

Background: Squalene monooxygenase (SM), a rate-limiting cholesterol synthesis enzyme, is degraded in conditions of excess cholesterol.
Results: The cholesterol-regulatory domain of SM is membrane-associated by a re-entrant loop and undergoes a cholesterol-dependent conformational change.
Conclusion: A cholesterol-induced conformational change appears to allow SM to be targeted for degradation.
Significance: This is the first structural, mechanistic explanation for cholesterol-regulated SM degradation.
Keywords: cholesterol, cholesterol regulation, conformational change, membrane, protein turnover, membrane topology, squalene monooxygenase
Abstract
Squalene monooxygenase (SM) is an important control point in cholesterol synthesis beyond 3-hydroxy-3-methylglutaryl-CoA reductase. Although it is known to associate with the endoplasmic reticulum, its topology has not been determined. We have elucidated the membrane topology of the sterol-responsive domain of SM comprising the first 100 amino acids fused to GFP (SM N100-GFP) by determining the accessibility of 16 introduced cysteines to the cysteine-reactive, membrane-impermeable reagent PEG-maleimide. We have identified a region integrally associated with the endoplasmic reticulum membrane that is likely to interact with cholesterol or respond to cholesterol-induced membrane effects. By comparing cysteine accessibility with and without cholesterol treatment, we further present evidence to suggest that cholesterol induces a conformational change in SM N100-GFP. This change is likely to lead to its targeted degradation by the ubiquitin-proteasome system because degradation is blunted by treatment with the chemical chaperone glycerol, which retains SM N100-GFP in its native conformation. Furthermore, degradation can be disrupted by insertion of two N-terminal myc tags, implicating the N terminus in this process. Together, this information provides new molecular insights into the regulation of this critical control point in cholesterol synthesis.
Introduction
Squalene monooxygenase (SM)2 is a key control point in the cholesterol synthesis pathway (1). SM lies downstream of 3-hydroxy-3-methylglutaryl-CoA reductase, the classical rate-limiting step in cholesterol synthesis and the target of the statins. Both 3-hydroxy-3-methylglutaryl-CoA reductase and SM are degraded in a sterol-dependent manner by the ubiquitin-proteasome system (2, 3). However, unlike 3-hydroxy-3-methylglutaryl-CoA reductase where oxysterols and sterol intermediates are the trigger for degradation (4), cholesterol itself accelerates the degradation of SM (1). We have previously shown that this cholesterol-dependent degradation requires the first 100 amino acids (SM N100), conserved in vertebrates but lacking in lower organisms (1), and is mediated by the E3 ubiquitin ligase membrane-associated RING-CH protein 6 (MARCH6) (5, 6). The structure of SM N100 and nature of its association with the membrane is of critical importance for understanding its regulatory mechanism. This region of the 574-amino acid protein is therefore the focus of our study.
Although there is evidence to suggest that SM interacts directly with cholesterol (7), studies using the enantiomer of cholesterol suggest that cholesterol can also accelerate degradation of SM through indirect membrane effects (8). In a previous attempt to identify a cholesterol-binding domain, we mutated a predicted cholesterol recognition amino acid consensus domain in the first 100 amino acids; however, SM remained sterol-responsive (1). It therefore remains unclear how SM N100 responds to changes in cellular cholesterol. Furthermore, there is no structural data available for SM. The PhosphoSite database (9) indicates a ubiquitination site at Lys-90, detected by mass spectrometry, suggesting that this residue is cytosolically exposed, but no other post-translational modifications are known. Here we characterize the membrane topology of SM N100 and investigate how this region senses cholesterol.
Previously, we showed that SM N100 is not expressed unless fused to a soluble protein like GFP or GST. In initial experiments, we also found that SM N100 and a truncation mutant of full-length SM missing the first 100 amino acids are both membrane-associated (1). In the present study, we have demonstrated that SM N100-GFP is indeed a membrane protein and define the nature of this association. Using substituted cysteine mutagenesis followed by chemical modification with a cysteine-reactive, membrane-impermeable reagent, PEG-maleimide (mPEG), we revealed that SM N100 is integrally associated with the ER membrane by a single re-entrant loop. Moreover, cholesterol induced a conformational change at the boundaries of this loop, suggesting that this region senses cholesterol either directly or indirectly. Furthermore, a conformational change in SM likely leads to its cholesterol-dependent degradation as SM N100-GFP degradation could be blunted by retaining SM in its native conformation using the chemical chaperone glycerol (10).
Experimental Procedures
Cell Culture
CHO-7 cells were gifts from Drs Michael S. Brown and Joseph L. Goldstein (University of Texas Southwestern, Dallas, TX) and were cultured in 5% (v/v) lipoprotein-deficient serum/DMEM/Ham's F-12 (Life Technologies) supplemented with penicillin (100 units/ml) and streptomycin (100 μg/ml) (Life Technologies). HEK-293 cells stably overexpressing SM N100-GFP-V5 (6) (HEK-SM N100-GFP cells) were maintained in 10% (v/v) FCS/DMEM (Life Technologies) with high glucose and penicillin (100 units/ml) and streptomycin (100 μg/ml) under selection with hygromycin B (200 μg/ml) (Life Technologies). All transfections were performed in media without penicillin (100 units/ml) and streptomycin (100 μg/ml).
Plasmids
Fig. 1 shows a schematic representation of various SM constructs used in this study. The pTK-SM N100-GFP-V5 and pTK-SM N100-GST-V5 plasmids (Fig. 1, a and b) have been described previously (1). For PEGylation experiments, the pTK-SM N100-GFP-V5 (WT) (1) was used to generate a cysteine-free mutant, SM N100 C31S,C46S (C-null) and C46S or C31S mutants (designated 31C and 46C, respectively). The C-null plasmid was used to generate single cysteine mutants with either T3C, T9C, T11C, G18C, S37C, V41C, S43C, S59C, G61C, L65C, S67C, S71C, S83C, or S87C mutations using megaprimer site-directed mutagenesis (11). The pCMV-Insig-6xmyc (Insig-myc) (12) plasmid used for differential solubilization assays was a generous gift from Drs Michael S. Brown and Joseph L. Goldstein (University of Texas Southwestern, Dallas, TX). The N- and C-terminally tagged SM N100-GST construct (pTK-2xmyc-SM N100-GST-V5; Fig. 1c) was prepared by overlap extension cloning site-directed mutagenesis (13) of pTK-SM N100-GST-V5 using tandem myc sequences amplified from Insig-myc. The pTK-SM-N49-GFP-V5 (SM N49-GFP; Fig. 1d) construct was generated using polymerase incomplete primer extension (14). The SM-FLAG constructs (pCMV-SM(FLAG53/54)-V5 and pCMV-SM(FLAG93/94)-V5) (Fig. 1, e and f) have FLAG tags inserted between residues 53 and 54 or 93 and 94, respectively. These constructs were generated using two-stage QuikChange mutagenesis (15). All primer sequences are available upon request.
FIGURE 1.
Schematic representation of SM constructs. Illustrated (a–f) are SM N100 fusion constructs fused to either GFP or GST, an SM N49 construct fused to GFP, and two full-length SM-V5 constructs with FLAG tag insertions.
Transfection and Cell Sterol Treatments
CHO-7 and HEK-SM N100-GFP cells were transfected with DNA and Lipofectamine-LTX transfection reagent (Life Technologies) in a 1:4 ratio. For membrane differential solubilization experiments, HEK-SM N100-GFP cells were transfected in 14.5-cm dishes for 24 h with 3 μg of Insig-myc and 12 μg of empty vector. For protease protection assays and PEGylation experiments, CHO-7 cells in 14.5-cm dishes were transfected for 24 h with 15 μg of the appropriate expression plasmid. For sterol regulation experiments, CHO-7 cells were transfected for 24 h in 6-well plates with 0.25 μg of expression plasmid and 0.75 μg of empty vector. For PEGylation assays and cholesterol regulation assays, cells were further treated overnight with compactin (5 μm) and mevalonate (50 μm) to reduce basal cholesterol status. For cell sterol regulation assays, sterols were complexed with methyl-β-cyclodextrin (CD) as described previously (16), and treatment was for 8 h (20 μg of sterol/ml).
Microsomal Harvest
Microsomal membranes containing SM N100-GFP were harvested as described (17).
Membrane Differential Solubilization
To determine the nature of the protein-membrane interaction, microsomes were subjected to the indicated dissociation reagents as described (17). Membrane pellets were resuspended in 200 μl of Buffer B (Buffer A containing 100 mm NaCl), and the supernatant was designated the soluble fraction.
Protease Protection Assay
To establish the membrane orientation of the N and C termini and to aid in determining the membrane topology of the first 100 amino acids of SM, protease protection assays were performed (adapted from Ref. 18). CHO-7 cells were transfected with an SM N100-GST construct tagged at both the N and C termini with 2xmyc and V5, respectively (pTK-2xmyc-SM N100-GST-V5; Fig. 1c), or with SM-FLAG(53/54)-V5 or SM-FLAG(93/94)-V5 constructs (Fig. 1, e and f) as indicated in the figure legends. Microsomes were harvested without protease inhibitors. Microsomal protein (50 μg) was suspended in a total volume of 50 μl in Buffer A and was incubated with the indicated amounts of trypsin for 30 min at 30 °C. Reactions were terminated by the addition of loading buffer and heat inactivation at 95 °C for 10 min.
PEGylation Assays
The PEGylation method was adapted (19, 20). Microsomal protein (40 μg) was suspended in a total volume of 54 μl of Buffer A containing protease inhibitors. For in vitro sterol treatments, microsomes were treated with either cholesterol complexed with CD (Chol·CD), lanosterol·CD, 25-hydroxycholesterol·CD, or β-sitosterol·CD (20 μg of sterol/ml) for 30 min at room temperature (24 °C) before being pelleted (20 000 × g, 20 min, 4 °C) to remove excess cholesterol and resuspended in Buffer A. Microsomes were treated with or without 1% (v/v) Triton X-100 as indicated. Microsomes were then treated with or without mPEG (catalog number 10406, Quanta Biodesign) (5 mm, 30 min) at 4 °C, room temperature, or 37 °C as indicated. Reactions were quenched with dithiothreitol (DTT) (10 mm, 10 min, room temperature). Samples were subjected to 12% (w/v) SDS-PAGE followed by Western blotting.
Glycerol Treatments
HEK-SM N100-GFP cells were seeded in 6-well plates for 24 h to ∼80% confluence. Cells were then pretreated overnight with compactin (5 μm), mevalonate (50 μm), and either 0, 0.1, 0.25, or 0.5 m glycerol as indicated in 10% (v/v) fetal calf lipoprotein-deficient serum/DMEM high glucose. Cells were treated with Chol·CD (20 μg of cholesterol/ml) for 8 h with compactin and mevalonate, with or without glycerol, in 10% (v/v) fetal calf lipoprotein-deficient serum/DMEM high glucose. Protein was harvested by scraping in 2% SDS lysis buffer (2% (w/v) SDS, 10 mm Tris-HCl, pH 7.6, 100 mm NaCl) with protease inhibitors. Cells were passed 20 times through a 21-gauge needle and then vortexed for 20 min. Protein concentration was quantified using the bicinchoninic acid assay (Thermo Fisher) and normalized.
Western Blotting
Samples were boiled for 5 min and then subjected to 10 or 12% (w/v) SDS-PAGE as indicated. Proteins were transferred to nitrocellulose membranes and then immunoblotted with either mouse anti-V5 tag (1:5000; Life Technologies), mouse anti-myc tag (1:1000; Santa Cruz Biotechnology), rabbit anti-sterol regulatory element-binding protein cleavage-activating protein (Scap) R139 (25 μg/ml) (21), a generous gift from Drs Michael S. Brown and Joseph L. Goldstein (University of Texas Southwestern, Dallas, TX), mouse anti-FLAG tag (1:10,000; Sigma), mouse anti-α-tubulin (1:10,000; Sigma), or rabbit anti-GAPDH (1:2000; Cell Signaling Technology) as indicated. Proteins were visualized using the ImageQuant LAS 500 (GE Healthcare). Band intensities obtained from a linear exposure range were quantified using the ImageJ software package.
Bioinformatics Predictions
An in silico approach was used to predict SM N100 membrane topology using the first 100 amino acids of the human SM sequence (UniProt Q14534). Prediction of transmembrane domains (TMDs) were performed using Kyte and Doolittle hydropathy plots (22) with a window size of 19, SPLIT 4.0 (23), TMHMM 2.0 (transmembrane hidden Markov models) (24), MEMSAT3 (25), MEMSAT-SMV (26), ΔG predictor server v1.0 (27), TOPCONS (28) and MemPype (29). Predictions of post-translational modifications that mediate membrane attachment including myristoylation, glycophosphatidylinositol anchors, prenylation, and palmitoylation were made using Myristoylator (30), NMT (31), Big-PI Predictor (32), MemPype (29), PrePS (33), and CSS-Palm (34). Secondary structure was predicted using PSIPRED (35).
Results
SM N100 Is an Integral Membrane Protein in the ER
SM N100-GFP, like the full-length enzyme, is localized to the ER but appears to lack any classical α-helical TMDs (1). However, given the apparent interaction of SM with cholesterol, directly (7) and indirectly (8), and our hypothesis that this occurs in the cholesterol-responsive N100, it is important to characterize the nature of the interaction between SM N100-GFP and the ER membrane. Thus, we performed membrane differential solubilization with cells stably expressing SM N100-GFP-V5 (6) (Fig. 2A). Transient expression of myc-tagged Insig, a known integral membrane protein associated with the ER (18), served as a control. SM N100 was dissociated from the membrane in the presence of a strong detergent, SDS (lanes 1 and 2), but not in aqueous Buffer A (lanes 3 and 4) or in conditions of high salt (lanes 5 and 6) or high pH (lanes 7 and 8), conditions known to remove peripheral membrane proteins (18, 36). Therefore, SM N100 is integrally associated with the ER membrane.
FIGURE 2.

SM N100-GFP is an integral membrane protein with predicted TMDs. A, membrane differential solubilization of SM N100. HEK-SM N100-GFP cells were transiently transfected with Insig-myc, a known integral membrane protein, for 24 h. Microsomal membranes were isolated, and proteins were solubilized in the indicated buffers for 30 min at 4 °C with end over end mixing and then centrifuged at 100,000 × g for 30 min at 4 °C. The supernatant (S), representing the cytoplasm, and pellet (P), representing the membrane, were collected. Proteins were separated by 10% SDS-PAGE and immunoblotted with anti-V5 or anti-myc antibodies. Data are representative of n ≥ 4 experiments. B, TMD predictions for SM N100. The SM N100 sequence (first 100 residues of UniProt Q14534) was used to make predictions of TMDs (indicated in red). Cytosolic regions are designated in blue and luminal regions are designated in yellow. White regions have unassigned orientation. Shown are the predictions from a Kyte and Doolittle hydropathy plot (22) with a window size of 19, the SPLIT 4.0 algorithm (23), TMHMM 2.0 (24), MEMSAT3 (25), MEMSAT-SVM (26), the ΔG predictor server (27) with predictions based on ΔGmi, TOPCONS consensus (57), and the MemPype ENSEMBLE prediction (37). TMD1 and TMD2 (shown in green) label the two regions encompassing the TMDs predicted by the six programs. C, Kyte and Doolittle hydropathy plot returned by ProtScale with a window size of 19. The recommended threshold for TMDs is 1.6 (indicated by the dotted red line). The grand average hydropathy score of 0.03 is indicated by a dotted green line. D, putative TMDs based on ΔGmi predictions returned by the ΔG predictor server (27). E, protease protection assays using N- and C-terminally tagged SM N100. CHO-7 cells were transfected with 2xmyc-SM N100-GST-V5 for 24 h. Microsomal membranes were isolated and digested with the indicated amount of trypsin. Proteins were separated by 10% SDS-PAGE and immunoblotted with anti-V5, anti-myc, and anti-Scap R139 antibodies as indicated. * denotes a nonspecific band. Data are representative of n ≥ 3 experiments.
In Silico Analysis Predicts Two TMDs in SM N100
Given that SM N100-GFP is an integral membrane protein (Fig. 2A), it follows that the protein must have at least one hydrophobic region anchoring it to the membrane. In searching for such anchoring points, we first utilized a number of bioinformatics prediction programs that use different algorithms to predict potential TMDs and/or re-entrant loops. Of the eight programs used, all predicted one TMD in the region spanning residues 20–50 (TMD1; Fig. 2B), and three predicted an additional TMD in the region between residues 60 and 80 (TMD2; Fig. 2B).
The Kyte and Doolittle method for predicting TMDs is based on hydrophobicity (22). The resultant hydropathy plot returned by ProtScale (Fig. 2C) shows a positive grand average hydropathy score of 0.03 (dotted green line), indicating that SM N100 is likely to be an integral membrane protein (22). The plot shows two peaks representing two hydrophobic regions. However, only the peak corresponding to TMD1 (Fig. 2B) has a score above the recommended threshold of 1.6 for TMDs (dotted red line) (22). Similarly, SPLIT 4.0 (23), which predicts membrane topology using charge bias and amino acid membrane position preferences, predicted just TMD1 (Fig. 2B). TMHMM 2.0 (24), which uses a hidden Markov model and multiple sequence alignments to predict membrane topology, also predicted TMD1 alone (Fig. 2B). TOPCONS (28), which gives the consensus of several prediction algorithms, predicted TMD1.
TMD1 was predicted by MEMSAT3 (25), which combines amino acid position preferences with signal peptide prediction and sequence profiles (Fig. 2B). In contrast, MEMSAT-SMV (26), which uses support vector machines (SVMs) to predict pore-lining transmembrane helices, predicted two TMDs, TMD1 and TMD2 (Fig. 2B). MEMSAT-SVM can also differentiate signal peptides from TMDs; however, none were predicted in SM N100. Likewise, the ΔG predictor server (27), which calculates the free energy for membrane insertion via the Sec61 translocon (ΔGmi), predicted both TMD1 and TMD2 (Fig. 2, B and D). However, of the two, only TMD1 had a negative ΔGmi (−1.727) compared with TMD2, which had a ΔGmi of 1.889, making it less likely to be a TMD. However, MemPype (29), which uses ENSEMBLE (37) to predict α-helical membrane regions, also predicted TMD2 in addition to TMD1 (Fig. 2B).
OCTOPUS (38), one of the TOPCONS algorithms, is also able to predict re-entrant loops and transmembrane hairpins but predicted none. In fact, neither OCTOPUS nor SCAMPI (39), another algorithm included in TOPCONS, predicted any TMDs at all (data not shown). MEMSAT-SVM can similarly predict re-entrant loops but predicted none.
We also used in silico methods to predict signal peptides and post-translational modifications that can anchor proteins to the membrane, including glycophosphatidylinositol anchors, myristoylation, prenylation, and palmitoylation. MemPype can predict signal peptides and glycophosphatidylinositol anchors, and TOPCONS and MEMSAT-SVM can predict signal peptides. However, neither was predicted in SM N100. Big-PI Predictor (32) similarly did not detect any glycophosphatidylinositol anchors. Myristoylator (30) and NMT (31) predicted no myristoylation, PrePS (33) predicted no prenylation, and CSS-Palm (34) predicted no palmitoylation.
PSIPRED (35) was used to predict protein secondary structure based on position-specific scoring matrices. It predicted a β-strand between residues 3 and 15 and α-helices spanning residues 19–43 (aligning with putative TMD1), 62–71, and 89–98 (data not shown).
Both the N and C Termini of SM N100 Have a Cytosolic Orientation
Having made predictions of SM N100 membrane topology (Fig. 2, B, C, and D), we next sought to experimentally validate these models. To establish the orientation of the N and C termini of SM N100, we transfected CHO-7 cells with an SM N100-GST construct tagged with two N-terminal myc tags and a C-terminal V5 tag. SM N100 alone is not expressed, so to assist in folding and expression, SM N100 was fused to either GST or GFP; both constructs exhibit normal cholesterol regulation (1). Intact microsomes were harvested and treated with increasing concentrations of trypsin, digesting regions exposed to the cytosol but not regions buried in or on the luminal side of the ER membrane. The integrity of the membrane vesicles was confirmed by immunoblotting with an antibody against the R139 epitope of a luminal loop of endogenous hamster Scap (21). This protected luminal fragment, ∼35 kDa in size, appears after digestion of the cytosolic trypsin-susceptible regions of full-length Scap (Fig. 2E, lanes 2–4) and remains resistant to trypsin cleavage (16). In contrast, both N-terminal 2xmyc and C-terminal V5 tags were digested by trypsin (Fig. 2E, lanes 2–4), and no low molecular weight fragments were visible (data not shown), indicating that both the N and C termini of SM N100-GST have a cytosolic orientation.
In Vitro PEGylation Assays Indicate SM N100 Has One Re-entrant Loop
The extreme hydrophobic nature of membrane proteins like SM makes it difficult to obtain their crystal structures. We instead took a biochemical approach to elucidate SM N100-GFP membrane topology. Individual residues were systematically mutated to cysteine, and their relative membrane positions were determined by their accessibility to mPEG using PEGylation assays (19). As mPEG is membrane-impermeable, it cannot react with residues buried within or on the luminal side of microsomal membranes. Instead it reacts only with the extramicrosomal (topologically cytosolic) residues and can be detected by Western blotting based on the 2.3-kDa size difference between PEGylated and unPEGylated protein. Microsomes treated with Triton X-100, a non-denaturing detergent, were used as a positive control by solubilizing the membrane, exposing any membrane-buried residues on the surface of the protein. For each cysteine mutant, the Triton X-100 condition indicates the level of PEGylation expected for an mPEG-accessible residue in the absence of membrane, so to assess membrane topology of individual residues, the densitometric values for the PEG band are given relative to the Triton X-100 PEG band.
Single cysteine mutants were generated by first mutating the native cysteines to serine. We then mutated all serines and threonines (because of their chemical similarity to cysteine) as well as some residues predicted to border TMDs. These cysteine mutants were tested for cholesterol regulation, and although expression levels fluctuated slightly, all retained cholesterol-responsiveness (Fig. 3). We then performed a proof-of-principle PEGylation experiment at room temperature using WT SM N100-GFP (Fig. 1a), a cysteineless SM N100-GFP mutant (C-null), and two single cysteine mutants, 31C and 46C, the native cysteines of SM N100 (Fig. 4A). Triton X-100 treatment of WT SM N100-GFP (Fig. 4A, lane 4) confirmed that it has two cysteines, indicated by the two PEG bands above the unPEGylated band (labeled 1 PEG and 2 PEG). However, only one PEG band is seen without Triton X-100 (lane 3). Therefore, one residue is membrane-buried, and one is exposed. PEGylation of the single cysteine constructs for each of the native cysteines, 31C and 46C, revealed that Cys-31 is membrane-buried, whereas Cys-46 is cytosol-exposed (Fig. 4A, lanes 8–13). GFP also contains two cysteines, which are buried in its β-barrel (40). PEGylation assays using the C-null mutant (Fig. 4A, lanes 5–7), which lacks the native Cys-31 and Cys-46 residues, showed no PEGylation even in the presence of Triton X-100. This demonstrates that the two GFP cysteines are inaccessible to mPEG in non-denaturing conditions and do not interfere with our assay.
FIGURE 3.

Single cysteine mutants retain cholesterol regulation. A–E, CHO-7 cells were transfected for 24 h with the indicated single cysteine mutants. Cells were then pretreated overnight with compactin (5 μm) and mevalonate (50 μm) and then treated for 8 h with Chol·CD (20 μg of cholesterol/ml), compactin, and mevalonate. Protein was harvested, separated by 10% SDS-PAGE, and then immunoblotted with anti-V5 using GAPDH as a loading control. F, densitometry from A–E. Data are normalized to the untreated condition for each mutant, which is set to 1 and indicated with a dotted line. Data are presented as mean ± S.E. (error bars) from n ≥ 3 experiments.
FIGURE 4.

Determination of SM N100 membrane topology using PEGylation assays. A, CHO-7 cells were transfected with the indicated constructs for 24 h, and then microsomal membranes were harvested. Microsomal proteins were treated with or without mPEG (5 mm) or Triton X-100 (1%, v/v) for 40 min at room temperature and quenched with DTT (10 mm) for 10 min at room temperature, and then proteins were separated by 12% SDS-PAGE. Membranes were immunoblotted with anti-V5 antibody. UnPEGylated protein is indicated by 0 PEG, singly PEGylated protein is indicated by 1 PEG, and doubly PEGylated protein is indicated by 2 PEG. Data are representative of n ≥ 3 taken from separate blots as indicated by separating lines. B, representative immunoblotting data for PEGylation of the indicated cysteine mutants at 4 °C, room temperature (denoted as Room temp.), or 37 °C performed as in A. C, average densitometries for the PEGylated bands of the indicated cysteine mutants at 4 °C, room temperature (denoted as RT), or 37 °C showing PEGylation relative to the Triton X-100-treated condition, which is set to 1. The increasing background gradient represents an increasing level of cytosolic exposure as indicated by the arrow. Data are presented as mean ± S.E. (error bars) from n ≥ 3 experiments. D, working model of SM N100-GFP-V5 based on densitometry values from C.
After establishing that SM N100-GFP is an integral membrane protein (Fig. 2A) and that both N and C termini are cytosolic (Fig. 2E), we used PEGylation assays with 16 single cysteine mutants to identify which regions of SM N100 are embedded in the ER membrane or within the ER lumen. PEGylation assays were carried out at 4 °C, room temperature, and 37 °C to account for any thermodynamic effects on SM N100 membrane topology (41). At 4 °C, there should be very little membrane fluidity, and SM N100 likely remains in a relatively fixed conformation, whereas at physiological temperatures, SM is expected to be more flexible, particularly around the membrane-cytosol interface.
To ascertain the relative PEGylation of each residue, PEGylated bands (Fig. 4B) were analyzed using densitometry (Fig. 4C). For each cysteine mutant, values are given relative to the PEGylated band in the Triton X-100-treated condition at each temperature. Values approaching 0 indicate membrane-buried or lumen-facing residues, and values close to 1 indicate full extramicrosomal (cytosolic) exposure. Moderate PEGylation (values less than ∼0.5) indicate that the residue is likely to be at the membrane-cytosolic interface. The fluidity of the membrane and flexibility of the protein mean that, for residues close to the membrane-cytosolic interface, at any given time a portion of the cysteines in the sample will be exposed, whereas others are buried, giving these intermediate values.
Although cysteine exposure tended to increase with increasing temperature as clearly seen at residue 18, the pattern of relative exposure between the residues at 4 °C, room temperature, and 37 °C was largely alike (Fig. 4C). These results were used to develop a model of SM N100 membrane topology (Fig. 4D) and indicate that SM N100 is embedded in the ER membrane somewhere in the region between residue 18 and residue 41. As this is the only membrane-buried region in SM N100, it is therefore the region integrally associated with the ER.
SM N100 Membrane Topology Resembles That of Full-length SM
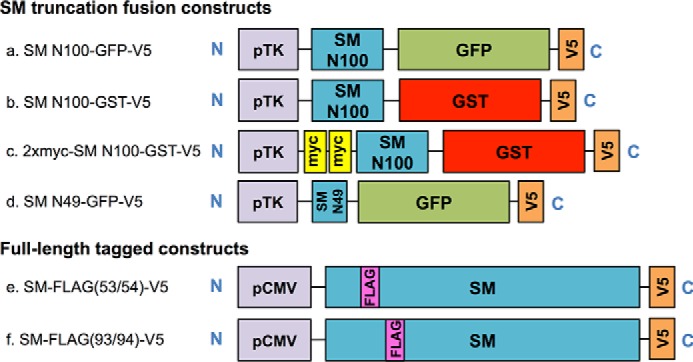
To ensure that the cytoplasmic orientation of the SM N100-GFP C terminus (Figs. 2E and 4) was not an artifact of its fusion to the large, soluble GFP, we performed protease protection assays on two full-length SM constructs with FLAG tag insertions between residues 53/54 and 93/94 (Figs. 1, e and f, and 5). Consistent with the membrane topology data generated from PEGylation assays (Fig. 4), both FLAG tags between residues 53/54 and 93/94 were cytosolically exposed as they were digested by trypsin, and no low molecular weight fragments were visible. As with Fig. 2E, the membrane-buried, trypsin-resistant Scap fragment was not digested, demonstrating that the microsomes were intact.
FIGURE 5.
Protease protection assays in full-length SM support SM N100 topology model. Protease protection assays were carried out using full-length SM with FLAG tags inserted between residues 53/54 and 93/94. CHO-7 cells were transfected with SM-FLAG(53/54)-V5 or with SM-FLAG(93/94)-V5 for 24 h. Microsomal membranes were isolated and digested with the indicated amount of trypsin. Proteins were separated by 10% SDS-PAGE and immunoblotted with anti-FLAG and anti-Scap R139 antibodies as indicated. * denotes a nonspecific band. Data are representative of n ≥ 3 experiments.
Glycerol Rescues SM N100 from Cholesterol-dependent Degradation
In conditions of excess cholesterol, SM N100 is targeted for proteasomal degradation by the E3 ubiquitin ligase MARCH6 (1, 5, 6). One mechanism for this could be via a conformational change in SM N100 either by direct binding of cholesterol to SM or through bulk membrane effects that expose a specific binding site for MARCH6. Alternatively, MARCH6 may recognize the altered structure of SM N100, such as general properties of a misfolded quality control substrate, rather than a sequence motif as is the case for yeast 3-hydroxy-3-methylglutaryl-CoA reductase (42). In either case, glycerol, a chemical chaperone that causes proteins to favor their native conformation (43, 44), should prevent this conformational change and thereby rescue SM N100 from cholesterol-dependent degradation. Glycerol was previously used to rescue yeast 3-hydroxy-3-methylglutaryl-CoA reductase from E3 ligase-dependent degradation where it was demonstrated that the glycerol effects were due to stabilization of its native protein conformation (10).
We treated HEK-SM N100-GFP cells with increasing concentrations of glycerol (Fig. 6). At low glycerol concentrations, little protection was offered. However, both 0.25 and 0.5 m glycerol increased SM N100-GFP stability in the absence of cholesterol with 0.5 m glycerol increasing SM N100-GFP levels by at least 4-fold and providing ∼70% protection from cholesterol-dependent degradation (Fig. 6B), consistent with the hypothesis that cholesterol-dependent degradation of SM N100 relies on a conformational change.
FIGURE 6.

Glycerol reduces cholesterol-dependent degradation of SM N100. A, HEK-SM N100-GFP cells were pretreated overnight with compactin (5 μm), mevalonate (50 μm), and the indicated concentration of glycerol. Cells were treated with cholesterol (Chol·CD; 20 μg of cholesterol/ml) for 8 h with compactin, mevalonate, and glycerol. Protein was harvested, separated by 10% SDS-PAGE, and then immunoblotted with anti-V5 using α-tubulin as a loading control. B, densitometry from A. Data are normalized to the 0.5 m glycerol treatment, which is set to 1, and are presented as mean ± S.E. (error bars) from n ≥ 3 experiments.
Cholesterol Alters the Conformation of SM N100
After observing the protective effect of glycerol on cholesterol-dependent degradation of SM N100-GFP, we further investigated the hypothesis that MARCH6 is able to target SM N100 via a cholesterol-induced conformational change in SM N100. It seemed likely that the effect would be best observed at residues flanking the putative membrane-cytosol interface, particularly those that show marked temperature sensitivity (Fig. 4C). We therefore focused initially on residues T3C, T9C, T11C, and G18C in PEGylation assays where microsomes were incubated with Chol·CD prior to PEGylation and compared their exposure relative to microsomes PEGylated without prior cholesterol treatment. Experiments were performed at 4 °C, room temperature, and 37 °C. We found that at all three temperatures T11C tended to be less exposed to the cytosol when treated with cholesterol, and this decrease was significant at 37 °C (Fig. 7, A and B). This, together with the fact that 37 °C is physiological, led us to perform these experiments at 37 °C for the remaining cysteine mutants (Fig. 7C) where significant cholesterol-induced conformational changes were also observed at T9C (increased exposure) and V41C (decreased exposure) (Fig. 7D). These data provide further evidence for our hypothesized mechanism of a conformational change underlying the cholesterol-dependent degradation of SM.
FIGURE 7.

Cholesterol induces a conformational change in SM N100. CHO-7 cells were transfected with the indicated constructs for 24 h, and then microsomal membranes were harvested. Microsomes were treated with or without cholesterol (Chol·CD; 20 μg of cholesterol/ml) for 30 min at room temperature. Excess cholesterol was removed by centrifugation prior to PEGylation. A, example blot for T11C. B, average densitometry values for T11C PEGylated at 4 °C, room temperature (denoted as RT), or 37 °C after cholesterol treatment. Values were normalized to the non-cholesterol-treated condition, which is set to 0. This line indicates no cholesterol-induced effect. Data are presented as mean ± S.E. (error bars) from n ≥ 3 experiments. C, representative immunoblotting data for PEGylation of the indicated cysteine mutants performed at 37 °C with or without cholesterol pretreatment. D, average densitometry values for PEGylation of Chol·CD-treated (20 μg of cholesterol/ml) microsomes from the indicated single cysteine mutants PEGylated at 37 °C and additional PEGylation of mutants T9C and T11C pretreated with lanosterol·CD, 25-hydroxycholesterol·CD (denoted 25-HC), or β-sitosterol·CD (20 μg of sterol/ml). Values were normalized as in B. *, values significantly different from PEGylation without prior cholesterol treatment (p < 0.05). Data are presented as mean ± S.E. (error bars) from n ≥ 3 experiments. E, representative immunoblot and average densitometry values for sterol treatments. CHO-7 cells were transiently transfected with SM N100-GFP for 24 h. Cells were pretreated overnight with compactin (5 μm) and mevalonate (50 μm) and then treated with the indicated sterols (20 μg of sterol/ml) for 8 h with compactin and mevalonate. Protein was harvested, separated by 10% SDS-PAGE, and then immunoblotted with anti-V5 using α-tubulin as a loading control. *, values significantly different from the untreated control (WT) (p < 0.001). Data are presented as mean ± S.E. (error bars) from n ≥ 3 experiments.
Sterol Specificity of Conformational Change and Degradation of SM N100-GFP
To determine whether other sterols could also induce a conformational change in SM N100, we further PEGylated T9C and T11C mutants after pretreating microsomes with a cholesterol pathway intermediate, lanosterol; an oxysterol, 25-hydroxycholesterol; or a plant sterol, β-sitosterol, all complexed to CD. These sterols neither had a significant effect on PEGylation (Fig. 7D) nor did they induce SM N100-GFP degradation (Fig. 7E), suggesting that the effect of cholesterol is relatively specific.
N-terminal Tags Disrupt Cholesterol Regulation
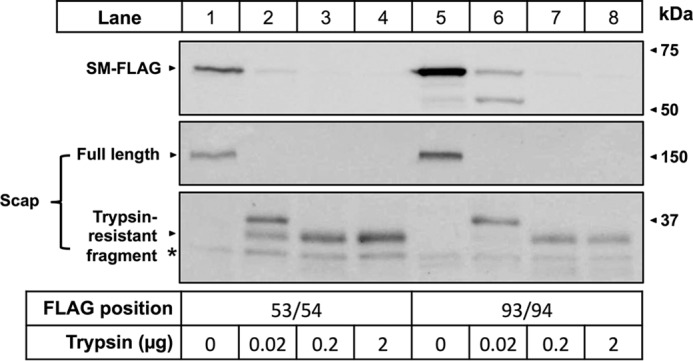
To investigate the possibility that cholesterol regulation occurs at the N terminus of SM N100, we tested the cholesterol regulation of the construct tagged at the N terminus with two myc tags, pTK-2xmyc-SM N100-GST-V5 (Fig. 1c), and compared it with a construct tagged at the C terminus only, pTK-SM N100-GST-V5 (Fig. 1b). When treated with cholesterol, C-terminally tagged SM N100-GST showed normal cholesterol regulation (Fig. 8A, lanes 2 and 3), whereas dramatically reduced degradation was observed with the construct tagged at the N terminus (Fig. 8A, lanes 4 and 5). Additionally, basal expression of the N-tagged construct was higher than that of the C-tagged construct, suggesting that the N terminus plays a role in basal protein turnover.
FIGURE 8.
Epitope tags inserted at the N terminus disrupt cholesterol-dependent degradation. A, CHO-7 cells were transfected for 24 h with either empty vector (EV), SM N100-GST-V5, or 2xmyc-SM N100-GST-V5. Cells were pretreated overnight with compactin (5 μm) and mevalonate (50 μm) and then treated for 8 h with Chol·CD (20 μg of cholesterol/ml), compactin, and mevalonate. Protein was harvested, separated by 10% SDS-PAGE, and then immunoblotted with anti-V5 using α-tubulin as a loading control. B, CHO-7 cells were transfected with either SM N100-GFP-V5 or SM N49-GFP-V5 and then pretreated and treated as in B. Data are representative of n = 3 experiments for each panel.
The N-terminal Half of SM N100 Alone Does Not Confer Cholesterol Regulation
To further determine the specific region of SM N100 required for cholesterol-dependent regulation, we tested the cholesterol responsiveness of a mutant expressing only the first 49 amino acids of SM fused to GFP (SM N49-GFP; Fig. 1d). We hypothesized that this region would retain cholesterol regulation. However, this region was not cholesterol-responsive. Furthermore, this mutant showed higher basal expression compared with the WT (Fig. 8B), suggesting that the C-terminal half of SM N100 is also required for cholesterol regulation.
Discussion
We have characterized the membrane topology of the cholesterol-regulatory domain of SM and shown that it undergoes a cholesterol-dependent conformational change. We first established that SM N100-GFP is an integral membrane protein (Fig. 2A) with no predicted membrane-anchoring post-translational modifications and up to two predicted TMDs (Fig. 2B). To identify the region(s) anchoring SM N100 to the ER membrane, we utilized PEGylation assays using 16 single cysteine mutants to map SM N100 membrane topology.
In contrast to several of the in silico predictions, we found that SM N100 has only one membrane-buried region, between residues 18 and 41 (Fig. 4, C and D), corresponding to the predicted TMD1 (Fig. 2B). Importantly, we have shown that both the N and C termini of SM N100 have a cytosolic orientation (Fig. 2E), consistent with results shown for the N terminus of full-length SM (1). We also inserted FLAG tags between residues 53/54 and 93/94 in a full-length SM construct and used protease protection assays to show that both sites were cytosolically exposed (Fig. 5). This validates the PEGylation data, which indicate that residues beyond Cys-46 lie within the cytosol, and shows that this is not simply due to the presence of the large and soluble fused GFP. In addition, the FAD-binding domain of full-length SM is predicted to be between residues 126 and 153 (45), also consistent with the C terminus of SM N100 being cytosolically oriented as is the Lys-90 ubiquitination site (9).
Of note is that PEGylation experiments revealed that residues prior to 18, as well as those beyond residue 41, are cytosolically exposed (Fig. 4C). Given that a typical TMD requires a minimum of 18–20 residues (22) and that the maximum span of the SM N100 membrane-buried region is ∼20 residues, SM N100 cannot possibly possess the two TMDs required for both N and C termini to have a cytosolic orientation. We therefore propose that SM N100 possesses a re-entrant loop (46, 47), entering the membrane just beyond residue 18 and resurfacing on the cytosolic face around residue 41.
As SM N100-GFP is associated with the ER membrane at just one region (Fig. 4, C and D), it is likely that this region contains the site that interacts with cholesterol or responds to cholesterol-mediated membrane effects (7, 8), causing SM degradation in conditions of excess cholesterol. We and others have shown that this cholesterol-dependent degradation is mediated by MARCH6 (5, 6). However, whether this occurs through a direct interaction between MARCH6 and SM and by what mechanism has not been established. We propose that it occurs via a conformational change in SM N100 induced by excess cholesterol that either exposes a buried MARCH6 target motif or alters the folding state such that it is recognized by MARCH6 (42). Treating HEK-SM N100-GFP cells with the chemical chaperone glycerol prevented SM N100 degradation. Glycerol at a concentration of 0.5 m stabilized SM N100 and provided ∼70% protection from cholesterol-dependent degradation (Fig. 6B). Although higher concentrations of glycerol may have offered better protection, 1 m glycerol was cytotoxic (data not shown). Glycerol has previously been shown to prevent proteins from changing conformation (43, 44). Hence, this protection offered by glycerol is evidence that SM N100 requires a conformational change to be targeted for degradation in conditions of excess cellular cholesterol.
Further evidence for this cholesterol-induced conformational change comes from PEGylation assays using microsomes treated with or without cholesterol. We found that cysteine accessibility changed subtly but significantly at residues bordering the apparent membrane-cytosol interface, increasing at T9C and decreasing at T11C and V41C (Fig. 7D). Moreover, temperature responsiveness appeared greatest in the membrane-cytosol boundary regions of SM N100 (Fig. 4C). We also found that this effect is apparently specific to cholesterol as treating microsomes with other sterols had no significant effect on the exposures of residues T9C or T11C (Fig. 7D). Furthermore, these sterols did not cause SM N100 degradation (Fig. 7E), consistent with observations for full-length SM (1).
Although relatively specific for cholesterol, it remains unclear whether this conformational change is a result of direct binding or bulk membrane effects. It is likely that SM both responds to membrane effects and has a discrete cholesterol-binding site as indicated by the direct binding of full-length SM to a sterol probe in a mass screen (7) and by the intermediate effect of the enantiomer of cholesterol on SM degradation (8). In an attempt to find a cholesterol-binding site, we previously mutated the critical tyrosine in the putative cholesterol recognition amino acid consensus domain, which bridges the re-entrant loop and the cytosolically exposed region between residues Leu-40 and Arg-49. However, cholesterol-dependent degradation of SM N100 was unaffected, so this is clearly not a functional motif (1).
Integral membrane association, a cholesterol-dependent conformational change, and disruption of cholesterol-dependent degradation through N-terminal tag insertion all occur in the first half of SM N100, suggesting that this region mediates cholesterol regulation. However, we showed that an SM N49-GFP construct (Fig. 1d) was not cholesterol-regulated (Fig. 8B) and had a higher basal level of expression compared with WT. Clearly, the cytosolically exposed region beyond residue 49 is also important for cholesterol regulation. In addition, this region appears to be important for basal degradation. It may be that the Lys-90 ubiquitination site (9) is involved in basal degradation rather than cholesterol-dependent degradation as lysineless SM N100 mutants remain cholesterol-regulated (1).
In this study, we attempted to locate potential cholesterol-sensing sites using scanning alanine mutagenesis. However, all 32 screened mutants, covering the length of SM N100, retained cholesterol regulation (data not shown). Cysteine mutants used in PEGylation assays likewise retained cholesterol regulation (Fig. 3). It is likely that several stretches of sequence are required for the proper local fold and for the conformational change even if they are not individually essential for cholesterol sensing. Instead of or in addition to mutagenesis, a clickable, photoreactive sterol probe (7) might be used in conjunction with mass spectrometry to identify peptides that bind to cholesterol.
It is also worth noting that the conformational change observed in response to cholesterol treatment (Fig. 7D) appears to alter the cytosolic accessibility of just a few amino acids. This favors the hypothesis that MARCH6 recognizes a cholesterol-induced change in folding state, rather than recognizing a newly exposed sequence motif, to target SM for degradation. This may include recognition of conformational changes within the membrane, which cannot be detected by the PEGylation assay.
There is evidence to suggest that SM N100-GFP degradation is critically dependent on its N terminus. Although Lys-90 is known to be ubiquitinated (9), when all lysines in SM N100 were simultaneously mutated, SM N100 still displayed cholesterol-dependent regulation (1). Additionally, the insertion of 2xmyc tags at the N terminus of SM N100-GST led to a near complete loss of cholesterol-dependent degradation (Fig. 8A). This is consistent with our previous study (1) that found 30-amino acid GA repeats inserted at both the N and C termini of full-length SM severely blunted cholesterol-dependent degradation, whereas a mutant expressing the insertion at the C terminus alone retained normal cholesterol-regulation. Together, these findings point to the N terminus playing a key role in degradation.
Recently, MARCH6 (also known as Teb4) was shown to recognize N-acetylated proteins and target them for degradation (48). The N-terminal acetyltransferase NatC is known to acetylate Met-Trp- sequences (for a review, see Ref. 49) present at the N terminus of SM. Therefore, SM could potentially be N-terminally acetylated, although this may not be relevant to cholesterol-regulated degradation of SM because this modification is co-translational.
Alternatively, SM could be ubiquitinated at the N terminus as suggested by the decreased degradation seen with N-terminal insertions (Fig. 8A and Ref. 1). Such additions can stabilize proteins that are normally N-terminally ubiquitinated (50). Ubiquitination usually occurs at Lys residues on the target protein but can also occur at Ser, Thr, Cys, or the N terminus (51–53). It is possible that SM N100 is ubiquitinated on one or more of these non-canonical sites. RING-type E3 ligases, such as MARCH6, catalyze the transfer of ubiquitin directly from the E2 ubiquitin-conjugating enzyme (for a review, see Ref. 54). Ube2w was recently identified as an E2 ubiquitin-conjugating enzyme that ubiquitinates N-terminal residues and can do so with just an E3 RING domain lacking any substrate-binding sites (55, 56). So far Ube2w is the only known E2 able to ubiquitinate the N terminus of proteins, and it is not known whether this E2 interacts with MARCH6. However, it is entirely possible that MARCH6 interacts with another E2 able to catalyze N-terminal ubiquitination. If MARCH6 does ubiquitinate the N terminus, this provides more evidence to suggest that it does so by detecting a change in conformation rather than by recognizing the appearance of previously buried residues as accessibility of residue T3C, which is very close to the N terminus, does not change in response to increased cholesterol (Fig. 7D). However, it remains possible that, in the absence of cholesterol, SM N100 conformation is such that ubiquitination is sterically hindered, but PEGylation by the much smaller mPEG molecule can still occur. Therefore, MARCH6 recognition via a change in exposure of the buried N terminus cannot be ruled out.
In conclusion, SM N100 is an integral membrane protein that is membrane-associated via a re-entrant loop and contains no TMDs. This regulatory domain undergoes a conformational change in response to cholesterol, which is also suggested by blunting of this degradation by the molecular chaperone glycerol. Furthermore, degradation can be disrupted by insertion of two N-terminal myc tags, implicating the N terminus in this process. Further work is required to determine the precise mechanism for cholesterol-dependent degradation of SM and whether MARCH6 interacts with SM directly or whether it acts via adaptor proteins.
Author Contributions
V. H. helped design the experiments, generated molecular tools, and acquired the vast majority of the data. She also analyzed and interpreted the data as well as drafted the article. N. K. C. performed cholesterol regulation assays on alanine and cysteine mutants. J. S. helped in the conception of this project and interpretation of data, performed preliminary experimental work, generated molecular tools, and critically revised the article. A. J. B. helped in the conception of this project, in the interpretation of the data, and in the drafting and critical revision of the article. All authors approved the final version of this article.
Acknowledgments
We thank the members of the Brown laboratory for critically reviewing this manuscript and Lisa Phan and Cyril Bur for contributions to preliminary experimental work.
This work was funded by National Health and Medical Research Council Grant 1060515. The authors declare that they have no conflicts of interest with the contents of this article.
- SM
- squalene monooxygenase
- CD
- methyl-β-cyclodextrin
- Chol·CD
- cholesterol complexed with CD
- ER
- endoplasmic reticulum
- MARCH6
- membrane-associated RING-CH protein 6
- mPEG
- polyethylene glycol-maleimide
- SVM
- support vector machine
- TMD
- transmembrane domain
- Scap
- sterol regulatory element-binding protein cleavage-activating protein.
References
- 1.Gill S., Stevenson J., Kristiana I., and Brown A. J. (2011) Cholesterol-dependent degradation of squalene monooxygenase, a control point in cholesterol synthesis beyond HMG-CoA reductase. Cell Metab. 13, 260–273 [DOI] [PubMed] [Google Scholar]
- 2.Sever N., Song B. L., Yabe D., Goldstein J. L., Brown M. S., and DeBose-Boyd R. A. (2003) Insig-dependent ubiquitination and degradation of mammalian 3-hydroxy-3-methylglutaryl-CoA reductase stimulated by sterols and geranylgeraniol. J. Biol. Chem. 278, 52479–52490 [DOI] [PubMed] [Google Scholar]
- 3.Sharpe L. J., Cook E. C., Zelcer N., and Brown A. J. (2014) The UPS and downs of cholesterol homeostasis. Trends Biochem. Sci. 39, 527–535 [DOI] [PubMed] [Google Scholar]
- 4.Song B.-L., Javitt N. B., and DeBose-Boyd R. A. (2005) Insig-mediated degradation of HMG CoA reductase stimulated by lanosterol, an intermediate in the synthesis of cholesterol. Cell Metab. 1, 179–189 [DOI] [PubMed] [Google Scholar]
- 5.Foresti O., Ruggiano A., Hannibal-Bach H. K., Ejsing C. S., and Carvalho P. (2013) Sterol homeostasis requires regulated degradation of squalene monooxygenase by the ubiquitin ligase Doa10/Teb4. Elife 2, e00953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zelcer N., Sharpe L. J., Loregger A., Kristiana I., Cook E. C., Phan L., Stevenson J., and Brown A. J. (2014) The E3 ubiquitin ligase MARCH6 degrades squalene monooxygenase and affects 3-hydroxy-3-methyl-glutaryl coenzyme A reductase and the cholesterol synthesis pathway. Mol. Cell. Biol. 34, 1262–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hulce J. J., Cognetta A. B., Niphakis M. J., Tully S. E., and Cravatt B. F. (2013) Proteome-wide mapping of cholesterol-interacting proteins in mammalian cells. Nat. Methods 10, 259–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kristiana I., Luu W., Stevenson J., Cartland S., Jessup W., Belani J. D., Rychnovsky S. D., and Brown A. J. (2012) Cholesterol through the looking glass: ability of its enantiomer also to elicit homeostatic responses. J. Biol. Chem. 287, 33897–33904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hornbeck P. V., Zhang B., Murray B., Kornhauser J. M., Latham V., and Skrzypek E. (2015) PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gardner R. G., Shearer A. G., and Hampton R. Y. (2001) In vivo action of the HRD ubiquitin ligase complex: mechanisms of endoplasmic reticulum quality control and sterol regulation. Mol. Cell. Biol. 21, 4276–4291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanchis J., Fernández L., Carballeira J. D., Drone J., Gumulya Y., Höbenreich H., Kahakeaw D., Kille S., Lohmer R., Peyralans J. J., Podtetenieff J., Prasad S., Soni P., Taglieber A., Wu S., Zilly F. E., and Reetz M. T. (2008) Improved PCR method for the creation of saturation mutagenesis libraries in directed evolution: application to difficult-to-amplify templates. Appl. Microbiol. Biotechnol. 81, 387–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yabe D., Brown M. S., and Goldstein J. L. (2002) Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc. Natl. Acad. Sci. U.S.A. 99, 12753–12758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stevenson J., Krycer J. R., Phan L., and Brown A. J. (2013) A practical comparison of ligation-independent cloning techniques. PLoS One 8, e83888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klock H. E., Koesema E. J., Knuth M. W., and Lesley S. A. (2008) Combining the polymerase incomplete primer extension method for cloning and mutagenesis with microscreening to accelerate structural genomics efforts. Proteins Struct. Funct. Genet. 71, 982–994 [DOI] [PubMed] [Google Scholar]
- 15.Wang W., and Malcolm B. A. (1999) Two-stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuikChangeTM site-directed mutagenesis. BioTechniques 26, 680–682 [DOI] [PubMed] [Google Scholar]
- 16.Brown A. J., Sun L., Feramisco J. D., Brown M. S., and Goldstein J. L. (2002) Cholesterol addition to ER membranes alters conformation of SCAP, the SREBP escort protein that regulates cholesterol metabolism. Mol. Cell 10, 237–245 [DOI] [PubMed] [Google Scholar]
- 17.Zerenturk E. J., Sharpe L. J., and Brown A. J. (2014) DHCR24 associates strongly with the endoplasmic reticulum beyond predicted membrane domains: implications for the activities of this multi-functional enzyme. Biosci. Rep. 34, 107–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feramisco J. D., Goldstein J. L., and Brown M. S. (2004) Membrane topology of human insig-1, a protein regulator of lipid synthesis. J. Biol. Chem. 279, 8487–8496 [DOI] [PubMed] [Google Scholar]
- 19.Sun L.-P., Seemann J., Goldstein J. L., and Brown M. S. (2007) Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Insig renders sorting signal in Scap inaccessible to COPII proteins. Proc. Natl. Acad. Sci. U.S.A. 104, 6519–6526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y., Toei M., and Forgac M. (2008) Analysis of the membrane topology of transmembrane segments in the C-terminal hydrophobic domain of the yeast vacuolar ATPase subunit a (Vph1p) by chemical modification. J. Biol. Chem. 283, 20696–20702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakai J., Nohturfft A., Cheng D., Ho Y. K., Brown M. S., and Goldstein J. L. (1997) Identification of complexes between the COOH-terminal domains of sterol regulatory element-binding proteins (SREBPs) and SREBP cleavage-activating protein. J. Biol. Chem. 272, 20213–20221 [DOI] [PubMed] [Google Scholar]
- 22.Kyte J., and Doolittle R. (1982) A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105–132 [DOI] [PubMed] [Google Scholar]
- 23.Juretić D., Zoranić L., and Zucić D. (2002) Basic charge clusters and predictions of membrane protein topology. J. Chem. Inf. Comput. Sci. 42, 620–632 [DOI] [PubMed] [Google Scholar]
- 24.Krogh A., Larsson B., von Heijne G., and Sonnhammer E. L. (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580 [DOI] [PubMed] [Google Scholar]
- 25.Jones D. T. (2007) Improving the accuracy of transmembrane protein topology prediction using evolutionary information. Bioinformatics 23, 538–544 [DOI] [PubMed] [Google Scholar]
- 26.Nugent T., and Jones D. T. (2012) Detecting pore-lining regions in transmembrane protein sequences. BMC Bioinformatics 13, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hessa T., Meindl-Beinker N. M., Bernsel A., Kim H., Sato Y., Lerch-Bader M., Nilsson I., White S. H., and von Heijne G. (2007) Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature 450, 1026–1030 [DOI] [PubMed] [Google Scholar]
- 28.Tsirigos K. D., Peters C., Shu N., Käll L., and Elofsson A. (2015) The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res. 43, W401–W407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pierleoni A., Indio V., Savojardo C., Fariselli P., Martelli P. L., and Casadio R. (2011) MemPype: a pipeline for the annotation of eukaryotic membrane proteins. Nucleic Acids Res. 39, W375–W380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bologna G., Yvon C., Duvaud S., and Veuthey A. L. (2004) N-terminal myristoylation predictions by ensembles of neural networks. Proteomics 4, 1626–1632 [DOI] [PubMed] [Google Scholar]
- 31.Maurer-Stroh S., Eisenhaber B., and Eisenhaber F. (2002) N-terminal N-myristoylation of proteins: prediction of substrate proteins from amino acid sequence. J. Mol. Biol. 317, 541–557 [DOI] [PubMed] [Google Scholar]
- 32.Eisenhaber B., Bork P., and Eisenhaber F. (1998) Sequence properties of GPI-anchored proteins near the ω-site: constraints for the polypeptide binding site of the putative transamidase. Protein Eng. 11, 1155–1161 [DOI] [PubMed] [Google Scholar]
- 33.Maurer-Stroh S., and Eisenhaber F. (2005) Refinement and prediction of protein prenylation motifs. Genome Biol. 6, R55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ren J., Wen L., Gao X., Jin C., Xue Y., and Yao X. (2008) CSS-Palm 2.0: an updated software for palmitoylation sites prediction. Protein Eng. Des. Sel. 21, 639–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones D. T. (1999) Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292, 195–202 [DOI] [PubMed] [Google Scholar]
- 36.Fujiki Y., Hubbard A. L., Fowler S., and Lazarow P. B. (1982) Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J. Cell Biol. 93, 97–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martelli P. L., Fariselli P., and Casadio R. (2003) An ENSEMBLE machine learning approach for the prediction of all-α membrane proteins. Bioinformatics 19, Suppl. 1, i205–i211 [DOI] [PubMed] [Google Scholar]
- 38.Viklund H., and Elofsson A. (2008) OCTOPUS: improving topology prediction by two-track ANN-based preference scores and an extended topological grammar. Bioinformatics 24, 1662–1668 [DOI] [PubMed] [Google Scholar]
- 39.Bernsel A., Viklund H., Falk J., Lindahl E., von Heijne G., and Elofsson A. (2008) Prediction of membrane-protein topology from first principles. Proc. Natl. Acad. Sci. U.S.A. 105, 7177–7181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aronson D. E., Costantini L. M., and Snapp E. L. (2011) Superfolder GFP is fluorescent in oxidising environments when targeted via the sec translocon. Traffic 12, 543–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lenaz G. (1987) Lipid fluidity and membrane protein dynamics. Biosci. Rep. 7, 823–837 [DOI] [PubMed] [Google Scholar]
- 42.Gardner R., Cronin S., Leader B., Rine J., Hampton R., and Leder B. (1998) Sequence determinants for regulated degradation of yeast 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol. Biol. Cell 9, 2611–2626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gekko K., and Timasheff S. (1981) Mechanism of protein stabilization by glycerol: preferential hydration in glycerol-water mixtures? Biochemistry 20, 4667–4676 [DOI] [PubMed] [Google Scholar]
- 44.Vagenende V., Yap M. G., and Trout B. L. (2009) Mechanisms of protein stabilization and prevention of protein aggregation by glycerol. Biochemistry 48, 11084–11096 [DOI] [PubMed] [Google Scholar]
- 45.Sakakibara J., Watanabe R., Kanai Y., and Ono T. (1995) Molecular cloning and expression of rat squalene epoxidase. J. Biol. Chem. 270, 17–20 [DOI] [PubMed] [Google Scholar]
- 46.Viklund H., Granseth E., and Elofsson A. (2006) Structural classification and prediction of reentrant regions in α-helical transmembrane proteins: application to complete genomes. J. Mol. Biol. 361, 591–603 [DOI] [PubMed] [Google Scholar]
- 47.Yan C., and Luo J. (2010) An analysis of reentrant loops. Protein J. 29, 350–354 [DOI] [PubMed] [Google Scholar]
- 48.Park S.-E., Kim J.-M., Seok O.-H., Cho H., Wadas B., Kim S.-Y., Varshavsky A., and Hwang C.-S. (2015) Control of mammalian G protein signaling by N-terminal acetylation and the N-end rule pathway. Science 347, 1249–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Starheim K. K., Gevaert K., and Arnesen T. (2012) Protein N-terminal acetyltransferases: when the start matters. Trends Biochem. Sci. 37, 152–161 [DOI] [PubMed] [Google Scholar]
- 50.Fajerman I., Schwartz A. L., and Ciechanover A. (2004) Degradation of the Id2 developmental regulator: targeting via N-terminal ubiquitination. Biochem. Biophys. Res. Commun. 314, 505–512 [DOI] [PubMed] [Google Scholar]
- 51.Cadwell K., and Coscoy L. (2005) Ubiquitination on nonlysine residues by a viral E3 ubiquitin ligase. Science 309, 127–130 [DOI] [PubMed] [Google Scholar]
- 52.Herr R. A., Harris J., Fang S., Wang X., and Hansen T. H. (2009) Role of the RING-CH domain of viral ligase mK3 in ubiquitination of non-lysine and lysine MHC I residues. Traffic 10, 1301–1317 [DOI] [PubMed] [Google Scholar]
- 53.Breitschopf K., Bengal E., Ziv T., Admon A., and Ciechanover A. (1998) A novel site for ubiquitination: the N-terminal residue, and not internal lysines of MyoD, is essential for conjugation and degradation of the protein. EMBO J. 17, 5964–5973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pickart C. M. (2001) Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70, 503–533 [DOI] [PubMed] [Google Scholar]
- 55.Scaglione K. M., Basrur V., Ashraf N. S., Konen J. R., Elenitoba-Johnson K. S., Todi S. V., and Paulson H. L. (2013) The ubiquitin-conjugating enzyme (E2) ube2w ubiquitinates the N terminus of substrates. J. Biol. Chem. 288, 18784–18788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vittal V., Shi L., Wenzel D. M., Scaglione K. M., Duncan E. D., Basrur V., Elenitoba-Johnson K. S., Baker D., Paulson H. L., Brzovic P. S., and Klevit R. E. (2015) Intrinsic disorder drives N-terminal ubiquitination by Ube2w. Nat. Chem. Biol. 11, 83–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bernsel A., Viklund H., Hennerdal A., and Elofsson A. (2009) TOPCONS: consensus prediction of membrane protein topology. Nucleic Acids Res. 37, W465–W468 [DOI] [PMC free article] [PubMed] [Google Scholar]