

Background: Compounds that inhibit spliceosomes are of very limited availability.
Results: A high-throughput screen of a marine bacteria natural products library identified a promising lead compound that inhibits pre-mRNA splicing in vitro.
Conclusion: N-palmitoyl-l-leucine is a new splicing inhibitor that affects a late stage of spliceosome assembly.
Significance: Natural products have great potential as small molecule tools for studying spliceosome function.
Keywords: drug screening, inhibitor, natural product, RNA splicing, spliceosome
Abstract
The spliceosome is a dynamic complex of five structural RNAs and dozens of proteins, which assemble together to remove introns from nascent eukaryotic gene transcripts in a process called splicing. Small molecules that target different components of the spliceosome represent valuable research tools to investigate this complicated macromolecular machine. However, the current collection of spliceosome inhibitors is very limited. To expand the toolkit we used a high-throughput in vitro splicing assay to screen a collection of pre-fractions of natural compounds derived from marine bacteria for splicing inhibition. Further fractionation of initial hits generated individual peaks of splicing inhibitors that interfere with different stages of spliceosome assembly. With additional characterization of individual peaks, we identified N-palmitoyl-l-leucine as a new splicing inhibitor that blocks a late stage of spliceosome assembly. Structure-activity relationship analysis of the compound revealed that length of carbon chain is important for activity in splicing, as well as for effects on the cytological profile of cells in culture. Together these results demonstrate that our combination of in vitro splicing analysis with complex natural product libraries is a powerful strategy for identifying new small molecule tools with which to probe different aspects of spliceosome assembly and function.
Introduction
Natural products are a rich source for bioactive compounds, many of which have been important tools in research. For example, a diverse collection of antibiotics was critical to dissecting the structure and function of the ribosome, the complex macromolecular machine responsible for protein translation (1). The same type of small molecule tools is needed to understand another complicated cellular ribonucleoprotein assembly called the spliceosome. This dynamic macromolecular complex catalyzes pre-mRNA splicing, an essential step in eukaryotic gene expression. During splicing, intervening intron sequences are removed from nascent gene transcripts while exon sequences are joined to generate messenger RNA (mRNA) for protein translation. For each splicing event, spliceosomes assemble from five U-rich small ribonucleoproteins (snRNPs)5 and dozens of additional proteins in a dynamic progression of intermediate splicing complexes. Small molecules that inhibit different spliceosome components could be powerful implements to arrest the complex at different assembly stages for structural and biochemical studies (2). Those same inhibitors would also aid in assigning specific functions to spliceosome proteins, which is currently lacking for the majority of them.
Only a handful of spliceosome inhibitors are currently available, and the most potent of these are natural products. The bacterially derived compounds spliceostatin A (SSA) and pladienolide B have been used to study early spliceosome assembly, as well as to modulate splicing in cells (3–5). However, there are several stages of the spliceosome that we cannot arrest, and likely many more intermediate states that are currently unknown and cannot yet be captured or analyzed. Therefore, there is a great need to identify additional chemical tools that interfere with splicing.
Until now, screening efforts to identify new splicing inhibitors have mainly focused on “simple” small molecule libraries, despite the fact that the most potent known splicing inhibitors are natural products (6–10). Here, we report a screen of a natural product library containing a great diversity of chemical scaffolds using a high-throughput splicing assay. The library is part of a discovery pipeline engineered to facilitate both structure determination and production of specific natural product inhibitors (11). The pipeline allowed us to identify N-palmitoyl-l-leucine as a new splicing inhibitor that stalls spliceosomes at a late assembly stage. Structure-activity relationship analysis of the compound revealed that each part of the relatively simple structure is important for splicing inhibition, which parallels its bioactivity in cells.
Experimental Procedures
In Vitro Splicing Reactions
[32P]UTP body-labeled G(5′)ppp(5′)G-capped pre-mRNA substrate consisting of a single intron flanked by two exons was generated from an optimized adenovirus major late template (12) by standard T7 run-off transcription followed by gel purification. Nuclear extract was prepared from HeLa cells grown in MEM/F12 1:1 and 5% (v/v) newborn calf serum and dialyzed into 20 mm Tris, pH 7.9, 0.1 m KCl, 0.2 mm EDTA, 20% (v/v) glycerol, 0.5 mm DTT (13). For splicing reactions (10 μl), substrate pre-mRNA was incubated at 5 nm concentration in 60 mm potassium glutamate, 2 mm magnesium acetate, 2 mm ATP, 5 mm creatine phosphate, 0.05 mg ml−1 tRNA, and 50% (v/v) HeLa nuclear extract at 30 °C for 0–30 min. When testing compounds, which are dissolved in DMSO, compound volume is calculated to ensure the final DMSO concentration in the reaction is less than 2% (v/v) because DMSO is inhibitory at higher concentrations. For DTT rescue experiments, a final concentration of 20 mm DTT was included at the beginning of the reaction.
High-Throughput Screening
Screening was carried out at the UC Santa Cruz Chemical Screening Center as described in Ref. 7. Briefly, liquid handling robots were used to set up 10-μl in vitro splicing reactions in 384-well plates. Reaction conditions were as described above with the exceptions that 10 nm pre-mRNA substrate was unlabeled and 0.2 μl of library pre-fractions were also included. Following incubation, reactions were diluted 1:2 with water and 15 nl were transferred as template to 5 μl of TaqMan® One-Step RT-PCR (Applied Biosystems) reactions. Reverse and forward RT-qPCR primers were directed to the exons of the pre-mRNA substrate and the TaqMan® probe to the exon junction sequence produced by splicing.
Denaturing Gel Analysis
RNA was isolated from in vitro splicing reactions by phenol:chloroform extraction and ethanol precipitation. Pellets were resuspended in 6 μl of loading buffer (95% (v/v) formamide, 20 mm EDTA, 0.01% (w/v) bromphenol blue, 0.01% (w/v) cyan blue), from which 2 μl were loaded on a denaturing 7 m urea, 15% (v/v) polyacrylamide gel in 1× TBE (45 mm Tris borate/1 mm EDTA) running buffer. Gels were run at 30 watts for 2 h and then and exposed to phosphorimaging screens, which were digitized with a Typhoon Scanner (Molecular Dynamics). 32P-labeled RNA species were quantified with ImageQuant software (Molecular Dynamics). Splicing efficiency was calculated as the amount of spliced mRNA relative to total RNA and normalized to a DMSO control reaction. IC50 values represent the concentration of inhibitor that decrease splicing efficiency by 50% and were estimated from plots of splicing efficiency versus compound concentration for triplicate reactions.
Native Gel Analysis
Splicing reactions were set up at 10 μl as described above. Following incubation, one volume of native loading buffer (20 mm Trizma base, 20 mm glycine, 25% (v/v) glycerol, 0.05% (w/v) cyan blue, 0.05% (w/v) bromphenol blue, 1 mg ml−1 heparin sulfate) was added to the reactions. After incubation for 5 min at room temperature, 2 μl of the samples were loaded on a horizontal 2.1% (w/v) low-melt agarose gel in 20 mm Tris, 20 mm glycine running buffer. Gels were run at 72 V for 3 h 50 min, vacuum-dried onto Whatman paper for 45 min at 65 °C, and exposed to phosphorimaging screens, which were digitized with a Typhoon Scanner (Molecular Dynamics). For native complexes that were challenged with heparin, the 2× native gel loading buffer contained between 1 and 10 mg ml−1 heparin sulfate.
Cytological Profiling Assay
Cytological profiling was performed at the UC Santa Cruz Chemical Screening Center. For the analysis data, HeLa cells were treated with 33.3–133 μm of N-palmitoyl-l-leucine or analogs for 20 h, followed by staining, automated imaging, and image processing as described in Ref. 14.
Isolation, Cultivation, and Extraction of Streptomyces sp
RL10–300-HVF-B— Strain RL10–300-HVF-B was isolated on HVF solid agar medium from a marine sediment sample collected in Dolan Canyon, CA, at a depth of 15 m using SCUBA. The pure culture (Streptomyces sp. RL10–300-HVF-B) was cultivated in four 2.8 liters Fernbach flasks, each containing 1 liter of modified SYP fermentation medium (31.2 g Instant Ocean, 10.0 g soluble starch, 4.0 g peptone, 2.0 g yeast extract, 1.0 liters Milli-Q water, pH 7.3), a stainless steel spring, and 20.0 g of Amberlite XAD-16 adsorbent resin. The culture was shaken at 200 rpm for 7 days. At the end of the fermentation period, the cells and resin were removed by vacuum filtration using Whatman glass microfiber filters and washed with deionized H2O. The resin and cells from each culture flask were extracted with 300 ml of 1:1 MeOH/CH2Cl2. The organic extract was removed from the cells and resin by vacuum filtration, concentrated in vacuo, and coded as RLUS1543.
Preparation of Pre-fraction RLUS1543E
The organic extract RLUS1543 was subjected to solid-phase extraction using a Supelco-Discovery C18 cartridge (4 × 5 g) and eluted using a MeOH/H2O step gradient (100 ml; 10% (v/v) MeOH, 20% (v/v) MeOH, 40% (v/v) MeOH, 60% (v/v) MeOH, 80% (v/v) MeOH, 100% MeOH, 100% EtOAc) to afford seven fractions. The 10% (v/v) MeOH fraction was discarded, and the remaining six (fractions A-F) were concentrated to dryness in vacuo. The fraction RLUS1543E (100% MeOH) was subjected to further bioassay-guided fractionation using RP-HPLC.
Peak-Library Preparation of RLUS1543E
RLUS1543E was subjected to reversed-phase (RP)-LCMS (Phenomenex Synergi Fusion-RP 10 μm, 250 × 4.6 mm, 30% (v/v) MeOH/H2O + 0.02% (v/v) formic acid isocratic for 4 min, 30–100% (v/v) for 80 min, 2 ml min−1 flow rate) with automated collection of the eluent in 1 min intervals directly into a 96-well plate to create the peak-library. The plate was dried in vacuo and 20 μl of DMSO was added to each well for secondary screening to identify the active constituent.
Bioassay-guided Fractionation of RLUS1543E
The pre-fraction RLUS1543E was fractionated using RP-HPLC (Phenomenex Synergi Fusion-RP, 10 μm, 250 × 4.6 mm, 90–100% MeOH/H2O + 0.02% (v/v) formic acid over 10 min, 2 ml min−1 flow rate), and 1 min intervals of the eluent were collected (from tR = 6.00 min to 14.00 min) and concentrated, and their in vitro splicing activity was assayed as described above, identifying the fraction RLUS1543E2 (tR = 7.00 min to 8.00 min) as bioactive.
Determination of Absolute Configuration
To a solution of RLUS1543E2 (100 μg) in DMF (100 μl) at 25 °C was added 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, followed by 1-hydroxy-7-azabenzotriazole. The reaction mixture was then treated with l-alanine tert-butyl ester hydrochloride, and stirred for 18 h. The reaction mixture was partitioned between ethyl acetate and H2O, and the organic extract was concentrated in vacuo. Analysis by UPLC-MS and comparison to authentic standards of N-palmitoyl-l-leucyl-l-alanine tert-butyl ester and N-palmitoyl-d-leucyl-l-alanine tert-butyl ester confirmed the identity and absolute configuration of the natural product as N-palmitoyl-l-leucine.
Preparation of N-acyl Amino Acids
Representative synthetic procedure: N-Palmitoyl-d-Leucine. To a suspension of d-leucine (528 mg, 4.02 mmol) in water (16 ml) and tetrahydrofuran (8 ml) at 0 °C was added sodium bicarbonate (1.01 g, 12.06 mmol). The reaction mixture was treated with dropwise addition of palmitoyl chloride (1.8 ml, 6.03 mmol). The solvent was removed under reduced pressure and the residue was purified by flash chromatography (SiO2, 5–12% (v/v) acetone-CH2Cl2 gradient) to afford N-palmitoyl-d-leucine (700 mg, quant.) as a white solid with spectra matching those previously reported (15, 16).
Dynamic Light Scattering (DLS)
DLS measurements were carried out by using a Protein Solution Dynapro temperature controlled microsampler with N-palmitoyl-l-leucine dissolved in the same media used for the in vitro splicing assay (60 mm potassium glutamate, 2 mm magnesium acetate, 5 mm creatine phosphate). The data were analyzed and nonlinear regression analysis preformed using GraphPad Prism 6 (GraphPad Software, Inc.).
General Experimental Procedures
NMR spectra were acquired on a Varian Inova 600 MHz spectrometer equipped with a 5 mm HCN triple resonance cryoprobe and referenced to residual solvent proton and carbon signals. HRMS data were acquired on an Agilent 6230 TOF mass spectrometer equipped with a Jetstream ESI source and an Agilent 1260 uPLC. Optical rotations were measured on a Jasco P-2000 polarimeter using a 10-mm path length cell at 589 nm. Solvents used for HPLC and LCMS chromatography were HPLC grade and were used without further purification.
Results
High Throughput Screen of Pre-fractioned Natural Products Library for Splicing Inhibitors
To identify novel natural products that inhibit the human spliceosome, we employed a library consisting 5304 pre-fractions of crude bacteria lysates. This library was derived from liquid cultures of marine-derived Actinobacteria that were isolated from sediment samples collected predominantly on the Western coast of the United States by hand using SCUBA. These environmental isolates were subjected to a standard liquid culture, extraction and fractionation protocol to afford DMSO stock solutions for subsequent chemical screening (for protocol, see Isolation, Cultivation, and Extraction of Streptomyces sp. RL10–300-HVF-B and Preparation of Pre-fraction RLUS1543E under “Experimental Procedures”). The pre-fractions are complex mixtures of typically 3–30 compounds such that the entire library comprises an estimated ∼50,000 natural products. The library is incorporated into a screening pipeline engineered for rapid isolation of hit compounds and regrowth from the original source organism. This design allows for active natural products to be isolated on the tens to hundreds of milligram scale, ensuring a supply of hit compounds for further biological evaluation.
We screened the library for splicing inhibitors with a high-throughput assay that directly quantifies the amount of spliced mRNA produced by in vitro splicing of a model pre-mRNA substrate consisting of an intron flanked by two exons (Fig. 1A) (7, 12). Briefly, we used liquid handling robots to combine splicing substrate, HeLa nuclear extract and library compounds in splicing reactions in 384-well plates. Following the reaction, we transferred a small amount of the splicing reaction as template into a reverse transcriptase (RT)-qPCR plate. In this assay, the amount of mRNA product is determined using a TaqMan® probe labeled with reporter and quencher dyes that is complementary to the splice junction created by splicing (Fig. 1A). When hybridized to spliced cDNA sequence, the probe is degraded by an exonuclease activity encoded in the TaqMan® polymerase, which releases the reporter dye into solution. The thermocycler instrument measures the fluorescence of the released reporter during the amplification process and reports the cycle at which a fluorescence threshold is reached (CT). In the context of our splicing assay, the CT value is inversely proportional to the amount of mRNA produced by splicing. An increase of CT relative to a control reaction reflects less mRNA production and thus splicing inhibition. In a primary screen of our bacterially derived natural products library, most pre-fractions did not affect splicing relative to control, and yielded CT values of ∼20 (Fig. 1B). However, 108 pre-fractions (2.4%) “hits” did cause a significant increase in CT compared with uninhibited control reactions.
FIGURE 1.

High-throughput screening of a complex natural product library identified pre-fractions with splicing inhibitory activity. A, left panel: schematic of the two-step splicing reaction used to screen for inhibitors. Right panel: schematic of the RT-qPCR reaction used to measure the amount of spliced mRNA product. B, histogram shows the number of pre-fractions that yielded the indicated CT value in RT-qPCR analysis of in vitro splicing reactions. The table below summarizes the pre-fraction screening results. C, top panel: representative denaturing gel analysis of RNA isolated from 10-μl splicing reactions into which 1 μl of DMSO or the indicated pre-fractions at 0.04×, 0.2×, and 1× was included. Identities of the bands are schematized to the left as (from top to bottom) lariat-intermediate, pre-mRNA, mRNA, 5′exon intermediate. Average normalized splicing efficiency from triplicate splicing reactions is shown at the bottom of each lane. Bottom panel: native gel analysis of spliceosome assembly using the same reactions as in the top panel. Complex identity is indicated on the left, with complexes assembling in the following order: H/E → A → B → C. D, denaturing gel analysis of in vitro splicing reactions as described in C containing DMSO or indicated pre-fractions at 1× in both the presence (+) and absence (−) of 20 mm DTT. Bands are schematized as in C. Normalized splicing efficiency is shown at the bottom of each lane.
Initial Characterization of the Active Pre-Fraction Hits
To confirm that the decrease in detected mRNA for the initial pre-fraction hits was due to splicing inhibition, we used a well-established gel-based assay to visualize the intermediates and products of splicing chemistry (17). Briefly, we performed the same in vitro splicing assays, now with a radiolabeled pre-mRNA substrate, and increasing concentrations of pre-fractions hits and then separated the resulting RNA species by denaturing PAGE. Twenty-three of the pre-fractions hits (0.6%) inhibited mRNA production. In some cases, 1st step intermediates still accumulated, indicating inhibition occurred between 1st and 2nd step splicing chemistry. Representative data for the three active pre-fraction hits discussed further in this report (RLUS1543E, RLUS1618E, and RLUS1547E) is shown in Fig. 1C (top panel). When compared with the results of a representative screening library containing individual compounds in each well (NCI diversity library) using the same assay, we obtained similar initial hit rates (2.4 versus 3%) and similar number of confirmed hits (0.06 versus 0.01%). This result demonstrates that our high-throughput splicing assay works robustly and reliably when screening complex mixtures of natural products.
We also determined the effect of pre-fraction hits on spliceosome assembly. For each splicing event, spliceosomes form as over 100 components join together in an ordered progression of intermediate complexes (H/E → A → B → C). In in vitro splicing reactions, the assembly process can be qualitatively assessed with native gel analysis by the upward shift of radiolabeled pre-mRNA in stereotypical bands (18). Fig. 1C (bottom panel) shows native gel results for pre-fractions RLUS1543E, RLUS1618E and RLUS1547E, which appear to interfere with higher order spliceosome assembly stages after A complex formation. This result shows that these pre-fractions do not generally disturb all components of the extract, because early stages of spliceosome assembly (E/H and A complex) are maintained.
Our previous splicing inhibitor screen also identified compounds that inhibit at later assembly stages (7). We determined that those molecules inhibit splicing by generating reactive oxygen species (ROS) (7). To rule out ROS as the mechanism of action for pre-fraction hits, we performed rescue experiments with the reducing agent DTT, which will neutralize ROS. Even with 20 mm DTT, which alone does not interfere with splicing, splicing was not restored for any of the confirmed pre-fraction hits, indicating that splicing inhibition is not caused by ROS. Representative data for pre-fractions RLUS1543E, RLUS1618E, and RLUS1547E is shown in Fig. 1D.
Isolating Active Compounds in Pre-fraction Hits
Because each pre-fraction comprises a complex mixture of natural products (typically 3 to 30 compounds), we needed to further partition our hits to identify the specific compound responsible for splicing inhibition. We fractionated each pre-fraction hit by automated time-based reversed-phase HPLC into “one-compound-per-well” peak-libraries. During the fractionation, we also generated UV-visual absorbance profiles and mass spectrometric data for each peak-library well to aid in compound identification and structure determination. We then screened each of the peak-library plates with the same high-throughput splicing assay. For 18 of 21 peak-libraries screened, we detected splicing inhibitor activity in single or adjacent wells of the peak-library plate, indicating that inhibition is likely due to an individual compound. To prioritize the peak-library hits, we matched the active wells with the UV profiles generated during the fractionation process. In six cases, we could match the inhibitory well directly to a distinct peak in the UV profile.
We were particularly interested in the active wells from peak-libraries generated from three pre-fraction hits: RLUS1543E, RLUS1547E, and RLUS1618E. Although the parent pre-fractions of these active wells were derived from three different bacterial strains originating from distant locations, the active wells in their peak-libraries exhibited very similar retention times and UV profiles, suggesting that they contained the same or very closely related compounds (Fig. 2A). This supposition was verified by 1H NMR of the active well component(s), which yielded three nearly identical spectra (Fig. 2B). As each of the three organisms appeared to be producing the same natural product at similar titer levels, we chose a single strain for large-scale cultivation, isolation of the bioactive natural product, and structure elucidation to simplify the overall workflow.
FIGURE 2.
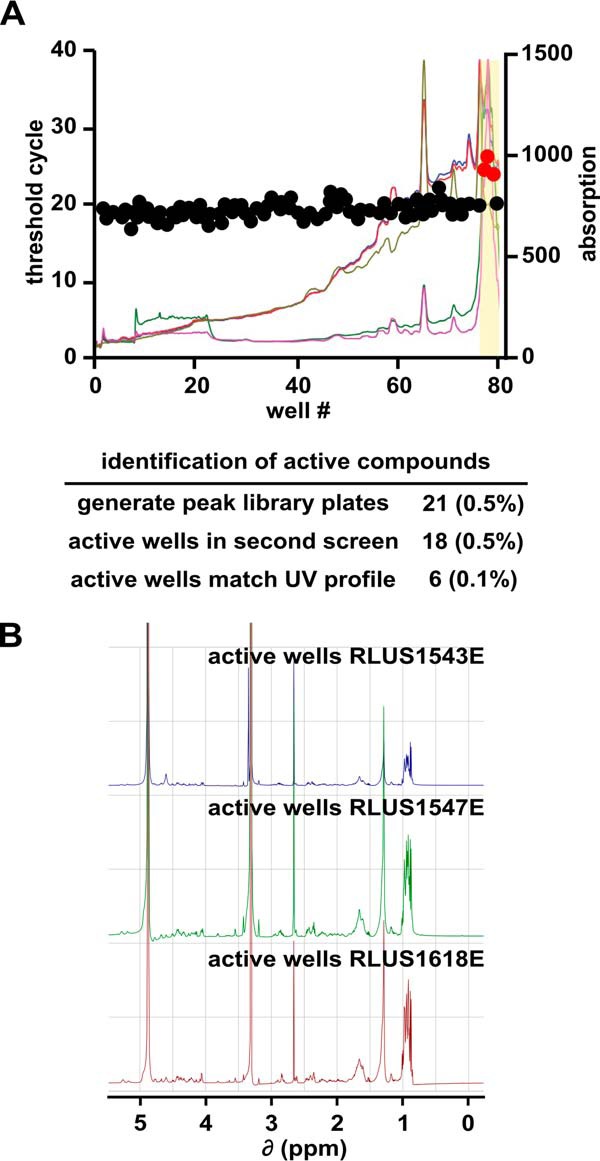
Separation of active components from pre-fractions. A, well number plotted versus CT value (left axis) and UV-visual absorption spectra (right axis) for a representative peak-library plate. The active region of the peak-library is highlighted in yellow. The table below summarizes the screening results for all peak-libraries. B, NMR spectra of active peak-library wells derived from the indicated bacterial strains.
In the analysis of the effect of the RLUS1543E, RLUS1547E, and RLUS1618E pre-fractions on in vitro splicing described earlier, there were some differences in their apparent potency relative to splicing inhibition and spliceosome assembly. These differences may be due to variations of the active compound, or may reflect differences in concentration of the active molecule(s) in the pre-fraction mixture and/or the presence of other interfering compounds.
Identification of N-palmitoyl-l-leucine as Splicing Inhibitor
To isolate more of the active compound, we chose RLUS1543 for large-scale culture followed by bioassay-guided fractionation. Examination of the purified active natural product again revealed a similar 1H NMR profile similar to those seen in the peak-library active wells (Fig. 3B). However, at this point the natural product extract was recalcitrant to further efforts to isolate material of sufficient purity for structural elucidation via standard two-dimensional NMR experiments. Given this situation, we proceeded with an unusual structural elucidation approach combining tandem mass spectrometry, lipidomics, and synthetic preparation of the putative natural product.
FIGURE 3.

Identification of N-palmitoyl-l-leucine as an active pre-fraction component. A, MS/MS spectra of an isolated splicing inhibitor. The chemical structure of the deduced active compound N-palmitoyl-l-leucine is shown in the inset. B, NMR spectra for purified active natural product (top) and synthetic compound (bottom). Peak assignments are shown in the inset table. C, UPLC-MS analysis of chiral derivatization of natural product mixture; natural product mixture alone (top), coinjection of natural product mixture with N-palmitoyl-d-leucyl-l-alanine tert-butyl ester (middle), coinjection of natural product mixture with N-palmitoyl-l-leucyl-l-alanine tert-butyl ester (bottom).
Our initial analysis of the 1H NMR of the active natural product indicated the presence of a long methylene chain and also resonances indicating more polar functionalities, suggesting the presence of a lipid type molecule. Because mass spectrometry is widely used in the analysis of complex mixtures of lipids, we employed MS/MS to analyze the active natural product (Fig. 3A). A database search through the LIPID MAPS Lipidomics Gateway (National Institute of General Medical Sciences) identified MS/MS spectra for N-palmitoyl-leucine (19), which correlated well with the MS/MS spectra for our unknown natural product. Comparison of published NMR data for N-palmitoyl-leucine to that of the active natural product was inconclusive; therefore, we prepared N-palmitoyl-leucine through chemical synthesis, and the synthetic material matched to the active natural product by 1H NMR (Fig. 3B). As the active natural product was insufficiently pure to employ optical rotation to determine the absolute configuration at the amino acid alpha position, a chiral derivatization of the both the natural product mixture and each synthetic enantiomer of N-palmitoyl-leucine was performed followed by co-injection using TOF/UPLC/MS. Results from this analysis revealed that the active natural product possessed the L configuration (Fig. 3C).
To verify that N-palmitoyl-l-leucine was indeed the active compound in the original pre-fractions, we repeated the in vitro splicing assays with the synthetic material. Denaturing PAGE analysis of N-palmitoyl-l-leucine titrations showed that the compound indeed inhibits splicing with an IC50 of 35 μm, as determined by quantification of the splicing efficiency as percent of pre-mRNA converted to mRNA relative to DMSO control (Fig. 4, A and B). Similar to the originating pre-fraction, we see loss of mRNA product before loss of 1st step intermediates as concentration of the inhibitor increases (compare 5–7th lanes in Fig. 4A and the 3rd and 4th lanes of the denaturing gel in Fig. 1C (top panel)). This result indicates that that 2nd step splicing chemistry is more sensitive to the N-palmitoyl-l-leucine relative to 1st step, which correlates to the late block in spliceosome assembly described below.
FIGURE 4.

N-palmitoyl-l-leucine inhibits splicing of a variety of substrates and interferes with late spliceosome assembly. A, representative denaturing gel analysis of RNA isolated from DMSO-treated control splicing reactions or reactions containing indicated concentrations of N-palmitoyl-l-leucine. Identities of the bands are schematized as (from top to bottom) lariat-intermediate, pre-mRNA, free lariat, mRNA, free intron, 5′ exon intermediate. Normalized splicing efficiency is shown at the bottom of each lane. B, average normalized splicing efficiency plotted versus N-palmitoyl-l-leucine concentration for four pre-mRNA substrates with different branch region sequences from triplicate splicing reactions. C, native gel analysis of ”kinetically trapped“ spliceosomes assembled in DMSO (first lane of left gel) and spliceosomes assembled in the presence of 200 μm N-palmitoyl-l-leucine (first lane of middle gel) or 1 μm SSA and pladienolide B (PB), which are splicing inhibitors that arrest at early assembly stages (right gels). The 2nd-5th lanes of the first gels show complexes challenged with increasing concentrations of heparin (hep) at the indicated concentration. D, dynamic light scattering analysis of N-palmitoyl-l-leucine plotting scattering intensity versus increasing compound concentration.
Native gel analysis revealed that the synthetic material interferes with later higher order spliceosome assembly, with an apparent stall at a B-like complex (compare the first lanes for DMSO and 200 μm comp. in Fig. 4C). There appears to be some difference in the effect of compound on assembly relative to the original pre-fraction, which accumulates lower, but still detectable, amounts of higher order spliceosome complexes at concentrations that inhibit splicing chemistry (compare the first lane for 200 μm comp. in Fig. 4C with the 3rd and 4th lanes of the native gels in Fig. 1C (bottom panel)). We hypothesize that the variation reflects differences in concentration of the active molecule(s) in the pre-fraction mixture and/or the presence of other interfering compounds. Therefore, with the overall similarities both in chemical features and bioactivity, we are confident that we have correctly identified the active constituent from these natural product extracts. Notably, in both cases, the stall in assembly is distinct from the previously reported splicing inhibitors pladienolide B and spliceostatin A, both of which block at an A-like complex and do not support any higher order complex formation (see the last two lanes of Fig. 4C).
N-Palmitoyl-l-leucine Blocks Spliceosome Progression
To begin defining the mechanism of N-palmitoy-l-leucine splicing inhibition, we asked whether the compound is specific to our model AdML splicing substrate, or also inhibits splicing of pre-mRNA substrates with different splicing signal sequences. Comparing three additional substrates with different splicing sequence variants (branch point yeast consensus, beta globin and RBM5 intron 16), we found that the compound inhibit their splicing to the same extent (Fig. 4B). This result indicates that in our in vitro assay, N-palmitoy-l-leucine is not specific to one substrate, and may be a general splicing inhibitor.
Previous studies with SSA and pladienolide B showed that they work, in part, by destabilizing spliceosome assembly at an early assembly stage (3, 4). We examined the stability of the B-like complex that accumulates in the presence of N-palmitoy-l-leucine by challenging it with increasing concentrations of heparin (0.5–5 mg ml−1), which disrupts weak protein/nucleic acid interactions (Fig. 4C). As a control with DMSO treatment, we “kinetically arrested” spliceosome assembly at B complex by stopping the splicing reaction after a short incubation time (4 min), which is sufficient to build up some B complex, but not yet to transition to C complex (20). In the presence of N-palmitoyl-l-leucine, the arrested spliceosomes appear to be no less stable than those treated with DMSO. This result means that the compound does not simply destabilize assembled complexes, but, instead, likely interferes with a specific interaction required for the continuation of spliceosome assembly.
With its hydrophilic head group and its hydrophobic tail, N-palmitoyl-l-leucine has an amphiphilic character that likely allows micelle formation in aqueous solution. To rule out that splicing inhibition is caused by micelle formation we determined the critical micellular concentration (CMC) of N-palmitoyl-l-leucine. Under splicing assay conditions, the CMC is 550 μm, which is more than 10-fold higher than the IC50 (Fig. 4D). We concluded that micelle formation is unrelated to the mode of action for this compound.
Similarities and Difference between in Vitro Splicing and Cytological Profile Structure-Activity Relationships
The simple structure of N-palmitoyl-l-leucine allows for straightforward synthesis of the original compound as well as multiple analogs. We prepared a series of synthetic versions of the natural product to explore structure-activity relationships (SAR). We altered three distinct features of the molecule: 1) the amino acid head group, 2) acyl chain length, and 3) the absolute configuration of the amino acid.
For inhibition of in vitro splicing, the most important feature was the length of the acyl chain (Fig. 5). Elongation of the original C16 palmitoyl group to a C18 stearoyl group decreased the IC50 ∼7-fold, and decreasing the length to C12 with a lauroyl group decreased the IC50 ∼2.5-fold. Incorporation of shorter octanoyl (C8) or butanoyl (C4) groups completely abolished splicing inhibition. Altering the amino acid head group was better tolerated, as demonstrated by a series of palmitoyl acylated amino acids (Glu, Ala), which resulted in a ∼4.5-fold decrease in IC50. The exception was replacing the head group with phenylalanine, which inactivated the compound. Inverting the amino acid configuration from l to d caused an ∼8-fold decrease in the ability of the original leucine analog to inhibit splicing, but had a smaller effect for most other amino acids tested. Together, these results show that while the IC50 of N-palmitoyl-l-leucine is relatively modest, its ability to inhibit splicing is clearly structure-dependent and sensitive to subtle substitutions.
FIGURE 5.
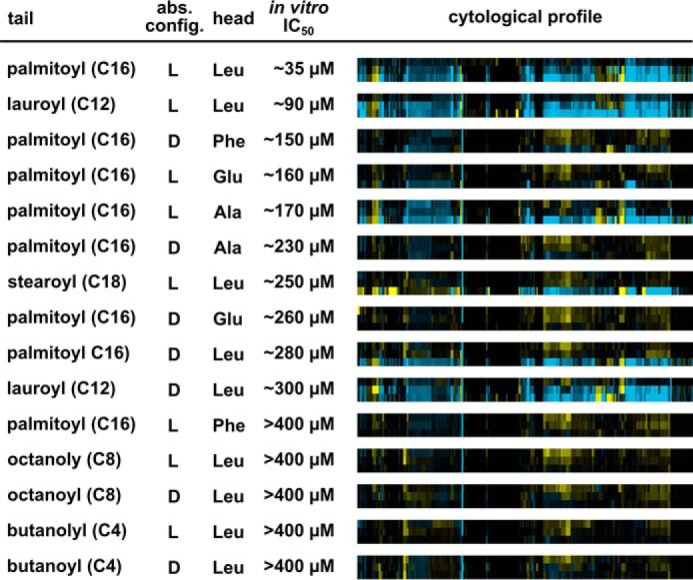
Structure-activity relationship analysis of N-palmitoyl-l-leucine analogs demonstrates parallel trends for the importance of functional groups for in vitro splicing and cell growth. In vitro IC50 values were determined by quantifying splicing efficiencies based on denaturing gel analysis of in vitro splicing reactions. Cytological profiles in which rows show results for a 2-fold dilution series (33, 66 and 133 μm from top to bottom) of the indicated compound, and columns represent the change in 244 cell parameters relative to DMSO control wells. The parameters are derived from staining patterns of actin, tubulin, phosphohistone H3, and 5-ethynyl-2′-deoxyuridine, which provides information pertaining to cell number, cytoskeletal structure, nuclear size, and morphology, DNA replication, and mitosis. Blue indicates an increase in the parameter, yellow indicates a decrease, and black indicates no difference. Color intensity represents the magnitude of the difference.
To investigate how N-palmitoyl-l-leucine and its analogs affect cells, we used a cytological profiling assay (11, 14). For the assay, HeLa cells cultured in 384-well plates were treated with test compounds or with DMSO alone for 20 h, after which they were fixed and stained with fluorescent probes targeting actin (phalloidin), tubulin (α-tub), total DNA (Hoechst), newly synthesized DNA (EdU), and phosphohistone H3 (α-pHH3). The cells were imaged through automated microscopy and a phenotypic profile for each compound was created using an algorithm that measured 244 cellular features based on the staining patterns. These features provide information pertaining to cell number, cytoskeletal structure, nuclear size and morphology, DNA replication and mitosis (11). The magnitude of differences in feature value relative to DMSO treatment is then visualized as color intensity versus compound concentration. The result is a cytological profile that represents the effects of test compounds on cell phenotype.
At higher concentrations, N-palmitoyl-l-leucine produced changes in many cellular features as indicated by the intense blue and yellow features observed in its cytological profile (Fig. 5). Although higher concentrations of compounds are required for effects in cells relative to our in vitro splicing system, the structure-activity relationships show interesting parallels. Acyl tail length again was important. C8 and C4 compounds have little to no cellular activity, as evidenced by their muted and largely featureless cytological profiles (Fig. 5). Amino acid head group identity had an effect in the cell-based assay, because compounds with glutamic acid or phenylalanine showed some in vitro activity but were essentially inactive in cells. The altered chemical properties of the head group may impair their ability to penetrate the cell membrane. When looking at the head-tail linkage, the comparison is harder to make. For example, N-palmitoyl-d-alanine is less active in cells than the in vitro data would predict, and N-lauroyl-d-leucine is more active. A possible explanation for these differences is that different pre-mRNA splicing events show different sensitivities for compound inhibition. Although our in vitro assays do not indicate substrate specificity, the situation in cells is much more complex with alternative splicing. Indeed, in cells, only a few splicing events appear to be sensitive to the early splicing inhibitors SSA and pladienolide B (4, 21). Still, as a whole, the cytological profiling data enforce that N-palmitoyl-l-leucine has a clear link to splicing inhibition with a sharply defined SAR both in vitro and in cells.
Discussion
In the search for new spliceosome inhibitors, it is important to probe a wide expanse of chemical space. Natural product libraries represent a rich source for chemically diverse bioactive compounds, but challenges associated with efficiently identifying and obtaining significant quantities of “hits” has limited their potential for high throughput screening. In this study, we applied a high-throughput splicing assay to screen complex pre-fractions of bacterial origin and exploited a robust discovery pipeline engineered to isolate specific bioactive compounds. We succeeded in identifying and determining the structure of a new splicing inhibitor, N-palmitoyl-l-leucine. The compound has a surprisingly simple structure that allowed us to delve into SAR analysis relative to its effects on spliceosome assembly and cellular phenotype.
N-palmitoyl-l-leucine selectively affects a discrete stage of spliceosome assembly and inhibits splicing of introns with different sequences, although the IC50 is relatively high. SAR analysis revealed strong parallels between in vitro and cell activity, with acyl tail length having the most impact. The cytological profile produced by N-palmitoyl-l-leucine is distinct from that of the well-characterized effects shared by the well-known splicing inhibitors spliceostatin A and pladienolide B (21), which raises the possibility we are observing a new splicing phenotype. Notably, those compounds target an early stage of spliceosome assembly, while N-palmitoyl-l-leucine interferes with spliceosome activity at a much later stage. Furthermore, cytological profiling data indicates that N-palmitoyl-l-leucine is able to penetrate cell membranes, which is the requirement for future cell-based investigations. At the top of the list is to examine changes in splicing of cellular transcripts to understand the pathways affected by the compound. Also, the same structural simplicity and chemical tractability of this natural product that led to rapid access of a series of analogs to better understand its SAR makes N-palmitoyl-l-leucine particularly promising as a tool for biological investigations. It should be straightforward to generate modified compounds functionalized with various chemical handles for experiments aimed at target identification.
Author Contributions
M. S. J., K. A. E., R. C. J., and R. G. L. designed the study and wrote the paper. K. A. E. carried out the high-throughput screen and performed in vitro splicing assays on hits with V. K. U. B. K. D. prepared pre-fraction and peak libraries. R. C. J. analyzed hits by HPLC and mass spectrometry and carried out NMR, dynamic light scattering assays, and chemical syntheses. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We thank R. S. Lokey, W. Bray, and the UCSC Chemical Screening Center, supported by NIH 1S10RR022455 and grants from the California Institute for Quantitative Biosciences (QB3) and the United States Department of State for assistance with cytological profiling measurements.
This work was supported by the Santa Cruz Cancer Benefit Society and National Institutes of Health Grant R01CA136762 (to M. S. J.). The authors declare that they have no conflicts of interest with the contents of this article.
- snRNP
- small ribonucleoprotein
- RT-qPCR
- reverse transcriptase-quantitative polymerase chain reaction
- CT
- threshold cycle
- SAR
- structure-activity relationship
- MS
- mass spectrometry
- NMR
- nuclear magnetic resonance
- CMC
- critical micellular concentration
- TOF
- time of flight
- UPLC
- ultra performance liquid chromatography
- HPLC
- high pressure liquid chromatography
- ROS
- reactive oxygen species
- DMSO
- dimethyl sulfoxide
- SSA
- spliceostatin A.
References
- 1.Wilson D. N. (2009) The A-Z of bacterial translation inhibitors. Crit. Rev. Biochem. Mol. Biol. 44, 393–433 [DOI] [PubMed] [Google Scholar]
- 2.Jurica M. S. (2008) Searching for a wrench to throw into the splicing machine. Nat. Chem. Biol. 4, 3–6 [DOI] [PubMed] [Google Scholar]
- 3.Folco E. G., Coil K. E., and Reed R. (2011) The anti-tumor drug E7107 reveals an essential role for SF3b in remodeling U2 snRNP to expose the branch point-binding region. Genes Dev. 25, 440–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Corrionero A., Miñana B., and Valcárcel J. (2011) Reduced fidelity of branch point recognition and alternative splicing induced by the anti-tumor drug spliceostatin A. Genes Dev. 25, 445–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roybal G. A., and Jurica M. S. (2010) Spliceostatin A inhibits spliceosome assembly subsequent to prespliceosome formation. Nucleic Acids Res. 38, 6664–6672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pawellek A., McElroy S., Samatov T., Mitchell L., Woodland A., Ryder U., Gray D., Lührmann R., and Lamond A. I. (2014) Identification of small molecule inhibitors of pre-mRNA splicing. J. Biol. Chem. 289, 34683–34698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Effenberger K. A., Perriman R. J., Bray W. M., Lokey R. S., Ares M. Jr., and Jurica M. S. (2013) A High-Throughput Splicing Assay Identifies New Classes of Inhibitors of Human and Yeast Spliceosomes. J. Biomol. Screen. 18, 1110–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Samatov T. R., Wolf A., Odenwälder P., Bessonov S., Deraeve C., Bon R. S., Waldmann H., and Lührmann R. (2012) Psoromic acid derivatives: a new family of small-molecule pre-mRNA splicing inhibitors discovered by a stage-specific high-throughput in vitro splicing assay. Chembiochem 13, 640–644 [DOI] [PubMed] [Google Scholar]
- 9.Berg M. G., Wan L., Younis I., Diem M. D., Soo M., Wang C., and Dreyfuss G. (2012) A quantitative high-throughput in vitro splicing assay identifies inhibitors of spliceosome catalysis. Mol. Cell. Biol. 32, 1271–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O'Brien K., Matlin A. J., Lowell A. M., and Moore M. J. (2008) The biflavonoid isoginkgetin is a general inhibitor of Pre-mRNA splicing. J. Biol. Chem. 283, 33147–33154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schulze C. J., Bray W. M., Woerhmann M. H., Stuart J., Lokey R. S., and Linington R. G. (2013) ”Function-first“ lead discovery: mode of action profiling of natural product libraries using image-based screening. Chem. Biol. 20, 285–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reed R. (1989) The organization of 3′ splice-site sequences in mammalian introns. Genes Dev. 3, 2113–2123 [DOI] [PubMed] [Google Scholar]
- 13.Dignam J. D., Lebovitz R. M., and Roeder R. G. (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11, 1475–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Woehrmann M. H., Bray W. M., Durbin J. K., Nisam S. C., Michael A. K., Glassey E., Stuart J. M., and Lokey R. S. (2013) Large-scale cytological profiling for functional analysis of bioactive compounds. Mol. Biosyst. 9, 2604–2617 [DOI] [PubMed] [Google Scholar]
- 15.Beller M., Eckert M., Vollmuller F., Bogdanovic S., and Geissler H. (1997) Palladium-catalyzed amidocarbonylation - a new, efficient synthesis of N-acyl amino acids. Angew. Chem. Int. Ed. 36, 1494–1496 [Google Scholar]
- 16.Takehara M., Yoshimura I., Takizawa K., and Yoshida R. (1972) Surface-active N-acylglutamate. I. Preparation of long-chain N-acylglutamic acid. J. Am. Oil Chem. Soc. 49, 157–161 [Google Scholar]
- 17.Padgett R. A., Hardy S. F., and Sharp P. A. (1983) Splicing of adenovirus RNA in a cell-free transcription system. Proc. Natl. Acad. Sci. U.S.A. 80, 5230–5234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Das R., and Reed R. (1999) Resolution of the mammalian E complex and the ATP-dependent spliceosomal complexes on native agarose mini-gels. RNA 5, 1504–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan B., O'Dell D. K., Yu Y. W., Monn M. F., Hughes H. V., Burstein S., and Walker J. M. (2010) Identification of endogenous acyl amino acids based on a targeted lipidomics approach. J. Lipid Res. 51, 112–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deckert J., Hartmuth K., Boehringer D., Behzadnia N., Will C. L., Kastner B., Stark H., Urlaub H., and Lührmann R. (2006) Protein composition and electron microscopy structure of affinity-purified human spliceosomal B complexes isolated under physiological conditions. Mol. Cell. Biol. 26, 5528–5543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Effenberger K. A., Anderson D. D., Bray W. M., Prichard B. E., Ma N., Adams M. S., Ghosh A. K., and Jurica M. S. (2014) Coherence between cellular responses and in vitro splicing inhibition for the anti-tumor drug pladienolide B and its analogs. J. Biol. Chem. 289, 1938–1947 [DOI] [PMC free article] [PubMed] [Google Scholar]