

ABSTRACT
Influenza A virus (IAV) undergoes RNA transcription by a unique capped-mRNA-dependent transcription, which is carried out by the viral RNA-dependent RNA polymerase (RdRp), consisting of the viral PA, PB1, and PB2 proteins. However, how the viral RdRp utilizes cellular factors for virus transcription is not clear. Previously, we conducted a genome-wide pooled short hairpin RNA (shRNA) screen to identify host factors important for influenza A virus replication. Ribosomal RNA processing 1 homolog B (RRP1B) was identified as one of the candidates. RRP1B is a nucleolar protein involved in ribosomal biogenesis. Upon IAV infection, part of RRP1B was translocated from the nucleolus to the nucleoplasm, where viral RNA synthesis likely takes place. The depletion of RRP1B significantly reduced IAV mRNA transcription in a minireplicon assay and in virus-infected cells. Furthermore, we showed that RRP1B interacted with PB1 and PB2 of the RdRp and formed a coimmunoprecipitable complex with RdRp. The depletion of RRP1B reduced the amount of capped mRNA in the RdRp complex. Taken together, these findings indicate that RRP1B is a host factor essential for IAV transcription and provide a target for new antivirals.
IMPORTANCE Influenza virus is an important human pathogen that causes significant morbidity and mortality and threatens the human population with epidemics and pandemics every year. Due to the high mutation rate of the virus, antiviral drugs targeting viral proteins might ultimately lose their effectiveness. An alternative strategy that explores the genetic stability of host factors indispensable for influenza virus replication would thus be desirable. Here, we characterized the rRNA processing 1 homolog B (RRP1B) protein as an important cellular factor for influenza A virus transcription. We showed that silencing RRP1B hampered viral RNA-dependent RNA polymerase (RdRp) activity, which is responsible for virus transcription and replication. Furthermore, we reported that RRP1B is crucial for RdRp binding to cellular capped mRNA, which is a critical step of virus transcription. Our study not only provides a deeper understanding of influenza virus-host interplay, but also suggests a potential target for antiviral drug development.
INTRODUCTION
Influenza virus is an important pathogen that threatens human public health and the global economy on an annual basis. Influenza A viruses (IAV), which belong to the family Orthomyxoviridae, are enveloped RNA viruses with eight segments of single-stranded, negative-sense RNA encoding up to 12 viral proteins (1). Based on the antigenicity of their hemagglutinin (HA) and neuraminidase (NA) molecules, they have been classified into combinations of 18 HA subtypes (H1 to H18) and 11 NA subtypes (N1 to N11) (2).
At the beginning of infection, the viral HA protein binds to sialic acid-containing receptors on the host cell surface and elicits endocytosis of the viral particle, thereby allowing viral entry into the cell. Subsequently, the engulfed viral particle fuses with the late endosome to release viral ribonucleoprotein (vRNP), which is composed of viral RNA (vRNA) associated with the nucleoprotein (NP) and viral RNA-dependent RNA polymerase (RdRp). The viral RdRp is composed of polymerase acidic protein (PA), polymerase basic protein 1 (PB1), and polymerase basic protein 2 (PB2) (3). Subsequently, vRNP is translocated to the nucleus for transcription and replication of the influenza virus genome, which are both carried out by RdRp (4). IAV RNA transcription undergoes a unique cap-snatching process, in which PB2 first binds to the 5′ cap of the host mRNA (5, 6), PA subsequently cleaves the host mRNA to produce capped RNA fragments with lengths of 10 to 13 nucleotides (7), and PB1 finally elongates the viral mRNA by RNA polymerization using the viral negative-sense RNA as a template (8). Of note, production of the primer RNA requires PB1 binding to the 5′ and 3′ end sequences of vRNA (9). As for viral RNA replication, RdRp first uses the viral RNA as a template to generate a replicative intermediate, named cRNA, and then uses cRNA as the template to produce vRNA in a primer-independent process (10). In the late stage of the viral life cycle, vRNP, M1, and viral envelope proteins assemble the virion particles, which are then released from the cell surface to produce viral progeny.
Productive infection of the influenza virus requires the cooperation of host proteins. Accordingly, identification of the host factors involved in viral replication is of interest to understand the mechanisms of the viral life cycle. To identify potential cellular factors crucial for viral replication, several genome-wide RNA interference (RNAi) library screens have been conducted (11). Unfortunately, relatively few overlaps among the candidates were found. We therefore carried out a modified pooled genome-wide RNAi library screen (12). In total, 38 candidate genes, hit by two unique short hairpin RNAs (shRNAs) showing at least 40% reduction of viral replication, were identified as hits. To explore the detailed mechanisms of influenza virus transcription, we focused on the candidate host proteins with potential functions in the regulation of cellular transcription or RNA processing, since influenza virus transcription relies on host transcription (13). From the list, we focused on the rRNA processing 1 homolog B (RRP1B) gene, which was previously identified as a susceptibility gene for breast cancer progression and metastasis (14). RRP1B is a multifunctional protein that has been implicated in several biological processes. It has been reported to modulate transcription and chromatin structure via interaction with transcription factors and nucleosome-binding proteins (15), as well as to regulate the expression of mRNA isoforms via interactions with serine/arginine-rich splicing factor 1 (SRSF1) (16). Additionally, RRP1B is associated with pre-60S ribosomal subunits and predicted to modulate ribosome biogenesis (17). Due to its potential function in the regulation of transcription or RNA processing, the protein was singled out for further studies to understand its role in IAV replication. In this study, we found that upon IAV infection, RRP1B is translocated to the nucleoplasm and facilitates the binding of RdRp to capped mRNAs to undergo virus transcription.
MATERIALS AND METHODS
Cell culture.
Human lung adenocarcinoma epithelial cells (A549) were maintained in F-12K medium (Gibco) supplemented with 10% fetal bovine serum (FBS) (Thermo Scientific) and antibiotics (100 U/ml penicillin and 100 μg/ml streptomycin). Madin-Darby canine kidney (MDCK) cells and human embryonic kidney (HEK293T) cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Gibco) supplemented with 10% FBS and antibiotics (100 U/ml penicillin G and 100 g/ml streptomycin). All the cells were cultured under 5% CO2 at 37°C.
Plasmids and viruses.
All the plasmids required for lentivirus production were provided by the National RNAi Core Facility, Academia Sinica, Taiwan. The identifier (ID) numbers of five shRNA clones used for knockdown of RRP1B were as follows: TRCN0000130433 (shRRP1B no. 1), TRCN0000131097 (shRRP1B no. 2), TRCN0000129332 (shRRP1B no. 3), TRCN0000130351 (shRRP1B no. 4), and TRCN0000131094 (shRRP1B no. 5). The ID number of the shLacZ clone is TRCN0000072240. To generate the pLAS3w-RRP1B-flag.bsd plasmid for expression of Flag-tagged RRP1B, the open reading frame of RRP1B was amplified by PCR from pBluescript-RRP1B (purchased from Thermo Fisher Scientific Inc.) and inserted into the NheI and PmeI sites of the pLAS3w.Pbsd lentivirus-based vector (provided by the National RNAi Core Facility, Academia Sinica, Taiwan). To deliver the shRNA, the lentiviruses carrying the distinct shRNA were prepared by cotransfection with a packaging construct (pCMVΔR8.91), an envelope construct (pMD.G), and different shRNA constructs using TransIT-LT1 transfection reagent (Mirus Bio) according to the protocol on the RNAi Core website. The influenza A/WSN/33 virus (WSN33) was prepared by using reverse genetics (18) and mainly used in the studies. Influenza A/New Caledonia/20/1999 (NC99) and A/Wisconsin/67/2005 (W10) viruses were kindly provided by Che Ma, Academia Sinica, Taiwan.
Generation of the knockdown stable cell line and the RRP1B-overexpressing cell line and IAV infection.
To generate the RRP1B or LacZ knockdown (KD) cells, A549 or HEK293T cells were transduced with the lentivirus carrying shRNAs and subsequently selected with 3 μg/ml puromycin (Sigma) for 10 days to generate the stable cell lines. For the RRP1B-overexpressing cell line, A549 cells were transduced with the lentivirus carrying Flag-tagged RRP1B and subsequently selected with 10 μg/ml blasticidin (Invitrogen) for 10 days. To perform IAV infection, the cells were washed with phosphate-buffered saline (PBS) and then infected with IAV in MEM-alpha medium (Gibco) containing 0.5 μg/ml TPCK (tosylsulfonyl phenylalanyl chloromethyl ketone)-trypsin (Sigma) at the indicated multiplicity of infection (MOI) for 1 h. Afterward, the cells were washed twice with 1× PBS to remove excess IAV and then placed in complete growth medium. The IAV-infected cells were harvested at the indicated time points for further analysis.
Plaque assay.
MDCK cells were infected with serial 10-fold dilutions of IAV for 1 h, washed twice with PBS, and then overlaid with 0.5% agarose-containing MEM-alpha medium. After 2 days, the cells were fixed with 10% formaldehyde and stained with 0.1% crystal violet solution.
Antibodies and reagents.
Anti-HA, anti-7-methylguanylate (m7G) cap antibody, and anti-β-actin antibodies were purchased from Millipore (no. 04-902, MABE419, and MAB1501). Anti-IAV NP, anti-PB1, anti-PB2, and anti-PA antibodies were purchased from GeneTex (no. GTX629633, GTX125923, GTX125925, and GTX118991). Anti-IAV matrix protein (M1) antibody was purchased from AbDSerotec (no. MCA401). Anti-RRP1B antibody was obtained from Santa Cruz Biotechnology (no. sc-83327). Anti-Flag antibody was purchased from Cell Signaling (no. 2368S). Anti-nucleolin antibody was procured from Enzo Life Sciences (no. KAM-CP100). All Alexa Fluor-conjugated secondary antibodies used for immunofluorescence were procured from Molecular Probes (Invitrogen). DAPI (4′,6′-diamidino-2-phenylindole dihydrochloride) and cycloheximide (CHX) were purchased from Sigma-Aldrich. M-PER mammalian protein extraction reagent and anti-HA agarose were obtained from Thermo Scientific.
Western blot analysis and immunoprecipitation.
Cell lysates were prepared using M-PER mammalian protein extraction reagent with additional protease inhibitors, subjected to SDS-PAGE, and transferred onto a Hybond-P membrane (Amersham Biosciences). The membrane was probed with the indicated primary and appropriate secondary antibodies, detected using an enhanced chemiluminescence detection kit (Thermo Scientific), and then imaged with ImageQuant LAS4000 (GE Healthcare Life Sciences). For immunoprecipitation assays, the cell lysates were incubated with anti-HA agarose at 4°C overnight and washed with wash buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 0.1% Triton X-100). The immunoprecipitated proteins were subjected to Western blot analysis.
Immunofluorescence microscopy.
For indirect immunofluorescence staining, cells were fixed with paraformaldehyde, permeabilized with Triton X-100, blocked with bovine serum albumin, and then probed with the indicated primary antibody. A secondary antibody was applied with appropriate Alexa Fluor-conjugated antibody. To visualize DNA, cells were counterstained with DAPI. Images were acquired using a Leica SP2 spectral confocal microscope.
qRT-PCR.
Total cellular RNA was extracted using a High Pure RNA isolation kit (Roche Diagnostics) according to the manufacturer's protocol. cDNA was synthesized by using the SuperScript III first-strand synthesis system (Invitrogen). The primers for reverse transcription (RT) of mRNA, vRNA, and U2 small nuclear RNA (snRNA) were oligo(dT)20, an IAV-specific RT primer (uni-12; 5′-AGCAAAAGCAGG-3′), and a U2 snRNA-specific RT primer (5′-CTGGAGGTACTGCAATACCAG-3′), respectively. For most quantitative RT (qRT)-PCR analyses, we followed the standard TaqMan method with the Universal Probe Library System. GAPDH (glyceraldehyde 3-phosphate dehydrogenase) was used as a control for the normalization of cellular mRNA and intracellular viral RNA. The primers and probes were as follows: IAV_NP segment, sense (5′-GATGGAGACTGATGGAGAACG-3′) and antisense (5′-TCATTTTTCCGACAGATGCTC-3′) with universal probe no. 59; RRP1B, sense (5′-AGACAAAAGTGGGTGATGGTG-3′) and antisense (5′-CAGCTCTTCTCAAGGATACCTCA-3′ with universal probe no. 73; GAPDH, sense (5′-AGCCACATCGCTCAGACAC-3′) and antisense (5′-GCCCAATACGACCAAATCC-3′) with universal probe no. 60; and actin, sense (5′-GGTGTATCTCTGCCTTACAGATCA-3′) and antisense (5′-TCCATCACGATGCCAGTG-3′) with universal probe no. 64. For quantification of U2 snRNA, SYBR green dye was used. The primers for U2 snRNA were as follows: sense, 5′-ATCGCTTCTCGGCCTTTTGG-3′, and antisense, 5′-CTGGAGGTACTGCAATACCAG-3′.
Minireplicon assay.
HEK293T cells were cotransfected with pPolI-Luc, a Renilla luciferase-expressing plasmid, and four plasmids for expression of the viral proteins PA, PB1, PB2, and NP. Renilla luciferase activity was used as an internal control to normalize transfection efficiency. At 48 h posttransfection, cells were collected, and the luciferase activity was measured by using Dual-Glo luciferase (Promega) according to the manufacturer's protocol.
Primer extension assay.
Primer extension assays were performed by using a primer extension system avian myeloblastosis virus (AMV) reverse transcriptase kit (Promega), as described previously (19). Five micrograms of total RNA was mixed with 0.5 pmol (each) of two DNA primers, labeled at the 5′ end with [γ-32P]ATP and T4 polynucleotide kinase (Promega). The mixture was heated at 50°C for 2 h, followed by cooling at room temperature for 10 min. Primer extensions were performed after the addition of 1 U of avian myeloblastosis virus reverse transcriptase (Promega) to the reaction buffer provided with the enzyme for 2 h at 42°C. Two NA-specific primers were used in the same reverse transcription reaction: 5′-TGGACTAGTGGGAGCATCAT-3′ to detect vRNA and 5′-TCCAGTATGGTTTTGATTTCCG-3′ to detect cRNA and mRNA. The sequence 5′-TCCCAGGCGGTCTCCCATCC-3′ was used as a primer to detect 5S rRNA. Transcription products were analyzed on 6% polyacrylamide gels containing 7 M urea in Tris-borate-EDTA (TBE) buffer and detected by autoradiography.
RIP assay.
RNA-binding protein immunoprecipitation (RIP) assays were performed using the Magna RIP kit (Millipore) according to the manufacturer's instructions.
Briefly, HEK293T knockdown cells were transfected in 10-cm dishes using TransIT-LT1 transfection reagent (Mirus Bio) and lysed with 100 μl of RIP lysis buffer at 24 h posttransfection. The cell lysates (50 μl) were incubated with 450 μl RIP buffer containing anti-HA agarose and rotated overnight at 4°C. Samples were washed four times with RIP wash buffer, and 50 μl out of 500 μl of suspension beads was analyzed by immunoblotting using the indicated antibodies to check immunoprecipitation efficiency. The remnant samples were incubated with proteinase K buffer at 55°C for 1 h to digest the protein. RNAs were extracted by using a standard phenol-chloroform protocol and subjected to qRT-PCR for relative quantification.
RNA-protein pulldown assay.
RNA-protein pulldown assays were performed using a Pierce magnetic RNA-protein pulldown kit (Thermo Scientific) according to the manufacturer's instructions. The pGEM vector was first linearized with XbaI and then used for in vitro transcription with T7 RNA polymerase using a RiboMax large-scale RNA production kit (Promega) to produce 18-nucleotide RNA (uncapped RNA [5′-UCUGGUGUUGCCAAAGGG-3′]). The in vitro-transcribed uncapped RNA was purified using a Direct-zol RNA miniprep kit (Zymo Research) and then capped with a 7-methylguanylate structure using a vaccinia virus capping system and mRNA cap 2′-O-methyltransferase (New England BioLabs) to produce capped RNA (5′-m7G-UCUGGUGUUGCCAAAGGG-3′). 5′ vRNA, which covers 12 nucleotides of the 5′ terminus of vRNA (5′-AGUAGAAACAAGG-3′), and poly(A)25 were obtained from a commercial company (AllBio). Subsequently, RNA molecules were desthiobiotinylated using a Pierce RNA 3′ end desthiobiotinylation kit (Thermo Scientific) and captured by streptavidin magnetic beads. Two hundred micrograms of cell lysates was incubated with desthiobiotinylated RNA-captured streptavidin magnetic beads at 4°C for 2 h. The pulldown proteins were eluted using biotin elution buffer and subjected to Western blot analysis.
RESULTS
Identification of RRP1B as an important cofactor for influenza A virus replication.
We previously carried out an RNA interference (RNAi) screen and identified RRP1B as one of the candidates potentially affecting IAV replication (12). To verify its role in IAV replication, A549 cells were first transduced with lentiviruses expressing the shRNAs targeting RRP1B and then infected with influenza A/WSN/33 virus for 6 h. Compared to the shLacZ control, shRRP1B no. 3 and no. 5 clones had significantly smaller amounts of RRP1B protein and RNA; correspondingly, the amounts of viral NP in the two clones were also significantly smaller, as evidenced by both Western blot (Fig. 1A) and qRT-PCR (Fig. 1B) analyses. Due to the high knockdown efficiencies of the shRRP1B no. 3 and no. 5 clones, the two clones were selected for further experiments. To ascertain the importance of RRP1B in IAV replication, we examined the silencing effect of RRP1B on IAV production. The progeny virus titer, as determined by PFU assay, was reduced up to 10-fold in RRP1B knockdown cells compared to that from the control cells at various time points of the viral life cycle (Fig. 1C). To determine the universality of these findings, we also tested the RRP1B knockdown effects on other influenza virus strains, A/New Caledonia/20/1999 (H1N1) (NC99) and A/Wisconsin/67/2005 (H3N2) (W10) viruses. Knockdown of RRP1B also had the same suppression effects on viral NP or virus production (Fig. 1D, E, and F), suggesting that dependence on RRP1B may be conserved across IAV subtypes.
FIG 1.

Silencing RRP1B reduces influenza A virus replication. (A and B) To prepare A549 knockdown cells, A549 cells were transduced with lentiviruses carrying shRNA as described in Materials and Methods. The A549 knockdown cells were infected with influenza A/WSN/33 virus at an MOI of 1. At 6 h p.i., the cells were harvested. (A) The cellular lysates were subjected to Western blot analysis using anti-nucleoprotein (α NP) and anti-RRP1B antibodies. Actin was used as a loading control. (B) Cellular RNA was extracted and measured by qRT-PCR. The viral RNA (Flu_NP) level was determined by detection of NP-specific vRNA. The levels of viral RNA and RRP1B mRNA were normalized by GAPDH mRNA. The values represent the means ± standard deviations (SD) of the results of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001 compared with controls (n = 3). (C) A549 knockdown cells were infected with influenza A/WSN/33 virus at an MOI of 0.01. The supernatants were collected at the indicated time points and used for determining the viral titer by plaque assay in MDCK cells. The values represent the means ± SD of the results of two independent experiments. (D, E, and F) Experiments were performed the same way as for panels A to C, except that influenza A/New Caledonia/20/1999 (NC99) and A/Wisconsin/67/2005 (W10) viruses were used in place of influenza A/WSN/33 virus. (F) To determine the knockdown effect on NC99 and W10 virus production, the supernatants were collected at 24 h p.i. and used for plaque assays.
RRP1B is required for influenza A virus RdRp activity.
To address whether RRP1B plays a role in the early stages of the viral life cycle, RRP1B knockdown cells were infected with IAV for 3 h and assessed by immunofluorescence staining of NP, which is localized mainly in the nucleus early in infection. As shown in Fig. 2A, silencing RRP1B dramatically reduced NP staining signals, suggesting that RRP1B is involved in the early stages of the viral life cycle, which include virus entry, uncoating, nuclear import of vRNP, genome transcription/replication, and translation. To further dissect the role of RRP1B in early viral infection, we examined whether RRP1B participates in virus entry. For this purpose, A549 knockdown cells were incubated with IAV for 1 h at 4°C or 37°C and then washed with acidic PBS to remove the attached viruses. At 4°C, the virus can bind to the cell surface but not internalize; however, the virus can bind and internalize at 37°C. The internalized virus particles were detected by immunoblotting of viral M1 protein, which is the most abundant and sensitive marker protein of IAV. As shown in Fig. 2B, M1 signals were faint with incubation at 4°C (lanes 1 to 3), suggesting that most of the attached viruses were removed by the acidic treatment. Compared to the control shLacZ cells, no significant change in the amount of internalized M1 could be observed in RRP1B knockdown cells, indicating that virus entry was not affected (Fig. 2B, lanes 4 to 6). We next investigated whether RRP1B is involved in the regulation of IAV transcription/replication. A well-established minireplicon assay was applied to examine the silencing effect of RRP1B on RdRp activity, which is responsible for viral transcription and replication (20). RRP1B knockdown cells were cotransfected with plasmids for expression of the viral PB1, PB2, PA, and NP proteins; Renilla luciferase; and the reporter pPolI-Luc. Transfection of pPolI-Luc produces a modified influenza virus vRNA in which the coding region was replaced with the firefly luciferase coding sequences; thus, the firefly luciferase levels reflect the overall transcription and replication activities of the viral polymerase complex. Compared to the shLacZ control, silencing RRP1B reduced RdRp activity by approximately 70% (Fig. 2C). Furthermore, we performed qRT-PCR analysis of various viral RNA species in IAV-infected cells. The results showed that both viral mRNA and vRNA levels were reduced in RRP1B knockdown cells, corresponding to the reduced RRP1B RNA level at 3 h postinfection (p.i.) (Fig. 2D). Additionally, primer extension studies measuring vRNA, cRNA, and mRNA of the viral NA segment in the IAV-infected cells (19) also showed that silencing RRP1B significantly diminished mRNA, cRNA, and vRNA levels (Fig. 2E). Taken together, these data suggested that in both the minireplicon system and the IAV-infected cells, RRP1B had been demonstrated to be required for IAV RdRp activities involved in transcription or replication.
FIG 2.

Silencing RRP1B inhibits IAV RdRp activity. (A) A549 knockdown cells were infected with influenza A/WSN/33 virus at an MOI of 5 for 3 h and processed for immunofluorescence staining with anti-NP antibody (green) and DAPI. (B) A549 knockdown cells were infected with influenza A/WSN/33 virus at an MOI of 10 for 1 h, washed with acidic PBS (pH 1.3) to remove the attached but not yet internalized virions, and harvested. Internalized virus particles were analyzed by Western blotting with anti-M1 antibody. Actin was used as an internal control. (C) 293T knockdown cells were cotransfected with pPolI-Luc and plasmids for the expression of the viral PB1, PB2, PA, and NP. Renilla luciferase was used as an internal control. At 48 h posttransfection, luciferase activity was determined. The data represent the respective values relative to the value detected in control shLacZ knockdown cells. **, P < 0.01, and ***, P < 0.001 compared with controls (n = 3). (D) A549 knockdown cells were infected with influenza A/WSN/33 virus at an MOI of 5. At 3 h p.i., the cellular RNA was extracted. The levels of viral mRNA, vRNA, and RRP1B mRNA were measured by qRT-PCR and normalized by GAPDH mRNA. The values represent the means ± SD of the results of two independent experiments. **, P < 0.01, and ***, P < 0.001 compared with controls (n = 2). (E) A549 knockdown cells were infected with influenza A/WSN/33 virus at an MOI of 5. At 3 h p.i., the cellular RNA was extracted. The mRNA, cRNA, and vRNA of the viral NA segment were detected by primer extension assay. 5S rRNA was used as an internal control. The band intensities were quantified, and the mRNA/5S rRNA and vRNA/5S rRNA ratios are shown below.
RRP1B participates in influenza A virus transcription.
Because IAV uses distinctly different strategies for transcription and for RNA replication, we next investigated the effects of RRP1B knockdown on viral RNA transcription and replication separately. Given that IAV RNA replication (but not transcription) requires newly synthesized viral proteins (21), CHX, a translation inhibitor, was used to block the synthesis of viral proteins, thereby inhibiting the synthesis of cRNA and the replication of viral RNA. As shown in Fig. 3A, in the absence of CHX, the reduction of RRP1B relative to the control cells was correlated with the reduction of both viral mRNA and vRNA levels (Fig. 3A, left), consistent with the results described above. Interestingly, in the presence of CHX, upon RRP1B knockdown, the IAV mRNA levels were reduced in correlation with the reduction of RRP1B; in contrast, the vRNA levels remained unaffected or even increased slightly in RRP1B knockdown cells (Fig. 3A, right). These results were further confirmed in IAV infection of RRP1B knockdown cells by primer extension assay of intracellular RNA species. The results showed that the IAV mRNA levels were reduced in RRP1B knockdown cells in both the presence and absence of CHX (Fig. 3B, lanes 2 to 7). On the other hand, the vRNA levels were low but equivalent in all cells in the presence of CHX (Fig. 3B, lanes 5 to 7). These vRNAs likely represent the entering viral RNA or the residual amount of viral RNA replication. These results suggest that RRP1B is directly involved in mRNA transcription, but it is not certain whether it also participates in RNA replication.
FIG 3.

Decrease of viral mRNA levels in RRP1B KD cells in the presence of cycloheximide. A549 knockdown cells were treated with cycloheximide (100 μg/ml) for 1 h and then infected with influenza A/WSN/33 virus at an MOI of 5. At 3 h p.i., the cellular RNA was extracted. (A) The levels of mRNA and vRNA of the viral NP segment and RRP1B mRNA were measured by qRT-PCR and normalized by GAPDH mRNA. The values represent the means ± SD of the results of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001 compared with controls (n = 3). (B) The mRNA, cRNA, and vRNA of the viral NA segment were detected by primer extension assay. 5S rRNA was used as an internal control. The band intensities were quantified, and the mRNA/5S rRNA and vRNA/5S rRNA ratios are shown below.
Redistribution of RRP1B upon influenza A virus infection.
To further explore the role of RRP1B in IAV, we next examined its expression and localization early in infection. As shown in Fig. 4A, the expression levels of RRP1B were similar in IAV-infected and uninfected A549 cells at 3 h p.i., indicating that IAV infection did not enhance the expression of RRP1B. In the absence of viral infection, all the RRP1B colocalized with nucleolin, indicating that it is a nucleolar protein, consistent with its purported role in ribosome biogenesis (17). Interestingly, upon virus infection, some RRP1B was detected in the nucleoplasm (Fig. 4B). Furthermore, RRP1B showed colocalization with NP (Fig. 4C), strengthening the redistribution of RRP1B upon virus infection. Taken together, these findings support a potential role of RRP1B in virus transcription, which takes place in the nucleus.
FIG 4.

Protein levels and distribution of RRP1B in IAV-infected cells. (A) A549 cells were infected (+) or not (−) with influenza A/WSN/33 virus at an MOI of 5 for 3 h and then harvested for Western blot analysis with antibodies against RRP1B and NP. Actin was used as a loading control. (B and C) A549 cells were transduced with lentiviruses carrying Flag-tagged RRP1B cDNA. The RRP1B-Flag-overexpressing A549 cells were infected with influenza A/WSN/33 virus at an MOI of 5. At 3 h p.i., the cells were processed for immunofluorescence with anti-Flag antibody (green), anti-nucleolin antibody (B), or anti-NP antibody (C) (red), and DAPI.
RRP1B associates with influenza A virus RdRp.
The redistribution of RRP1B into the nucleoplasm upon IAV infection offered the possibility that RRP1B might interact with IAV. To address this possibility, Flag-tagged RRP1B and HA-tagged IAV proteins, excluding HA, NA, and NS2, were coexpressed in 293T cells and used for coimmunoprecipitation assays. As shown in Fig. 5A, among all the viral proteins, PB1, PB2, and NP coprecipitated Flag-tagged RRP1B, indicating their potential for interaction. To demonstrate that the interaction between RRP1B and IAV proteins indeed occurs in natural viral infections, A549 cells were infected with IAV, and the lysates harvested at 3 h p.i. were used for a coimmunoprecipitation assay. Anti-RRP1B antibody immunoprecipitated RRP1B and all three components of RdRp, but not NS1; in contrast, the IgG control did not precipitate RdRp (Fig. 5B), supporting the conclusion that RRP1B can interact with RdRp and that RRP1B is present in, or associated with, IAV RdRp.
FIG 5.
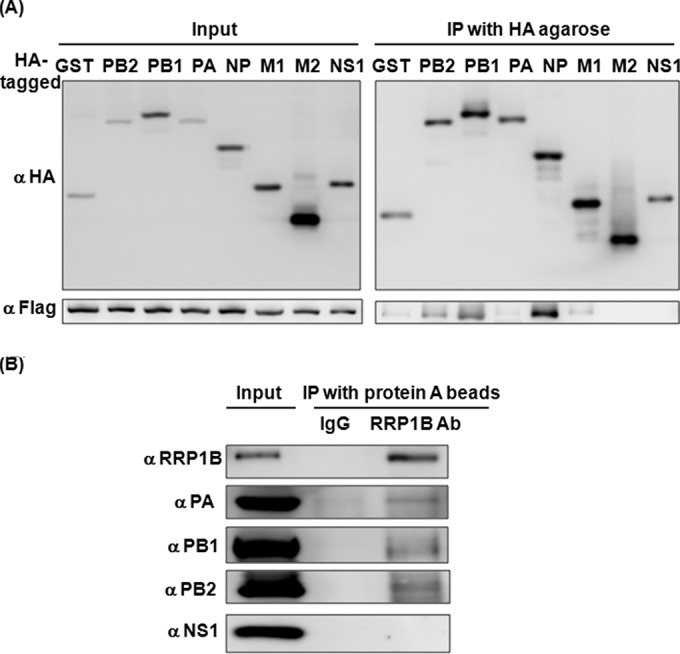
RRP1B interacts with IAV. (A) HEK293T cells were cotransfected with Flag-tagged RRP1B and the indicated HA-tagged plasmids. The cell lysates were prepared and used for immunoprecipitation with anti-HA agarose. The cell lysates (Input) and the immunoprecipitates (IP) were resolved on SDS-PAGE and immunoblotted with anti-HA or anti-Flag antibody. (B) A549 cells were infected with influenza A/WSN/33 virus at an MOI of 5. At 3 h p.i., the cell lysates were prepared and used for immunoprecipitation with rabbit IgG or anti-RRP1B antibody. The cell lysates (Input) and the immunoprecipitates were resolved on SDS-PAGE and immunoblotted with antibodies (Ab) against RRP1B, PA, PB1, PB2, and NS1, as indicated.
RRP1B affects the capped-RNA-binding ability of RdRp.
The three components of IAV RdRp have distinct functions. PB2 is for binding of host capped mRNA, PA is for endonuclease activity, and PB1 is for RNA polymerization (4). Notably, upon virus transcription, PB1 binds to the 5′ and 3′ termini of vRNA to activate RdRp activity (9). Since RRP1B interacts with PB2 and PB1 (Fig. 5A), we speculated that RRP1B may affect the RNA-binding activity of RdRp. To test this hypothesis, RRP1B knockdown 293T cells were cotransfected with HA-tagged PB1, PB2, and PA, and the lysates were used for immunoprecipitation of the RdRp-RNA complex using anti-m7G cap antibody, which specifically recognizes m7G-capped RNA, and then immunoblotted with anti-HA antibody to detect RdRp. As shown in Fig. 6A, RdRp could be coimmunoprecipitated by anti-m7G cap antibody (lane 7), but not by the IgG control (lane 4); importantly, anti-m7G cap antibody coprecipitated less RdRp from the RRP1B knockdown cell lysates than from the control cells (lanes 7 to 9), suggesting that RRP1B is essential for RdRp binding to capped mRNA. Previously, it was shown that the N-terminal region of PB1 interacts with PA, whereas the C-terminal region of PB1 interacts with PB2 (22); thus, RdRp exists as a tripartite complex. It was noted that since PA, PB1, and PB2 have similar molecular weights, the RdRp complex was often detected as two bands, as seen in our Western blot analysis.
FIG 6.
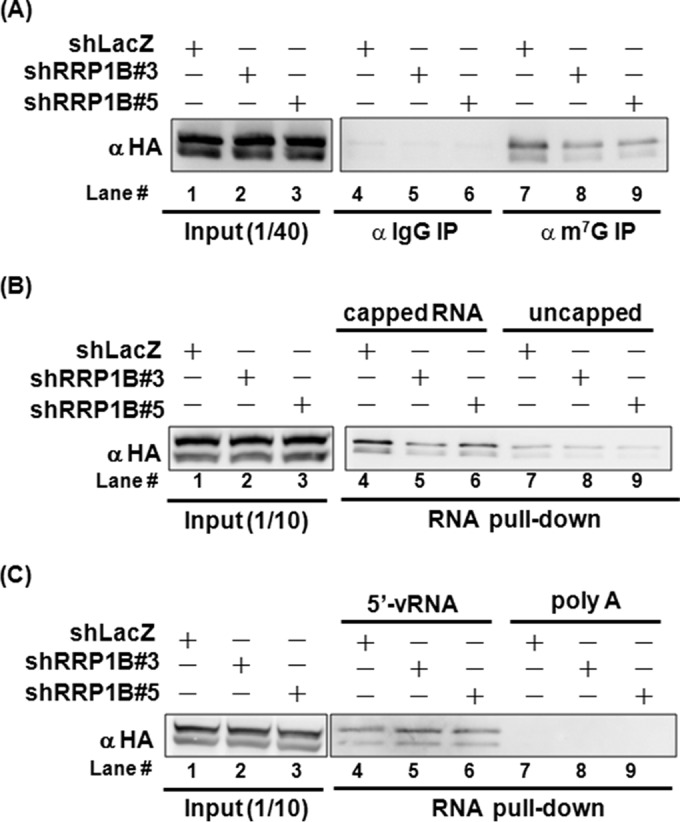
Silencing RRP1B reduces RdRp binding to capped RNA. HEK293T knockdown cells were cotransfected with plasmids expressing HA-tagged PB1, PB2, and PA. At 48 h posttransfection, the cells were harvested. (A) Cell lysates were prepared using RIP lysis buffer and used for immunoprecipitation with mouse IgG or anti-m7G antibody. The cell lysates (Input) and the immunoprecipitates were resolved on SDS-PAGE and immunoblotted with anti-HA antibody. (B and C) Cell lysates were prepared using MPER buffer and used for RNA pulldown assays with desthiobiotinylated RNA-captured streptavidin beads as described in Materials and Methods. Four kinds of RNA, i.e., capped RNA, uncapped RNA, the 5′ terminus of vRNA, and poly(A), were used for the assay. The cell lysates (Input) and pulldown proteins were resolved on SDS-PAGE and immunoblotted with anti-HA antibody.
To establish further that the RRP1B-affected RNA recruitment in the RdRp complex was indeed the capped RNA, RRP1B knockdown 293T cells were cotransfected with HA-tagged PB1, PB2, and PA. The lysates were incubated with various RNA species-conjugated beads, and the captured proteins were immunoblotted with anti-HA antibody to detect RdRp. Four kinds of RNA, including capped and uncapped nonspecific RNA, 5′ termini of vRNA, and poly(A), were used. The uncapped RNA was prepared by in vitro transcription of the pGEM plasmid and then capped with 7-methylguanylate structure to produce capped RNA. As shown in Fig. 6B, a significant amount of RdRp was captured by capped RNA (lanes 4 to 6), and the amounts of captured RdRp recovered from the lysates of RRP1B knockdown cells were less than that from the control cells (lanes 4 to 6). In contrast, the amounts of RdRp bound to the uncapped RNA were very small and were not affected by RRP1B knockdown (lanes 7 to 9), strengthening the evidence that RRP1B is critical for the binding of RdRp to capped RNA. We further demonstrated that 5′-vRNA, which presumably bound to PB1 (9), binds to RdRp as a whole, whereas poly(A) did not bind, indicating some RNA specificity in the RNA-RdRp-RRP1B interactions (Fig. 6C). However, the amount of RdRp captured by 5′-vRNA was not affected by the depletion of RRP1B (Fig. 6C, lanes 4 to 6). These results likely suggest that RRP1B primarily assists RdRp in binding to capped mRNA.
RRP1B influences RdRp binding to mRNA in vivo.
Since the above-mentioned evidence implied that RRP1B is involved in the binding of the RdRp complex to capped RNA in vitro, we next examined whether RRP1B knockdown could affect the binding of RdRp to cellular mRNA in vivo by performing an RIP assay. RRP1B knockdown cells were cotransfected with plasmids expressing HA-tagged RdRp, NP protein, and NP-specific vRNA and then immunoprecipitated using HA agarose. As shown in Fig. 7A, the same amounts of RdRp were immunoprecipitated from the lysates of the shLacZ control and RRP1B knockdown cells, suggesting that RRP1B does not alter the formation of the RdRp protein complex. The immunoprecipitated complex was then examined for the presence of cellular capped mRNA by using reverse transcription with oligo(dT) primer and qPCR with gene-specific primers. Since it has been demonstrated that the majority of the cap-snatching host primers originate from the most abundant cellular genes (23), we chose two housekeeping genes, the GAPDH and β-actin genes, to monitor the amount of mRNA coimmunoprecipitated with RdRp. Compared to the control cells, silencing RRP1B did not affect the expression levels of GAPDH and β-actin RNAs (Fig. 7B and C, left). However, the amounts of GAPDH and actin mRNAs coimmunoprecipitated with RdRp from the RRP1B knockdown cells were smaller than those from the control cells (Fig. 7B and C, right), suggesting the importance of RRP1B in RdRp binding to mRNA. Recently, small nuclear RNAs and small nucleolar RNAs have been shown to be abundant capped leaders for influenza virus transcription (24). We thus assessed whether RRP1B affects RdRp binding to U2 snRNA. We found that knockdown of RRP1B slightly reduced RdRp binding to U2 snRNA (Fig. 7D), suggesting that the capped U2 snRNA may be recruited into the viral RdRp through another cellular factor or directly by interacting with viral RdRp.
FIG 7.

Silencing RRP1B reduces RdRp binding to mRNA in vivo. HEK293T knockdown cells were cotransfected with plasmids expressing HA-tagged PB1, HA-tagged PB2, HA-tagged PA, and pFlu-NP, which produces NP protein and NP vRNA. At 24 h posttransfection, the cells were harvested for RIP assays as described in Materials and Methods. The cell lysates were subjected to immunoprecipitation using anti-HA agarose to isolate the RdRp-mRNA complex. (A) The cell lysates and immunoprecipitated proteins were resolved on SDS-PAGE and immunoblotted with anti-HA antibody. (B, C, and D) The levels of input (left) and coprecipitated (right) cellular mRNAs of GAPDH (B), β-actin (C), and U2 snRNA (D) were determined by qRT-PCR. The values represent the means ± SD of the results of three independent experiments. *, P < 0.05, and **, P < 0.01 compared with controls.
DISCUSSION
It has been widely recognized that influenza virus transcription, replication, splicing, and nuclear export of viral messenger RNPs (mRNPs) depend on the host cell nuclear environment. Here, we have identified RRP1B as such a molecule. RRP1B has been demonstrated to regulate the expression of alternative mRNA isoforms through interactions with a splicing factor, SRSF1 (16). Therefore, we tried to address whether RRP1B plays a role in splicing of viral RNAs and found that silencing RRP1B did not change the ratio of spliced M2 to unspliced M1 (data not shown), implying that RRP1B may not participate in splicing of viral RNA. Moreover, RRP1B has the property of binding chromatin by interaction with many nucleosome-binding factors (15), and the RRP1B-binding sites have recently been found to be related to transcriptional repression, which is associated with the TRIM28/HP1α heterochromatin complex (25). In this study, we identified RRP1B as a critical factor in regulating IAV cap-dependent transcription. We proposed that IAV infection causes RRP1B to dissociate from the heterochromatin complex and instead form a viral RNA transcription complex. Indeed, upon IAV infection, RRP1B redistributed in the nuclei (Fig. 4B) and formed a coimmunoprecipitable complex with RdRp (Fig. 5B), supporting this hypothesis. It is worth mentioning that although RRP1B interacts with PB2 and PB1, but not PA (Fig. 5A), in IAV-infected cells, PA was coimmunoprecipitated by RRP1B, since PA forms a complex with PB1 and PB2 (Fig. 5B).
Several host factors have been identified as crucial for influenza virus RdRp activity using functional assays or genome-wide RNAi screening (11, 26). However, only a few of these host factors have been shown to participate in viral cap-dependent transcription. The viral RdRp has been found to interact with the C-terminal domain (CTD) of the largest subunit of cellular RNA polymerase II (Pol II), and viral transcription has been demonstrated to depend on cellular Pol II (27, 28). Furthermore, cyclin T1/CDK9 is proposed to recruit vRNP to hyperphosphorylated Pol II for cap snatching and to facilitate viral mRNA transcription (29). Additionally, splicing factor proline-glutamine rich (SFPQ/PSF) has been found to increase the efficiency of viral mRNA polyadenylation (30). In this study, we identified RRP1B as another important host factor for influenza virus transcription. Although we presently cannot rule out the possibility that RRP1B may also affect viral RNA replication, we clearly showed that RRP1B participates in viral transcription by mediating the capped-mRNA-binding ability of viral RdRp (Fig. 6). Since RRP1B facilitated RdRp binding to mRNA (Fig. 6 and 7), we hypothesized that RRP1B might bind to mRNA to serve as a carrier for delivering capped mRNA to the RdRp complex. Therefore, we examined the mRNA-binding ability of RRP1B using an RNA-protein pulldown assay and found that RRP1B alone, without RdRp, could not bind to capped mRNA or uncapped mRNA (data not shown), suggesting that RRP1B facilitates RdRp binding to capped mRNA indirectly by promoting RNA-PB1 or RNA-PB2 binding.
It remains unclear how the influenza virus RdRp is committed to either transcription or replication on the same vRNA template. Various models of the switch of transcription and replication in influenza A virus infections have been proposed. Some mechanisms are assumed to be controlled by viral factors, whereas others are related to host factors. Regarding viral factors, NP, which is involved in encapsulation of the vRNA, was identified as a key player because of its RNA binding, which might modify the panhandle structure for the switch (31). Phosphorylation of NP has been investigated to regulate the polymerization state of NP, and ubiquitination of NP has been demonstrated to mediate the RNA-binding ability of NP (19, 32). We previously reported that ubiquitination of the NP protein supports IAV RNA replication, but not transcription (19), indicating the two processes are clearly separable. Moreover, the interaction between NP and the polymerase complex was also proposed to potentiate unprimed RNA replication via inhibiting cap-snatching activity (33). Furthermore, expression of the viral NS2/NEP protein affected viral RNA levels by reducing transcription and increasing replication, suggesting NS2/NEP is associated with the regulation of viral replication and transcription (34). Also, influenza virus-generated small RNAs were reported to regulate switching from transcription to replication through interactions with the viral polymerase machinery (35). Host factors, such as UAP56 (36) and Tat-SF1 (37), can bind NP and facilitate NP-RNA interaction; thus, both potentially mediate the switch. In addition, the availability of capped RNA primers and ribonucleoside triphosphates (rNTPs) are also important for the switch (38, 39). Given that RRP1B could associate with not only RdRp, but also NP (Fig. 5A), and mediate RdRp binding to capped mRNA (Fig. 6), we speculate that RRP1B may have the potential to participate in the switch of influenza virus transcription and replication. It will be interesting to investigate this possibility.
In conclusion, we propose that upon influenza virus infection, RRP1B will be redistributed in the nucleus to access viral RdRp. The interaction of RRP1B may cause a conformational change of PB1 or PB2 or stabilize the RdRp complex, thereby facilitating RdRp binding to capped mRNA and eventually enhancing viral transcription. It will be of interest to understand how RRP1B mediates RdRp binding to capped mRNA.
ACKNOWLEDGMENTS
This study was supported by grant MOST 103-2320-B-039-013-MY2 from the Ministry of Science and Technology, Taiwan.
We thank the National RNAi Core Facility, Academia Sinica, Taiwan, for providing the lentivector and lentiviral shRNAs and technical support. We also thank Che Ma for providing the influenza A/New Caledonia/20/1999 (NC99) and A/Wisconsin/67/2005 (W10) viruses and Muzzammil Shittu for editing the manuscript.
REFERENCES
- 1.Bouvier NM, Palese P. 2008. The biology of influenza viruses. Vaccine 26(Suppl 4):D49–D53. doi: 10.1016/j.vaccine.2008.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tong S, Zhu X, Li Y, Shi M, Zhang J, Bourgeois M, Yang H, Chen X, Recuenco S, Gomez J, Chen LM, Johnson A, Tao Y, Dreyfus C, Yu W, McBride R, Carney PJ, Gilbert AT, Chang J, Guo Z, Davis CT, Paulson JC, Stevens J, Rupprecht CE, Holmes EC, Wilson IA, Donis RO. 2013. New World bats harbor diverse influenza A viruses. PLoS Pathog 9:e1003657. doi: 10.1371/journal.ppat.1003657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lakadamyali M, Rust MJ, Zhuang X. 2004. Endocytosis of influenza viruses. Microbes Infect 6:929–936. doi: 10.1016/j.micinf.2004.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fodor E. 2013. The RNA polymerase of influenza A virus: mechanisms of viral transcription and replication. Acta Virol 57:113–122. doi: 10.4149/av_2013_02_113. [DOI] [PubMed] [Google Scholar]
- 5.Ulmanen I, Broni BA, Krug RM. 1981. Role of two of the influenza virus core P proteins in recognizing cap 1 structures (m7GpppNm) on RNAs and in initiating viral RNA transcription. Proc Natl Acad Sci U S A 78:7355–7359. doi: 10.1073/pnas.78.12.7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blaas D, Patzelt E, Kuechler E. 1982. Identification of the cap binding protein of influenza virus. Nucleic Acids Res 10:4803–4812. doi: 10.1093/nar/10.15.4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dias A, Bouvier D, Crepin T, McCarthy AA, Hart DJ, Baudin F, Cusack S, Ruigrok RW. 2009. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 458:914–918. doi: 10.1038/nature07745. [DOI] [PubMed] [Google Scholar]
- 8.Braam J, Ulmanen I, Krug RM. 1983. Molecular model of a eucaryotic transcription complex: functions and movements of influenza P proteins during capped RNA-primed transcription. Cell 34:609–618. [DOI] [PubMed] [Google Scholar]
- 9.Li ML, Ramirez BC, Krug RM. 1998. RNA-dependent activation of primer RNA production by influenza virus polymerase: different regions of the same protein subunit constitute the two required RNA-binding sites. EMBO J 17:5844–5852. doi: 10.1093/emboj/17.19.5844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shapiro GI, Krug RM. 1988. Influenza virus RNA replication in vitro: synthesis of viral template RNAs and virion RNAs in the absence of an added primer. J Virol 62:2285–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watanabe T, Watanabe S, Kawaoka Y. 2010. Cellular networks involved in the influenza virus life cycle. Cell Host Microbe 7:427–439. doi: 10.1016/j.chom.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Su WC, Chen YC, Tseng CH, Hsu PW, Tung KF, Jeng KS, Lai MM. 2013. Pooled RNAi screen identifies ubiquitin ligase Itch as crucial for influenza A virus release from the endosome during virus entry. Proc Natl Acad Sci U S A 110:17516–17521. doi: 10.1073/pnas.1312374110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engelhardt OG, Fodor E. 2006. Functional association between viral and cellular transcription during influenza virus infection. Rev Med Virol 16:329–345. doi: 10.1002/rmv.512. [DOI] [PubMed] [Google Scholar]
- 14.Crawford NP, Qian X, Ziogas A, Papageorge AG, Boersma BJ, Walker RC, Lukes L, Rowe WL, Zhang J, Ambs S, Lowy DR, Anton-Culver H, Hunter KW. 2007. Rrp1b, a new candidate susceptibility gene for breast cancer progression and metastasis. PLoS Genet 3:e214. doi: 10.1371/journal.pgen.0030214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crawford NP, Yang H, Mattaini KR, Hunter KW. 2009. The metastasis efficiency modifier ribosomal RNA processing 1 homolog B (RRP1B) is a chromatin-associated factor. J Biol Chem 284:28660–28673. doi: 10.1074/jbc.M109.023457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee M, Dworkin AM, Gildea D, Trivedi NS, NISC Comparative Sequencing Program, Moorhead GB, Crawford NP. 2014. RRP1B is a metastasis modifier that regulates the expression of alternative mRNA isoforms through interactions with SRSF1. Oncogene 33:1818–1827. doi: 10.1038/onc.2013.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chamousset D, De Wever V, Moorhead GB, Chen Y, Boisvert FM, Lamond AI, Trinkle-Mulcahy L. 2010. RRP1B targets PP1 to mammalian cell nucleoli and is associated with Pre-60S ribosomal subunits. Mol Biol Cell 21:4212–4226. doi: 10.1091/mbc.E10-04-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoffmann E, Neumann G, Kawaoka Y, Hobom G, Webster RG. 2000. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc Natl Acad Sci U S A 97:6108–6113. doi: 10.1073/pnas.100133697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liao TL, Wu CY, Su WC, Jeng KS, Lai MM. 2010. Ubiquitination and deubiquitination of NP protein regulates influenza A virus RNA replication. EMBO J 29:3879–3890. doi: 10.1038/emboj.2010.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Z, Watanabe T, Hatta M, Watanabe S, Nanbo A, Ozawa M, Kakugawa S, Shimojima M, Yamada S, Neumann G, Kawaoka Y. 2009. Mutational analysis of conserved amino acids in the influenza A virus nucleoprotein. J Virol 83:4153–4162. doi: 10.1128/JVI.02642-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barrett T, Wolstenholme AJ, Mahy BW. 1979. Transcription and replication of influenza virus RNA. Virology 98:211–225. doi: 10.1016/0042-6822(79)90539-7. [DOI] [PubMed] [Google Scholar]
- 22.Ohtsu Y, Honda Y, Sakata Y, Kato H, Toyoda T. 2002. Fine mapping of the subunit binding sites of influenza virus RNA polymerase. Microbiol Immunol 46:167–175. doi: 10.1111/j.1348-0421.2002.tb02682.x. [DOI] [PubMed] [Google Scholar]
- 23.Sikora D, Rocheleau L, Brown EG, Pelchat M. 2014. Deep sequencing reveals the eight facets of the influenza A/HongKong/1/1968 (H3N2) virus cap-snatching process. Sci Rep 4:6181. doi: 10.1038/srep06181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koppstein D, Ashour J, Bartel DP. 2015. Sequencing the cap-snatching repertoire of H1N1 influenza provides insight into the mechanism of viral transcription initiation. Nucleic Acids Res 43:5052–5064. doi: 10.1093/nar/gkv333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee M, Dworkin AM, Lichtenberg J, Patel SJ, Trivedi NS, Gildea D, Bodine DM, Crawford NP. 2014. Metastasis-associated protein ribosomal RNA processing 1 homolog B (RRP1B) modulates metastasis through regulation of histone methylation. Mol Cancer Res 12:1818–1828. doi: 10.1158/1541-7786.MCR-14-0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagata K, Kawaguchi A, Naito T. 2008. Host factors for replication and transcription of the influenza virus genome. Rev Med Virol 18:247–260. doi: 10.1002/rmv.575. [DOI] [PubMed] [Google Scholar]
- 27.Engelhardt OG, Smith M, Fodor E. 2005. Association of the influenza A virus RNA-dependent RNA polymerase with cellular RNA polymerase II. J Virol 79:5812–5818. doi: 10.1128/JVI.79.9.5812-5818.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chan AY, Vreede FT, Smith M, Engelhardt OG, Fodor E. 2006. Influenza virus inhibits RNA polymerase II elongation. Virology 351:210–217. doi: 10.1016/j.virol.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 29.Zhang J, Li G, Ye X. 2010. Cyclin T1/CDK9 interacts with influenza A virus polymerase and facilitates its association with cellular RNA polymerase II. J Virol 84:12619–12627. doi: 10.1128/JVI.01696-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Landeras-Bueno S, Jorba N, Perez-Cidoncha M, Ortin J. 2011. The splicing factor proline-glutamine rich (SFPQ/PSF) is involved in influenza virus transcription. PLoS Pathog 7:e1002397. doi: 10.1371/journal.ppat.1002397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fodor E, Pritlove DC, Brownlee GG. 1994. The influenza virus panhandle is involved in the initiation of transcription. J Virol 68:4092–4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chenavas S, Estrozi LF, Slama-Schwok A, Delmas B, Di Primo C, Baudin F, Li X, Crepin T, Ruigrok RW. 2013. Monomeric nucleoprotein of influenza A virus. PLoS Pathog 9:e1003275. doi: 10.1371/journal.ppat.1003275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Newcomb LL, Kuo RL, Ye Q, Jiang Y, Tao YJ, Krug RM. 2009. Interaction of the influenza a virus nucleocapsid protein with the viral RNA polymerase potentiates unprimed viral RNA replication. J Virol 83:29–36. doi: 10.1128/JVI.02293-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robb NC, Smith M, Vreede FT, Fodor E. 2009. NS2/NEP protein regulates transcription and replication of the influenza virus RNA genome. J Gen Virol 90:1398–1407. doi: 10.1099/vir.0.009639-0. [DOI] [PubMed] [Google Scholar]
- 35.Perez JT, Varble A, Sachidanandam R, Zlatev I, Manoharan M, Garcia-Sastre A, ten Oever BR. 2010. Influenza A virus-generated small RNAs regulate the switch from transcription to replication. Proc Natl Acad Sci U S A 107:11525–11530. doi: 10.1073/pnas.1001984107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Momose F, Basler CF, O'Neill RE, Iwamatsu A, Palese P, Nagata K. 2001. Cellular splicing factor RAF-2p48/NPI-5/BAT1/UAP56 interacts with the influenza virus nucleoprotein and enhances viral RNA synthesis. J Virol 75:1899–1908. doi: 10.1128/JVI.75.4.1899-1908.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Naito T, Kiyasu Y, Sugiyama K, Kimura A, Nakano R, Matsukage A, Nagata K. 2007. An influenza virus replicon system in yeast identified Tat-SF1 as a stimulatory host factor for viral RNA synthesis. Proc Natl Acad Sci U S A 104:18235–18240. doi: 10.1073/pnas.0705856104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Resa-Infante P, Jorba N, Coloma R, Ortin J. 2011. The influenza virus RNA synthesis machine: advances in its structure and function. RNA Biol 8:207–215. doi: 10.4161/rna.8.2.14513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vreede FT, Gifford H, Brownlee GG. 2008. Role of initiating nucleoside triphosphate concentrations in the regulation of influenza virus replication and transcription. J Virol 82:6902–6910. doi: 10.1128/JVI.00627-08. [DOI] [PMC free article] [PubMed] [Google Scholar]