

Abstract
Recent studies documented that the selective estrogen receptor modulator tamoxifen prevents follicle loss and promotes fertility following in vivo exposure of rodents to irradiation or ovotoxic cancer drugs, cyclophosphamide and doxorubicin. In an effort to characterize the ovarian-sparing mechanisms of tamoxifen in preantral follicle classes, cultured neonatal rat ovaries (Day 4, Sprague Dawley) were treated for 1–7 days with active metabolites of cyclophosphamide (i.e., 4-hydroxycyclophosphamide; CTX) (0, 1, and 10 μM) and tamoxifen (i.e., 4-hydroxytamoxifen; TAM) (0 and 10 μM) in vitro, and both apoptosis and follicle numbers were measured. CTX caused marked follicular apoptosis and follicular loss. TAM treatment decreased follicular loss and apoptosis from CTX in vitro. TAM alone had no effect on these parameters. IGF-1 and IGF-1 receptor were assessed in ovarian tissue showing no impact of TAM or CTX on these endpoints. Targeted mRNA analysis during follicular rescue by TAM revealed decreased expression of multiple genes related to inflammation, including mediators of lipoxygenase and prostaglandin production and signaling (Alox5, Pla2g1b, Ptgfr), cytokine binding (Il1r1, Il2rg ), apoptosis (Tnfrsf1a), second messenger signaling (Mapk1, Mapk14, Plcg1), as well as tissue remodeling and vasodilation (Bdkrb2, Klk15). The results suggest that TAM protects the ovary from CTX-mediated toxicity through direct ovarian actions that oppose follicular loss.
Keywords: environmental contaminants and toxicants, ovary, premature ovarian failure, toxicology
INTRODUCTION
Tamoxifen (ICI46,474) was initially developed as a contraceptive selective estrogen receptor modulator (SERM) [1] prodrug and was repurposed for treatment and prevention of breast cancer [2]. Tamoxifen is a commonly used cancer drug that has been tested extensively in combination with other antineoplastic agents [3, 4]. Additionally, tamoxifen and other SERMs are being developed as protectants against neurologic and brain injury [5, 6] and potentiators of cancer treatments [7–11].
Tamoxifen exerts differential effects across tissues expressing estrogen receptors (ERs). The selectivity of tamoxifen is complex and is determined by the interplay of ER abundance and isotype, concentrations of endogenous estrogens, and availability or occupancy of ER cofactors and corepressors [2, 12]. Estrogen signaling in breast tissue is generally blocked by tamoxifen [13, 14]. Skeletal ER remains functional, avoiding osteoporosis [2]. Ovarian effects of tamoxifen are less clear and can involve both neuroendocrine and local ovarian actions [15]. Broadly speaking, tamoxifen is used for its tissue-specific effects, and ovarian protection may be one such selective action.
Tamoxifen prevents follicle loss following exposure to two widely used and ovotoxic cancer drugs, cyclophosphamide and doxorubicin, in preclinical in vivo models [16]. Additionally, recent work has shown similar prevention of radiation-mediated ovarian toxicity using tamoxifen as a countermeasure [6, 7]. It is unclear from previous work whether the protective effects of tamoxifen against cyclophosphamide toxicity in the ovary are direct ovarian actions or actions via such indirect mechanisms as alterations in gonadotropin release or hepatic drug metabolism [15].
Mahran [17] implicated IGF-1 signaling as a protective ovarian mechanism activated by tamoxifen during protection from radiation-mediated injury. IGF-1 and its receptor (IGF-1R) are important factors regulating ovarian cells proliferation and differentiation as well as follicular development and ovulation [18, 19]. Moreover previous studies showed that tamoxifen exerts a direct regulatory effect on IGF-1 secretion and modulates the IGF-1/IGF-1R system differentially across tissues [20, 21]. The present study was designed to determine the in vitro effects of active metabolites of tamoxifen and cyclophosphamide during their actions on follicle numbers, apoptosis, IGF-1 signaling, and inflammatory gene expression in neonatal rat ovaries.
MATERIALS AND METHODS
Reagents
Reagents used in this study were purchased from the following sources. Penicillin/streptomycin, ascorbic acid, bovine serum albumin, transferrin, phosphate buffered saline (PBS, pH 7.1), and 4-hydroxytamoxifen (TAM) were from Sigma-Aldrich; Ham F12/Dulbecco-modified Eagle medium (1:1) with GlutaMax, Albumax II, and fetal bovine serum were from Gibco; 4-hydroxycyclophosphamide (mafosfamide; CTX) was from IIT GmbH/Niomech; and Millicell-CM filter inserts were from Millipore. RNA was isolated using the Trizol reagent, and cDNA was generated using High-Capacity Reverse Transcription Kit (Life Technologies).
Animals
Female Sprague-Dawley rats (timed pregnancy) were purchased from Harlan Laboratories. All the animals were housed one per cage under 12L:12D (0600–1800 h) and controlled temperature (23°C ± 2°C) and humidity. The animals were provided with standard food (Purina Rodent Chow; Ralston Purina Co.) with ad libitum access to food and water and allowed to give birth. All the experimental procedures have been approved by the University of Kansas Medical Center Institutional Animal Care and Use Committee.
In Vitro Ovarian Culture
Postnatal Day 4 female Sprague-Dawley rats were euthanized, and each ovary was removed with oviduct and bursal tissue trimmed away under a dissection microscope in PBS. Ovaries were cultured as described previously [22, 23] in Ham F12/Dulbecco-modified Eagle medium (1:1) with GlutaMax medium containing 1 mg/ml bovine serum albumin, 5% fetal bovine serum, 1 mg/ml Albumax II, 50 μg/ml ascorbic acid, 2.75 μg/ml transferrin, 5 units/ml penicillin, and 5 μg/ml streptomycin. The neonatal ovary culture model is commonly used to study ovotoxicity and its prevention [24, 25] because it focuses on direct treatment effects on the primordial and primary follicle pool, a major determinant of reproductive lifespan in women [26].
Culture medium (500 μl per well) was added to 24-well culture plates, and Millicell-CM filter inserts were placed on top of the medium. Plates were preequilibrated for 1 h at 37°C before ovaries were placed on the top of the floating filters and covered with a drop of medium. Ovaries were cultured at 37°C in a humidified atmosphere, containing 5% CO2. Ovaries were treated with vehicle (control: 0.1% dimethyl sulfoxide plus ethanol), TAM (10 μM), and/or two doses of CTX (1 and 10 μM) for 24 h (gene expression), 48 h (gene expression and apoptosis), or 7 days (follicle counts). For cultures lasting more than 48 h, the medium was replaced and fresh medium and treatments were added every 2 days. Different exposure time (24 and 48 h and 7 days) were chosen based on past work in our laboratory [16] and others [27]. Doses were based on human pharmacokinetic concentrations [28, 29] as well as past work in culture systems [21, 27, 30].
Ovarian Follicle Counts
Following 7 days of culture, ovaries (n = 5–6/group) were placed in 4% paraformaldehyde overnight, transferred through graded ethanol, embedded in paraffin, serially sectioned (6 μm), and stained with hematoxylin and eosin. Follicles were counted in every fifth section with observers being blind to the treatment group. The numbers of follicles present in the marked sections were multiplied by five to calculate total numbers per ovary as described previously [31]. Follicle classification was according to published criteria [32–34]. Follicle numbers were compared between treatment groups using ANOVA followed by least significant difference post hoc test. Differences with a probability of P < 0.05 were considered statistically significant.
Apoptosis
Cultured ovaries (n = 6–8/group; 48 h culture) were fixed as described above. We used terminal deoxynucleotydyl transferase-mediated deoxyuridine triphosphate nick end-labeling (TUNEL) with ApoAlert DNA Fragmentation Assay Kit (Clontech) following the manufacturer's instructions [35, 36] for the detection of apoptotic follicles. TUNEL-positive follicles were visualized in three equidistant midsaggital sections per ovary using Zeiss LSM Pascal confocal microscopy as described previously [16]. The microscopists were blind to the treatment group. Follicles were defined as apoptotic if the majority of granulosal or pregranulosal cells or the oocyte were TUNEL positive. Apoptosis (TUNEL-positive follicles) were compared using ANOVA followed by the least significant difference post hoc test. Differences with a probability of P < 0.05 were considered statistically significant.
Inflammatory Gene Expression
The effect of TAM on gene expression in CTX-treated ovaries was compared using targeted real-time PCR arrays in three independent experiments. Rat ovaries (Day 4, n = 30/group) were cultured as two to three ovaries/well for 48 h with CTX (10 μM) or with CTX (10 μM) + TAM (10 μM) and flash frozen in 10–20 μl PBS and stored at −80°C until ready for RNA isolation. RNA was isolated from three pooled samples of 10 ovaries each for each treatment group using Trizol reagent, and cDNA was generated using the High-Capacity Reverse Transcription Kit. Complementary DNA was quantified at 260 nm using a spectrophotometer (ND-1000; NanoDrop). Real-time PCR analysis was conducted using the TaqMan Array Rat Inflammation Plate (4414081; Life Technologies) that contains 92 assays to inflammation-associated genes and four housekeeping genes. The inflammation-associated genes fall into four classes: channels (L-type calcium, ligand gated), enzymes and inhibitors (lipases, kinases, nitric oxide synthase, phosphodiesterase, proteases), regulators of prostaglandin metabolism and factors (tumor necrosis factors, nuclear factor Kappa-Beta, interleukin, annexins, kininogens), and receptors (G protein coupled receptors, interleukin receptor family, adhesion molecules, tumor necrosis factor family receptors, nuclear receptors). The reaction mix consisted of TaqMan Master Mix, water, and cDNA (50 ng per well) per manufacturer's instructions. For each treatment plate, 20 μl of its respective reaction mix was placed into each well. The plates were run using an ABI PRISM 7000 Sequence Detection System (Life Technologies) with an initial start of 50°C for 2 min and 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min. Nontemplate reactions were used as controls for contamination. The expression of housekeeping genes was not affected by treatments and did not vary significantly between TaqMan Array Plates used in the experiment. Gene expression was expressed using the comparative Ct method utilizing an average Ct value for the four housekeeping genes [37, 38], and the effect of TAM on inflammatory gene expression in CTX-treated ovaries was analyzed by Student t-test. Data are presented as fold change for TAM+CTX versus CTX. Differences with a probability of P < 0.05 were considered statistically significant.
IGF-1 and IGF-1 Receptor
Neonatal ovaries (n = 12/group) were treated for 24 or 48 h with CTX and TAM, alone and in combination as for apoptosis and follicular count endpoints. RNA was isolated from two pooled ovaries per well and processed for real-time PCR as described above. Commercially available prevalidated primers for rat Igf-1 and its receptor were used (RN00710306 and RN00583837, respectively; Life Technologies). Protein was extracted from separate experimental replicates (n = 12/group) using two pooled ovaries per well for IGF-1 (MG-100 kit; R&D Systems). Protein concentrations were determined by Bradford assay and diluted 1:5 for assay of 50 μl diluted homogenate. The results were analyzed and expressed as nanograms IGF-1/mg tissue. Data were analyzed by multifactorial ANOVA with CTX, TAM, and time as the main effects. Differences with a probability of P < 0.05 were considered statistically significant.
RESULTS
Follicle Counts and Apoptosis
Follicle populations were homogenously primordial or primary in these neonatal ovaries, and the results are presented as total follicle numbers (Figs. 1 and 2). Active metabolite of CTX decreased the number of ovarian follicles at 7 days of culture in a dose-dependent fashion, with the highest dose of CTX depleting ovarian follicles to approximately 25% of controls (Fig. 1). TAM, while not altering follicle numbers when added alone, was associated with significantly greater follicular reserves when coadministered with CTX. This is reminiscent of the in vivo follicular rescue by TAM previously documented [16]. These changes in follicle counts were paralleled in the apoptosis data at 48 h of culture (Figs. 3 and 4), with high dose CTX markedly inducing TUNEL staining of ovarian follicles and TAM cotreatment reversing this effect.
FIG. 1.
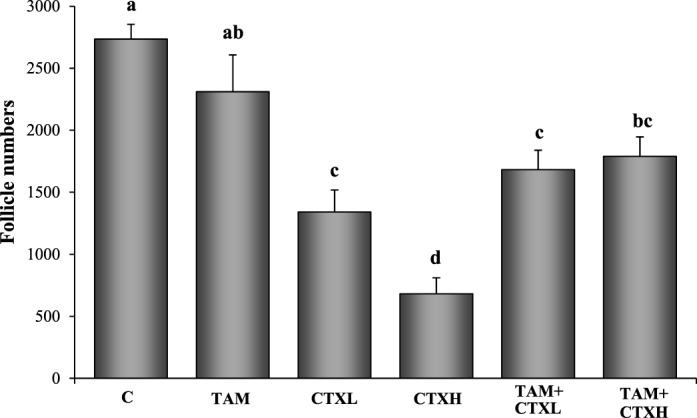
Effects of 4-hydroxycyclophosphamide (CTX) and 4-hydroxytamoxifen (TAM, 10 μM) on the total number of follicles (primordial and primary). C, control; CTXL, 1 μM CTX; CTXH, 10 μM CTX. Values are mean ± SEM follicles counted per ovary (n = 5–8). Different letters indicate significant differences among treatment groups (P < 0.05).
FIG. 2.

Representative effects of 4-hydroxycyclophosphamide (CTX) and 4-hydroxytamoxifen (TAM, 10 μM) on ovarian morphology. Cultured neonatal rat ovaries are stained with hematoxylin and eosin. C, control; CTXL, 1 μM CTX; CTXH, 10 μM CTX. Bar = 200 μm.
FIG. 3.

Representative effects of 4-hydroxycyclophosphamide (CTX) and 4-hydroxytamoxifen (TAM, 10 μM) on the number of apoptotic follicles (n = 6–8 ovaries/group). TUNEL-positive (apoptotic) follicles are labeled with fluorescein isothiocyanate (green fluorescence). C, control; CTXL, 1 μM CTX; CTXH, 10 μM CTX. Bar = 50 μm.
FIG. 4.

Effects of 4-hydroxycyclophosphamide (CTX) and 4-hydroxytamoxifen (TAM, 10 μM) on the number of apoptotic follicles. Values represent mean ± SEM apoptotic follicles counted per ovary (n = 6–8 ovaries/group). C, control; CTXL, 1 μM CTX; CTXH, 10 μM CTX. Bars with different letters denote significant differences among treatment groups (P < 0.05).
Inflammatory Gene Expression
Genes significantly regulated by TAM are shown in Table 1. Overall, TAM modestly but significantly downregulated multiple genes relating to inflammation in ovaries cotreated with a toxic dose of CTX. Messenger RNAs decreased by TAM included mediators of lipoxygenase and prostaglandin production and signaling (5-lipoxygenase, Alox5; phospholipase A2 g1b, Pla2g1b; prostaglandin F receptor, Ptgfr), cytokine binding (interleukin 1 receptor type I, Il1r1; interleukin 2 receptor gamma, Il2rg), apoptosis (tumor necrosis factor receptor superfamily member 1A, Tnfrsf1a), second messenger signaling (mitogen activated protein kinase 1 and 14, Mapk1, Mapk14; phospholipase C gamma 1, Plcg1), as well as tissue remodeling and vasodilation (bradykinin receptor B2, Bdkrb2; kallikrein-15, Klk15). Several additional genes appeared to be downregulated by TAM but did not reach the level of significance (0.06 < P > 0.10; data not shown). These included caspase-1 (Casp1), intercellular adhesion molecule 1/CD54 (Icam1), beta 1 integrin (Itgb1), phospholipase A2 g5 (Pla2g5), and phospholipase C b3 (Plcb3). Surprisingly, both Cox-2 (prostaglandin-endoperoxide synthase, Ptgs2) and Cd40 (TNF receptor superfamily member 5) were significantly upregulated by TAM (Table 1). A gene network of known relationships among the inflammatory genes that were found to be differentially expressed between CTX and CTX+TAM treated ovaries cultured for 48 h is shown in Figure 5.
TABLE 1.
Effect of 4-hydroxycyclophosphamide (TAM) on inflammatory gene expression in 4-hydroxycyclophosphamide (CTX)-treated rat ovaries after 48 h of culture (n = 3 replicates; 10 ovaries/group per replicate).
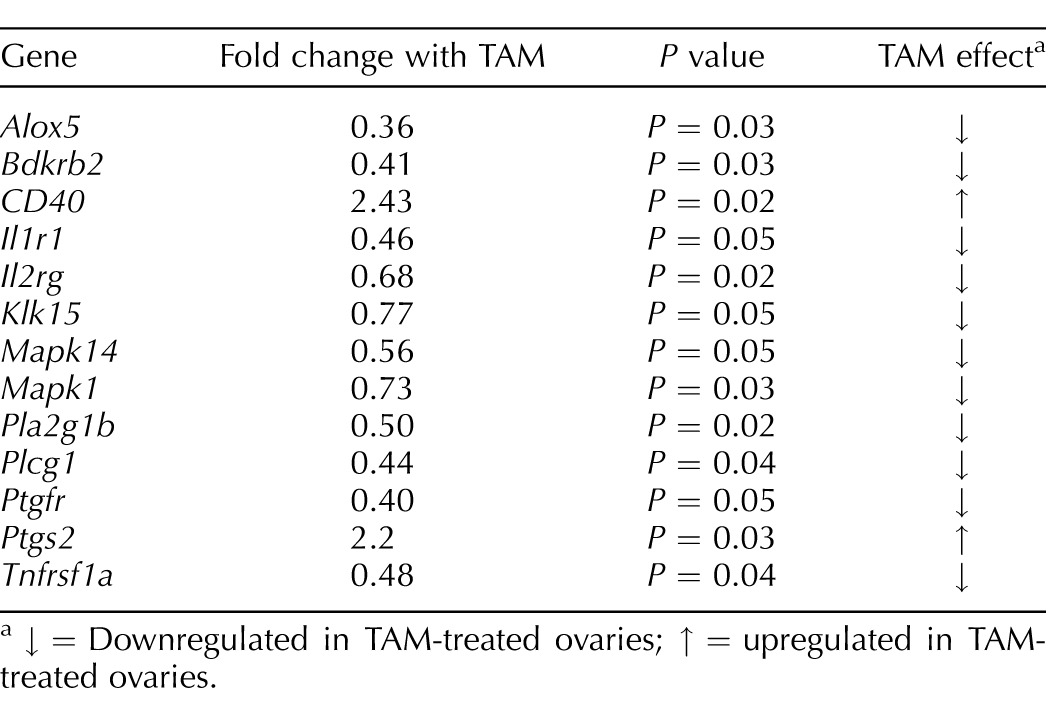
↓ = Downregulated in TAM-treated ovaries; ↑ = upregulated in TAM-treated ovaries.
FIG. 5.

A gene network of known relationships among the inflammatory genes found to be differentially expressed between CTX (4-hydroxycyclophosphamide) and CTX+TAM (4-hydroxytamoxifen) treated ovaries. Green denotes downregulated genes while red symbols are associated with increased gene expression.
IGF-1 and IGF-1 Receptor
While there was a trend (P = 0.12) for Igf-1 mRNA and protein as well as Igf-1r mRNA to increase from 24 to 48 h of culture, there was no significant effect of TAM or CTX on these endpoints (Fig. 6). Neither was there detectable interaction between TAM and CTX for these biomarkers of IGF-1 signaling.
FIG. 6.

Effects of 4-hydroxycyclophosphamide (CTX, 10 μM) and 4-hydroxytamoxifen (TAM, 10 μM) on Igf-1 (A) and Igf-1r (B) gene expression (n = 3; mean ± SEM) and IGF-1 protein (C) concentrations (n = 3; mean ± SEM) in neonatal rat ovaries treated for 24 or 48 h. CONT, control.
DISCUSSION
The current in vitro study follows an initial investigation that showed follicle rescue and improved fertility in rats receiving tamoxifen as a countermeasure against ovarian toxicity from 7,12-dimethylbenz [α] anthracene, cyclophosphamide, and doxorubicin in vivo [16]. We assessed direct actions of TAM and CTX on the rat ovary in vitro using both follicle numbers, confocal (TUNEL) staining for apoptosis, and mRNA expression of genes involved in inflammation. CTX induced marked follicular apoptosis and decreased follicular reserve. TAM alone did not alter basal apoptosis but decreased CTX-induced apoptosis and significantly increased follicle numbers in CTX-treated animals, in agreement with previous in vivo studies. Results of the current study suggest that a major component of follicular rescue by TAM involves direct ovarian interactions rather than extraovarian actions, such as altered neuroendocrine function leading to ovarian quiescence.
In a parallel study, another group found that tamoxifen prevented follicle loss from gamma irradiation in rats and correlated tamoxifen administration with increased IGF-1R [17]. We failed to observe similar upregulation of IGF-1 protein or Igf-1 and Igf-1r gene expression when TAM was used alone or in combination with CTX. This discrepancy may be due to differences between the models, particularly because this work was in vitro. Additionally, upregulation of IGF-1 signaling appears to be an endogenous protective mechanism that is activated by irradiation [39, 40], in contrast to the chemotherapy exposure used in the current study.
TAM did acutely downregulate expression of multiple genes related to inflammation in CTX-treated ovaries. These included mediators of lipoxygenase (Alox5), prostaglandin (Pla2g1b, Ptgfr) and cytokine (Il1r1, Il1r2) production and action, as well as inflammatory second messengers (Mapk1, Mapk14, Plcg1) and genes involved in tissue remodeling and vasodilation (Bdkrb2, Klk15). Indeed, there appeared to be a broad effect on mRNAs related to inflammation, suggesting that TAM may rescue follicles in part by alleviating these ovarian pathways of tissue damage following cyclophosphamide exposure. Additionally, TNF receptor (Tnfrsf1a), a central mediator of apoptosis, was suppressed by TAM in agreement with decreased TUNEL staining seen during follicular rescue with the SERM.
In vitro and in vivo studies in rodents have identified primordial and small primary follicles to be extremely sensitive targets of cyclophosphamide and its metabolites [41, 42]. Similar to the effect of cyclophosphamide observed in our experiment, others also reported increase of DNA degradation markers (TUNEL) in oocytes and granulosa cells of primordial and small primary follicles [25, 27]. In contrast, a recently published in vivo study [43] shows that cyclophosphamide does not induce apoptosis of primordial follicles but activates growth of the quiescent primordial follicle population in mice. This discrepancy may be due to difference between models and species-sensitivity to cyclophosphamide. Interestingly, cyclophosphamide-induced activation of primordial follicles was prevented by AS101, a nontoxic immunomodulatory compound. Although the authors did not focus on the immune effects of AS101, we find it very interesting in the context of our results indicating that TAM modulates ovarian genes related to inflammation.
Taken together the results of our study clearly demonstrate that TAM increases follicle numbers and decreases CTX-induced apoptosis in rat ovaries in vitro. We also propose, based on the PCR array data, that the mechanism of TAM action on CTX-treated ovaries involves changes in the expression of genes modulating inflammatory response and pathways. However, PCR array results should be interpreted with some caution because the mRNA changes observed in our study were modest, if significant, and protein correlates remain to be examined. Additionally, the in vitro system used here, while allowing isolation of ovarian effects of TAM and CTX, does exclude contributions of nonresident immune cells to this interaction. Therefore, further experiments are necessary and should specifically focus on the influence of TAM and CTX on inflammatory pathways in the ovary.
Avoiding ovarian toxicities from cancer chemotherapy is particularly difficult because the anticancer actions must remain intact, that is, protective effects must be selective. Tamoxifen is used as an antihormonal therapy for early ER-positive breast cancer with clinical benefit similar to cytotoxic chemotherapy in these patients [44–46]. In a single study, sequential tamoxifen was found to be modestly more beneficial than concurrent tamoxifen and chemotherapy for ER-positive disease [47]. Several other studies have found no difference in clinical outcome between TAM administered before or after breast cancer chemotherapy or radiation treatment versus simultaneously with chemotherapy [4, 48]. Similarly, Albain and colleagues [49] recently found statistically similar disease-free survival and overall survival for simultaneous versus sequential tamoxifen with cyclophosphamide-based chemotherapy of ER-positive breast cancer, although the overall hazard ratio reflecting side effects was greater for the combined treatment. In contrast with this lack of therapeutic interaction between tamoxifen and therapies for ER-positive breast cancer, at least one clinical trial [50] and several preclinical studies [51, 52] have suggested interactions between tamoxifen and ER- and progesterone receptor-negative breast cancer treatment, leading to current recommendations for sequential rather than simultaneous antihormonal and cytotoxic chemotherapy for breast cancers.
Among nonhormonal cancers, tamoxifen has been tested as part of frontline lymphoma therapy because of the ability of tamoxifen to downregulate drug resistance pathways in some cancer models [53, 54]. Tamoxifen has little effect of the pharmacokinetics or toxicities of chemotherapy in lymphoma patients [53–55], although increased myelosuppression was seen in a phase I trial of tamoxifen plus cyclophosphamide/doxorubicin/vincristine/prednisone/etoposide chemotherapy [3]. Overall, past human work suggests that tamoxifen can be safely combined with other drugs for many cancers without reducing their therapeutic ability. Data from our group and others suggest the possibility of eventual repurposing of tamoxifen for ovarian protection from toxicants [39] after appropriate translational studies.
ACKNOWLDGMENT
Authors would like to thank Dr. David Albertini and Ms. Darlene Limback for assistance with confocal microscopy. Ms. Teresa Phillips assisted with PCR arrays.
Footnotes
Financial support was provided by the NIH Center for Biomedical Research Excellence at University of Kansas Medical Center.
REFERENCES
- Harper MJ, Walpole AL. A new derivative of triphenylethylene: effect on implantation and mode of action in rats. J Reprod Fertil. 1967;13:101–119. doi: 10.1530/jrf.0.0130101. [DOI] [PubMed] [Google Scholar]
- Jordan VC. Tamoxifen (ICI46,474) as a targeted therapy to treat and prevent breast cancer. Br J Pharmacol. 2006;147((Suppl 1)):S269–S276. doi: 10.1038/sj.bjp.0706399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DC, Trump DL. A phase I trial of high-dose oral tamoxifen and CHOPE. Cancer Chemother Pharmacol. 1995;36:65–68. doi: 10.1007/BF00685734. [DOI] [PubMed] [Google Scholar]
- Pierce LJ, Hutchins LF, Green SR, Lew DL, Gralow JR, Livingston RB, Osborne CK, Albain KS. Sequencing of tamoxifen and radiotherapy after breast-conserving surgery in early-stage breast cancer. J Clin Oncol. 2005;23:24–29. doi: 10.1200/JCO.2005.01.198. [DOI] [PubMed] [Google Scholar]
- Guptarak J. The cancer drug tamoxifen–a potential therapeutic treatment for spinal cord injury. J Neurotrauma. 2014;31:268–283. doi: 10.1089/neu.2013.3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakade C, Wiktorowicz JE, Sadygov RG, Zivadinovic D, Paulucci-Holthauzen AA, Vergara L, Nesic O. Tamoxifen neuroprotection in cerebral ischemia involves attenuation of kinase activation and superoxide production and potentiation of mitochondrial superoxide dismutase. Endocrinology. 2008;149:367–379. doi: 10.1210/en.2007-0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandes LJ. N,N-diethyl-2-[4-(phenylmethyl) phenoxy] ethanamine (DPPE; tesmilifene), a chemopotentiating agent with hormetic effects on DNA synthesis in vitro, may improve survival in patients with metastatic breast cancer. Hum Exp Toxicol. 2008;27:143–147. doi: 10.1177/0960327108090751. [DOI] [PubMed] [Google Scholar]
- Ferguson PJ, Brisson AR, Koropatnick J, Vincent MD. Enhancement of cytotoxicity of natural product drugs against multidrug resistant variant cell lines of human head and neck squamous cell carcinoma and breast carcinoma by tesmilifene. Cancer Lett. 2009;274:279–289. doi: 10.1016/j.canlet.2008.09.021. [DOI] [PubMed] [Google Scholar]
- Chatterjee M, Harris AL. Reversal of acquired resistance to adriamycin in CHO cells by tamoxifen and 4-hydroxy tamoxifen: role of drug interaction with alpha 1 acid glycoprotein. Br J Cancer. 1990;62:712–717. doi: 10.1038/bjc.1990.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonessa F, Jacobson M, Boyle B, Lippman J, McGarvey M, Clarke R. Effect of tamoxifen on the multidrug-resistant phenotype in human breast cancer cells: isobologram, drug accumulation, and M(r) 170,000 glycoprotein (gp170) binding studies. Cancer Res. 1994;54:441–447. [PubMed] [Google Scholar]
- Mao Z, Zhou J, Luan J, Sheng W, Shen X, Dong X. Tamoxifen reduces P-gp-mediated multidrug resistance via inhibiting the PI3K/Akt signaling pathway in ER-negative human gastric cancer cells. Biomed Pharmacother. 2014;68:179–183. doi: 10.1016/j.biopha.2013.10.003. [DOI] [PubMed] [Google Scholar]
- Jordan VC, O'Malley BW. Selective estrogen-receptor modulators and antihormonal resistance in breast cancer. J Clin Oncol. 2007;25:5815–5824. doi: 10.1200/JCO.2007.11.3886. [DOI] [PubMed] [Google Scholar]
- Clarke M, Coates AS, Darby SC, Davies C, Gelber RD, Godwin J, Goldhirsch A, Gray R, Peto R, Pritchard KI, Wood WC. Adjuvant chemotherapy in oestrogen-receptor-poor breast cancer: patient-level meta-analysis of randomised trials. Lancet. 2008;371:29–40. doi: 10.1016/S0140-6736(08)60069-0. [DOI] [PubMed] [Google Scholar]
- Clarke RB, Laidlaw IJ, Jones LJ, Howell A, Anderson E. Effect of tamoxifen on Ki67 labelling index in human breast tumours and its relationship to oestrogen and progesterone receptor status. Br J Cancer. 1993;67:606–611. doi: 10.1038/bjc.1993.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inal MM, Incebiyik A, Sanci M, Yildirim Y, Polat M, Pilanci B, Nayki C, Camuzcuoglu H. Ovarian cysts in tamoxifen-treated women with breast cancer. Eur J Obstet Gynecol Reprod Biol. 2005;120:104–106. doi: 10.1016/j.ejogrb.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Ting AY, Petroff BK. Tamoxifen decreases ovarian follicular loss from experimental toxicant DMBA and chemotherapy agents cyclophosphamide and doxorubicin in the rat. J Assist Reprod Genet. 2010;27:591–597. doi: 10.1007/s10815-010-9463-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahran YF, El-Demerdash E, Nada AS, Ali AA, Abdel-Naim AB. Insights into the protective mechanisms of tamoxifen in radiotherapy-induced ovarian follicular loss: impact on insulin-like growth factor 1. Endocrinology. 2013;154:3888–3899. doi: 10.1210/en.2013-1214. [DOI] [PubMed] [Google Scholar]
- Baumgarten SC, Convissar SM, Fierro MA, Winston NJ, Scoccia B, Stocco C. IGF1R signaling is necessary for FSH-induced activation of AKT and differentiation of human Cumulus granulosa cells. J Clin Endocrinol Metab. 2014;99:2995–3004. doi: 10.1210/jc.2014-1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Baumgarten SC, Wu Y, Bennett J, Winston N, Hirshfeld-Cytron J, Stocco C. IGF-I signaling is essential for FSH stimulation of AKT and steroidogenic genes in granulosa cells. Mol Endocrinol. 2013;27:511–523. doi: 10.1210/me.2012-1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak MN, Huynh HT, Lefebvre SP. Tamoxifen reduces serum insulin-like growth factor I (IGF-I) Breast Cancer Res Treat. 1992;22:91–100. doi: 10.1007/BF01833337. [DOI] [PubMed] [Google Scholar]
- Ramachandran C, Khatib Z, Petkarou A, Fort J, Fonseca HB, Melnick SJ, Escalon E. Tamoxifen modulation of etoposide cytotoxicity involves inhibition of protein kinase C activity and insulin-like growth factor II expression in brain tumor cells. J Neurooncol. 2004;67:19–28. doi: 10.1023/b:neon.0000021738.77612.1b. [DOI] [PubMed] [Google Scholar]
- Parrott JA, Skinner MK. Kit-ligand/stem cell factor induces primordial follicle development and initiates folliculogenesis. Endocrinology. 1999;140:4262–4271. doi: 10.1210/endo.140.9.6994. [DOI] [PubMed] [Google Scholar]
- Keating AF, Sen N, Sipes IG, Hoyer PB. Dual protective role for glutathione S-transferase class pi against VCD-induced ovotoxicity in the rat ovary. Toxicol Appl Pharmacol. 2010;247:71–75. doi: 10.1016/j.taap.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine PJ, Sipes IG, Skinner MK, Hoyer PB. Characterization of a rat in vitro ovarian culture system to study the ovarian toxicant 4-vinylcyclohexene diepoxide. Toxicol Appl Pharmacol. 2002;184:107–115. [PubMed] [Google Scholar]
- Petrillo SK. Detection of DNA damage in oocytes of small ovarian follicles following phosphoramide mustard exposures of cultured rodent ovaries in vitro. Toxicol Appl Pharmacol. 2011;253:94–102. doi: 10.1016/j.taap.2011.03.012. [DOI] [PubMed] [Google Scholar]
- Morgan S, Anderson AR, Gourley C, Wallace WH, Spears N. How do chemotherapeutic agents damage the ovary? Hum Reprod Update. 2012;18:525–535. doi: 10.1093/humupd/dms022. [DOI] [PubMed] [Google Scholar]
- Desmeules P, Devine PJ. Characterizing the ovotoxicity of cyclophosphamide metabolites on cultured mouse ovaries. Toxicol Sci. 2006;90:500–509. doi: 10.1093/toxsci/kfj086. [DOI] [PubMed] [Google Scholar]
- Pollack IF, DaRosso RC, Robertson PL, Jakacki RL, Mirro JR, Jr, Blatt J, Nicholson S, Packer RJ, Allen JC, Cisneros A, Jordan VC. A phase I study of high-dose tamoxifen for the treatment of refractory malignant gliomas of childhood. Clin Cancer Res. 1997;3:1109–1115. [PubMed] [Google Scholar]
- Pollack IF, Randall MS, Kristofik MP, Kelly RH, Selker RG, Vertosick FT., Jr Effect of tamoxifen on DNA synthesis and proliferation of human malignant glioma lines in vitro. Cancer Res. 1990;50:7134–7138. [PubMed] [Google Scholar]
- Chen L, Yu LJ, Waxman DJ. Potentiation of cytochrome P450/cyclophosphamide-based cancer gene therapy by coexpression of the P450 reductase gene. Cancer Res. 1997;57:4830–4837. [PubMed] [Google Scholar]
- Flaws JA, Abbud R, Mann RJ, Nilson JH, Hirshfield AN. Chronically elevated luteinizing hormone depletes primordial follicles in the mouse ovary. Biol Reprod. 1997;57:1233–1237. doi: 10.1095/biolreprod57.5.1233. [DOI] [PubMed] [Google Scholar]
- Flaws JA, Salyers KL, Sipes IG, Hoyer PB. Reduced ability of rat preantral ovarian follicles to metabolize 4-vinyl-1-cyclohexene diepoxide in vitro. Toxicol Appl Pharmacol. 1994;126:286–294. doi: 10.1006/taap.1994.1118. [DOI] [PubMed] [Google Scholar]
- Pedersen T, Peters H. Proposal for a classification of oocytes and follicles in the mouse ovary. J Reprod Fertil. 1968;17:555–557. doi: 10.1530/jrf.0.0170555. [DOI] [PubMed] [Google Scholar]
- Devine PJ, Sipes IG, Skinner MK, Hoyer PB. Characterization of a rat in vitro ovarian culture system to study the ovarian toxicant 4-vinylcyclohexene diepoxide. Toxicol Appl Pharmacol. 2002;184:107–115. [PubMed] [Google Scholar]
- Dhanabal M, Ramchandran R, Waterman MJ, Lu H, Knebelmann B, Segal M, Sukhatme VP. Endostatin induces endothelial cell apoptosis. J Biol Chem. 1999;274:11721–11726. doi: 10.1074/jbc.274.17.11721. [DOI] [PubMed] [Google Scholar]
- Verma NK, Davies AM, Long A, Kelleher D, Volkov Y. STAT3 knockdown by siRNA induces apoptosis in human cutaneous T-cell lymphoma line Hut78 via downregulation of Bcl-xL. Cell Mol Biol Lett. 2010;15:342–355. doi: 10.2478/s11658-010-0008-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes Genome Biol 2002. 3 Research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleicher N, Kushnir VA, Barad DH. Therapeutic interventions into early stages of follicle maturation: a new treatment paradigm after over 50 years of modern infertility therapy. Endocrinology. 2013;154:3498–3501. doi: 10.1210/en.2013-1679. [DOI] [PubMed] [Google Scholar]
- Cosaceanu D, Budiu RA, Carapancea M, Castro J, Lewensohn R, Dricu A. Ionizing radiation activates IGF-1R triggering a cytoprotective signaling by interfering with Ku-DNA binding and by modulating Ku86 expression via a p38 kinase-dependent mechanism. Oncogene. 2007;26:2423–2434. doi: 10.1038/sj.onc.1210037. [DOI] [PubMed] [Google Scholar]
- Plowchalk DR, Mattison DR. Phosphoramide mustard is responsible for the ovarian toxicity of cyclophosphamide. Toxicol Appl Pharmacol. 1991;107:472–481. doi: 10.1016/0041-008x(91)90310-b. [DOI] [PubMed] [Google Scholar]
- Meirow D, Lewis H, Nugent D, Epstein M. Subclinical depletion of primordial follicular reserve in mice treated with cyclophosphamide: clinical importance and proposed accurate investigative tool. Hum Reprod. 1999;14:1903–1907. doi: 10.1093/humrep/14.7.1903. [DOI] [PubMed] [Google Scholar]
- Kalich-Philosoph L, Roness H, Carmely A, Fishel-Bartal M, Ligumsky H, Paglin S, Wolf I, Kanety H, Sredni B, Meirow D. Cyclophosphamide triggers follicle activation and “burnout”; AS101 prevents follicle loss and preserves fertility Sci Translat Med 2013. 5. 185ra162. [DOI] [PubMed] [Google Scholar]
- Oktay K, Sönmezer M. Fertility issues and options in young women with cancer. Recent Results Cancer Res. 2008;178:203–224. doi: 10.1007/978-3-540-71274-9_18. [DOI] [PubMed] [Google Scholar]
- Falcone T, Attaran M, Bedaiwy MA, Goldberg JM. Ovarian function preservation in the cancer patient. Fertil Steril. 2004;81:243–257. doi: 10.1016/j.fertnstert.2003.06.031. [DOI] [PubMed] [Google Scholar]
- Early Breast Cancer Trialists' Collaborative Group. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365:1687–1717. doi: 10.1016/S0140-6736(05)66544-0. [DOI] [PubMed] [Google Scholar]
- Fodor J. Interactions between radiation and hormonal therapy in breast cancer: simultaneous or sequential treatment [in Hungarian] Orv Hetil. 2006;147:121–125. [PubMed] [Google Scholar]
- Harris EE, Christensen VJ, Hwang WT, Fox K, Solin LJ. Impact of concurrent versus sequential tamoxifen with radiation therapy in early-stage breast cancer patients undergoing breast conservation treatment. J Clin Oncol. 2005;23:11–16. doi: 10.1200/JCO.2005.09.056. [DOI] [PubMed] [Google Scholar]
- Albain KS, Barlow WE, Ravdin PM, Farrar WB, Burton GV, Ketchel SJ, Cobau CD, Levine EG, Ingle JN, Pritchard KI, Lichter AS, Schneider DJ, et al. Adjuvant chemotherapy and timing of tamoxifen in postmenopausal patients with endocrine-responsive, node-positive breast cancer: a phase 3, open-label, randomised controlled trial. Lancet. 2009;374:2055–2063. doi: 10.1016/S0140-6736(09)61523-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher B, Redmond C, Brown A, Fisher ER, Wolmark N, Bowman D, Plotkin D, Wolter J, Bornstein R, Lequalt-Poisson S, et al. Adjuvant chemotherapy with and without tamoxifen in the treatment of primary breast cancer: 5-year results from the National Surgical Adjuvant Breast and Bowel Project Trial. J Clin Oncol. 1986;4:459–471. doi: 10.1200/JCO.1986.4.4.459. [DOI] [PubMed] [Google Scholar]
- Hug V, Hortobagyi GN, Drewinko B, Finders M. Tamoxifen-citrate counteracts the antitumor effects of cytotoxic drugs in vitro. J Clin Oncol. 1985;3:1672–1677. doi: 10.1200/JCO.1985.3.12.1672. [DOI] [PubMed] [Google Scholar]
- Osborne CK, Kitten L, Arteaga CL. Antagonism of chemotherapy-induced cytotoxicity for human breast cancer cells by antiestrogens. J Clin Oncol. 1989;7:710–717. doi: 10.1200/JCO.1989.7.6.710. [DOI] [PubMed] [Google Scholar]
- Berman E, McBride M, Lin S, Menedez-Botet C, Tong W. Phase I trial of high-dose tamoxifen as a modulator of drug resistance in combination with daunorubicin in patients with relapsed or refractory acute leukemia. Leukemia. 1995;9:1631–1637. [PubMed] [Google Scholar]
- Ezzat AA, Ibrahim EM, Stuart RK, Ajarim D, Bazarbashi S, El-Foudeh MO, Rahal M, Al-Sayed A, Berry J. Adding high-dose tamoxifen to CHOP does not influence response or survival in aggressive non-Hodgkin's lymphoma: an interim analysis of a randomized phase III trial. Med Oncol. 2000;17:39–46. doi: 10.1007/BF02826215. [DOI] [PubMed] [Google Scholar]
- El-Yazigi A, Berry J, Ezzat A, Wahab FA. Effect of tamoxifen on the pharmacokinetics of doxorubicin in patients with non-Hodgkin's lymphoma. Ther Drug Monit. 1997;19:632–636. doi: 10.1097/00007691-199712000-00005. [DOI] [PubMed] [Google Scholar]