

Abstract
Humans are ubiquitously exposed to di(2-ethylhexyl) phthalate (DEHP), which is an environmental toxicant present in common consumer products. DEHP potentially targets the ovary through its metabolite mono(2-ethylhexyl) phthalate (MEHP). However, the direct effects of MEHP on ovarian folliculogenesis and steroidogenesis, two processes essential for reproductive and nonreproductive health, are unknown. The present study tested the hypotheses that MEHP directly accelerates early folliculogenesis via overactivation of phosphatidylinositol 3-kinase (PI3K) signaling, a pathway that regulates primordial follicle quiescence and activation, and inhibits the synthesis of steroid hormones by decreasing steroidogenic enzyme levels. Neonatal ovaries from CD-1 mice were cultured for 6 days with vehicle control, DEHP, or MEHP (0.2–20 μg/ml) to assess the direct effects on folliculogenesis and PI3K signaling. Further, antral follicles from adult CD-1 mice were cultured with vehicle control or MEHP (0.1–10 μg/ml) for 24–96 h to establish the temporal effects of MEHP on steroid hormones and steroidogenic enzymes. In the neonatal ovaries, MEHP, but not DEHP, decreased phosphatase and tensin homolog levels and increased phosphorylated protein kinase B levels, leading to a decrease in the percentage of germ cells and an increase in the percentage of primary follicles. In the antral follicles, MEHP decreased the mRNA levels of 17alpha-hydroxylase-17,20-desmolase, 17beta-hydroxysteroid dehydrogenase, and aromatase leading to a decrease in testosterone, estrone, and estradiol levels. Collectively, MEHP mediates the effect of DEHP on accelerated folliculogenesis via overactivating PI3K signaling and inhibits steroidogenesis by decreasing steroidogenic enzyme levels.
Keywords: follicle, folliculogenesis, mono(2-ethylhexyl) phthalate, ovary, phthalate, steroidogenesis
INTRODUCTION
Di(2-ethylhexyl) phthalate (DEHP) is a widely used synthetic plasticizer incorporated in the manufacturing of common polyvinyl chloride consumer, medical, and building products in order to impart flexibility [1, 2]. DEHP is produced in vast quantities, up to 300 million pounds annually, to account for its high demand in products ranging from carpeting and roofing to food and beverage packaging [2]. This is of concern because DEHP is noncovalently bound to the plastic, meaning DEHP frequently leaches from the plastics into the environment and products that humans consume [1]. Based on its high production volume, use in common items, and ability to leach out from the items, humans are exposed to DEHP on a daily basis via oral ingestion, inhalation, and dermal contact [1]. Specifically, the estimated range of daily human exposure to DEHP is 3–30 μg/kg/day [3, 4]. Exposure to DEHP represents a public health concern because DEHP and its metabolite, mono(2-ethylhexyl) phthalate (MEHP), have been identified as top contaminants in human tissues such as blood [5, 6], urine [1, 5–9], amniotic fluid [10–12], umbilical cord blood [13, 14], breast milk [6], and ovarian follicular fluid [15]. Important from a toxicological standpoint, it is hypothesized that the toxicity of DEHP is mediated by the bioactive metabolite MEHP in many organ systems, including the ovary [16–21].
Also important for public health, DEHP and MEHP are known endocrine-disrupting chemicals and reproductive toxicants that potentially target the ovary [1, 2, 16–21]. Understanding the effects of these chemicals on normal ovarian function is crucial because the ovary is responsible for the maturation of follicles for ovulation, a process termed folliculogenesis, and for the synthesis of sex steroid hormones, a process termed steroidogenesis [22]. Defects in folliculogenesis and steroidogenesis, including accelerated depletion of follicles within the ovary and decreased estradiol production, can cause reproductive and nonreproductive complications, such as infertility, premature ovarian failure, cardiovascular disease, osteoporosis, mood disorders, and even premature death [23–34].
Interestingly, DEHP and MEHP have been shown to disrupt ovarian folliculogenesis. Specifically, we have previously reported that oral exposure to DEHP in adult mice accelerates primordial follicle recruitment following 10 and 30 days of dosing [35]. Primordial follicle recruitment is a gonadotropin-independent process involving strict regulation of intrinsic ovarian growth factors [36–39]. Factors within the phosphatidylinositol 3-kinase (PI3K)-signaling pathway have recently been identified as regulators of primordial follicle survival, dormancy, and recruitment, where inhibition of PI3K signaling maintains primordial follicle dormancy and overactivation of signaling leads to primordial follicle recruitment [38, 40–45]. We have also previously reported that the DEHP-induced acceleration of primordial follicle recruitment is likely via overactivation of PI3K signaling, indicated by a decrease in ovarian phosphatase and tensin homolog (PTEN) levels, an inhibitor of PI3K signaling that maintains primordial follicle dormancy, and an increase in ovarian phosphorylated protein kinase B (pAKT) levels, a second messenger of PI3K signaling that promotes primordial follicle recruitment [35]. However, in previous studies, we did not determine whether the effects on primordial follicle recruitment and PI3K signaling were direct effects on the ovary or whether the effects were a secondary response caused by toxicity in other organ systems following exposure in vivo.
Additionally, DEHP and MEHP have been shown to disrupt ovarian steroidogenesis. Specifically, DEHP exposure has been shown to decrease estradiol levels and aromatase levels in vivo [46–48] and in vitro [16, 49, 50]. We have reported that the inhibition of steroidogenesis may be via a direct effect on antral follicle functionality because DEHP exposure in cultured mouse antral follicles directly decreased the availability of precursor sex steroid hormone levels at time points prior to decreasing estradiol levels [51]. This toxicity of DEHP may be mediated by MEHP, and MEHP exposure has also been shown to inhibit steroidogenesis in vitro [19–21, 52, 53]. Specifically, we have reported that MEHP exposure directly decreased the levels of estradiol and the mRNA levels of aromatase, Cyp19a1, in cultured mouse antral follicles following 96 h of exposure [16]. Steroidogenesis is primarily conducted by the mature antral follicle and is a step-wise process involving the enzymatic conversion of cholesterol to estradiol and other necessary precursor steroid hormones that are essential for reproductive and nonreproductive health [54, 55]. However, few studies have investigated the effects of MEHP on the precursor steroid hormones and the steroidogenic enzymes upstream of estradiol and aromatase. Additionally, the temporal effects by which MEHP directly inhibits steroidogenesis in the antral follicle remain unknown.
The present study was designed to investigate the direct effects of DEHP and MEHP on early folliculogenesis and to compare the mechanisms by which these chemicals alter folliculogenesis in vivo and in vitro. Further, the present study was designed to establish a time course of the initial and time-specific effects of MEHP on antral follicle steroidogenesis upstream of estradiol and aromatase and to compare the mechanisms by which MEHP and DEHP inhibit steroidogenesis in cultured antral follicles. Specifically, we tested the hypotheses that MEHP mediates the DEHP-induced acceleration of primordial follicle recruitment and does so via a mechanism involving overactivation of PI3K signaling, which is the mechanism observed in vivo. Additionally, we hypothesized that MEHP directly inhibits the synthesis of sex steroid hormones to ultimately lead to a decrease in estradiol levels.
MATERIALS AND METHODS
Chemicals
DEHP (99% purity) was purchased from Sigma-Aldrich (St. Louis, MO), and MEHP (99% purity) was purchased from AccuStandard (New Haven, CT). Stock solutions of DEHP and MEHP were prepared using dimethylsulfoxide (DMSO) (Sigma-Aldrich) as the vehicle in various concentrations (0.133, 0.267, 1.33, 2.67, 13.3, and 26.7 mg/ml). This allowed for an equal volume of each stock to be added to the culture wells to control for vehicle concentration. Final concentrations of DEHP in culture were 0.2, 2, and 20 μg/ml, which is approximately equivalent to 0.54, 5.4, and 54 μM, respectively. Final concentrations of MEHP in culture were 0.1, 0.2, 1, 2, 10, and 20 μg/ml, which is approximately equivalent to 0.34, 0.68, 3.4, 6.8, 34, and 68 μM, respectively.
The concentrations of DEHP and MEHP were chosen based on their ability to disrupt ovarian steroidogenesis and folliculogenesis. Specifically, concentrations of these chemicals cause inhibition of antral follicle growth, decreased estradiol production from antral follicles, decreased antral follicle Cyp19a1 expression, and increased antral follicle atresia [16, 17, 56]. We have also previously determined that oral exposure to DEHP for 10 and 30 days accelerates primordial follicle recruitment at doses ranging from 200 μg/kg/day to 20 mg/kg/day [35]. These doses are approximately equivalent to 0.492–49.2 μM, which encompass the doses ranging from 0.2 to 20 μg/ml. The concentrations of DEHP and MEHP selected in these experiments have also been determined to be clinically relevant [20, 52, 57–59]. Importantly, plasma concentrations of DEHP and MEHP in healthy women have been reported to be 0.18 μg/ml and 0.58 μg/ml, respectively, and peritoneal fluid concentrations of DEHP and MEHP in these women were reported to be 0.46 μg/ml and 0.37 μg/ml, respectively, which encompasses the lower selected concentrations [60]. In addition, the extensive use of DEHP in medical equipment leads to markedly higher levels of DEHP and MEHP in patients undergoing consistent medical care when compared to healthy people [61, 62]. Neonatal patients in intensive care units that receive blood transfusions have plasma levels of DEHP and MEHP at 11.1 μg/ml and 15.1 μg/ml, respectively, which encompass the higher selected concentrations [63]. Further, the Agency for Toxic Substances and Disease Registry reported in their 2002 report that the lowest-observed adverse-effect level of DEHP is 140 mg/kg/day, which is approximately equivalent to 344.6 μM [2]. However, more recent studies have shown effects at lower doses of DEHP [64]. The selected concentrations of DEHP and MEHP in these experiments fall below the lowest-observed adverse-effect level concentration reported by the Agency for Toxic Substances and Disease Registry.
Animals
For the experiments investigating the direct effects of DEHP and MEHP on early folliculogenesis, cycling, adult CD-1 female mice (40–50 days of age) and mature, adult CD-1 male mice (40–50 days of age) were obtained from Charles River Laboratories (Wilmington, MA). Breeding groups (one male with two females) were housed in the College of Veterinary Medicine Animal Facility at the University of Illinois at Urbana-Champaign to serve as a source for Postnatal Day 4 (PND4) ovaries for culture. Once pregnant, the mice were individually placed in separate cages and allowed to give birth. On PND4, female pups were removed from the dam and used in the neonatal ovary cultures described below. For the experiments investigating the effects of MEHP on antral follicles, cycling, adult CD-1 female mice (35–39 days of age) were obtained from Charles River Laboratories. The mice were housed in groups of four in the College of Veterinary Medicine Animal Facility at the University of Illinois at Urbana-Champaign and were allowed to acclimate to the facility prior to experimentation. All the animals in these experiments were housed in a controlled animal room environment (temperature at 22°C ± 1°C and 12L:12D cycles) and were provided food and water ad libitum. The Institutional Animal Use and Care Committee at the University of Illinois at Urbana-Champaign approved all the procedures involving animal care, euthanasia, and tissue collection.
In Vitro Neonatal Ovary Cultures
Whole ovaries were aseptically collected from CD-1 pups on PND4 and were used for ovarian culture as described previously [65, 66]. Briefly, each ovary was removed on PND4 and cleaned of excess fat and oviductal tissue under a light microscope. The ovaries were placed in media droplets on pieces of Millicell culture plate inserts (Millipore, Billercia, MA) that were floating on 500 μl of equilibrated (37°C) supplemented ovary culture media in a four-well culture plate. The treatment groups for the neonatal ovary culture experiments included vehicle control (DMSO), DEHP (0.2, 2, and 20 μg/ml), and MEHP (0.2, 2, and 20 μg/ml). The supplemented media contained Dulbecco-modified Eagle medium/Ham F12 minus phenol red medium (Life Technologies, Grand Island, NY) with 1 mg/ml bovine serum albumin (Sigma-Aldrich), 1 mg/ml Albumax (Life Technologies), 50 μg/ml ascorbic acid (Sigma-Aldrich), 5 units/ml penicillin plus 5 μg/ml streptomycin (Life Technologies), and 27.5 μg/ml transferrin (Sigma-Aldrich) per well. Each ovary was cultured for 6 days until PND10 in an incubator at 37°C supplying 5% CO2. The media were replaced daily with fresh, treated, supplemented media, and one drop of medium was placed on the ovary to prevent the ovary from drying. Following culture, one ovary was collected for histological evaluation of follicle numbers, and the other ovary was subjected to immunohistochemistry as described below.
The neonatal ovary culture system and PND4–PND10 time points were chosen because the neonatal ovary is enriched in primordial and primary follicles that are naturally undergoing the initial stages of folliculogenesis [67]. Because DEHP exposure (approximately equivalent to 0.492–49.2 μM or 0.2–20 μg/ml) for 10 and 30 days accelerates primordial follicle recruitment in vivo [35], this culture system provides a direct tool to assess the effects of DEHP and MEHP on early folliculogenesis. Specifically, at PND4, the neonatal ovary is completing germ cell nest breakdown; therefore, the ovary contains a population of follicles that are primarily in the primordial stage [22, 67]. At PND10, some primordial follicles naturally begin to undergo recruitment to the primary stage of development both in vivo and in vitro, and these follicles are structurally and functionally identical to those in the adult [22, 67].
Histological Evaluation of Follicle Numbers
Following 6 days of culture, some PND10 neonatal ovaries were fixed in Dietrich fixative for at least 24 h for histological evaluation of follicle numbers (n = 4–6 ovaries/group). The ovaries were then transferred to 70% ethanol, embedded in paraffin wax, and serial sectioned (5 μm) using a microtome. The serial sections were mounted on glass slides and stained with hematoxylin and eosin. The numbers of oocyte containing follicles and germ cells were counted in every fifth serial section using a light microscope, and the percentage of each follicle type was calculated by dividing the number of follicles of each specific type by the total number of follicles counted and multiplying that value by 100. Stage of follicular development was assessed using previously defined criteria [68, 69]. Briefly, germ cells were identified by the presence of a single oocyte or group of oocytes without a defined granulosa cell layer encapsulating them, primordial follicles contained the oocyte surrounded by a single layer of squamous granulosa cells, and primary follicles contained the oocyte surrounded by a single layer of cuboidal granulosa cells. Very few preantral and antral follicles are present at PND10; therefore, these follicle types were not included in the analysis. All germ cells and primordial and primary follicles with oocytes, regardless of nuclear material in the oocytes, were counted.
Immunohistochemistry
Following 6 days of culture, some PND10 neonatal ovaries were fixed in 4% paraformaldehyde overnight and transferred to 70% ethanol for immunohistochemical analysis for PTEN and pAKT (Ser473) (the active form of the protein) as described previously (n = 3/group) [35]. Briefly, the ovaries were embedded in paraffin wax, serial sectioned (5 μm), and three representative sections that span the entire length of the ovary were mounted on glass slides. Some tissue sections were subjected to staining for PTEN (1:200) (Cell Signaling Technology, Inc., Boston, MA), while other tissue sections were subjected to staining for pAKT (1:50) (Cell Signaling Technology, Inc.). The tissues were then incubated with a secondary biotinylated goat anti-rabbit antibody (Vector Laboratories, Inc., Burlingame, CA), followed by an incubation with an avidin biotin complex solution (Vector Laboratories, Inc.). ImmPACT NovaRED peroxidase substrate solution (Vector Laboratories, Inc.) was then applied until the color optimally developed. Each sample was exposed to the chromogen for equal amounts of time. The slides were then rinsed, counterstained with a 1:10 dilution of hematoxylin, and coverslipped. A negative control was used in each experiment and was subjected to the same methods listed above, except instead of primary antibody, the negative control tissues were incubated with a negative control rabbit immunoglobulin fraction (Dako, Carpinteria, CA) at the same concentration of each primary antibody. Negative controls were confirmed to have no positive staining after exposure to the chromogen for equal amounts of time as the samples. Both PTEN and pAKT were chosen because they are integral regulators of PI3K signaling and have been associated with the regulation of primordial follicle recruitment [38, 43, 70–72]. Specifically, PTEN inhibits PI3K signaling and ultimately maintains primordial follicle dormancy [38, 43, 70–72]. Additionally, pAKT is a second messenger of PI3K signaling and ultimately promotes primordial follicle recruitment [38, 43, 70–72]. Further, 10 day oral exposure to DEHP decreases ovarian PTEN and increases ovarian pAKT, potentially driving the acceleration of primordial follicle recruitment that was observed in that study [35].
Analysis of the Levels of Protein Staining
Following immunohistochemistry, the levels of PTEN and pAKT staining in the neonatal ovaries were quantified using published methods [35, 73–75]. Briefly, images of ovaries were digitally captured using a Leica DFC 290 camera and analyzed using the ImageJ software (http://rsb.info.nih.gov/nih-image/). For analysis, three representative sections from each ovary were assessed from three separate animals per treatment group. Digital images were converted to 8-bit grayscale images and then converted to pseudocolored images. Colors were based on relative stain intensity, as defined digitally. Areas with no staining appeared black and blue, while areas with the most intense staining appeared deep yellow to orange. Percentages of pixels corresponding to positive staining of PTEN and pAKT in the whole ovary and percentages of positively stained PTEN and pAKT primordial follicle oocytes were compared across treatment groups. For whole ovarian analysis, the pixels of positively stained PTEN and pAKT areas were divided by the total amount of pixels in the whole ovary, and that value was multiplied by 100. For primordial follicle oocyte analysis, positively stained PTEN and pAKT primordial follicle oocytes were quantified, divided by the total amount of primordial follicle oocytes, and that value was multiplied by 100.
In Vitro Antral Follicle Cultures
Unprimed, female CD-1 mice were euthanized and their ovaries were aseptically removed for antral follicle isolation. Antral follicles (250–400 μm) were isolated from the ovary and were cleaned of interstitial tissue using watchmaker's forceps [76, 77]. At least two to three mice were used in each experiment, in which approximately 20–30 antral follicles per mouse were obtained. Each treatment group contained 8–16 follicles (n = 8–16 pooled follicles from individual wells from three to nine separate experiments). Isolated antral follicles were randomly and individually plated in wells of a 96-well culture plate containing unsupplemented α-minimal essential medium (α-MEM, Life Technologies) prior to treatment.
The treatment groups for the follicle culture experiments included vehicle control (DMSO) and MEHP (0.1, 1, and 10 μg/ml), and they were prepared in supplemented α-MEM. Supplemented α-MEM was prepared as described previously [76, 77]. Briefly, supplemented α-MEM contained 10 ng/ml insulin, 5.5 ng/ml transferrin, 5.5 ng/ml selenium (Sigma-Aldrich), 100 units/ml penicillin (Sigma-Aldrich), 100 mg/ml streptomycin (Sigma-Aldrich), 5 international units/ml human recombinant follicle-stimulating hormone (FSH; Dr. A.F. Parlow, National Hormone and Peptide Program, Harbor-UCLA Medical Center, Torrance, CA), and 5% fetal calf serum (Atlanta Biologicals, Lawrenceville, GA). An equal volume of chemical (0.75 μl/ml of media) was added for each dose to control that the amount of vehicle in each preparation was 0.075%. Each follicle was cultured in 150 μl of medium for 24–96 h in an incubator at 37°C supplying 5% CO2. Following each 24 h time point, some cultures were ended, and media from cultured follicles were collected for measurements of the levels of sex steroid hormones. In addition, cultured follicles were collected, snap frozen, and stored at −80°C for gene expression analysis. A time course was conducted to observe when the effects of MEHP on steroidogenesis begin and to determine which hormone/enzyme was causing the reduction of estradiol production from antral follicles following 96 h as reported previously [16].
Analysis of Sex Steroid Hormone Levels
Following each 24 h antral follicle culture window, culture media were subjected to enzyme-linked immunosorbent assays (ELISAs) for the measurements of the levels of sex steroid hormones. Levels of progesterone, dehydroepiandrosterone (DHEA), androstenedione, testosterone, estrone, and 17β-estradiol were measured following the manufacturer's protocol using ELISA kits purchased from Diagnostics Research Group (Mountainside, NJ) (n = 8–16 wells of pooled media containing follicles from three to nine separate experiments). The analytical sensitivity of each kit was 0.044 μg/ml for DHEA, 0.1 ng/ml for progesterone, 0.019 ng/ml for androstenedione, 0.083 ng/ml for testosterone, 6.3 pg/ml for estrone, and 9.71 pg/ml for estradiol. All the samples were run in duplicates, and all intra- and interassay coefficients of variability were less than 10%. Some samples were diluted to match the dynamic range of each ELISA kit. Mean values for each sample were used in this analysis.
Analysis of Antral Follicle Gene Expression
Following each 24 h antral follicle culture window, antral follicles were frozen in liquid nitrogen, and stored at −80°C for quantitative real-time polymerase chain reaction (qPCR) analysis (n = 8–16 follicles/3–9 separate experiments). Total RNA (200 ng) was extracted from the follicles using the RNeasy Micro Kit (Qiagen, Inc., Valencia, CA) according to the manufacturer's protocol, and was then reverse transcribed to cDNA using the iScript RT kit (Bio-Rad Laboratories, Inc., Hercules, CA) according to the manufacturer's protocol. Prior to qPCR analysis, each cDNA sample was diluted 1:4 using nuclease-free water. The CFX96 Real-Time PCR Detection System (Bio-Rad Laboratories, Inc.) and accompanying CFX Manager Software was used for analysis of qPCR according to the manufacturer's protocol. All the qPCR reactions were done in triplicate using 1 μl cDNA, forward and reverse primers (5 pmol), and a SsoFastEvaGreen Supermix for a final reaction volume of 10 μl. A standard curve was generated from six serial dilutions of three samples spanning different treatment groups to calculate the efficiencies of each primer set. Specific qPCR primers (Integrated DNA Technologies, Inc., Coralville, IA) for the genes of interest as well as the reference gene, beta-actin (Actb), can be found in Table 1. Beta-actin was chosen as the reference gene because its expression did not differ across treatment groups. The genes tested were steroidogenic acute regulatory protein (Star), cytochrome P450 cholesterol side-chain cleavage (Cyp11a1), 3β-hydroxysteroid dehydrogenase 1 (Hsd3b1), 17α-hydroxylase-17,20-desmolase (Cyp17a1), 17β-hydroxysteroid dehydrogenase1 (Hsd17b1), and aromatase (Cyp19a1). These genes were chosen because they synthesize estradiol and its precursor sex steroid hormones in the ovary.
TABLE 1.
Sequence of primer sets used for gene expression analysis.

The CFX96 machine quantifies the amount of PCR product generated by measuring SsoFastEvaGreen dye (Bio-Rad Laboratories, Inc.) that fluoresces when bound to double-stranded DNA. The qPCR program consisted of an enzyme activation step (95°C for 1 min), an amplification and quantification program (40 cycles of 95°C for 10 sec, 60°C for 10 sec, single fluorescence reading), a step of 72°C for 5 min, a melt curve (65°C−95°C heating 0.5°C per sec with continuous fluorescence readings), and a final step at 72°C for 5 min as per the manufacturer's protocol. Expression data were generated using the mathematical standard comparative (ΔΔCt) method. The ΔCt was calculated by subtracting the Actb Ct value from the gene of interest Ct value. The ΔΔCt was calculated as the difference between the ΔCt between the treatment groups and the DMSO groups. The relative fold-change of expression was then equaled to 2(−ΔΔCt) for each sample.
Statistical Analysis
Analysis of data was conducted using SPSS statistical software (SPSS, Inc., Chicago, IL). Data were expressed as means and error bars represent the SEM. Multiple comparisons between normally distributed experimental groups were made using one-way ANOVA followed by Tukey posthoc comparison. Multiple comparisons between nonnormally distributed experimental groups were made using Kruskal Wallis tests when appropriate. Statistical significance was assigned at P ≤ 0.05.
RESULTS
Effect of DEHP and MEHP on Early Folliculogenesis
Proper regulation of ovarian folliculogenesis is essential for fertility and the maintenance of appropriately timed reproductive senescence. Previously, we have shown that daily oral exposure to DEHP for 10 and 30 days disrupts folliculogenesis by accelerating primordial follicle recruitment to the primary stage of development [35]. However, that study utilized an in vivo dosing regimen and was unable to test if the effects on primordial follicle recruitment were due to direct ovarian toxicity caused by DEHP or the bioactive metabolite MEHP. Therefore, the direct effects of DEHP and MEHP on follicular dynamics were evaluated in this study. Following 6 days of culture, DEHP-exposed ovaries had similar percentages of germ cells, primordial follicles, and primary follicles counted when compared to the vehicle control group (Fig. 1A, n = 5–6/group). Similar effects were observed when quantifying total counts of germ cells and follicles in response to DEHP treatment (Supplemental Fig. S1A, n = 5–6/group; Supplemental Data are available online at www.biolreprod.org). However, following 6 days of culture, MEHP exposure significantly decreased the percentage of germ cells counted at all selected doses when compared to the vehicle control group (Fig. 1B, n = 4–6/group, P ≤ 0.05). MEHP exposure did not significantly alter the percentage of primordial follicles counted, but it significantly increased the percentage of primary follicles counted at all selected doses when compared to the vehicle control group (Fig. 1B, n = 4–6/group, P ≤ 0.05). Similar effects were observed when quantifying total counts of germ cells and follicles in response to MEHP treatment (Supplemental Fig. S1B, n = 4–6/group, P ≤ 0.05).
FIG. 1.
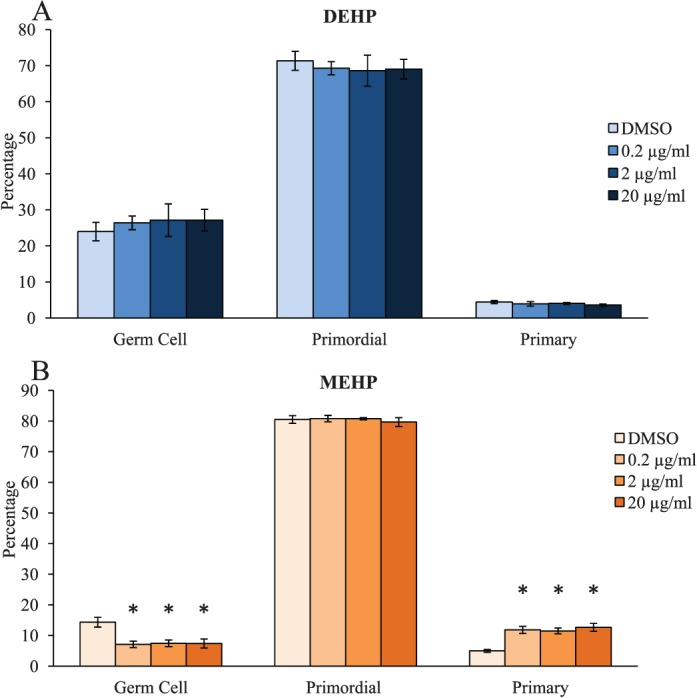
Effect of DEHP and MEHP on early folliculogenesis. Ovaries from CD-1 mice were cultured from PND4 to PND10 with vehicle (DMSO), DEHP (0.2–20 μg/ml; A), or MEHP (0.2–20 μg/ml; B). Following culture, ovaries were processed for histological evaluation of total germ cell and follicle counts. Percentages of each stage of folliculogenesis were calculated and compared in each treatment group. Graph represents means ± SEM (n = 4–6 ovaries/treatment group). Although slight, panel A and panel B have different scales. Asterisks (*) represent significant difference from vehicle control (P ≤ 0.05).
Effect of DEHP and MEHP on Protein Levels of PI3K-Signaling Factors
We have previously shown that the DEHP-induced acceleration of primordial follicle recruitment following 10 and 30 days of exposure in vivo may involve overactivation of the PI3K-signaling pathway [35], but it was unknown if the DEHP-induced dysregulation of PI3K signaling was due to a direct ovotoxic effect of DEHP and/or MEHP. Thus, this study tested whether DEHP or MEHP directly altered the levels of PTEN and/or pAKT in the neonatal ovaries. Qualitatively, it appears that MEHP exposure decreased the staining of PTEN in the whole neonatal ovary (Supplemental Fig. 2A–D), decreased the percentage of PTEN-positive oocytes (Supplemental Fig. S2, E and F), and increased the percentage of pAKT-positive oocytes (Supplemental Fig. S2, G and H). When quantified, MEHP significantly decreased the levels of PTEN staining in the whole neonatal ovary at the 2 and 20 μg/ml doses when compared to the vehicle control group (Fig. 2A, n = 3/group, P ≤ 0.05), while all selected doses of DEHP had comparable levels of PTEN staining to the vehicle control group (Fig. 2A, n = 3/group). Conversely, both MEHP and DEHP had comparable levels of pAKT staining in the whole ovary when compared to the vehicle control group (Fig. 2A, n = 3/group). To ensure that the levels of the PI3K proteins were altered in the follicles of interest, the percentages of positively stained PTEN and pAKT oocytes in primordial follicles were quantified. MEHP significantly decreased the percentage of PTEN-positive oocytes and significantly increased the percentage of pAKT-positive oocytes in primordial follicles at all selected doses of MEHP when compared to the vehicle control group (Fig. 2B, n = 3/group, P ≤ 0.05). Meanwhile, DEHP had comparable percentages of PTEN- and pAKT-positive oocytes to the vehicle control group (Fig. 2B, n = 3/group).
FIG. 2.
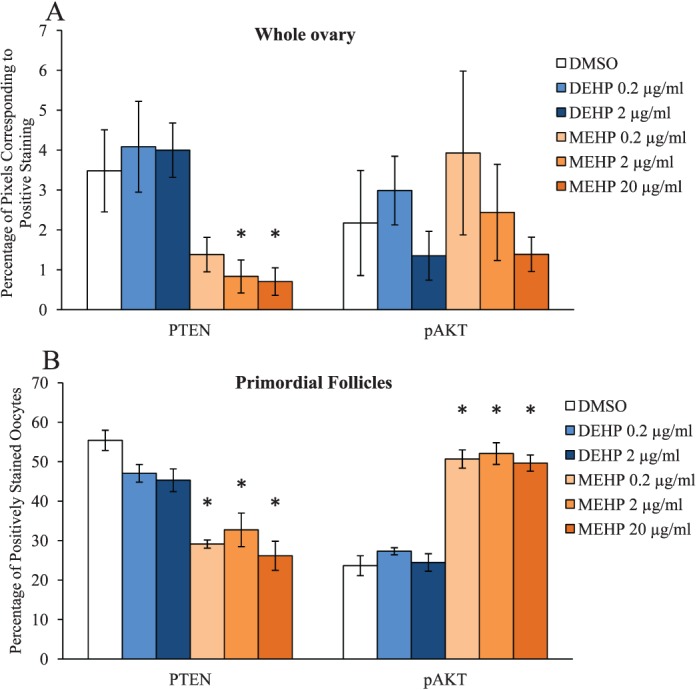
Effect of DEHP and MEHP on protein levels of PI3K-signaling factors. Ovaries from CD-1 mice were cultured from PND4 to PND10 with vehicle (DMSO), DEHP (0.2–2 μg/ml), or MEHP (0.2–20 μg/ml). Following culture, ovaries were subjected to immunohistochemistry for quantification of PTEN (the left side of A and B) and pAKT (the right side of A and B) staining. Digital images were converted to 8-bit grayscale images and then converted to pseudocolored images. Colors were based on relative stain intensity. Percentage of pixels corresponding to positive staining in the whole ovary (A), and percentage of positively stained oocytes in primordial follicles (B) were calculated and compared in each treatment group. Graph represents means ± SEM (n = 3/group). Asterisks (*) represent significant difference from vehicle control (P ≤ 0.05).
Effect of MEHP on Sex Steroid Hormone Production by Antral Follicles
Previous studies have determined that DEHP exposure decreases estradiol levels in vivo [46–48] and in cultured ovarian cell types and follicles in vitro [16, 49–51]. Likewise, MEHP exposure has also been shown to decrease estradiol levels in cultured ovarian cell types and follicles in vitro [16, 19–21, 53]. However, the mechanism by which MEHP decreases estradiol levels is unknown. We have previously reported that MEHP exposure for 96 h decreases estradiol levels produced by cultured mouse antral follicles [16], but the direct effects of MEHP on antral follicle steroidogenesis at earlier time points of exposure and on the precursor steroid hormones upstream of estradiol are unknown. Thus, the temporal effects of MEHP on the levels of sex steroid hormones produced by the antral follicle were investigated utilizing time course culture experiments. Beginning with the most upstream precursor steroid hormones measured, MEHP exposure did not significantly alter the levels of progesterone or DHEA at any time point tested when compared to the vehicle control group (Fig. 3, A and B, n = 3–9/group). However, MEHP exposure significantly decreased the levels of androstenedione at the 10 μg/ml dose following 24 and 72 h of exposure when compared to the vehicle control group (Fig. 3C, n = 3–9/group, P ≤ 0.05). Further, MEHP exposure significantly decreased the levels of testosterone at the 10 μg/ml dose following 24 h of exposure when compared to the vehicle control group (Fig. 3D, n = 3/group, P ≤ 0.05). Likewise, MEHP exposure significantly decreased the levels of testosterone at all selected doses following 96 h of exposure when compared to the vehicle control group (Fig. 3D, n = 4–9/group, P ≤ 0.05). Further down the steroidogenesis pathway, MEHP exposure significantly decreased the levels of estrone at the 0.1 and 10 μg/ml doses following 96 h of exposure when compared to the vehicle control group (Fig. 3E, n = 4–9/group, P ≤ 0.05). Additionally, MEHP exposure significantly decreased the levels of estradiol at all selected doses following 96 h of exposure when compared to the vehicle control group (Fig. 3F, n = 4–9/group, P ≤ 0.05).
FIG. 3.

Effect of MEHP on the antral follicle-produced sex steroid hormone levels. Antral follicles were isolated from adult CD-1 mice and were cultured with vehicle (DMSO) or MEHP (0.1–10 μg/ml) for 24–96 h. Following each 24 h time point, media were pooled per treatment group and were subjected to ELISAs for the measurements of progesterone (A), dehydroepiandrosterone (DHEA) (B), androstenedione (C), testosterone (D), estrone (E), and estradiol (F). The graph represents means ± SEM from three to nine separate experiments with media from 8 to 16 wells/treatment group in each experiment. ND indicates that hormone levels were not detectable because they were below the threshold level for detection. Asterisks (*) represent significant difference from vehicle control (P ≤ 0.05).
Effect of MEHP on Steroidogenic Enzyme Gene Expression in Antral Follicles
Previous studies have suggested that one potential mechanism by which DEHP and MEHP decrease estradiol levels is through a decrease in the ovarian mRNA levels of Cyp19a1, also known as aromatase, following exposure in vivo [46, 78] and in vitro [16, 49–51]. However, not much is known about the direct effects of MEHP on the steroidogenic enzyme levels upstream of Cyp19a1 in the antral follicle. Further, not much is known about the direct effects of MEHP on the enzyme levels prior to 96 h of exposure. In addition, MEHP decreased the levels of sex steroid hormones upstream of estradiol across multiple time points in the present study (Fig. 3). Thus, the temporal effects and potential mechanisms by which MEHP inhibits steroidogenesis were investigated. Following 48 h of exposure, MEHP significantly decreased the mRNA levels of Star and Cyp11a1 at the 1 and 10 μg/ml doses when compared to the vehicle control group (Fig. 4, A and B, n = 3–5/group, P ≤ 0.05). Following 24 h of exposure, MEHP significantly decreased the mRNA levels of Hsd3b1 at the 10 μg/ml dose when compared to the vehicle control group (Fig. 4C, n = 3/group, P ≤ 0.05). Following 96 h of exposure, MEHP significantly decreased the mRNA levels of Cyp17a1 at all selected doses when compared to the vehicle control group (Fig. 4D, n = 4–9/group, P ≤ 0.05). Following 72 h of exposure, MEHP significantly increased the mRNA levels of Hsd17b1 at all selected doses when compared to the vehicle control group (Fig. 4E, n = 4–9/group, P ≤ 0.05). However, following 96 h of exposure, MEHP significantly decreased the mRNA levels of Hsd17b1 at all selected doses when compared to the vehicle control group (Fig. 4E, n = 4–9/group, P ≤ 0.05). MEHP exposure significantly increased the mRNA levels of Cyp19a1 at the 0.1 and 1 μg/ml doses following 48 h of exposure and at all selected doses following 72 h of exposure when compared to the vehicle control group (Fig. 4F, n = 3–9/group, P ≤ 0.05). However, following 96 h of exposure, MEHP significantly decreased the mRNA levels of Cyp19a1 at all selected doses when compared to the vehicle control group (Fig. 4F, n = 4–9/group, P ≤ 0.05).
FIG. 4.

Effect of MEHP on the antral follicle mRNA levels of the steroidogenic enzymes. Antral follicles were isolated from adult CD-1 mice and were cultured with vehicle (DMSO) or MEHP (0.1–10 μg/ml) for 24–96 h. Following each 24 h time point, antral follicles were pooled per treatment group and were subjected to qPCR for the measurements of the mRNA levels of Star (A), Cyp11a1 (B), Hsd3b1 (C), Cyp17a1 (D), Hsd17b1 (E), and Cyp19a1 (F). All values were normalized to Actb. Graph represents means ± SEM from three to nine separate experiments, with 8–16 follicles/treatment group in each experiment. Asterisks (*) represent significant difference from vehicle control (P ≤ 0.05).
DISCUSSION
We utilized a neonatal whole ovarian culture system and an antral follicle culture system to investigate the direct effects of DEHP and MEHP on early folliculogenesis and to investigate the direct and temporal effects of MEHP on steroidogenesis. Our main findings suggest that MEHP, but not DEHP, has a direct effect on overactivating PI3K signaling in the ovary to potentially lead to the acceleration of primordial follicle recruitment. Additionally, we have shown that MEHP directly inhibits the production of estradiol and upstream sex steroid hormones and alters the mRNA levels of Cyp19a1 and upstream estradiol biosynthesis enzymes. We have also established the initial and time-specific effects of MEHP on antral follicle steroidogenesis. Importantly, we have determined that DEHP and MEHP have differential effects on the initial stages of folliculogenesis and on steroidogenesis.
The use of the whole neonatal ovarian culture system is essential for understanding the direct effects of chemicals on the earliest stages of folliculogenesis [65, 67]. Previous studies have shown that DEHP and MEHP accelerate primordial follicle recruitment in vivo [35, 79, 80]. The limitation of these studies is that they did not determine if these chemicals act directly on the ovary to elicit this response. A further study has shown that DEHP also affects early folliculogenesis in vitro, but that study only focused on germ cell nest breakdown and primordial follicle assembly [81]. Although these studies are influential in understanding the effects of DEHP and MEHP on early folliculogenesis, the effects of these chemicals on the activation of the dormant and already established primordial follicle pool remain unknown. Thus, we investigated the effects of DEHP and MEHP from PND4 to PND10 on the neonatal ovary, a time span when the follicular reserve is nearly established and primordial follicles naturally begin to undergo activation to the primary stage of development [22, 67]. Importantly, at PND10, the neonatal ovary contains a population of follicles that are primarily in the primordial and primary stages of development. Thus, we were able to primarily focus on the populations of follicles in which we observed DEHP-induced effects in vivo [35].
In the present study, MEHP, but not DEHP, directly decreased the percentage of germ cells counted and increased the percentage of primary follicles counted when compared to the vehicle control group. We have previously reported that oral exposure to DEHP for 10 and 30 days in adult mice decreased the percentage of primordial follicles counted and increased the percentage of primary follicles counted at doses similar to the 0.492–49.2 μM range (0.2–20 μg/ml) used in this study [35]. Although we did not observe a change in primordial follicle numbers in the present study, we likely missed the time point in which MEHP altered primordial follicle numbers. Our follicle count data represent a single snapshot following 6 days of exposure; therefore, future studies should investigate the effects of MEHP on primordial follicle numbers prior to and after 6 days of exposure. However, our primary follicle count data correlate to those obtained in the in vivo studies [35]. Thus, these data suggest that MEHP has a direct effect on the ovary, indicating that the observed effect in vivo is likely via a direct mechanism of action.
We have previously reported that the DEHP-induced acceleration of primordial follicle recruitment in vivo is likely via overactivation of the PI3K-signaling pathway [35]. Primordial follicle recruitment is a tightly regulated process involving a balance between the factors that maintain primordial follicle quiescence and promote primordial follicle activation [36–39]. Interestingly, factors within the PI3K-signaling pathway regulate primordial follicle survival, quiescence, and recruitment. Specifically, PTEN, an inhibitor of PI3K signaling, maintains primordial follicle quiescence, and pAKT, a stimulator of PI3K signaling, promotes development to the primary stage [38, 40–45]. Following 10 days of oral exposure to adult mice, DEHP at similar micromolar doses to those used in this study decreased the staining of PTEN in whole ovary and increased the staining of pAKT in the whole ovary and in the primordial and primary follicles [35]. Similarly, DEHP exposure decreased the mRNA levels of Pten in the whole adult ovary following 30 days of oral exposure [35]. In the present study, DEHP had no statistically significant effect on follicle numbers or PI3K-signaling factors. However, MEHP exposure directly decreased PTEN staining in the whole ovary, decreased the percentage of PTEN-positive primordial follicle oocytes, and increased the percentage of pAKT-positive primordial follicle oocytes, again at doses that mimic those used in the in vivo studies. Thus, MEHP has a direct effect on overactivating ovarian PI3K signaling to promote an environment conducive for the acceleration of primordial follicle recruitment, and this mechanism is similar to the mechanism by which DEHP accelerates primordial follicle recruitment in vivo [35]. Collectively, these data suggest that DEHP must be converted to MEHP in vivo to accelerate primordial follicle recruitment.
The antral follicle culture system used for the steroidogenesis experiments is an essential method to investigate the direct effects of MEHP on the antral follicle, which is the most mature follicle type capable of ovulation and is the major producer of sex steroid hormones in the female. Previous studies have determined that MEHP exposure decreases estradiol levels in vitro; however, these studies utilized secondary follicles or murine/human granulosa cells [19–21, 53]. Ovarian steroidogenesis is primarily conducted by the mature antral follicle, and it involves the strict coordination of theca and granulosa cells. Thus, the antral follicle culture system used in the present study expands upon the previously published data by using a more direct and sensitive approach [58, 82].
We have previously reported that MEHP exposure decreases estradiol levels and the mRNA levels of Cyp19a1 in cultured mouse antral follicles following 96 h of exposure. However, the temporal effects of MEHP upstream of estradiol and CYP19A1 and the mechanism by which MEHP decreases estradiol levels were not known. Further, we have previously reported that DEHP exposure decreases estradiol levels following 72 and 96 h of exposure in cultured mouse antral follicles via a mechanism involving depletion of available precursor steroid hormones beginning at 48 h of exposure [51]. However, it was unknown if DEHP and MEHP exhibited similar mechanisms of steroidogenesis inhibition.
The initial effect on steroidogenesis was observed following 24 h of exposure, where MEHP decreased the levels of androstenedione and testosterone at the 10 μg/ml dose, and this is likely attributable to the decrease in the mRNA levels of Hsd3b1 at that same time point and dose. Interestingly, these effects were ablated and no changes in the levels of steroid hormones were observed following 48 h of exposure. However, MEHP decreased the mRNA levels of Star and Cyp11a1 at that time point, and a compensatory increase in the mRNA levels of Cyp19a1 was observed. The compensatory increase in Cyp19a1 levels persisted following 72 h of exposure, and also at this time point, a compensatory increase in the levels of Hsd17b1 was observed. Perhaps these compensatory increases in the levels of steroidogenic enzymes are present to combat the toxicity of MEHP; however, these increases do not lead to increases in sex steroid hormone levels. The increase in the levels of Hsd17b1 at the 72 h time point likely explains why we do not observe a statistically significant decrease in testosterone levels following 72 h of exposure. Following 96 h of exposure, the compensatory increases in the steroidogenic enzymes were ablated, and MEHP exposure decreased the mRNA levels of Cyp17a1, Hsd17b1, and Cyp19a1. It is likely this decrease in steroidogenic enzyme levels led to the decrease in the levels of testosterone, estrone, and estradiol following 96 h of exposure. Thus, MEHP decreased the levels of immediate precursor hormones and estradiol via a mechanism involving depletion of the enzymes responsible for generating these hormones.
These findings on steroidogenesis also add to the existing literature that MEHP disrupts antral follicle functionality. Normal follicle function includes antral follicle growth, survival from apoptosis-induced atresia, and production of steroid hormones [22]. Previously, MEHP exposure for 72 and 96 h has been shown to inhibit antral follicle growth in vitro, likely via disruption of cell cycle regulators [16, 17]. Additionally, MEHP exposure for 96 h induced antral follicle atresia in vitro, likely via disruption of pro- and antiapoptotic factors following 48 h of exposure [56]. Further, MEHP exposure for 72 h decreased the activity of glutathione peroxidase in antral follicles, which led to oxidative stress in the cultured follicles [17]. In the present study, it appears that that major defects in steroidogenesis begin following 96 h of exposure. Thus, the defects in antral follicle growth, atresia, and oxidative stress prior to 96 h of exposure may promote the compensatory increases in Hsd17b1 and Cyp19a1. Further, it appears that these defects in antral follicle functionality precede and may cause the decreases in Cyp17a1, Hsd17b1, and Cyp19a1, ultimately leading to the decreases in testosterone, estrone, and estradiol levels. Specifically, the MEHP-induced apoptosis in the steroidogenically active cells may contribute to the inhibition of steroidogenesis. Future studies should address how MEHP interferes with the cross talk between antral follicle growth, atresia, and steroidogenesis.
Interestingly, DEHP and MEHP appear to exert toxicity differently depending on the system used. For the folliculogenesis studies, MEHP, but not DEHP, had a direct effect on accelerating primordial follicle recruitment via overactivation of the PI3K-signaling pathway. For the steroidogenesis studies, both DEHP and MEHP have direct effects on the antral follicle. Specifically, we previously reported that DEHP exposure directly decreases precursor steroid hormones following 48 h of exposure, and it is likely this decrease in the availability of precursor hormones that lead to the decrease in estradiol levels following 72 and 96 h of exposure [51]. This is because the levels of the steroidogenic enzymes are relatively unchanged in response to 24–96 h DEHP treatment [51]. However, in the present study, MEHP exposure decreased the mRNA levels of the steroidogenic enzymes following 96 h of exposure, likely leading to the decrease in steroid hormone levels. We hypothesize that the differences between DEHP and MEHP on folliculogenesis and steroidogenesis are attributed to ovarian metabolism of the chemicals.
The ovary contains enzymes, specifically aldehyde dehydrogenases, alcohol dehydrogenases, and lipoprotein lipases, responsible for converting DEHP to MEHP and MEHP to further metabolic derivatives [83–87]. Importantly, the levels and activity of these metabolic enzymes increase with age, meaning the metabolic capacity of the neonatal ovary is much lower than the adult [85–88]. Thus, the reason that we likely do not observe direct effects of DEHP on folliculogenesis in our neonatal ovary culture system is because the neonatal ovaries do not have the metabolic capacity or possess the enzymes needed to convert DEHP to the toxic metabolite MEHP. However, these metabolic enzymes are present in the antral follicles [85–88], and perhaps differential metabolism explains why we observe differences in the DEHP and MEHP steroidogenesis studies. Thus, the composition of the mixture between parent compound and metabolites is likely different between our DEHP and MEHP treated follicles, and it is possible that this composition dictates toxicity in the antral follicle. However, it is plausible that the parent compound and metabolite simply exert toxicity in different ways. Future studies should investigate the metabolic capacity of the antral follicle to shed insight into the differential mechanisms of DEHP- and MEHP-induced inhibition of steroidogenesis.
In conclusion, we utilized neonatal ovary and antral follicle culture systems to elucidate the mechanisms by which MEHP disrupts ovarian folliculogenesis and steroidogenesis. Collectively, our results indicate that MEHP has a direct effect on the neonatal ovary to accelerate primordial follicle recruitment, potentially via overactivation of ovarian PI3K signaling. Importantly, this mechanism in vitro is similar to the mechanism by which DEHP accelerates primordial follicle recruitment in vivo [35]. Additionally, our results indicate that MEHP has a direct effect on decreasing the mRNA levels of the steroidogenic enzymes responsible for generating estradiol and its immediate precursor steroid hormones. Interestingly, this mechanism is different than the mechanism by which DEHP inhibits steroidogenesis in vitro [51]. The results also indicate that MEHP has a nonmonotonic dose response for the folliculogenesis and steroidogenesis endpoints tested; however, this is a common observation with endocrine-disrupting chemicals such as phthalates [89, 90]. Overall, these effects of MEHP on folliculogenesis and steroidogenesis are of concern because ovarian-controlled reproductive and nonreproductive processes have the potential to be compromised.
ACKNOWLEDGMENT
The authors thank Dr. Ayelet Ziv-Gal, Liying Gao, and Shreya Patel for assistance with follicle isolations, and all the members of Dr. Flaws' laboratory for technical assistance.
Footnotes
This work was supported by the National Institute of Environmental Health grant R01ES019178 (J.A.F.) and an Interdisciplinary Environmental Toxicology Program Fellowship (P.R.H.).
REFERENCES
- Heudorf U, Mersch-Sundermann V, Angerer J. Phthalates: toxicology and exposure. Int J Hyg Environ Health. 2007;210:623–634. doi: 10.1016/j.ijheh.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Toxicological Profile for Di(2-Ethylhexyl)Phthalate (DEHP) [Internet] U.S. Department of Health and Human Services, Public Health Service, Agency for Toxic Substances & Disease Registry http://www.atsdr.cdc.gov/ToxProfiles/tp9.pdf. Accessed 16 January 2015. [PubMed] [Google Scholar]
- Doull J, Cattley R, Elcombe C, Lake BG, Swenberg J, Wilkinson C, Williams G, van Gemert M. A cancer risk assessment of di(2-ethylhexyl)phthalate: application of the new U.S. EPA Risk Assessment Guidelines. Regul Toxicol Pharmacol. 1999;29:327–357. doi: 10.1006/rtph.1999.1296. [DOI] [PubMed] [Google Scholar]
- Kavlock R, Boekelheide K, Chapin R, Cunningham M, Faustman E, Foster P, Golub M, Henderson R, Hinberg I, Little R, Seed J, Shea K, et al. NTP Center for the Evaluation of Risks to Human Reproduction: phthalates expert panel report on the reproductive and developmental toxicity of di(2-ethylhexyl) phthalate. Reprod Toxicol. 2002;16:529–653. doi: 10.1016/s0890-6238(02)00032-1. [DOI] [PubMed] [Google Scholar]
- Kato K, Silva MJ, Reidy JA, Hurtz D, III, , Malek NA, Needham LL, Nakazawa H, Barr DB, Calafat AM. Mono(2-ethyl-5-hydroxyhexyl) phthalate and mono-(2-ethyl-5-oxohexyl) phthalate as biomarkers for human exposure assessment to di-(2-ethylhexyl) phthalate. Environ Health Perspect. 2004;112:327–330. doi: 10.1289/ehp.6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogberg J, Hanberg A, Berglund M, Skerfving S, Remberger M, Calafat AM, Filipsson AF, Jansson B, Johansson N, Appelgren M, Hakansson H. Phthalate diesters and their metabolites in human breast milk, blood or serum, and urine as biomarkers of exposure in vulnerable populations. Environ Health Perspect. 2008;116:334–339. doi: 10.1289/ehp.10788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva MJ, Barr DB, Reidy JA, Malek NA, Hodge CC, Caudill SP, Brock JW, Needham LL, Calafat AM. Urinary levels of seven phthalate metabolites in the U.S. population from the National Health and Nutrition Examination Survey (NHANES) 1999–2000. Environ Health Perspect. 2004;112:331–338. doi: 10.1289/ehp.6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker K, Seiwert M, Angerer J, Heger W, Koch HM, Nagorka R, Rosskamp E, Schluter C, Seifert B, Ullrich D. DEHP metabolites in urine of children and DEHP in house dust. Int J Hyg Environ Health. 2004;207:409–417. doi: 10.1078/1438-4639-00309. [DOI] [PubMed] [Google Scholar]
- Marsee K, Woodruff TJ, Axelrad DA, Calafat AM, Swan SH. Estimated daily phthalate exposures in a population of mothers of male infants exhibiting reduced anogenital distance. Environ Health Perspect. 2006;114:805–809. doi: 10.1289/ehp.8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang PC, Kuo PL, Chou YY, Lin SJ, Lee CC. Association between prenatal exposure to phthalates and the health of newborns. Environ Int. 2009;35:14–20. doi: 10.1016/j.envint.2008.05.012. [DOI] [PubMed] [Google Scholar]
- Silva MJ, Reidy JA, Herbert AR, Preau JL, Jr, Needham LL, Calafat AM. Detection of phthalate metabolites in human amniotic fluid. Bull Environ Contam Toxicol. 2004;72:1226–1231. doi: 10.1007/s00128-004-0374-4. [DOI] [PubMed] [Google Scholar]
- Wittassek M, Angerer J, Kolossa-Gehring M, Schafer SD, Klockenbusch W, Dobler L, Gunsel AK, Muller A, Wiesmuller GA. Fetal exposure to phthalates–a pilot study. Int J Hyg Environ Health. 2009;212:492–498. doi: 10.1016/j.ijheh.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Latini G, De Felice C, Presta G, Del Vecchio A, Paris I, Ruggieri F, Mazzeo P. Exposure to di(2-ethylhexyl)phthalate in humans during pregnancy. A preliminary report. Biol Neonate. 2003;83:22–24. doi: 10.1159/000067012. [DOI] [PubMed] [Google Scholar]
- Lin L, Zheng LX, Gu YP, Wang JY, Zhang YH, Song WM. Levels of environmental endocrine disruptors in umbilical cord blood and maternal blood of low-birth-weight infants. Zhonghua Yu Fang Yi Xue Za Zhi. 2008;42:177–180. [in Chinese] [PubMed] [Google Scholar]
- Krotz SP, Carson SA, Tomey C, Buster JE. Phthalates and bisphenol do not accumulate in human follicular fluid. J Assist Reprod Genet. 2012;29:773–777. doi: 10.1007/s10815-012-9775-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RK, Singh JM, Leslie TC, Meachum S, Flaws JA, Yao HH. Di-(2-ethylhexyl) phthalate and mono-(2-ethylhexyl) phthalate inhibit growth and reduce estradiol levels of antral follicles in vitro. Toxicol Appl Pharmacol. 2010;242:224–230. doi: 10.1016/j.taap.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Craig ZR, Basavarajappa MS, Hafner KS, Flaws JA. Mono-(2-ethylhexyl) phthalate induces oxidative stress and inhibits growth of mouse ovarian antral follicles. Biol Reprod. 2012;87:152. doi: 10.1095/biolreprod.112.102467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovekamp-Swan T, Davis BJ. Mechanisms of phthalate ester toxicity in the female reproductive system. Environ Health Perspect. 2003;111:139–145. doi: 10.1289/ehp.5658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BJ, Weaver R, Gaines LJ, Heindel JJ. Mono-(2-ethylhexyl) phthalate suppresses estradiol production independent of FSH-cAMP stimulation in rat granulosa cells. Toxicol Appl Pharmacol. 1994;128:224–228. doi: 10.1006/taap.1994.1201. [DOI] [PubMed] [Google Scholar]
- Lovekamp TN, Davis BJ. Mono-(2-ethylhexyl) phthalate suppresses aromatase transcript levels and estradiol production in cultured rat granulosa cells. Toxicol Appl Pharmacol. 2001;172:217–224. doi: 10.1006/taap.2001.9156. [DOI] [PubMed] [Google Scholar]
- Lovekamp-Swan T, Jetten AM, Davis BJ. Dual activation of PPARalpha and PPARgamma by mono-(2-ethylhexyl) phthalate in rat ovarian granulosa cells. Mol Cell Endocrinol. 2003;201:133–141. doi: 10.1016/s0303-7207(02)00423-9. [DOI] [PubMed] [Google Scholar]
- Hirshfield AN. Development of follicles in the mammalian ovary. Int Rev Cytol. 1991;124:43–101. doi: 10.1016/s0074-7696(08)61524-7. [DOI] [PubMed] [Google Scholar]
- Bagur AC, Mautalen CA. Risk for developing osteoporosis in untreated premature menopause. Calcif Tissue Int. 1992;51:4–7. doi: 10.1007/BF00296207. [DOI] [PubMed] [Google Scholar]
- Cooper GS, Sandler DP. Age at natural menopause and mortality. Ann Epidemiol. 1998;8:229–235. doi: 10.1016/s1047-2797(97)00207-x. [DOI] [PubMed] [Google Scholar]
- Hu FB, Grodstein F, Hennekens CH, Colditz GA, Johnson M, Manson JE, Rosner B, Stampfer MJ. Age at natural menopause and risk of cardiovascular disease. Arch Intern Med. 1999;159:1061–1066. doi: 10.1001/archinte.159.10.1061. [DOI] [PubMed] [Google Scholar]
- Christiansen C. Prevention and treatment of osteoporosis with hormone replacement therapy. Int J Fertil Menopausal Stud. 1993;38((Suppl 1)):45–54. [PubMed] [Google Scholar]
- Bush TL, Barrett-Connor E, Cowan LD, Criqui MH, Wallace RB, Suchindran CM, Tyroler HA, Rifkind BM. Cardiovascular mortality and noncontraceptive use of estrogen in women: results from the Lipid Research Clinics Program Follow-up Study. Circulation. 1987;75:1102–1109. doi: 10.1161/01.cir.75.6.1102. [DOI] [PubMed] [Google Scholar]
- Dennerstein L, Lehert P, Burger H, Mood Dudley E. and the menopausal transition. J Nerv Ment Dis. 1999;187:685–691. doi: 10.1097/00005053-199911000-00006. [DOI] [PubMed] [Google Scholar]
- Couse JF, Korach KS. Exploring the role of sex steroids through studies of receptor deficient mice. J Mol Med (Berl) 1998;76:497–511. doi: 10.1007/s001090050244. [DOI] [PubMed] [Google Scholar]
- Britt KL, Findlay JK. Estrogen actions in the ovary revisited. J Endocrinol. 2002;175:269–276. doi: 10.1677/joe.0.1750269. [DOI] [PubMed] [Google Scholar]
- Findlay JK, Britt K, Kerr JB, O'Donnell L, Jones ME, Drummond AE, Simpson ER. The road to ovulation: the role of oestrogens. Reprod Fertil Dev. 2001;13:543–547. doi: 10.1071/rd01071. [DOI] [PubMed] [Google Scholar]
- Fauser BC, Laven JS, Tarlatzis BC, Moley KH, Critchley HO, Taylor RN, Berga SL, Mermelstein PG, Devroey P, Gianaroli L, D'Hooghe T, Vercellini P, et al. Sex steroid hormones and reproductive disorders: impact on women's health. Reprod Sci. 2011;18:702–712. doi: 10.1177/1933719111405068. [DOI] [PubMed] [Google Scholar]
- Bhattacharya P, Keating AF. Impact of environmental exposures on ovarian function and role of xenobiotic metabolism during ovotoxicity. Toxicol Appl Pharmacol. 2012;261:227–235. doi: 10.1016/j.taap.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig ZR, Wang W, Flaws JA. Endocrine-disrupting chemicals in ovarian function: effects on steroidogenesis, metabolism and nuclear receptor signaling. Reproduction. 2011;142:633–646. doi: 10.1530/REP-11-0136. [DOI] [PubMed] [Google Scholar]
- Hannon PR, Peretz J, Flaws JA. Daily exposure to di(2-ethylhexyl) phthalate alters estrous cyclicity and accelerates primordial follicle recruitment potentially via dysregulation of the phosphatidylinositol 3-kinase signaling pathway in adult mice. Biol Reprod. 2014;90:136. doi: 10.1095/biolreprod.114.119032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortune JE. The early stages of follicular development: activation of primordial follicles and growth of preantral follicles. Anim Reprod Sci. 2003;78:135–163. doi: 10.1016/s0378-4320(03)00088-5. [DOI] [PubMed] [Google Scholar]
- Oktem O, Urman B. Understanding follicle growth in vivo. Hum Reprod. 2010;25:2944–2954. doi: 10.1093/humrep/deq275. [DOI] [PubMed] [Google Scholar]
- Reddy P, Zheng W, Liu K. Mechanisms maintaining the dormancy and survival of mammalian primordial follicles. Trends Endocrinol Metab. 2010;21:96–103. doi: 10.1016/j.tem.2009.10.001. [DOI] [PubMed] [Google Scholar]
- Skinner MK. Regulation of primordial follicle assembly and development. Hum Reprod Update. 2005;11:461–471. doi: 10.1093/humupd/dmi020. [DOI] [PubMed] [Google Scholar]
- Kim JY. Control of ovarian primordial follicle activation. Clin Exp Reprod Med. 2012;39:10–14. doi: 10.5653/cerm.2012.39.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Kawamura K, Cheng Y, Liu S, Klein C, Liu S, Duan EK, Hsueh AJ. Activation of dormant ovarian follicles to generate mature eggs. Proc Natl Acad Sci U S A. 2010;107:10280–10284. doi: 10.1073/pnas.1001198107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy P, Adhikari D, Zheng W, Liang S, Hamalainen T, Tohonen V, Ogawa W, Noda T, Volarevic S, Huhtaniemi I, Liu K. PDK1 signaling in oocytes controls reproductive aging and lifespan by manipulating the survival of primordial follicles. Hum Mol Genet. 2009;18:2813–2824. doi: 10.1093/hmg/ddp217. [DOI] [PubMed] [Google Scholar]
- Reddy P, Liu L, Adhikari D, Jagarlamudi K, Rajareddy S, Shen Y, Du C, Tang W, Hamalainen T, Peng SL, Lan ZJ, Cooney AJ, et al. Oocyte-specific deletion of Pten causes premature activation of the primordial follicle pool. Science. 2008;319:611–613. doi: 10.1126/science.1152257. [DOI] [PubMed] [Google Scholar]
- John GB, Gallardo TD, Shirley LJ, Castrillon DH. Foxo3 is a PI3K-dependent molecular switch controlling the initiation of oocyte growth. Dev Biol. 2008;321:197–204. doi: 10.1016/j.ydbio.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Nagaraju G, Liu Z, Liu K. Functional roles of the phosphatidylinositol 3-kinases (PI3Ks) signaling in the mammalian ovary. Mol Cell Endocrinol. 2012;356:24–30. doi: 10.1016/j.mce.2011.05.027. [DOI] [PubMed] [Google Scholar]
- Davis BJ, Maronpot RR, Heindel JJ. Di-(2-ethylhexyl) phthalate suppresses estradiol and ovulation in cycling rats. Toxicol Appl Pharmacol. 1994;128:216–223. doi: 10.1006/taap.1994.1200. [DOI] [PubMed] [Google Scholar]
- Hirosawa N, Yano K, Suzuki Y, Sakamoto Y. Endocrine disrupting effect of di-(2-ethylhexyl)phthalate on female rats and proteome analyses of their pituitaries. Proteomics. 2006;6:958–971. doi: 10.1002/pmic.200401344. [DOI] [PubMed] [Google Scholar]
- Ma M, Zhang Y, Pei X, Duan Z. Effects of di-(2-ethylhexyl) phthalate exposure on reproductive development and PPARs in prepubertal female rats. Wei Sheng Yan Jiu. 2011;40:688–692. [in Chinese] [PubMed] [Google Scholar]
- Laskey JW, Berman E. Steroidogenic assessment using ovary culture in cycling rats: effects of bis(2-diethylhexyl)phthalate on ovarian steroid production. Reprod Toxicol. 1993;7:25–33. doi: 10.1016/0890-6238(93)90006-s. [DOI] [PubMed] [Google Scholar]
- Svechnikova I, Svechnikov K, Soder O. The influence of di-(2-ethylhexyl) phthalate on steroidogenesis by the ovarian granulosa cells of immature female rats. J Endocrinol. 2007;194:603–609. doi: 10.1677/JOE-07-0238. [DOI] [PubMed] [Google Scholar]
- Hannon PR, Brannick KE, Wang W, Gupta RK, Flaws JA. Di(2-ethylhexyl) phthalate inhibits antral follicle growth, induces atresia, and inhibits steroid hormone production in cultured mouse antral follicles. Toxicol Appl Pharmacol. 2015;284:42–53. doi: 10.1016/j.taap.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinsberg J, Wegener-Toper P, van der Ven K, van der Ven H, Klingmueller D. Effect of mono-(2-ethylhexyl) phthalate on steroid production of human granulosa cells. Toxicol Appl Pharmacol. 2009;239:116–123. doi: 10.1016/j.taap.2009.05.022. [DOI] [PubMed] [Google Scholar]
- Inada H, Chihara K, Yamashita A, Miyawaki I, Fukuda C, Tateishi Y, Kunimatsu T, Kimura J, Funabashi H, Miyano T. Evaluation of ovarian toxicity of mono-(2-ethylhexyl) phthalate (MEHP) using cultured rat ovarian follicles. J Toxicol Sci. 2012;37:483–490. doi: 10.2131/jts.37.483. [DOI] [PubMed] [Google Scholar]
- Hanukoglu I. Steroidogenic enzymes: structure, function, and role in regulation of steroid hormone biosynthesis. J Steroid Biochem Mol Biol. 1992;43:779–804. doi: 10.1016/0960-0760(92)90307-5. [DOI] [PubMed] [Google Scholar]
- Miller WL. Molecular biology of steroid hormone synthesis. Endocr Rev. 1988;9:295–318. doi: 10.1210/edrv-9-3-295. [DOI] [PubMed] [Google Scholar]
- Craig ZR, Singh J, Gupta RK, Flaws JA. Co-treatment of mouse antral follicles with 17beta-estradiol interferes with mono-2-ethylhexyl phthalate (MEHP)-induced atresia and altered apoptosis gene expression. Reprod Toxicol. 2014;45:45–51. doi: 10.1016/j.reprotox.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillum N, Karabekian Z, Swift LM, Brown RP, Kay MW, Sarvazyan N. Clinically relevant concentrations of di (2-ethylhexyl) phthalate (DEHP) uncouple cardiac syncytium. Toxicol Appl Pharmacol. 2009;236:25–38. doi: 10.1016/j.taap.2008.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenie S, Smitz J. Steroidogenesis-disrupting compounds can be effectively studied for major fertility-related endpoints using in vitro cultured mouse follicles. Toxicol Lett. 2009;185:143–152. doi: 10.1016/j.toxlet.2008.12.015. [DOI] [PubMed] [Google Scholar]
- Mlynarcikova A, Nagyova E, Fickova M, Scsukova S. Effects of selected endocrine disruptors on meiotic maturation, cumulus expansion, synthesis of hyaluronan and progesterone by porcine oocyte-cumulus complexes. Toxicol in Vitro. 2009;23:371–377. doi: 10.1016/j.tiv.2008.12.017. [DOI] [PubMed] [Google Scholar]
- Cobellis L, Latini G, De Felice C, Razzi S, Paris I, Ruggieri F, Mazzeo P, Petraglia F. High plasma concentrations of di-(2-ethylhexyl)-phthalate in women with endometriosis. Hum Reprod. 2003;18:1512–1515. doi: 10.1093/humrep/deg254. [DOI] [PubMed] [Google Scholar]
- Pak VM, Nailon RE, McCauley LA. Controversy: neonatal exposure to plasticizers in the NICU. MCN Am J Matern Child Nurs. 2007;32:244–249. doi: 10.1097/01.NMC.0000281965.45905.c0. [DOI] [PubMed] [Google Scholar]
- Kamrin MA. Phthalate risks, phthalate regulation, and public health: a review. J Toxicol Environ Health B Crit Rev. 2009;12:157–174. doi: 10.1080/10937400902729226. [DOI] [PubMed] [Google Scholar]
- Sjoberg PO, Bondesson UG, Sedin EG, Gustafsson JP. Exposure of newborn infants to plasticizers. Plasma levels of di-(2-ethylhexyl) phthalate and mono-(2-ethylhexyl) phthalate during exchange transfusion. Transfusion. 1985;25:424–428. doi: 10.1046/j.1537-2995.1985.25586020115.x. [DOI] [PubMed] [Google Scholar]
- Hannon PR, Flaws JA. The effects of phthalates on the ovary. Front Endocrinol. 2015;6:8. doi: 10.3389/fendo.2015.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine PJ, Sipes IG, Skinner MK, Hoyer PB. Characterization of a rat in vitro ovarian culture system to study the ovarian toxicant 4-vinylcyclohexene diepoxide. Toxicol Appl Pharmacol. 2002;184:107–115. [PubMed] [Google Scholar]
- Greenfeld CR, Roby KF, Pepling ME, Babus JK, Terranova PF, Flaws JA. Tumor necrosis factor (TNF) receptor type 2 is an important mediator of TNF alpha function in the mouse ovary. Biol Reprod. 2007;76:224–231. doi: 10.1095/biolreprod.106.055509. [DOI] [PubMed] [Google Scholar]
- Eppig JJ, O'Brien MJ. Development in vitro of mouse oocytes from primordial follicles. Biol Reprod. 1996;54:197–207. doi: 10.1095/biolreprod54.1.197. [DOI] [PubMed] [Google Scholar]
- Pedersen T, Peters H. Proposal for a classification of oocytes and follicles in the mouse ovary. J Reprod Fertil. 1968;17:555–557. doi: 10.1530/jrf.0.0170555. [DOI] [PubMed] [Google Scholar]
- Flaws JA, Doerr JK, Sipes IG, Hoyer PB. Destruction of preantral follicles in adult rats by 4-vinyl-1-cyclohexene diepoxide. Reprod Toxicol. 1994;8:509–514. doi: 10.1016/0890-6238(94)90033-7. [DOI] [PubMed] [Google Scholar]
- Adhikari D, Liu K. Molecular mechanisms underlying the activation of mammalian primordial follicles. Endocr Rev. 2009;30:438–464. doi: 10.1210/er.2008-0048. [DOI] [PubMed] [Google Scholar]
- Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- Stokoe D. The phosphoinositide 3-kinase pathway and cancer. Expert Rev Mol Med. 2005;7:1–22. doi: 10.1017/S1462399405009361. [DOI] [PubMed] [Google Scholar]
- Masters RA, Crean BD, Yan W, Moss AG, Ryan PL, Wiley AA, Bagnell CA, Bartol FF. Neonatal porcine endometrial development and epithelial proliferation affected by age and exposure to estrogen and relaxin. Domest Anim Endocrinol. 2007;33:335–346. doi: 10.1016/j.domaniend.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Barnett KR, Tomic D, Gupta RK, Miller KP, Meachum S, Paulose T, Flaws JA. The aryl hydrocarbon receptor affects mouse ovarian follicle growth via mechanisms involving estradiol regulation and responsiveness. Biol Reprod. 2007;76:1062–1070. doi: 10.1095/biolreprod.106.057687. [DOI] [PubMed] [Google Scholar]
- Paulose T, Hernandez-Ochoa I, Basavarajappa MS, Peretz J, Flaws JA. Increased sensitivity of estrogen receptor alpha overexpressing antral follicles to methoxychlor and its metabolites. Toxicol Sci. 2011;120:447–459. doi: 10.1093/toxsci/kfr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RK, Miller KP, Babus JK, Flaws JA. Methoxychlor inhibits growth and induces atresia of antral follicles through an oxidative stress pathway. Toxicol Sci. 2006;93:382–389. doi: 10.1093/toxsci/kfl052. [DOI] [PubMed] [Google Scholar]
- Miller KP, Gupta RK, Greenfeld CR, Babus JK, Flaws JA. Methoxychlor directly affects ovarian antral follicle growth and atresia through Bcl-2- and Bax-mediated pathways. Toxicol Sci. 2005;88:213–221. doi: 10.1093/toxsci/kfi276. [DOI] [PubMed] [Google Scholar]
- Pocar P, Fiandanese N, Secchi C, Berrini A, Fischer B, Schmidt JS, Schaedlich K, Borromeo V. Exposure to di(2-ethyl-hexyl) phthalate (DEHP) in utero and during lactation causes long-term pituitary-gonadal axis disruption in male and female mouse offspring. Endocrinology. 2012;153:937–948. doi: 10.1210/en.2011-1450. [DOI] [PubMed] [Google Scholar]
- Zhang XF, Zhang LJ, Li L, Feng YN, Chen B, Ma JM, Huynh E, Shi QH, De Felici M, Shen W. Diethylhexyl phthalate exposure impairs follicular development and affects oocyte maturation in the mouse. Environ Mol Mutagen. 2013;54:354–361. doi: 10.1002/em.21776. [DOI] [PubMed] [Google Scholar]
- Moyer B, Hixon ML. Reproductive effects in F1 adult females exposed in utero to moderate to high doses of mono-2-ethylhexylphthalate (MEHP) Reprod Toxicol. 2012;34:43–50. doi: 10.1016/j.reprotox.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Li L, Qin XS, Zhou Y, Zhang XF, Wang LQ, De Felici M, Chen H, Qin GQ, Shen W. Di-(2-ethylhexyl) phthalate and bisphenol A exposure impairs mouse primordial follicle assembly in vitro. Environ Mol Mutagen. 2014;55:343–353. doi: 10.1002/em.21847. [DOI] [PubMed] [Google Scholar]
- Cortvrindt RG, Smitz JE. Follicle culture in reproductive toxicology: a tool for in-vitro testing of ovarian function? Hum Reprod Update. 2002;8:243–254. doi: 10.1093/humupd/8.3.243. [DOI] [PubMed] [Google Scholar]
- Wittassek M, Angerer J. Phthalates: metabolism and exposure. Int J Androl. 2008;31:131–138. doi: 10.1111/j.1365-2605.2007.00837.x. [DOI] [PubMed] [Google Scholar]
- Ito Y, Kamijima M, Hasegawa C, Tagawa M, Kawai T, Miyake M, Hayashi Y, Naito H, Nakajima T. Species and inter-individual differences in metabolic capacity of di(2-ethylhexyl)phthalate (DEHP) between human and mouse livers. Environ Health Prev Med. 2014;19:117–125. doi: 10.1007/s12199-013-0362-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alnouti Y, Klaassen CD. Tissue distribution, ontogeny, and regulation of aldehyde dehydrogenase (Aldh) enzymes mRNA by prototypical microsomal enzyme inducers in mice. Toxicol Sci. 2008;101:51–64. doi: 10.1093/toxsci/kfm280. [DOI] [PubMed] [Google Scholar]
- Messiha FS. The gender of alcohol and aldehyde dehydrogenases. Neurobehav Toxicol Teratol. 1983;5:205–210. [PubMed] [Google Scholar]
- Semenkovich CF, Chen SH, Wims M, Luo CC, Li WH, Chan L. Lipoprotein lipase and hepatic lipase mRNA tissue specific expression, developmental regulation, and evolution. J Lipid Res. 1989;30:423–431. [PubMed] [Google Scholar]
- Kawai T, Mihara T, Kawashima I, Fujita Y, Ikeda C, Negishi H, Richards JS, Shimada M. Endogenous acetaldehyde toxicity during antral follicular development in the mouse ovary. Reprod Toxicol. 2012;33:322–330. doi: 10.1016/j.reprotox.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beausoleil C, Ormsby JN, Gies A, Hass U, Heindel JJ, Holmer ML, Nielsen PJ, Munn S, Schoenfelder G. Low dose effects and non-monotonic dose responses for endocrine active chemicals: science to practice workshop: workshop summary. Chemosphere. 2013;93:847–856. doi: 10.1016/j.chemosphere.2013.06.043. [DOI] [PubMed] [Google Scholar]
- Vandenberg LN. Non-monotonic dose responses in studies of endocrine disrupting chemicals: bisphenol A as a case study. Dose Response. 2014;12:259–276. doi: 10.2203/dose-response.13-020.Vandenberg. [DOI] [PMC free article] [PubMed] [Google Scholar]