

Abstract
Production of a vesicular stomatitis virus spike protein G (VSVG)-pseudotyped lentiviral expression vector in HEK293 cells decreased on overexpression of low-density lipoprotein receptor (LDLR) but not that of ICAM1 or TfR1. Reverse transcription-quantitative PCR (RT-qPCR) revealed a reduction in vector RNA as a function of LDLR expression. Decreased syncytium formation suggested diminished surface expression of VSVG. Intracellular VSVG granules colocalized with LDLR, ER-Golgi intermediate compartment protein 53 (ERGIC53), LAMP2, and vimentin but not with GM130 or calnexin, suggesting that VSVG interacts with LDLR within the ERGIC, resulting in rerouting into the aggresome/autophagosome pathway.
TEXT
Gene-therapeutic approaches seek to alleviate inherited familial hypercholesterolemia by complementing the defective low-density lipoprotein receptor (LDLR) gene with its functional homologue. One much-sought avenue toward this aim involves transduction with a replication-defective pseudotyped lentiviral expression vector carrying the gene encoding LDLR (reviewed in reference 1). Along these lines, it had been attempted to produce such lentiviral vectors via third-generation equine infectious anemia virus (EIAV)-based packaging in human embryonic kidney (HEK293T) cells. However, initial attempts at producing vesicular stomatitis virus spike protein G (VSVG)-pseudotyped lentivirus by using such a three-plasmid transient-cotransfection protocol demonstrated a dramatic decrease in vector yield on increased cytomegalovirus (CMV)-promoter-driven LDLR expression (F. A. Al-Allaf, unpublished observation).
To confirm the above observation and determine the underlying reason, virus was produced in HEK293 cells (2.5 × 106/10-cm2 plate) via transient transfection with the self-inactivating vector construct pHR-CMV-EGFP (3.33 μg; Addgene 14858) (2), as well as the packaging construct psPAX2 (5 μg; Addgene 12260) and the envelope construct pMD2.G, encoding the VSVG surface protein (1.66 μg; Addgene 12259) (both a kind gift from Didier Trono). To prevent eventual SV40ori-based replication of plasmids mediated by large-T antigen, we used HEK293 cells in all experiments; despite the absence of the large-T antigen, we observed high expression of the transgenes. Cells were then cotransfected with different amounts of plasmids encoding either LDLR (Origene RC200006) or, as controls, human intercellular adhesion molecule 1 (ICAM1; Origene RC200714) and human transferrin receptor (TfR1; Origene RC200980), all carrying the sequence coding for the C-terminal peptide DYKDDDDK (DDK tag). The total DNA transfected was kept constant by adding herring sperm DNA to make up 11.66 μg. The growth medium (Dulbecco's modified Eagle's medium [DMEM], 10% fetal calf serum [FCS]) was replaced and supplemented with 5 mM Na-butyrate at 24 h posttransfection (hpt). The cell supernatant was harvested at 48 hpt and filtered through a membrane (0.22-μm pore size), and the lentivirus vector was pelleted by ultracentrifugation for 1.5 h at 4°C and 40,000 × g in a Ti70.1 Beckman rotor. Titer was assessed via infection of HeLa cells grown in 12-well plates at 37°C in 5% CO2; the medium was replaced with 500 μl infection medium (DMEM, 2% FCS), and then aliquots of the resuspended pellets described above were added. After incubation for 30 min, 1 ml infection medium supplemented with Polybrene (final concentration, 8 μg/ml) was added and incubation continued for 72 h. The percentage of fluorescent cells—as a consequence of transduction with the enhanced green fluorescent protein (EGFP)-encoding viral vector—was determined by flow cytometry in a BD FACSCalibur using CellQuest software, and the number of transducing units (TU) per milliliter was calculated. The number of TU per milliliter in the concentrated viral vector suspensions from the positive control (i.e., HEK293 cells transfected with pHR-CMV-EGFP, psPAX2, and pMD2.G) was only slightly higher than that obtained upon cotransfection with different amounts of plasmids encoding ICAM1 and TfR1, respectively. However, as previously observed, cotransfection with increasing amounts of the LDLR-encoding plasmid resulted in a gradual decrease of TU per milliliter (data not shown).
The flow cytometry experiment described above measures the number of cells transduced as a consequence of infection with vector particles encoding EGFP (biological titer). It does not take into account physical vector particles that might be defective in any process related to transgene expression, including binding, entry, or uncoating, and other factors like genome integration. Previously, Finkelshtein and colleagues demonstrated that LDLR acts as host cell receptor for VSV and VSVG-pseudotyped lentiviruses (3). Hence, we asked whether LDLR might be incorporated into the lentiviral membrane like host factors detected in other enveloped viruses (4), which would result in blockage of VSVG at the viral surface and prevent binding to and infection of the host cell. Consequently, we assessed the copy number of HIV-1 packaging sequences (psi) present in the concentrated virus in a reverse transcription-quantitative PCR (RT-qPCR) assay by using psi-specific primers (5′-TGAACCGCATCGAGCTGAAG-3′ and 5′-TAGACGTTGTGGCTGTTGTAGTTG-3′). The plasmid pHR-CMV-EGFP was used as a standard. Contamination with pHR-CMV-EGFP eventually carried over from transfection was removed via DNase I digestion prior to RT-qPCR. From the decreased copy number of psi RNA present in virus released from transfected cells as a function of LDLR expression (Fig. 1A), we deduce that the total number of vector particles, and not the specific transduction efficiency, is affected by LDLR expression. The Western blot depicted in Fig. 1B and the plot shown in Fig. 1C support the inverse relationship between LDLR expression and the lentiviral particles obtained from the transfected HEK293 cells.
FIG 1.

Lentiviral copy number increases with decreasing amount of LDLR-encoding plasmid cotransfected with the virus-producing plasmids used in the positive control. (A) Viral RNA in the supernatant of HEK293 cells was determined by RT-qPCR. The negative controls (NegC) were psPAX2 and pMD2.G, and the positive controls (PosC) were pHR-CMV-EGFP, psPAX2, and pMD2.G. Data are the arithmetic means of 3 biological replicates ± standard errors of the means (SEM). The presence of LDLR decreases the copy number significantly [one-way analysis of variance (ANOVA), F(4, 2) = 356.18], and the copy number is inversely related to the concentration of LDLR [one-way ANOVA, F(3, 2) = 373.33]. Wedges indicate the amount of the LDLR-encoding plasmid relative to pHR-CMV-EGFP used in the transfection (LDLR, 1:2 to 1:16). ***, P < 0.0005. (B) Western blot analysis of tagged LDLR present in lysates of the transfected HEK293 cells from panel A. Scanned film was automatically white corrected using GIMP software. (C) Correlation between relative LDLR expression (data derived from panel B via the GelAnalyzer plugin of ImageJ software) and copy number (data from panel A). Note the very high correlation of R2 = 0.9881.
To assess whether the LDLR-dependent decrease of the vector count in the cell supernatant could be due to a block in vector release via interaction of host LDLR with virus-associated VSVG, or vice versa, at the plasma membrane, we determined the fractions of viral RNA in the cell supernatant (S), at the cell surface (W), and inside the transfected cells (C) again by RT-qPCR. After harvesting of fraction S, the cells were detached with 10 ml phosphate-buffered saline (PBS)–10 mM EGTA, which destroys the structural integrity of LDLR (5) and thus releases attached ligands. Cells were pelleted, collected in DMEM–2% FCS, and counted prior to RNA isolation and RT-qPCR as described above. The data depicted in Fig. 2A suggest that the recombinant lentiviral vector neither substantially accumulates at the cell surface nor accumulates inside the cells in response to LDLR cotransfection. As for the TU (as described above) and the vector RNA in the cell supernatant (Fig. 1A), the sum of the RNA copy numbers (from fractions S, W, and C) significantly dropped on increased LDLR expression (Fig. 2A). The decrease in total viral RNA copy numbers could result from reduced long terminal repeat (LTR)-driven transcription or reuptake of vector particles via ligand-mediated internalization of LDLR. In case of the latter, they might initiate an infection cycle or become routed to lysosomes for degradation.
FIG 2.
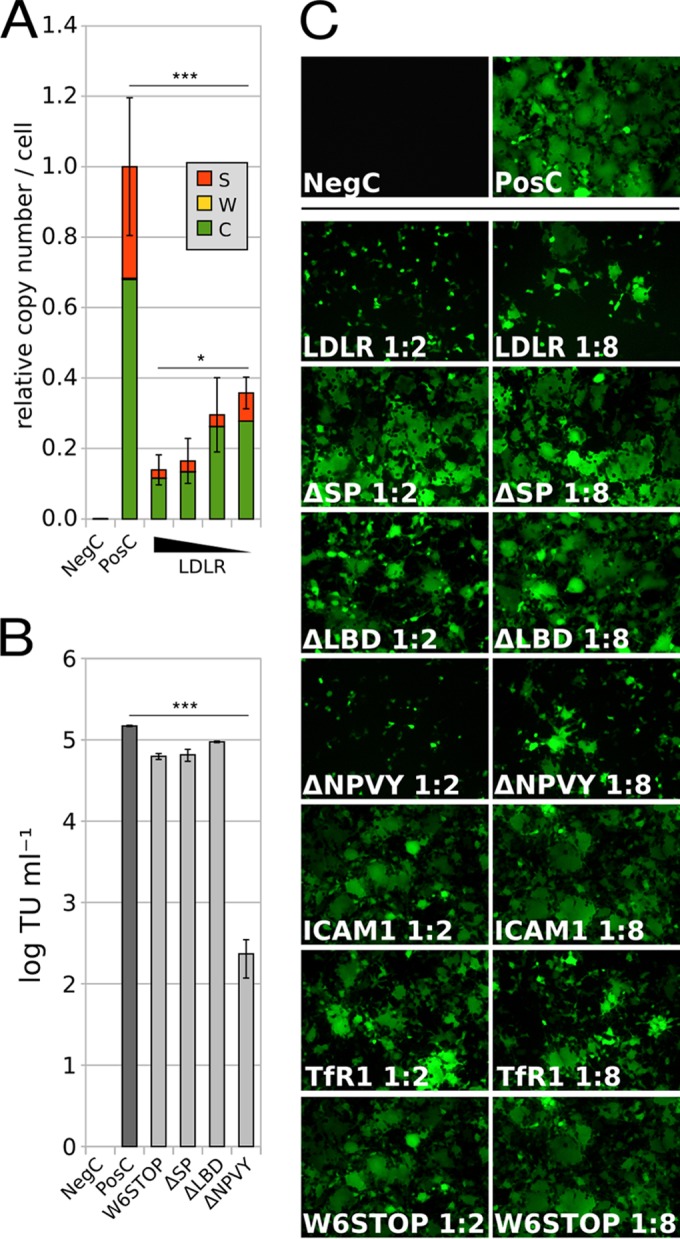
Upon LDLR expression, the lentiviral vector is neither trapped inside the cell nor at the cell surface. After transfection of HEK293 cells with the plasmids as in Fig. 1, the supernatant (fraction S) was saved, and cells were washed with EGTA buffer (fraction W) and pelleted (fraction C). Plasmid ratios are indicated (see the legend to Fig. 1). (A) The copy number of viral RNA was determined by RT-qPCR in each fraction (S, W, and C) relative to the positive control (roughly 2,000 copies per cell). The presence of LDLR decreases the total amount of viral RNA in fractions S, C, and W significantly [one-way ANOVA, F(4, 2) = 42.19; P < 0.0005], and the copy number is dependent on the concentration of LDLR [one-way ANOVA, F(3, 2) = 5.10; P < 0.05]. Data are the arithmetic means from 3 biological replicates ± SEM. (B) TU obtained upon cotransfection with the indicated LDLR mutants. Note the strong decrease in TU in the presence of ΔNPVY compared to the other mutants [one-way ANOVA, F(4, 2) = 37.22; P < 0.0005]. Data are the arithmetic means from triplicate measurements ± 2 SD. (C) Live-cell fluorescence microscopy of HEK293 cells transfected with the “virus-producing plasmids” together with the mutated plasmids, as indicated, during virus production. Note the virtual absence of syncytia even at small amounts of transfected wild-type LDLR and ΔNPVY plasmids. *, P < 0.05; **, P < 0.005; ***, P < 0.0005.
To test the above hypotheses, LDLR deletion constructs either lacking the signal peptide (ΔSP), the ligand binding domain (ΔLBD), or the internalization signal (ΔNPVY) were prepared to interfere with subcellular localization, binding VSVG, and endocytosis, respectively, in addition to a 6-residue LDLR nonsense mutant (W6STOP). These were used in cotransfection experiments as described above. TU were indiscernible from those of the positive control in W6STOP-, ΔSP-, and ΔLBD-cotransfected cells, but a strong decrease of TU was found upon ΔNPVY cotransfection (Fig. 2B). Live-cell imaging revealed that syncytium formation, which occurs upon cell surface expression of VSVG on acidification (6), but under certain conditions also at neutral pH (7), was indiscernible from the positive control (no cotransfection) in cells cotransfected with ΔLBD and ΔSP and from cells cotransfected with W6STOP, ICAM1, or TfR1 (Fig. 2C). However, syncytia were virtually absent in cells cotransfected with ΔNPVY as well as wild-type LDLR-encoding plasmids. This confirms that the ligand-binding domain of LDLR interferes with the cell surface targeting of VSVG. Thus, internalization-related processes most likely do not account for reduced vector production, which is supported by the absence of degradation products of VSVG on a Western blot developed with two different rabbit antisera directed against a C-terminal peptide (Thermo Scientific PA1-30138, Abcam 83196) (data not shown). In parallel, indirect immunofluorescence microscopy of HEK293 cells expressing VSVG and/or LDLR was carried out by using primary antibodies directed against VSVG (Thermo Scientific PA1-30138), early endosome antigen 1 (EEA1; BD 610456), Golgi matrix protein 130 (GM130; BD 610822), ER-Golgi intermediate compartment protein 53 (ERGIC53; Alexis Biochemicals no. ALX-804-602), lysosome-associated membrane protein 2 (LAMP2; BD 555803), vimentin (Dako M0725), calnexin (BD 610524), and the DDK tag of LDLR (Origene TA50011). Cells were fixed with paraformaldehyde (PFA [2%]), permeabilized with saponin (0.2%), and after incubation with primary and Alexa 488- or Alexa 555-conjugated secondary antibodies, counterstained with Hoechst 33342. VSVG colocalized with LDLR in dense intracellular granules, which were distinct from EEA1-positive endosomes, making internalization-related processes unlikely (data not shown). VSVG granules were spatially unrelated to GM130 (Fig. 3A), suggesting that LDLR expression impairs anterograde transport of VSVG to the Golgi apparatus. Expression of LDLR alone does not lead to granule formation (not shown). Colocalization of VSVG with ERGIC53 (Fig. 3B), LAMP2 (Fig. 3C), and vimentin (Fig. 3D) indicates an alternative transport route to autophagosomal/lysosomal (8) and/or aggresomal compartments (9). In accordance with reference 10, disruption of microtubules via nocodazole (50 ng/ml) for 24 h did not influence aggresome formation (not shown). We did not find colocalization of VSVG with calnexin (not shown); therefore, we can exclude that VSVG is sequestered in the ER. The previous finding that receptor-associated protein (RAP) dissociates from LDLR family members in the ERGIC due to acidic pH (11) provides an explanation for why a putative VSVG-LDLR interaction takes place in the ERGIC, where LDLR is rendered binding competent for VSVG, leading to their aggregation; thus, these aggregates do not move further to the Golgi apparatus.
FIG 3.

Indirect immunofluorescence microscopy demonstrates that LDLR expression induces rerouting of VSVG rather than progression to the Golgi apparatus. HEK293 cells were transfected with psPAX2 and pMD2.G with or without RC200006 and probed for VSVG and GM130 (A). Note colocalization in the absence of LDLR (yellow). (B) Cells probed for VSVG and ERGIC53. Note colocalization in the presence of LDLR. (C) Cells probed for VSVG and LAMP2. Note the VSVG-granules enclosed by LAMP2-positive membranes in the presence of LDLR. (D) Cells probed for VSVG and vimentin. Note the VSVG-granules surrounded by cages of collapsed vimentin in the presence of LDLR. In any case, prominent surface expression of VSVG was only seen in the absence of LDLR expression. The micrographs in panels A, B, and C were taken using a Zeiss Axioplan 2 microscope at 100× magnification and deconvolved using the deconvolution filter of the G'MIC plugin package for GIMP software. The micrographs in panel D were acquired by using a confocal microscope at 100× magnification. Scale bars, 10 μm.
According to our model in Fig. 4, complex formation between LDLR and VSVG in the ERGIC results in aggregation and presumably in rerouting into an aggresomal/autophagic pathway. To test this model biochemically, transfected HEK293 cells coexpressing VSVG and LDLR were sorted on a BD FACSAria cell sorter. Double-positive cells were collected and compared with cells transfected with the LDLR expression plasmid only, on a Western blot (Fig. 4, inset). The absence of the O-glycosylated 160-kDa LDLR species (12) in VSVG/LDLR-coexpressing cells indicates that VSVG-LDLR complexes are retained in the ERGIC and do not reach the Golgi apparatus. The nonproductive transport of VSVG at high LDLR expression levels provides no direct explanation for the reduced copy number of lentiviral RNA. Since mRNA transcription levels of VSVG and GAPDH were unaffected, as determined via RT-qPCR (not shown), it seems that only LTR-driven transcription might be affected by a so far unknown mechanism.
FIG 4.

Model of rerouting of LDLR-VSVG complexes. Following translation of VSVG (red) and LDLR (green) at the rough ER and their transport to the ERGIC (step 1), they acquire their respective native protein conformation, allowing for complex formation as soon as the pH drops. ERGIC-initiated signals impair anterograde transport of the complexes to the Golgi apparatus (step 2) and subsequent progression of bulk VSVG through the secretory pathway. Instead, VSVG-LDLR complexes are rerouted to aggresomal/autophagosomal compartments (step 3) at the ERGIC. The Western blot (inset) shows the absence of the DDK-tagged 160-kDa LDLR species in cell lysates from LDLR/VSVG-coexpressing cells, indicating that LDLR-VSVG complexes become sequestered upstream of the Golgi apparatus. The 250-kDa band present in cells expressing LDLR only presumably originates from dimers generated by the PFA fixation.
Our findings call for care when attempting production of pseudotyped lentiviral vectors encoding foreign membrane proteins since their premature interaction with viral envelope proteins might prevent successful packaging. Additionally, the results reveal consequences of intracellular VSVG-LDLR interactions, including exit from the secretory pathway at the ERGIC, which might apply to other envelope protein host cell receptor pairs as well.
ACKNOWLEDGMENTS
This work was funded by the Saudi Arabian government, Umm Al-Qura University, grant no. UQU 150411. Faisal A. Al-Allaf received funding from the King Abdulaziz City for Science and Technology (KACST), grant no. (KACST AR-30-125).
We thank Sandra Kleinberger for great help during the initial phase of the project, Ivan Yudushkin and Heinrich Kowalski for advice, Margret Kilian and Claudine Kraft for critically reading the manuscript, and Thomas Sauer for FACS.
REFERENCES
- 1.Al-Allaf FA, Coutelle C, Waddington SN, David AL, Harbottle R, Themis M. 2010. LDLR-gene therapy for familial hypercholesterolaemia: problems, progress, and perspectives. Int Arch Med 3:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miyoshi H, Takahashi M, Gage FH, Verma IM. 1997. Stable and efficient gene transfer into the retina using an HIV-based lentiviral vector. Proc Natl Acad Sci U S A 94:10319–10323. doi: 10.1073/pnas.94.19.10319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finkelshtein D, Werman A, Novick D, Barak S, Rubinstein M. 2013. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc Natl Acad Sci U S A 110:7306–7311. doi: 10.1073/pnas.1214441110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paquette J-M, Fortin J-F, Blanchard L, Tremblay MJ. 1998. Level of ICAM-1 surface expression on virus producer cells influences both the amount of virion-bound host ICAM-1 and human immunodeficiency virus type 1 infectivity. J Virol 72:9329–9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao Z, Michaely P. 2009. The role of calcium in lipoprotein release by the LDL receptor. Biochemistry 48:7313–7324. doi: 10.1021/bi900214u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun X, Belouzard S, Whittaker GR. 2008. Molecular architecture of the bipartite fusion loops of vesicular stomatitis virus glycoprotein G, a class III viral fusion protein. J Biol Chem 283:6418–6427. doi: 10.1074/jbc.M708955200. [DOI] [PubMed] [Google Scholar]
- 7.Roberts PC, Kipperman T, Compans RW. 1999. Vesicular stomatitis virus g protein acquires pH-independent fusion activity during transport in a polarized endometrial cell line. J Virol 73:10447–10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eskelinen E-L, Illert AL, Tanaka Y, Schwarzmann G, Blanz J, von Figura K, Saftig P. 2002. Role of LAMP-2 in lysosome biogenesis and autophagy. Mol Biol Cell 13:3355–3368. doi: 10.1091/mbc.E02-02-0114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garcia-Mata R, Gao Y-S, Sztul E. 2002. Hassles with taking out the garbage: aggravating aggresomes. Traffic 3:388–396. doi: 10.1034/j.1600-0854.2002.30602.x. [DOI] [PubMed] [Google Scholar]
- 10.Harada M, Sakisaka S, Terada K, Kimura R, Kawaguchi T, Koga H, Kim M, Taniguchi E, Hanada S, Suganuma T, Furuta K, Sugiyama T, Sata M. 2001. A mutation of the Wilson disease protein, ATP7B, is degraded in the proteasomes and forms protein aggregates. Gastroenterology 120:967–974. doi: 10.1053/gast.2001.22543. [DOI] [PubMed] [Google Scholar]
- 11.Bu G, Geuze HJ, Strous GJ, Schwartz AL. 1995. 39 kDa receptor-associated protein is an ER resident protein and molecular chaperone for LDL receptor-related protein. EMBO J 14:2269–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshimura A, Yoshida T, Seguchi T, Waki M, Ono M, Kuwano M. 1987. Low binding capacity and altered O-linked glycosylation of low density lipoprotein receptor in a monensin-resistant mutant of Chinese hamster ovary cells. J Biol Chem 262:13299–13308. [PubMed] [Google Scholar]