

ABSTRACT
Innate immunity is the first line of host defense against infections. Many oncogenic viruses can deregulate several immune-related pathways to guarantee the persistence of the infection. Here, we show that the cutaneous human papillomavirus 38 (HPV38) E6 and E7 oncoproteins suppress the expression of the double-stranded DNA sensor Toll-like receptor 9 (TLR9) in human foreskin keratinocytes (HFK), a key mediator of the antiviral innate immune host response. In particular, HPV38 E7 induces TLR9 mRNA downregulation by promoting accumulation of ΔNp73α, an antagonist of p53 and p73. Inhibition of ΔNp73α expression by antisense oligonucleotide in HPV38 E6/E7 HFK strongly rescues mRNA levels of TLR9, highlighting a key role of ΔNp73α in this event. Chromatin immunoprecipitation experiments showed that ΔNp73α is part of a negative transcriptional regulatory complex with IκB kinase beta (IKKβ) that binds to a NF-κB responsive element within the TLR9 promoter. In addition, the Polycomb protein enhancer of zeste homolog 2 (EZH2), responsible for gene expression silencing, is also recruited into the complex, leading to histone 3 trimethylation at lysine 27 (H3K27me3) in the same region of the TLR9 promoter. Ectopic expression of TLR9 in HPV38 E6/E7 cells resulted in an accumulation of the cell cycle inhibitors p21WAF1 and p27Kip1, decreased CDK2-associated kinase activity, and inhibition of cellular proliferation. In summary, our data show that HPV38, similarly to other viruses with well-known oncogenic activity, can downregulate TLR9 expression. In addition, they highlight a new role for TLR9 in cell cycle regulation.
IMPORTANCE The mucosal high-risk HPV types have been clearly associated with human carcinogenesis. Emerging lines of evidence suggest the involvement of certain cutaneous HPV types in development of skin squamous cell carcinoma, although this association is still under debate. Oncogenic viruses have evolved different strategies to hijack the host immune system in order to guarantee the persistence of the infection. Their capability to evade the immune system is as important as their ability to promote cellular transformation. Therefore, understanding the viral mechanisms involved in viral persistence is a valid tool to evaluate their potential role in human carcinogenesis. Here, we show that E6 and E7 oncoproteins from the cutaneous HPV38 downregulate the expression of the double-stranded DNA sensor TLR9 of innate immunity. We also present evidence that the HPV38-mediated downregulation of TLR9 expression, in addition to its potential impact on the innate immune response, is linked to cell cycle deregulation.
INTRODUCTION
In addition to the well-characterized mucosal high-risk human papillomaviruses (HPV), a subgroup of cutaneous HPV types belonging to the genus beta of the HPV phylogenetic tree appears to be associated with human carcinogenesis (1–3). These HPV types are suspected to be involved together with UV radiation in the development of nonmelanoma skin cancer (4, 5). Beta HPV types were originally isolated in patients suffering from a rare autosomal recessive cancer-prone genetic disorder, epidermodysplasia verruciformis (EV), and are consistently detected in nonmelanoma skin cancer from EV patients and immunocompromised and healthy individuals (1). More than 40 different beta HPV types have been identified so far, but only a few have been studied for the characterization of their biological properties (6). In particular, several studies have demonstrated that E6 and E7 oncoproteins from beta HPV 38 (HPV38) display transforming activities in in vitro and in vivo experimental models (7–12). The transforming activity of HPV38 is explained partly by the ability of E7 to induce the accumulation of ΔNp73α, which antagonizes p53 functions in activating the transcription of genes encoding cell cycle inhibitors or proapoptotic regulators (9, 10). HPV38 E7 induces the accumulation of IκB kinase beta (IKKβ) in the nucleus, where it, in turn, binds and phosphorylates the ΔNp73α protein at serine 422 (S422), resulting in a large increase in the half-life of ΔNp73α (10). The IKKβ/ΔNp73α complex binds p53 responsive elements together with two epigenetic enzymes, DNA methyltransferase 1 (DNMT1) and enhancer of zeste homolog 2 (EZH2), and inhibits the expression of some p53-regulated genes, such as the PIG3 gene (13).
Studies with transgenic mice expressing HPV38 E6 and E7 in the basal layer of the epidermis further highlighted its transforming properties. In fact, these transgenic animals, upon chronic UV irradiation, developed actinic keratosis-like lesions, which are considered precursors of squamous cell carcinomas (SCC) in humans, and subsequently SCC. In contrast, wild-type animals subjected to identical treatments did not develop any type of skin lesions (12).
However, despite the well-characterized oncogenic properties of HPV38 in in vivo and in vitro experimental models, its role in human carcinogenesis remains to be proven.
In addition to their ability to promote cellular transformation, human cancer-associated viruses deregulate pathways linked to the host immune response, thus favoring the persistence of the infection, which is an essential condition for cancer development (14–16). Mucosal high-risk HPV16, Epstein-Barr virus (EBV), Merkel cell polyomavirus, and hepatitis B virus alter the expression of Toll-like receptors (TLRs), which are fundamental players in the innate immune response, acting as pattern recognition receptors (PRRs) (17, 18). In particular, all four of these oncogenic viruses, using distinct mechanisms, downregulate the transcription of TLR9, which resides in the endosomal compartments of the cell and senses viral double-stranded DNA (16, 19–24). To gain further insights into the possible role of HPV38 in human carcinogenesis, in this study, we investigated whether HPV38 E6 and E7 have the ability to deregulate TLR9 expression. Our data show that HPV38, similarly to the well-established oncogenic viruses, efficiently downregulated the expression of TLR9.
MATERIALS AND METHODS
Plasmid constructs.
The genes of interest were expressed in the following expression vectors: pcDNA3 (Invitrogen), pLXSN (Clontech, Le Pont-de-Claix, France), and pBabe (25). The pLXSN-HPV38 E6/E7, pLXSN-HPV16 E6/E7, pcDNA3 HA-ΔNp73α, and pBabe-puro-ΔN-IκBα (lacking the first 36 N-terminal amino acids) constructs and the TLR9 promoter luciferase construct, full-length (−3227/−1) or with deletions (−1017/−1 and −290/−1), have been previously described (7, 10, 16, 24). The pBabe-puro-TLR9 construct was generated during this study using standard molecular biology techniques.
Cell culture, retroviral infection, and treatment.
Cell culture, antibiotic selection, and generation of high-titer retroviral supernatants were carried out as previously described (7). For treatment, cells were incubated in medium containing SB203580 (Sigma) with a final concentration of 5 μM for 3 h. For fluorescence-activated cell sorter (FACS) staining, cells were collected, washed twice in PBS, and then stained with propidium iodide (PI) at a final concentration of 5 μg/ml. Subsequently, cells were analyzed by FACS CANTO (Becton Dickinson).
Gene silencing.
Downregulation of ΔNp73α was achieved by transfecting the antisense (AS) oligonucleotide as previously described (9). Silencing of HPV38 E6E7 gene expression was obtained using the pRetroSuper construct (pRS), expressing small hairpin RNAs (shRNAs) for the polycistronic HPV38 E6 and E7 mRNA (pRS 38E6/E7) as previously described (9, 13). Gene silencing of p65 and IKKβ was achieved using synthetic small interfering RNA (siRNA) (Table 1). siRNA or scrambled RNA at a concentration of 50 nM was transfected using Lipofectamine 2000 according to the standard protocol (Invitrogen). EZH2 and p38 silencing was performed using specific stealth siRNA as previously described (13).
TABLE 1.
Sequences of different siRNAs used for gene silencing
| Target | siRNA sequence or description (source) |
|---|---|
| Scrambled (negative control) | 5′-GGUGGAAGAGGUGGUGAGC-3′ |
| IKKβ | 5′-CGUACGCGGAAUACUUCGA-3′ |
| EZH2 | 5′-AGUGGUGCUGAAGCCUCAAUGUUUA-3′ |
| p65 | siGenome SMART pool M-003533-02-0005, human RELA, NM_021975 (Thermo Scientific) |
| p38 (MAPK14) | Silencer Select predesigned siRNA, catalog no. 4392420, IDa s3587 (Life Technologies) |
| S ΔNp73α | 5′-accgACGTACAGcatg-3′b |
| AS ΔNp73α | 5′-ccatGCTGTACGtcggT-3′b |
ID, identification number.
For the ΔNp73α sense (S) and antisense (AS) oligonucleotides, the capital letters indicate phosphorothioate nucleotides.
ELISA.
To measure secreted cytokines, HPV16 E6/E7 or HPV38 E6/E7 human foreskin keratinocytes (HFK) were seeded at 4 × 104 cells/96 wells in 200 μl of growth medium. The next day, cells were washed with PBS, and Gpc or CpG 2006 was added. Twenty-four hours later, supernatants were collected for analysis of interleukin 8 (IL-8) and MIP3α secretion using Quantikine enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems) as previously described (22).
Luciferase assay.
Transient transfections were conducted using X-tremeGene 9 (Roche) according to the manufacturer's protocol. RPMI 8226 cells were cotransfected with firefly luciferase TLR9 promoter vector (0.5 μg) and the vector of interest. pRL-TK Renilla reporter vector (15 ng) was used as an internal control. After 48 h, cells were lysed and luciferase activity was measured using a dual-luciferase reporter assay system (Promega). The expression of firefly luciferase relative to that of Renilla luciferase was expressed in relative luminescence units (RLU).
Reverse transcription-PCR (RT-PCR) and qPCR.
Total RNA was extracted using the Absolutely RNA Miniprep kit (Stratagene). The obtained RNA was reverse transcribed to cDNA with the RevertAid H Minus Moloney murine leukemia virus (M-MuLV) reverse transcriptase kit (Fermentas) according to the manufacturer's instructions. Quantitative PCR (qPCR) was performed using the MesaGreen qPCR MasterMix Plus for SYBR Assay (Eurogentec) with the primers listed in Table 2.
TABLE 2.
Sequences of primers used for RT-PCR analyses, ChIP, and oligonucleotide pulldown
| Promoter or gene | Primer sequencea |
|---|---|
| TLR9 promoter NF-κB site A | F: 5′-TGGGTCTGTACCTGTGTGTGCA-3′ |
| R: 5′-TTCATTCCCTCCATCCACCTC-3′ | |
| TLR9 promoter NF-κB site B | F: 5′-AGGAGCTCAGGAGTGCCAG-3′ |
| R: 5′-TGGGATGTGCTGTTCCCTC-3′ | |
| TLR9 promoter NF-κB site C | F: 5′-GAGAGCACTCAGGGGAACAG-3′ |
| R: 5′-GGTCACATTCAGCCCCTAGA-3′ | |
| TLR9 promoter NF-κB site D | F: 5′-AGGCCCTGCAGAACTTGGAG-3′ |
| R: 5′-TCAGGCAGAGAGCAGGGAGA-3′ | |
| GAPDH | F: 5′-AAGGTGGTGAAGCAGGCGT-3′ |
| R: 5′-GAGGAGTGGGTGTCGCTGTT-3′ | |
| TLR9 | F: 5′-CGTCTTGAAGGCCTGGTGTTGA-3′ |
| R: 5′-CTGGAAGGCCTTGGTTTTAGTGA-3′ | |
| IKKβ | F: 5′-GCTGCAACTGATGCTGATGT-3′ |
| R: 5′-TGTCACAGGGTAGGTGTGGA-3′ | |
| p65 | F: 5′-GTCACCGGATTGAGGAGAAA-3′ |
| R: 5′-GCTCAGGGATGACGTAAAGG-3′ | |
| EZH2 | F: 5′-ACGTCAGATGGTGCCAGCAATA-3′ |
| R: 5′-CCCTGACCTCTGTCTTACTTGTGGA-3′ | |
| Probes for oligonucleotide pulldown assay | F: 5′-Btn-GAGAGCACTCAGGGGAACAG-3′ |
| R: 5′-GGTCACATTCAGCCCCTAGA-3′ |
F, forward; R, reverse.
IB.
Total protein extraction, sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE), and immunoblotting (IB) were conducted as described previously (9). Antibodies to the following antibodies were used: β-actin (C4; MP Biomedicals), TLR9 (2254; Cell Signaling), p73 (OP108; Calbiochem), hemagglutinin (HA; 3F10; Roche), IKKβ (2684; Cell Signaling), NF-κB p65 (3034; Cell Signaling), p21WAF/Cip1 (2946; Cell Signaling), p27Kip1 (2552; Cell Signaling), p38 mitogen-activated protein kinase (MAPK; 9212; Cell Signaling), and phospho-p38 MAPK Thr180/Tyr182 (9211; Cell Signaling). Images were taken using the ChemiDoc XRS imaging system (Bio-Rad).
ChIP.
Chromatin immunoprecipitation (ChIP) was performed with Shearing ChIP and OneDay ChIP kits (Diagenode) according to the manufacturer's protocols. Briefly, cells were sonicated to obtain DNA fragments of 200 to 500 bp. Sheared chromatin was immunoprecipitated with isotype control IgG or antibodies to the following: EZH2 (AC22; Cell Signaling), p73 (OP108; Calbiochem), IKKβ (2684; Cell Signaling), DNMT1 (60B1220; Abnova), H3Lys27me3 (4039; Epigentek), and NF-κB p65 (3034; Cell Signaling). The eluted DNA was used as a template for qPCR. Primers for qPCR are listed in Table 2.
Oligonucleotide pulldown.
Cells were lysed and sonicated in HKMG buffer (10 mM HEPES [pH 7.9], 100 mM KCl, 5 mM MgCl2, 10% glycerol, 1 mM dithiothreitol [DTT], and 0.5% Nonidet P-40) containing protease and phosphatase inhibitors. After centrifugation at 12,000 × g for 10 min, protein extracts were precleared with streptavidin-agarose beads. The TLR9 promoter was used as a template to amplify the NF-κB RE region containing wild-type or mutated NF-κB RE. PCR amplification was performed using a biotinylated forward primer and a nonbiotinylated reverse primer (listed in Table 2). Amplicons were extracted from an agarose gel by using the MinElute gel extraction kit (Qiagen) and quantified. Then, 2 mg of prepared protein extract was incubated with 1 μg of biotin-TLR9 promoter probes and 10 μg of poly(dI-dC)-poly(dI-dC) for 16 h at 4°C. DNA-bound proteins were collected with streptavidin-agarose beads for 1 h and washed five times with HKMG buffer. DNA-bound proteins were then analyzed by IB.
Colony formation assay.
For colony formation assays, the different types of cells were seeded at different dilutions (104, 103, and 102) and cultured for 7 days. Cells were then stained with crystal violet, and the average numbers of cells per colony were determined.
Time course.
As cells were split for selection, 105 cells were seeded in 6-well plates and allowed to grow for 4 days. The growth was monitored daily by cell trypsinization and counting with trypan blue. Double determinations were performed in each independent experiment.
Kinase assay.
CDK2 kinase assays were performed using 1 mg of keratinocyte lysate. CDK2 complexes were immunoprecipitated using an anti-CDK2 antibody (sc-748; Santa Cruz). Immunopellets were washed three times in lysis buffer and twice in kinase buffer (50 mM Tris [pH 7.5], 10 mM MgCl2, 1 mM DTT, and 100 μM ATP). Samples were then resuspended in 10 μl of kinase buffer containing 10 μCi of [γ-32P]ATP (PerkinElmer) and incubated for 15 min at room temperature in the presence of 2 μg of histone H1 (Millipore). Samples were subsequently analyzed by SDS-PAGE (12%), transferred to nitrocellulose, and visualized on X-ray films.
Statistical analysis.
Statistical significance was determined by the Student t test. Statistically significant P values (P < 0.05 [*] and P < 0.01 [**]) for each experiment are indicated in the corresponding figure legends. Error bars in the graphs represent the standard deviations.
RESULTS
Beta HPV38 E6 and E7 downregulate TLR9 expression.
We aimed to test our hypothesis that, as previously shown for many other viral oncoproteins, beta HPV38 E6 and E7 have the ability to inhibit TLR9 promoter activity in RPMI 8226 human myeloma cells. These cells express high levels of TLR9 and have previously been used to demonstrate the ability of E6 and E7 from mucosal HPV16 to inhibit TLR9 promoter activity (22). We found that HPV38 E6 and E7 inhibited TLR9 promoter activity in transiently transfected RPMI 8226 cells with an efficiency similar to that of human papillomavirus 16 (HPV16) E6 and E7 (Fig. 1A). Next, we analyzed TLR9 mRNA and protein levels in human keratinocytes, the natural host of the virus. Primary human foreskin keratinocytes (HFK) were transduced with a recombinant retrovirus expressing HPV38 E6 and E7 oncoproteins or empty retrovirus (pLXSN), and TLR9 mRNA or protein levels were determined by quantitative RT-PCR or immunoblotting, respectively. TLR9 expression was decreased in HPV38 E6/E7 HFK compared with pLXSN HFK (Fig. 1B). When expressed alone, both oncoproteins were able to inhibit TLR9 expression, although E7 appeared to be more efficient than E6 (Fig. 1C). Finally, we checked whether the viral proteins could inhibit TLR9 functionality. pLXSN and HPV38 E6/E7 HFK were exposed to a TLR9 synthetic ligand (CpG oligodeoxynucleotide 2006) for 24 h, and the concentrations of secreted cytokines, IL-8 and MIP3α, were determined by ELISA. The secretion of both cytokines was significantly inhibited in HPV38 E6/E7 HFK compared with control cells (Fig. 1D). These data show that, similar to the case with several oncogenic viruses, HPV38 E6 and E7 block TLR9 function by inhibiting its transcription.
FIG 1.

HPV38 E6 and E7 downregulate TLR9 expression. (A) RPMI 8226 cells were cotransfected with the TLR9 promoter construct (−3227/−1) fused with the luciferase reporter gene and with pLXSN empty vector (pLXSN), pLXSN HPV16 E6/E7 (HPV16), or HPV38 E6/E7 (HPV38). After 48 h, cells were harvested and luciferase activity was measured. Data are the means from three independent experiments performed in triplicate. **, P < 0.01. (B) Total RNA and total proteins from HFK and HPV38 E6/E7 HFK were extracted, and TLR9 expression levels were measured by RT-qPCR and normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) levels (left); in addition, TLR9 protein levels were determined by immunoblotting (right). Error bars on the left side represent standard deviations from three biological replicates. **, P < 0.01. (C) Total RNA from control HFK or the indicated transduced HFK was extracted. TLR9 expression levels were measured by RT-qPCR. Error bars represent standard deviations from three biological replicates. (D) HFK transduced with pLXSN, HPV38 E6/E7, or HPV16 E6/E7 were treated with GpC or CpG 2006. After 24 h, the supernatants were collected to measure IL-8 or MIP3α secretion. Data are the means from three independent experiments performed in triplicate. **, P < 0.01.
ΔNp73α is directly involved in TLR9 downregulation.
HPV38 E7 oncoprotein promotes the accumulation of the p53 antagonist ΔNp73α, repressing the transcription of p53-regulated genes (9, 10). Recent studies have demonstrated that TLR9 expression can be positively regulated by the p53 transcription factor (26, 27). Therefore, we next determined whether ΔNp73α can modulate TLR9 transcription. ΔNp73α expression inhibited TLR9 promoter activity in transient-transfection experiments performed with RPMI 8226 cells (Fig. 2A). In addition, downregulation of ΔNp73α expression in HPV38 E6/E7 HFK using an antisense (AS) oligonucleotide led to an increase of both mRNA and protein levels of TLR9 (Fig. 2B and C). Finally, downregulation of viral oncoproteins by shRNA resulted in an increase of TLR9 mRNA levels (Fig. 2D).
FIG 2.
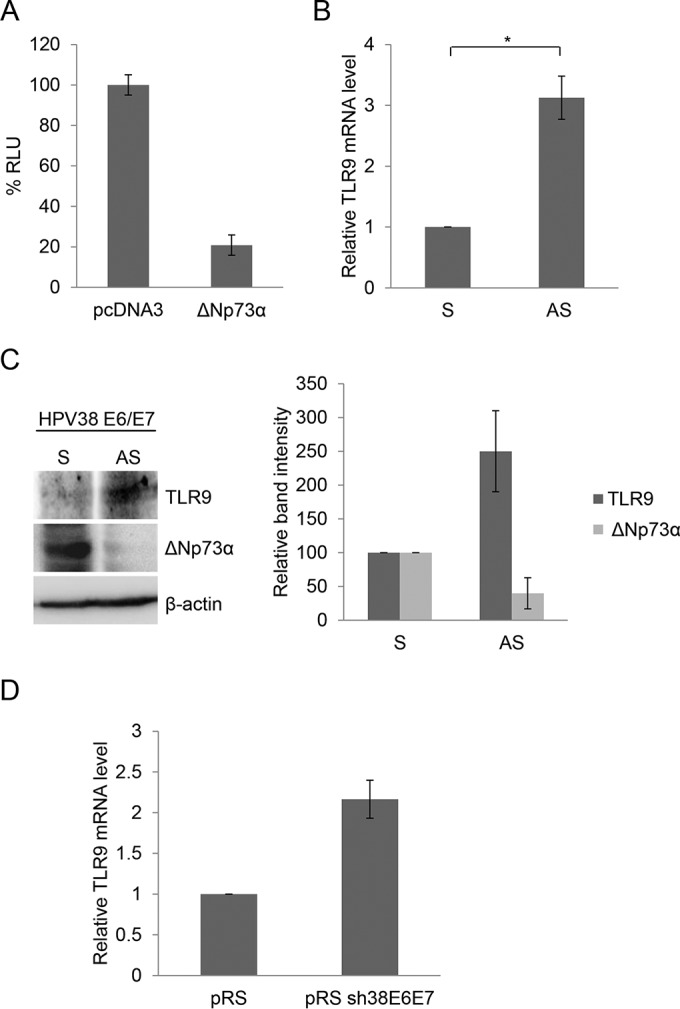
ΔNp73α negatively regulates TLR9 expression. (A) RPMI 8226 cells were cotransfected with the TLR9 promoter construct (−3227/−1) cloned in front of the luciferase reporter gene with pcDNA3 empty vector or expressing ΔNp73α. After 48 h, cells were harvested and luciferase activity was measured. Data are the means from three independent experiments performed in triplicate. (B and C) HPV38 E6/E7 HFK were transfected with ΔNp73α sense (S) and antisense (AS) oligonucleotides. (B) Expression of TLR9 and GAPDH was determined by RT-qPCR. Results are representative of those from three independent experiments. *, P < 0.05. (C) Protein extracts (40 μg) were analyzed by immunoblotting (IB) with indicated antibodies (left side). Band intensities were quantified and normalized to β-actin levels (right side). Data are the means from three independent experiments. (D) HFK and HPV38 E6/E7 HFK transduced with pRetroSuper (pRS) or shRNA-HPV38E6E7 (pRS sh38E6E7) were processed for RNA extraction. The mRNA levels of TLR9 were determined by RT-qPCR.
Previous studies have shown that ΔNp73α also has the ability to modulate the activity of NF-κB (28), a key transcription factor involved in the regulation of TLR9 expression (16, 22, 29). We therefore determined by chromatin immunoprecipitation (ChIP) experiments whether ΔNp73α is recruited to or near to the four characterized NF-κB responsive elements (RE), termed A, B, C, and D (16, 22, 29). We found that ΔNp73α binds mainly the promoter region near NF-κB RE C (Fig. 3A). To corroborate these findings, we performed oligonucleotide pulldown experiments using biotinylated DNA fragments of the TLR9 promoter (−1150/−1024) that encompass wild-type or mutated NF-κB RE C. The two biotinylated DNA probes were incubated with cellular extracts of HPV38 E6/E7 HFK overexpressing hemagglutinin (HA)-tagged ΔNp73α. The TLR9 promoter fragment containing the wild-type NF-κB RE C coprecipitated with ΔNp73α and IKKβ as well as EZH, which was previously shown to be part of the ΔNp73α repressive transcriptional complex (Fig. 3B) (13). In contrast, mutation of NF-κB RE C significantly decreased the binding of the ΔNp73α/IKKβ/ΕΖΗ2 complex to the DNA fragment, indicating that the DNA binding region of ΔNp73α coincides with or partially overlaps with NF-κB RE C. Transient-transfection experiments using two mutants of the TLR9 promoter cloned in front of the luciferase reporter gene showed that deletion of the region that includes NF-κB RE C abolished the inhibitory function of ΔNp73α (Fig. 3C and D). Point mutations of RE C had the same effect (Fig. 3E). Finally, downregulation of ΔNp73α by antisense oligonucleotide and/or deletion of promoter region containing RE C resulted in the loss of HPV38 E7-mediated inhibition of TLR9 promoter activity (Fig. 3F). Together, these findings highlight the crucial role of ΔNp73α in the HPV38-induced downregulation of TLR9 transcription.
FIG 3.

ΔNp73α binds to TLR9 promoter on NF-κB site C. (A) ChIP was performed in HPV38 E6/E7 HFK using anti-ΔNp73α antibody. Simultaneously, part of the total chromatin fraction (1/10) was used as input. qPCR was performed using specific primers flanking the NF-κB RE within the TLR9 promoter. The histogram shows the relative amount of the promoter bound by ΔNp73α after subtraction of the background of nonspecific IgG control expressed as a percentage of the input. Data are the means from three independent experiments. **, P < 0.01. (B) HPV38 E6/E7 HFK transiently expressing HA-ΔNp73α was processed for the oligonucleotide pulldown assay. Cell lysate was incubated with biotinylated (btn) probes containing the NF-κB RE of the TLR9 promoter, either wild type (pTLR9wt) or mutated (pTLR9mut). DNA-associated proteins were recovered by precipitation with streptavidin beads and analyzed by IB (left side). The intensity of the protein bands in three independent experiments was quantified (right side). *, P < 0.05; **, P < 0.01. (C) Scheme of the TLR9 promoter luciferase constructs: the full-length (−3227/−1) construct containing the 4 NF-κB RE (sites A, B, C, and D) and the deletion (−1017/−1 and −290/−1) constructs. (D) RPMI 8226 cells were cotransfected with TLR9 promoter constructs (−3227/−1, −1017/−1, and −290/−1) cloned in front of the luciferase reporter gene with pcDNA3 empty vector or expressing ΔNp73α. After 48 h, cells were harvested and luciferase activity was measured. Data are the means from three independent experiments performed in triplicate. **, P < 0.01. (E) RPMI 8226 cells were cotransfected with TLR9 promoter constructs wild-type (−3227/−1) or NF-κB point mutated in site C (mutC) cloned in front of the luciferase reporter gene with pcDNA3 empty vector or expressing ΔNp73α. After 48 h, cells were harvested and luciferase activity was measured. Data are the means from three independent experiments performed in triplicate. **, P < 0.01. (F) RPMI 8226 cells were cotransfected with TLR9 promoter constructs (−3227/−1 or −1017/−1) cloned in front of the luciferase reporter gene with pLXSN empty vector or pLXSN HPV38E7 construct and ΔNp73α sense (S) or AS oligonucleotides. After 48 h, cells were harvested and luciferase activity was measured. Data are the means from three experiments. **, P < 0.01.
ΔNp73α and NF-κB p65 inversely regulate TLR9 expression.
We recently showed that ΔNp73α forms a complex with the Polycomb group (PcG) 2 member EZH2 and DNA methyltransferase DNMT1 to repress the transcription of p53-regulated genes (13). Therefore, we performed ChIP experiments to determine which cellular proteins were recruited to the TLR9 promoter region near NF-κB RE C. We observed that in addition to ΔNp73α and IKKβ, the subunit of NF-κB transcription complex p65 as well as DMNT1 and EZH2 was associated with the TLR9 promoter in the NF-κB RE C region (Fig. 4A). Consistent with the recruitment of EZH2, histone 3 was found to be trimethylated at lysine 27 (H3K27me3) in the same region of the TLR9 promoter (Fig. 4A). Downregulation of ΔNp73α by AS oligonucleotide experiments resulted in a significant decrease in the amount of IKKβ and EZH2 recruited to the TLR9 promoter as well as of H3K27 (Fig. 4B). The recruitment of DMNT1 appeared to be less affected by the decrease in ΔNp73α levels, suggesting that it may also be associated with another, as-yet-unidentified complex. Similarly to the downregulation of ΔNp73α, silencing the expression of EZH2 increased TLR9 mRNA levels (Fig. 4C). All these findings show that ΔNp73α plays a key role in the inhibition of TLR9 expression by promoting the formation of a repressive transcriptional complex.
FIG 4.

ΔNp73α and p65 compete for binding to the TLR9 promoter. (A) HPV38 E6/E7 HFK were processed for ChIP using ΔNp73α, IKKβ, DNMT1, EZH2, H3K27me3, and p65 antibodies. qPCR was performed using specific primers flanking NF-κB RE site C within the TLR9 promoter. Data are the means from three independent experiments. All the bindings shown are statistically significant compared to the negative control, with a P value of <0.01. (B) HPV38 E6/E7 HFK were transfected with ΔNp73α S and AS oligonucleotides, and the levels of indicated cellular proteins were determined by IB (right side). Total cellular extracts were processed for ChIP with the indicated antibodies. Data are from one representative experiment of two independent experiments. **, P < 0.01. (C) HPV38 E6/E7 HFK were transfected with a control siRNA (scramble) or siRNA specific for EZH2 (siEZH2). After 72 h, total RNA was extracted and EZH2 or TLR9 mRNA levels were analyzed by RT-qPCR. Error bars represent standard deviations from three biological replicates. **, P < 0.01. The inset shows the EZH2 protein levels determined by IB. (D) HPV38 E6/E7 HFK were transfected with a control siRNA (scramble) or siRNA specific for p65 (sip65), and p65 as well as ΔNp73a levels were determined by IB (inset). Total cellular extracts were processed for ChIP using anti-ΔNp73α antibody. The results were analyzed by qPCR. Data are the means from three independent experiments. **, P < 0.01.
We also observed that higher levels of p65 were associated with the NF-κB RE C region when ΔNp73α was downregulated, indicating that ΔNp73α and p65 compete in binding the same or a partially overlapping DNA region within the TLR9 promoter (Fig. 4B). Consistent with these conclusions, downregulation of p65 by siRNA increased the recruitment of ΔNp73α in the same promoter region (Fig. 4D). Experiments with HPV38 E6/E7 HFK in which the ΔNp73α levels were downregulated provided evidence for the positive role of p65 in TLR9 transcriptional regulation. In fact, although the decrease in ΔNp73α protein levels is associated with reactivation of TLR9 expression, the simultaneous silencing of p65 expression repressed TLR9 transcription (Fig. 5A). Consistent with these findings, inhibition of NF-κB signaling in HPV38 E6/E7 HFK by overexpressing a nondegradable deletion mutant of IκBα (ΔN-IκBα) that lacks the first 36 amino acids at the N terminus containing the IKK-phosphorylated amino acid further decreased TLR9 mRNA levels (Fig. 5B). Similar results were obtained with cells expressing p65 or IKKβ siRNA (Fig. 5C).
FIG 5.

ΔNp73α and p65 inversely regulate TLR9 expression. (A) HPV38 E6/E7 HFK were cotransfected with 1 μg of ΔNp73α S or AS oligonucleotides and 50 pmol of a control siRNA (scramble) or siRNA specific for p65 (sip65). After 36 h, total RNA was extracted and analyzed by RT-qPCR. Data are the means from three independent experiments. **, P < 0.01. (B) HPV38 E6/E7 HFK were transduced with pBabe or pBabe-ΔN-IκBα superrepressor (ΔN-IκBα). Total RNA was extracted and TLR9 levels were analyzed by RT-qPCR. Data are the means from three independent experiments. **, P < 0.01. (C) HPV38 E6/E7 HFK were transfected with scramble or p65 siRNA (left) or with scramble or IKKβ siRNA (right). After 40 h, total RNA was extracted and TLR9 mRNA levels were analyzed by RT-qPCR. Data are the means from three independent experiments. *, P < 0.05; **, P < 0.01.
In summary, these findings showed that p65 and ΔNp73α compete for binding to the TLR9 promoter, leading to an inverse regulation of its activity.
TLR9 ectopic expression affects cellular proliferation in HPV38 E6/E7 HFK.
To gain more insights on the biological significance of HPV38-mediated downregulation of TLR9 transcription, we reexpressed TLR9 in HPV38 E6/E7 HFK using a retrovirus expression system. As expected, TLR9 mRNA and protein were detected only in cells transduced with TLR9 recombinant retrovirus (Fig. 6A). No changes in viral gene expression were observed in mock-treated cells or TLR9 HPV38 E6/E7 HFK (Fig. 6A). We first determined whether TLR9 expression affected the proliferation of HPV38 E6/E7 HFK by performing a colony formation assay. Although we detected approximately the same numbers of colonies in TLR9-expressing and control cells, the colony size was significantly decreased in the presence of TLR9 expression (Fig. 6B). The negative impact of TLR9 overexpression on the proliferation of HPV38 E6/E7 HFK was also observed in a short-term growth curve (Fig. 6C). Analysis of cell cycle profile by flow cytometry of HPV38 E6/E7 HFK overexpressing TLR9 revealed a significant decrease of cells in S and G2/M phase compared to the value for the mock-treated cells (Fig. 6D). Immunoblotting for many cell cycle regulators showed that the cell cycle inhibitors p21WAF1 and p27Kip1 are strongly accumulated in TLR9-expressing HPV38 E6/E7 HFK (Fig. 6E).
FIG 6.

TLR9 expression in HPV38 E6/E7 HFK inhibits cellular proliferation by the accumulation of p21WAF1 and p27Kip1. (A) HPV38 E6/E7 HFK were transduced with pBabe empty vector or expressing TLR9. The efficiency of transduction was verified by RT-PCR (left) and IB (right). Data on the left are the means from three independent experiments. **, P < 0.01. The bottom right shows the levels of HPV38 E6 and E7 mRNA determined by qPCR. (B) Colony formation assay. After 6 to 8 days of culture in puromycin-containing medium, colonies were fixed in 20% methanol and stained with crystal violet. The number of cells per colony was determined by cell counting. Results are the mean counts from 10 colonies randomly selected from three independent experiments. Double-blind counting was performed. **, P < 0.01. (C) Growth time course. The growth of all cell populations was monitored for 3 days as described in Materials and Methods. The growth was assessed by counting live cells with trypan blue. Data represent the means from two independent experiments, each performed in duplicate. **, P < 0.01. (D) Flow cytometry analysis of cells described for panel C (day 3) stained by propidium iodide. Data are from one representative experiment of three independent experiments. *, P < 0.05. (E) Total proteins (20 μg) from HPV38 E6/E7 HFK transduced with pBabe or pBabe-TLR9 were analyzed by IB (left) and band intensities quantified and normalized to β-actin levels (right). Data are the means from three independent experiments. **, P < 0.01.
Consistent with p21WAF1 levels, lower CDK2-associated kinase activity was detected in these cells than in mock-treated cells (Fig. 7A). However, TLR9 expression did not lead to significant changes in p21WAF1 or p27Kip1 mRNA levels (Fig. 7B). Based on these data, we investigated whether posttranslational mechanisms are implicated in the TLR9-induced accumulation of p21WAF1 and p27Kip1. Interestingly, it is known that p38 MAPK is activated by TLR9 signaling and positively regulate the p21WAF1 protein levels by posttranslational mechanisms (30). Accordingly, we observed that the phosphorylated form (Thr180/Tyr182) of p38 MAPK was strongly accumulated in TLR9-expressing HPV38 E6/E7 HFK (Fig. 7C). Moreover, both p21WAF1 and p27Kip1 levels were reduced in these cells treated with a chemical inhibitor of p38 MAPK, SB203580 (Fig. 7D), or with a specific siRNA against p38 (Fig. 7E). Taken together, these findings highlight a novel function of TLR9 in inhibiting cellular proliferation via accumulation of the cell cycle inhibitors p21WAF1 and p27Kip1.
FIG 7.

TLR9-induced accumulation of p21WAF1 and p27Kip1 is mediated by activation of p38 signaling. (A) Kinase assay. CDK2 was immunoprecipitated from 1 mg of total proteins in whole-cell lysate. As a control, an identical reaction was carried out with a negative IgG antibody. Immunocomplexes were then incubated with purified histone H1 in the presence of [γ-32P]ATP. After SDS-PAGE followed by autoradiography (top), band intensity was quantified (bottom). Data are the means from three independent experiments. *, P < 0.05. (B) Total RNA from indicated cell lines was extracted and p21WAF1 and p27Kip1 expression levels were measured by RT-qPCR. Error bars represent standard deviations from four biological replicates. (C) Total proteins (20 μg) from HPV38 E6/E7 HFK transduced with pBabe or pBabe-TLR9 were analyzed by IB (left). The intensity of the protein bands in three independent experiments was quantified (right). **, P < 0.01. (D) TLR9-expressing HPV38 E6/E7 HFK were treated with the p38 inhibitor SB203580 (5 μM) or dimethyl sulfoxide (DMSO). After 3 h, total proteins were extracted and analyzed by IB using the indicated antibodies (top). The intensity of the protein bands in three independent experiments was quantified (bottom). *, P < 0.05. (E) TLR9-expressing HPV38 E6/E7 HFK were transfected with siRNA scramble or siRNA specific for p38 (sip38). After 48 h, total proteins were extracted and analyzed by IB (left). Band intensity was quantified in three independent experiments and is shown as a histogram (right). **, P < 0.01.
DISCUSSION
Previous studies demonstrated that several oncogenic viruses use different mechanisms to inhibit the expression of TLR9 (16, 19–24). Here, we describe a novel mechanism of beta HPV38 in repressing TLR9 expression that is mediated by ΔNp73α, a well-characterized antagonist of p53. Thus, beta HPV38, in addition to its ability to promote cellular transformation, also shares with oncogenic viruses the property of repressing the TLR9 signaling pathway. The high conservation of this property among human oncogenic viruses underlines the importance of downregulation of TLR9 expression in virus-mediated carcinogenesis. It is likely that loss of the functionality of TLR9 signaling has a positive impact on completion of the viral life cycle and/or evasion of the host immune response. However, the fact that the inhibition of TLR9 gene transcription occurs after the translocation of viral DNA into the nucleus and is dependent on the expression of the viral oncoproteins does not fully support an exclusive role of the event in the evasion of the innate immune response to viral infection. In fact, at an early stage of infection and before viral gene expression, TLR9 is functional and is activated by nonmethylated CpG islands present in the viral genome (16, 22). A possible explanation is that by inhibiting TLR9 expression, oncogenic viruses prevent its further activation by the microflora present at specific anatomical sites, which may have a negative impact on viral infection. In agreement with this hypothesis, it has been reported that commensal bacterial subspecies could be protective against HPV infection (31). In addition, it is plausible that inactivation of TLR9 by oncogenic viruses may play an additional role in infected cells. Previous findings from our group showed that EBV infection of primary B cells resulted in a rapid decrease of TLR9 mRNA levels, which became more pronounced upon immortalization, suggesting that the decrease in TLR9 expression may be linked to cellular transformation (19).
In this study, we provided evidence for a novel function of TLR9 in controlling cellular proliferation. Reexpression of TLR9 in HPV38 E6/E7 HFK resulted in a strong accumulation of the cell cycle inhibitors p21WAF1 and p27Kip1 and a clear decrease in cellular proliferation. It is likely that the inhibitory role of TLR9 on cellular proliferation is a response to the cellular stress induced by the expression of the viral oncogenes. The precise mechanism of this novel TLR9 function is still unknown. Ongoing studies by our group are focused on determining whether the TLR9-induced p21WAF1 accumulation requires TLR9 engagement with endogenous ligands, for example, damage-associated molecular patterns (DAMP) generated during virus-induced stress. In support of the hypothesis that TLR9 promotes antiproliferative events in response to cellular stresses, it has recently been shown that TLR9 expression is strongly activated via p53 in primary human blood lymphocytes and alveolar macrophages upon exposure to different types of DNA-damaging insults (27). Based on these findings, it is likely that UV irradiation, the main risk factor for the development of skin squamous cell carcinoma, can induce TLR9 upregulation via p53 activation in keratinocytes. The fact that HPV38 E7 induces ΔNp73α accumulation may prevent activation of TLR9 in cells exposed to UV irradiation. This model is in line with the concept of a synergy between UV irradiation and beta HPV38 in skin carcinogenesis (12).
In conclusion, we have demonstrated that beta HPV38, as observed for several well-established oncogenic viruses, has the ability to inhibit the expression of TLR9. Interestingly, the HPV38 mechanism elucidated here differs substantially from those previously characterized for other oncogenic viruses. Most importantly, our study highlights a novel function of TLR9 in negatively regulated cellular proliferation via accumulation of the cell cycle inhibitors p21WAF1 and p27Kip1.
ACKNOWLEDGMENTS
We are grateful to all members of the Infections and Cancer Biology Group for their support, Isabelle Rondy for her help with preparation of the manuscript, and Karen Müller and Jessica Cox for editing the manuscript.
REFERENCES
- 1.Tommasino M. 2014. The human papillomavirus family and its role in carcinogenesis. Semin Cancer Biol 26:13–21. doi: 10.1016/j.semcancer.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 2.Smola S. 2014. Human papillomaviruses and skin cancer. Adv Exp Med Biol 810:192–207. [PubMed] [Google Scholar]
- 3.Accardi R, Gheit T. 2014. Cutaneous HPV and skin cancer. Press Med doi: 10.1016/j.lpm.2014.08.008. [DOI] [PubMed] [Google Scholar]
- 4.Bouvard V, Gabet AS, Accardi R, Sylla SB, Tommasino M. 2006. The cutaneous human papillomavirus types and non-melanoma-skin cancer, p 269–277. In Campo MS. (ed), Papillomavirus research: from natural history to vaccine and beyond. Caister Academic Press, Norfolk, United Kingdom. [Google Scholar]
- 5.Pfister H, Fuchs PG, Majewski S, Jablonska S, Pniewska I, Malejczyk M. 2003. High prevalence of epidermodysplasia verruciformis-associated human papillomavirus DNA in actinic keratoses of the immunocompetent population. Arch Dermatol Res 295:273–279. doi: 10.1007/s00403-003-0435-2. [DOI] [PubMed] [Google Scholar]
- 6.Bernard HU, Burk RD, Chen Z, van Doorslaer K, zur Hausen H, de Villiers EM. 2010. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 401:70–79. doi: 10.1016/j.virol.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caldeira S, Zehbe I, Accardi R, Malanchi I, Dong W, Giarre M, de Villiers EM, Filotico R, Boukamp P, Tommasino M. 2003. The E6 and E7 proteins of the cutaneous human papillomavirus type 38 display transforming properties. J Virol 77:2195–2206. doi: 10.1128/JVI.77.3.2195-2206.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gabet AS, Accardi R, Bellopede A, Popp S, Boukamp P, Sylla BS, Londono-Vallejo JA, Tommasino M. 2008. Impairment of the telomere/telomerase system and genomic instability are associated with keratinocyte immortalization induced by the skin human papillomavirus type 38. FASEB J 22:622–632. doi: 10.1096/fj.07-8389com. [DOI] [PubMed] [Google Scholar]
- 9.Accardi R, Dong W, Smet A, Cui R, Hautefeuille A, Gabet AS, Sylla BS, Gissmann L, Hainaut P, Tommasino M. 2006. Skin human papillomavirus type 38 alters p53 functions by accumulation of deltaNp73. EMBO Rep 7:334–340. doi: 10.1038/sj.embor.7400615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Accardi R, Scalise M, Gheit T, Hussain IS, Yue J, Carreira C, Collino A, Indiveri C, Gissmann L, Sylla BS, Tommasino M. 2011. IκB kinase β promotes cell survival by antagonizing p53 functions through ΔNp73α phosphorylation and stabilization. Mol Cell Biol 31:2210–2226. doi: 10.1128/MCB.00964-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muench P, Probst S, Schuetz J, Leiprecht N, Busch M, Wesselborg S, Stubenrauch F, Iftner T. 2010. Cutaneous papillomavirus E6 proteins must interact with p300 and block p53-mediated apoptosis for cellular immortalization and tumorigenesis. Cancer Res 70:6913–6924. doi: 10.1158/0008-5472.CAN-10-1307. [DOI] [PubMed] [Google Scholar]
- 12.Viarisio D, Mueller-Decker K, Kloz U, Aengeneyndt B, Kopp-Schneider A, Grone HJ, Gheit T, Flechtenmacher C, Gissmann L, Tommasino M. 2011. E6 and E7 from beta HPV38 cooperate with ultraviolet light in the development of actinic keratosis-like lesions and squamous cell carcinoma in mice. PLoS Pathog 7:e1251002. doi: 10.1371/journal.ppat.1002125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saidj D, Cros MP, Hernandez-Vargas H, Guarino F, Sylla B, Tommasino M, Accardi R. 4 September 2013. The E7 oncoprotein from beta human papillomavirus type 38 induces the formation of an inhibitory complex for a subset of p53-regulated promoters. J Virol doi: 10.1128/JVI.01047-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hirsch I, Caux C, Hasan U, Bendriss-Vermare N, Olive D. 2010. Impaired Toll-like receptor 7 and 9 signaling: from chronic viral infections to cancer. Trends Immunol 31:391–397. doi: 10.1016/j.it.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 15.Sathish N, Yuan Y. 2011. Evasion and subversion of interferon-mediated antiviral immunity by Kaposi's sarcoma-associated herpesvirus: an overview. J Virol 85:10934–10944. doi: 10.1128/JVI.00687-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hasan UA, Zannetti C, Parroche P, Goutagny N, Malfroy M, Roblot G, Carreira C, Hussain I, Muller M, Taylor-Papadimitriou J, Picard D, Sylla BS, Trinchieri G, Medzhitov R, Tommasino M. 2013. The human papillomavirus type 16 E7 oncoprotein induces a transcriptional repressor complex on the Toll-like receptor 9 promoter. J Exp Med 210:1369–1387. doi: 10.1084/jem.20122394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawai T, Akira S. 2007. Antiviral signaling through pattern recognition receptors. J Biochem 141:137–145. doi: 10.1093/jb/mvm032. [DOI] [PubMed] [Google Scholar]
- 18.Takeda K, Akira S. 2005. Toll-like receptors in innate immunity. Int Immunol 17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- 19.Fathallah I, Parroche P, Gruffat H, Zannetti C, Johansson H, Yue J, Manet E, Tommasino M, Sylla BS, Hasan UA. 2010. EBV latent membrane protein 1 is a negative regulator of TLR9. J Immunol 185:6439–6447. doi: 10.4049/jimmunol.0903459. [DOI] [PubMed] [Google Scholar]
- 20.van Gent M, Griffin BD, Berkhoff EG, van Leeuwen D, Boer IG, Buisson M, Hartgers FC, Burmeister WP, Wiertz EJ, Ressing ME. 2011. EBV lytic-phase protein BGLF5 contributes to TLR9 downregulation during productive infection. J Immunol 186:1694–1702. doi: 10.4049/jimmunol.0903120. [DOI] [PubMed] [Google Scholar]
- 21.Martin HJ, Lee JM, Walls D, Hayward SD. 2007. Manipulation of the Toll-like receptor 7 signaling pathway by Epstein-Barr virus. J Virol 81:9748–9758. doi: 10.1128/JVI.01122-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hasan UA, Bates E, Takeshita F, Biliato A, Accardi R, Bouvard V, Mansour M, Vincent I, Gissmann L, Iftner T, Sideri M, Stubenrauch F, Tommasino M. 2007. TLR9 expression and function is abolished by the cervical cancer-associated human papillomavirus type 16. J Immunol 178:3186–3197. doi: 10.4049/jimmunol.178.5.3186. [DOI] [PubMed] [Google Scholar]
- 23.Vincent IE, Zannetti C, Lucifora J, Norder H, Protzer U, Hainaut P, Zoulim F, Tommasino M, Trepo C, Hasan U, Chemin I. 2011. Hepatitis B virus impairs TLR9 expression and function in plasmacytoid dendritic cells. PLoS One 6:e26315. doi: 10.1371/journal.pone.0026315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shahzad N, Shuda M, Gheit T, Kwun HJ, Cornet I, Saidj D, Zannetti C, Hasan U, Chang Y, Moore PS, Accardi R, Tommasino M. 2013. The T antigen locus of Merkel cell polyomavirus downregulates human Toll-like receptor 9 expression. J Virol 87:13009–13019. doi: 10.1128/JVI.01786-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morgenstern JP, Land H. 1990. Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res 18:3587–3596. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shatz M, Menendez D, Resnick MA. 2012. The human TLR innate immune gene family is differentially influenced by DNA stress and p53 status in cancer cells. Cancer Res 72:3948–3957. doi: 10.1158/0008-5472.CAN-11-4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Menendez D, Shatz M, Azzam K, Garantziotis S, Fessler MB, Resnick MA. 2011. The Toll-like receptor gene family is integrated into human DNA damage and p53 networks. PLoS Genet 7:e1001360. doi: 10.1371/journal.pgen.1001360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tanaka Y, Ota K, Kameoka M, Itaya A, Yoshihara K. 2006. Up-regulation of NFkappaB-responsive gene expression by DeltaNp73alpha in p53 null cells. Exp Cell Res 312:1254–1264. doi: 10.1016/j.yexcr.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 29.Takeshita F, Suzuki K, Sasaki S, Ishii N, Klinman DM, Ishii KJ. 2004. Transcriptional regulation of the human TLR9 gene. J Immunol 173:2552–2561. doi: 10.4049/jimmunol.173.4.2552. [DOI] [PubMed] [Google Scholar]
- 30.Kim GY, Mercer SE, Ewton DZ, Yan Z, Jin K, Friedman E. 2002. The stress-activated protein kinases p38 alpha and JNK1 stabilize p21(Cip1) by phosphorylation. J Biol Chem 277:29792–29802. doi: 10.1074/jbc.M201299200. [DOI] [PubMed] [Google Scholar]
- 31.Gillet E, Meys JF, Verstraelen H, Bosire C, De SP, Temmerman M, Broeck DV. 2011. Bacterial vaginosis is associated with uterine cervical human papillomavirus infection: a meta-analysis. BMC Infect Dis 11:10. doi: 10.1186/1471-2334-11-10. [DOI] [PMC free article] [PubMed] [Google Scholar]