

Background: Osteogenic differentiation is initiated by transcriptional and post-transcriptional epigenetic mechanisms.
Results: Inhibition of H3K27 methyltransferase EZH2 enhances osteogenic commitment of human mesenchymal progenitors, and its depletion in mouse mesenchymal cells causes multiple skeletal abnormalities.
Conclusion: EZH2 is required for skeletal patterning and bone formation.
Significance: EZH2-dependent epigenetic mechanisms control osteogenesis both in vitro and in vivo.
Keywords: adipogenesis, bone, epigenetics, histone methylation, mesenchymal stem cells (MSCs), osteoblast, Ezh2, osteogenesis
Abstract
Epigenetic control of gene expression is critical for normal fetal development. However, chromatin-related mechanisms that activate bone-specific programs during osteogenesis have remained underexplored. Therefore, we investigated the expression profiles of a large cohort of epigenetic regulators (>300) during osteogenic differentiation of human mesenchymal cells derived from the stromal vascular fraction of adipose tissue (AMSCs). Molecular analyses establish that the polycomb group protein EZH2 (enhancer of zeste homolog 2) is down-regulated during osteoblastic differentiation of AMSCs. Chemical inhibitor and siRNA knockdown studies show that EZH2, a histone methyltransferase that catalyzes trimethylation of histone 3 lysine 27 (H3K27me3), suppresses osteogenic differentiation. Blocking EZH2 activity promotes osteoblast differentiation and suppresses adipogenic differentiation of AMSCs. High throughput RNA sequence (mRNASeq) analysis reveals that EZH2 inhibition stimulates cell cycle inhibitory proteins and enhances the production of extracellular matrix proteins. Conditional genetic loss of Ezh2 in uncommitted mesenchymal cells (Prrx1-Cre) results in multiple defects in skeletal patterning and bone formation, including shortened forelimbs, craniosynostosis, and clinodactyly. Histological analysis and mRNASeq profiling suggest that these effects are attributable to growth plate abnormalities and premature cranial suture closure because of precocious maturation of osteoblasts. We conclude that the epigenetic activity of EZH2 is required for skeletal patterning and development, but EZH2 expression declines during terminal osteoblast differentiation and matrix production.
Introduction
The skeleton, which is critical for several processes including locomotion and protection of vital organs, is generated by endochondral bone formation and intramembranous ossification (1). Osteoblasts, the bone-building cells, originate from immature mesenchymal cells during skeletal development (2). Phenotypic commitment of these progenitor cells is initiated and sustained by both transcriptional and post-transcriptional epigenetic mechanisms (3, 4). Bone production is controlled by gene expression programs in osteoblasts that mediate deposition of collagens and noncollagenous proteins into an extracellular matrix (ECM)3 that ossifies through nucleation of minerals (e.g. hydroxyapatite) (5). The adult skeleton is constantly remodeled to accommodate physiological demands and restore bone fractures. Alterations in normal skeletal development or maintenance can impact health and reduce quality of life.
Osteoporosis is a skeletal disease characterized by a loss of bone mineral density. It has a major impact on public health and economy because of increased fracture risk (6–8). Bone loss reflects an imbalance between bone formation and its resorption during remodeling. Currently, both anti-resorptive and pro-anabolic drug classes have been used or are being considered for the prevention and treatment of osteoporosis (9).
There is a clinical need for new bone anabolic agents. The current treatment options target signaling pathways (teriparatide, denosumab, and selective estrogen receptor modulators) and/or induce apoptosis of osteoclasts by creating a drug-rich environment (bisphosphonates) (10–18). A greater understanding of mechanisms that control bone formation is critical for the development of novel approaches to prevent or reverse osteoporosis. In addition, new anabolic strategies could also be applied to treat nonhealing fractures (19). One attractive strategy is to target epigenetic mechanisms that comprise heritable changes in gene expression that are not linked to modifications in DNA sequence. This nongenetic cellular memory, which is sensitive to environmental cues, controls cellular development and maintains phenotypes in multicellular organisms by modulating gene expression through post-translational modifications of histones, genomic DNA methylation (e.g. CpG dinucleotides), and small noncoding RNAs (e.g. miRNAs) (20, 21).
Polycomb group proteins, which assemble into polycomb-repressive complex 1 and 2 (PRC1 and PRC2), are conserved genes that suppress gene expression to ensure proper embryonic development, stem cell maintenance, and differentiation (22). PRC1 hinders the ATP activity of SWI-SNF complex at the nucleosome and induces chromatin compaction (23). The PRC2 complex possesses histone methyltransferase activity and is responsible for the methylation of histone 3 at lysine 27 (H3K27) (22). EZH2, the enzymatic subunit of PRC2, catalyzes mono-, di-, and trimethylation of H3K27; reduces chromatin accessibility; and promotes gene silencing (24–27). An alternative PRC2 complex that contains EZH1 (enhancer of zeste homolog 1) exists. The PRC1-EZH1 complex has low histone methylation activity and is believed to compact the chromatin through a different mechanism (22). Thus, PRC1 and PRC2 complexes are critical in the establishment of compacted chromatin and together suppress gene expression.
Next generation proteomic and genomic technologies have enabled the genomic mapping of histones (28), DNA methylation regions (29), as well as chromatin conformations (30). DNA methylation in bone has been demonstrated to control the transcription of key osteogenic genes (31, 32). Also, critical osteogenic transcription factors have been shown to interact with histone modifying enzymes (33–35). Furthermore, studies have shown that alterations in the epigenetic code can be harnessed to enhance osteogenic differentiation. Early studies demonstrated that WDR5, an epigenetic protein involved in histone methylation, accelerates osteoblast differentiation (36, 37). More recently, studies have demonstrated that inhibition of histone deacetylases can enhance osteogenic differentiation of mesenchymal stem cells, primary osteoblasts, MC3T3 osteoblasts, and ex vivo calvarial cultures (38–41). Encouraged by these earlier results, we have initiated studies that examine epigenetic mechanisms controlling osteoblast differentiation.
An advantage of epigenetic regulators is that they have enzymatically active sites and/or well defined interaction domains that are amenable to drug inhibition. In this study, we therefore performed an expression screen using a large cohort of epigenetic regulators (>300) to define candidate drug-sensitive molecular targets that control osteogenic differentiation of human AMSCs. We show that EZH2 is down-regulated during osteogenic commitment of mesenchymal progenitors and that inhibition of this epigenetic regulator promotes osteogenic differentiation of AMSCs. Furthermore, we find that loss of functional Ezh2 in the mesenchymal lineage results in skeletal patterning and bone defects in mice.
Experimental Procedures
Cell Culture
AMSCs were derived from lipo-aspirates obtained from consenting healthy donors with approval from the Mayo Clinic Institutional Review Board as previously described (42, 43). Fat tissue was enzymatically digested using 0.075% type I collagenase (Worthington Biochemicals) for 1.5 h at 37 °C. Adipocytes were separated from the stromal vascular fraction by low speed centrifugation (400 × g for 5 min). The adipose supernatant was removed, and the cell pellet was rinsed with PBS and passed through 70- and 40-μm cell strainers (BD Biosciences). The resulting AMSC cell fraction was maintained in Advanced minimum essential media containing 5% PLTMax (a clinical grade commercial platelet lysate product [MillCreekLifeSciences]), 2 mm GlutaMAX (Invitrogen), 2 units/ml heparin (hospital pharmacy), 100 units/ml penicillin, and 100 μg/ml streptomycin (Cellgro) as described previously (42).
mRNA Quantitative Real Time Reverse Transcriptase PCR (RT-qPCR)
RNA was isolated using the miRNeasy kit (Qiagen). Isolated RNA was reverse transcribed into cDNA using the SuperScript III first strand synthesis system (Invitrogen). Gene expression was quantified using real time PCR whereby each reaction was performed with 10 ng of cDNA per 10 μl, QuantiTect SYBR Green PCR kit (Qiagen), and the CFX384 real time system machine (Bio-Rad). Transcript levels were quantified using the 2ΔΔCt method and normalized to the housekeeping gene GAPDH/Gapdh (set at 100). Gene specific primers are shown in supplemental Table S1.
MTS Activity Assay
AMSCs were plated in 96-well plates in maintenance medium (5,000 cells/well). The following day, vehicle (DMSO) or EZH2 inhibitor (GSK126) in fresh maintenance medium were added to the cells. Three days later, MTS activity assay was performed according to manufacturer's protocol (Promega). Absorbance was measured at 490 nm using a SpectraMAX Plus (Molecular Devices) spectrophotometer.
Western Blotting
AMSCs (4,000 cells/cm2) were plated in 6-well plates in maintenance medium. Cells were treated with vehicle or EZH2 inhibitors (GSK126) as described. Cells were lysed in radioimmunoprecipitation buffer (150 mm NaCl, 50 mm Tris, pH 7.4, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 1% Triton X-100) supplemented with protease inhibitor mixture (Sigma) and phenylmethylsulfonyl fluoride (Sigma). Lysates were cleared by centrifugation. Protein concentrations were determined by the DC Protein Assay (Bio-Rad). Proteins were resolved by SDS-PAGE and transferred to polyvinylidene difluoride membranes. After blocking in 5% nonfat dry milk for 45 min at room temperature, primary antibodies were added overnight at 4 °C, followed by secondary antibodies for 1 h at room temperature. Proteins were visualized using an ECL Prime detection kit. Primary antibodies used were: Actin (1:10,000; sc-1616; Santa Cruz), H3 (1:10,000; 05-928; Millipore), H3K27me3 (1:5,000; 17-622; Millipore), and EZH2 (1:10,000; 5246; Cell Signaling).
Osteogenic Differentiation
AMSCs were plated in 6-well plates in maintenance medium (4,000 cells/cm2). The following day, maintenance medium was replaced with osteogenic medium (maintenance medium supplemented with 50 μg/ml ascorbic acid, 10 mm β-glycerol phosphate, and 10 nm dexamethasone) containing vehicle (DMSO) or 2 μm GSK126. Three days later, GSK126 and vehicle were removed, and fresh osteogenic medium added. The media were changed every 3 days. RNA was isolated at indicated times. For part of this study, 50 ng/ml of BMP2 (R&D Systems) was added to the osteogenic mixture as described in the figure. On day 6, cells were fixed in 10% neutral buffered formalin and stained with 5-bromo-4-chloro-3-indolyl-phosphate/nitro blue tetrazolium to monitor the enzymatic activity of alkaline phosphatase (Promega). On day 14, cells were fixed in 10% neutral buffered formalin and stained with 2% alizarin red to visualize calcium deposition.
Adipogenic Differentiation
AMSCs were plated in 6-well plates in maintenance medium (4,000 cells/cm2). The following day, vehicle (DMSO) or 2 μm GSK126 were added to cells in maintenance medium. Three days later, GSK126 and vehicle were removed, and adipogenesis was induced (adipogenic supplement (R&D Systems) in maintenance medium). The media were changed every 3 days. RNA was isolated at indicated times. On day 14, cells were fixed in 10% neutral buffered formalin and stained with Oil Red O (Sigma), which binds to lipids and neutral triglycerides. Oil Red O stain was dissolved in isopropyl alcohol and optical density measured at 490 nm using a spectrophotometer.
Ezh2 Knockdown and Osteogenic Differentiation
MC3T3 cells were seeded in 6- or 12-well plates in maintenance medium (10,000 cells/cm2). The following day, siRNA transfections with control (D-001810-10-20; GE Healthcare) and mouse Ezh2 (L-040882-00; GE Healthcare) ON-TARGETplus siRNA smartpools were performed using RNAiMAX as instructed by manufacturer (Invitrogen). The next day, MC3T3 osteogenic medium was added, and were cells cultured as described.
Animal Welfare
All animal studies were conducted according to guidelines provided by the National Institutes of Health and the Institute of Laboratory Animal Resources, National Research Council. The Mayo Clinic Institutional Animal Care and Use Committee approved all animal studies. Animals were housed in an accredited facility under a 12-h light/dark cycle and provided water and food (PicoLab Rodent Diet 20, LabDiet) ad libitum.
Deletion of Function Ezh2 in Mesenchymal Lineage
Mice containing a conditional Ezh2fl/fl allele (44) and harboring two loxP sites flanking the SET domain were obtained from the Mutant Mouse Regional Resource Center (B6;129P2-Ezh2tm1Tara/Mmnc; University of North Carolina, Chapel Hill, NC). Ezh2 function was conditionally ablated in uncommitted mesenchymal cells by mating with mice expressing Cre recombinase from the Prrx1 enhancer (45). These crosses generated WT (Ezh2wt/wt: Prrx1-Cre+) and conditional knock-out (cKO, Ezh2fl/fl: Prrx1-Cre+) animals. The mice are on the C57Bl/6 genetic background. The following primers were used for genotyping: Ezh2 (forward, TGTCATGTCTGGGTCTAATGCTAC, and reverse, GGAACCTCGCTATGTGTAACCA) and Cre (forward, TCCAATTTACTGACCGTACACCAA, and reverse, CCTGATCCTGGCAATTTCGGCTA).
Whole Mount Staining
Skeletons from 1-day-old female mouse pups were dissected and fixed overnight in ethanol. Cartilage was stained with a 0.2% Alcian blue dye (dissolved in 80% ethanol and 20% glacial acetic acid) for 24 h. Skeletons were washed twice with 95% ethanol and then placed in 2% KOH until the remaining soft tissues were dissolved. Bones were stained with 75 μg/ml alizarin red (Sigma) in 1% KOH overnight and destained (20% glycerol, 1% KOH) for 2 weeks, with daily solution changes. Skeletons were transferred to a 20% glycerol, 20% ethanol solution overnight and then stored in a 50% glycerol, 50% ethanol solution. Images of tissues were obtained using a Wild M420 macroscope (Wild Heerbrugg) and ProGres C3 camera (Jenoptik).
Histological Assessments
Tibias from 1-day-old male or 3-week-old female WT or Ezh2 cKO mice were fixed in 10% neutral buffered formalin, decalcified in 15% EDTA for 7 days, paraffin-embedded, sectioned, and stained with Alcian blue (1% Alcian blue, 3% acetic acid) and eosin. The distance from the epiphysis to the hypertrophic zone in 1-day-old mice was assessed by taking the average of four measurements from each mouse using ImageJ software. The proliferative area of these mice was also assessed. Total, proliferative, and hypertrophic growth plate depths from 3-week-old animals were determined by taking the average of 15 measurements across the imaged growth plate from each mouse using ImageJ software. Images from the midshaft marrow cavity were also collected.
Microcomputed Tomography Analysis
Bone architecture of 3-week-old mice was evaluated in the proximal tibia and skull using ex vivo microcomputed tomography (microCT). Bones were scanned in 70% ethanol on a μCT35 scanner (Scanco Medical AG, Basserdorf, Switzerland) at 7-μm voxel size (tibias) or 20-μm voxel size (skulls) using an energy setting of 70 kVp and an integration time of 300 ms. For the proximal tibia scans, a region of interest spanning from 17 to 20% of total bone length (relative to the proximal epiphysis) was analyzed in each mouse (threshold = 220). Bone parameters as presented in the figures were computed using the manufacturer's software (46). Skulls were reconstructed using a threshold value of 125 for gross observation of calvarial and suture morphology.
High Throughput RNA Sequencing and Bioinformatic Analysis
mRNASeq was performed on the following samples: 1) RNA from confluent AMSCs, 2) RNA from AMSCs grown on plastic or titanium disks during osteogenic differentiation, 3) RNA from AMSCs treated with vehicle or 2 μm GSK126 in osteogenic mixture for 3 days, 4) RNA from 3-day-old female WT or Ezh2 cKO (pooled RNA from three WT and two cKO animals), and 5) RNA from AMSCs in adipogenic mixture for 6 days. High throughput mRNA sequencing and bioinformatic analyses (mRNASeq) were performed as previously reported (47). Gene expression is expressed in reads per kilobase pair per million mapped reads). The mRNASeq is available under accession number GSE73075 at the Gene Expression Omnibus.
Statistics
Statistical analysis was performed with unpaired Student's t test. Significance is noted in the figures, when applicable (*, p < 0.05; **, p < 0.01; and ***, p < 0.001).
Results
Identification of EZH2 as an Epigenetic Regulator of Mesenchymal Cell Differentiation
To understand which regulatory proteins generate, interpret, and edit the epigenetic landscape during bone formation, we performed a mRNA expression screen using RT-qPCR for more than 300 histone code writers, readers, and erasers (48) during osteogenic differentiation in immature mesenchymal cells (Fig. 1a). Studies on osteogenic differentiation of mesenchymal cells typically use cells derived from bone marrow. However, these cells are difficult to harvest from human patients (i.e. iliac crest aspiration) and are isolated with low yields. Our studies use AMSCs (47, 49) that circumvent these limitations of bone marrow mesenchymal stem cells. We have demonstrated that clinical grade AMSCs, which are currently being used in multiple patient trials at our institution, have the capacity for trilineage differentiation (49). Hence, the AMSCs used in this work have potential in bone tissue engineering strategies and are a valid model for examining epigenetic mechanisms of osteogenic differentiation.
FIGURE 1.

Identification of EZH2 as an inhibitor of osteogenic differentiation. a, epigenetic regulators exhibiting modulations in mRNA levels during osteogenic differentiation of clinical grade AMSCs were identified by expression screening using a semiautomated RT-qPCR platform with >300 primer pairs. Sorted expression ratios for 322 mRNAs for epigenetic proteins were determined using differentiating cells for 11 days (D11) versus undifferentiated cells (D0). Although >200 mRNAs are elevated, EZH2 is 1 of 14 genes that are suppressed by more than 2-fold. b, expression pattern of 12 down-regulated epigenetic regulators with robust expression (i.e. 0.1 arbitrary units where GAPDH = 100 units) and eight other representative genes during osteogenic differentiation of AMSCs. c and d, RT-qPCR (c) and Western blot analyses (d) show down-regulation of, respectively, EZH2 mRNA and protein at different days of osteogenic differentiation. e and f, levels of EZH2 and other representative mRNAs (ALPL, OPG, SOX9, RUNX2, and JHDM1D) were validated by mRNASeq from AMSCs undergoing an osteogenic time course using standard osteogenic medium on plastic cell culture vessels (e) or porous sintered titanium inserts (f). Graphs show expression values (reads per kilobase pair per million mapped reads (RKPM)) at different days of culture (D-1 is 1 day prior to osteogenic induction at D0).
The epigenetic expression screen compared triplicate AMSC cultures prior to administration of an osteogenic differentiation mixture (D0) and 11 days after osteogenic induction (D11). The values were expressed as the fold change between these 2 days. The results reveal that many epigenetic proteins (n = 201) are up-regulated, but only 14 factors are down-regulated by at least 2-fold during osteogenic differentiation (Fig. 1a). Of the 14 down-regulated genes, 12 are robustly expressed by RT-qPCR and are persistently suppressed during the differentiation time course of AMSCs (Fig. 1b). One prominently down-regulated mRNA encodes the epigenetic regulator EZH2 (Fig. 1c). Reduction in EZH2 mRNA expression correlates with reduced EZH2 protein levels, as was confirmed by immunoblotting (Fig. 1d). Changes in EZH2 expression were validated by next generation RNA sequencing (mRNASeq) performed on RNA samples of AMSCs during osteogenic differentiation on two different substrates: standard tissue culture plastic (Fig. 1e) and porous sintered titanium discs (Fig. 1f) as a model for differentiation on orthopedic implants. Osteogenic differentiation on both substrates is evident by increased expression of established osteoblastic markers (e.g. ALPL, OPG/TNFRSF11B, and RUNX2) and reduced levels of the chondrogenic markers (e.g. SOX9) (Fig. 1, e and f). As EZH2 mRNA levels decrease during osteogenic differentiation, the H3K27 demethylase JHDM1D maintains robust expression (Fig. 1, e and f). Because EZH2 is an epigenetic enzyme with focused activity that modifies a single histone residue (H3K27), we considered it a promising target for the development of novel bone anabolic strategies.
EZH2 Inhibition Enhances Osteogenic Differentiation of AMSCs
We investigated whether enzymatic inhibition of EZH2, and therefore suppression of H3K27 trimethylation (H3K27me3) may promote lineage commitment of AMSCs. GSK126, a specific EZH2 inhibitor, exhibits overt toxicity only at high concentrations (6 μm and higher) (Fig. 2a), yet this drug effectively attenuates H3K27me3 at lower doses (2 μm) (Fig. 2b). These inhibitory effects of GSK126 (2 μm) are observed as early as 6 h and persist for at least 72 h after drug administration (Fig. 2c). The observation that global levels of H3K27me3 are decreased as early as 6 h upon treatment with GSK126 suggests that one or more of the corresponding demethylases (i.e. JHDM1D, KDM6A, and KDM6B) may be very active; indeed, mRNAs for all three genes are robustly expressed in AMSC cultures.4 Because GSK126 effectively attenuates H3K27me3 formation, we conclude that EZH2 is a principal mediator of global H3K27me3 levels in AMSCs, consistent with current models for EZH2 function (50).
FIGURE 2.

EZH2 inhibition enhances osteogenic differentiation of AMSCs. a, toxicity profile of GSK126 based on MTS activity in AMSCs for 3 days (n = 3). b and c, dose-dependent (b) and time-dependent (c) modulation of H3K27me3 by GSK126 in AMSCs. Cells were treated with different concentrations of GSK126 1 day after plating and harvested 3 days later for Western blotting (b) or treated with 2 μm GSK126 and protein lysates collected at the specified times (c). d, diagram of the experimental protocol for treatment of AMSCs with GSK126 (2 μm) shown in panels e–h. V, vehicle; G, GSK126. e–g, alkaline phosphatase staining (e) and quantitation (f) or alizarin-red staining (g) for AMSCs treated with vehicle or GSK126. h, mRNA analysis by RT-qPCR of selected bone-related markers and ACTB as indicated by the respective gene symbols (n = 3).
To assess the effects of EZH2 inhibition on lineage commitment of progenitors, GSK126 was added to the cultures for the first 3 days of osteogenic differentiation of AMSCs (Fig. 2d). This regimen was selected because EZH2 is robustly expressed in uncommitted AMSCs and is down-regulated within 1 week of osteogenic differentiation. We reasoned therefore that short term administration of GSK126 would accelerate the natural biological loss of EZH2, while generating epigenetic alterations that may accelerate osteogenic differentiation and perhaps persist for the duration of the culturing period. The results show that EZH2 inhibition enhances osteogenic AMSC differentiation as evidenced by increased staining for alkaline phosphatase (Fig. 2, e and f) and alizarin red (Fig. 2g). We note that alizarin red staining under our control (vehicle) conditions typically is not robust until after 21 days of differentiation. Hence, GSK126 accelerates differentiation such that strong alizarin red staining already is observed well before it becomes apparent in the control cultures. At day 6 of osteogenic differentiation, EZH2 inhibition enhanced the expression of ALPL, IBSP, OPG, and RUNX2, which are key osteogenic markers (Fig. 2h). Reflecting the selectivity of the changes in osteoblast-related gene expression, EZH2 inhibition has no effect on COL1A1 and ATF4, whereas it suppresses the expression of the fibroblast marker TWIST2.
To ensure the specificity of the biological effects of GSK126 as an inhibitor of EZH2, we treated AMSCs with a siRNA for EZH2 (Fig. 3a); the siRNA we used is a “smart pool siRNA” with modifications that reduce off-targets (GE Healthcare). Two days after treatment, we observed specific knockdown of EZH2 mRNA (Fig. 3b) and protein (Fig. 3c). Similar to enzymatic inhibition, the knockdown of EZH2 enhances the expression of several osteogenic markers including RUNX2, ALPL, OPG, and SATB2 after 1 week of osteogenic differentiation (Fig. 3d). Together, our results indicate that EZH2 inhibition, and therefore suppression of H3K27me3, promotes osteogenic lineage-commitment of AMSCs.
FIGURE 3.

Down-regulation of EZH2 promotes osteogenic differentiation of AMSCs. a, diagram of the experimental protocol for treatment of AMSCs with siRNA for EZH2. Cells were plated (D-1) and transfected (Tf) on the next day (D0, ∼50% confluent), followed by cell harvest for protein and RNA analysis on the indicated days. b and c, RT-qPCR analysis of EZH2 mRNA (b) and Western blotting of EZH2 protein relative to β-actin (c) 2 days after transfection. d, RT-qPCR analysis of selected bone-related markers for AMSCs treated with control nonsilencing RNA or EZH2 siRNA on indicated days after transfection.
EZH2 Inhibition Suppresses Adipogenic Differentiation of AMSCs
To determine the effects of EZH2 inhibition on the differentiation of human AMSCs into adipocytes grown in platelet lysate, cells were treated with 2 μm GSK126 and vehicle for the first 3 days of adipogenic differentiation (Fig. 4a). On day 12 of adipogenic differentiation, EZH2 inhibition suppresses the expression of PPARγ, FABP4, and ADIPOQ, which are representative adipogenic genes (Fig. 4b). EZH2 inhibition significantly reduces Oil Red O staining after 14 days of adipogenic differentiation (Fig. 4, c and d). Thus, our data indicate that EZH2 inhibition suppresses adipogenic differentiation of AMSCs.
FIGURE 4.

EZH2 inhibition suppresses adipogenic differentiation of AMSCs. a, diagram of the experimental protocol for treatment and adipogenic differentiation of AMSCs with GSK126 (2 μm) shown in panels b–d. V, vehicle; G, 2 μm GSK126. b, RT-qPCR analysis of representative adipogenic genes at day 9 of differentiation (n = 3). c and d, representative images of Oil Red O staining (10× magnification on right) (c) and quantification (n = 3) (d) after 14 days of adipogenic differentiation.
EZH2 Inhibition Induces Expression of Bone ECM Proteins and Reduces Proliferation-related Gene Expression in AMSCs
Analysis of AMSCs treated with the EZH2 inhibitor GSK126 by mRNASeq was performed to identify molecular pathways altered by EZH2 inhibition. Gene ontology analysis demonstrates that EZH2 inactivation primarily stimulates expression of genes encoding ECM proteins (Fig. 5a), while suppressing genes involved in cell cycle progression and microtubule-processing (Fig. 5b). Hence, EZH2 inhibition of AMSCs reinforces a nonproliferative and ECM-anabolic biological state. Furthermore, EZH2 inhibition induces many genes encoding transcriptional regulators (e.g. EGR1, HEY1, MSX2, and NKX3-2) involved in osteogenic cell fate commitment and lineage progression (Fig. 5, c and d), potentially clarifying why EZH2 inhibition enhances osteogenic differentiation and inhibits adipogenic lineage commitment of AMSCs. Therefore, AMSC lineage commitment is affected in part by direct and indirect alterations of EZH2-dependent gene regulatory programs.
FIGURE 5.

Changes in AMSC gene expression after EZH2 inhibitor treatment. a and b, functional gene annotation clusters (Top 20) for genes up-regulated (a) or down-regulated (b) >1.4-fold change in AMSCs 3 days after treatment with 2 μm GSK126 during osteogenic differentiation. Bar graphs in a show preferential up-regulation of genes promoting a cellular anabolic state (e.g. ECM genes), whereas b shows inhibition of genes required for mitosis and cell cycle progression. c, unsupervised hierarchical clustering of >900 transcriptional regulators in AMSCs during adipogenic, osteogenic, and undifferentiated states, showing substantial changes in transcriptional profiles. EZH2 inhibitor treatment markedly changes the expression of transcriptional regulators in AMSCs, providing a possible mechanism by which EZH2 inhibition enhances osteogenesis. d, GSK126 (2 μm) responsiveness of mRNAs for representative transcription factors in osteogenic medium (n = 3).
Mesenchymal Deletion of Functional Ezh2 Is Associated with Skeletal Malformation in Mice
Because the epigenetic function of EZH2 attenuates osteogenic differentiation of uncommitted mesenchymal stem cells in culture, we addressed whether loss of EZH2 would have consequences for skeletal development and mesenchymal cell differentiation in vivo. Mice with a constitutive Ezh2 null mutation perish during early embryonic development (24). Therefore we combined a conditional knock-out mouse model containing a conditional Ezh2fl/fl allele (44) with a Cre transgene under control of the Prrx1 enhancer (45). In the resulting Ezh2 cKO mice, Ezh2 function is ablated in uncommitted Prrx1-expressing mesenchymal cells. Ezh2 cKO mice are smaller than WT mice 1 day after birth (Fig. 6a), and the size difference is further enhanced at 3 weeks of age (Fig. 6c). In addition to a smaller overall body size, Ezh2 cKO mice are characterized by shorter limbs, especially the front legs as demonstrated by whole mount staining 1 day after birth (Fig. 6b).The smaller body size of cKO mice is exemplified by the total body weight (Fig. 6d), the size of front limbs (Fig. 6e), spine (Fig. 6f), and femurs (Fig. 6g); for example, the height of the lumbar vertebrae show a 27% reduction in height. The Ezh2 cKO animals exhibit clinodactyly, which is apparent on radiographs showing abnormal bending of the digits (Fig. 6, e and h). These animals also exhibit a domed head that is apparent through photography (Fig. 6a) and whole mount staining (Fig. 6b) 1 day after birth and x-ray analysis in 3 week-old mice (Fig. 6h). These mutant mice have clavicles, albeit much shorter than in WT animals (Fig. 6i). Hence, loss of Ezh2 in mesenchymal progenitors disrupts intramembranous bone formation as demonstrated by abnormalities in clavicle and cranial bone development.
FIGURE 6.
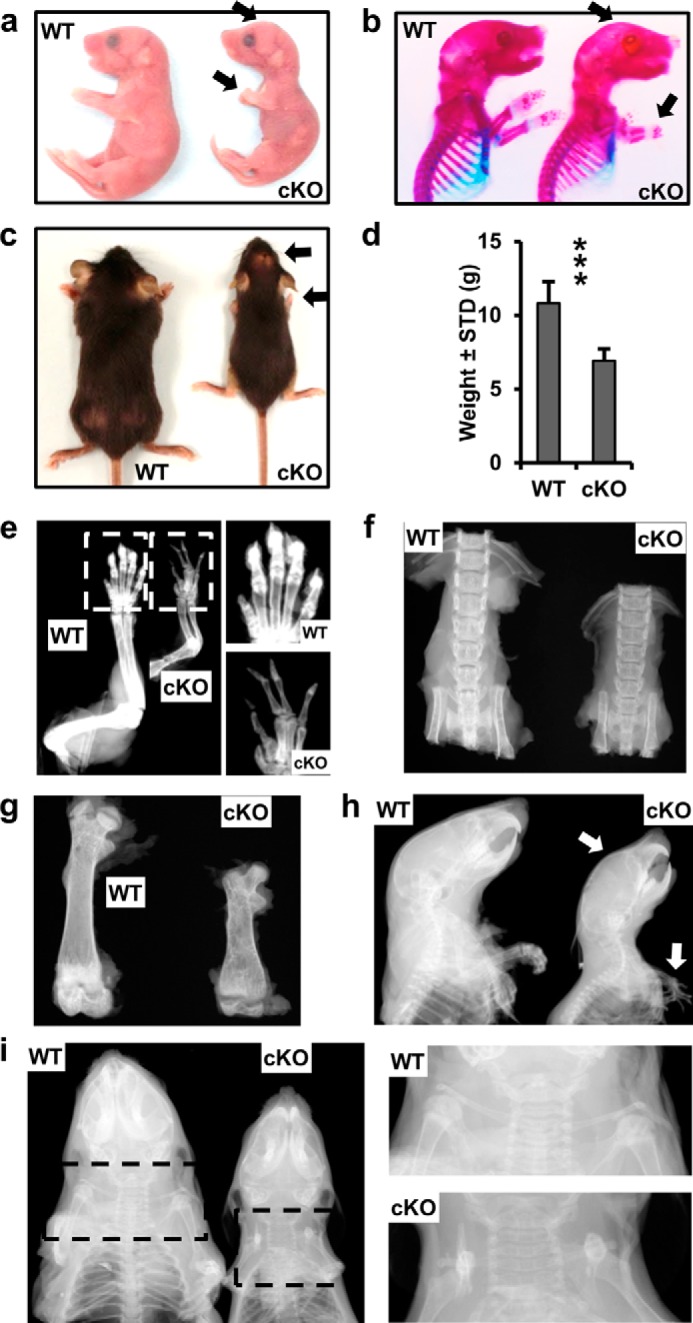
Skeletal defects upon conditional ablation of functional Ezh2 in mesenchymal precursor cells. a and b, images (a) and whole mount staining (b) reveal a short stature, as well as a domed head and shortened limbs (arrows) in newborn Ezh2 cKO mice. c–i, phenotype of 3-week-old WT and Ezh2 cKO female animals. c and d, representative picture (c) and weight of WT (n = 14) and cKO (n = 7) (d) animals. e–g, radiographic images of right front limbs (e), vertebrae (f), and right femurs (g); paw enlargements (selection area in e). h and i, x-ray images of the cephalad half of WT and cKO mice; clavicle enlargement (selection area in i). X-ray analysis demonstrates the presence of clavicles in cKO (albeit shorter than in WT). Arrows in the images point to phenotypic changes (short forelimbs, domed head, and clinodactyly) in Ezh2 cKO mice.
Mesenchymal Deletion of Functional Ezh2 Leads to Abnormal Growth Plate Development
Endochondral ossification is the process by which the majority of bones are formed in the skeleton. This is an orchestrated process that includes mesenchymal condensation, chondrogenesis, chondrocyte maturation, chondrocyte hypertrophy, vascularization, and ultimately the recruitment of osteoblast progenitors. The smaller stature and shorter long bones observed in Ezh2 cKO animals encouraged the evaluation of proximal tibial growth plate structure. Histological analysis of 1-day-old and 3-week-old mice revealed ordinary matrix staining and columnar organization of the growth plate in Ezh2 cKO animals (Fig. 7, a and c). Measurements of 1-day-old growth plate show reduced distance from the articular surface to the hypertrophic zone (Fig. 7, a and b), which may be indicative of accelerated hypertrophy or reduced proliferation. No changes in BrdU staining were observed in Ezh2 cKO animals (data not shown), so the reduced distance from epiphysis to hypertrophic zone may result from accelerated hypertrophy. Histologic analysis reveal reduced proliferative area in the 1-day-old Ezh2 cKO pups (Fig. 7b), especially within the periarticular region (Fig. 7a). At 3 weeks of age, significant reduction in growth plate depth is observed in Ezh2 cKO animals (Fig. 7, c and d), which parallels decreased limb length and vertebral height mentioned earlier. In addition, alcian blue staining demonstrates reduced hypertrophic zone depth in 3-week-old Ezh2 cKO animals (Fig. 7d). Finally, histological analysis revealed an increased number of adipocytes in the tibial midshaft from 3-week-old Ezh2 cKO animals (Fig. 7e). These data demonstrate that depletion of functional Ezh2 in the mesenchymal lineage alters growth plate development and enhances adipocyte formation in the bone marrow cavity in mice.
FIGURE 7.

Abnormal growth plate development in Ezh2 cKO mice. a, proximal tibial growth plates of 1-day-old male WT and cKO mice. b, distance from the epythysis to hypertrophy (left panel) and proliferative area (right panel) in 1-day-old male WT (n = 4) and cKO (n = 3) mice. The distance (arrow) and area (yellow tracing) were measured as depicted in a. c, proximal tibial growth plates of 3-week-old female WT and cKO mice. d, growth plate depth (left panel) and hypertrophic depth (right panel) in 3-week-old female WT and cKO mice (n = 4). The growth plate depth (dashed arrow) and hypertrophic depth (solid arrow) were measured as depicted in c (solid arrow). e, representative images of the midshaft marrow cavity from 3-week-old female WT and cKO mice.
Mesenchymal Deletion of Functional Ezh2 Causes Craniosynostosis and Low Trabecular Bone Volume
The doming of the head in Ezh2 cKO animals prompted microCT analysis of skulls from 3-week-old WT and Ezh2 cKO animals. The microCT scans revealed thin and porous calvarial bones in the Ezh2 cKO mice (Fig. 8, a and b). The microCT images demonstrated premature fusion of coronal and frontal sutures and a widening of the lambdoid suture (Fig. 8a). The microCT analysis confirmed the doming of the skull (Fig. 8b) and shortened clavicles (Fig. 8c), as suggested earlier (Fig. 6). The premature suture fusion and corresponding doming of the head suggests that depletion of functional Ezh2 in the mesenchyme leads to craniosynostosis, a human disease hallmarked by premature fusion of the sutures in the skull.
FIGURE 8.

Craniosynostosis and decreased bone volume in Ezh2 cKO mice. a–d, representative microCT reconstructions of skulls (a and b), skull and clavicles (c), and trabecular bone in tibia (d) from 3-week-old female WT and cKO mice. e, structural parameters including tibia length (mm), trabecular bone volume fraction (BV/TV, %), trabecular number (Tb.N, mm−1), trabecular separation (Tb.Sp, mm), connectivity density (ConnD, 1/mm3), and structure model index (SMI) of 3-week-old mice (n = 4).
To assess bone volume, microCT analysis was performed on tibias from 3-week-old WT and Ezh2 cKO mice. The scans revealed a significant reduction in tibial trabecular bone volume fraction in Ezh2 cKO animals (Fig. 8, d and e). Similar to x-ray scans of femurs and vertebrae (Fig. 6), microCT examination showed there is a significant reduction in tibia length in Ezh2 cKO when compared with WT animals (Fig. 8e). Our analysis did not show a difference in trabecular thickness, trabecular spacing, and SMI (a measure that describes the shape of trabeculae) between the two genotypes, but it revealed a significant reduction in connectivity density, a measure of how well trabeculae are connected to each other, in Ezh2 cKO animals (Fig. 8e). These microCT data demonstrate that depletion of functional Ezh2 in the mesenchymal cells decreases the length and trabecular bone volume.
Deletion of Functional Ezh2 Accelerates Expression of Osteogenic Genes in Calvaria
To understand the molecular mechanisms by which Ezh2 loss alters differentiation of cranial osteogenic precursor cells, we performed mRNASeq analysis of cranial bone tissue (calvaria) from cKO and WT mice (Fig. 9). Raw RNA reads were mapped to 23,355 mouse genes and filtered for normalized read counts. We selected a value of reads per kilobase pair per million mapped reads of >0.3 in either sample as an absolute cutoff as previously suggested (25) and defined genes with a fold change of four (“Up”; 1,273 genes) or 0.25-fold (“Down”, 559 genes) in the cKO animals (Fig. 9a). Each set of genes was subjected to gene ontology analysis using DAVID 6.7, and the resulting annotation clusters were sorted by enrichment scores (ES) (Fig. 9, b and c). For the 1273 up-regulated genes, gene ontology analysis revealed 238 potential annotation clusters (Fig. 9b). Up-regulated proteins are most prominently enriched for proteins associated with ribosomal function (ES > 40) or mitochondrial function (ES > 10). These two categories account for approximately one-quarter of all genes up-regulated by 4-fold upon Ezh2 inactivation. We also observed 14 up-regulated DNA binding proteins, 10 of which are transcription factors (TFs) with known functions in development.
FIGURE 9.

Differential expression of osteogenic genes between primary WT and Ezh2 cKO calvaria. To understand changes in calvarial anatomy (craniosynostosis and doming of the skull), we performed mRNASeq analysis to assess gene expression changes caused by inactivation of Ezh2 in calvarial osteoblasts in 3-day-old mice. a–c, up-regulated and down-regulated genes in cKO mice calvaria. a, of 23,355 mouse genes, 1273 genes are up-regulated and 559 genes are down-regulated (criteria: fold change >4, RPKM >0.3 in either genotype). b and c, DAVID 6.7 ES for the top 10 clusters of the 1273 up-regulated genes (b) and 559 down-regulated genes (c). d and e, expression of osteogenic markers (e.g. Bglap and Sparc) (d) and several cyclin-dependent kinase inhibitors (e) is enhanced in the calvaria of cKO mice. f, expression of Hox gene clusters, which are known to be regulated by Ezh2 and H3K27me3, are highly up-regulated in calvaria derived from cKO mice.
Down-regulated genes were categorized by 150 possible annotation clusters (Fig. 9c). The top category is metal ion binding proteins (ES > 6; n = 153), and many of these are known zinc finger proteins (n = 53). The second category encompasses proteins with an EGF-like region (ES > 4). The third category is enriched for secreted proteins (n = 44) and/or proteins with an extracellular region (ES > 3, n = 53). Most of the latter proteins are structural components of the cell surface and/or the ECM, but none of these secreted proteins represent a well established paracrine developmental signaling molecule (e.g. BMP, WNT, or FGF). Furthermore, Ezh2 loss results in down-regulation of a large number of TFs (>70) including at least 17 of the Zn finger proteins discussed above. The number of down-regulated TFs far exceeds the number of TFs (n = 10) that are up-regulated by the loss of Ezh2. Hence, the presence of Ezh2 normally supports the expression of a broad spectrum of TFs that may perform functions in immature osteogenic cells during calvarial development.
Four down-regulated proteins represent histone modifying enzymes, including the H3K36 methyltransferase Ash1l, the H3K9 demethylase Jmjd1c, and the H3K4 methyltransferases Mll3 and Mll5. Their collective loss predicts that Ezh2 inactivation, which results in reduction of the repressive mark H3K27me3, may also results in more of the repressive K9me3 mark and less of the active marks H3K4me3 and H3K36me3. Taken together, Ezh2 loss may enhance both the protein synthesis capacity and mitochondrial metabolic activity, as well as alter gene regulatory pathways controlled by select epigenetic regulators and Zn-finger related transcription factors.
Similar to mRNASeq data for human AMSCs treated with the EZH2 inhibitor GSK126 (Fig. 5), loss of Ezh2 in mouse calvarial cells increases expression of bone-related ECM proteins, including Sparc, Spp1, Ibsp, Bglap, and Alpl (Fig. 9d). The up-regulation of such noncollagenous ECM proteins is expected from accelerated maturation of preosteoblastic progenitors, and these findings corroborate our data obtained with human AMSCs. Other prominently up-regulated non-ECM genes are known Ezh2 target genes, including the senescence-associated cyclin-dependent kinase (CDK) inhibitory protein p16/CDKN2A (26) and the CDK inhibitor p57/CDKN1C (27) (Fig. 9e), as well as homeobox transcription factors (51) (Fig. 9f). Consequently, Ezh2 inactivation in calvarial cells induces a postproliferative state concomitant with increased production of a bone-related mineralizing ECM.
Distinct Biological Effects of BMP2 and EZH2
To assess the osteogenic potential of EZH2 inhibition in the presence of BMP2, a well characterized pro-osteogenic molecule, AMSCs were differentiated in the presence or absence of GSK126 and BMP2 (Fig. 10a). The expression of MKI67, a proliferation marker, and EZH2 is suppressed during the osteogenic differentiation time course (compare D2 to D6) (Fig. 10b). The reduction in EZH2 compliments earlier mRNA and protein analysis of this epigenetic regulator (Fig. 1). BMP2 enhances the expression of ID1 (inhibitor of DNA binding 1) (Fig. 10b), a well characterized target of BMP2 (52). The expression of osteogenic genes (ATF4, ALPL, and OPG) is induced over time (compare D2 with D6), suggesting that AMSCs are differentiating into the osteogenic lineage (Fig. 10c). Corroborating other findings (Fig. 1), EZH2 inhibition enhances the expression of osteogenic markers (e.g. ALPL and OPG) (Fig. 10c). BMP2 significantly enhances the expression of OPG (D6) and does not appear to significantly alter the expression of other osteogenic genes (RUNX2, ATF4, and ALPL). Interestingly, BMP2 blunts the enhanced expression of ALPL by EZH2 inhibition (D6, G versus B+G). These data suggest that EZH2 inhibition and BMP2 appear to independently enhance the expression of some osteogenic genes (e.g. OPG), although these two treatment strategies may co-regulate the expression of other osteogenic genes (e.g. ALPL).
FIGURE 10.

Biological differences upon co-administration of an EZH2 inhibitor and BMP2 during osteogenic differentiation of AMSCs. Diagram of the experimental protocol for treatment and osteogenic differentiation of AMSCs with GSK126 (2 μm) in the presence or absence of BMP2 (50 ng/ml) shown in b and c. a, B, 50 ng/ml BMP2; V, vehicle; G, 2 μm GSK126). b and c, mRNA analysis by RT-qPCR of proliferation (MKI67), EZH2, and BMP2-reponsive gene (ID1) (n = 3) (b) and several representative osteogenic genes (c).
Discussion
In this study, we investigated epigenetic regulators during osteogenic differentiation of mesenchymal stromal cells (i.e. AMSCs) to identify critical regulators of osteoblast differentiation. Expression and genetic analyses revealed that EZH2 is one of the most prominently down-regulated epigenetic enzymes during osteogenic differentiation of mesenchymal stromal cells (i.e. AMSCs) and is functionally required in mesenchymal precursor cells that mediate normal skeletal development and bone formation. Because EZH2 is highly expressed during early stages of osteogenic differentiation, we reasoned that inactivation of EZH2 would enhance the commitment of mesenchymal stromal cells into the osteogenic lineage. Indeed, small molecule inhibition and siRNA knockdown of EZH2 enhances osteogenic differentiation while suppressing adipogenic differentiation of mesenchymal stromal cells. These findings are particularly interesting, because epigenetic drugs are currently used in the treatment of various cancers (53, 54) and other medical conditions including mood disorders and epilepsy (5, 55), Alzheimer's disease (56), pain (57), and autoimmune diseases (58). Our study suggests that inhibition EZH2 could be leveraged to alter the epigenetic landscape of mesenchymal stromal cells to control bone-specific gene expression and osteoblast differentiation for bone anabolic therapeutic applications.
In support of our main biological finding that loss of EZH2 functionally accommodates osteoblast differentiation, several studies have provided important clues for the molecular mechanisms by which EZH2 may be down-regulated. For example, a recent study has demonstrated that suppression of Ezh2 expression is potentially controlled by up-regulation of Runx2 in mature osteoblasts (59). Furthermore, the long noncoding RNA LncRNA-ANCR targets EZH2, and this inhibition of EZH2 supports expression of RUNX2 (60). Thus, EZH2 expression may be reciprocally interlocked with the activity of the bone-related master regulator Runx2 through a LncRNA-ANCR/EZH2/RUNX2 feedback loop. However, RUNX2 depletion by siRNA treatment does not affect EZH2 levels in transiently transfected AMSCs (data not shown), suggesting that RUNX2 is not rate-limiting for EZH2 expression under our experimental conditions. It is possible that EZH2 may also be targeted more directly by miR-101, which is known to interact with the 3′-untranslated region of EZH2 mRNA (55). Expression of miR-101 is dramatically induced during osteoblast differentiation in a manner reciprocal with EZH2 protein levels.4
During the course of our studies, Hemming et al. (61) demonstrated that H3K27me3, controlled by the activity of EZH2 and the corresponding demethylase KDM6A, controls lineage commitment of mesenchymal progenitor cells. In addition, CDK1-induced phosphorylation of EZH2 modifies the epigenetic landscape of H3K27me3 by mediating the dissociation of EZH2 from the PRC2 complex, thus enhancing osteogenic differentiation of human mesenchymal progenitor cells (62). EZH2 has also been shown to inhibit osteogenic differentiation of human dental pulp cells (63). Taken together, our current work and previous studies firmly suggest that EZH2 is a negative regulator of osteogenic differentiation, and inhibition of this enzyme accelerates the commitment of progenitors into osteoblasts.
Our finding that EZH2 attenuates osteoblastogenesis is relevant to skeletal deformities seen in congenital human disorders and consistent with accruing data on epigenetic regulation of osteoblast differentiation (22, 31–41, 59). Our initial studies with mice lacking functional Ezh2 expression in mesenchymal progenitor cells show premature closure of the cranial sutures characteristic of craniosynostosis. These and other skeletal abnormalities may occur because Ezh2 ablation reduces the proliferation of mesenchymal cells and promotes their osteogenic differentiation. In vivo, this would cause those mesenchymal stem/progenitor cells to differentiate prematurely, resulting in a depletion of the progenitor pool. This depletion is a plausible reason for the osteopenic phenotype of mice in which Ezh2 is depleted in mesenchymal cells.
While our studies were in progress, Schwarz et al. (64) reported that craniofacial bone and cartilage are absent in mice where Ezh2 is ablated in neural crest cells. We find that beyond a dysmorphic skull, loss of Ezh2 in mesenchymal cells also causes clinodactyly. Similar skull and hand abnormalities are seen in patients with Weaver syndrome that have hypomorphic germline mutations in EZH2 (65–67). Furthermore, limb and spine abnormalities we observe in mice with a mesenchymal deletion of Ezh2 reflect growth plate abnormalities that are reminiscent of human osteochondrodysplasias. Interestingly, our mRNASeq analysis of Ezh2 cKO calvaria demonstrates that abnormalities observed in the mice could be a result of altered expression of TFs important for proper development, including Hox and zinc finger genes. Along these lines, Wyngaarden et al. (68) showed that Ezh2 is a key regulator of anterior and posterior prepatterning during embryonic stages of mouse development by maintaining late phase Hox gene expression. Thus, the genetic evidence in both human and mouse models indicates that EZH2 is important for skeletal patterning and early stages of bone formation.
To understand the downstream effects of Ezh2 loss on the accelerated progression of osteoblast differentiation, we performed next generation mRNASeq analysis using calvarial tissues from both WT and Ezh2 cKO mice. Deletion of functional Ezh2 accelerates expression of a broad range of genes encoding principal developmental gene regulators (e.g. Hox proteins) and bone-related ECM proteins (e.g. Bglap, Sparc, Spp1, and Ibsp). We also observed that loss of Ezh2 induces the expression of multiple CDK inhibitors (e.g. Cdkn1c/p57, Cdkn2a/p16) which are key regulators capable of maintaining a postproliferative cell growth arrest. The mRNASeq data collectively suggest that loss of Ezh2 is bone anabolic because it activates the expression of genes that reduce cell cycle progression, as well as up-regulate the production of bone ECM proteins required for a mineralized matrix. We note that EZH2 inhibition and BMP2 have distinct biological effects on the expression of bone-specific genes. It remains to be established whether EZH2 inhibitions may be able to synergize with other anabolic strategies for promoting osteogenic differentiation.
Based on the evidence presented in this paper, it is apparent that inhibition of EZH2 function accelerates commitment of mesenchymal progenitors and preferentially enhances their osteogenic differentiation. In vivo, we find that conditional deletion of functional Ezh2 in the mesenchymal lineage modifies skeletal development and results in skeletal patterning defects. We conclude that EZH2 is a key regulator of osteogenic differentiation and skeletal development.
Author Contributions
A. D., E. T. C., F. X., S. M. R., G. S. S., M. A. M., J. J. W., and A. J. V. W. designed the study; A. D., E. T. C., F. X., S. M. R., M. E. M.-L., E. W. B., C. R. P., R. T., and E. A. L. performed the experiments; D. R. D., A. N. L., and S. M. R. assisted in the interpretation of skeletal phenotypes and human bone disorders; D. G. provided expertise for studies using porous sintered titanium scaffolds; A. B. D. generated clinical grade adipose tissue-derived mesenchymal stem cells and provided expertise on their characterization and use. A. D., E. T. C., S. M. R., G. S. S., M. A. M., J. J. W., and A. J. V. W. wrote the paper with comments from all authors.
Supplementary Material
Acknowledgments
We thank Bridget Stensgard, David Razidlo, Xiaodong Li, and Oksana Pichurin for technical support and the members of our laboratory (especially Martina Gluscevic, Catalina Galeano-Garces, Dakota Lee Jones, Paul Siegert, and Markus Schreiner), as well as our colleagues Sundeep Khosla, John R. Hawse, Malayannan Subramaniam, Anne Gingery, Alexey Leontovich, Sanjeev Kakar, Jay Smith, Aaron Krych, John Sperling, Wenchun Qu, Jane Lian, and Janet Stein for sharing reagents and/or discussions. We also acknowledge the support of Asha Nair and Jared Evans from the Bioinformatics Core, Medical Genome Facility, and Biomaterials Characterization & Quantitative Histomorphometry Core Facility at the Mayo Clinic.
This work was supported, in whole or in part, by National Institutes of Health Grants F32 AR066508 (to A. D.), R01 AR049069 (to A. J. V. W.), R01 AR068103 (to J. J. W.), R01 DE020194 (to J. J. W.), T32 AR056950 (to J. J. W. and E. A. L.), R03 AR065753 (to D. R. D.), R03 AR066342 (to A. N. L.), K01 AR065397 (to E. W. B.), and R01 AR039588 (to G. S. S.). Additional support was provided by Center for Clinical and Translational Science Grant UL1 TR000135 and intramural grants from the Center for Regenerative Medicine at Mayo Clinic, as well as Fondo de Financiamiento de Centros de Investigación en Áreas Prioritarias Grant 15090007 (to M. A. M.) and the generous philanthropic support of William H. and Karen J. Eby and the charitable foundation in their names. Dr. Allan Dietz has a commercial interest in Mill Creek Life Sciences, which manufactures the clinical grade commercial platelet lysate product used for maintaining adipose tissue-derived mesenchymal stem cells. Dr. David Lewallen reports personal fees and other from Stryker Mako, Pipeline Biomedical, Zimmer, and Ketai Medical Devices. In addition, Dr. David Lewallen has patents on selected hip and knee implants with royalties paid by Zimmer and is employed part time as the Medical Director for the American Joint Replacement registry.

This article contains supplemental Table S1.
A. Dudakovic, E. T. Camilleri, F. Xu, S. M. Riester, M. E. McGee-Lawrence, E. W. Bradley, C. R. Paradise, E. A. Lewallen, R. Thaler, D. R. Deyle, A. N. Larson, D. G. Lewallen, A. B. Dietz, G. S. Stein, M. A. Montecino, J. J. Westendorf, and A. J. van Wijnen, unpublished data.
- ECM
- extracellular matrix
- AMSC
- adipose mesenchymal stromal cell
- mRNASeq
- high throughput RNA sequence
- PRC
- polycomb-repressive complex
- RT-qPCR
- quantitative real time reverse transcriptase-PCR
- microCT
- microcomputed tomography
- ES
- enrichment score
- TF
- transcription factor
- CDK
- cyclin-dependent kinase.
References
- 1.Yang Y. (2013) Skeletal Morphogenesis and Embryonic Development. In Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, pp. 1–14, John Wiley & Sons, Inc. [Google Scholar]
- 2.Jiang Y., Jahagirdar B. N., Reinhardt R. L., Schwartz R. E., Keene C. D., Ortiz-Gonzalez X. R., Reyes M., Lenvik T., Lund T., Blackstad M., Du J., Aldrich S., Lisberg A., Low W. C., Largaespada D. A., and Verfaillie C. M. (2002) Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 418, 41–49 [DOI] [PubMed] [Google Scholar]
- 3.Pike J. W. (2011) Genome-scale techniques highlight the epigenome and redefine fundamental principles of gene regulation. J. Bone Miner Res. 26, 1155–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lian J. B., Stein G. S., van Wijnen A. J., Stein J. L., Hassan M. Q., Gaur T., and Zhang Y. (2012) MicroRNA control of bone formation and homeostasis. Nat. Rev. Endocrinol. 8, 212–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McGee-Lawrence M. E., and Westendorf J. J. (2011) Histone deacetylases in skeletal development and bone mass maintenance. Gene 474, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(1993) Consensus development conference: diagnosis, prophylaxis, and treatment of osteoporosis. Am. J. Med. 94, 646–650 [DOI] [PubMed] [Google Scholar]
- 7.Burge R., Dawson-Hughes B., Solomon D. H., Wong J. B., King A., and Tosteson A. (2007) Incidence and economic burden of osteoporosis-related fractures in the United States, 2005–2025. J. Bone Miner. Res. 22, 465–475 [DOI] [PubMed] [Google Scholar]
- 8.Dennison E., and Cooper C. (2000) Epidemiology of osteoporotic fractures. Horm. Res. 54, 58–63 [DOI] [PubMed] [Google Scholar]
- 9.(2006) Management of osteoporosis in postmenopausal women: 2006 position statement of the North American Menopause Society. Menopause 13, 340–367; quiz 368–349 [DOI] [PubMed] [Google Scholar]
- 10.Ettinger B., Black D. M., Mitlak B. H., Knickerbocker R. K., Nickelsen T., Genant H. K., Christiansen C., Delmas P. D., Zanchetta J. R., Stakkestad J., Glüer C. C., Krueger K., Cohen F. J., Eckert S., Ensrud K. E., Avioli L. V., Lips P., and Cummings S. R. (1999) Reduction of vertebral fracture risk in postmenopausal women with osteoporosis treated with raloxifene: results from a 3-year randomized clinical trial: Multiple Outcomes of Raloxifene Evaluation (MORE) Investigators. JAMA 282, 637–645 [DOI] [PubMed] [Google Scholar]
- 11.Silverman S. L., Christiansen C., Genant H. K., Vukicevic S., Zanchetta J. R., de Villiers T. J., Constantine G. D., and Chines A. A. (2008) Efficacy of bazedoxifene in reducing new vertebral fracture risk in postmenopausal women with osteoporosis: results from a 3-year, randomized, placebo-, and active-controlled clinical trial. J. Bone Miner. Res. 23, 1923–1934 [DOI] [PubMed] [Google Scholar]
- 12.Bekker P. J., Holloway D. L., Rasmussen A. S., Murphy R., Martin S. W., Leese P. T., Holmes G. B., Dunstan C. R., and DePaoli A. M. (2004) A single-dose placebo-controlled study of AMG 162, a fully human monoclonal antibody to RANKL, in postmenopausal women. J. Bone Miner. Res. 19, 1059–1066 [DOI] [PubMed] [Google Scholar]
- 13.Cummings S. R., San Martin J., McClung M. R., Siris E. S., Eastell R., Reid I. R., Delmas P., Zoog H. B., Austin M., Wang A., Kutilek S., Adami S., Zanchetta J., Libanati C., Siddhanti S., and Christiansen C. (2009) Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N. Engl. J. Med. 361, 756–765 [DOI] [PubMed] [Google Scholar]
- 14.Canalis E. (2010) Update in new anabolic therapies for osteoporosis. J. Clin. Endocrinol. Metab. 95, 1496–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neer R. M., Arnaud C. D., Zanchetta J. R., Prince R., Gaich G. A., Reginster J. Y., Hodsman A. B., Eriksen E. F., Ish-Shalom S., Genant H. K., Wang O., and Mitlak B. H. (2001) Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N. Engl. J. Med. 344, 1434–1441 [DOI] [PubMed] [Google Scholar]
- 16.Reszka A. A., and Rodan G. A. (2004) Nitrogen-containing bisphosphonate mechanism of action. Mini Rev. Med. Chem. 4, 711–719 [PubMed] [Google Scholar]
- 17.Papapoulos S. E. (2006) Bisphosphonate actions: physical chemistry revisited. Bone 38, 613–616 [DOI] [PubMed] [Google Scholar]
- 18.Bellido T., and Plotkin L. I. (2011) Novel actions of bisphosphonates in bone: preservation of osteoblast and osteocyte viability. Bone 49, 50–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rodriguez-Merchan E. C., and Forriol F. (2004) Nonunion: general principles and experimental data. Clin. Orthop. Relat. Res. 4–12 [PubMed] [Google Scholar]
- 20.Gibney E. R., and Nolan C. M. (2010) Epigenetics and gene expression. Heredity 105, 4–13 [DOI] [PubMed] [Google Scholar]
- 21.Zaidi S. K., Grandy R. A., Lopez-Camacho C., Montecino M., van Wijnen A. J., Lian J. B., Stein J. L., and Stein G. S. (2014) Bookmarking target genes in mitosis: a shared epigenetic trait of phenotypic transcription factors and oncogenes? Cancer Res. 74, 420–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marchesi I., Giordano A., and Bagella L. (2014) Roles of enhancer of zeste homolog 2: from skeletal muscle differentiation to rhabdomyosarcoma carcinogenesis. Cell Cycle 13, 516–527 [DOI] [PubMed] [Google Scholar]
- 23.Francis N. J., Kingston R. E., and Woodcock C. L. (2004) Chromatin compaction by a polycomb group protein complex. Science 306, 1574–1577 [DOI] [PubMed] [Google Scholar]
- 24.O'Carroll D., Erhardt S., Pagani M., Barton S. C., Surani M. A., and Jenuwein T. (2001) The polycomb-group gene Ezh2 is required for early mouse development. Mol. Cell. Biol. 21, 4330–4336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramsköld D., Wang E. T., Burge C. B., and Sandberg R. (2009) An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Comput. Biol. 5, e1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cakouros D., Isenmann S., Cooper L., Zannettino A., Anderson P., Glackin C., and Gronthos S. (2012) Twist-1 induces Ezh2 recruitment regulating histone methylation along the Ink4A/Arf locus in mesenchymal stem cells. Mol. Cell. Biol. 32, 1433–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang X., Karuturi R. K., Sun F., Aau M., Yu K., Shao R., Miller L. D., Tan P. B., and Yu Q. (2009) CDKN1C (p57) is a direct target of EZH2 and suppressed by multiple epigenetic mechanisms in breast cancer cells. PLoS One 4, e5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rando O. J., and Chang H. Y. (2009) Genome-wide views of chromatin structure. Annu. Rev. Biochem. 78, 245–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laird P. W. (2010) Principles and challenges of genomewide DNA methylation analysis. Nat. Rev. Genet. 11, 191–203 [DOI] [PubMed] [Google Scholar]
- 30.de Wit E., and de Laat W. (2012) A decade of 3C technologies: insights into nuclear organization. Genes Dev. 26, 11–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Villagra A., Gutiérrez J., Paredes R., Sierra J., Puchi M., Imschenetzky M., Wijnen Av A., Lian J., Stein G., Stein J., and Montecino M. (2002) Reduced CpG methylation is associated with transcriptional activation of the bone-specific rat osteocalcin gene in osteoblasts. J. Cell. Biochem. 85, 112–122 [PubMed] [Google Scholar]
- 32.Lee J. Y., Lee Y. M., Kim M. J., Choi J. Y., Park E. K., Kim S. Y., Lee S. P., Yang J. S., and Kim D. S. (2006) Methylation of the mouse DIx5 and Osx gene promoters regulates cell type-specific gene expression. Mol. Cells 22, 182–188 [PubMed] [Google Scholar]
- 33.Westendorf J. J., Zaidi S. K., Cascino J. E., Kahler R., van Wijnen A. J., Lian J. B., Yoshida M., Stein G. S., and Li X. (2002) Runx2 (Cbfa1, AML-3) interacts with histone deacetylase 6 and represses the p21(CIP1/WAF1) promoter. Mol. Cell. Biol. 22, 7982–7992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schroeder T. M., Kahler R. A., Li X., and Westendorf J. J. (2004) Histone deacetylase 3 interacts with runx2 to repress the osteocalcin promoter and regulate osteoblast differentiation. J. Biol. Chem. 279, 41998–42007 [DOI] [PubMed] [Google Scholar]
- 35.Kang J. S., Alliston T., Delston R., and Derynck R. (2005) Repression of Runx2 function by TGF-β through recruitment of class II histone deacetylases by Smad3. EMBO J. 24, 2543–2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gori F., Divieti P., and Demay M. B. (2001) Cloning and characterization of a novel WD-40 repeat protein that dramatically accelerates osteoblastic differentiation. J. Biol. Chem. 276, 46515–46522 [DOI] [PubMed] [Google Scholar]
- 37.Gori F., Friedman L. G., and Demay M. B. (2006) Wdr5, a WD-40 protein, regulates osteoblast differentiation during embryonic bone development. Dev. Biol. 295, 498–506 [DOI] [PubMed] [Google Scholar]
- 38.Schroeder T. M., and Westendorf J. J. (2005) Histone deacetylase inhibitors promote osteoblast maturation. J. Bone Miner. Res. 20, 2254–2263 [DOI] [PubMed] [Google Scholar]
- 39.Di Bernardo G., Squillaro T., Dell'Aversana C., Miceli M., Cipollaro M., Cascino A., Altucci L., and Galderisi U. (2009) Histone deacetylase inhibitors promote apoptosis and senescence in human mesenchymal stem cells. Stem Cells Dev. 18, 573–581 [DOI] [PubMed] [Google Scholar]
- 40.Lee S., Park J. R., Seo M. S., Roh K. H., Park S. B., Hwang J. W., Sun B., Seo K., Lee Y. S., Kang S. K., Jung J. W., and Kang K. S. (2009) Histone deacetylase inhibitors decrease proliferation potential and multilineage differentiation capability of human mesenchymal stem cells. Cell Prolif. 42, 711–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dudakovic A., Evans J. M., Li Y., Middha S., McGee-Lawrence M. E., van Wijnen A. J., and Westendorf J. J. (2013) Histone deacetylase inhibition promotes osteoblast maturation by altering the histone H4 epigenome and reduces Akt phosphorylation. J. Biol. Chem. 288, 28783–28791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crespo-Diaz R., Behfar A., Butler G. W., Padley D. J., Sarr M. G., Bartunek J., Dietz A. B., and Terzic A. (2011) Platelet lysate consisting of a natural repair proteome supports human mesenchymal stem cell proliferation and chromosomal stability. Cell Transplant. 20, 797–811 [DOI] [PubMed] [Google Scholar]
- 43.Mader E. K., Butler G., Dowdy S. C., Mariani A., Knutson K. L., Federspiel M. J., Russell S. J., Galanis E., Dietz A. B., and Peng K. W. (2013) Optimizing patient derived mesenchymal stem cells as virus carriers for a phase I clinical trial in ovarian cancer. J. Transl. Med. 11, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Su I. H., Basavaraj A., Krutchinsky A. N., Hobert O., Ullrich A., Chait B. T., and Tarakhovsky A. (2003) Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat. Immunol. 4, 124–131 [DOI] [PubMed] [Google Scholar]
- 45.Logan M., Martin J. F., Nagy A., Lobe C., Olson E. N., and Tabin C. J. (2002) Expression of Cre recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis 33, 77–80 [DOI] [PubMed] [Google Scholar]
- 46.Bouxsein M. L., Boyd S. K., Christiansen B. A., Guldberg R. E., Jepsen K. J., and Müller R. (2010) Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J. Bone Miner. Res. 25, 1468–1486 [DOI] [PubMed] [Google Scholar]
- 47.Dudakovic A., Camilleri E., Riester S. M., Lewallen E. A., Kvasha S., Chen X., Radel D. J., Anderson J. M., Nair A. A., Evans J. M., Krych A. J., Smith J., Deyle D. R., Stein J. L., Stein G. S., Im H. J., Cool S. M., Westendorf J. J., Kakar S., Dietz A. B., and van Wijnen A. J. (2014) High-resolution molecular validation of self-renewal and spontaneous differentiation in clinical-grade adipose-tissue derived human mesenchymal stem cells. J. Cell. Biochem. 115, 1816–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu L., Zhen X. T., Denton E., Marsden B. D., and Schapira M. (2012) ChromoHub: a data hub for navigators of chromatin-mediated signalling. Bioinformatics 28, 2205–2206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dudakovic A., Camilleri E. T., Lewallen E. A., McGee-Lawrence M. E., Riester S. M., Kakar S., Montecino M., Stein G. S., Ryoo H. M., Dietz A. B., Westendorf J. J., and van Wijnen A. J. (2015) Histone deacetylase inhibition destabilizes the multi-potent state of uncommitted adipose-derived mesenchymal stromal cells. J. Cell. Physiol. 230, 52–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Margueron R., Li G., Sarma K., Blais A., Zavadil J., Woodcock C. L., Dynlacht B. D., and Reinberg D. (2008) Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell 32, 503–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cao R., Wang L., Wang H., Xia L., Erdjument-Bromage H., Tempst P., Jones R. S., and Zhang Y. (2002) Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science 298, 1039–1043 [DOI] [PubMed] [Google Scholar]
- 52.López-Rovira T., Chalaux E., Massagué J., Rosa J. L., and Ventura F. (2002) Direct binding of Smad1 and Smad4 to two distinct motifs mediates bone morphogenetic protein-specific transcriptional activation of Id1 gene. J. Biol. Chem. 277, 3176–3185 [DOI] [PubMed] [Google Scholar]
- 53.Issa J. P., and Kantarjian H. M. (2009) Targeting DNA methylation. Clin. Cancer Res. 15, 3938–3946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Khan O., and La Thangue N. B. (2012) HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol. Cell Biol. 90, 85–94 [DOI] [PubMed] [Google Scholar]
- 55.Chuang D. M. (2005) The antiapoptotic actions of mood stabilizers: molecular mechanisms and therapeutic potentials. Ann. N.Y. Acad. Sci. 1053, 195–204 [DOI] [PubMed] [Google Scholar]
- 56.Chouliaras L., Rutten B. P., Kenis G., Peerbooms O., Visser P. J., Verhey F., van Os J., Steinbusch H. W., and van den Hove D. L. (2010) Epigenetic regulation in the pathophysiology of Alzheimer's disease. Prog. Neurobiol. 90, 498–510 [DOI] [PubMed] [Google Scholar]
- 57.Chiechio S., Zammataro M., Morales M. E., Busceti C. L., Drago F., Gereau R. W. 4th, Copani A., and Nicoletti F. (2009) Epigenetic modulation of mGlu2 receptors by histone deacetylase inhibitors in the treatment of inflammatory pain. Mol. Pharmacol. 75, 1014–1020 [DOI] [PubMed] [Google Scholar]
- 58.Hewagama A., and Richardson B. (2009) The genetics and epigenetics of autoimmune diseases. J. Autoimmun. 33, 3–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu H., Whitfield T. W., Gordon J. A., Dobson J. R., Tai P. W., van Wijnen A. J., Stein J. L., Stein G. S., and Lian J. B. (2014) Genomic occupancy of Runx2 with global expression profiling identifies a novel dimension to control of osteoblastogenesis. Genome Biol. 15, R52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhu L., and Xu P. C. (2013) Downregulated LncRNA-ANCR promotes osteoblast differentiation by targeting EZH2 and regulating Runx2 expression. Biochem. Biophys. Res. Commun. 432, 612–617 [DOI] [PubMed] [Google Scholar]
- 61.Hemming S., Cakouros D., Isenmann S., Cooper L., Menicanin D., Zannettino A., and Gronthos S. (2014) EZH2 and KDM6A act as an epigenetic switch to regulate mesenchymal stem cell lineage specification. Stem Cells 32, 802–815 [DOI] [PubMed] [Google Scholar]
- 62.Wei Y., Chen Y. H., Li L. Y., Lang J., Yeh S. P., Shi B., Yang C. C., Yang J. Y., Lin C. Y., Lai C. C., and Hung M. C. (2011) CDK1-dependent phosphorylation of EZH2 suppresses methylation of H3K27 and promotes osteogenic differentiation of human mesenchymal stem cells. Nat Cell Biol. 13, 87–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hui T., A P., Zhao Y., Wang C., Gao B., Zhang P., Wang J., Zhou X., and Ye L. (2014) EZH2, a potential regulator of dental pulp inflammation and regeneration. J. Endod. 40, 1132–1138 [DOI] [PubMed] [Google Scholar]
- 64.Schwarz D., Varum S., Zemke M., Scholer A., Baggiolini A., Draganova K., Koseki H., Schubeler D., and Sommer L. (2014) Ezh2 is required for neural crest-derived cartilage and bone formation. Development 141, 867–877 [DOI] [PubMed] [Google Scholar]
- 65.Tatton-Brown K., Hanks S., Ruark E., Zachariou A., Duarte Sdel V., Ramsay E., Snape K., Murray A., Perdeaux E. R., Seal S., Loveday C., Banka S., Clericuzio C., Flinter F., Magee A., McConnell V., Patton M., Raith W., Rankin J., Splitt M., Strenger V., Taylor C., Wheeler P., Temple K. I., Cole T., Childhood Overgrowth Collaboration,Douglas J., and Rahman N. (2011) Germline mutations in the oncogene EZH2 cause Weaver syndrome and increased human height. Oncotarget 2, 1127–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weaver D. D., Graham C. B., Thomas I. T., and Smith D. W. (1974) A new overgrowth syndrome with accelerated skeletal maturation, unusual facies, and camptodactyly. J. Pediatr. 84, 547–552 [DOI] [PubMed] [Google Scholar]
- 67.Gibson W. T., Hood R. L., Zhan S. H., Bulman D. E., Fejes A. P., Moore R., Mungall A. J., Eydoux P., Babul-Hirji R., An J., Marra M. A., FORGE Canada Consortium, Chitayat D., Boycott K. M., Weaver D. D., and Jones S. J. (2012) Mutations in EZH2 cause Weaver syndrome. Am. J. Hum. Genet. 90, 110–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wyngaarden L. A., Delgado-Olguin P., Su I. H., Bruneau B. G., and Hopyan S. (2011) Ezh2 regulates anteroposterior axis specification and proximodistal axis elongation in the developing limb. Development 138, 3759–3767 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.