

ABSTRACT
UL36p (VP1/2) is the largest protein encoded by herpes simplex virus 1 (HSV-1) and resides in the innermost layer of the viral tegument, lying between the capsid and the envelope. UL36p performs multiple functions in the HSV life cycle, including an essential role in cytoplasmic envelopment. We earlier described the isolation of a virion-associated cytoplasmic membrane fraction from HSV-infected cells. Biochemical and ultrastructural analyses showed that the organelles in this buoyant fraction contain enveloped infectious HSV particles in their lumens and naked capsids docked to their cytoplasmic surfaces. These organelles can also recruit molecular motors and transport their cargo virions along microtubules in vitro. Here we examine the properties of these HSV-associated organelles in the absence of UL36p. We find that while capsid envelopment is clearly defective, a subpopulation of capsids nevertheless still associate with the cytoplasmic faces of these organelles. The existence of these capsid-membrane structures was confirmed by subcellular fractionation, immunocytochemistry, lipophilic dye fluorescence microscopy, thin-section electron microscopy, and correlative light and electron microscopy. We conclude that capsid-membrane binding can occur in the absence of UL36p and propose that this association may precede the events of UL36p-driven envelopment.
IMPORTANCE Membrane association and envelopment of the HSV capsid are essential for the assembly of an infectious virion. Envelopment involves the complex interplay of a large number of viral and cellular proteins; however, the function of most of them is unknown. One example of this is the viral protein UL36p, which is clearly essential for envelopment but plays a poorly understood role. Here we demonstrate that organelles utilized for HSV capsid envelopment still accumulate surface-bound capsids in the absence of UL36p. We propose that UL36p-independent binding of capsids to organelles occurs prior to the function of UL36p in capsid envelopment.
INTRODUCTION
Herpesviruses replicate their genomes and assemble DNA-packaged capsids in the cell nucleus. It is then generally accepted that capsids bud into the inner nuclear membrane to generate primary enveloped perinuclear virions that subsequently fuse with the outer nuclear membrane, releasing mature nucleocapsids (“naked” capsids) into the cytoplasm (1–3). These naked cytoplasmic capsids subsequently undergo secondary envelopment at a postnuclear organelle to assemble the mature, infectious virion (4–9). Completion of cytoplasmic envelopment requires the coordinated interaction of multiple virally encoded proteins (8, 10–13) and the participation of components of the cellular endosomal sorting complexes required for transport (ESCRT) machinery (14–19).
UL36p (VP1/2) is the largest structural protein encoded by the herpesviridae (20, 21). It is a major constituent of the innermost layer of tegument, the complex protein layer between the capsid and the inner surface of the envelope, and connects the capsid to multiple outer tegument proteins (22–30). During entry, UL36p recruits dynein/dynactin (31–34) to mediate retrograde traffic of capsids to the microtubule organizing center (35) and nuclear pores (36–38). UL36p also plays essential but poorly understood roles in assembly and egress: transporting capsids to their site of envelopment (34, 39, 40), helping drive envelopment (30, 39, 41), and also supporting subsequent traffic of enveloped particles (42). Ultrastructural analysis of HSV assembly and egress in a strain lacking the UL36 gene revealed an accumulation of nonenveloped cytoplasmic capsids and a defect in capsid trafficking within the cytoplasm (39, 41). Similar results were reported for the swine alphaherpesvirus pseudorabies virus (PRV) (43).
For both HSV and PRV, a key role for UL36p is to recruit UL37p to the capsid (23, 39, 43–45). UL37p may then mimic cellular multisubunit tethering complexes (26) to mediate capsid docking to organelles, including the trans-Golgi network (TGN) (25), perhaps via membrane anchors such as the heterodimer glycoprotein K (gK)/UL20p (46). UL36p and UL37p can also associate with organelles in the absence of capsids, since they are assembled into L particles (enveloped structures lacking capsids) in a mutually interdependent fashion (47–49) and because UL36p can mediate capsid-independent targeting of UL37p to the TGN (50). Loss of UL37p and UL36p results in similar phenotypes: accumulation of nonenveloped capsids in the nucleus and cytosol (27, 41, 51, 52) and a decrease in microtubule-mediated transport of capsids (39, 40, 42, 53). The interplay between UL36p/UL37p, the ESCRT apparatus, and other virally encoded proteins during capsid-membrane association and envelopment (8, 10, 46, 54) is not well understood.
The first UL36-null HSV strain was generated and characterized by the Desai laboratory. This virus, KΔUL36GFP, was found to be noninfectious in noncomplementing cell lines, was defective in cytoplasmic envelopment, and accumulated naked cytoplasmic capsids (41). In later studies using subcellular fractionation, ultrastructural analysis, and light microscopy, we obtained essentially identical results, except that in addition to naked cytoplasmic capsids we also observed a subpopulation of noninfectious but membrane-associated capsids present in the cytoplasm (42). Unlike wild-type enveloped virions, these buoyant membrane-associated capsids also showed diminished attachment to microtubules in vitro and were severely impaired in their ability to traffic along them (42). One possible explanation for the difference between our data and those of the Desai laboratory is that the original studies of KΔUL36GFP were carried out using cell extracts prepared by several freeze-thaw cycles followed by sonication (41), whereas we broke cells by shearing in a syringe needle without freezing (42). We proposed that in the absence of UL36p some capsids are able to attach to membranes (though no envelopment occurs) but that vigorous conditions of cell breakage might detach the capsids from their docking site (42).
A complication regarding analysis of UL36p function is that KΔUL36GFP contains an internal deletion of the UL36 gene; this removes codons 362 to 1555 from the 3,164-codon UL36 open reading frame (ORF) and frameshifts the downstream codons, generating 42 missense codons followed by a stop codon (41). As a result, the engineered locus carries an ORF of 403 codons, the first 361 of which correspond to the 5′ end of the normal UL36 gene. It is now clear that this amino-terminal coding region actually expresses an ∼43-kDa capsid-binding polypeptide in KΔUL36GFP-infected cells (47). Interestingly, the protein resembles the ∼47-kDa amino-terminal fragment of UL36p that is generated in wild-type HSV-infected cells and has ubiquitin-specific cysteine protease activity (55–57). Hence, it is possible that the amino-terminal portion of UL36p is providing a function that is normally present in wild-type cells and may be sufficient to enable association of HSV capsids with membranes.
In this study, to better characterize the role of UL36p in capsid-membrane interaction, we used biochemical fractionation, immunocytochemistry, lipophilic fluorescent dye staining, and ultrastructural imaging to test the association of HSV capsids with membranes in the presence and absence of any UL36 coding sequences. As expected, capsid envelopment was completely defective in the absence of UL36p; however, a subpopulation of membrane-bound capsids were still present. This suggests the existence of additional docking mechanisms for attachment of capsids to cytoplasmic organelles, perhaps as a precursor to envelopment.
MATERIALS AND METHODS
Cells and viruses.
Vero cells were maintained in Dulbecco modified Eagle's medium (DMEM) supplemented with 10% newborn calf serum (NCS) and 1% penicillin-streptomycin (PS) (Gibco Laboratories). HS30 cells (41) were grown in DMEM supplemented with 1% PS, 10% fetal calf serum (FCS) (Gibco Laboratories), and 100 μg/ml Geneticin (Life Technologies). Strains HSV1-K26GFP (58) and HSV1-GS2491 were grown, and titers were determined by plaque assay on Vero and HS30 cell monolayers as previously described (59).
Isolation of membrane-associated HSV.
Vero or HS30 cells were infected with K26GFP or HSV1-GS2491 at a multiplicity of infection (MOI) of 20 for 1 h at 37°C. Infected cells were then overlaid with fresh prewarmed media and incubated at 37°C for 16 h. Cells were then washed once with ice-cold MEPS buffer (5 mM MgSO4, 5 mM EGTA, 0.25 M sucrose, 35 mM PIPES, pH 7.1) [where PIPES is piperazine-N,N′-bis (2-ethanesulfonic acid)], scraped up, collected, and resuspended in MEPS buffer containing 2 mM phenylmethylsulfonyl fluoride (PMSF), 2% (vol/vol) protease inhibitor cocktail (Sigma), and 4 mM dithiothreitol (DTT). A postnuclear supernatant (PNS) and a membrane-associated fraction were then isolated as previously described (42, 60).
Western blotting and antibodies.
Membrane-associated fractions were isolated from K26GFP- and HSV1-GS2491-infected cells, and Western blotting was performed as previously described (42). Primary antibodies were as follows: VP5 was obtained from Genetech, VP16 was obtained from Santa Cruz Biotechnology, vhs antiserum was raised as previously described (61), and anti-VP22 rabbit antiserum was raised against a synthetic peptide corresponding to the carboxy-terminal 21 amino acids of HSV-1 VP22 (S. Smith and D. W. Wilson, unpublished data). Anti-UL37p and anti-UL36p rabbit antisera were kind gifts from Richard Courtney and John Wills. Secondary antibodies were alkaline phosphatase-conjugated goat anti-rabbit (Chemicon, Pittsburgh, PA) or alkaline phosphatase-conjugated goat anti-mouse (Antibodies Incorporated, Davis, CA) antibodies, as previously described (62).
Fluorescence and immunofluorescence microscopy.
All imaging was performed in the Analytical Imaging Facility of the Albert Einstein College of Medicine. Immunofluorescence studies were performed as previously described (16, 63). Primary antibodies were as follows: mouse monoclonal anti-Rab4 and anti-Rab5 (Transduction Laboratories), rabbit polyclonal anti-Rab7 (Santa Cruz Biotechnology), and affinity-purified sheep anti-TGN46 antiserum (Serotec). Secondary antibodies were Alexa Fluor 633-conjugated goat anti-mouse, goat anti-rabbit, or donkey anti-sheep IgG or Cy5-conjugated goat anti-sheep IgG (all from Molecular Probes).
For fluorescent dye staining of membranes, PNS preparations were diluted in staining buffer (SB; consisting of 5 mM MgSO4, 1 mM EGTA, 0.5 mM EDTA, 4 mM DTT, 2 mg/ml ascorbic acid, 35 mM PIPES [pH 7.1]) and attached to coverslips using 100 μg/ml poly-l-lysine (Sigma). They were then incubated with or without 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine perchlorate (DiD) or 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine-5,5′-disulfonic acid [DilC18(5)-DS] (Life Technologies) diluted in SB. After 45 min at room temperature, unbound dye was washed away with SB and samples were fixed with 4% paraformaldehyde in SB for 20 min at room temperature and then washed with SB. Coverslips were mounted with ProLong Gold antifade reagent (Life Technologies) for imaging. Images were processed using Volocity software V 2.2.2 (PerkinElmer).
Transmission electron microscopy.
Thin-section electron microscopy of isolated cytoplasmic organelles was as described earlier (60); membrane fractions were isolated from sucrose float-up gradients and diluted 5-fold to reduce the sucrose concentration, and organelles were collected by pelleting at 75,000 × g. The pellet was fixed with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, postfixed with 1% osmium tetroxide followed by 2% uranyl acetate, dehydrated through a graded series of ethanol, and embedded in LX112 resin (LADD Research Industries, Burlington, VT). Ultrathin sections were cut on a Reichert Ultracut UCT, stained with uranyl acetate followed by lead citrate, and viewed on a JEOL 1200EX transmission electron microscope at 80 kV.
CLEM.
For correlative light and electron microscopy (CLEM), PNS samples were prepared, attached, dye stained, and fixed as in our standard fluorescence studies described above but using MatTek dishes (MatTek Corporation) and then imaged using a Zeiss AxioObserver microscope and Axiovision “Shuttle & Find” software to mark regions of interest (ROI). After fluorescence imaging, samples were fixed with 2.5% glutaraldehyde, dehydrated in ethanol, critical point dried using a Tousimis Samdri 790, and then coated with chromium using an EMS 150T-ES sputter coater. The earlier-identified ROIs were automatically located in a Zeiss Supra 40 field emission scanning electron microscope (SEM) and imaged with a secondary electron detector. Fluorescence and SEM images were correlated with Zeiss AxioVision software (V 4.8).
RESULTS
Growth properties of a complete UL36 deletion strain on complementing and noncomplementing cells.
Strain HSV1-GS2491 carries a complete deletion of the UL36 gene and an in-frame fusion of green fluorescent protein (GFP) to the small capsid subunit VP26, similar to that originally described by the Desai laboratory (58; Gregory A. Smith, personal communication). To confirm the properties of this virus, we tested its replication in Vero cells and in the UL36p-expressing Vero-derived cell line HS30 (41). Vero and HS30 cells were infected with HSV1-GS2491 and the control UL36p-expressing, GFP-VP26-expressing HSV strain K26GFP (58) at an MOI of 10. After 1 h at 37°C, cells were acid washed as previously described (42) and then harvested immediately or after 12 or 18 h, and titers were determined by plaque assay on HS30 cell monolayers as previously described (59). HSV1-GS2491 replicated to an ∼4-log-higher titer on UL36p-expressing cells than on noncomplementing Vero cells, whereas the K26GFP virus replicated similarly on the two cell types (Fig. 1A). We also infected Vero and HS30 cells with HSV1-GS2491 or K26GFP at an MOI of 0.01 and harvested cells at full cytopathic effect after 72 h. When the titers of the extracts were determined on monolayers of HS30 cells, K26GFP plaqued at similar efficiencies whether it had been grown in Vero or HS30 cells. In contrast, the titer of HSV1-GS2491 was approximately 105-fold lower when grown on Vero than on HS30 cells (data not shown). Western blotting of infected Vero cell extracts with an anti-UL36p polyclonal antiserum confirmed that K26GFP, but not HSV1-GS2491, expressed the UL36p polypeptide (Fig. 1B).
FIG 1.

Preliminary characterization of a UL36-null HSV strain. (A) Single-step growth curves of the UL36-null HSV1-GS2491 on Vero and complementing HS30 cells. Vero (V) or HS30 (H) cells were infected with HSV-1 strain K26GFP (K26) or HSV1-GS2491 (GS) at an MOI of 10. After 1 h, they were acid washed to inactivate unpenetrated virus, and then cells were harvested immediately or after an additional 12 or 18 h at 37°C. Cell extracts were prepared, and their titers were determined on HS30 cells. Plotted values and error bars represent the means and standard deviations from the means. (B) Vero cells were infected with K26GFP or HSV1-GS2491 exactly as described for panel A. After 16 h of infection, total cell extracts were prepared and then subjected to SDS-PAGE and immunoblotted for UL36p. (C) PNS and membrane-associated fractions were prepared from K26GFP- or HSV1-GS2491-infected Vero or HS30 cells. They were then subjected to SDS-PAGE and immunoblotted for the major capsid protein VP5 and for tegument protein vhs, VP16, VP22, or UL37p (as indicated on the left). In panels B and C, the lanes are labeled at the top according to the virus strain used and the cell line infected, as described for panel A.
HSV1-GS2491 capsids and outer tegument proteins associate with membranes in the absence of UL36p.
We tested whether HSV capsid and tegument proteins were associated with membranes in the absence of UL36p. HSV-infected cells were gently broken by passage through a syringe needle, and a postnuclear supernatant was prepared. The PNS was then loaded at the bottom of a sucrose float-up gradient as previously described (60). Following centrifugation, soluble cytoplasmic proteins and dense structures, such as naked HSV capsids or ribosome-bound rough endoplasmic reticulum, remain at the bottom of the gradient. In contrast, cellular organelles, including endosomes, lysosomes, and the Golgi complex, float to a 0.8 M/1.2 M sucrose interface (60). This fraction contains the organelles to which HSV capsids traffic from the nucleus during a single synchronized wave of egress and is the first compartment at which fully enveloped infectious virions accumulate in the cytoplasm (60). HSV capsids enveloping in this gradient fraction also acquire molecular motors and become capable of traffic along microtubules in vitro (16, 42, 63). Biochemical analyses and electron microscopy revealed that this region of the gradient contains fully enveloped HSV particles within organellar lumens, and nonenveloped capsids docked to the cytoplasmic faces of the organelles (60).
The PNS and membrane-associated fractions of K26GFP and HSV1-GS2491 were prepared from either Vero or HS30 cells and then subjected to SDS-PAGE and immunoblotted for tegument proteins VP16, vhs, VP22, UL37p, and the major capsid protein VP5 (Fig. 1C). Levels of each of these antigens in the PNS were similar for all viral strain and cell line combinations tested (left half of Fig. 1C), confirming equivalent overall levels of expression of these proteins. Similarly, there were comparable amounts of VP5 and each of the tested tegument proteins in the membrane-associated fraction (right half of Fig. 1C). These results are identical to those we previously reported for HSV expressing the amino-terminal portion of UL36p (42). We conclude that, under these conditions, HSV capsid-membrane association occurs with similar efficiency in the presence or absence of UL36p.
Capsids colocalize with organellar antigens despite the absence of UL36p.
As an alternative test for the presence and origin of capsid-associated membranes, we examined cytoplasmic viral particles by immunocytochemistry. Membrane-associated fractions were prepared from K26GFP- or HSV1-GS2491-infected Vero or HS30 cells as described for Fig. 1C. They were then attached to coverslips and incubated with antibodies against the trans-Golgi network-localized integral membrane protein TGN46 or the membrane-anchored Rab proteins Rab4/Rab5 (early endosomes) and Rab7 (late endosomes). Representative immunostained and control fields and quantitation of the percentage of colocalization of capsids with each organellar marker are shown in Fig. 2A to C). As in our earlier studies (63), approximately 50% of immunostained K26GFP HSV capsids were TGN46 positive, and a smaller number were stained with anti-Rab antibodies. The UL36-null strain gave results similar to those of K26GFP when prepared from complementing HS30 cells. However, when the UL36-null strain was isolated from Vero cells, there was an ∼30% decrease in the numbers of capsids that colocalized with TGN46 and a corresponding increase in localization to Rab5/7-positive endosomes (Fig. 2C). Though modest, this change may suggest that the absence of UL36p results in some organellar mislocalization of capsids.
FIG 2.

HSV capsids colocalize with organellar markers in the absence of UL36p. Membrane-associated fractions were isolated from K26GFP or HSV1-GS2491-infected Vero or HS30 cells. They were then attached to coverslips, immunostained with anti-TGN46, anti-Rab4, anti-Rab5, or anti-Rab7 antibodies and appropriate secondary antibodies, and visualized in the green (capsid) and Cy5 (antibody) channels. (A) Representative field of HSV1-GS2491 (UL36-null) capsid-associated particles stained with anti-TGN46 antibodies. White arrowheads indicate colocalization of capsids (green) and TGN46 immunoreactivity (blue). The inset at the top right corner is a 3-fold magnification of the boxed region in the lower part of the panel. (B) Field similar to that in panel A but omitting anti-TGN46 antibodies. (C) Determination of percent colocalization based on the total number of VP26-GFP-positive K26GFP or HSV1-GS2491 particles compared to the number staining with each antiorganellar antibody (indicated below the graph). Viral strain and cell type are indicated in the key at the top left, labeled as in Fig. 1. The numbers of individual particles counted (n) for each sample were as follows: K26-V, n = 1,326; GS-V, n = 517; GS-H, n = 1,518. Plotted values represent means and standard deviations from the means.
Quantitating membrane association of capsids by staining with fluorescent lipophilic dyes.
To determine the numbers of cytoplasmic capsids that colocalized with biological membranes, we used two different fluorescent lipophilic dyes: DiD and DilC18(5)-DS. A PNS was prepared from infected cells, attached to polylysine-coated coverslips, and then incubated with DiD or DilC18(5)-DS or mock treated. Samples were then fixed and imaged in the green (VP26-GFP capsid subunit) or Cy5 (infrared-emitting lipophilic dye) channels as we described earlier (16). Figure 3 shows representative fields of DilC18(5)-DS-stained PNS particles from HSV1-GS2491-infected Vero (Fig. 3A) and HS30 cells (Fig. 3B). All fields contained a mixture of GFP-fluorescing particles of various sizes. Although occasional large GFP-fluorescent structures were visible, the majority of particles were in the range of ∼500 to 1,000 nm in apparent (fluorescent) diameter, and a subset of them stained with either lipophilic dye. To quantitate these membrane-associated, capsid-containing structures in the presence and absence of UL36p, we counted the DiD-positive and DilC18(5)-DS-positive GFP-positive particles in microscopic fields, and the data are summarized in Tables 1 and 2. Background fluorescence in the absence of dye was negligible, confirming that all fluorescence in the Cy5 channel was dye specific (Tables 1 and 2). For HSV K26GFP, approximately 20% of the fluorescent capsid-containing structures stained with either of the lipophilic dyes. Essentially identical results were obtained for the UL36-null HSV1-GS2491 virus, whether grown in Vero or UL36p-expressing HS30 cells. This suggests that membrane association of a subpopulation of HSV capsids can occur even in the absence of UL36p, consistent with the Western blot and immunostaining data in Fig. 1C and 2.
FIG 3.

Representative fields of fluorescent dye-stained cytoplasmic HSV particles. PNS was prepared from HSV1-GS2491-infected Vero (A) or HS30 (B) cells, and then virions and organelles were attached to a microscope slide and stained with the fluorescent lipophilic dye DilC18(5)-DS. After fixation, fields of particles were imaged in the green (VP26-GFP) and Cy5 (infrared, dye) channels; merged images are shown. GFP-positive/dye-positive and GFP-positive/dye-negative particles were counted to generate the data in Tables 1 and 2. Bar, 10 μm.
TABLE 1.
Quantitation of DiD lipophilic dye-stained GFP-capsid-associated particles in the PNS of HSV-infected cellsa
| Virus-cell (concn of DiD [μM])b | Mean no. (± SD) of particles per field |
DiD-positive green particles as % of total | |
|---|---|---|---|
| Total green | DiD-positive green | ||
| K26-V (0) | 1,108 ± 178 | 13 ± 3 | 1.2 ± 0.3 |
| K26-V (10) | 1,211 ± 286 | 226 ± 65 | 18.7 ± 4.7 |
| K26-H (0) | 1,131 ± 96 | 14 ± 5 | 1.2 ± 0.4 |
| K26-H (10) | 1,195 ± 143 | 257 ± 27 | 21.5 ± 2.9 |
| GS-V (0) | 1,062 ± 125 | 15 ± 4 | 1.4 ± 0.4 |
| GS-V (10) | 1,073 ± 240 | 228 ± 49 | 21.2 ± 5.6 |
| GS-H (0) | 1,085 ± 102 | 14 ± 4 | 1.3 ± 0.4 |
| GS-H (10) | 1,086 ± 160 | 216 ± 46 | 19.9 ± 4.5 |
For each concentration of dye, counting was done in 12 independent microscopic fields from K26-V and K26-H and in 11 fields from GS-V and GS-H to determine the mean particle number per field.
V, Vero cells; H, HS30 cells; K26, HSV-1 strain K26GFP; GS, HSV1-GS2491.
TABLE 2.
Quantitation of DilC18(5)-DS (Dil) lipophilic dye-stained GFP-capsid-associated particles in the PNS of HSV-infected cellsa
| Virus-cell (concn of Dil [μM])b | Mean no. (± SD) of particles per field |
Dil-positive green particles as % of total | |
|---|---|---|---|
| Total green | Dil-positive green | ||
| K26-V (0) | 1,189 ± 64 | 14 ± 3 | 1.2 ± 0.3 |
| K26-V (10) | 1,194 ± 86 | 333 ± 52 | 27.9 ± 3.5 |
| K26-H (0) | 1,220 ± 77 | 16 ± 4 | 1.3 ± 0.4 |
| K26-H (10) | 1,314 ± 120 | 389 ± 45 | 29.6 ± 3.1 |
| GS-V (0) | 1,195 ± 23 | 18 ± 2 | 1.5 ± 0.2 |
| GS-V (10) | 1,254 ± 112 | 354 ± 36 | 28.2 ± 2.8 |
| GS-H (0) | 1,221 ± 73 | 17 ± 4 | 1.4 ± 0.3 |
| GS-H (10) | 1,221 ± 79 | 336 ± 48 | 27.5 ± 3.4 |
For each concentration of dye, counting was done in five independent microscopic fields from K26-V and K26-H and in seven fields from GS-V and GS-H to determine the mean particle number per field.
V, Vero cells; H, HS30 cells; K26, HSV-1 strain K26GFP; GS, HSV1-GS2491.
Examination of the morphology of capsid-membrane-containing cytoplasmic particles by light microscopy.
Because of the limited resolving power of light microscopy, the numbers of dye-positive capsid-associated particles shown in Tables 1 and 2 may be an overestimate. Capsids in close proximity to dye-staining membranes may exhibit overlapping fluorescence signals even if not physically attached. To further refine our analysis, we prepared fields of DiD- or DilC18(5)-DS-stained PNS, imaged individual capsid-containing particles, and determined the intensity of each pixel in the green and Cy5 channels. For particles exhibiting at least one dually fluorescent pixel, we calculated a Pearson product-moment correlation coefficient for the GFP and DiD or DilC18(5)-DS fluorescence signals (64). Galleries of representative DiD and DilC18(5)-DS-stained particles and their calculated Pearson values are shown in Fig. 4A and C, respectively. A Pearson value of zero or less indicates no positive correlation between the locations of the capsid and dye fluorescence signals. Pearson coefficients in the range of ∼0.4 to 1.0 indicate a relatively high correlation between the lipid dye and capsid fluorescence signals; these particles predominantly exhibited a green core with a lipid dye-staining halo. Particles with Pearson coefficients between 0 and ∼0.39 commonly displayed adjacent poles of dye and capsid fluorescence, with various degrees of overlap at the “equator.” We counted approximately 1,000 to 2,000 individual particles for each combination of cell line and virus strain (see Fig. 4 legend for details). The distribution of particle numbers for various values of the Pearson coefficient is shown for DiD- and DilC18(5)-DS-stained structures in Fig. 4B and D, respectively.
FIG 4.

Appearance of fluorescent cytoplasmic HSV capsid-membrane particles in the presence and absence of UL36p. PNS samples were isolated from K26GFP- or HSV1-GS2491-infected Vero or HS30 cells (labeled according to virus strain and cell line as in Fig. 1), and then fields of HSV particles were stained with fluorescent lipophilic dyes and imaged. Pearson correlation values were determined for the capsid and lipophilic dye fluorescence of individual particles. (A) Representative gallery of DiD-stained particles imaged in the green (capsid) and Cy5 (infrared, DiD) channels. In each panel, the Pearson value of the particle is shown in the upper right, and a 500-nm vertical scale bar in the lower right. (B) Numbers of particles falling within each of three different Pearson value ranges (shown below the graph) plotted as a percentage of the total. Plotted values are means and standard deviations from the means. Numbers of individual particles counted were 1,457 (GS-V), 1,363 (GS-H), 1,083 (K26-V), and 1,229 (K26-H). The numbers of HSV1-GS2491 particles with Pearson values less than or equal to zero and also those with values between 0.4 and 1.0 differed significantly depending upon whether they had been isolated from Vero or HS30 cells (P < 0.0001 and P < 0.001, respectively, for the two Pearson ranges). (C, D) Similar to the data in panels A and B but using the lipophilic dye DilC18(5)-DS. Numbers of individual particles counted were 1,917 (GS-V), 2,392 (GS-H), 1,704 (K26-V), and 1,801 (K26-H). As described for panel B, the distribution of Pearson values was significantly different for HSV1-GS2491 particles depending on whether they originated from Vero or HS30 cells (P < 0.05 for Pearson values less than or equal to zero, P < 0.01 for values in the range of 0.4 to 1.0).
The numbers of particles with relatively low Pearson correlations (0.01 to 0.39) were similar for K26GFP and HSV1-GS2491, whether they had replicated in Vero or HS30 cells (Fig. 4B and D). In contrast, when HSV1-GS2491 was grown in Vero cells, in the absence of UL36p, the number of moderate and strong Pearson correlation particles (0.4 to 1.0) was markedly increased. This change was reversed when the UL36-deleted strain was grown in complementing HS30 cells. Identical results were seen for both lipophilic dyes (Fig. 4B and D). We consider possible explanations for these data in Discussion.
Ultrastructural examination of the morphology of capsid-membrane-containing cytoplasmic particles.
To better understand the structure of the capsid-membrane complexes, we performed transmission electron microscopic analysis of the buoyant organellar fraction known to contain assembling and mature virions (60). HSV-associated organelles were prepared on gradients from HSV1-GS2491-infected Vero or HS30 cells as in Fig. 1C and then pelleted, fixed, and sectioned. As shown in Fig. 5A to K, B and C capsids were readily observed on the external surfaces of membranes prepared from Vero cells but were never found enveloped or in the organellar lumen. In contrast, enveloped luminal virions were present when organelles were isolated from infected HS30 cells (Fig. 5L to N).
FIG 5.

Transmission electron microscopy of membrane-associated HSV particles. Vero (A to K) or HS30 cells (L to N) were infected with UL36-null HSV1-GS2491, and then cytoplasmic buoyant virus-associated organelles were prepared as described for Fig. 1C. The organelle-containing fraction was pelleted, fixed, and processed for thin-section electron microscopy. White and black arrowheads in each panel indicate B and C capsids, respectively. All images are at the same magnification. Bar in panel A, 100 nm.
We next repeated our DilC18(5)-DS staining and imaging of cytoplasmic virions as described for Fig. 4C but in the presence of 1-μm- and 0.5-μm-diameter fluorescent beads. These beads, visible in both fluorescence and scanning electron microscopy, were then used to align the light and SEM images, to perform correlative light and electron microscopy (Fig. 6). Both dye-negative (Fig. 6A to D) and dye-positive (Fig. 6E to J) green fluorescent particles were imaged and aligned. From these images, it appears that dye-negative green particles (Fig. 6A to D) align with discrete spheres that presumably represent HSV capsids. In contrast, dye-positive, GFP-positive particles with lower Pearson values (Fig. 6E to G) correspond to capsids attached to the tips of membrane tubules or the edges of membrane sheets. Capsid-membrane complexes with higher Pearson values (Fig. 6H to J) appear to be derived from capsids more centrally located on the surfaces of membrane tubules or sheets. Note that in panel 6H, while some capsids are centrally located on the dye-stained organelle and contribute to a yellow fluorescence signal, other, apparently unattached, capsids lie to the lower right of the structure. The latter capsids give rise to a mainly green, but still partially overlapping, area of fluorescence. Importantly, the Pearson values summarized in Fig. 4 were calculated from fields of particles attached to microscope slides at a 20- to 25-fold-lower concentration than in our CLEM studies, to reduce the likelihood of unattached particles coming to lie in close proximity to one another. Nevertheless, because of the limited resolving power of light microscopy, the possibility of false colocalization signals remains. Possible explanations for why the presence or absence of UL36p influences the distribution of fluorescent particle types (Fig. 4) are considered in Discussion.
FIG 6.
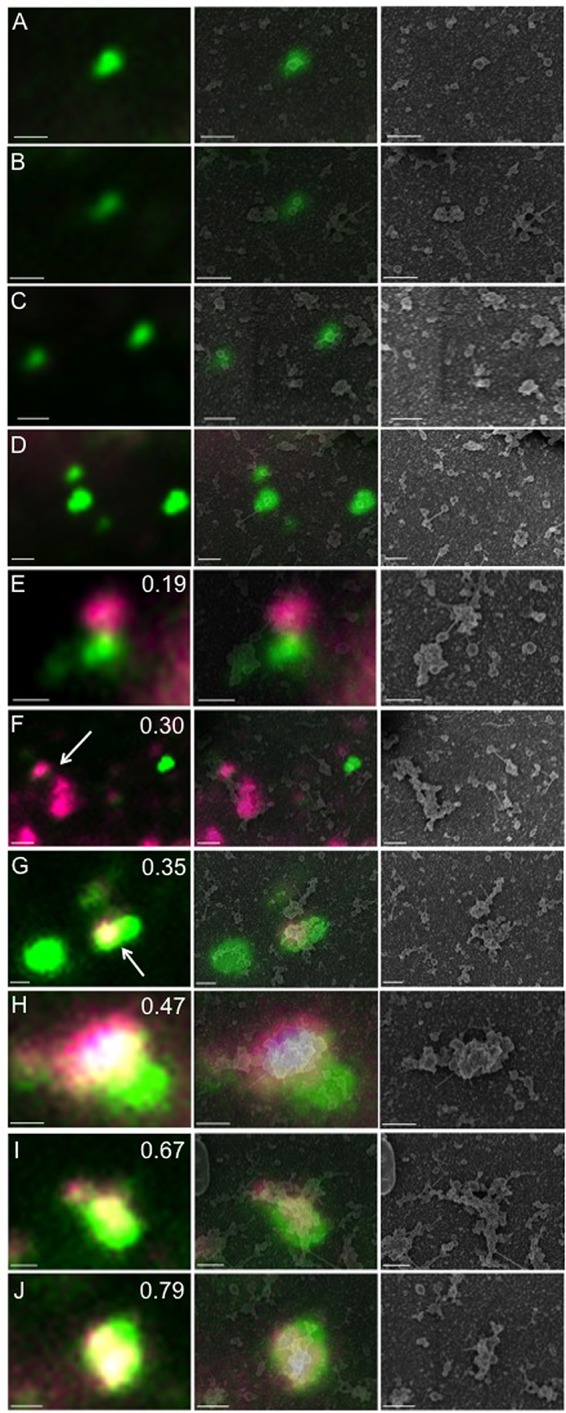
Correlative light and electron microscopic analysis of cytoplasmic HSV particles. Cytoplasmic particles from HSV1-GS2491-infected Vero cells were prepared, stained with DilC18(5)-DS, and fixed as described for Fig. 3 and 4. After fluorescence imaging, the sample was processed for scanning electron microscopy (SEM), and then SEM and fluorescence images were aligned. In each horizontal row, the fluorescence image is shown in the left-hand panel, an alignment of the fluorescence and SEM images is in the middle panel, and the SEM image alone is in the right-hand panel. Bar, 500 nm. (A to D) Dye-negative green fluorescent particles. (E to J) Lipophilic dye-positive green fluorescent particles. The Pearson correlation coefficient is shown in the upper right of each left-hand panel. In panels F and G, the white arrow indicates the particle for which a Pearson value was determined.
DISCUSSION
We have previously developed techniques for the isolation of enveloped and membrane-associated HSV particles from the perinuclear space (65) and cytoplasm (60, 63) of infected cells, based on standard methodologies of subcellular fractionation. Using this approach, we earlier reported that membrane-associated capsids were present in cytoplasmic extracts of cells (42) infected by the prototypical UL36-null strain K26ΔUL36 (41). Although membrane associated, these viral particles were clearly noninfectious (42), consistent with the finding that HSV capsids fail to envelope when the UL36 gene is partially or completely deleted (39, 41). Membrane-associated capsids could also be visualized in the cytoplasm of intact cells infected by this UL36-null virus, though naked cytoplasmic capsids were most commonly seen (42), as others have observed (39, 41). Capsids peripherally associated with organelles may be difficult to identify by transmission electron microscopy, since they will appear “naked” unless the microscopic section intersects both capsid and organelle. In contrast, fully enveloped cytoplasmic capsids are surrounded by two lipid bilayers (the envelope and bounding membrane of the surrounding organelle) in most or all sections. Furthermore, capsid-membrane-docked complexes may not be abundant structures if they are generated slowly or consumed rapidly during virion assembly.
Since the UL36-null strain used in our earlier study is now known to express an amino-terminally derived fragment of UL36p (41, 47), we here tested whether capsid-membrane association (as distinct from envelopment) could also occur in the complete absence of any UL36-derived gene products and further investigated the morphology and composition of the viral particles that are formed. To examine the structure of cytoplasmic HSV particles in the presence and absence of UL36p, we used gentle cell breakage followed by density gradient isolation of a defined cytoplasmic organellar fraction. We earlier showed that this fraction contains the organelles to which HSV capsids traffic and at which they become enveloped, during a single synchronized wave of egress (60). In a wild-type HSV infection, this region of the gradient contains nonenveloped capsids docked to the cytoplasmic faces of organelles and fully enveloped, infectious HSV particles within organellar lumens (60). Whether UL36p was present or absent, Western blotting confirmed the accumulation of similar levels of the major capsid protein VP5 in this organellar gradient fraction (Fig. 1C), suggesting that the association of HSV capsids with the surface of membranes does not absolutely require UL36p.
The presence of outer tegument components in this region of the gradient (Fig. 1C) even in the absence of UL36p is to be expected. These proteins associate with the cytoplasmic faces of cellular organelles through multiple mechanisms, including interaction with envelope protein tails, and can often occur independently of the presence of capsids (47, 62, 66–71). It also appears that the inner tegument component UL37p was able to assemble onto membranes normally in the absence of UL36p (Fig. 1C). UL37p-membrane binding could be by direct interaction between UL37p and the HSV capsid or outer tegument, as suggested by yeast two-hybrid analysis (24); however, UL37p recruitment to capsids is generally thought to be dependent upon UL36p (22, 39, 43). UL37p-membrane association in the absence of UL36p is therefore most likely mediated by other UL37p-binding partners such as the membrane protein complex gK/UL20p (46) or the cellular protein dystonin/BPAG1 (53), known to be involved in Golgi complex organization and dynamics (72).
To examine the organelle-associated capsids further, we immunostained fields of virally associated cytoplasmic particles. Capsid-associated structures could be stained by antibodies that recognize the trans-Golgi network marker TGN46 and (to a lesser extent) the endosomal antigens Rab4/5/7, as we reported earlier (63). In the absence of UL36p, the association of capsids with these organellar antigens underwent a modest shift; fewer capsids colocalized with the trans-Golgi network and more with endosomes. The reasons for this change are unclear, but the absence of UL36p from capsids would result in failure to recruit UL37p, which is a putative viral mimic of cellular membrane-tethering complexes (26). Perhaps UL36p/UL37p-null capsids are unable to target to the proper membrane prior to envelopment and mislocalize to incorrect organellar surfaces.
Our immunolocalization approach limited our imaging to those capsid-organelle structures that contain a particular antigen. To obtain a more comprehensive view of membrane-associated capsids, we examined microscopic fields of viral particles stained with the fluorescent lipophilic dyes DiD and DilC18(5)-DS. PNS preparations contained abundant numbers of small GFP-positive particles and less numerous, larger heterogeneous structures (Fig. 3). The former resemble in size and appearance the assembly intermediates that we have described earlier, a subset of which are motile along microtubules in vitro (16). The latter, which are more abundant in Vero (Fig. 3A) than HS30 cells (Fig. 3B) may correspond to the naked HSV capsids that accumulate in the absence of UL36p, often in large particulate clusters (39, 41). We did not attempt to quantitate these less abundant, large heterogenous structures but counted the smaller, more numerous GFP-positive particles and determined how many were lipophilic dye positive or dye negative. In either the presence of absence of UL36p, approximately 20% of these capsid-associated particles stained with each dye, suggesting that they were in close proximity to membranes; the remaining 80% of the capsids appeared to be naked (Tables 1 and 2).
We next attempted to investigate the structure of the DiD (Fig. 4A and B) and DilC18(5)-DS (Fig. 4C and D) dye-stained capsid-associated particles. Though we are limited by the resolving power of light microscopy, we found that the particles could be usefully categorized by measuring the degree to which membrane and capsid-associated fluorescence colocalized using a Pearson correlation coefficient. In even UL36p-expressing K26GFP-infected cells, the degree of colocalization of capsid and membrane-derived fluorescence varied from relatively low (Pearson values in the range of 0 to 0.39) to high (Pearson values between 0.4 and 1.0) suggesting that a variety of structures are present during the course of a normal infection (Fig. 4B and D). When UL36p was absent, the number of particles with moderate-to-strong Pearson correlation values (0.4 to 1.0) increased compared to those of the K26GFP virus, and the number of structures showing adjacent but nonoverlapping fluorescence decreased for both lipophilic dyes. This distribution was corrected back to that of K26GFP by restoring UL36p with the complementing cell line HS30 (Fig. 4B and D).
In order to better understand the structural implications of these Pearson values, we used correlative light and electron microscopy (CLEM). This enabled us to align the fluorescence image, with its known Pearson value, to the same structure visualized by scanning electron microscopy. Following CLEM, dye-negative green particles (Fig. 6A to D) were found to correspond to discrete spheres with a mean diameter of 139 nm (average of 9 capsids counted from 4 different fields; standard deviation, 14.1). This size corresponds well to the expected 125-nm diameter of the naked HSV capsid plus an approximately 16-nm-thick chromium sputter coating deposited during SEM preparation. In contrast, consistent with our Western blotting and thin-section EM studies, dye-stained capsids appeared as green spheres associated with tubules and sheets of membrane (Fig. 6E to J).
Alphaherpesvirus capsid-membrane association and envelopment are driven by numerous cooperative, and in some cases redundant, interactions between multiple tegument and envelope proteins (8–13, 73) and the cellular ESCRT machinery (14–19). While it is clear that UL36p is essential for envelopment, the simplest interpretation of our data is that the association of some HSV capsids with the surface of cytoplasmic organelles does not absolutely require UL36p. The apparatus that could mediate UL36p-independent capsid-membrane docking is unknown, but one attractive candidate is the Golgi network-localized membrane-anchored UL11p-UL16p-UL21p complex (74–76). This heterotrimer facilitates cytoplasmic envelopment in both HSV and PRV (8, 73, 77) perhaps via recruitment of envelope proteins and outer tegument components, including gE and VP22 (54, 78). Importantly, UL16p assembles onto HSV capsids in the absence of UL36p (79), raising the possibility that UL11p-UL16p-UL21p could connect capsids to the envelopment membrane and outer tegument independently of UL36p/UL37p function. In this model, UL36p might cooperate with UL11p-UL16p-UL21p to mediate initial capsid-membrane association and then play an essential role downstream, such as integrating the viral tegument/envelope protein envelopment machinery with the cellular ESCRT/Vps4 budding/scission apparatus (14–16). Alternatively, or in addition, UL36p may be required to target capsids to the correct organelle for subsequent envelope assembly, perhaps by virtue of its association with the putative membrane-tethering factor UL37p (26) and the integral membrane heterodimer gK/UL20p (46). In this case, when UL36p is absent, capsids might associate with organelles or organellar membrane subdomains, unsuitable for ESCRT-driven envelopment. This possibility is consistent with organellar mistargeting of capsids in the absence of UL36p (Fig. 2) and provides a working hypothesis to explain the particle structural differences seen by fluorescence microscopy (Fig. 4). Our CLEM analysis suggests that capsid-membrane complexes with higher Pearson values (structures that are enriched when UL36p is absent [Fig. 4]) correspond to capsids more centrally located on the surfaces of membrane tubules or sheets (Fig. 6H to J). Conversely, particles with lower Pearson values (in the majority when UL36p is present [Fig. 4]) represent more-peripheral capsids, docked at the tips of membrane tubules or at the edges of membrane sheets (Fig. 6E to G). Such regions of high membrane curvature are enriched in lipids that are favorable to vesicle budding and may be preferred sites for UL36p-driven capsid envelopment (80, 81). Alternatively, or in addition, the propensity of UL36-null HSV capsids to form clusters in the cytoplasm (41) might be shared by these membrane-docked capsids; in the absence of UL36p, capsids on the organellar surface might associate with one another, forming bright GFP-fluorescent foci on the dye-stained membranes and giving rise to the higher Pearson value particles that we observe.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health Grant AI083285 (to D.W.W.).
We thank Gregory A. Smith for the HSV UL36-null strain HSV1-GS2491 and for helpful discussions. HSV strain K26GFP and the HS30 cell line were generously provided by Prashant Desai. We thank Leslie Gunther-Cummins, Frank Macaluso, and the Analytical Imaging Facility of the Albert Einstein College of Medicine for help with CLEM and Erik Snapp and the Liver Center Imaging and Cell Structure Core (P30 DK041296) for support and advice.
REFERENCES
- 1.Mettenleiter TC, Muller F, Granzow H, Klupp BG. 2013. The way out: what we know and do not know about herpesvirus nuclear egress. Cell Microbiol 15:170–178. doi: 10.1111/cmi.12044. [DOI] [PubMed] [Google Scholar]
- 2.Bigalke JM, Heuser T, Nicastro D, Heldwein EE. 2014. Membrane deformation and scission by the HSV-1 nuclear egress complex. Nat Commun 5:4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bigalke JM, Heldwein EE. 2015. The great (nuclear) escape: new insights into the role of the nuclear egress complex of herpesviruses. J Virol 89:9150–9153. doi: 10.1128/JVI.02530-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Browne H, Bell S, Minson T, Wilson DW. 1996. An endoplasmic reticulum-retained herpes simplex virus glycoprotein H is absent from secreted virions: evidence for reenvelopment during egress. J Virol 70:4311–4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skepper JN, Whiteley A, Browne H, Minson A. 2001. Herpes simplex virus nucleocapsids mature to progeny virions by an envelopment –> deenvelopment –> reenvelopment pathway. J Virol 75:5697–5702. doi: 10.1128/JVI.75.12.5697-5702.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mettenleiter TC, Klupp BG, Granzow H. 2006. Herpesvirus assembly: a tale of two membranes. Curr Opin Microbiol 9:423–429. doi: 10.1016/j.mib.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 7.Turcotte S, Letellier J, Lippe R. 2005. Herpes simplex virus type 1 capsids transit by the trans-Golgi network, where viral glycoproteins accumulate independently of capsid egress. J Virol 79:8847–8860. doi: 10.1128/JVI.79.14.8847-8860.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mettenleiter TC, Klupp BG, Granzow H. 2009. Herpesvirus assembly: an update. Virus Res 143:222–234. doi: 10.1016/j.virusres.2009.03.018. [DOI] [PubMed] [Google Scholar]
- 9.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol 9:382–394. doi: 10.1038/nrmicro2559. [DOI] [PubMed] [Google Scholar]
- 10.Kopp M, Granzow H, Fuchs W, Klupp B, Mettenleiter TC. 2004. Simultaneous deletion of pseudorabies virus tegument protein UL11 and glycoprotein M severely impairs secondary envelopment. J Virol 78:3024–3034. doi: 10.1128/JVI.78.6.3024-3034.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Browne H, Bell S, Minson T. 2004. Analysis of the requirement for glycoprotein M in herpes simplex virus type 1 morphogenesis. J Virol 78:1039–1041. doi: 10.1128/JVI.78.2.1039-1041.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foster TP, Chouljenko VN, Kousoulas KG. 2008. Functional and physical interactions of the herpes simplex virus type 1 UL20 membrane protein with glycoprotein K. J Virol 82:6310–6323. doi: 10.1128/JVI.00147-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chouljenko DV, Kim IJ, Chouljenko VN, Subramanian R, Walker JD, Kousoulas KG. 2012. Functional hierarchy of herpes simplex virus 1 viral glycoproteins in cytoplasmic virion envelopment and egress. J Virol 86:4262–4270. doi: 10.1128/JVI.06766-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crump CM, Yates C, Minson T. 2007. Herpes simplex virus type 1 cytoplasmic envelopment requires functional Vps4. J Virol 81:7380–7387. doi: 10.1128/JVI.00222-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pawliczek T, Crump CM. 2009. Herpes simplex virus type 1 production requires a functional ESCRT-III complex but is independent of TSG101 and ALIX expression. J Virol 83:11254–11264. doi: 10.1128/JVI.00574-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kharkwal H, Smith CG, Wilson DW. 2014. Blocking ESCRT-mediated envelopment inhibits microtubule-dependent trafficking of alphaherpesviruses in vitro. J Virol 88:14467–14478. doi: 10.1128/JVI.02777-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee CP, Liu PT, Kung HN, Su MT, Chua HH, Chang YH, Chang CW, Tsai CH, Liu FT, Chen MR. 2012. The ESCRT machinery is recruited by the viral BFRF1 protein to the nucleus-associated membrane for the maturation of Epstein-Barr Virus. PLoS Pathog 8:e1002904. doi: 10.1371/journal.ppat.1002904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fraile-Ramos A, Pelchen-Matthews A, Risco C, Rejas MT, Emery VC, Hassan-Walker AF, Esteban M, Marsh M. 2007. The ESCRT machinery is not required for human cytomegalovirus envelopment. Cell Microbiol 9:2955–2967. doi: 10.1111/j.1462-5822.2007.01024.x. [DOI] [PubMed] [Google Scholar]
- 19.Tandon R, AuCoin DP, Mocarski ES. 2009. Human cytomegalovirus exploits ESCRT machinery in the process of virion maturation. J Virol 83:10797–10807. doi: 10.1128/JVI.01093-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mocarski ES., Jr 2007. Comparative analysis of herpesvirus-common proteins. In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, England. [PubMed] [Google Scholar]
- 21.Kelly BJ, Fraefel C, Cunningham AL, Diefenbach RJ. 2009. Functional roles of the tegument proteins of herpes simplex virus type 1. Virus Res 145:173–186. doi: 10.1016/j.virusres.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 22.Ko DH, Cunningham AL, Diefenbach RJ. 2010. The major determinant for addition of tegument protein pUL48 (VP16) to capsids in herpes simplex virus type 1 is the presence of the major tegument protein pUL36 (VP1/2). J Virol 84:1397–1405. doi: 10.1128/JVI.01721-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vittone V, Diefenbach E, Triffett D, Douglas MW, Cunningham AL, Diefenbach RJ. 2005. Determination of interactions between tegument proteins of herpes simplex virus type 1. J Virol 79:9566–9571. doi: 10.1128/JVI.79.15.9566-9571.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee JH, Vittone V, Diefenbach E, Cunningham AL, Diefenbach RJ. 2008. Identification of structural protein-protein interactions of herpes simplex virus type 1. Virology 378:347–354. doi: 10.1016/j.virol.2008.05.035. [DOI] [PubMed] [Google Scholar]
- 25.Pasdeloup D, Beilstein F, Roberts AP, McElwee M, McNab D, Rixon FJ. 2010. Inner tegument protein pUL37 of herpes simplex virus type 1 is involved in directing capsids to the trans-Golgi network for envelopment. J Gen Virol 91:2145–2151. doi: 10.1099/vir.0.022053-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pitts JD, Klabis J, Richards AL, Smith GA, Heldwein EE. 2014. Crystal structure of the herpesvirus inner tegument protein UL37 supports its essential role in control of viral trafficking. J Virol 88:5462–5473. doi: 10.1128/JVI.00163-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kelly BJ, Bauerfeind R, Binz A, Sodeik B, Laimbacher AS, Fraefel C, Diefenbach RJ. 2014. The interaction of the HSV-1 tegument proteins pUL36 and pUL37 is essential for secondary envelopment during viral egress. Virology 454–455:67–77. [DOI] [PubMed] [Google Scholar]
- 28.Mijatov B, Cunningham AL, Diefenbach RJ. 2007. Residues F593 and E596 of HSV-1 tegument protein pUL36 (VP1/2) mediate binding of tegument protein pUL37. Virology 368:26–31. doi: 10.1016/j.virol.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 29.Fan WH, Roberts AP, McElwee M, Bhella D, Rixon FJ, Lauder R. 2015. The large tegument protein pUL36 is essential for formation of the capsid vertex-specific component at the capsid-tegument interface of herpes simplex virus 1. J Virol 89:1502–1511. doi: 10.1128/JVI.02887-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schipke J, Pohlmann A, Diestel R, Binz A, Rudolph K, Nagel CH, Bauerfeind R, Sodeik B. 2012. The C terminus of the large tegument protein pUL36 contains multiple capsid binding sites that function differently during assembly and cell entry of herpes simplex virus. J Virol 86:3682–3700. doi: 10.1128/JVI.06432-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sodeik B, Ebersold MW, Helenius A. 1997. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J Cell Biol 136:1007–1021. doi: 10.1083/jcb.136.5.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Radtke K, Kieneke D, Wolfstein A, Michael K, Steffen W, Scholz T, Karger A, Sodeik B. 2010. Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PLoS Pathog 6:e1000991. doi: 10.1371/journal.ppat.1000991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zaichick SV, Bohannon KP, Hughes A, Sollars PJ, Pickard GE, Smith GA. 2013. The herpesvirus VP1/2 protein is an effector of dynein-mediated capsid transport and neuroinvasion. Cell Host Microbe 13:193–203. doi: 10.1016/j.chom.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wolfstein A, Nagel CH, Radtke K, Dohner K, Allan VJ, Sodeik B. 2006. The inner tegument promotes herpes simplex virus capsid motility along microtubules in vitro. Traffic 7:227–237. doi: 10.1111/j.1600-0854.2005.00379.x. [DOI] [PubMed] [Google Scholar]
- 35.Abaitua F, Hollinshead M, Bolstad M, Crump CM, O'Hare P. 2012. A nuclear localization signal in herpesvirus protein VP1-2 is essential for infection via capsid routing to the nuclear pore. J Virol 86:8998–9014. doi: 10.1128/JVI.01209-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jovasevic V, Liang L, Roizman B. 2008. Proteolytic cleavage of VP1-2 is required for release of herpes simplex virus 1 DNA into the nucleus. J Virol 82:3311–3319. doi: 10.1128/JVI.01919-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ojala PM, Sodeik B, Ebersold MW, Kutay U, Helenius A. 2000. Herpes simplex virus type 1 entry into host cells: reconstitution of capsid binding and uncoating at the nuclear pore complex in vitro. Mol Cell Biol 20:4922–4931. doi: 10.1128/MCB.20.13.4922-4931.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Batterson W, Furlong D, Roizman B. 1983. Molecular genetics of herpes simplex virus. VIII. Further characterization of a temperature-sensitive mutant defective in release of viral DNA and in other stages of the viral reproductive cycle. J Virol 45:397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sandbaumhuter M, Dohner K, Schipke J, Binz A, Pohlmann A, Sodeik B, Bauerfeind R. 2013. Cytosolic herpes simplex virus capsids not only require binding inner tegument protein pUL36 but also pUL37 for active transport prior to secondary envelopment. Cell Microbiol 15:248–269. doi: 10.1111/cmi.12075. [DOI] [PubMed] [Google Scholar]
- 40.Luxton GW, Lee JI, Haverlock-Moyns S, Schober JM, Smith GA. 2006. The pseudorabies virus VP1/2 tegument protein is required for intracellular capsid transport. J Virol 80:201–209. doi: 10.1128/JVI.80.1.201-209.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Desai PJ. 2000. A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J Virol 74:11608–11618. doi: 10.1128/JVI.74.24.11608-11618.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shanda SK, Wilson DW. 2008. UL36p is required for efficient transport of membrane-associated herpes simplex virus type 1 along microtubules. J Virol 82:7388–7394. doi: 10.1128/JVI.00225-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fuchs W, Klupp BG, Granzow H, Mettenleiter TC. 2004. Essential function of the pseudorabies virus UL36 gene product is independent of its interaction with the UL37 protein. J Virol 78:11879–11889. doi: 10.1128/JVI.78.21.11879-11889.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Klupp BG, Fuchs W, Granzow H, Nixdorf R, Mettenleiter TC. 2002. Pseudorabies virus UL36 tegument protein physically interacts with the UL37 protein. J Virol 76:3065–3071. doi: 10.1128/JVI.76.6.3065-3071.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bottcher S, Granzow H, Maresch C, Mohl B, Klupp BG, Mettenleiter TC. 2007. Identification of functional domains within the essential large tegument protein pUL36 of pseudorabies virus. J Virol 81:13403–13411. doi: 10.1128/JVI.01643-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jambunathan N, Chouljenko D, Desai P, Charles AS, Subramanian R, Chouljenko VN, Kousoulas KG. 2014. Herpes simplex virus 1 protein UL37 interacts with viral glycoprotein gK and membrane protein UL20 and functions in cytoplasmic virion envelopment. J Virol 88:5927–5935. doi: 10.1128/JVI.00278-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roberts AP, Abaitua F, O'Hare P, McNab D, Rixon FJ, Pasdeloup D. 2009. Differing roles of inner tegument proteins pUL36 and pUL37 during entry of herpes simplex virus type 1. J Virol 83:105–116. doi: 10.1128/JVI.01032-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Szilagyi JF, Cunningham C. 1991. Identification and characterization of a novel non-infectious herpes simplex virus-related particle. J Gen Virol 72:661–668. doi: 10.1099/0022-1317-72-3-661. [DOI] [PubMed] [Google Scholar]
- 49.McLauchlan J, Liefkens K, Stow ND. 1994. The herpes simplex virus type 1 UL37 gene product is a component of virus particles. J Gen Virol 75:2047–2052. doi: 10.1099/0022-1317-75-8-2047. [DOI] [PubMed] [Google Scholar]
- 50.Desai P, Sexton GL, Huang E, Person S. 2008. Localization of herpes simplex virus type 1 UL37 in the Golgi complex requires UL36 but not capsid structures. J Virol 82:11354–11361. doi: 10.1128/JVI.00956-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Desai P, Sexton GL, McCaffery JM, Person S. 2001. A null mutation in the gene encoding the herpes simplex virus type 1 UL37 polypeptide abrogates virus maturation. J Virol 75:10259–10271. doi: 10.1128/JVI.75.21.10259-10271.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klupp BG, Granzow H, Mundt E, Mettenleiter TC. 2001. Pseudorabies virus UL37 gene product is involved in secondary envelopment. J Virol 75:8927–8936. doi: 10.1128/JVI.75.19.8927-8936.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pasdeloup D, McElwee M, Beilstein F, Labetoulle M, Rixon FJ. 2013. Herpesvirus tegument protein pUL37 interacts with dystonin/BPAG1 to promote capsid transport on microtubules during egress. J Virol 87:2857–2867. doi: 10.1128/JVI.02676-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Han J, Chadha P, Meckes DG Jr, Baird NL, Wills JW. 2011. Interaction and interdependent packaging of tegument protein UL11 and glycoprotein E of herpes simplex virus. J Virol 85:9437–9446. doi: 10.1128/JVI.05207-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kattenhorn LM, Korbel GA, Kessler BM, Spooner E, Ploegh HL. 2005. A deubiquitinating enzyme encoded by HSV-1 belongs to a family of cysteine proteases that is conserved across the family Herpesviridae. Mol Cell 19:547–557. doi: 10.1016/j.molcel.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 56.Schlieker C, Korbel GA, Kattenhorn LM, Ploegh HL. 2005. A deubiquitinating activity is conserved in the large tegument protein of the herpesviridae. J Virol 79:15582–15585. doi: 10.1128/JVI.79.24.15582-15585.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schlieker C, Weihofen WA, Frijns E, Kattenhorn LM, Gaudet R, Ploegh HL. 2007. Structure of a herpesvirus-encoded cysteine protease reveals a unique class of deubiquitinating enzymes. Mol Cell 25:677–687. doi: 10.1016/j.molcel.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Desai P, Person S. 1998. Incorporation of the green fluorescent protein into the herpes simplex virus type 1 capsid. J Virol 72:7563–7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Church GA, Wilson DW. 1997. Study of herpes simplex virus maturation during a synchronous wave of assembly. J Virol 71:3603–3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harley CA, Dasgupta A, Wilson DW. 2001. Characterization of herpes simplex virus-containing organelles by subcellular fractionation: role for organelle acidification in assembly of infectious particles. J Virol 75:1236–1251. doi: 10.1128/JVI.75.3.1236-1251.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee GE, Church GA, Wilson DW. 2003. A subpopulation of tegument protein vhs localizes to detergent-insoluble lipid rafts in herpes simplex virus-infected cells. J Virol 77:2038–2045. doi: 10.1128/JVI.77.3.2038-2045.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mukhopadhyay A, Lee GE, Wilson DW. 2006. The amino terminus of the herpes simplex virus 1 protein Vhs mediates membrane association and tegument incorporation. J Virol 80:10117–10127. doi: 10.1128/JVI.00744-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee GE, Murray JW, Wolkoff AW, Wilson DW. 2006. Reconstitution of herpes simplex virus microtubule-dependent trafficking in vitro. J Virol 80:4264–4275. doi: 10.1128/JVI.80.9.4264-4275.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bolte S, Cordelieres FP. 2006. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 65.Padula ME, Sydnor ML, Wilson DW. 2009. Isolation and preliminary characterization of herpes simplex virus 1 primary enveloped virions from the perinuclear space. J Virol 83:4757–4765. doi: 10.1128/JVI.01927-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stylianou J, Maringer K, Cook R, Bernard E, Elliott G. 2009. Virion incorporation of the herpes simplex virus type 1 tegument protein VP22 occurs via glycoprotein E-specific recruitment to the late secretory pathway. J Virol 83:5204–5218. doi: 10.1128/JVI.00069-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chi JH, Harley CA, Mukhopadhyay A, Wilson DW. 2005. The cytoplasmic tail of herpes simplex virus envelope glycoprotein D binds to the tegument protein VP22 and to capsids. J Gen Virol 86:253–261. doi: 10.1099/vir.0.80444-0. [DOI] [PubMed] [Google Scholar]
- 68.Gross ST, Harley CA, Wilson DW. 2003. The cytoplasmic tail of herpes simplex virus glycoprotein H binds to the tegument protein VP16 in vitro and in vivo. Virology 317:1–12. doi: 10.1016/j.virol.2003.08.023. [DOI] [PubMed] [Google Scholar]
- 69.O'Regan KJ, Bucks MA, Murphy MA, Wills JW, Courtney RJ. 2007. A conserved region of the herpes simplex virus type 1 tegument protein VP22 facilitates interaction with the cytoplasmic tail of glycoprotein E (gE). Virology 358:192–200. doi: 10.1016/j.virol.2006.08.024. [DOI] [PubMed] [Google Scholar]
- 70.Brignati MJ, Loomis JS, Wills JW, Courtney RJ. 2003. Membrane association of VP22, a herpes simplex virus type 1 tegument protein. J Virol 77:4888–4898. doi: 10.1128/JVI.77.8.4888-4898.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rixon FJ, Addison C, McLauchlan J. 1992. Assembly of enveloped tegument structures (L particles) can occur independently of virion maturation in herpes simplex virus type 1-infected cells. J Gen Virol 73:277–284. doi: 10.1099/0022-1317-73-2-277. [DOI] [PubMed] [Google Scholar]
- 72.Ryan SD, Bhanot K, Ferrier A, De Repentigny Y, Chu A, Blais A, Kothary R. 2012. Microtubule stability, Golgi organization, and transport flux require dystonin-a2-MAP1B interaction. J Cell Biol 196:727–742. doi: 10.1083/jcb.201107096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fulmer PA, Melancon JM, Baines JD, Kousoulas KG. 2007. UL20 protein functions precede and are required for the UL11 functions of herpes simplex virus type 1 cytoplasmic virion envelopment. J Virol 81:3097–3108. doi: 10.1128/JVI.02201-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Loomis JS, Courtney RJ, Wills JW. 2003. Binding partners for the UL11 tegument protein of herpes simplex virus type 1. J Virol 77:11417–11424. doi: 10.1128/JVI.77.21.11417-11424.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yeh PC, Meckes DG Jr, Wills JW. 2008. Analysis of the interaction between the UL11 and UL16 tegument proteins of herpes simplex virus. J Virol 82:10693–10700. doi: 10.1128/JVI.01230-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Harper AL, Meckes DG Jr, Marsh JA, Ward MD, Yeh PC, Baird NL, Wilson CB, Semmes OJ, Wills JW. 2010. Interaction domains of the UL16 and UL21 tegument proteins of herpes simplex virus. J Virol 84:2963–2971. doi: 10.1128/JVI.02015-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kopp M, Granzow H, Fuchs W, Klupp BG, Mundt E, Karger A, Mettenleiter TC. 2003. The pseudorabies virus UL11 protein is a virion component involved in secondary envelopment in the cytoplasm. J Virol 77:5339–5351. doi: 10.1128/JVI.77.9.5339-5351.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Starkey JL, Han J, Chadha P, Marsh JA, Wills JW. 2014. Elucidation of the block to herpes simplex virus egress in the absence of tegument protein UL16 reveals a novel interaction with VP22. J Virol 88:110–119. doi: 10.1128/JVI.02555-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Meckes DG Jr, Marsh JA, Wills JW. 2010. Complex mechanisms for the packaging of the UL16 tegument protein into herpes simplex virus. Virology 398:208–213. doi: 10.1016/j.virol.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Callan-Jones A, Sorre B, Bassereau P. 2011. Curvature-driven lipid sorting in biomembranes. Cold Spring Harb Perspect Biol 3(2):a004648. doi: 10.1101/cshperspect.a004648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McMahon HT, Gallop JL. 2005. Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature 438:590–596. doi: 10.1038/nature04396. [DOI] [PubMed] [Google Scholar]