

Abstract
In nonprimate species, it has been well established that prostaglandin F2 alpha (PGF2alpha) initiates luteolysis. Changes in intracellular cholesterol concentrations caused by modulation of cholesterol uptake and efflux may mediate PGF2alpha-induced luteolysis. These changes in cholesterol efflux and uptake are controlled, in part, by the liver x receptors (LXR) alpha (NR1H3) and beta (NR1H2), nuclear receptors that increase expression of genes necessary for cholesterol efflux or limiting cholesterol uptake. Therefore, we hypothesized that PGF2alpha reduces expression of cholesterol uptake and increases expression of cholesterol efflux genes, mediated in part by enhanced LXR activity. To test this hypothesis, an induced luteolysis model was used whereby ewes were treated during their midluteal phase with saline or PGF2alpha and corpora lutea (CL) collected 12, 24, or 48 h later for determination of mRNA and protein concentrations by quantitative real-time PCR and Western blot analysis, respectively. As a complementary approach, CL undergoing spontaneous luteolysis were compared to midluteal phase CL. The lipoprotein receptors responsible for cholesterol uptake were significantly decreased in both luteolysis models. Expression of the LXR target gene ATP binding cassette subfamily A1 (ABCA1), an important mediator of cholesterol efflux, was significantly increased in both experimental models. Chromatin immunoprecipitation confirmed that PGF2alpha treatment resulted in enhanced NR1H3 and NR1H2 binding to the ABCA1 promoter. Qualitative changes in lipid droplet distribution were also observed following PGF2alpha treatment. These data support the hypothesis that reduced cholesterol uptake and increased efflux mediate luteolysis in sheep, which is partially controlled by PGF2alpha stimulation of LXR activity.
Keywords: cholesterol, corpus luteum, liver x receptor, luteolysis, ovine, prostaglandin F2 alpha
INTRODUCTION
The corpus luteum (CL) is a transient endocrine gland that forms from the antecedent ovulatory follicle. It is responsible for the synthesis of progesterone, a hormone absolutely required for maintenance of early pregnancy in all mammalian species [1]. Timely regression of the CL (luteolysis) at the end of nonfertile cycles is necessary to allow initiation of the next ovarian cycle, whereas luteolysis must be prevented if conception occurs. Numerous studies have demonstrated that prostaglandin F2 alpha (PGF2α) of uterine origin initiates luteolysis in nonprimate species [2, 3]. Hysterectomy in primates, however, does not extend the normal lifespan of the CL, indicating that the luteolytic signal is not of uterine origin and may be an intraovarian mechanism [4–6]. Prostaglandin F2α may still have a luteolytic role in primates as intraluteal infusion of PGF2α analogs results in premature menses in monkeys and women [5, 7]. However, intraluteal administration of a prostaglandin synthesis inhibitor in macaques shortened CL lifespan rather than extending it as was predicted [8]. Thus, the mechanisms causing luteolysis of the primate CL are not known.
It has been previously reported that increased efflux and reduced uptake of cholesterol within the primate CL may mediate luteolysis [9, 10]. These changes are controlled, in part, by the liver x receptors (LXR) α (NR1H3) and/or β (NR1H2), which are members of the nuclear hormone receptor superfamily [11]. These receptors bind various precursors or derivatives of cholesterol and thereby serve as cholesterol sensors [12]. They increase the expression of numerous genes involved in reverse cholesterol transport and can inhibit cholesterol uptake by decreasing lipoprotein receptors [13, 14], which ultimately causes a reduction in intracellular cholesterol concentrations [11]. As cholesterol is the precursor to all steroids. including progesterone, reducing luteal cholesterol may inhibit progesterone production and cause luteolysis. In ruminants, including sheep and cattle, where uterine-derived PGF2α is the known luteolysin, the role of cholesterol uptake in luteolysis has received limited attention. While PGF2α inhibits lipoprotein-stimulated steroidogenesis, it is believed that this effect is not at the level of cholesterol uptake [2]. Furthermore, the role of cholesterol efflux in PGF2α-induced luteolysis has not been evaluated. Given that PGF2α is known to cause luteolysis in nonprimate species and PGF2α may play a role in luteolysis of the primate CL, our objective is to determine if changes in expression of cholesterol efflux and uptake pathway members, controlled in part via the LXRs, are associated with PGF2α-induced and spontaneous luteolysis in an ovine model.
In the primate CL during spontaneous luteolysis, there is increased expression of genes encoding the cholesterol efflux proteins ATP binding cassette subfamilies A1 (ABCA1) and G1 (ABCG1), while the lipoprotein receptors that are responsible for extracellular cholesterol uptake, including scavenger receptor class B member 1 (SCARB1) and low-density lipoprotein receptor (LDLR), decrease in expression [10, 15]. In addition to ABCA1 and ABCG1, another LXR target gene includes myosin regulatory light chain interacting protein (MYLIP) that encodes an ubiquitin protein ligase involved in LDLR degradation [14]. MYLIP was increased in primate luteal cells by a synthetic LXR agonist [9] and thus may play a role as the intermediate between LXR and reduced LDLR protein. Furthermore, NR1H3 itself is an LXR target gene [16, 17], and its expression is increased during spontaneous luteolysis of the primate CL [10]. We hypothesize that PGF2α will enhance LXR activity resulting in increased expression of the LXR targets NR1H3, ABCA1, ABCG1, and MYLIP, while SCARB1 and LDLR will decrease during PGF2α-induced and spontaneous luteolysis in sheep.
MATERIALS AND METHODS
CL Collection
All the procedures involving animals were approved by the University of Arizona Institutional Animal Care and Use Committee. Western range ewes (Columbia-Rambouillet cross, 2- to 5-yr of age) were used in this study. Estrous cycles were synchronized with combinations of controlled internal drug release devices (CIDR; Pfizer Animal Health, New York, NY) and Lutalyse (Pfizer). For the induced luteolysis model, at approximately Day 8 of the synchronized luteal phase, ewes were treated with either saline or PGF2α (Lutalyse, 15 mg intramuscular) and serum collected via jugular venipuncture at 0, 2, 4, 8, 12, 24, 36, and 48 h postinjection or until the time of CL recovery. Corpora lutea were collected at 12, 24, or 48 h postinjection. For the spontaneous luteolysis model, CL were collected from ewes between Days 17–19 post-CIDR removal (approximately Days 14–16 of the luteal phase) and compared to midluteal phase CL collected from saline-treated ewes in the induced luteolytic model (approximately Days 9–10 of the luteal phase when CL were recovered). A series of blood samples were collected from ewes beginning 3 days after CIDR removal and continuing until the day of CL recovery with a serum progesterone level of <1 ng/ml on the day of recovery used as the criteria for identifying CL undergoing luteolysis. The CL (n = 5–6 for each treatment at each time point) were sectioned with one piece embedded in optimal cutting temperature media. All the sections were snap-frozen in liquid nitrogen and stored at −80°C until analyzed.
Progesterone Extractions and Assay
Progesterone was extracted from serum using a 10-fold volume of ethyl ether. The solution was vortexed for 1 min, incubated at room temperature for 5 min, and frozen in a dry ice and acetone bath. The organic layer (supernatant) was transferred to a new tube and the procedure was repeated once more. Ether extracts were dried under a stream of nitrogen in a water bath set at 30°C to compensate for evaporative cooling. Extracts were resuspended in assay buffer—0.1 M potassium phosphate, pH 7.4, 400 mM NaCl, 1 mM ethylenediaminetetraacetic acid, 0.1% (w:v) bovine serum albumin, and 0.01% (w:v) NaN3—by vortexing for 1 min followed by a 5 min incubation at room temperature, repeated three times total.
The assay for progesterone was a colorimetric enzyme immunoassay performed in 96 well plates. Briefly, clear high-binding 96 well plates (Greiner Bio-One, Monroe, NC) were coated with 500 ng/well of recombinant protein A/G in 150 μl of PBS overnight at 4°C. The wells were washed three times with PBS using an ELx50 automatic plate washer (Bio-Tek, Winooski, VT), and wells blocked with 5% nonfat dry milk (w:v) in PBS for 1 h at room temperature. Wells were washed again with PBS, and standards (2-fold dilutions of progesterone ranging from 80 to 0.625 pg/well), samples, and controls diluted in assay buffer were added. A progesterone-acetylcholinesterase conjugate (Cayman Chemical, Ann Arbor, MI) in assay buffer was used as the tracer. Rabbit anti-progesterone-11-bovine serum albumin serum (P132; Cal Bioreagents, San Mateo, CA) was added at a 1:400 000 final dilution in assay buffer. After addition of the antibody, plates were incubated for 1 h at room temperature with constant agitation. Wells were washed three times with wash buffer, that is, 0.01 M potassium phosphate, pH 7.4, and 0.05% (v:v) Tween 20, and Ellman reagent containing acetylthiocholine substrate (Cayman Chemical) diluted in water was added and incubated for 1–1.5 h at room temperature with constant agitation. Absorbance at 415 nm was determined with a Synergy H1 Hybrid plate reader (Bio-Tek). Background-adjusted absorbance values were determined for each standard, sample, and control, and concentrations of progesterone determined by extrapolation from the standard curve. Quality controls (QC) consisted of charcoal-stripped ovine serum with three different amounts of progesterone added and assayed at dilutions necessary to create low (60%–80% bound/maximum binding or B/B0), medium (40%–60% B/B0), and high (20%–40% B/B0) concentrations of progesterone. The sensitivity (80% B/B0) of the assay was 2.6 pg/well. The intraassay coefficients of variation were 9.1%, 6.1%, and 6.6% for the low, medium, and high QCs, respectively. The interassay coefficients of variation were 9.4%, 7.0%, and 10.7% for the low, medium, and high QCs, respectively.
Quantitative Real-Time PCR
Messenger RNA was extracted from frozen CL with Trizol reagent (Life Technologies Inc., Grand Island, NY) and further purified over RNeasy columns according to manufacturer's instructions. Messenger RNA purity was determined via 260/280 nm spectrophotometry ratios, and RNA concentrations determined with a fluorometric assay (Life Technologies) following the manufacturer's instructions. The RNA was transcribed into cDNA with the High Capacity cDNA Reverse Transcription kit (Life Technologies) following the manufacturer's instructions.
Quantitative real-time PCR (QPCR) was performed for NR1H3, NR1H2, ABCA1, ABCG1, SCARB1, LDLR, and MYLIP using Taqman probes as described previously [18] with mitochondrial ribosomal protein S10 (MRPS10) as the housekeeping gene. The corresponding forward and reverse primer and Taqman probe sequences are listed in Supplemental Table S1 (Supplemental data are available online at www.biolreprod.org). The reaction was performed with a StepOne Plus Real-Time PCR system (Life Technologies). Relative mRNA abundance was determined by extrapolation of threshold (Ct) values from a standard curve of serial cDNA dilutions and normalized to MRPS10.
Western Blot Analysis
Rabbit monoclonal antibodies against SCARB1 (ab52629), NR1H3 (ab176323), and a rabbit polyclonal NR1H2 antibody (ab28479) were purchased from Abcam (Cambridge, MA). A mouse monoclonal antibody against ABCA1 (sc-58219) and polyclonal rabbit β tubulin (TUBB) (sc-9104) were both purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Monoclonal mouse TUBB (T8328) was purchased from Sigma-Aldrich (St. Louis, MO).
Frozen CL were ground in RIPA buffer containing a protease and phosphatase inhibitor cocktail (Thermo Scientific Pierce, Rockford, IL), and incubated on an end-over-end rotator at 4°C for 2 h. The lysates were cleared by centrifugation at 10 000 × g for 20 min at 4°C, and the supernatant transferred to a new tube. Protein concentrations were determined by a bicinchoninic acid protein assay (Thermo Scientific Pierce) following the manufacturer's recommendations. Proteins (30 μg each) were resolved with 4%–15% gradient polyacrylamide TGX gels (Bio-Rad, Hercules, CA) and transferred to nitrocellulose membranes using a Bio-Rad Trans-Blot Turbo. Primary antibodies were used at the following concentrations: anti-NR1H3, 0.03 μg/ml; anti-SCARB1, 0.02 μg/ml; and anti-NR1H2, anti-ABCA1, rabbit anti-TUBB, and mouse anti-TUBB were used at 0.5 μg/ml. A two-color detection scheme was used whereby membranes were simultaneously probed for a target protein (NR1H2, NR1H3, ABCA1, or SCARB1) and the housekeeping protein TUBB with primary antibodies produced in different host species. Appropriate secondary antibodies conjugated to IRDye 680RD or IRDye 800CW (LI-COR, Inc., Lincoln, NE) were used at 1:2500 to 1:4000 dilutions to detect bound primary antibodies. Membranes were imaged and signals quantified with a Li-COR Odyssey CLx and Image Studio software. Data were normalized to TUBB for each target protein.
Chromatin Immunoprecipitation
Corpora lutea collected at 48 h in the induced luteolysis model as well as late luteal phase CL were pulverized while frozen and the tissue pieces incubated in 9 ml of PBS with 1% (w:v) formaldehyde on an end-over-end rotator at room temperature for 10 min. Glycine was added to a 125 mM final concentration and incubated an additional 5 min. The tissue pieces were pelleted by centrifugation at 1000 × g for 5 min, supernatant was aspirated, and the tissue was washed twice with 10 ml of cold PBS. The tissue pieces were homogenized in 1 ml of RIPA buffer containing fresh protease and phosphatase inhibitor cocktail, and lysates were incubated on ice for 10 min. The lysates were sonicated in an ice water bath using a Branson 250 digital sonifier programmed to cycle at a 20% amplitude with 5 sec on followed by 45 sec off and a 3 min total sonication time. The sonicated lysates were centrifuged at 8000 × g for 1 min and the supernatant recovered. An aliquot of cleared lysate was treated with 25 μg RNase A and 50 μg proteinase K in a buffer containing 250 mM NaCl, and heated at 65°C for 1.5 h to reverse cross-links. The DNA concentration in this aliquot was determined using a fluorometric assay (Life Technologies) following the manufacturer's recommendations. Lysates were snap-frozen and stored at −80°C until chromatin immunoprecipitation (ChIP) was performed. A total of 5 μg (DNA concentration) was used in each ChIP reaction, and 0.5 μg was saved for input control. Chromatin was diluted to a 500 μl final volume in binding/wash buffer (50 mM Tris, 150 mM NaCl, 1% Triton X-100) containing protease and phosphatase inhibitors, and 3 μg of antibody was added per reaction. Tubes were incubated on an end-over-end rotator at 4°C overnight. Antibodies used for ChIP were mouse monoclonal immunoglobulin G2a (IgG2a), including anti-NR1H3 (clone PPZ0412), anti-NR1H2 (clone K8917), and nonspecific control IgG2a (clone MOPC-173). The following day, 25 μl of protein G-conjugated Dynabeads (Life Technologies) were added and incubated for 1 h at 4°C. The beads were recovered using a magnet and washed three times at 4°C for 5 min each wash with 1 ml of binding/wash buffer. The beads were then washed once in final wash buffer (50 mM Tris, 500 mM NaCl, 1% Triton X-100). Elutions were performed with 130 μl elution buffer (100 mM NaHCO3, 250 mM NaCl, 1% SDS) containing 20 μg RNase A and 40 μg proteinase K, and ChIP reactions and input controls were incubated on a thermal mixer at 65°C for 1.5 h. The supernatant was recovered and DNA purified using PCR purification columns (Life Technologies) following the manufacturer's recommendations. Purified DNA was analyzed by QPCR in a duplex reaction with primers and a Taqman probe (Supplemental Table S1) specific for the region spanning the LXR response element (LXRE) on the ABCA1 gene, and MRPS10 was quantified in the same reaction as a control for nonspecific DNA carryover. Serial dilutions of purified sonicated DNA were included to verify linear amplification and PCR efficiency, and Ct values from ChIP reactions and input controls were used to calculate the percentage of DNA recovered relative to the input. The percentage recovery of the anti-NR1H2 and NR1H3 ChIP reactions was then divided by the corresponding IgG2a control value for each primer and probe combination to determine fold enrichment relative to nonspecific IgG2a at each locus, and the ratio of ABCA1 LXRE to MRPS10 was calculated to determine specific enrichment of the LXRE on the ABCA1 promoter.
Neutral Lipid Staining
Corpora lutea frozen in optimal cutting temperature media were cut in 10 μm slices, adhered to glass slides, and stored at −80°C. Frozen slides (n = 3–4 animals per treatment and time point) were fixed in 50 mM Tris, 150 mM NaCl, pH 7.4 with 1% calcium chloride and 4% formaldehyde (w:v) for 15 min at room temperature. Slides were washed twice with PBS and permeabilized for 10 min at room temperature in PBS with 0.1% saponin (w:v). Slides were washed twice again with PBS and stained for 30 min at room temperature with LipidTox Red (1:100; Life Technologies) and HCS Cell Mask Blue (1:1000; Life Technologies) diluted in PBS. Slides were washed twice with PBS, cover slips mounted using ProLong Gold reagent (Life Technologies), and cured for 24 h at room temperature. Control slides were included that had been treated with methanol after formaldehyde fixation to extract the lipids. Images were captured using an EVOS fluorescent microscope (Life Technologies) equipped with 20× (numerical aperture or NA = 0.45) and 40× (NA = 0.65) objectives and fluorescent light cubes for blue (peak excitation/emission 357/447) and red (excitation/emission 585/624) fluorescence.
Statistical Analysis
For the induced luteolysis model, the progesterone data were analyzed by mixed effects regression analysis to account for repeated measures. Treatment (coded 0 for saline and 1 for PGF2α), hours postinjection, and treatment × time interaction were included as fixed effects in the regression model, with ewe identification as the random effect. Differences between treatments at each time point were determined from the treatment × time interaction coefficient. Normalized QPCR and Western blot data were analyzed using linear regression analysis. Hours postinjection and treatment × time interaction were included as predictors in the regression model. Ratios for specific ABCA1 LXRE enrichment as determined by ChIP were compared by Student t-test. Statistical comparisons in the spontaneous luteolysis model were made by t-test. Statistical analyses were performed with Stata Version 11 (College Station, TX), and differences were considered significant at P < 0.05.
RESULTS
Serum Progesterone Concentrations
In the induced luteolysis model, serum progesterone remained relatively constant in saline-treated ewes throughout the collection period while it decreased significantly by 4 h in PGF2α-treated ewes and remained significantly lower than saline-treated ewes for the duration of the collection period (P < 0.05, Fig. 1A). For CL collected during the late luteal phase for analysis of the effects of spontaneous luteolysis, progesterone concentrations were lowest at 3 days post-CIDR removal and then rose steadily to a peak at 11 days before declining thereafter. Serum progesterone was less than 1 ng/ml on the day of CL recovery (Fig. 1B).
FIG. 1.

Serum concentrations of progesterone in both luteolytic models. A) Progesterone concentrations during induced luteolysis. Error bars indicate ± one SEM. Asterisk denotes significant (P < 0.05) difference between saline and PGF2α treatments at the corresponding time point. B) Mean progesterone concentrations during the luteal phase leading up to collection of late luteal phase CL for analysis of the effects of spontaneous luteolysis.
QPCR
There were no significant differences in mRNA concentrations of NR1H2 or NR1H3 between saline- and PGF2α-treated samples during induced luteolysis, while there was a significant decrease in NRIH3 but not NRIH2 expression during spontaneous luteolysis (Fig. 2). Furthermore, no difference was detected in ABCG1 mRNA in either experimental model (Fig. 2). There was no significant difference between saline and PGF2α in ABCA1 mRNA 12 h after injection. However, ABCA1 mRNA was greater in PGF2α-treated CL after 24 and 48 h as well as in late-stage CL undergoing spontaneous luteolysis (P < 0.05, Fig. 2).
FIG. 2.

Effect of luteolysis on mRNA of genes involved in cholesterol efflux. Results from both the induced and spontaneous luteolysis models are plotted within each chart. The x axis has been separated between the two luteolysis models. Error bars indicate one SEM, and asterisk denotes significant difference between saline and PGF2α treatments at the corresponding time point (induced luteolysis) or between mid- and late luteal phase CL (spontaneous luteolysis).
Concentrations of SCARB1 were significantly lower in PGF2α-treated CL at 12 and 48 h posttreatment and in late-stage CL (P < 0.05, Fig. 3). Concentrations of LDLR were significantly lower (P < 0.05) in PGF2α compared to saline treatments at all time points in the induced luteolysis model as well as in spontaneous luteolysis (Fig. 3). No significant differences in MYLIP mRNA were found in either luteolysis model (Fig. 3).
FIG. 3.

Effect of luteolysis on mRNA of genes involved in cholesterol uptake or limiting cholesterol uptake. The lipoprotein receptors SCARB1 and LDLR are responsible for cholesterol uptake, while MYLIP encodes a protein that can restrict cholesterol uptake via LDLR. Error bars indicate one SEM, and asterisk denotes significant difference between saline and PGF2α treatments at the corresponding time point or between mid- and late luteal phase CL.
Western Blot Analysis
There were no significant differences in NR1H2 protein concentrations in the induced luteolytic model while there was a significant decrease during spontaneous luteolysis (P < 0.05, Fig. 4). Concentrations of NR1H3 protein were significantly lower at 48 h postinjection in PGF2α-treated CL and during spontaneous luteolysis (P < 0.05, Fig. 4). PGF2α increased ABCA1 protein significantly (P < 0.05) by 48 h postinjection during induced luteolysis; however, ABCA1 did not significantly increase in spontaneous luteolysis (Fig. 4). All three time points had significantly lower SCARB1 in PGF2α-treated CL compared to saline, and late-stage CL had significantly lower SCARB1 concentrations compared to midluteal phase CL (P < 0.05, Fig. 4).
FIG. 4.

Effect of luteolysis on protein concentrations of the LXRs, ABCA1, and SCARB1. A) Western blot images of pooled lysates for the induced and spontaneous luteolysis models with TUBB used as a loading control. B) Charts plot results from densitometry analysis of individual replicates and contain values from both induced and spontaneous luteolysis comparisons. The x axis has been separated between the two luteolysis models with each model having its own y axis. Error bars indicate one SEM, and asterisk denotes significant difference between saline and PGF2α treatments at the corresponding time point or between mid- and late luteal phase CL.
ChIP
Samples from the 48 h time point of the induced luteolysis model were selected for ChIP because there was a significant increase in ABCA1 mRNA and protein at this time. Late-stage CL were also included in ChIP analyses. The anti-NR1H3 antibody selected has previously been validated for ChIP [19] and reacted with the ovine protein as determined by Western blot analysis (data not shown). The anti-NR1H2 antibody also recognized the ovine protein by Western blot analysis and immunoprecipitation (data not shown). There was a significant increase (P < 0.05) in enrichment of the ABCA1 LXRE in PGF2α compared to saline-treated ewes caused by ChIP with either NR1H2 or NR1H3 antibodies (Fig. 5). There was no increase in enrichment of the ABCA1 LXRE in late-stage CL associated with either LXR isoform (data not shown).
FIG. 5.

Effect of PGF2α on binding of each LXR isoform to the LXRE on the ABCA1 gene promoter. The IgG2a-normalized ratio of the ABCA1 LXRE to MRPS10 (negative control region) as determined by ChIP with antibodies specific for NR1H2 or NR1H3 is plotted. Error bars indicate one SEM, and asterisk denotes significant difference between saline and PGF2α treatments for the corresponding LXR isoform.
Neutral Lipid Staining
The cytoplasm and nuclei of cells were stained for visualization of luteal structure and to provide a background against which to overlay neutral lipid staining. Lipid droplets displayed punctate, spherical staining. Additional diffuse fluorescence was observed in the cytoplasm of what are presumably large steroidogenic luteal cells based on their apparent size relative to neighboring cells. Distinct droplets were often distinguishable within the diffuse cytoplasmic staining, so it is not clear if the diffuse cytoplasmic fluorescence is due to specific binding of neutral lipids located throughout the cytoplasm or if it is nonspecific cytoplasmic background fluorescence. Corpora lutea from saline-treated ewes displayed numerous punctate cytoplasmic droplets as well as droplets lining the plasma membrane of individual cells (Fig. 6 and Supplemental Figs. S1–S3). Slices of CL from PGF2α-treated ewes were not noticeably different at 12 and 24 h postinjection. At 48 h postinjection, three of four PGF2α-treated CL that were analyzed appeared to have less abundant punctate cytoplasmic droplets, with the remaining sample displaying abnormally large cytoplasmic droplets. All four samples from PGF2α-treated ewes at 48 h postinjection also displayed numerous lipid deposits that appeared to be in extracellular regions and were not lining the plasma membrane of individual cells as was observed in saline-treated CL. Late luteal phase CL undergoing spontaneous luteolysis displayed a similar distribution of lipid deposits as compared to PGF2α-treated CL at 48 h (Fig. 6 and Supplemental Figs. S1–S4).
FIG. 6.
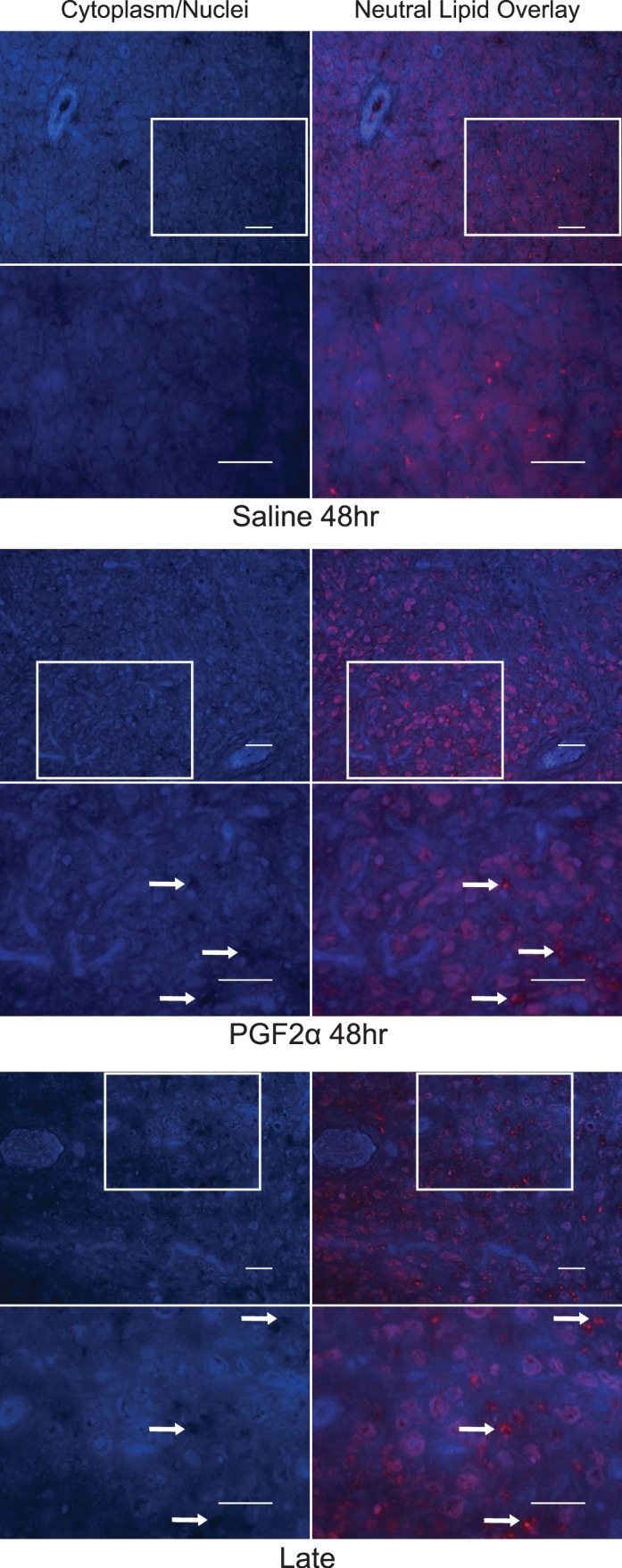
Qualitative changes in neutral lipid distribution in ovine luteal tissue during luteolysis. For each treatment, the left column shows cytoplasmic and nuclear staining to provide a view of the tissue structure, while the right column overlays neutral lipid staining. Bars = 50 μm. The boxes in the top row outline the approximate region that is shown at a higher magnification (×400) in the bottom row. Arrows in the PGF2α-treated and late luteal phase CL designate some of the potential extracellular regions that contain neutral lipid deposits.
DISCUSSION
In the spontaneous luteolysis model, serum progesterone concentrations during the cycle of tissue collection confirmed that the CL were of the expected age and were currently or had recently undergone luteolysis. In the induced luteolysis model, progesterone concentrations confirmed that luteolysis was achieved or initiated in PGF2α-treated ewes, while saline-treated ewes had functional CL throughout the study period. The drop in serum progesterone occurred quickly with a significant (P < 0.05) decrease in PGF2α-treated animals observed as early as 4 h after injection. Because the LXRs are nuclear transcription factors that initiate changes in gene transcription, their effects would be expected to manifest more gradually and are not consistent with the rapid drop in progesterone caused by PGF2α. However, subluteolytic doses of exogenous PGF2α caused serum progesterone concentrations to decline transiently and then return to preinjection concentrations without a shortening of CL lifespan [20, 21]. Therefore, other processes may be necessary to continue suppressing progesterone production and cause luteolysis in response to longer PGF2α exposure. Indeed, spontaneous luteolysis in cattle requires multiple pulses of PGF2α over a 24 h or longer period [22]. Initial pulses of PGF2α result in transient declines in progesterone followed by returns to prepulse concentrations. As luteolysis proceeds, this rebound effect is diminished so that progesterone secretion continues to decrease after each PGF2α pulse but does not fully recover [23, 24]. This indicates that progesterone suppression during luteolysis consists of both short-term (immediate decline following PGF2α pulse) and long-term (loss of rebound capability due to a loss of steroidogenic potential) effects. Thus, slower mechanisms such as changes in gene expression may mediate the gradual loss of steroidogenic capacity during spontaneous luteolysis.
Reduced extracellular cholesterol uptake would be necessary to deplete intracellular cholesterol stores. Induced and spontaneous luteolysis decreased the expression of both SCARB1 and LDLR mRNA, and SCARB1 protein. Protein concentrations of LDLR were not determined because a suitable antibody was not identified. It has been previously reported that PGF2α decreased LDLR mRNA in sheep [25] and SCARB1 mRNA in rats [26], which are consistent with the current findings. However, in vitro and in vivo studies have not identified an effect of PGF2α in reducing extracellular lipoprotein binding or cholesterol uptake, which has cast doubt on decreased cholesterol uptake being involved in PGF2α-induced luteolysis [2]. While lipoprotein uptake was not directly measured in the current study, the dramatic decreases in expression of both lipoprotein receptors would likely impair cholesterol uptake in the regressing CL. Only limited evidence supports the possibility that the reduction in expression of SCARB1 and LDLR mRNA is related to enhanced LXR activity. Synthetic LXR agonists decreased SCARB1 and LDLR mRNA in human adrenal cells [13]. However, it is not known if this was a direct transcriptional effect of the LXRs or mediated by an indirect mechanism because LDLR itself has been reported to be an LXR target gene whose transcription is modestly increased by an LXR agonist [27]. The LXRs can reduce LDLR protein through induction of the LXR target gene MYLIP, which specifically targets the LDLR for proteolytic digestion [14]. However, MYLIP mRNA remained unchanged in both experimental models, indicating that LXR-mediated MYLIP expression will not cause decreased cholesterol uptake during ovine luteolysis.
There were no increases in expression of the LXR target genes NR1H3 and ABCG1 associated with luteolysis, and surprisingly there was a significant decrease in NR1H3 mRNA during spontaneous luteolysis. However, ABCA1 mRNA increased in both induced and spontaneous luteolysis, which is consistent with enhanced LXR activity mediating some of the effects of PGF2α to cause luteolysis. Higher ABCA1 concentrations may promote cholesterol efflux thus contributing to luteal regression by depleting cholesterol stores that are needed for progesterone synthesis. Although increased ABCA1 expression is consistent with enhanced transcription by the LXRs in response to PGF2α, the lack of increases in the other LXR target genes NR1H3, ABCG1, and MYLIP casts doubt on this assumption. Therefore, ChIP was performed to directly test whether enhanced binding of the LXRs to the ABCA1 gene promoter occurred during luteolysis. There was no increase in binding of either LXR isoform to the ABCA1 LXRE in late compared to midluteal CL. However, concentrations of NR1H2 and NR1H3 protein were also significantly lower in late compared to midluteal phase CL as determined by Western blots, which may explain the lack of a difference between the two groups in absolute quantities of LXR bound to the ABCA1 promoter. In the induced luteolysis model, NR1H2 protein concentrations were unchanged and NR1H3 was only modestly reduced in PGF2α- versus saline-treated CL. In this comparison, CL from PGF2α-treated ewes had significantly more binding of both LXR isoforms to the LXRE of the ABCA1 gene compared to saline-treated CL at 48 h as determined by ChIP. This provides direct evidence supporting the conclusion that PGF2α enhanced LXR activity.
Qualitative evidence for a functional shift in lipid transport during luteolysis was obtained through neutral lipid staining. Numerous cytoplasmic and plasma membrane localized droplets were observed in CL from saline-treated ewes. In CL from PGF2α-treated ewes at 48 h postinjection, punctate cytoplasmic droplets appeared either less abundantly or were abnormally large in size. Additionally, numerous aggregates of lipid droplets were observed in PGF2α-treated CL at 48 h, which appeared to be in extracellular regions and were not specifically localized along the plasma membrane as was observed in saline-treated CL. Sections from CL undergoing spontaneous luteolysis looked similar to the PGF2α-induced luteolysis CL at 48 h posttreatment. Although not a quantitative analysis, this provides support for the possibility that a functional change occurs in lipid droplet localization and storage during luteolysis. Some differences between our observations and previous reports of lipid droplets in the ovine CL during the luteal phase and luteolysis are worth noting. It has been reported that in contrast to other species, lipid droplets are not normally observed in ovine luteal cells during periods of high progesterone production, but lipid droplets begin to appear in luteal cells during both natural [28] and PGF2α-induced [29] luteolysis. These observations were based on high magnification light images obtained using electron microscopy, whereas in the present study, we utilized lower magnification with fluorescent detection of neutral lipids. Thus, the methodological differences may underlie some of the discrepancies between our observations and previous studies. We detected numerous lipid droplets in CL from saline-treated animals (functional CL), and detected an increase in size of cytoplasmic lipid droplets in only one out of four PGF2α-treated CL at 48 h posttreatment, while the remaining three PGF2α-treated specimens and the late luteal phase CL appeared to have less abundant cytoplasmic droplets. We observed an apparent increase in accumulation of extracellular neutral lipid deposits following induced and spontaneous luteolysis. Large deposits of lipids have been observed at similar magnifications and with similar detection methods as used in the present report in regressing rat [30] and monkey [10] CL.
These data support the hypothesis that luteolysis of the ovine CL is associated with reduced luteal extracellular cholesterol uptake and enhanced cholesterol efflux. This could reduce intracellular cholesterol concentrations and contribute to suppression of luteal progesterone production caused by PGF2α. One possible mechanism for enhanced cholesterol efflux is by LXR-induced transcription of ABCA1. The actual mechanism whereby PGF2α may increase LXR activity is unknown but may involve increased LXR ligand concentrations and/or posttranslational modifications of the LXRs and/or proteins interacting with the LXRs. However, it does not appear to involve PGF2α-stimulation of LXR expression as neither isoform increased during luteolysis but rather decreased in spontaneous luteolysis and in the case of NR1H3, during induced luteolysis as well. Further research is needed to elucidate the mechanism whereby PGF2α may enhance LXR activity and whether changes in cholesterol supply via enhanced efflux and/or reduced uptake can restrict progesterone production, leading to luteolysis.
In contrast to PGF2α, prostaglandin E2 (PGE2) is luteoprotective and may play a critical role in the prevention of luteolysis during early pregnancy [2, 31]. In ruminants the conceptus releases interferon tau, which causes a shift in endometrial production of prostaglandins from PGF2α to PGE2 [32]. Furthermore, uterine-derived PGE2 is transported directly to the CL via the utero-ovarian plexus where it stimulates luteal PGE2 synthesis [33]. This may result in an amplification of the PGE2 signal and prevention of luteolysis via autocrine and/or paracrine mechanisms [33]. Therefore, future studies are needed to determine if PGE2 promotes cholesterol uptake and suppresses LXR-mediated cholesterol efflux as part of its antiluteolytic mechanism.
ACKNOWLEDGMENT
The authors would like to thank the University of Arizona Campus Agricultural Center Sheep Unit (Dan Kiesling, Manager), Octavio Mendivil, Mandie Dunham, and Dr. Peder Cuneo for assistance with animal care and surgical procedures. Contributions by the following undergraduates are also appreciated: Brianna Gruenwald, Alyxandra Beveridge, Courtney Moore, Sophia Leone, Michael Ross, and Devon Kartchner.
Footnotes
This research was supported by the Eunice Kennedy Shriver National Institute of Child Health & Human Development, National Institutes of Health, Award Number R00 HD067678 to R.L.B.; and by start-up funds to R.L.B. from the University of Arizona College of Agriculture and Life Sciences. N.L.S. was supported in part by Howard Hughes Medical Institute Grant 52006942 to the Undergraduate Biology Research Program at the University of Arizona.
REFERENCES
- Hodgen G, Itskovitz J. Recognition and maintenance of pregnancy. Knobil E, Neill J, editors. The Physiology of Reproduction, vol. 2. New York: Raven Press; 1988:1995–2021. In. (eds.) [Google Scholar]
- Niswender GD, Juengel JL, Silva PJ, Rollyson MK, McIntush EW. Mechanisms controlling the function and life span of the corpus luteum. Physiol Rev. 2000;80:1–29. doi: 10.1152/physrev.2000.80.1.1. [DOI] [PubMed] [Google Scholar]
- Stouffer RL. Structure, function, and regulation of the corpus luteum In Neill JD (ed.) Knobil and Neill's Physiology of Reproduction, vol. 1, 3rd ed. New York: Academic Press; 2006. 475 526 [Google Scholar]
- Castracane VD, Moore GT, Shaikh AA. Ovarian function of hysterectomized Macaca fascicularis. Biol Reprod. 1979;20:462–472. doi: 10.1095/biolreprod20.3.462. [DOI] [PubMed] [Google Scholar]
- Auletta FJ, Speroff L, Caldwell BV. Prostaglandin F-2 induced steroidogenesis and luteolysis in the primate corpus luteum. J Clin Endocrinol Metab. 1973;36:405–407. doi: 10.1210/jcem-36-2-405. [DOI] [PubMed] [Google Scholar]
- Ranney B, Abu-Ghazaleh S. The future function and fortune of ovarian tissue which is retained in vivo during hysterectomy. Am J Obstet Gynecol. 1977;128:626–634. doi: 10.1016/0002-9378(77)90208-3. [DOI] [PubMed] [Google Scholar]
- Wentz AC, Jones GS. Transient luteolytic effect of prostaglandin F2alpha in the human. Obstet Gynecol. 1973;42:172–181. [PubMed] [Google Scholar]
- Sargent EL, Baughman WL, Novy MJ, Stouffer RL. Intraluteal infusion of a prostaglandin synthesis inhibitor, sodium meclofenamate, causes premature luteolysis in rhesus monkeys. Endocrinology. 1988;123:2261–2269. doi: 10.1210/endo-123-5-2261. [DOI] [PubMed] [Google Scholar]
- Bogan RL, Debarber AE, Hennebold JD. Liver x receptor modulation of gene expression leading to proluteolytic effects in primate luteal cells. Biol Reprod. 2012;86:89. doi: 10.1095/biolreprod.111.096347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogan RL, Hennebold JD. The reverse cholesterol transport system as a potential mediator of luteolysis in the primate corpus luteum. Reproduction. 2010;139:163–176. doi: 10.1530/REP-09-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repa JJ, Mangelsdorf DJ. The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu Rev Cell Dev Biol. 2000;16:459–481. doi: 10.1146/annurev.cellbio.16.1.459. [DOI] [PubMed] [Google Scholar]
- Wojcicka G, Jamroz-Wisniewska A, Horoszewicz K, Beltowski J. Liver X receptors (LXRs). Part I: structure, function, regulation of activity, and role in lipid metabolism. Postepy Hig Med Dosw (Online) 2007;61:736–759. [PubMed] [Google Scholar]
- Nilsson M, Stulnig TM, Lin CY, Yeo AL, Nowotny P, Liu ET, Steffensen KR. Liver X receptors regulate adrenal steroidogenesis and hypothalamic-pituitary-adrenal feedback. Mol Endocrinol. 2007;21:126–137. doi: 10.1210/me.2006-0187. [DOI] [PubMed] [Google Scholar]
- Zelcer N, Hong C, Boyadjian R, Tontonoz P. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science. 2009;325:100–104. doi: 10.1126/science.1168974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogan RL, Murphy MJ, Hennebold JD. Dynamic changes in gene expression that occur during the period of spontaneous functional regression in the rhesus macaque corpus luteum. Endocrinology. 2009;150:1521–1529. doi: 10.1210/en.2008-1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laffitte BA, Joseph SB, Walczak R, Pei L, Wilpitz DC, Collins JL, Tontonoz P. Autoregulation of the human liver X receptor alpha promoter. Mol Cell Biol. 2001;21:7558–7568. doi: 10.1128/MCB.21.22.7558-7568.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney KD, Watson MA, Goodwin B, Galardi CM, Maglich JM, Wilson JG, Willson TM, Collins JL, Kliewer SA. Liver X receptor (LXR) regulation of the LXRalpha gene in human macrophages. J Biol Chem. 2001;276:43509–43515. doi: 10.1074/jbc.M106155200. [DOI] [PubMed] [Google Scholar]
- Bogan RL, Murphy MJ, Stouffer RL, Hennebold JD. Systematic determination of differential gene expression in the primate corpus luteum during the luteal phase of the menstrual cycle. Mol Endocrinol. 2008;22:1260–1273. doi: 10.1210/me.2007-0484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann R, Fischer C, Kodelja V, Behrens S, Haas S, Vingron M, Timmermann B, Geikowski A, Sauer S. Genome-wide analysis of LXRalpha activation reveals new transcriptional networks in human atherosclerotic foam cells. Nucleic Acids Res. 2013;41:3518–3531. doi: 10.1093/nar/gkt034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinckley ST, Milvae RA. Endothelin-1 mediates prostaglandin F(2alpha)-induced luteal regression in the ewe. Biol Reprod. 2001;64:1619–1623. doi: 10.1095/biolreprod64.6.1619. [DOI] [PubMed] [Google Scholar]
- Juengel JL, Haworth JD, Rollyson MK, Silva PJ, Sawyer HR, Niswender GD. Effect of dose of prostaglandin F(2alpha) on steroidogenic components and oligonucleosomes in ovine luteal tissue. Biol Reprod. 2000;62:1047–1051. doi: 10.1095/biolreprod62.4.1047. [DOI] [PubMed] [Google Scholar]
- Ginther OJ, Beg MA. Dynamics of circulating progesterone concentrations before and during luteolysis: a comparison between cattle and horses. Biol Reprod. 2012;86:170. doi: 10.1095/biolreprod.112.099820. [DOI] [PubMed] [Google Scholar]
- Ginther OJ, Shrestha HK, Fuenzalida MJ, Shahiduzzaman AK, Beg MA. Characteristics of pulses of 13,14-dihydro-15-keto-prostaglandin f2alpha before, during, and after spontaneous luteolysis and temporal intrapulse relationships with progesterone concentrations in cattle. Biol Reprod. 2010;82:1049–1056. doi: 10.1095/biolreprod.109.081976. [DOI] [PubMed] [Google Scholar]
- Ginther OJ, Shrestha HK, Fuenzalida MJ, Shahiduzzaman AK, Hannan MA, Beg MA. Intrapulse temporality between pulses of a metabolite of prostaglandin F 2alpha and circulating concentrations of progesterone before, during, and after spontaneous luteolysis in heifers. Theriogenology. 2010;74:1179–1186. doi: 10.1016/j.theriogenology.2010.05.018. [DOI] [PubMed] [Google Scholar]
- Tandeski TR, Juengel JL, Nett TM, Niswender GD. Regulation of mRNA encoding low density lipoprotein receptor and high density lipoprotein-binding protein in ovine corpora lutea. Reprod Fertil Dev. 1996;8:1107–1114. doi: 10.1071/rd9961107. [DOI] [PubMed] [Google Scholar]
- McLean MP, Sandhoff TW. Expression and hormonal regulation of the high-density lipoprotein (HDL) receptor scavenger receptor class B type I messenger ribonucleic acid in the rat ovary. Endocrine. 1998;9:243–252. doi: 10.1385/ENDO:9:3:243. [DOI] [PubMed] [Google Scholar]
- Ishimoto K, Tachibana K, Sumitomo M, Omote S, Hanano I, Yamasaki D, Watanabe Y, Tanaka T, Hamakubo T, Sakai J, Kodama T, Doi T. Identification of human low-density lipoprotein receptor as a novel target gene regulated by liver X receptor alpha. FEBS Lett. 2006;580:4929–4933. doi: 10.1016/j.febslet.2006.08.010. [DOI] [PubMed] [Google Scholar]
- Gemmell RT, Stacy BD, Thorburn GD. Morphology of the regressing corpus luteum in the ewe. Biol Reprod. 1976;14:270–279. doi: 10.1095/biolreprod14.3.270. [DOI] [PubMed] [Google Scholar]
- Stacy BD, Gemmell RT, Thorburn GD. Morphology of the corpus luteum in the sheep during regression induced by prostaglandin F2alpha. Biol Reprod. 1976;14:280–291. doi: 10.1095/biolreprod14.3.280. [DOI] [PubMed] [Google Scholar]
- Ning G, Fujimoto T, Koike H, Ogawa K. Cholesterol ester in corpus luteum of rat observed by analytical color fluorescence electron microscopy. J Histochem Cytochem. 1993;41:617–625. doi: 10.1177/41.4.8450201. [DOI] [PubMed] [Google Scholar]
- McCracken JA, Custer EE, Lamsa JC. Luteolysis: a neuroendocrine-mediated event. Physiol Rev. 1999;79:263–323. doi: 10.1152/physrev.1999.79.2.263. [DOI] [PubMed] [Google Scholar]
- Hansen TR, Austin KJ, Perry DJ, Pru JK, Teixeira MG, Johnson GA. Mechanism of action of interferon-tau in the uterus during early pregnancy. J Reprod Fertil Suppl. 1999;54:329–339. [PubMed] [Google Scholar]
- Lee J, McCracken JA, Stanley JA, Nithy TK, Banu SK, Arosh JA. Intraluteal prostaglandin biosynthesis and signaling are selectively directed towards PGF2alpha during luteolysis but towards PGE2 during the establishment of pregnancy in sheep. Biol Reprod. 2012;87:97. doi: 10.1095/biolreprod.112.100438. [DOI] [PubMed] [Google Scholar]