

Objective:
To evaluate whether once daily (q.d.) lopinavir/ritonavir is noninferior to twice daily (b.i.d.) dosing in children.
Design:
International, multicentre, phase II/III, randomized, open-label, noninferiority trial (KONCERT/PENTA18/ANRS150).
Setting:
Clinical centres participating in the PENTA, HIV-NAT and PHPT networks.
Participants:
Children/adolescents with HIV-1 RNA viral load less than 50 copies/ml for at least 24 weeks on lopinavir/ritonavir-containing antiretroviral therapy.
Intervention:
Children were randomized to continue lopinavir/ritonavir b.i.d. or change to q.d.
Main outcome measure:
Confirmed viral load ≥50 copies/ml by 48 weeks (12% noninferiority margin).
Results:
One hundred seventy-three children were randomized in the KONCERT trial (86 q.d., 87 b.i.d.); 46% men, median (IQR) age 11 (9–14) years, CD4% 33 (27–38)%. By week 48, 97 and 98% of time was spent on q.d. and b.i.d., respectively (one q.d. child lost at week 4). Twelve q.d. vs. seven b.i.d. children had confirmed viral load ≥50 copies/ml within 48 weeks; estimated difference in percentage with viral load rebound 6% [90% CI (–2, 14)]. Numbers of children with grade 3/4 adverse events (11 vs. 7) or major resistance mutations (3 vs. 2) were similar, q.d. vs. b.i.d. (both P > 0.3). Among 26 children in an intrasubject lopinavir/ritonavir pharmacokinetic substudy, lower daily exposure (AUC0–24 161 h.mg/l vs. 224 h.mg/l) and lower Clast (1.03 mg/l vs. 5.69 mg/l) were observed with q.d. vs. b.i.d. dosing.
Conclusion:
Noninferiority for viral load suppression on q.d. vs. b.i.d. lopinavir/ritonavir was not demonstrated. Although results, therefore, do not support routine use of q.d. lopinavir/ritonavir, lack of safety concerns or resistance suggest that q.d. dosing remains an option in selected, adherent children, with close viral load monitoring.
Keywords: children, HIV-1, lopinavir/ritonavir, once daily, randomized, trial
Introduction
Antiretroviral drugs have changed HIV-1 infection from a life-threatening disease to a chronic infection. However, adherence to therapy remains a key determinant of disease outcome. For perinatally HIV-infected children, who face a lifetime on treatment, maintaining long-term adherence is often a challenge. Simplification of treatment, including decreasing the frequency of dosing, is likely to increase convenience and enhance adherence to antiretroviral therapy (ART) [1]. Although several q.d. regimens have been shown to have noninferior efficacy and safety in adults [2], resulting in Food and Drug Administration (FDA) and European Medicines Agency (EMA) approval and widespread use in clinical practice, fewer antiretroviral drugs are licensed to be taken q.d. by children.
Protease inhibitors are potential candidates for q.d. dosing. They have a high genetic barrier to development of resistance [3] and when coadministered with ritonavir, resulting in increased absorption and/or prolonged terminal elimination half-life, have increasing potential for decreased dosing frequency. The coformulation of ritonavir-boosted lopinavir (lopinavir/r) in one tablet (also available as a smaller paediatric formulation) also enhances convenience of dosing. Various studies have supported the licensing of q.d. dosing of lopinavir/r for HIV-infected adults [2,4–7]. However, based on the currently available evidence in children, paediatric treatment guidelines recommend lopinavir/r to be taken twice daily (b.i.d.) [8,9]. Small studies using q.d. lopinavir/r oral solution or soft gel capsules in children showed high interpatient variability in lopinavir pharmacokinetic parameters and low trough levels [10,11]. Reduced variability in lopinavir pharmacokinetic in adults and children has been observed after administration of the tablet formulation, suggesting that this formulation could be more appropriate for q.d. dosing [12,13]. Here we report the results of KONCERT (PENTA18/ANRS150), the first randomized controlled trial evaluating the safety, efficacy and pharmacokinetic of lopinavir/r tablets dosed q.d. vs. b.i.d. following FDA body-weight band dosing guidelines in virologically suppressed ART-experienced children and adolescents.
Methods
Study design and participants
KONCERT was an open-label, multicentre, randomized trial (ISRCTN 02452400, EudraCT 2009-013648-35) in HIV-infected children aged below 18 years who had a stable CD4+ cell count on combination ART containing b.i.d. lopinavir/r and had been virologically suppressed (viral load <50 copies/ml) for at least 24 weeks (single viral load <400 copies/ml allowed). In addition, eligibility required that children had viral load less than 50 copies/ml at screening, weighed ≥15 kg and were able to swallow tablets. Kaletra tablets were used throughout. If required, lopinavir/r dose was adjusted at screening in line with the US FDA dosing guidelines based on body-weight band [total daily dose: 400/100 mg lopinavir/r (15 to ≤25 kg), 600/150 mg (25 to ≤35 kg) or 800/200 mg (>35 kg)] [14]. Children were randomized 1 : 1 to continue taking lopinavir/r b.i.d. or to take their total daily lopinavir/r in a single dose. Parents/guardians and adolescents provided written consent, younger children gave assent according to their age and knowledge of HIV status. The study received approval from ethics committees and regulatory bodies in each participating country and clinical site.
Randomization was stratified by weight band (as above) and participation in the pharmacokinetic substudy. The computer-generated sequentially numbered randomization list (with variable block sizes) was preprepared by the trial statistician and securely incorporated within the database at the Trials Unit. Randomization was undertaken via a web service accessed by the clinician or Trials Unit, who could access the next allocation but not the whole list.
Outcome measures
The primary endpoint was a viral load at least 50 copies/ml (confirmed within 4 weeks) within the first 48 weeks of follow-up. Primary endpoints for the pharmacokinetic substudies were pharmacokinetic parameters of lopinavir/r [area under the curve (AUC), Cmax, Clast] [1], comparing b.i.d. (week 0) to historical pharmacokinetic data, and [2] comparing q.d. (week 4) to b.i.d. (week 0) in the same children. Analysis of endpoint [1] has previously been described [15]. Secondary outcomes included the following: viral load at least 400 copies/ml (confirmed) within 48 weeks; number of major HIV-1 RNA mutations in those with viral rebound; change in CD4+ cell count/percentage from baseline to 48 weeks; adherence to, acceptability of, and changes made to the ART regimen; ART-related grades 3 and 4 clinical or laboratory adverse events [16,17].
Data collection and follow-up procedures
Follow-up visits were scheduled at weeks 4, 8 and 12, then 12 weekly until the last child reached week 48 (Fig. 1). Viral load was measured at each study visit; children with viral load at least 50 copies/ml returned within 4 weeks for retest of viral load. Assessment of adherence to treatment and a resistance test were requested when children had a confirmed viral load at least 50 copies/ml. T-cell lymphocyte subsets were performed at all visits; biochemistry and haematology were performed 12-weekly; blood lipids were measured at weeks 0, 24 and 48; adherence questionnaires were given to carers and children at weeks 0, 4, 12, 24 and 48; acceptability questionnaires were completed at baseline and if children switched from q.d. to b.i.d. dosing. At each study visit, a plasma sample was stored for subsequent assessment of population lopinavir/r pharmacokinetic.
Fig. 1.
Trial profile.
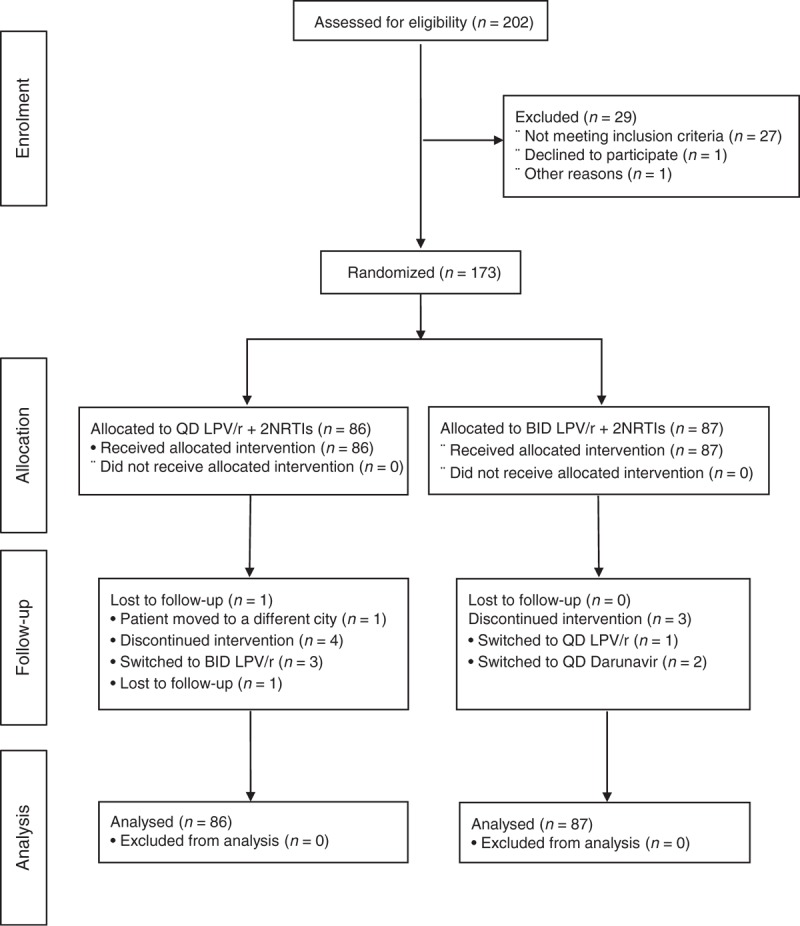
LPV, lopinavir; NRTI, nucleoside reverse-transcriptase inhibitor.
Pharmacokinetic substudy
Children who consented were enrolled in a pharmacokinetic substudy, until a minimum of 16 children in each stratification weight band had evaluable pharmacokinetic data. Children with nonevaluable pharmacokinetic results were followed within the main study, but excluded from the pharmacokinetic analysis. Lopinavir pharmacokinetics were determined at week 0 in both arms, and at week 4 if randomized to q.d. dosing. Prior to the day of pharmacokinetic assessment, children took the paediatric lopinavir/r tablet (100/25 mg) formulation for at least seven days, following the FDA-recommended weight band-based dosing. On the pharmacokinetic assessment day, 2 ml of blood was taken before observed intake of lopinavir/r in the morning (t = 0) and at 2, 4, 6, 8 and 12 (week 0, b.i.d.) or 24 h (week 4, q.d.) after the dose. Plasma concentrations were determined using a validated ultrahigh performance liquid chromatography assay with UV detection derived from a previously published assay [18]. Lopinavir pharmacokinetic parameters were determined using noncompartmental analysis (WinNonlin/Phoenix version 6.3; Pharsight Corporation, Mountain View, California, USA): AUC0–24 [area under the plasma concentration–time curve calculated (linear up-log down method) over a dosing interval from time 0 to 24 h after dosing], Cmax (maximum observed plasma concentration), Tmax (time of maximum observed plasma concentration), Clast (last observed drug concentration) and clearance (CL/F). The intensive pharmacokinetic analyses were performed at the Department of Pharmacy, Radboud University Medical Center, Nijmegen, the Netherlands.
Lopinavir concentrations were also determined on available stored plasma samples at the screening visit and at weeks 0, 4, 8, 12, 24, 36 and 48 on all children. This was done to investigate the effect of having lopinavir plasma concentration below the lower limit of quantification (LLOQ = 0.10 mg/l) at any visit on virological rebound. Pharmacokinetic analyses for these stored samples were performed at Radboud University Medical Center, except for samples in Thailand which were performed at the PHPT-AMS laboratory, Chiang Mai University, Thailand. Both laboratories participate in an international interlaboratory quality control programme for therapeutic drug monitoring of antiretroviral drugs [19].
Statistical analyses
A target enrolment of 160 children (80 in each arm) provided at least 80% power to exclude a noninferiority margin of 12% for the difference between the two arms in the proportion of children reaching the primary endpoint, assuming a 10% virological rebound rate and one-sided α = 0.05. An Independent Data Monitoring Committee reviewed interim data for safety and efficacy three times during the study.
All comparisons between randomized arms (q.d. vs. b.i.d.) were intention-to-treat, with follow-up censored at week 52 or last follow-up date (if before the week 48 visit). The proportion of children experiencing virological rebound by week 48 in each arm was estimated using the Kaplan–Meier method, with 90% confidence intervals (CIs) for the difference in proportions calculated using bootstrap standard errors [20]. Two prespecified sensitivity analyses of the primary outcome were completed: adjusting for baseline stratification factors, and censoring follow-up at the time of lopinavir/r treatment modification (change in dose, >7-day interruption or permanent discontinuation; a ‘per-protocol’ analysis). A post-hoc analysis adjusting for chance imbalance between arms in viral load and CD4% at baseline was also performed.
Change in CD4% and other continuous laboratory outcomes from baseline to 48 weeks were analyzed using normal regression, adjusting for the baseline measurement and stratification factors. Major resistance mutations known to confer resistance to antiretroviral drugs not seen in any pretrial resistance tests were summarized by drug class. Categorical variables were compared using Fisher's exact tests; rates were estimated using Poisson regression. All P values were two sided and all statistical calculations were performed using STATA (Stata Statistical Software, Release 13; StataCorp LP, College Station, Texas, USA).
All paired evaluable pharmacokinetic assessments [on b.i.d. (week 0) and q.d. (week 4)] in children randomized to q.d. were included. Within subject ratios of AUC0–24, clearance (Cl/F/kg), Cmax and Clast for q.d. vs. b.i.d. dosing were calculated. AUC0–24 for b.i.d. dosing was calculated as 2∗AUC0–12. An overall geometric mean ratio (GMR) for each pharmacokinetic parameter was calculated after log-transformation of the within-subject ratios; 90% CIs were calculated (using the t-distribution) using the bioequivalence crossover design tool approach within the Phoenix WinNonlin software package (with fixed effects in the model specification). A GMR with a 90% CI including 1.0 and falling entirely within 0.80–1.25 was considered as bioequivalence for AUC0–24 and Cmax. Relative risk ratios were calculated comparing the likelihood of virological rebound for children with at least one sample with lopinavir concentration levels below LLOQ to those children with all samples ≥ LLOQ.
Results
Baseline characteristics
Between August 2010 and August 2012, 173 children were randomized (86 allocated to q.d., 87 to b.i.d.) (Fig. 1); 80 children from Europe, 59 from Thailand and 34 from South America; participants were from 49 clinical centres in 12 countries. Fifty-three took part in the pharmacokinetic substudy, 27 randomized to the q.d. arm; 46, 50 and 77 children were in the 15 to 25kg, >25 to 35kg, >35kg weight bands, respectively.
Baseline demographics were similar in the two arms (Table 1); median (IQR) age was 11.0 (8.7, 14.3) years and 94 (54%) were female. More children in the q.d. arm had advanced HIV disease, lower CD4% and a viral load at least 50 copies/ml at baseline (Table 1). Pretrial ART exposure was comparable between arms; 35 (20%) children were on their first-line regimen at baseline, and half had been exposed to three different antiretroviral drug classes. The children were on a variety of NRTI backbones at baseline (44% zidovudine + lamivudine or emtricitabine, 20% abacavir + lamivudine or emtricitabine, 16% tenofovir + any other NRTI, 20% other); 29% of backbone NRTIs were taken as q.d. dosing (28% q.d. arm, 30% b.i.d. arm); this proportion increased over the time of the trial.
Table 1.
Baseline characteristics.
| Once daily | Twice daily | Total | |
| Children randomized: n | 86 | 87 | 173 |
| Men: n (%) | 41 (48) | 38 (44) | 79 (46) |
| Age (years): median (IQR) [range] | 10.8 (8.7, 14.2) [4.3, 17.6] | 11.2 (9.0, 14.5) [3.8, 17.7] | 11.0 (8.7, 14.3) [3.8, 17.7] |
| Ethnic origin: n (%) | |||
| White | 27 (31) | 17 (20) | 44 (25) |
| Black: African or other | 17 (20) | 29 (33) | 46 (27) |
| Mixed black/white | 5 (6) | 6 (7) | 11 (6) |
| Asian/Thai | 31 (36) | 30 (34) | 61 (35) |
| Other | 6 (7) | 5 (6) | 11 (6) |
| Vertically infected: n (%) | 86 (100) | 84 (97) | 170 (98) |
| CDC stage: n (%) | |||
| N or A | 28 (33) | 39 (45) | 67 (38) |
| B or C | 58 (68) | 48 (55) | 106 (61) |
| Viral load (HIV-1 RNA) ≥50 copies/ml at randomizationa: | |||
| n (%) | 12 (14) | 4 (5) | 16 (9) |
| Median [range] | 120 [51, 91 201] | 135 [57, 270] | 120 [51, 91 201] |
| CD4%: mean (SD) | 32.0 (6.5) | 33.9 (8.6) | 32.9 (7.7) |
| Weight (kg): median (IQR) | 33.3 (24.6, 42.0) | 32.2 (23.9, 43.8) | 33.1 (24.6, 42.6) |
| [Range] | [15.0, 72.5] | [15.6, 68.9] | [15.0, 72.5] |
| Baseline ART first regime: n (%) | 18 (21) | 17 (20) | 35 (20) |
| Exposed to three classes of ART: n (%) | 41 (48) | 46 (53) | 87 (50) |
aAll <50 copies/ml at screening.
Follow-up and antiretroviral therapy received
One q.d. child withdrew consent at week 4; all other children completed 48 weeks follow-up and are included in all analyses. In total, 98 and 97% of follow-up time was spent on q.d. and b.i.d. dosing of lopinavir/r in the q.d. and b.i.d. arms, respectively. Twenty-nine (17%) children made changes to their ART regimen in the first 48 weeks of follow-up [20 (23%) q.d., 9 (10%) b.i.d.]. In the q.d. arm, two children switched back to b.i.d. lopinavir/r dosing (at week 1 and 39), 17 children changed their NRTI backbone (66% to q.d. regimens), and one child did both at week 8. In the b.i.d. arm, one child switched to q.d. lopinavir/r dosing at week 38 and eight children changed their NRTI backbone.
Primary outcome
Nineteen children (12 q.d., seven b.i.d.) experienced confirmed viral rebound at least 50 copies/ml during 48 weeks of follow-up; all but one rebound (q.d.) was considered by the treating clinician to be adherence related. The estimated percentage of children with viral rebound by 48 weeks was 14% [95% CI (8, 24%)] in the q.d. arm vs. 8% [(95% CI (4, 16%)] in the b.i.d. arm, an estimated difference between arms of 6% [(90% CI (-2, 14%), bootstrap P = 0.19] (Fig. 2). The upper 90% confidence limit of 14% was greater than the predefined noninferiority margin of 12%.
Fig. 2.
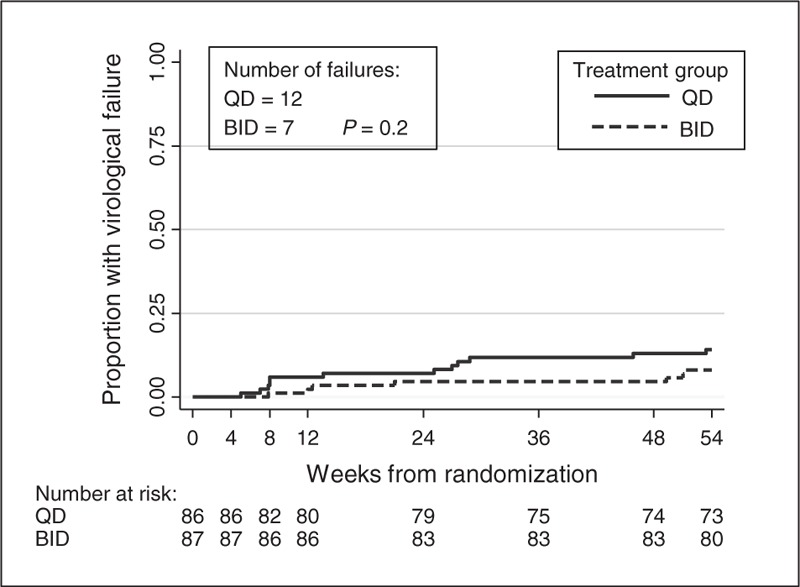
Time to virological failure.
Results were similar after adjustment for stratification factors – estimated difference between arms of 6% [90% CI (–2%, 14%), bootstrap P = 0.20] – and for per-protocol analyses wherein follow-up for eight children was censored as a result of treatment modification – estimated difference between arms of 5% [90% CI (–3%, 13%), bootstrap P = 0.27]. A post-hoc analysis adjusting for the chance imbalance between arms in viral rebound at baseline, reduced the estimated difference in proportion rebounding to 4% [90% CI (–4, 11%), bootstrap P = 0.39], bringing the upper 90% confidence limit just within the noninferiority margin.
Fifteen children [nine (75%) q.d., six (86%) b.i.d.] remained on the same dosing regimen of lopinavir/r after rebound, the majority of whom went on to resuppress [seven (78%) q.d., four (67%) b.i.d.]. Two children (q.d.) returned to b.i.d. dosing (one of whom resuppressed during follow-up) and two (one q.d., one b.i.d.) discontinued lopinavir/r after rebound (both resuppressed).
Secondary outcomes
Viral rebound defined as at least 400 copies/ml was observed in 11 children (eight q.d., three b.i.d.); the estimated difference between arms in the probability of rebounding by 48 weeks was 6% [90% CI (0, 12%), P = 0.10].
Genotypic resistance tests were available in 18 (12 q.d., 6 b.i.d.) children with rebound within 48 weeks; major new resistance-associated mutations were detected in five (three q.d., two b.i.d.; one b.i.d. did not resupress <50 copies/ml during follow-up). Major protease inhibitor mutations were detected in none of the children on q.d. vs. two children on b.i.d. lopinavir/r (L90M, M46I + V82A). The M184V mutation was detected in one child from each arm; three children (two q.d., one b.i.d.) had at least one thymidine-associated mutation.
Mean changes in CD4% from baseline to week 48 were similar in both arms: 0.4% for the q.d. arm and 0.1% for the b.i.d. arm [difference 0.3%, 95% CI (–1.0, 1.7%), P = 0.61]. Changes in biochemistry, haematology and lipid measurements were also minimal and comparable (data not shown).
There were no new CDC stage C events or deaths reported during the trial. Three new stage B events were reported (two q.d.: pneumonia and herpes zoster; one b.i.d.: cholecystitis).
There were no significant differences between the trial arms for any of the clinical safety endpoints (Table 2). Fifteen serious adverse events in 14 children occurred during the first 48 weeks of the trial [episodes (children): nine q.d. [8], six b.i.d. [6], Fisher's exact test: P = 0.6], none of which were fatal or life threatening. All reported serious adverse events were as a result of hospitalization; only one event, diarrhoea reported during the first week of the trial in a child taking q.d. lopinavir/r, was considered possibly related to lopinavir/r by the treating clinician. The incident risk ratio for q.d. relative to b.i.d. was 1.72 [95% CI (0.63, 4.66), GEE Poisson regression P = 0.29].
Table 2.
Summary of adverse events to week 48 assessment.
| Once-daily episodes (children) | Twice-daily episodes (children) | Total episodes (children) | P value* | |
| Total adverse events | 271 (73) | 232 (76) | 503 (149) | 0.7 |
| Grades 1 and 2 adverse events | 256 (70) | 222 (76) | 478 (146) | 0.3 |
| Grades 3 and 4 adverse events | 13 (11) | 9 (7) | 22 (18) | 0.3 |
| Gastrointestinal disorders | 2 (2) | 2 (1) | 4 (3) | 1.0 |
| Infections and infestations | 4 (4) | 3 (3) | 7 (7) | 0.7 |
| Laboratory investigations† | 5 (4) | 3 (2) | 8 (6) | 0.4 |
| Blood and lymphatic system disorders | 0 (0) | 1 (1) | 1 (1) | 1.0 |
| Hepatobiliary disorders | 1 (1) | 0 (0) | 1 (1) | 0.5 |
| Nervous system disorders | 1 (1) | 0 (0) | 1 (1) | 0.5 |
| Adverse events leading to treatment modification | 4 (2) | 1 (1) | 5 (3) | 0.6 |
| Serious adverse events | 9 (8) | 6 (6) | 15 (14) | 0.6 |
| Gastrointestinal disorders | 2 (2) | 1 (1) | 3 (3) | 0.6 |
| Infections and infestations | 5 (5) | 4 (4) | 9 (9) | 0.75 |
| Respiratory, thoracic and mediastinal disorders | 0 (0) | 1 (1) | 1 (1) | 1.0 |
| Hepatobiliary disorders | 1 (1) | 0 (0) | 1 (1) | 0.5 |
| Surgical and medical procedures | 1 (1) | 0 (0) | 1 (1) | 0.5 |
| SAE rate per 100 person-years (95% CI) | 9.5 (4.7, 19.0) | 6.7 (3.0, 14.9) | 8.0 (4.8, 13.6) | 0.6** |
SAE, serious adverse event.
*Fisher's exact test.
**Poisson regression.
†Abnormal laboratory values without reported associated clinical symptoms.
Twenty-two grade 3 or 4 clinical or laboratory adverse events in 18 children were reported: [episodes (children): 13 [11] q.d., 9 [7] b.i.d., Fisher's exact test: P = 0.3]. Three children experienced adverse events that led to treatment modification: two children on q.d. with nausea and vomiting changed back to b.i.d. dosing at weeks 1 and 8; one child on b.i.d. had neutropenia at week 4 and substituted abacavir for zidovudine.
Both children and carers reported a preference for q.d. dosage of lopinavir/r; 120 of 140 (86%) children and 128 of 144 (89%) carers completing the acceptability questionnaire at trial enrolment thought q.d. dosing would be easier than b.i.d. dosing. This preference persisted at the end of trial, when 50 of 68 (74%) of children and 45 of 64 (70%) of carers reported a preference. Combining responses to adherence questionnaires completed by children or carers at each trial visit during the first 48 weeks (89% completion rate), missing a dose within 3 days of the clinic visit was only reported on 20 occasions [14 (3.5%) q.d. vs. 6 (1.5%) b.i.d., GEE logistic regression: P = 0.2].
Pharmacokinetic analysis
Intraindividual, paired comparison of lopinavir twice-daily and once-daily dosing
Twenty-six out of 27 children randomized to the q.d. arm in the pharmacokinetic substudy had evaluable full pharmacokinetic at weeks 0 and 4. Table 3a and b shows child demographic data and pharmacokinetic parameters for lopinavir, respectively. Fifteen (58%) children on q.d. dosing at week 4 compared with all children on b.i.d. dosing at baseline had a Clast above 1.0 mg/l, a measurement associated with optimal virological response in b.i.d. regimens [9]. The GMR (90% CI), q.d. vs. b.i.d., of lopinavir AUC0–24 and lopinavir Cmax were calculated as 0.72 (0.62, 0.83) and 1.13 (1.00, 1.26), respectively. Neither falling within the 80–125% limits required for bioequivalence.
Table 3.
Within-children pharmacokinetic substudy in 26 children randomized to once-daily arm.
| (a) Baseline characteristics | ||||
| Weight band | ||||
| ≥15 to ≤25 kg | >25 to ≤35 kg | >35 kg | Total | |
| Children: n | 7 | 8 | 11 | 26 |
| Men: n (%) | 4 (57) | 5 (63) | 3 (27) | 12 (46) |
| Age (years): median (IQR) [range] | 7.1 (6.7, 8.7) [4.4, 8.9] | 10.6 (9.5, 15.0) [6.3, 16.0] | 14.3 (13.5, 15.4) [12.7, 16.8] | 12.8 (8.7, 14.7) [4.4, 16.8] |
| Weight (kg): median (IQR) [range] | 19.4 (19.0, 23.1) [15.0, 24.1] | 30.7 (29.8, 32.1) [26.4, 33.8] | 42.0 (38.5, 49.5) [36.0, 72.5] | 32.1 (24.1, 41.0) [15.0, 72.5] |
| BMI (kg/m2): median (IQR) [range] | 15.1 (14.4, 15.7) [11.5,15.8] | 15.7 (14.8, 18.0) [14.5, 19.4] | 17.7 (17.4, 20.6) [16.0, 27.6] | 16.5 (15.1, 18.6) [11.5, 27.6] |
| Vertically infected: n (%) | 7 (100) | 8 (100) | 11 (100) | 26 (100) |
| Ethnic origin: n (%) | ||||
| White | 0 (0) | 1 (13) | 1 (9) | 2 (8) |
| Black: African or other | 2 (29) | 3 (38) | 2 (18) | 7 (27) |
| Mixed black/white | 1 (14) | 1 (13) | 0 (0) | 2 (8) |
| Asian/Thai | 4 (57) | 3 (38) | 7 (64) | 14 (54) |
| Other | 0 (0) | 0 (0) | 1 (9) | 1 (4) |
| (b) Pharmacokinetic parameters for lopinavir on once-daily (week 4) and twice-daily (week 0) dosing | ||||
| n | Once-daily geometric mean (95% CI)c | Twice-daily geometric mean (95% CI)c | Once-daily/twice-daily geometric mean ratio (90% CI) | |
| AUC0–24 (h*mg/l)a | ||||
| Total: | 26 | 160.9 (138.4, 187.0) | 223.9 (194.8, 257.4) | 0.72 (0.62, 0.83) |
| Weight: | ||||
| ≥15 to ≤25 kg | 7 | 172.6 (121.3, 245.7) | 232.1 (153.3, 351.4) | |
| >25 to ≤35 kg | 8 | 159.3 (120.6, 210.5) | 256.8 (209.3, 315.2) | |
| >35 kg | 11 | 155.0 (116.8, 205.6) | 198.1 (159.8, 245.5) | |
| Cmax (mg/l) | ||||
| Total: | 26 | 14.0 (12.7, 15.6) | 12.5 (11.1, 14.0) | 1.13 (1.00, 1.26) |
| Weight: | ||||
| ≥15 to ≤25 kg | 7 | 15.5 (12.4, 19.4) | 13.5 (9.8, 18.7) | |
| >25 to ≤35 kg | 8 | 15.0 (12.2, 18.5) | 14.1 (11.4, 17.3) | |
| >35 kg | 11 | 12.5 (10.7, 14.7) | 10.9 (9.3, 12.6) | |
| Clast (mg/l) | ||||
| Total: | 26 | 1.03 (0.61, 1.75) | 5.69 (4.58, 7.07) | 0.18 (0.12, 0.27) |
| Weight: | ||||
| ≥15 to ≤25 kg | 7 | 0.91 (0.27, 3.07) | 4.92 (2.65, 9.16) | |
| >25 to ≤35 kg | 8 | 0.93 (0.38, 2.26) | 6.65 (5.22, 8.47) | |
| >35 kg | 11 | 1.20 (0.42, 3.44) | 5.57 (3.73, 8.32) | |
| Clearance (l/(h*kg))b | ||||
| Total: | 26 | 0.115 (0.099, 0.134) | 0.084 (0.074, 0.095) | 1.37 (1.19, 1.57) |
| Weight: | ||||
| ≥15 to ≤25 kg | 7 | 0.112 (0.076, 0.165) | 0.085 (0.062, 0.117) | |
| >25 to ≤35 kg | 8 | 0.120 (0.091, 0.158) | 0.076 (0.062, 0.094) | |
| >35 kg | 11 | 0.114 (0.086, 0.150) | 0.089 (0.071, 0.113) | |
| Tmax(h)c | ||||
| Total: | 26 | 4.0 (2.0, 8.0) | 3.5 (0.0, 12.0) | |
| Weight: | ||||
| ≥15 to ≤25 kg | 7 | 4.0 (2.0, 8.0) | 3.8 (0.0, 4.1) | |
| >25 to ≤35 kg | 8 | 4.0 (2.0, 6.0) | 2.8 (1.7, 4.0) | |
| >35 kg | 11 | 4.0 (2.0, 8.0) | 3.4 (1.7, 12.0) | |
aAUC0–24 for b.i.d. dosing = AUC0–12*2.
bClearance calculated as Cl/F/kg = dose(mg)/[AUC0–24(h*mg/l)*body weight (kg)].
cFor Tmax median values (minimum, maximum) are reported.
Routine measurement of lopinavir plasma concentrations
Most children (76 q.d., 74 b.i.d.) had eight samples available during the initial 48 weeks of follow-up for determination of lopinavir plasma concentration (19 children had seven samples, 1 had six, 2 had five, 1 had three). Overall, 28 (16.2%) children had at least one lopinavir concentration that was below the LLOQ of 0.10 mg/l: 21 (24.4%) q.d. vs. 7 (8.0%) b.i.d., Fisher's exact test P = 0.004. A higher proportion of children reaching the primary endpoint of viral rebound had at least one lopinavir plasma concentration <LLOQ [11 (57.9%) at least 1 sample <LLOQ vs. 8 (42.1%) no samples <LLOQ: 9 q.d. 2 b.i.d., Fisher's exact test P = 0.03].
The overall relative risk (95% CI) of viral rebound, stratified by randomized arm, given at least one lopinavir concentration <LLOQ was 7.61 (2.95, 19.69). A trend was observed of an increasing proportion experiencing virological rebound when the number of samples with concentrations <LLOQ increased: 5.5% with no samples <LLOQ, 21.4% with one sample <LLOQ and 57.1% with two or more samples <LLOQ.
Discussion
KONCERT is the first randomized controlled trial in children and adolescents to investigate the safety and efficacy of q.d. vs. b.i.d. dosing of lopinavir/r. Children from a wide age-range were included, and all main ethnic groups were represented. The rate of virological rebound, defined as confirmed viral load at least 50 copies/ml at any time within 48 weeks, was low in both arms. However, noninferiority of lopinavir/r q.d. vs. lopinavir/r b.i.d. dosing, when taken as part of combination ART, was not demonstrated; 6% more children in the q.d. arm experienced viral load rebound within the first 48 weeks, and the upper bound of the CI of 14% was outside the predetermined noninferiority bound of 12%. This difference was partially explained by the chance imbalance between arms in viral rebound which occurred between screening and baseline. However, even after adjustment the upper bound of the CI was 11%, only just within the predefined 12% margin of noninferiority. No significant safety issues were demonstrated and there were no differences between arms in development of resistance mutations.
The within patient pharmacokinetic substudy showed that administration of lopinavir/r paediatric tablets q.d. resulted in lower daily exposure to lopinavir and a lower Clast compared with b.i.d. dosing in the same child. In adults, higher exposure (AUC0–24 206.5 μg.h/ml), but comparable Cmax (14.8 μg/ml) has been observed after q.d. dosing of 800/200 mg lopinavir [21]. Elimination half-life (t1/2) was comparable with values found in adults: mean t1/2 (SD) in our study was 6.0 h (3.0 h) for q.d. and 7.7 h (3.0 h) for b.i.d., compared with 6.1 h (2.5 h) and 8.6 h (4.2 h) in adults, respectively [5]. Previous smaller paediatric studies have reported that the AUC0–24 of lopinavir after q.d. dosing of lopinavir/r using various formulations (solution, soft-gel capsules and tablets) lies between 150 and 215 h∗mg/l, and Clast between 1.6 and 5.8 mg/l [10–12,22–25]. In our larger study, the AUC was at the lower end of this range and Clast below it. This cannot be explained by lower dose, as the median lopinavir dose received by children in the pharmacokinetic study was 19.0 mg/kg or 537 mg/m2 q.d., which is comparable or higher than the doses received by children in the other studies. In addition, exposure to lopinavir from tablets in adults was shown to be significantly higher than from soft-gel capsules, although the 90% CI of the GMR was reported to be within the bioequivalence range [13].
Additional findings from this trial reflect ‘real life’ dosing, as not only were formal ‘within-child’ pharmacokinetic studies undertaken, but also sparse random sampling in all children attending clinic throughout the 48 weeks. We demonstrated that more children in the q.d. treatment group had at least one undetectable (<LLOQ) lopinavir plasma concentration: 24.4% q.d. vs. 8.0% b.i.d. Further we observed a pharmacokinetic/pharmacodynamics relationship, with the overall risk of viral load rebound being over seven-fold greater among children with at least one lopinavir concentration <LLOQ, and twice as high in q.d. vs. b.i.d. children (9.3 vs. 4.6). These findings together with the results of the within-child pharmacokinetic show that lopinavir is less forgiving when children are dosed q.d., and thus if children are nonadherent, there is a higher chance of virological rebound. Despite this during the trial, nine out of 12 children on q.d. who rebounded later resuppressed, and seven of the nine remained on q.d. lopinavir/r. Although drug concentration measurements demonstrated that missed q.d. doses had a greater risk of viral rebound, reassuringly due to the relatively high resistance barrier of ritonavir-boosted lopinavir, development of new mutations remained low, and similar to b.i.d. dosing.
Both children and carers reported a preference for taking lopinavir/r q.d., but data from the adherence questionnaires suggests that a small number of children may miss more doses on q.d.
In resource-rich countries, other q.d. boosted protease inhibitor treatments are now widely available for children, but in resource poor situations, which carry the burden of the epidemic, lopinavir/r remains the mainstay of paediatric protease inhibitor based therapy (Habiyambere V, WHO ARV use survey, 2014, personal communication) [26], and the findings of the trial are particularly relevant to these settings.
In conclusion, based on the combination of viral load rebound and pharmacokinetic results in the KONCERT trial, q.d. lopinavir/r cannot be routinely recommended as a simplification option for children with suppressed viral load on b.i.d. lopinavir/r. However, among selected adherent children for whom regular viral load monitoring is available, q.d. dosing remains an option, as we have demonstrated that it is both safe and not associated with any increased risk of developing resistance mutations.
Acknowledgements
The authors would like to thank all of the children, families, and staff from the centres participating in the KONCERT trial.
Writing Committee: H. Lyall, J. Inshaw, Y. Saïdi, T.R. Cressey, S. Forcat, Y. Riault, D. Bastiaans, D. Burger, C. Königs, S. Kanjanavanit, D. Nayagam, T. Bunupuradah, R. Hugo Oliveira, M. della Negra, C. Giaquinto, A.G. Babiker, A. Compagnucci, D.M. Gibb, R.L. Goodall.
H.L., A.G.B., A.C., D. Bu, Y.S., C.G. and D.M.G. conceived and designed the study. H.L., C.K., S.K., D.N., T.B., R.H.O. and M.dN. enrolled patients. S.F., Y.R., Y.S., T.C. oversaw data entry and undertook trial management. A.G.B., J.I., D. Ba and R.L.G. assisted with verification, analysis and interpretation of the data. R.L.G., J.I., S.F. and D. Ba drafted the original version of the manuscript. D. Ba and D. Bu were responsible for measurement of lopinavir drug levels and pharmacokinetic analyses. All authors participated in the writing of the manuscript, and read and approved the final version.
PENTA Steering Committee: J.-P. Aboulker, J. Ananworanich, A. Babiker, E. Belfrage, S. Bernardi, R. Bologna, D. Burger, K. Butler, G. Castelli-Gattinara, P. Clayden, A. Compagnucci, T. Cressey, J.H. Darbyshire, M. Debré, R. de Groot, M. Della Negra, A. De Rossi, A. Di Biagio, D. Duicelescu (deceased), A. Faye, V. Giacomet, C. Giaquinto (Chairperson), D.M. Gibb, I. Grosch-Wörner, M. Hainault, L. Harper, N. Klein, M. Lallemant, J. Levy, H. Lyall, M. Marczynska, M. Mardarescu, L. Marques, M.J. Mellado Peña, L. Meyer, D. Nadal, E. Nastouli, L. Naver, T. Niehues, C. Peckham, D. Pillay, J. Popieska, J.T. Ramos Amador, P. Rojo Conejo, L. Rosado, V. Rosenfeldt (deceased), R. Rosso (deceased), C. Rudin, Y. Saïdi, M. Sharland, H.J. Scherpbier, C. Thorne, P.A. Tovo, G. Tudor-Williams, A. Turkova, N. Valerius, A. Volokha, A.S. Walker, S. Welch, U. Wintergerst.
KONCERT Trial Management Group: A. Babiker, D. Bastiaans, D. Burger, S. Chalermpantmetagul, A. Compagnucci, T. Cressey, S. Forcat, C. Giaquinto, D.M. Gibb, R. Goodall, L. Harper, J. Inshaw, H. Lyall, Y. Riault, Y. Saïdi.
Trials Units: Inserm SC10-US19, France: J.P. Aboulker, A. Compagnucci, G. Hadjou, S. Léonardo, L. Meyer, M. Ndiaye, Y. Riault, Y. Saïdi. ANRS: F. Cardon, S. Couffin-Cadiergues, A. Diallo, S. Lemestre, C. Paul. MRC Clinical Trials Unit at UCL, UK: A. Babiker, J. Bleier, H. Castro, D.M. Gibb, S. Forcat, R. Goodall, L. Harper, L. Harrison, J. Inshaw, D. Johnson, J. Kenny, S. Montero, A. Turkova, A.S. Walker. PHPT, Thailand: S. Chalermpantmetagul, S. Chailert, T.R. Cressey, S. Jinasa, G. Jourdain, C. Kasemrat (L), W. Khamjakkaew, P. Krueduangkam, N. Kruenual, M. Lallemant, S. Le Coeur, N. Ngo-Giang-Huong, R. Peongjakta, K. Seubmongkolchai, W. Sripaoraya (L), P. Sukrakanchana, S. Thammajitsagul, K. Than-in-at, T. Thimakam, J. Thonglo, A. Upra (P).
Pharmacokinetics Laboratory Nijmegen: D. Burger, D. Bastiaans, A. Colbers.
Pharmacokinetics Laboratory Chiang Mai: T. Cressey, P. Punyati, Y. Tawon.
Pharmacology Advisory Group: D. Burger, T. Cressey, E. Jacqz-Aigrain, S. Khoo, J.M. Treluyer, M. Regazzi.
Immunology and Virology Advisory Group: D. Pillay, A. de Rossi, N. Klein, M.A. Munoz Fernandez, E. Nastouli, N. Ngo-Giang-Huong.
Independent Data Monitoring Committee: A. Pozniak (Chair), S. Vella, G. Chène, T. Vesikari.
Endpoint Review Committee: D. Gibb, H. Lyall, S. Montero.
PENTA Foundation (sponsor): L. Comacchio, C. Giaquinto.
Participating sites: Argentina: Hospital de Pediatría S.A.M.I.C. ‘Prof. Dr Juan P. Garrahan’, Buenos Aires: R. Bologna, D. Mecikovsky, N. Maita, N. Pineda, L. Sen (L), A. Mangano (L), M. Moragas (L); Fundación Helios Salud, Buenos Aires: R. Bologna, D. Mecikovsky, G. Sotera, J. Da Bouza, L. Ramos (L). Brazil: Instituto de Infectologia Emilio Ribas, Sao Paolo: M. della Negra, W. Queiroz, T. Zampa de Campos, P. Peraro Barbosa, L. Flora Figueira, D. Peluso Pacola, Y. Ching Lian, R. Alves Carneiro Júnior; IPPMG-UFRJ, Rio de Janeiro: R. Hugo Oliveira, M. Chermont, A. Marins Pala, V. Gomes Louro, H. Carreira Jr. (P), C. Buffoni (L); Universidade de Sao Paulo: H.H. de Sousa Marques, N. Keico Sakita, M.F. de Paula Ramos. France: Centre Hospitalier Universitaire Necker, Paris: S. Blanche, P. Frange, A. Mogenet, M. Diagne, S. Sotou Bere, C. Broissand (P); Hôpital d’Enfants Trousseau, Paris: C. Dollfus, G. Vaudre, E. Sigward (P), A. Schnuriger (L); Hôpital Jeanne de Flandre, Lille: F. Mazingue, F. Bertrand, S. Frade, C. Veron, S. Brice (P), B. Thielemans (P), F. Taillet (L), L. Bocket (L); Centre Hospitalier Universitaire Louis Mourier, Colombes: C. Floch-Tudal, L. Marty, V. Tournier (P), H. Ichou (L); Hôpital Robert Debré, Paris: A. Faye, Y. Wang, L. Toumi, C. Schwartz (P); Centre Hospitalier Universitaire Strasbourg: N. Entz-Werle, F. Uettwiller, A. Hutt-Clauss (P), M.J. Wendling (L), F. Jegou (L). Germany: J.W. Goethe University Frankfurt: C. Koenigs, B. Reimers, K. Beuckmann, M. Sturmer; Dr von Haunersches Kinderspital Munich: G. Notheis; Kinderklinik Dusseldorf: P. Lankisch, H. Laws, J. Neubert, E. Dellers; HELIOS Klinikum Krefeld: T. Niehues, G. Duckers, K. Siepermann; Universitätsklinikum Mannheim: B. Bucholz; Kinderklinik Berlin: C. Feiterna-Sperling; Medizinische Hochschule Hannover: U. Baumann; Universitätsklinikum Hamburg-Eppendorf: A. Brolund, R. Ganschow, R. Kobbe, M. Schaefer, A. Meyer-Bahlburg. Ireland: Our Lady's Children's Hospital, Dublin: K. Butler, A. Rochford, M. Goode, A. Walsh, E. Hyland, M. O’Connor. Italy: Università di Padova: O. Rampon, C. Giaquinto; Ospedale L. Sacco Milan: V. Giacomet; Ospedale del Bambino Gesù: S. Bernardi, G. Pontrelli. Netherlands: Emma Children's Hospital, Amsterdam: H. Scherpbier, H. Voogt, A. Weijsenfeld, M. de Jong; Radboud University Nijmegen: A. Warris, R. de Groot, M. Flier, M. Las, D. Bastiaans (P). Portugal: Hospital Maria Pia Porto: L. Marques, C. Teixeira, A.P. Castro (P), A.C. Mendes (P), T. Almeida (L). Romania: M. Stefan; Matei Bals National Institute Infectious Bucarest: M. Mardarescu, R. Neagu-Draghicenou, L. Alecsandru (P), D. Otelea (L); Victor Babes Hospital Infectious Diseases Bucarest: D. Duiculescu, L. Ene, F. Abaab (P), G. Tardei (L). Spain: I. Garcia Mellado; Hospital 12 de Octubre, Madrid: P. Rojo Conejo, M.I. Gonzalez Tomé; Hospital Sant Joan De Déu, Barcelona: C. Fortuny Guasch, A. Noguera Julian, P. Santin Riba, J. Vinent Genestar (P), A. Murciano Cabeza (P), C. Muñoz-Almagro (L); Hospital La Fe, Valencia: C. Otero Reigada, A. Orti, A. Perez Tamarit, F. Castera Brugada, E. Bellmunt Barreda (L), R. Amigo Moreno (L), A.B. Martin (L), M. Tordera Baviera (P); Hospital Carlos III, Madrid: M.J Mellado Peña, M. Garcia Lopez Hortelano, I. Jimenez Nachez (P), S. de Andres Morera (P); Biobanco Hospital Gregorio Marañon, Madrid: M.A. Muñoz Fernandez (L), J.L. Jimenez Fuentes (L), A. Garcia Torre (L); Hospital Miguel Servet, Zaragoza: L. Ciria Calavia, I. Gale Anso, S. Bernabe Antolin (P), A. Idoipe Tomas (P), A. Martinez Sapiña (L); Hospital Universitario de Getafe: J.T. Ramos Amador, T. Molina (P), P. Tejada Gonzalez (P). Thailand: The HIV Netherlands Australia Thailand Research Collaboration (HIV-NAT), the Thai Red Cross AIDS Research Centre: J. Ananworanich, T. Bunupuradah, W. Prasitsuebsai, T. Pitimahajanaka, N. Thammajaruk (L), Anuntaya Uanithirat (P); Phayao Provincial Hospital: P. Techakunakorn, P. Srisuwan, C. Ruklao, N. Palee (P), A. Maneekaew (L), B. Changlor (L), K. Thungkham (L); Chiangrai Prachanukroh Hospital: R. Hansudewechakul, P. Taeprasert, S. Denjanta (P), P. Thongsuk (L), W. Sangkaew (L), K. Preechapongmit, W. Srisuk; Chonburi Hospital: S. Hongsiriwon, D. Ekkomonrat, S. Soontaros (P), J. Sriyarak (L), J. Sriyarak (L), S. Matchua (L), U. Ruttanamora (L); Nakornping Hospital: S. Kanjanavanit, M. Anathanavanich, P. Sornchai, T. Namwong, S. Peangta, S. Chaisri, D. Suwan (P), D. Chutima (P), S. Roumsuk (L); Bhumibol Adulyadej Hospital: P. Layangool (MD), J. Mekmullica (MD), A. Binyasing (L), B. Tonboon (P), P. Riangrod; Hat Yai Hospital: B. Warachit, T. Borkird, U. Sukhaphan, P. Chanachaiwong (P), R. Jitsakulchaidej (L); Mahasarakam Hospital: S. Na-Rajsima (MD), A. Udomvised (L), N. Kunjaroenrut (P), T. Khayanchoomnoom. UK: Imperial College Hospital Healthcare Trust, London: H. Lyall, G. Tudor-Williams, C. Foster, D. Hamadache, A. Walley, A. Abdulla, D. Patel (P); Great Ormond Street Hospital, London: D. Shingadia, J. Flynn, M. Clapson, K. Parkes, L. Spencer-Walsh, M. Jagani (P); Evelina Children's Hospital, London: E. Menson, R. Cross, C. Duncan, V. Timms, E. Reus, E. Jones (P), M. Wan (P); King's College Hospital, London: C. Ball, D. Nayagam, E. Nsirim, S. Bi, S. Doshi (P); Institute of Child Health, London: H. Poulsom (L), N. Klein (L); Alder Hey Children's Hospital Liverpool: A. Riordan, C. Benson, C. Barker (P), D. Sharpe (P), P. Newland (L); Royal Infirmary Bristol: J. Bernatoniene, A. Finn, F. Manyika, L. Hutchison, K. Stevenson (L); Heartlands Hospital Birmingham: S. Welch, S. Hackett, G. Gilleran, J. Daglish, L. Horton (L), K. Gandhi (P); Leicester Royal Infirmary, S. Bandi, J. Philps, J. Bwire, J. Gardener; Queen's Medical Centre Nottingham: A. Smyth, J. Smith, L. Fear, S. Stafford, S. Hodgson (P), Y. Taha (L); John Radcliffe Hospital Oxford: A. Pollard, L. Willis, R. Howard, A. de Veciana (P), T. Dong (L).
L= laboratory, P=pharmacy
Financial and Drug Support: Paediatric European Network for Treatment of AIDS (PENTA) funded and coordinated the trial. Additional funding was received from the European Union Seventh Framework Program (FP7/2007–2013) under EuroCoord grant agreement number 260694 and from AbbVie Laboratories. INSERM SC10 and MRC CTU are supported by Inserm and the MRC, respectively. Inserm-ANRS supported the trial in France. Lopinavir/ritonavir tablets were provided to sites in Belgium, Brazil, France, Germany, Spain, Portugal, Romania and Thailand by AbbVie. GSK provided abacavir and lamivudine to participating sites in Thailand.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Mehta S, Moore RD, Graham NM. Potential factors affecting adherence with HIV therapy. AIDS 1997; 11:1665–1670. [DOI] [PubMed] [Google Scholar]
- 2.Molina JM, Podsadecki TJ, Johnson MA, Wilkin A, Domingo P, Myers R, et al. A lopinavir/ritonavir-based once-daily regimen results in better compliance and is noninferior to a twice-daily regimen through 96 weeks. AIDS Res Hum Retroviruses 2007; 23:1505–1514. [DOI] [PubMed] [Google Scholar]
- 3.Rabi SA, Laird GM, Durand CM, Laskey S, Shan L, Bailey JR, et al. Multistep inhibition explains HIV-1 protease inhibitor pharmacodynamics and resistance. J Clin Investig 2013; 123:3848–3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gathe J, da Silva BA, Cohen DE, Loutfy MR, Podzamczer D, Rubio R, et al. A once-daily lopinavir/ritonavir-based regimen is noninferior to twice-daily dosing and results in similar safety and tolerability in antiretroviral-naive subjects through 48 weeks. J Acquir Immune Defic Syndr 2009; 50:474–481. [DOI] [PubMed] [Google Scholar]
- 5.Johnson MA, Gathe JC, Jr, Podzamczer D, Molina JM, Naylor CT, Chiu YL, et al. A once-daily lopinavir/ritonavir-based regimen provides noninferior antiviral activity compared with a twice-daily regimen. J Acquir Immune Defic Syndr 2006; 43:153–160. [DOI] [PubMed] [Google Scholar]
- 6.Eron JJ, Feinberg J, Kessler HA, Horowitz HW, Witt MD, Carpio FF, et al. Once-daily versus twice-daily lopinavir/ritonavir in antiretroviral-naive HIV-positive patients: a 48-week randomized clinical trial. J Infect Dis 2004; 189:265–272. [DOI] [PubMed] [Google Scholar]
- 7.EMA. EPAR summary for the public: Kaletra (EMA/33611/2011). 2015 [23/03/15]; http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Summary_for_the_public/human/000368/WC500039044.pdf [Accessed 27 August 2015]. [Google Scholar]
- 8.Bamford A, Turkova A, Lyall H, Foster C, Klein N, Bastiaans D, et al. Paediatric European Network for Treatment of AIDS (PENTA) guidelines for treatment of paediatric HIV-1 infection 2015: optimizing health in preparation for adult life. HIV Med. 2015. Feb 3. doi: 10.1111/hiv.12217. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Working Group on Antiretroviral Therapy and Medical Management of HIV-Infected Children. Guidelines for the use of antiretroviral agents in pediatric HIV infection. Rockville: National Institutes of Health; 2008. [Google Scholar]
- 10.van der Lee M, Verweel G, de Groot R, Burger D. Pharmacokinetics of a once-daily regimen of lopinavir/ritonavir in HIV-1-infected children. Antivir Ther 2006; 11:439–445. [PubMed] [Google Scholar]
- 11.Rosso R, Di Biagio A, Dentone C, Gattinara GC, Martino AM, Vigano A, et al. Lopinavir/ritonavir exposure in treatment-naive HIV-infected children following twice or once daily administration. J Antimicrob Chemother 2006; 57:1168–1171. [DOI] [PubMed] [Google Scholar]
- 12.van der Flier M, Verweel G, van der Knaap LC, van Jaarsveld P, Driessen GJ, van der Lee M, et al. Pharmacokinetics of lopinavir in HIV type-1-infected children taking the new tablet formulation once daily. Antivir Ther 2008; 13:1087–1090. [PubMed] [Google Scholar]
- 13.Klein CE, Chiu YL, Awni W, Zhu T, Heuser RS, Doan T, et al. The tablet formulation of lopinavir/ritonavir provides similar bioavailability to the soft-gelatin capsule formulation with less pharmacokinetic variability and diminished food effect. J Acquir Immune Defic Syndr 2007; 44:401–410. [DOI] [PubMed] [Google Scholar]
- 14.AbbVieLimited. Highlights of prescribing information (Reference ID: 3089639) 2011; http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/021251s042,021906s035lbl.pdf [Accessed 23 March 2015] [Google Scholar]
- 15.Bastiaans DE, Forcat S, Lyall H, Cressey TR, Hansudewechakul R, Kanjanavanit S, et al. Pharmacokinetics of pediatric lopinavir/ritonavir tablets in children when administered twice daily according to FDA weight bands. Pediatr Infect Dis J 2014; 33:301–305. [DOI] [PubMed] [Google Scholar]
- 16.Division of AIDS. Division of AIDS table for grading the severity of adult and pediatric adverse events. Bethesda, MD: National Institutes of Health; 2004. [Google Scholar]
- 17.Centers for Disease Control and Prevention.. 1994 revised classification system for human immunodeficiency virus infection in children less than 13 years of age. MMWR 1994; 43 (RR-12):1–10. [Google Scholar]
- 18.Droste JA, Verweij-Van Wissen CP, Burger DM. Simultaneous determination of the HIV drugs indinavir, amprenavir, saquinavir, ritonavir, lopinavir, nelfinavir, the nelfinavir hydroxymetabolite M8, and nevirapine in human plasma by reversed-phase high-performance liquid chromatography. Ther Drug Monit 2003; 25:393–399. [DOI] [PubMed] [Google Scholar]
- 19.Burger D, Teulen M, Eerland J, Harteveld A, Aarnoutse R, Touw D. The International Interlaboratory Quality Control Program for Measurement of Antiretroviral Drugs in Plasma: a global proficiency testing program. Ther Drug Monit 2011; 33:239–243. [DOI] [PubMed] [Google Scholar]
- 20.Carpenter JB. J. Bootstrap confidence intervals: when, which, what? A practical guide for medical statisticians. Stat Med 2009; 19:1141–1164. [DOI] [PubMed] [Google Scholar]
- 21.AbbVie Limited. Kaletra 200 mg/50 mg film-coated tablets - Summary of Product Characteristics (SPC) 2014. http://www.medicines.org.uk/emc/medicine/18442 [Accessed 23 March 2015] [Google Scholar]
- 22.Chokephaibulkit K, Nuntarukchaikul M, Phongsamart W, Wittawatmongkol O, Lapphra K, Vanprapar N, et al. Once- versus twice-daily lopinavir/ritonavir tablets in virologically suppressed, HIV-infected, treatment-experienced children: comparative pharmacokinetics and virological outcome after switching to once-daily lopinavir/ritonavir. J Antimicrob Chemother 2012; 67:2927–2931. [DOI] [PubMed] [Google Scholar]
- 23.Foissac F, Urien S, Hirt D, Frange P, Chaix ML, Treluyer JM, et al. Pharmacokinetics and virological efficacy after switch to once-daily lopinavir-ritonavir in treatment-experienced HIV-1-infected children. Antimicrob Agents Chemother 2011; 55:4320–4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.La Porte C, van Heeswijk R, Mitchell CD, Zhang G, Parker J, Rongkavilit C. Pharmacokinetics and tolerability of once- versus twice-daily lopinavir/ritonavir treatment in HIV-1-infected children. Antivir Ther 2009; 14:603–606. [PubMed] [Google Scholar]
- 25.Verweel G, Burger DM, Sheehan NL, Bergshoeff AS, Warris A, van der Knaap LC, et al. Plasma concentrations of the HIV-protease inhibitor lopinavir are suboptimal in children aged 2 years and below. Antivir Ther 2007; 12:453–458. [PubMed] [Google Scholar]
- 26.Bunupuradah PT, Fahey P, Kariminia A, Yusoff NKN, Khanh TH, Sohn AH, et al. Second-line protease inhibitor-based HAART after failing nonnucleoside reverse transcriptase inhibitor-based regimens in Asian HIV-infected children. Antivir Ther 2013; 18:591–598. [DOI] [PMC free article] [PubMed] [Google Scholar]