK.219 cells were quantified by counting the cells in 4 random fields. (D) iSLK.219 cells were infected with scrambled control or ISG15 shRNA lentivirus for 72 h prior to reactivation with doxycycline. Cells were harvested 24 or 48 h after reactivation, and the RNA was isolated, reverse transcribed, and subjected to real-time qPCR to quantitate KSHV Orf57 message levels. (E) iSLK.219 cells were infected with scrambled control or ISG15 shRNA lentivirus and reactivated for 48 h. The cell supernatants were harvested and used to infect naive 293 cells, and GFP expression was quantitated 72 h postinfection. (F) GFP-expressing 293 cells were quantified by counting the cells in 5 randomly selected fields. (G) iSLK.219 cells were infected with scrambled control or ISG15 shRNA lentivirus and reactivated for 48 h. The cell supernatants were then used to infect naive 293 cells. The infected cells were harvested 48 h postinfection; DNA was isolated, and Orf57 DNA levels were compared to a standard curve to assess the absolute number of viral genomes by real-time qPCR. The data are representative of the results of four independent experiments, and the values represent the means ± the standard deviations of the means from triplicate biological replicates. *, P < 0.005; **, P < 0.05 (Student's t test).
To further examine the effect of ISG15 knockdown on the relative production of infectious virions, supernatants were collected from scrambled control and ISG15 shRNA lentivirus-infected and reactivated iSLK.219 cells and used to infect 293 cells. Higher numbers of GFP-positive 293 cells were seen after infection with supernatants from ISG15 knockdown expressing cells than with supernatants from the scrambled control (Fig. 8E and F). KSHV infection in 293 cells was quantified via real-time PCR for Orf57 DNA levels compared to a standard curve. The number of viral genomes was increased over 2-fold by supernatants from cells with knocked-down ISG15 levels compared to scrambled control cells (Fig. 8G). These data suggest that a reduction of ISG15 results in the production of more infectious virions during lytic reactivation and that ISG15 conjugation of proteins can suppress KSHV replication.
HERC5 knockdown prior to reactivation results in increased KSHV production.
As a reduction of ISG15 resulted in increased replication of KSHV following reactivation, we next asked if HERC5 was also necessary for maximal production of virions. iSLK.219 cells were infected with a scrambled or HERC5-targeting shRNA lentivirus. Efficient knockdown of HERC5 in iSLK.219 cells was detected by real-time PCR quantification of HERC5 message levels (Fig. 9A). Seventy-two hours after lentiviral infection, iSLK.219 cells were reactivated by addition of doxycycline and examined for reactivation by observing RFP expression using fluorescence microscopy. As expected, cells that were not reactivated exhibited no RFP expression (Fig. 9B). Depletion of HERC5 resulted in increased RFP expression following reactivation compared to cells expressing the nontargeting shRNA (Fig. 9B and C). Supernatants were collected from reactivated cells depleted of HERC5 or nontargeting control and used to infect 293 cells. Higher numbers of GFP-positive 293 cells were seen after infection with supernatants from HERC5 knockdown expressing cells than with supernatants from the scrambled control (Fig. 9D). KSHV infection in 293 cells was quantified via real-time PCR for Orf57 DNA levels compared to a standard curve. The number of viral genomes was increased nearly 7-fold by infection with supernatants from cells depleted of HERC5 compared to scrambled control cells (Fig. 9E). These data suggest that a reduction of HERC5, similar to ISG15, resulted in the production of more infectious virions during lytic reactivation.
FIG 9.

Knockdown of HERC5 increases KSHV replication upon reactivation. (A) iSLK.219 cells were infected with scrambled control or HERC5 shRNA lentivirus for 72 h prior to harvesting for detection of HERC5 knockdown through real-time qPCR. (B) Seventy-two hours after lentiviral infection with scrambled control or HERC5 shRNA lentivirus, iSLK.219 cells were reactivated for 48 h with doxycycline. Lytic virus was detected by RFP expression via fluorescence microscopy. (C) RFP-expressing iSLK.219 cells were quantified by counting the cells in 6 randomly selected fields. (D) iSLK.219 cells were infected with scrambled control or ISG15 shRNA lentivirus and reactivated for 48 h. The cell supernatants were harvested and used to infect naive 293 cells, and infection was detected by GFP expression via fluorescence microscopy 48 h postinfection. (E) iSLK.219 cells were infected with scrambled control or ISG15 shRNA lentivirus and reactivated for 48 h. The cell supernatants were then used to infect naive 293 cells. The infected cells were harvested 48 h postinfection; DNA was isolated, and Orf57 DNA levels were compared to a standard curve to assess the absolute number of viral genomes by real-time qPCR. The data are representative of the results of three independent experiments, and the values represent the means ± the standard deviations of the means from triplicate biological replicates. *, P < 0.01 (Student's t test).
DISCUSSION
To elucidate the mechanisms of KSHV vIRF1 inhibition of type I IFN, we performed an unbiased analysis of cellular proteins that interact with vIRF1. Mass spectrometry identified the cellular ISG15 E3 ligase, HERC5. Immunoprecipitation confirmed the interaction between vIRF1 and HERC5 and pinpointed the region between amino acids 224 and 349 of vIRF1 as the site of HERC5 association. Expression of vIRF1 resulted in a global decrease in ISGylation of cellular proteins, including decreased ISGylation of IRF3, which resulted in a decrease in IRF3 protein levels. Furthermore, we found that the ISG15 pathway, not previously studied in the context of KSHV infection, played a role during viral reactivation. ISG15 conjugation increased in response to reactivation, and knockdown of ISG15 resulted in increased levels of KSHV reactivation and production of infectious KSHV particles.
This is the first report to show the relationship between KSHV and ISG15, although the ISG15 pathway has been shown to play a role in viral replication during other viral infections. Knockdown of HERC5 in 293 cells resulted in an attenuated interferon response following Sendai virus (SeV) infection (8). Conversely, overexpression of HERC5 increased interferon responses mediated by SeV (8). Loss of Ube1L did not result in a change in Newcastle disease virus (NDV) infection, although Ube1L−/− cells produced higher levels of IFN-β transcripts (28). The NS1 viral protein of influenza virus is conjugated by ISG15, and this modification reduced influenza virus replication (26). Therefore, ISG15 modification of NS1 contributes to cellular antiviral activity (26). In sum, ISG15 expression and conjugation have been shown to inhibit the replication of SeV, Japanese encephalitis virus, NDV, avian sarcoma leucosis virus (ASLV), human papillomavirus (HPV), dengue virus, HIV, vaccinia virus, VSV, influenza virus, Ebola virus, HSV-1, and MHV-68 (29). Other viruses have developed mechanisms to evade ISG15-mediated antiviral responses. For example, porcine reproductive and respiratory syndrome virus (PRRSV) encodes nonstructural protein 2 (nsp2), which is an antagonist of ISG15 production and conjugation (30). Together, these data suggest that coevolution has occurred between viral evasion of ISG15 and IFN responses and host efforts to block viral infection and reproduction.
Here, we established that KSHV vIRF1 alters the cellular ISG15 posttranslational modification system. Other vIRFs have been shown to play a role in posttranslational modifications, as well. Specifically, expression of vIRF3 increases the levels of SUMO2 ubiquitination-modified promyelocytic leukemia (PML) gene product oncogenic domains (PODs) (31). Furthermore, vIRF3 is covalently conjugated to SUMO1 and SUMO2 in vitro and in KSHV latently infected B cells (32). vIRF3 is also capable of inhibiting SUMOylation of the pRb proteins pRb, p107, and p130 (33). We recently showed that vIRF1 affects phosphorylation of STING as well (34). These data suggest that one of the mechanisms of action of the vIRF proteins may be through alteration of cellular posttranslational modifications and that further study of all vIRFs in this area is warranted.
ACKNOWLEDGMENTS
We thank members of the Damania laboratory for their helpful discussions. We thank Robert Krug at the University of Texas at Austin for pcDNA3 Ube1L, UbcH8, HA-HERC5, HIS6-HA-ISG15, and HIS6-3×FLAG-ISG15 constructs. We thank Jae Jung for the vIRF1 expression plasmid as well as the TREx BCBL1 cell line.
S.R.J. was supported by grants 5T32-CA09156 and 5T32-AI007151. This work was supported by NIH grants DE018281, DE023946, AI107810, and AI109965 to B.D. B.D. is a Leukemia and Lymphoma Society Scholar and a Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Disease.
REFERENCES
- 1.Potter JL, Narasimhan J, Mende-Mueller L, Haas AL. 1999. Precursor processing of pro-ISG15/UCRP, an interferon-beta-induced ubiquitin-like protein. J Biol Chem 274:25061–25068. doi: 10.1074/jbc.274.35.25061. [DOI] [PubMed] [Google Scholar]
- 2.Der SD, Zhou A, Williams BR, Silverman RH. 1998. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci U S A 95:15623–15628. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Loeb KR, Haas AL. 1992. The interferon-inducible 15-kDa ubiquitin homolog conjugates to intracellular proteins. J Biol Chem 267:7806–7813. [PubMed] [Google Scholar]
- 4.Narasimhan J, Potter JL, Haas AL. 1996. Conjugation of the 15-kDa interferon-induced ubiquitin homolog is distinct from that of ubiquitin. J Biol Chem 271:324–330. doi: 10.1074/jbc.271.1.324. [DOI] [PubMed] [Google Scholar]
- 5.Hemelaar J, Borodovsky A, Kessler BM, Reverter D, Cook J, Kolli N, Gan-Erdene T, Wilkinson KD, Gill G, Lima CD, Ploegh HL, Ovaa H. 2004. Specific and covalent targeting of conjugating and deconjugating enzymes of ubiquitin-like proteins. Mol Cell Biol 24:84–95. doi: 10.1128/MCB.24.1.84-95.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang D, Zhang DE. 2011. Interferon-stimulated gene 15 and the protein ISGylation system. J Interferon Cytokine Res 31:119–130. doi: 10.1089/jir.2010.0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeon YJ, Yoo HM, Chung CH. 2010. ISG15 and immune diseases. Biochim Biophys Acta 1802:485–496. doi: 10.1016/j.bbadis.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi HX, Yang K, Liu X, Liu XY, Wei B, Shan YF, Zhu LH, Wang C. 2010. Positive regulation of interferon regulatory factor 3 activation by Herc5 via ISG15 modification. Mol Cell Biol 30:2424–2436. doi: 10.1128/MCB.01466-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lenschow DJ. 2010. Antiviral properties of ISG15. Viruses 2:2154–2168. doi: 10.3390/v2102154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao C, Collins MN, Hsiang TY, Krug RM. 2013. Interferon-induced ISG15 pathway: an ongoing virus-host battle. Trends Microbiol 21:181–186. doi: 10.1016/j.tim.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lenschow DJ, Lai C, Frias-Staheli N, Giannakopoulos NV, Lutz A, Wolff T, Osiak A, Levine B, Schmidt RE, Garcia-Sastre A, Leib DA, Pekosz A, Knobeloch KP, Horak I, Virgin HWT. 2007. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc Natl Acad Sci U S A 104:1371–1376. doi: 10.1073/pnas.0607038104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 13.Ablashi DV, Chatlynne LG, Whitman JE Jr, Cesarman E. 2002. Spectrum of Kaposi's sarcoma-associated herpesvirus, or human herpesvirus 8, diseases. Clin Microbiol Rev 15:439–464. doi: 10.1128/CMR.15.3.439-464.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schulz TF. 1998. Kaposi's sarcoma-associated herpesvirus (human herpesvirus-8). J Gen Virol 79:1573–1591. doi: 10.1099/0022-1317-79-7-1573. [DOI] [PubMed] [Google Scholar]
- 15.Jacobs SR, Damania B. 2011. The viral interferon regulatory factors of KSHV: immunosuppressors or oncogenes? Front Immunol 2:19. doi: 10.3389/fimmu.2011.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacobs SR, Gregory SM, West JA, Wollish AC, Bennett CL, Blackbourn DJ, Heise MT, Damania B. 2013. The viral interferon regulatory factors of Kaposi's sarcoma-associated herpesvirus differ in their inhibition of interferon activation mediated by Toll-like receptor 3. J Virol 87:798–806. doi: 10.1128/JVI.01851-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.West J, Damania B. 2008. Upregulation of the TLR3 pathway by Kaposi's sarcoma-associated herpesvirus during primary infection. J Virol 82:5440–5449. doi: 10.1128/JVI.02590-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao C, Denison C, Huibregtse JM, Gygi S, Krug RM. 2005. Human ISG15 conjugation targets both IFN-induced and constitutively expressed proteins functioning in diverse cellular pathways. Proc Natl Acad Sci U S A 102:10200–10205. doi: 10.1073/pnas.0504754102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dastur A, Beaudenon S, Kelley M, Krug RM, Huibregtse JM. 2006. Herc5, an interferon-induced HECT E3 enzyme, is required for conjugation of ISG15 in human cells. J Biol Chem 281:4334–4338. doi: 10.1074/jbc.M512830200. [DOI] [PubMed] [Google Scholar]
- 20.Nakamura H, Lu M, Gwack Y, Souvlis J, Zeichner SL, Jung JU. 2003. Global changes in Kaposi's sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J Virol 77:4205–4220. doi: 10.1128/JVI.77.7.4205-4220.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Myoung J, Ganem D. 2011. Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: maintenance of tight latency with efficient reactivation upon induction. J Virol Methods 174:12–21. doi: 10.1016/j.jviromet.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Durfee LA, Huibregtse JM. 2012. The ISG15 conjugation system. Methods Mol Biol 832:141–149. doi: 10.1007/978-1-61779-474-2_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin P, Hu SW, Chang TH. 2003. Correlation between gene expression of aryl hydrocarbon receptor (AhR), hydrocarbon receptor nuclear translocator (Arnt), cytochromes P4501A1 (CYP1A1) and 1B1 (CYP1B1), and inducibility of CYP1A1 and CYP1B1 in human lymphocytes. Toxicol Sci 71:20–26. doi: 10.1093/toxsci/71.1.20. [DOI] [PubMed] [Google Scholar]
- 24.Burysek L, Yeow WS, Lubyova B, Kellum M, Schafer SL, Huang YQ, Pitha PM. 1999. Functional analysis of human herpesvirus 8-encoded viral interferon regulatory factor 1 and its association with cellular interferon regulatory factors and p300. J Virol 73:7334–7342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin R, Genin P, Mamane Y, Sgarbanti M, Battistini A, Harrington WJ Jr, Barber GN, Hiscott J. 2001. HHV-8 encoded vIRF-1 represses the interferon antiviral response by blocking IRF-3 recruitment of the CBP/p300 coactivators. Oncogene 20:800–811. doi: 10.1038/sj.onc.1204163. [DOI] [PubMed] [Google Scholar]
- 26.Zhao C, Hsiang TY, Kuo RL, Krug RM. 2010. ISG15 conjugation system targets the viral NS1 protein in influenza A virus-infected cells. Proc Natl Acad Sci U S A 107:2253–2258. doi: 10.1073/pnas.0909144107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu FX, Cusano T, Yuan Y. 1999. Identification of the immediate-early transcripts of Kaposi's sarcoma-associated herpesvirus. J Virol 73:5556–5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim MJ, Hwang SY, Imaizumi T, Yoo JY. 2008. Negative feedback regulation of RIG-I-mediated antiviral signaling by interferon-induced ISG15 conjugation. J Virol 82:1474–1483. doi: 10.1128/JVI.01650-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morales DJ, Lenschow DJ. 2013. The antiviral activities of ISG15. J Mol Biol 425:4995–5008. doi: 10.1016/j.jmb.2013.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun Z, Li Y, Ransburgh R, Snijder EJ, Fang Y. 2012. Nonstructural protein 2 of porcine reproductive and respiratory syndrome virus inhibits the antiviral function of interferon-stimulated gene 15. J Virol 86:3839–3850. doi: 10.1128/JVI.06466-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marcos-Villar L, Lopitz-Otsoa F, Gallego P, Munoz-Fontela C, Gonzalez-Santamaria J, Campagna M, Shou-Jiang G, Rodriguez MS, Rivas C. 2009. Kaposi's sarcoma-associated herpesvirus protein LANA2 disrupts PML oncogenic domains and inhibits PML-mediated transcriptional repression of the survivin gene. J Virol 83:8849–8858. doi: 10.1128/JVI.00339-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marcos-Villar L, Campagna M, Lopitz-Otsoa F, Gallego P, Gonzalez-Santamaria J, Gonzalez D, Rodriguez MS, Rivas C. 2011. Covalent modification by SUMO is required for efficient disruption of PML oncogenic domains by Kaposi's sarcoma-associated herpesvirus latent protein LANA2. J Gen Virol 92:188–194. doi: 10.1099/vir.0.024984-0. [DOI] [PubMed] [Google Scholar]
- 33.Marcos-Villar L, Gallego P, Munoz-Fontela C, de la Cruz-Herrera CF, Campagna M, Gonzalez D, Lopitz-Otsoa F, Rodriguez MS, Rivas C. 2014. Kaposi's sarcoma-associated herpesvirus lana2 protein interacts with the pocket proteins and inhibits their sumoylation. Oncogene 33:495–503. doi: 10.1038/onc.2012.603. [DOI] [PubMed] [Google Scholar]
- 34.Ma Z, Jacobs SR, West JA, Stopford C, Zhang Z, Davis Z, Barber GN, Glaunsinger BA, Dittmer DP, Damania B. 2015. Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc Natl Acad Sci U S A 112:E4306-15. doi: 10.1073/pnas.1503831112. [DOI] [PMC free article] [PubMed] [Google Scholar]