

Background: The pathways governing biosynthesis of the Aspergillus fumigatus exopolysaccharide galactosaminogalactan are poorly understood.
Results: The structure of Sph3 revealed a (β/α)8 barrel fold. The enzyme hydrolyzes galactosaminogalactan and is required for the synthesis of this exopolysaccharide.
Conclusion: Sph3 is a glycoside hydrolase (GH) whose activity is essential for galactosaminogalactan biosynthesis.
Significance: Sph3 defines a new glycoside hydrolase superfamily, GH family 135.
Keywords: Aspergillus, biofilm, carbohydrate biosynthesis, crystal structure, glycoside hydrolase, in vivo imaging, mutagenesis in vitro, polysaccharide, galactosaminogalactan, Sph3
Abstract
Aspergillus fumigatus is the most virulent species within the Aspergillus genus and causes invasive infections with high mortality rates. The exopolysaccharide galactosaminogalactan (GAG) contributes to the virulence of A. fumigatus. A co-regulated five-gene cluster has been identified and proposed to encode the proteins required for GAG biosynthesis. One of these genes, sph3, is predicted to encode a protein belonging to the spherulin 4 family, a protein family with no known function. Construction of an sph3-deficient mutant demonstrated that the gene is necessary for GAG production. To determine the role of Sph3 in GAG biosynthesis, we determined the structure of Aspergillus clavatus Sph3 to 1.25 Å. The structure revealed a (β/α)8 fold, with similarities to glycoside hydrolase families 18, 27, and 84. Recombinant Sph3 displayed hydrolytic activity against both purified and cell wall-associated GAG. Structural and sequence alignments identified three conserved acidic residues, Asp-166, Glu-167, and Glu-222, that are located within the putative active site groove. In vitro and in vivo mutagenesis analysis demonstrated that all three residues are important for activity. Variants of Asp-166 yielded the greatest decrease in activity suggesting a role in catalysis. This work shows that Sph3 is a glycoside hydrolase essential for GAG production and defines a new glycoside hydrolase family, GH135.
Introduction
Aspergillus fumigatus is a ubiquitous filamentous fungus that causes invasive infections in immunocompromised patients (1). Even with currently available antifungal agents, the mortality of invasive aspergillosis remains over 50%, highlighting the need for new therapies that target A. fumigatus (2). Of the over 250 Aspergillus species identified to date, A. fumigatus is responsible for the majority of human infections, even though other Aspergillus species are more commonly recovered during environmental sampling (3, 4). These observations suggest that A. fumigatus expresses unique virulence factors that enhance its ability to infect human hosts.
Production of the exopolysaccharide galactosaminogalactan (GAG)6 by A. fumigatus has been recently identified as an important factor in the pathogenesis of invasive aspergillosis (5–7). GAG is a heteropolysaccharide composed of α-1,4-linked galactose, N-acetylgalactosamine, and galactosamine residues that is found in both a secreted form and bound to the cell wall of hyphae (5, 8, 9). A. fumigatus produces higher levels of cell wall-associated GAG than other Aspergillus species (8). GAG is required for biofilm formation and adherence to host cells, and it mediates resistance to killing by neutrophil extracellular traps (6, 10). GAG also functions as an immunosuppressive polysaccharide both indirectly, through masking β-glucan from dectin-1 recognition, and directly by inducing neutrophil apoptosis and secretion of the immunosuppressive cytokine interleukin 1 receptor antagonist (IL-1RA) (6, 7, 11). Given the multiple roles that GAG plays in pathogenesis, the biosynthetic pathways governing GAG synthesis represent promising targets for novel antifungal therapies.
A comparative transcriptomic analysis of transcription factor mutants with impaired GAG production identified a cluster of five co-regulated genes predicted to encode proteins required for GAG biosynthesis (Fig. 1A) (6, 12). This five-gene cluster shares compositional similarities with the exopolysaccharide biosynthesis operons found in bacteria as postulated previously (8, 13–15) (Fig. 1, A and B). The production of exopolysaccharides by both Gram-negative and Gram-positive bacteria has also been shown to increase adhesion, cohesion, and structural complexity within the biofilm (13, 16–19). The similarities between these bacterial exopolysaccharide systems and the components of the GAG cluster allow a model of the GAG biosynthetic pathway to be proposed (Fig. 1C). Two of the proteins encoded within the GAG cluster, Uge3 and Agd3, have been shown to be required for the synthesis of functional GAG (8, 20). Uge3 is a cytoplasmic UDP-glucose-4-epimerase that mediates the synthesis of the GAG precursors UDP-N-acetylgalactosamine and UDP-galactose from UDP-N-acetylglucosamine and UDP-glucose, respectively (6, 20). Agd3, a secreted protein, was found to be required for the deacetylation of the GalNAc residues within the newly synthesized GAG polymer (6, 8). Two of the three remaining genes located in the A. fumigatus cluster are predicted to encode enzymes involved in carbohydrate biosynthesis. The first of these genes, ega3 (Afu3g07890), is predicted to encode an α-1,4-galactosaminidase (6). Carbohydrate-cleaving enzymes are found within the operon of a number of bacterial exopolysaccharide secretion systems (13, 22, 24, 26, 28). The function of these enzymes is unclear, but they have been suggested to play a role in clearing the periplasm of residual polysaccharide and to constitute part of a trans envelope secretion complex (29, 30). The second uncharacterized gene within the cluster, gtb3 (Afu3g07860), is predicted to encode a large integral membrane glycosyltransferase. The presence of a synthase complex that polymerizes and transports the exopolysaccharide across the membrane is also a common feature of bacterial exopolysaccharide systems (Fig. 1C) (13, 31, 32).
FIGURE 1.
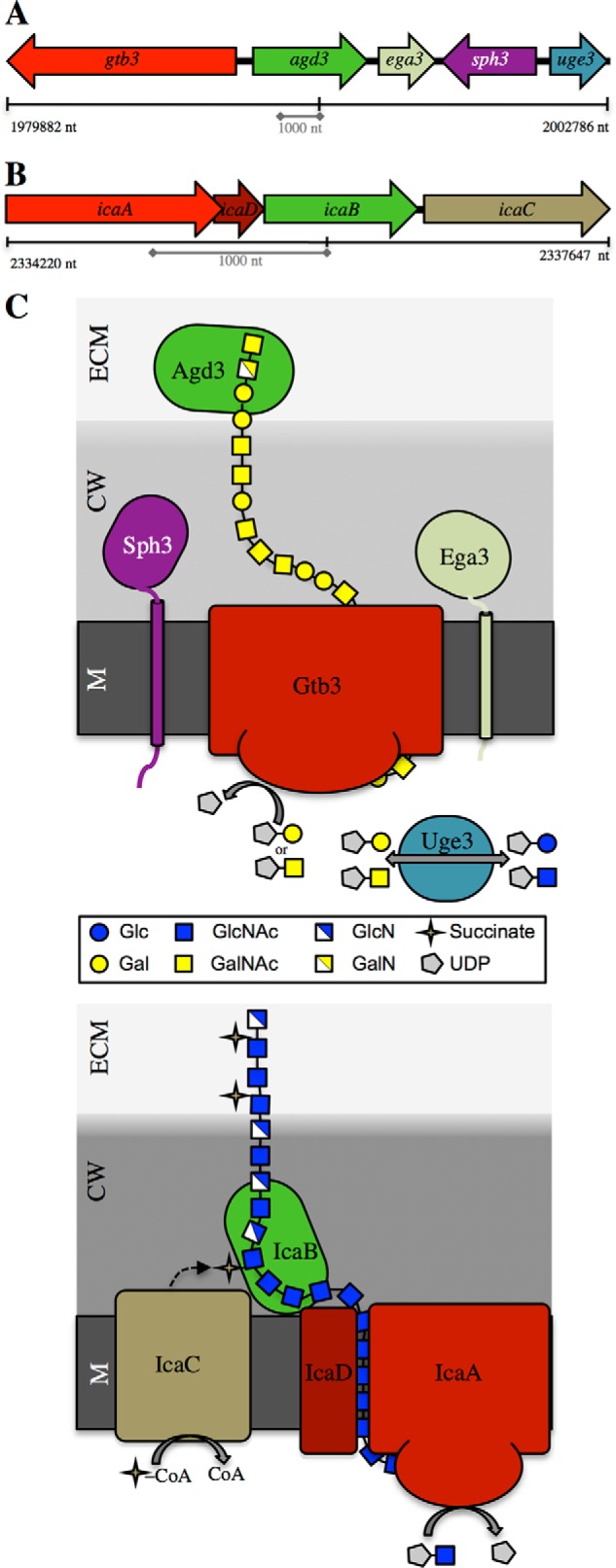
Proposed GAG biosynthetic system and current model of the Ica system of Staphylococcus epidermidis. A, diagram of the co-regulated five gene cluster located on chromosome 3 involved in GAG biosynthesis. B, schematic representation of the ica operon from S. epidermidis. C, comparative model of the biosynthetic pathways of GAG and PNAG/polysaccharide intercellular adhesin polysaccharides from A. fumigatus and S. epidermidis, respectively. M, membrane; CW, cell wall; ECM, extracellular matrix. The synthase complex is red; deacetylase is green; epimerase is teal, and other modifying enzymes are tan.
The final co-regulated gene contained within the cluster, sph3 (Afu3g07900), is annotated as a gene encoding a spherulin 4 protein, and primary sequence analysis reveals no conserved domains suggestive of a carbohydrate active enzyme. The spherulin 4 family was first identified through investigation of mRNAs specific to encystment in the slime mold Physarum polycephalum (33). The spherulin 4 mRNA was detected during late cyst formation and was suggested to be involved in maturation or the ability of the mold to germinate, although these hypotheses have never been investigated (33). Spherulin 4 genes have been found in multiple eukaryotic and prokaryotic species as well as a few in archaea, although their functions remain unknown.
In this report, the role of Sph3 in GAG biosynthesis was investigated using genetic, structural, and biochemical analysis. Structure-function studies demonstrated that sph3 encodes a glycoside hydrolase that can cleave both purified and cell wall-associated GAG. Sph3 activity was found to be necessary for GAG biosynthesis by A. fumigatus as deletion of sph3 or mutation of the key conserved residues of Sph3 result in a complete loss of detectable cell wall-associated and secreted GAG. Collectively, these results indicate that polysaccharide cleavage by Sph3 is required during GAG biosynthesis and biofilm formation by A. fumigatus.
Materials and Methods
Bioinformatics Analysis of Sph3
The amino acid sequence of Sph3 from A. fumigatus was retrieved from the Aspergillus Genome Database and was submitted to multiple web servers for analysis. The primary servers used were SignalP, BLASTP, Phyre2, TMHMM, and Phobius (34–38). ClustalW was used for sequence alignments (39).
Strains and Culture Conditions
A. fumigatus strain Af293 was used as the parent wild-type strain for all molecular manipulations. Unless otherwise noted, strains were grown and harvested on yeast extract peptone dextrose (YPD) agar (Fisher Scientific) at 37 °C as described previously (40). For growth in liquid medium, Brian's medium (5), Aspergillus minimum medium (AspMM) (41), and Roswell Park Memorial Institute (RPMI) 1640 medium (Wisent, Inc.) were used as indicated.
Deletion of sph3
The sph3 open reading frame (ORF) was replaced by the hygromycin resistance cassette following our standard disruption protocol (41) adapted to the Gateway® (Invitrogen) system (6). To generate the disruption constructs, ∼1 kb of sequence flanking the sph3 ORF was amplified by PCR from Af293 genomic DNA using primers S1, S2, and S3, S4 to generate fragments FS1 and FS4, respectively (Table 1). The resulting PCR products were then cloned into pENTR-D-TOPO® entry plasmid. An LR recombination allowed recombination of pENTR::FS1 with pHY, resulting in the fusion of FS1 upstream of the hph cassette, and of pENTR::FS4 with pYG, resulting in the fusion of FS4 downstream of the hph cassette. DNA fragments for transformation were then amplified by PCR, using the primers S1, HY with pHY::FS1, and S4, YG with pYG::FS4. Protoplasts of A. fumigatus Af293 were transformed with 5 μg of each DNA fragment, as described previously. Transformants were selected on minimal media 0.025% (w/v) hygromycin-enriched plates. Complete deletion of the sph3 ORF was confirmed by PCR using primers S-ext1, S-ext4, S-RT sense, HY, and YG, and by real time reverse transcriptase (RT)-PCR primers S-RT sense and S-RT antisense to ensure a complete absence of sph3 mRNA.
TABLE 1.
Strains and primers used
| Primers or strain | Sequence or description | Source or Ref. |
|---|---|---|
| S1 | CACCGCACAATACAGCAGGGT | This study |
| S2 | CAGAGGATCAAGGGGAAACG | This study |
| S3 | CACCTAAAATCCTGAACTGACC | This study |
| S4 | ATTGGATAGCAGGGAACCAG | This study |
| SgfI-S5 | GCGATCGCACTTGGGGGAAAAA | This study |
| PacI-S6 | TTAATTAACCATATTTTAGCAAATATACCTTAGGAAG | This study |
| HY | ATTGTTGGAGCCGAAATCC | This study |
| YG | AAGATCGTTATGTTTATCGGCACT | This study |
| S-ext1 | TGATCGCACTTGGGGGAAAAA | This study |
| S-ext4 | CATGGGCGGAGTTGGTTTCA | This study |
| S-RT sense | CAACCCGGCTCTAGCTGTTC | This study |
| S-RT antisense | TGAACCACGTAATGATCCGGC | This study |
| S-E216A-sense | TCGTGTTCGCAGCAACGTA | This study |
| S-E216A-antisense | CGTTGCTGCGAACACGA | This study |
| LE | GTTGACCAGTGCCGTTCC | This study |
| BL | GCTGATGAACAGGGTCACG | This study |
| T-trpC | AGAGCGGATTCCTCAGTCTC | This study |
| Sph3 52start | GGGCATATGTCCAAGGTCTTTGTGCCTCTCTATGTG | This study |
| Sph3 298stop | GGCTCGAGCTATTTTCCCATCAAATCCACAAACTC | This study |
| E222A Fwd | GGGCATATGTACAAGGTGGACATGCTCC | This study |
| E222A Rev | GGGCATATGCCCGATCCCGACGACGGCCT | This study |
| E222Q Fwd | CGTTGTTTTTCAAGCAACCTATGC | This study |
| E222Q Rev | GCATAGGTTGCTTGAAAAACAACG | This study |
| D166A Fwd | CTTTTTTGCAGAAACACCGCAGCAG | This study |
| D166A Rev | GTGTTTCTGCAAAAAAGATACCACG | This study |
| D166N Fwd | CTTTTTTAATGAAACACCGCAGCAG | This study |
| D166N Rev | GTGTTTCATTAAAAAAGATACCACG | This study |
| E167A Fwd | GTATCTTTTTTGATGCAACACCGCAG | This study |
| E167A Rev | CTGCGGTGTTGCATCAAAAAAGATAC | This study |
| E216A Fwd | CGTTCGTGTTCGCAGCAACGTACG | This study |
| E216A Rev | GGTGTCGTACGTTGCTGCGAACAC | This study |
| Af293 | Wild-type pathogenic strain of A. fumigatus | P. Magee |
| B834 Met− | E. coli laboratory expression strain: F− ompT hsdSB (rB− mB−) gal dcm met (DE3) | Novagen |
| TOP10 | E. coli cloning strain | Invitrogen |
| BL21 CodonPlus | E. coli laboratory expression strain: F− ompT hsdS (rB− mB−) dcm+ Tetr galλ (DE3) endA [argU proL Camr] | Stratagene |
Construction of sph3-complemented Strains
To verify the specificity of the mutant phenotype, the Δsph3 strain was complemented by reintroducing a wild-type copy of sph3 at its native locus (41). For this approach, a new plasmid, pTAPA, was first constructed from pUC19 (pUC ori, bla cassette) containing the following: a polylinker containing SgfI, PacI, and AscI restriction enzymes sites for cloning; the terminator of Aspergillus nidulans trpC gene (TtrpC); the ble gene encoding a phleomycin and bleomycin resistance protein driven by the Aspergillus oryzae thiA gene promotor (PthiA) followed by the Candida albicans CYC1 terminator (Tcyc1). The sph3 ORF and ∼1 kb of the upstream sequence were amplified by PCR using the primers SgfI-S5 and PacI-S6 and ligated between the SgfI and PacI sites of pTAPA to generate the plasmid pTAPA::sph3. DNA fragments for transformation were then produced from the pTAPA::sph3 vector by PCR using primers S1 and LE and primers BL and T-trpC. Protoplasts of the Δsph3 mutant were then transformed with 5 μg of each PCR product, as described previously (41). Transformants were selected on minimal media containing 0.015% (w/v) phleomycin. Re-integration of sph3 and sph3 expression was verified by real time RT-PCR using primers S-RT sense and S-RT antisense to ensure that sph3 mRNA production was restored. A mutant strain expressing an Sph3 protein with a glutamate residue replaced by an alanine residue at position 216, Sph3E216A, was also constructed. The sph3 ORF was modified using primer mutagenesis to introduce A:C mutation at position 951 in the sph3 ORF. Protoplasts of the Δsph3 mutant were then transformed with this construct as detailed above. The sph3 allele in the resulting strain was amplified and sequenced to confirm the successful integration of the point mutation, and gene expression was verified by real time RT-PCR.
Real Time RT-PCR
In vitro, the expression of the genes of interest was quantified by relative real time RT-PCR analysis as described previously (12). The primers used for each gene are shown in Table 1. First strand synthesis was performed from total RNA with QuantiTect reverse transcription kit (Qiagen) using random primers. Real time PCR was then performed using an ABI 7000 thermocycler (Applied Biosystems). Amplification products were detected with Maxima® SYBR Green quantitative PCR system (Fermentas). Fungal gene expression was normalized to A. fumigatus TEF1 expression, and relative expression was estimated using the formula 2−ΔΔCt, where ΔΔCt = ((Cttarget gene)sample − (CtTEF1)sample)/((Cttarget gene)reference − (CtTEF1)reference). To verify the absence of genomic DNA contamination, negative controls were used for each gene set in which reverse transcriptase was omitted from the mix.
Visualization of Cell Wall-associated GAG by Lectin Staining
Young hyphae were grown as above on tissue culture-treated coverslips, washed with PBS, and stained with 30 μg/ml of the GalNAc-specific lectin soybean agglutinin lectin (SBA) conjugated to fluorescein for 2 h at 4 °C. Samples were washed twice with PBS and fixed with 4% (w/v) paraformaldehyde for 10 min at 4 °C. Samples were washed once with PBS and counterstained with DRAQ5TM (1:1000 dilution) for 5 min at room temperature to visualize hyphae. Samples were mounted with SlowFade® mounting media, sealed, and imaged on a Zeiss confocal microscope with a 488- and 633-nm laser.
For samples treated with recombinant Sph3, conidia were grown for 9 h in Dulbecco's modified Eagle's medium (DMEM) at 37 °C, 5% CO2, washed once with PBS, and incubated with the indicated concentrations of Sph3 variants in Ham's F-12K (Kaighn's) medium for 3 h at 37 °C, 5% CO2. Samples were then washed with PBS, transferred to poly-d-lysine-coated coverslips, and stained with 30 μg/ml of the GalNAc-specific lectin SBA conjugated to fluorescein and DRAQ5TM as above.
Detection of Soluble GAG by Indirect Enzyme Immunoassay (EIA)
Liquid cultures of A. fumigatus (5 × 105 conidia/ml) were grown in Brian's medium for 24 h at 37 °C, and the resulting spent culture supernatants were collected using a 0.22-μm filter (SteritopTM, EMD Millipore). Supernatants were diluted using fresh Brian's medium and incubated for 1 h at 37 °C in a medium-binding multiwall assay plate (Greiner Bio-One) to allow binding of GAG. Wells were washed three times with PBS + 0.05% (v/v) Tween 20, blocked with PBS + 2% (v/v) Tween 20 + 1% (w/v) BSA and then incubated with mouse anti-GAG antibody for 1 h at room temperature. Wells were washed four times and incubated with sheep anti-mouse antibody conjugated to horseradish peroxidase (Jackson ImmunoResearch) for 1 h at room temperature. Wells were washed four times and developed using TMB substrate (Thermo Scientific). After the reaction was stopped using 2 m sulfuric acid, absorbance was read at 450 nm.
Scanning Electron Microscopy of Hyphae
Conidia were incubated for 12 h in phenol red-free RPMI 1640 medium at 37 °C, 5% CO2 on glass coverslips. Samples were fixed in 2.5% (w/v) glutaraldehyde in 0.1 m sodium cacodylate buffer at 4 °C overnight, sequentially dehydrated in ethanol, and critical-point dried. Samples were then sputter coated with Au-Pd and imaged with a field-emission scanning electron microscope (S-4700 FE-SEM, Hitachi).
Germination Assay
To determine whether there were differences in germination between strains, 1 × 105 conidia of the strain of interest were inoculated in 500 μl of RPMI 1640 medium in a 24-well tissue culture-treated microtiter plate and grown at 37 °C with 5% CO2. After 5 h, the growth of the fungus was assessed hourly by microscopy. Germination was defined as the emergence of a germ tube from the swollen conidia.
Radial Growth Assay
To test for differences in growth rate between the strains, 3 × 104 conidia were spot inoculated on AspMM agar plates containing a range of iron concentrations (0 and 30 μm) and varying pH values (5.4 to 9.0). The radial growth of the fungus was recorded daily for 6 days post-inoculation.
Aspergillus clavatus Sph3 Expression and Purification
A pUC57 plasmid containing a codon-optimized version of the gene encoding A. clavatus Sph3 (Sph3Ac) from amino acids 54–304 (Sph3Ac(54–304)) was obtained from BioBasic. The gene was subcloned into the pET28a vector between the NdeI and XhoI sites creating the expression plasmid pET28-Sph3Ac(54–304). This yielded a plasmid encoding a thrombin-cleavable N-terminal hexa-histidine tag fused to the C-terminal domain of Sph3Ac. The E222A, E222Q, D166A, D166N, and E167A mutants were created using the QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies) with the following forward and reverse primers: E222A Fwd and E222A Rev; E222Q Fwd and E222Q Rev; D166A Fwd and D166A Rev; D166N Fwd and D166N Rev; and E167A Fwd and E167A Rev (Table 1). Wild-type and all Sph3Ac variants were expressed and purified from Escherichia coli BL21 CodonPlus cells. Cells were transformed and grown at 37 °C in 1 liter Luria-Bertani (LB) media containing 50 μg/ml kanamycin until an absorbance of ∼0.4–0.5 at 600 nm (A600). Growth was continued at 18 °C with induction occurring at A600 of ∼0.6–0.7 using isopropyl d-1-thiogalactopyranoside to a final concentration of 0.5 mm. Cells were harvested 20 h post-induction by centrifugation at 3750 × g for 20 min. Cell pellets were resuspended in 25 ml of 50 mm HEPES, pH 8, with 300 mm NaCl, 5% (v/v) glycerol, 10 mm imidazole, 2 mm tris(2-carboxyethyl)phosphine (TCEP), and one complete mini protease inhibitor mixture tablet (Roche Applied Science). Resuspended cells were lysed with three passes through an Emulsiflex-c3 (Avestin) at 15,000 p.s.i. Cellular debris was separated using centrifugation at 31,000 × g for 30 min. The supernatant was applied to a gravity column containing nickel-nitrilotriacetic acid resin (Qiagen) that was pre-equilibrated with buffer A (20 mm HEPES, pH 8.0, 300 mm NaCl, 2% (v/v) glycerol, 1 mm TCEP, and 10 mm imidazole). The column was washed with 8 column volumes of buffer A followed by 4 column volumes of buffer A containing 20 mm imidazole. The bound protein was then eluted using buffer A containing 250 mm imidazole. The elution fraction was concentrated to 2 ml using a ultrafiltration device (Millipore) before further purification and buffer exchanged into buffer B (20 mm HEPES, pH 8.0, 250 mm NaCl, 2% (v/v) glycerol, 1 mm TCEP) by size-exclusion chromatography using a HiLoad 16/60 Superdex 200 prep-grade gel filtration column (GE Healthcare).
Se-Met protein was produced using the method described by Lee et al. (42) with B834 Met− E. coli cells (Novagen). Se-Met-containing protein was purified as described above.
Sph3Af Expression and Purification
The gene encoding A. fumigatus Sph3 amino acids 52–298 (Sph3Af(52–298)) was cloned from cDNA into a pET28a vector using primers Sph3 52start and Sph3 298end with NdeI and XhoI sites, respectively. This produced a plasmid analogous to the one outlined above for Sph3Ac. The QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies) was used to create the E216A mutant with forward and reverse primers E216A Fwd and E216A Rev (Table 1). Wild-type Sph3Af and the E216A variant were purified as described above for Sph3Ac with the following modifications: proteins were buffer-exchanged into buffer C (20 mm CHES, pH 9, 150 mm NaCl, and 2 mm dithiothreitol (DTT)) by gel filtration chromatography instead of buffer B.
Crystallization, Data Collection, and Structure Determination
Purified Sph3Ac(54–304) and Sph3Af(52–298) were concentrated to 10–11 mg/ml and screened for crystallization conditions using sitting-drop vapor diffusion at 20 °C. MCSG 1–4 sparse matrix screens (Microlytic) were set up using the Crystal Gryphon robot in Art Robbins Instruments Intelli-Plates 96-2 Shallow Well (Hampton). Initial Sph3Ac(54–304) crystals were formed in MCSG-3 number 59 (0.1 m Bistris propane-HCl, pH 7.0, 2.5 m ammonium sulfate) and used as a seed stock to grow optimized native and SeMet-incorporated crystals by rescreening. Crystals were reproduced using hanging-drop vapor diffusion with streak seeding. Crystal 1 (SeMet) was formed in MCSG-3 number 89 (0.2 m sodium chloride, 0.1 m Tris-HCl, pH 7.0, 1.0 m sodium citrate) and was cryoprotected by soaking in 0.84 m sodium citrate, 0.1 m Tris-HCl, pH 7.0, and 30% (v/v) ethylene glycol for 5 s before vitrification in liquid nitrogen. Crystal 2 (SeMet) was formed in MCSG-3 number 57 (0.09 m HEPES-NaOH, pH 7.5, 1.26 m sodium citrate, and 10% (v/v) glycerol) and was cryoprotected using 0.82 m sodium citrate, 0.09 m HEPES, pH 7.5, 10% (v/v) glycerol, and 20% (v/v) ethylene glycol. X-Ray diffraction data were collected from crystals 1 and 2 at the National Synchrotron Light Source using beamline X29 with an ADSC Quantum 315r detector. The data were indexed, integrated, and scaled using HKL2000 (Table 2) (43). Initial phases for crystal 1 were determined using selenium-single wavelength anomalous dispersion data in PHENIX AutoSol (44) and accessed through the SBGrid consortium (45), which located three selenium sites in the asymmetric unit. Density-modified phases were calculated using SOLVE/RESOLVE (46), and the resulting electron density map was of high quality and enabled PHENIX AutoBuild to build ∼95% of the protein. The structure was refined in PHENIX.REFINE (44) alternated with manual building in Coot (47). The TLSMD server was used to add TLS groups during the refinement stage. Phasing for crystal 2, which diffracted to a higher resolution, was performed using molecular replacement with PHENIX AutoMR (44). Model building and refinement were carried out as described above for crystal 1. The resulting model from crystal 2 encompassed residues 54–304 and three residues from the hexa-histidine tag. Hydrogen atoms were added during the final rounds of refinement using PHENIX.REFINE. Structural figures were generated in PYMOL Molecular Graphics System (DeLano Scientific). Structural analysis was completed using the DALI server (48), Consurf server (49–51), and APBS Tools (52). The Consurf server aligned 61 sequences with a maximal identity of 95% and minimum identity of 35% to create the conservation map.
TABLE 2.
Summary of data collection and refinement statistics
Values in parentheses correspond to the highest resolution shell. NSLS is National Synchrotron Light Source.
| Crystal 1 (SeMet) | Crystal 2 (SeMet) | Crystal 3 (native) | |
|---|---|---|---|
| Data collection | |||
| Beamline | NSLS X29 | NSLS X29 | 08B1-1 |
| Wavelength (Å) | 0.9792 | 1.075 | 0.9792 |
| Space group | P212121 | P212121 | P212121 |
| Unit cell parameters (Å,°) | a = 44.8, b = 60.2, c = 98.6, α = β = γ = 90.0 | a = 44.8, b = 60.3, c = 98.8, α = β = γ = 90.0 | a = 44.7, b = 58.9, c = 96.8, α = β = γ = 90.0 |
| Resolution (Å) | 50.00–1.76 (1.82–1.76) | 50.00–1.25 (1.29–1.25) | 37.39–1.93 (1.99–1.93) |
| Total no. of reflections | 355,299 | 141,016 | 95,941 |
| No. of unique reflections | 27,059 | 73,990 | 19,190 |
| Redundancy | 13.1 (12.9) | 7.7 (7.6) | 5.0 (4.9) |
| Completeness (%) | 99.9 (99.8) | 99.1 (97.9) | 96.6 (98.3) |
| Average I/σ(I) | 57.8 (20.8) | 52.5 (5.2) | 21.4 (3.9) |
| Rmerge (%)a | 6.7 (19.8) | 6.1 (59.7) | 6.2 (40.2) |
| Refinement | |||
| Rworkb/Rfreec | 14.8/16.6 | 17.0/21.7 | |
| No. of atoms | |||
| Protein | 1981 | 1973 | |
| Ethylene glycol | 8 | ||
| GalNAc | 16 | ||
| Water | 227 | 203 | |
| Average B-factors (Å2) d | 16.3 | 23.7 | |
| Protein | 15.10 | 22.8 | |
| Ethylene glycol | 20.20 | ||
| GalNAc | 50.4 | ||
| Water | 26.80 | 29.7 | |
| r.m.s.d. | |||
| Bond lengths (Å) | 0.008 | 0.007 | |
| Bond angles (°) | 1.22 | 1.00 | |
| Ramachandran plotd | |||
| Total favored (%) | 98 | 98 | |
| Total allowed (%) | 100 | 100 | |
| Coordinate error (Å)e | 0.11 | 0.19 | |
| PDB code | 5C5G | 5D6T | |
a Rmerge = ΣΣ|I(k) − 〈I〉|/ΣI(k), where I(k) and 〈I〉 represent the diffraction intensity values of the individual measurements and the corresponding mean values. The summation is over all unique measurements.
bRwork = Σ‖Fobs| − k|Fcalc‖/|Fobs|, where Fobs and Fcalc are the observed and calculated structure factors, respectively.
cRfree is the sum extended over a subset of reflections (2.7% crystal 2 and 5.0% crystal 3) excluded from all stages of the refinement.
dData are as calculated using MolProbity (25).
eMaximum-likelihood based coordinate Error, as determined by PHENIX (44).
Sph3-GalNAc Complex Structure Determination
Rescreening of Sph3Ac(54–304) in the presence of 1.9 m GalNAc yielded crystal 3 in MCSG-1 condition number 49 (0.1 m HEPES-NaOH, pH 7.5 and 2.0 m ammonium sulfate). Crystal 3 was vitrified in liquid nitrogen, and data were collected at the Canadian Light Source using beamline 08B1-1. Data were processed using Autoprocess (Table 2), and the phases were determined using the molecular replacement technique with Sph3Ac(54–304) (PDB code 5C5G) as the starting model. Refinement and model building were performed using PHENIX and Coot through SBGrid as described above for the apo-structure.
In Vitro Hydrolysis Assays
Sph3 glycoside hydrolase activity was first tested on para-nitrophenyl (pNP) glycoside substrates. Briefly, 100-μl reactions contained 45 mm HEPES, pH 7.0, 7 μm Sph3Af or buffer control, and 2.5 mm pNP-glycoside and were performed in triplicate. The absorbance was read at 13-s intervals for 10 min at 405 nm for each sample. This assay was performed with pNP-α-Gal, pNP-β-Gal, pNP-α-GalNAc, pNP-β-GalNAc, pNP-α-glucose, pNP-β-glucose, pNP-β-mannose, and pNP-β-GlcNAc.
Crude GAG was isolated from A. fumigatus biofilms using ethanol precipitation as described previously (6, 20). 200-μl GAG aliquots were centrifuged to pellet the gelatinous fraction. The pellets were washed twice with 350 μl of PBS. The wash procedure included vortexing for 5 min, sonicated in a bath for 3 min, and manual mixing by pipetting to reach homogeneity. The final pellet was resuspended in 200 μl of PBS. Samples were treated with 12 μm protein and incubated at 26 °C. Fractions were taken at 24 h. GAG hydrolysis was quantified using a reducing sugar assay as described previously by Anthon and Barrett (53) with slight modifications. Briefly, 20 μl of enzyme reaction was mixed with 20 μl of 0.5 m NaOH and 20 μl of 3-methyl-2-benzothiazolinone hydrazine/DTT solution (1.5 mg/liter 3-methyl-2-benzothiazolinone hydrazine and 0.5 mg/liter DTT). The samples were incubated at 80 °C for 15 min before the addition of 40 μl of acidic iron reagent (0.5% (FeNH4(SO4)2)·12H2O, 0.5% sulfamic acid, and 0.25 n HCl). Samples were diluted by half with water, before absorbance was quantified at 620 nm. Standards were created with known concentrations of GalNAc, and reducing sugar levels were determined using the same assay. The resulting standard curve was used to determine the approximate number of reducing ends produced.
Results
Sph3 Belongs to the Spherulin 4 Family
Bioinformatics analyses of A. fumigatus Sph3 using the Phobius (38) and TMHMM (37) servers suggested that Sph3 is a single span type-II integral membrane protein. The predicted transmembrane domain spanned residues 20–42 (Fig. 2A). The predicted extracellular domain, residues 52–297, was recognized by BLASTP (35) as belonging to the spherulin 4 family (Pfam12138). Sequence alignment found three highly conserved regions common to other spherulin 4 proteins (Fig. 2B). The presence of an arginine-glycine-proline (NPG) motif is canonical to the spherulin 4 family and is conserved in Sph3 (Fig. 2B). Structural modeling using Phyre2 suggested that the spherulin domain adopts a structure similar to that of (β/α)8 glycoside hydrolases (GH). The top hit was a hypothetical protein (tm1410) from Thermotoga maritima, which matched residues 83–278 with a confidence level of 96.9%. When submitted to the carbohydrate active enzyme toolbox (54), Sph3 is not recognized as a glycoside hydrolase or any other carbohydrate active enzyme.
FIGURE 2.

Sph3 contains a spherulin 4 family domain. A, schematic model of the Sph3 protein based on bioinformatics shows an N-terminal transmembrane domain and a C-terminal spherulin 4 domain. B, primary sequence alignment using ClustalW of spherulin 4 proteins from A. fumigatus, A. clavatus, Marssonina brunnea (Uniprot K1WTQ0), Ralstonia pickettii (Uniprot A0A080W3T6), and P. polycephalum (Lav6-1). Sequence identity as compared with Sph3Af is listed. Conservation of amino acids is shown using gray scale with darker columns indicating higher conservation.
Sph3 Is Required for GAG Production
To investigate the role of Sph3 and determine whether the protein is essential for GAG production, a deletion strain was constructed. The Δsph3 mutant displayed wild-type (WT) growth and germination levels (Fig. 3, A and B). Immunofluorescence lectin staining with GalNAc-specific SBA (Fig. 3C) could not detect any cell wall-associated GAG in the Δsph3 mutant. The loss of cell wall-associated GAG was confirmed by scanning electron microscopy, which revealed hyphae that lacked the GAG-dependent cell surface decorations typically found on wild-type hyphae (Fig. 3D) (6). To determine whether the loss of cell wall-associated GAG was a consequence of altered adherence of the polymer or impaired exopolysaccharide synthesis, the levels of GAG in the culture filtrate were quantified using an anti-GAG-specific antibody. Levels of GAG in the Δsph3 mutant culture were undetectable and comparable with those observed in the GAG-deficient Δuge3 mutant (Fig. 3E). Furthermore, no GAG could be detected after ethanol precipitation of culture supernatants from the Δsph3 mutant. These data suggest that Sph3 is required for the production and/or export of the polymer.
FIGURE 3.

Sph3 is required the production of secreted and cell wall-associated GAG. A, germination rate of the Δsph3 mutant as compared with wild-type A. fumigatus as determined by serial microscopy. B, radial growth rate of the Δsph3 mutant and wild-type A. fumigatus. C, cell wall-associated GAG production by the indicated strains was detected by SBA lectin staining (green). Hyphae were counterstained with DRAQ5 (red) for visualization in confocal images. D, scanning electron microscopy images of hyphae of each indicated strain. Wild-type A. fumigatus produces surface decorations associated with GAG production, whereas the Δsph3 mutant and GAG-deficient Δuge3 mutant lack these structures. E, quantification of secreted GAG in culture supernatants of the indicated strains using an indirect EIA.
Structure of Sph3 Reveals a (β/α)8 Fold
To shed light on the function of the protein, recombinant Sph3 lacking its predicted transmembrane domain was expressed in E. coli for structural determination by crystallographic methods. Sph3 from A. fumigatus was recalcitrant to crystallization, so an orthologue from A. clavatus (Sph3Ac), which exhibits 87% sequence identity to Sph3Af, was used. The annotated sequence for Sph3Ac contains only 251 amino acids and no predicted transmembrane domain. Analysis of the region upstream of the annotated sph3Ac gene revealed an alternative start site that would add 53 residues. These 53 residues contain the predicted transmembrane helix found in sph3Af. The sph3Ac gene is likely annotated incorrectly, and the amino acid numbering used herein is therefore based on our current analysis.
The predicted extracellular domain of Sph3Ac was purified to homogeneity and subjected to crystallization trials. Small irregular crystals formed in two conditions within 1 week of incubation. Multiple new hits were obtained after rescreening with streak seeding. All crystals indexed to space group P212121. The structure was solved using selenium-single wavelength anomalous dispersion technique and then used to phase and refine the higher 1.25 Å resolution data set (Table 2). Model building and refinement produced a final model with Rwork and Rfree of 14.8 and 16.6%, respectively.
The structure of Sph3 revealed a (β/α)8 barrel with a core of eight parallel β-strands surrounded by eight α-helices (Fig. 4A). There were two small additional helices, one on either end of α6. Surface representation of the conserved residues shows a well conserved groove along the C termini of the core β-strands, which defines the top face of the (β/α)8 barrel (Fig. 4B). The groove is lined with highly conserved tyrosine residues and three acidic residues around a central depression. Situated near these acidic residues is the NPG spherulin 4 motif. In the apo-structure an ethylene glycol molecule was bound in the central depression of the conserved groove.
FIGURE 4.

Structure of Sph3Ac(54–304) and investigation of conserved regions. A, Sph3Ac shown in schematic representation with transparent surface model reveals a (β/α)8 barrel fold. The surface β-strands (purple) and α-helices (blue) are labeled β1–8 and α1–8, respectively. Loop regions are shown in light gray, and the ethylene glycol molecules are depicted as orange sticks. B, representation of the conserved surface as calculated by the ConSurf server (49–51) reveals a conserved groove on the top-face (C termini of β-strands) of the (β/α)8 barrel with a small depression in the center. Conserved residues are shown in fuchsia and variable residues in teal. C, transparent surface representation of the Sph3Ac structure (teal) in complex with GalNAc (yellow). D, close-up of the |Fo − Fo| omit density map contoured around GalNAc at σ = 2.5. E, superposition of the (β/α)8 barrels of Sph3 (purple) and a representative member of GH18 (blue, PDB 4WIW), GH27 (yellow, PDB 1KTB), and GH84 (green, PDB 2CBJ) shows the similarity in the overall folds. Right panel, close-up of the active sites of these glycoside hydrolases reveals three acidic residues of Sph3 (Asp-166, Glu-167, and Glu-222) in the same vicinity. E, comparison of the ethylene glycol molecule (orange) found in the active site of the apo-structure (purple) with GalNAc (yellow) from the co-crystal structure (teal). Residues connected with black lines represent those within hydrogen bonding distance.
Sph3 Defines a New GH Family
The DALI server was used to search for structurally similar proteins and identified glycoside hydrolases from families 18 (GH18), 27 (GH27), and 84 (GH84). Sph3 does not contain the primary catalytic motifs of any of these families and is not recognized by CAZy as belonging to any GH family. Although the CAZy database creates families of carbohydrate hydrolases based on primary sequence, often there are correlations found with substrate specificity and tertiary structure within and between these families. Family GH18 and GH84 members display hydrolase activity on N-acetylglucosamine polymers, whereas GH27 members have galactosidase and N-acetylgalactosaminidase activities. The top DALI hit is a putative chitinase from Desulfitobacterium hafniense (PDB 4WIW) that belongs to GH18 and aligns with r.m.s.d. of 3.1 Å over 202-eq Cα atoms (Fig. 4E). NagJ (PDB 2X0Y), a GH84 enzyme from Clostridium perfringens, aligned to Sph3 with r.m.s.d. of 3.1 Å over 162 Cα atoms. Alignment with a GH27 member, chicken α-N-acetylgalactosaminidase, yielded r.m.s.d. of 3.2 Å over 190 Cα atoms (PDB 1KTB). All three DALI hits contain a secondary β-rich domain that is absent from Sph3. Sequence alignment showed amino acid identity of 17 and 19% with the D. hafniense chitinase and the chicken α-N-acetylgalactosaminidase, respectively. NagJ has slightly higher sequence identity at 24%. In Sph3Ac residues Asp-166 and Glu-167 are located at the end of strand β4 in a similar location to the DXE catalytic motif of the GH18 family member; however, Glu-167 is oriented away from the groove (Fig. 4E). Asp-166 and Glu-167 are also in a similar location to the DD catalytic motif of the GH84 member NagJ. The two aspartic acid catalytic residues of GH27 family members are typically located at the C terminus of strand β4 and β6. In α-N-acetylgalactosaminidase, the catalytic acid and base are Asp-140 and Asp-121, respectively, and these residues superimpose with Asp-166 and Glu-222 in Sph3Ac (Fig. 4E). These acidic residues in Sph3 line the central depression of the conserved groove. Asp-166, Glu-167, and Glu-222 as well as Asn-202 are part of the conserved spherulin 4 motif identified in the primary sequence alignments (Fig. 2B). Taken together, these structural similarities and unique primary sequence motifs place Sph3 in a new GH family, GH135.
Sph3 Hydrolyzes Isolated and Cell Wall-associated GAG Polysaccharide
To determine whether Sph3 functions as a GAG hydrolase, the ability of the soluble recombinant Sph3 to cleave purified A. fumigatus GAG was tested. Hydrolytic activity was initially assayed using various pNP glycosides; however, no activity could be detected. Thus, we attempted to monitor GAG hydrolase activity through the release of sugar reducing ends. Treatment of purified GAG with Sph3Ac(54–304) and Sph3Af(52–298) resulted in an increased amount of soluble sugar reducing ends over a 24-h reaction period as compared with untreated samples (Fig. 5A). No increase in reducing sugars was detected upon co-incubation of Sph3Ac(54–304) with chitosan (Fig. 5B), suggesting that the hydrolytic activity has specificity for GAG. Using a GalNAc standard curve, it was estimated that 12 μm Sph3Ac(54–304) produced 8.8 nm reducing ends after 24 h of incubation. Single point mutations of the conserved acidic residues, Asp-166, Glu-167, and Glu-222, decreased or abolished the activity of Sph3Ac. The D166A, D166N, and E167A variants displayed no significant activity against purified GAG. The E222A variant was not able to hydrolyze purified GAG, but the conservative E222Q variant retained 60% of wild-type activity. Similarly, the Sph3Af E216A mutant (equivalent to Sph3Ac E222A) displayed no significant activity against purified GAG polymers. The Asp-166 and Glu-167 Sph3Af mutants were not tested due to decreased protein stability as compared with Sph3Ac.
FIGURE 5.
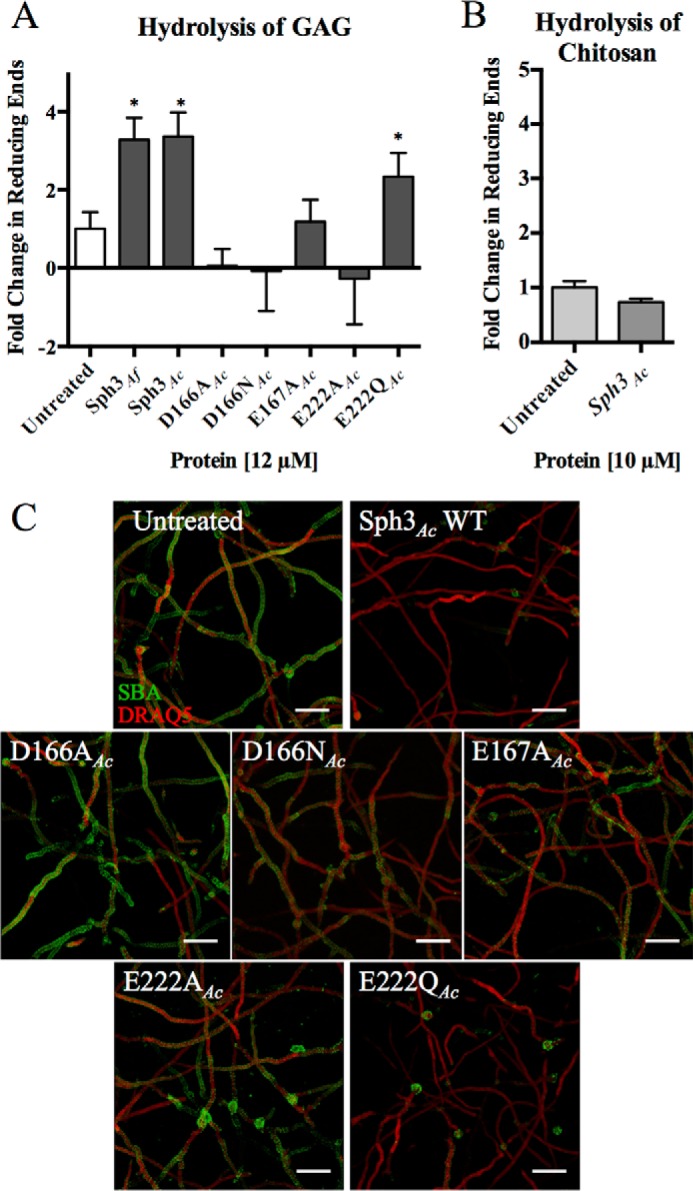
Sph3 hydrolyzes purified and cell wall-associated GAG. A, Sph3 hydrolysis of purified GAG as measured by the release of reducing sugars. Purified GAG was incubated with 12 μm of the indicated proteins for 24 h. Activity is shown as difference between untreated and treated samples. *, p < 0.05 as compared with untreated levels with n = 3. B, treatment of chitosan with 10 μm Sph3Ac for 24 h yielded no increase in reducing ends as compared with untreated polysaccharide. C, degradation of cell wall-associated GAG by the indicated Sph3 protein variants. A. fumigatus hyphae were treated with 0.05 μm of the indicated recombinant Sph3 variant for 3 h and then residual GAG was detected by SBA lectin staining (green). Bars represent means, and error bars are one S.D.
As purified GAG is relatively insoluble and to confirm that Sph3 was able to hydrolyze native cell wall-associated GAG, hyphae of wild-type A. fumigatus were grown for 9 h and then incubated with Sph3Ac(54–304) or catalytic variants for 3 h. The degree of cell wall-associated GAG was then visualized by staining with the GalNAc-specific lectin SBA. Treatment of hyphae with Sph3Ac(54–304) resulted in a complete loss of detectable GAG on the surface of hyphae (Fig. 5C). Treatment of hyphae with the catalytically inactive D166A variant had no effect on hyphal GAG levels compared with untreated hyphae (Fig. 5C). In this assay D166N, E167A, and E222A showed lower degradation levels than WT but higher than D166A. Collectively, these data confirm that Sph3 can hydrolyze both purified GAG and native cell wall-associated GAG on the hyphal cell wall.
GalNAc Binds to Sph3
Purified GAG polymer is insoluble and cannot be used for crystallographic studies, and shorter α1,4-linked GAG oligosaccharides are difficult to synthesize. Therefore, to gain insight into how the Sph3 may bind GAG, we attempted to co-crystallize the protein in the presence of each of the monosaccharide components of the polymer. Examination of the resulting difference electron density maps revealed the presence of a bound GalNAc molecule but not galactose or galactosamine in the structures obtained. The N-acetyl group of the GalNAc bound to Sph3 in the central depression of the conserved groove, in the same location as the ethylene glycol molecule found in the apo-structure (Fig. 4, C, D, and F). The N-acetyl group forms hydrogen bonds with residues Glu-222 and Tyr-65. Glu-222 is coordinated by Asn-202, which in turn is hydrogen bonded to Asp-166 (Fig. 4F). A water molecule bridges the hydrogen bond network between Glu-167, Asp-166, and the oxygen of the galactopyranose ring (Fig. 4F). The anomeric hydroxyl is coordinated by Asp-166. The ability of the protein to bind GalNAc at the active site, but not galactose or galactosamine, suggests that the enzyme may be specific for this monosaccharide.
Sph3 Hydrolytic Activity Is Required for GAG Biosynthesis
To examine the role of Sph3 enzymatic activity in GAG biosynthesis, the Δsph3 mutant was complemented with both wild-type sph3 and the sph3 E216A allele. This point mutant was chosen based on its in vitro stability and the low activity observed on purified GAG as well as the low activity of the Sph3Ac equivalent on cell wall-associated GAG. Complementation of the deletion strain with the wild-type allele rescued GAG expression as measured by lectin staining, S.E., and EIA of culture supernatants (Fig. 6, A–C). In contrast, complementation of the Δsph3 mutant with the sph3 E216A allele failed to restore GAG production. This suggests that the biosynthesis of GAG requires the enzymatic activity of Sph3.
FIGURE 6.

Sph3 activity is required for functional GAG production. A, levels of Sph3 mRNA were measured using RT quantitative PCR for each indicated strain. B, SBA (green) staining for GAG and DRAQ5 (red) for DNA on indicated fungal strains. Af293 and the Δsph3 images as seen in Fig. 3A have been reproduced here for ease of comparison. C, hyphal morphology shown using S.E. images of indicated fungal strains. For comparison, the S.E. images of Af293 and Δsph3 from Fig. 3B have been included. D, absorbance readings of an indirect EIA using the anti-GAG antibody indicate cell filtrate GAG levels. Bars represent means, and error bars are one S.D.
Discussion
Sph3 is predicted to be a membrane-anchored extracellular protein, and structural determination of the soluble domain revealed a (β/α)8 barrel (Fig. 4A). The extracellular domain shares structural similarity to members of glycoside hydrolase families 18, 27, and 84, but low primary sequence identity precludes classification of Sph3 as a member of any of these GH families (55, 56). The active members of GH families 18, 27, and 84 all use acidic residues to coordinate the substrate and catalyze the hydrolysis of the glycosidic bond (57–60). Structural alignment of representative members from these GH families identified three acidic amino acids in Sph3Ac, Asp-166, Glu-167, and Glu-222, that overlapped with active site residues from the various GH active sites suggesting that these may be involved in catalysis (Fig. 4E). These three acidic residues in Sph3Ac line the active site pocket and are conserved across Sph3 orthologues from Aspergillus species and other fungi (Fig. 2B). The co-crystal structure of Sph3 with GalNAc revealed the monosaccharide bound to this active site pocket in the same location as an ethylene glycol molecule in the apo-structure. Ethylene glycol has been found in previous studies to mimic sugar-binding sites (61, 62), an observation that is further supported by the GalNAc structure. The electron density for the N-acetyl group was better defined than the density for the galactopyranose ring (Fig. 4D). This observation suggests a potentially higher mobility for the galactose pyranose ring than for the N-acetyl group and/or some heterogeneity in its mode of binding. The heterogeneity observed in the galactose pyranose ring is not expected to be present when the GAG polymer binds to the protein. Binding of GAG would result in better defined sugar placement due protein-carbohydrate interactions with adjacent sugar moieties. A similar result has been observed for the C-terminal domain of PgaB, a putative poly-β-N-acetylglucosamine (PNAG) glycoside hydrolase, where GlcNAc and glucosamine monomers were bound in the same location but in slightly different conformations than the GlcNAc moieties in a PNAG hexamer (22). The density observed for the N-acetyl group supports the location of the active site where it is coordinated by Glu-222, which in turn hydrogen bonds Asn-202 (Fig. 4F). These data suggest that the spherulin 4 NPG motif may be important for activating the acidic residues in the catalytic mechanism.
Given the structural similarities of Sph3 with other glycoside hydrolases, we investigated whether it displayed enzymatic activity against GAG. Recombinant Sph3 exhibited hydrolase activity against both purified and cell-associated forms of GAG (Fig. 5, A and B). The activity against purified GAG was low in vitro, although this observation is likely a consequence of the inaccessibility of the substrate once purified. GAG becomes insoluble upon isolation from A. fumigatus culture supernatants, and this might preclude Sph3 binding. Despite suboptimal reaction conditions, Sph3 activity was specific to GAG as the protein did not hydrolyze the partially deacetylated GlcNAc-based polysaccharide, chitosan (Fig. 5B). The degradation of cell wall-associated GAG was more efficient because GAG could not be detected after treatment of hyphae with Sph3 for 3 h (Fig. 5C), suggesting that the substrate either contained more potential cleavage sites or was more accessible. The difference between cell wall-associated GAG and secreted GAG has not been analyzed, and the composition could be different as the characterization of GAG in the literature supports very heterogeneous length and monomer ratios between isolates (5). A similar substrate preference is even more pronounced in the exopolysaccharide system from Listeria monocytogenes (63), where the glycoside hydrolase, PssZ, is able to degrade cell wall-associated exopolysaccharide but displays no detectable activity against the ethanol-precipitated polymer (63). This phenomenon has also been seen for the cellulose glycoside hydrolase BcsZ, which only showed hydrolytic activity against carboxymethylcellulose when the polymer was embedded in agar plates. No activity could be detected when the polymer was in solution (64).
Mutagenesis of the conserved acidic residues identified only one residue, Asp-166 (Asp-167Af), that was necessary for enzymatic activity (Fig. 5, A and B). Although substitution of each of the glutamic acids (Glu-167 and Glu-222) with alanine caused reduced activity, replacement of Glu-222 with glutamine led to little change to in vitro activity levels. The E167A variant had some activity on cell wall-associated GAG thus suggesting neither Glu-222 nor Glu-167 are essential in the mechanism and likely play a role in substrate binding or positioning of the other residues (Fig. 5A). Coordination of the C1 hydroxyl of GalNAc in the co-crystal structure further supports the importance of Asp-166 in catalysis (Fig. 4F). The catalytic mechanism of both GH18 and GH84 enzymes requires only one catalytic acid and employs a substrate-assisted mechanism, but in these cases the sugar linkages are in the β configuration allowing for the attack of the glycosidic bond by the N-acetyl group of the central glycoside moiety (57–59). It is unlikely that Sph3 uses the same reaction mechanism due to the steric hindrance of the sugar linkages. More detailed assays to determine the catalytic mechanism and substrate specificity will require the availability of homogeneous and soluble substrates. The positioning of the active site in a long shallow conserved cleft (Fig. 4B) is suggestive of an endo-acting enzyme (65–67). The inability of Sph3 to hydrolyze p-nitrophenyl glycosides is consistent with other endo-acting enzymes, such as BcsZ (64), which similarly lack activity for these pseudo-substrates. Sph3 may thus bind and cleave the nascent polymer during secretion in vivo.
Complementation studies suggested that the enzymatic activity of Sph3 is required for the synthesis of functional GAG. Although this requirement of Sph3 enzymatic activity for functional GAG synthesis seems to represent a conundrum, similar observations have been reported in bacteria. The presence of a putative glycoside hydrolase or lyase has been found in the biosynthetic operons of many bacterial biofilm exopolysaccharides, including, but not limited to cellulose, alginate, Pel, Psl, PNAG, and L. monocytogenes systems (13, 23, 26, 27, 68). The alginate lyase, AlgL, from Pseudomonas fluorescens, has a role in clearing the periplasmic space of residual alginate, and it has also been shown to affect polymer length and modulate other proteins in the alginate system (29). Perhaps more similar to GAG is the exopolysaccharide system from L. monocytogenes because PssZ is also predicted to be membrane-bound and have a glycoside hydrolase domain on the extracellular surface (63). L. monocytogenes, like A. fumigatus, has only one cell membrane. Deletion of pssZ in L. monocytogenes led to reduced exopolysaccharide levels suggesting that PssZ is required for optimal polymer biosynthesis (63). A similar role for glycoside hydrolases in cellulose systems has also been suggested (5, 69). In Acetobacter xylinum and Agrobacterium tumefaciens, the cellulose endoglucanases enhance cellulose synthesis in vivo (69, 70). In Gluconacetobacter xylinus, the carboxylmethylcellulase, CMCax, is also extracellular and is thought to be essential for cellulose assembly (21). Nakai et al. (21) proposed that G. xylinus produces tangled cellulose in the absence of CMCax, which reduced the rate of synthesis and export. CMCax may clear this amorphous polymer from the synthase complex allowing for increased production (21). The in vivo functions of PssZ and the cellulose endoglucanase have not been determined, but it is clear that glycoside hydrolase function enhances exopolysaccharide production in multiple bacterial systems. In A. fumigatus, we have demonstrated that glycoside hydrolase activity of Sph3 is essential for GAG biosynthesis. It is possible that Gtb3, the proposed synthase-porin complex, easily stalls and that Sph3 cleaves the polymer allowing for synthesis to continue. If the glycan synthase complex is not abundant on the cell membrane, then stalled synthesis could lead to production of GAG levels too low to measure. Further experimentation is required to determine the function of Sph3 in the GAG system in vivo.
Interestingly, this study suggests that there are two glycoside hydrolases in the GAG system, Sph3 and Ega3. The loss of GAG production in the Δsph3 strain suggests that they do not have redundant roles. The activity of Ega3 has not been explored, and it may be inactive or not required for GAG biosynthesis. GAG is a heterogeneous polymer, which allows for hydrolases of different specificities to act on the same chain. Ega3 and Sph3 could preferentially cleave different substrates such as Gal-rich versus GalNAc/GalN-rich regions. The cleavage specificity of Sph3 could not be determined in this study due to lack of synthetic substrates and the insolubility of GAG. However, in vivo, deacetylation of GAG by Agd3 occurs in the extracellular matrix after secretion of the polymer (8). Sph3 is located at the cell surface, and it is therefore likely that it cleaves the fully acetylated polymer. The co-crystal structure supports Sph3 specificity for a GalNAc-containing substrate. Further study of Ega3 and Sph3 is required to determine the role of each protein in the system.
The data presented herein allow for an updated model of the GAG biosynthetic pathway in which the glycoside hydrolase Sph3 is required for exopolysaccharide biosynthesis. Sph3 is likely associated with or in proximity to Gtb3 to have access to the nascent polysaccharide. Classification of Sph3 as a glycoside hydrolase containing conserved catalytic motifs (Fig. 2B) led to the creation of a new glycoside hydrolase family GH135. These results suggest that the spherulin 4 family, which until now had no known function, may embody this novel GH family.
Author Contributions
N. C. B., B. D. S., P. L. H., and D. C. S. designed the study and wrote the paper. N. C. B., B. D. S., F. N. G., D. J. L., M. J. L., J. C. C., A. M. G., S. D. B., C. A. Z., and H. R. performed the experiments. N. C. B., B. D. S., F. N. G., D. J. L., M. J. L., C. A. Z., P. B., P. L. H., and D. C. S. analyzed the results. All authors approved the final version of the manuscript.
Acknowledgments
We thank P. Magee (University of Minnesota, St. Paul) for the Af293 strain, J. P. Latge (Institut Pasteur, Paris, France) for the anti-GAG antibody, and P. Yip (The Hospital for Sick Children, Toronto, Canada) for technical assistance. Beamline X29 at the National Synchrotron Light Source is supported by the United States Department of Energy Office and National Institutes of Health Grant P41RR012408 from NCRR and Grant P41GM103473 from NIGMS. Beamline 08B1-1 at the Canadian Light Source is supported by Natural Sciences and Engineering Research Council of Canada, Canadian Institutes of Health Research, the National Research Council Canada, the Province of Saskatchewan, Western Economic Diversification Canada, and the University of Saskatchewan.
This work was supported in part by Canadian Institutes of Health Research Operating Grants 81361 (to P. L. H. and D. C. S.) and 123306 (to D. C. S.) and by the Cystic Fibrosis Canada (to D. C. S. and P. L. H.). The authors declare that they have no conflicts of interest with the contents of this article.
This article was selected as a Paper of the Week.
The atomic coordinates and structure factors (codes 5C5G and 5D6T) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- GAG
- galactosaminogalactan
- Bistris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane
- TCEP
- tris(2-carboxyethyl)phosphine
- PDB
- Protein Data Bank
- r.m.s.d.
- root mean square deviation
- GH
- glycoside hydrolase
- SeMet
- selenomethionine
- Fwd
- forward
- Rev
- reverse
- CHES
- N-cyclohexyl-2-aminoethanesulfonic acid
- pNP
- para-nitrophenyl
- SBA
- soybean agglutinin lectin
- EIA
- enzyme immunoassay
- PNAG
- poly-β-N-acetylglucosamine.
References
- 1.Kaur S., and Singh S. (2014) Biofilm formation by Aspergillus fumigatus. Med. Mycol. 52, 2–9 [DOI] [PubMed] [Google Scholar]
- 2.Brown G. D., Denning D. W., Gow N. A., Levitz S. M., Netea M. G., and White T. C. (2012) Hidden killers: human fungal infections. Sci. Transl. Med. 4, 165rv13. [DOI] [PubMed] [Google Scholar]
- 3.Lewis R. E., Cahyame-Zuniga L., Leventakos K., Chamilos G., Ben-Ami R., Tamboli P., Tarrand J., Bodey G. P., Luna M., and Kontoyiannis D. P. (2013) Epidemiology and sites of involvement of invasive fungal infections in patients with haematological malignancies: a 20-year autopsy study. Mycoses 56, 638–645 [DOI] [PubMed] [Google Scholar]
- 4.Geiser D. M., Klich M. A., Frisvad J. C., Peterson S. W., Varga J., and Samson R. A. (2007) The current status of species recognition and identification in Aspergillus. Stud. Mycol. 59, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fontaine T., Delangle A., Simenel C., Coddeville B., van Vliet S. J., van Kooyk Y., Bozza S., Moretti S., Schwarz F., Trichot C., Aebi M., Delepierre M., Elbim C., Romani L., and Latgé J. P. (2011) Galactosaminogalactan, a new immunosuppressive polysaccharide of Aspergillus fumigatus. PLoS Pathog. 7, e1002372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gravelat F. N., Beauvais A., Liu H., Lee M. J., Snarr B. D., Chen D., Xu W., Kravtsov I., Hoareau C. M., Vanier G., Urb M., Campoli P., Al Abdallah Q., Lehoux M., Chabot J. C., et al. (2013) Aspergillus galactosaminogalactan mediates adherence to host constituents and conceals hyphal β-glucan from the immune system. PLoS Pathog. 9, e1003575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gresnigt M. S., Bozza S., Becker K. L., Joosten L. A., Abdollahi-Roodsaz S., van der Berg W. B., Dinarello C. A., Netea M. G., Fontaine T., De Luca A., Moretti S., Romani L., Latge J. P., and van de Veerdonk F. L. (2014) A polysaccharide virulence factor from Aspergillus fumigatus elicits anti-inflammatory effects through induction of interleukin-1 receptor antagonist. PLoS Pathog. 10, e1003936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee M. J., Geller A. M., Liu H., Gravelat F. N., Snarr B. D., Cerone R. P., Baptista S. D., Bamford N. C., Binogradov E., Fontaine T., Latge J., Stajich J., Howell P. L., Filler S. G., and Sheppard D. C. (2015) Deacetylation of galactosaminogalactan in Aspergillus fumigatus is a required post-synthesis modification for adherence and virulence. mBio [Google Scholar]
- 9.Loussert C., Schmitt C., Prevost M. C., Balloy V., Fadel E., Philippe B., Kauffmann-Lacroix C., Latgé J. P., and Beauvais A. (2010) In vivo biofilm composition of Aspergillus fumigatus. Cell. Microbiol. 12, 405–410 [DOI] [PubMed] [Google Scholar]
- 10.Lee M. J., Liu H., Barker B. M., Snarr B. D., Gravelat F. N., Al Abdallah Q., Gavino C., Xiao T., Ralph B., Solis N. V., Lehoux M., Baptista S. D., Thammahong A., Cerone R. P., Baistrocchi S. R., et al. (2015) Fungal exopolysaccharide galactosaminogalactan mediates virulence by enhancing resistance to neutrophil extracellular traps. PLoS Pathog. 10.1371/journal.ppat.1005187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robinet P., Baychelier F., Fontaine T., Picard C., Debré P., Vieillard V., Latgé J. P., and Elbim C. (2014) A polysaccharide virulence factor of a human fungal pathogen induces neutrophil apoptosis via NK cells. J. Immunol. 192, 5332–5342 [DOI] [PubMed] [Google Scholar]
- 12.Gravelat F. N., Doedt T., Chiang L. Y., Liu H., Filler S. G., Patterson T. F., and Sheppard D. C. (2008) In vivo analysis of Aspergillus fumigatus developmental gene expression determined by real time reverse transcription-PCR. Infect. Immun. 76, 3632–3639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whitney J. C., and Howell P. L. (2013) Synthase-dependent exopolysaccharide secretion in Gram-negative bacteria. Trends Microbiol. 21, 63–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vuong C., Kocianova S., Voyich J. M., Yao Y., Fischer E. R., DeLeo F. R., and Otto M. (2004) A crucial role for exopolysaccharide modification in bacterial biofilm formation, immune evasion, and virulence. J. Biol. Chem. 279, 54881–54886 [DOI] [PubMed] [Google Scholar]
- 15.Heilmann C., Schweitzer O., Gerke C., Vanittanakom N., Mack D., and Götz F. (1996) Molecular basis of intercellular adhesion in the biofilm-forming Staphylococcus epidermidis. Mol. Microbiol. 20, 1083–1091 [DOI] [PubMed] [Google Scholar]
- 16.Rohde H., Frankenberger S., Zähringer U., and Mack D. (2010) Structure, function and contribution of polysaccharide intercellular adhesin (PIA) to Staphylococcus epidermidis biofilm formation and pathogenesis of biomaterial-associated infections. Eur. J. Cell Biol. 89, 103–111 [DOI] [PubMed] [Google Scholar]
- 17.Parise G., Mishra M., Itoh Y., Romeo T., and Deora R. (2007) Role of a putative polysaccharide locus in Bordetella biofilm development. J. Bacteriol. 189, 750–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sloan G. P., Love C. F., Sukumar N., Mishra M., and Deora R. (2007) The Bordetella Bps polysaccharide is critical for biofilm development in the mouse respiratory tract. J. Bacteriol. 189, 8270–8276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sutherland I. (2001) Biofilm exopolysaccharides: a strong and sticky framework. Microbiology 147, 3–9 [DOI] [PubMed] [Google Scholar]
- 20.Lee M. J., Gravelat F. N., Cerone R. P., Baptista S. D., Campoli P. V., Choe S. I., Kravtsov I., Vinogradov E., Creuzenet C., Liu H., Berghuis A. M., Latgé J. P., Filler S. G., Fontaine T., and Sheppard D. C. (2014) Overlapping and distinct roles of Aspergillus fumigatus UDP-glucose 4-epimerases in galactose metabolism and the synthesis of galactose-containing cell wall polysaccharides. J. Biol. Chem. 289, 1243–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakai T., Sugano Y., Shoda M., Sakakibara H., Oiwa K., Tuzi S., Imai T., Sugiyama J., Takeuchi M., Yamauchi D., and Mineyuki Y. (2013) Formation of highly twisted ribbons in a carboxymethylcellulase gene-disrupted strain of a cellulose-producing bacterium. J. Bacteriol. 195, 958–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Little D. J., Li G., Ing C., DiFrancesco B. R., Bamford N. C., Robinson H., Nitz M., Pomès R., and Howell P. L. (2014) Modification and periplasmic translocation of the biofilm exopolysaccharide poly-β-1,6-N-acetyl-d-glucosamine. Proc. Natl. Acad. Sci. U.S.A. 111, 11013–11018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Little D. J., Milek S., Bamford N. C., Ganguly T., DiFrancesco B. R., Nitz M., Deora R., and Howell P. L. (2015) BpsB is a poly-β-1,6-N-acetyl-d-glucosamine deacetylase required for biofilm formation in Bordetella bronchiseptica. J. Biol. Chem. 290, 22827–22840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Itoh Y., Rice J. D., Goller C., Pannuri A., Taylor J., Meisner J., Beveridge T. J., Preston J. F. 3rd, and Romeo T. (2008) Roles of pgaABCD genes in synthesis, modification, and export of the Escherichia coli biofilm adhesin poly-β-1,6-N-acetyl-d-glucosamine. J. Bacteriol. 190, 3670–3680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Little D. J., Poloczek J., Whitney J. C., Robinson H., Nitz M., and Howell P. L. (2012) The structure- and metal-dependent activity of Escherichia coli PgaB provides insight into the partial de-N-acetylation of poly-β-1,6-N-acetyl-d-glucosamine. J. Biol. Chem. 287, 31126–31137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Colvin K. M., Alnabelseya N., Baker P., Whitney J. C., Howell P. L., and Parsek M. R. (2013) PelA deacetylase activity is required for Pel polysaccharide synthesis in Pseudomonas aeruginosa. J. Bacteriol. 195, 2329–2339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., and Terwilliger T. C. (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 29.Bakkevig K., Sletta H., Gimmestad M., Aune R., Ertesvåg H., Degnes K., Christensen B. E., Ellingsen T. E., and Valla S. (2005) Role of the Pseudomonas fluorescens alginate lyase (AlgL) in clearing the periplasm of alginates not exported to the extracellular environment. J. Bacteriol. 187, 8375–8384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jain S., and Ohman D. E. (2005) Role of an alginate lyase for alginate transport in mucoid Pseudomonas aeruginosa. Infection and immunity 73, 6429–6436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steiner S., Lori C., Boehm A., and Jenal U. (2013) Allosteric activation of exopolysaccharide synthesis through cyclic di-GMP-stimulated protein-protein interaction. EMBO J. 32, 354–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gerke C., Kraft A., Süssmuth R., Schweitzer O., and Götz F. (1998) Characterization of the N-acetylglucosaminyltransferase activity involved in the biosynthesis of the Staphylococcus epidermidis polysaccharide intercellular adhesin. J. Biol. Chem. 273, 18586–18593 [DOI] [PubMed] [Google Scholar]
- 33.Savard L., Laroche A., Lemieux G., and Pallotta D. (1989) Developmentally regulated late mRNAs in the encystment of Physarum polycephalum plasmodia. Biochim. Biophys. Acta 1007, 264–269 [DOI] [PubMed] [Google Scholar]
- 34.Petersen T. N., Brunak S., von Heijne G., and Nielsen H. (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786 [DOI] [PubMed] [Google Scholar]
- 35.Altschul S. F., Gish W., Miller W., Myers E. W., and Lipman D. J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410 [DOI] [PubMed] [Google Scholar]
- 36.Kelley L. A., and Sternberg M. J. (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4, 363–371 [DOI] [PubMed] [Google Scholar]
- 37.Krogh A., Larsson B., von Heijne G., and Sonnhammer E. L. (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580 [DOI] [PubMed] [Google Scholar]
- 38.Käll L., Krogh A., and Sonnhammer E. L. (2004) A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 338, 1027–1036 [DOI] [PubMed] [Google Scholar]
- 39.Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., and Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 40.Gravelat F. N., Ejzykowicz D. E., Chiang L. Y., Chabot J. C., Urb M., Macdonald K. D., al-Bader N., Filler S. G., and Sheppard D. C. (2010) Aspergillus fumigatus MedA governs adherence, host cell interactions and virulence. Cell. Microbiol. 12, 473–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gravelat F. N., Askew D. S., and Sheppard D. C. (2012) Targeted gene deletion in Aspergillus fumigatus using the hygromycin-resistance split-marker approach. Methods Mol. Biol. 845, 119–130 [DOI] [PubMed] [Google Scholar]
- 42.Lee J. E., Cornell K. A., Riscoe M. K., and Howell P. L. (2001) Structure of E. coli 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase reveals similarity to the purine nucleoside phosphorylases. Structure 9, 941–953 [DOI] [PubMed] [Google Scholar]
- 43.Otwinowski Z., and Minor W. (1997) Processing of x-ray diffraction data collection in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 44.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morin A., Eisenbraun B., Key J., Sanschagrin P. C., Timony M. A., Ottaviano M., and Sliz P. (2013) Collaboration gets the most out of software. eLife 2, e01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Terwilliger T. C. (2000) Maximum-likelihood density modification. Acta Crystallogr. D Biol. Crystallogr. 56, 965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 48.Holm L., and Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ashkenazy H., Erez E., Martz E., Pupko T., and Ben-Tal N. (2010) ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 38, W529–W533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Glaser F., Pupko T., Paz I., Bell R. E., Bechor-Shental D., Martz E., and Ben-Tal N. (2003) ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 19, 163–164 [DOI] [PubMed] [Google Scholar]
- 51.Landau M., Mayrose I., Rosenberg Y., Glaser F., Martz E., Pupko T., and Ben-Tal N. (2005) ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 33, W299–W302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baker N. A., Sept D., Joseph S., Holst M. J., and McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anthon G. E., and Barrett D. M. (2001) Colorimetric method for the determination of lipoxygenase activity. J. Agric. Food Chem. 49, 32–37 [DOI] [PubMed] [Google Scholar]
- 54.Park B. H., Karpinets T. V., Syed M. H., Leuze M. R., and Uberbacher E. C. (2010) CAZymes Analysis Toolkit (CAT): web service for searching and analyzing carbohydrate-active enzymes in a newly sequenced organism using CAZy database. Glycobiology 20, 1574–1584 [DOI] [PubMed] [Google Scholar]
- 55.Henrissat B. (1991) A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem. J. 280, 309–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Henrissat B., and Davies G. (1997) Structural and sequence-based classification of glycoside hydrolases. Curr. Opin. Struct. Biol. 7, 637–644 [DOI] [PubMed] [Google Scholar]
- 57.Papanikolau Y., Prag G., Tavlas G., Vorgias C. E., Oppenheim A. B., and Petratos K. (2001) High resolution structural analyses of mutant chitinase A complexes with substrates provide new insight into the mechanism of catalysis. Biochemistry 40, 11338–11343 [DOI] [PubMed] [Google Scholar]
- 58.van Aalten D. M., Komander D., Synstad B., Gåseidnes S., Peter M. G., and Eijsink V. G. (2001) Structural insights into the catalytic mechanism of a family 18 exo-chitinase. Proc. Natl. Acad. Sci. U.S.A. 98, 8979–8984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rao F. V., Dorfmueller H. C., Villa F., Allwood M., Eggleston I. M., and van Aalten D. M. (2006) Structural insights into the mechanism and inhibition of eukaryotic O-GlcNAc hydrolysis. EMBO J. 25, 1569–1578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guce A. I., Clark N. E., Salgado E. N., Ivanen D. R., Kulminskaya A. A., Brumer H. 3rd, and Garman S. C. (2010) Catalytic mechanism of human α-galactosidase. J. Biol. Chem. 285, 3625–3632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bourne Y., Roig-Zamboni V., Barre A., Peumans W. J., Astoul C. H., Van Damme E. J., and Rougé P. (2004) The crystal structure of the Calystegia sepium agglutinin reveals a novel quaternary arrangement of lectin subunits with a β-prism fold. J. Biol. Chem. 279, 527–533 [DOI] [PubMed] [Google Scholar]
- 62.Roberts S. M., and Davies G. J. (2012) Chapter 8: The crystallization and structural analysis of cellulases (and other glycoside hydrolases): strategies and tactics. Methods Enzymol. 510, 141–168 [DOI] [PubMed] [Google Scholar]
- 63.Köseoǧlu V. K., Heiss C., Azadi P., Topchiy E., Güvener Z. T., Lehmann T. E., Miller K. W., and Gomelsky M. (2015) Listeria monocytogenes exopolysaccharide: origin, structure, biosynthetic machinery and c-di-GMP-dependent regulation. Mol. Microbiol. 96, 728–743 [DOI] [PubMed] [Google Scholar]
- 64.Mazur O., and Zimmer J. (2011) Apo- and cellopentaose-bound structures of the bacterial cellulose synthase subunit BcsZ. J. Biol. Chem. 286, 17601–17606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kleywegt G. J., Zou J. Y., Divne C., Davies G. J., Sinning I., Stâhlberg J., Reinikainen T., Srisodsuk M., Teeri T. T., and Jones T. A. (1997) The crystal structure of the catalytic core domain of endoglucanase I from Trichoderma reesei at 3.6 A resolution, and a comparison with related enzymes. J. Mol. Biol. 272, 383–397 [DOI] [PubMed] [Google Scholar]
- 66.Payne C. M., Baban J., Horn S. J., Backe P. H., Arvai A. S., Dalhus B., Bjørås M., Eijsink V. G., Sørlie M., Beckham G. T., and Vaaje-Kolstad G. (2012) Hallmarks of processivity in glycoside hydrolases from crystallographic and computational studies of the Serratia marcescens chitinases. J. Biol. Chem. 287, 36322–36330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Davies G., and Henrissat B. (1995) Structures and mechanisms of glycosyl hydrolases. Structure 3, 853–859 [DOI] [PubMed] [Google Scholar]
- 68.Franklin M. J., Nivens D. E., Weadge J. T., and Howell P. L. (2011) Biosynthesis of the Pseudomonas aeruginosa extracellular polysaccharides, Alginate, Pel, and Psl. Front. Microbiol. 2, 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kawano S., Tajima K., Kono H., Erata T., Munekata M., and Takai M. (2002) Effects of endogenous endo-β-1,4-glucanase on cellulose biosynthesis in Acetobacter xylinum ATCC23769. J. Biosci. Bioeng. 94, 275–281 [DOI] [PubMed] [Google Scholar]
- 70.Koo H. M., Song S. H., Pyun Y. R., and Kim Y. S. (1998) Evidence that a β-1,4-endoglucanase secreted by Acetobacter xylinum plays an essential role for the formation of cellulose fiber. Biosci. Biotechnol. Biochem. 62, 2257–2259 [DOI] [PubMed] [Google Scholar]