

Background: TANK is a negative regulator of canonical NF-κB signaling.
Results: EMCV 3C cleaves TANK and regulates NF-κB activation.
Conclusion: EMCV 3C relieves TANK inhibitory effect on TRAF6-mediated NF-κB signaling pathway.
Significance: TANK is a novel target protein of viral proteases encoded by several positive RNA viruses.
Keywords: cell signaling, immunology, NF-kappa B (NF-KB), viral protease, virus
Abstract
TRAF family member-associated NF-κB activator (TANK) is a negative regulator of canonical NF-κB signaling in the Toll-like receptor- and B-cell receptor-mediated signaling pathways. However, functions of TANK in viral infection-mediated NF-κB activation remain unclear. Here, we reported that TANK was cleaved by encephalomyocarditis virus 3C at the 197 and 291 glutamine residues, which depends on its cysteine protease activity. In addition, encephalomyocarditis virus 3C impaired the ability of TANK to inhibit TRAF6-mediated NF-κB signaling. Interestingly, we found that several viral proteases encoded by the foot and mouth disease virus, porcine reproductive and respiratory syndrome virus, and equine arteritis virus also cleaved TANK. Our results suggest that TANK is a novel target of some viral proteases, indicating that some positive RNA viruses have evolved to utilize their major proteases to regulate NF-κB activation.
Introduction
Encephalomyocarditis virus (EMCV)4 infection causes myocarditis, encephalitis, neurological diseases, reproductive disorders, and diabetes in many mammalian species (1). EMCV belongs to the genus Cardiovirus within the picornaviridae family. Like other picornaviruses, EMCV is a small, non-enveloped virus containing single-stranded positive-sense RNA of ∼7.8 kb flanked by two untranslated regions (UTRs). The 5′UTR is 800–1,200 nucleotides long, whereas the 3′UTR is ∼120 nucleotides long with short stem-loop structures, followed by a poly(A) tail (1). Upon virus entry and uncoating, EMCV genomic RNA (vRNA) is released into the cytoplasm. Host proteins, including eukaryotic initiation factors, bind the viral internal ribosome entry site and initiate cap-independent translation. The EMCV genome is translated into two separate polyproteins through ribosome skipping (2). EMCV 3C protease (EMCV 3C) cleaves the two polyproteins to produce at least 13 mature viral proteins that are involved in genome replication, NLRP3-dependent inflammasome activation, and host innate immune responses (3, 4).
NF-κB activation is regulated by the IKK complex, a trimetric holoenzyme consisting of the following kinases: IKKα, IKKβ, and the regulatory subunit NEMO (also called IKKγ). In the canonical NF-κB signaling pathway, inhibitory IκB proteins (IκBs) bind NF-κB dimers and sequester NF-κB complexes in the cytoplasm (5). Viral infection and inflammatory cytokines elicit the degradation of the IκBs by the 26S proteasome following the phosphorylation of the IκBs. Free NF-κB dimers are transferred into the nucleus and stimulate the transcription of target genes encoding inflammatory and immunoregulatory molecules (6–8). The canonical NF-κB signaling pathway is also regulated by different physiological stimuli such as signals emanating from the interleukin-1 receptor (IL-1R), the tumor necrosis factor receptor (TNFR), and other cytokine receptors (5, 9, 10).
TRAF family member-associated NF-κB activator (TANK) was first identified as a TRAF-binding protein. A previous study revealed that TANK enhances NF-κB activation in cells expressing TRAF2. Therefore, TANK was considered as an NF-κB activator (11). However, TANK was also found to interact with the conserved TRAF-C domain of TRAFs, which inhibited NF-κB activation by impeding the interaction between TRAFs and their receptors (12). Additionally, TANK is functional in the inhibition of TRAF6-mediated NF-κB activation in TNFα-, IL-1-, and CD40-mediated signaling pathways (11, 12, 38, 50).
TRAF6 is unique among the seven TRAF family members, which is involved in a range of physiological processes, including innate immunity, adaptive immunity, and bone metabolism (13–16). Stimulation with IL-1 triggers recruitment of the adaptor MyD88 to the intracellular domain of the IL-1 receptor at the cell membrane, resulting in recruitment of IL-1 receptor-associated kinases and TRAF6 and subsequent activation of IKK (17). TRAF6 is also an E3 ubiquitin ligase, which is necessary for the polyubiquitination of its substrates and itself. It has been demonstrated that TRAF6 activates TAK1 and triggers the activation of both AP-1 and NF-κB (18, 19). Because of the important biological functions of NF-κB in the innate and adaptive immune responses, the transcriptional activity of nuclear NF-κB is tightly regulated through post-translational modifications at multiple levels by positive and negative regulatory elements (20). Recently, the IKK complex, its regulators, and the key gatekeepers of NF-κB signaling were reported to be targeted by different pathogens (8, 20).
Here, we report a novel post-translational modification of TANK. TANK is cleaved by EMCV 3C at the 197 and 291 glutamine residues which are dependent on its enzymatic activity. Cleavage of TANK by EMCV 3C disrupts the ability of TANK to inhibit TRAF6-mediated NF-κB signaling. Interestingly, we also found that other viral proteases encoded by FMDV, PRRSV, and EAV could cleave TANK in vitro, although the cleavage sites are different. To our knowledge, this is the first report to show that EMCV 3C specifically cleaves TANK in vitro. Our findings also reveal that several positive RNA viruses have adopted a novel mechanism to regulate NF-κB signaling by cleavage of TANK using their proteases.
Experimental Procedures
Cell Lines and Viruses
Human embryonic kidney 293T (HEK293T) and baby hamster kidney (BHK)-21 cells were purchased from the ATCC and cultured in Dulbecco's modified Eagle's medium (DMEM, Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Hyclone Laboratories, Inc., Logan, UT), 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C in a 5% CO2-humidified atmosphere. EMCV HB10 strain and FMDV (Asia 1/YS/CHA/05) were propagated in BHK-21 cells as described previously (21, 22). In brief, cellular supernatants of EMCV-infected BHK-21 cells were diluted 10-fold serially. BHK-21 cells in a 96-well plate were infected by 0.1 ml/well of serially diluted supernatant in quintuplicate. After incubating for 90 min at 37 °C, unattached virus was removed, and DMEM supplemented with 2% FBS was added into each well and incubated at 37 °C. Five days post-infection, 50% tissue culture infectious dose (TCID50) was determined by the Reed-Muench method. All data were shown as the means of three independent experiments. EMCV infection was performed as described previously (23). Other viruses and their cDNAs used in this study are listed in Table 1.
TABLE 1.
cDNA of viruses used in this study
| RNA virus | Virus | Strain | GenBankTM accession no. | Main protease | cDNA | |
|---|---|---|---|---|---|---|
| Picornaviridae | Encephalomyocarditis virus | EMCV | HB10 | JQ864080.1 | EMCV-3C | PCR |
| Foot and mouth disease virus | FMDV | Asia1/YS/CHA/05 | GU931682.1 | FMDV-3C | PCR | |
| Human enterovirus 71 | EV71 | Wuhan/3018/2010 | KF501389.1 | EV-71–3C | Synthesized | |
| Arteriviridae | Porcine reproductive and respiratory syndrome virus | PRRSV | HuN4 | EF635006.1 | PRRSV-NSP4 | PCR |
| Equine arteritis virus | EAV | Bucyrus | DQ846750.1 | EAV-NSP4 | PCR | |
| Flaviviridae | Classical swine fever virus | CSFV | Shimen | AF092448.2 | CSFV-NS3 | PCR |
| Japanese encephalitis virus | JEV | SA-14–14–2 | JN604986.1 | JEV-NS3 | PCR | |
| Coronaviridae | Severe Acute Respiratory Syndromes | SARS | SARS-CoV | AY278741 | SARS-NS3 | Synthesized |
| Transmissible gastroenteritis virus | TGEV | HX | KC962433.1 | TGEV-NS3 | PCR |
Plasmids, Antibodies, and Reagents
The plasmids expressing FLAG-tagged RIG-I, MDA5, MAVS, TANK, TBK1, IKKϵ, NEMO, IKKα, TRAF6, TRAF3, and IRF3 have been described previously (24). pGL3-NF-κB-Luc and pRL-SV40 plasmids were gifts from Hong Tang (Wuhan Institute of Virology, Wuhan, Hubei, China). To construct plasmids expressing HA-tagged EMCV proteins (VP1, VP2, VP3, L-VP4, 2A, 2B, 2C, 3AB, 3C, and 3D) and EMCV 3C intermediates, the cDNAs of EMCV were cloned into the pCAGGS-HA (pHA) vector. TANK cDNAs from human or other species (monkey, swine, and mouse) were amplified by standard RT-PCR using total RNA extracted from HeLa, Marc-145, PK-15, or Raw 264.7 cells as templates and cloned into the pCAGGS-FLAG (pFLAG) vector. Mutants of human TANK (Q182A, Q190A, Q197R, Q266A, Q291A, Q301A, and Q197R/Q291A) and EMCV 3C (H46A, C159A, and H46A/C159A) were constructed by site-directed mutagenesis or by overlap extension using Pfu DNA polymerase (Stratagene, La Jolla, CA). The cDNAs encoding deletion mutants of TANK, including 197N (1–197 amino acids), 197C (198–425 amino acids), 291N (1–291 amino acids), and 291C (292–425 amino acids), were cloned into the pFLAG or pHA vector. The cDNAs of TANK and EMCV 3C were also cloned into the pGEX-4T3 or pET-28a vectors to express and purify recombinant proteins. The cDNAs encoding other viral proteases (such as FMDV, PRRSV, and EAV) were cloned into the pHA vector, and the mutants of FMDV-3C and EV71–3C were constructed using site-directed mutagenesis. The primers used in this study are available upon request. All constructs were validated by DNA sequencing.
Antibodies against NF-κB-p65 (4764), p-NF-κB-p65 (3033S), and IκBα (IκBα) (4814) were purchased from Cell Signaling Technology, Inc. (Danvers, MA). PARP1 (13371-1-AP), anti-β-actin (66009-1-Ig), and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (60004-1-Ig) antibodies were purchased from Proteintech (Wuhan, China). Anti-TANK (sc-1998) and anti-caspase-3 (sc-271759) antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX). Antibodies against FLAG, HA, Myc, goat anti-mouse (A4416), or anti-rabbit (A0545) secondary antibodies were purchased from Sigma. Polyclonal and monoclonal antibodies against EMCV VP1 and 3C and FMDV VP2 were prepared by immunizing rabbits and mice using purified recombinant EMCV VP1, EMCV 3C, or FMDV VP2 proteins as immunogens. Goat anti-mouse fluorescein isothiocyanate (FITC)-labeled secondary antibody (F9006), the caspase inhibitor Z-VAD-FMK, the proteasome inhibitor MG132, and the lysosome inhibitor NH4Cl were purchased from Sigma.
Luciferase Reporter Gene Assays
Reporter assays were performed as described previously (25–27). In brief, 2.5 × 105 HEK293T cells were transfected with different concentrations of plasmids expressing the proteins as indicated, in combination with 100 ng/well pGL3-NF-κB-Luc and 5 ng/well of the pRL-SV40 plasmid (Promega Corp., Madison, WI), which was used as a control for transfection efficiency. In each experiment, the total amount of DNA in each sample was kept constant by supplementation with the appropriate empty parental expression vector. At 24 h post-transfection (hpt), the activities of Firefly and Renilla luciferase were measured using the Dual-Luciferase® reporter assay system (Promega Corp.) according to the manufacturer's instructions. These data represent relative Firefly luciferase activity normalized to Renilla luciferase activity and are representative of the independent experiments in triplicate. Data were presented as means ± S.D.
Expression of Recombinant Proteins and Cleavage Assay in Vitro
Purification of recombinant proteins and the cleavage assay were performed in vitro as described previously (28). In brief, His6-EMCV 3C protein was purified from clarified bacterial lysates by metal chelation chromatography. The GST-TANK protein was purified on a glutathione-agarose column. These proteins were dialyzed and stored at −80 °C. To examine TANK cleavage in vitro, aliquots of recombinant EMCV 3C (His6-EMCV 3C) and GST-TANK were incubated in Buffer A (50 mm Tris acetate, pH 8.5, 1 mm EDTA, 10% glycerol, and 1 mm dithiothreitol). After incubation at 30 °C for 20 min, the reactions were terminated by adding 1× loading buffer and then subjected to Western blot analysis.
Co-immunoprecipitation (Co-IP) and Western Blot Analysis
Co-IP and Western blot analysis were performed as described previously (28, 29). At 36–48 hpt, the cells transfected with the different plasmids were lysed with cell lysis buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Triton X-100, 5 mm MgCl2, 1 mm EDTA, and 10% glycerol) containing 1 mm PMSF and 1× protease inhibitor mixture (Roche Diagnostics). The cell lysates were incubated with anti-FLAG-agarose beads overnight at 4 °C on a roller. The immunoprecipitants were subjected to electrophoresis. For Western blot analysis, equal amounts of cell lysates and immunoprecipitants were resolved on 10–12% SDS-polyacrylamide gel and then transferred to polyvinylidine difluoride membrane (PVDF) (EMD Millipore Corp., Billerica, MA). After incubating with the indicated antibodies, the membranes were visualized using chemiluminescence (ECL; GE Healthcare, Buckinghamshire, UK).
Fluorescence Microscopy
HEK293T cells were transfected with a plasmid expressing FLAG-tagged TANK or its deletion mutants in the presence or absence of MG132 as indicated. At 12–24 hpt, the cells were fixed with 4% paraformaldehyde, permeabilized with 0.3% Triton X-100, and then probed with the anti-FLAG antibodies and 4′,6′-diamidino-2- phenylindole (DAPI). Samples were visualized with a Zeiss LSM-510 laser scanning fluorescence microscope (Carl Zeiss AG, Jena, Germany).
RNA Interference
The control shRNA (shNC) and shRNA targeting human TANK (shTANK) were purchased from GenePharma (Shanghai, China). shTANK sequence is UCACUUCAACAGACUAUUATT. To reduce TANK protein expression in HEK293T cells, shNC or shTANK was transfected into HEK293T cells with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. To test the shRNA knockdown effects, TANK protein levels were analyzed by Western blotting.
RNA Extraction and Quantitative Real Time RT-PCR
To detect human TNFα mRNA levels, total mRNA was extracted from the HEK293T cells infected with EMCV using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. Reverse transcription was carried out using PrimescrriptTM RT master mix (Takara Bio Inc., Japan). Quantitative real time PCR (qRT-PCR) was performed, and products were detected using an Agilent-Stratagene Mx Real-Time qPCR system according to SYBR Premix DimerEraser (catalog no. RR091A) instructions (Takara Bio Inc., Japan). Each sample was run in triplicate. The relative amount of mRNA for target gene was normalized to β-actin mRNA in the same sample. The target sequences were amplified by using the following primers: human TNFα RNA forward primer (5′-GCTGAGGAACAAGCACCG-3′) and reverse primer (5′-CCTCTCACATACTGACCCACG-3′); β-actin RNA forward primer (5′-CCTTCCTGGGCATGGAGTCCTG-3′) and reverse primer (5′-GGAGCAATGATCTTGATCTTC-3′).
50% Tissue Culture Infectious Dose (TCID50) Assay
Cellular supernatants from EMCV-infected HEK293T cells at different times were serially diluted 10-fold. HEK293T cells grown in a 96-well plate were infected with 0.1 ml/well of serially diluted supernatant in quintuplicate. After incubating for 90 min at 37 °C, unattached virus was removed, and DMEM supplemented with 2% FBS was added to the HEK293T cells prior to incubation at 37 °C. Five days post-infection, the TCID50 was determined by the Reed-Muench method. All data are shown as the means of three independent experiments.
Accession Numbers
The following information was arranged in the format of “species,” “symbol,” and “accession numbers” as follows: human RIG-I (O95786); human MDA5 (Q9BYX4); human MAVS (Q7Z434); human TBK1 (Q9UHD2); human IKKϵ (Q14164); human NEMO (Q9Y6K9); human IKKα (O15111); human IRF3 (Q14653); human TRAF3 (Q60803); human TRAF6 (Q9Y4K3); human TANK (Q6NW12); swine TANK (F1RPR5); monkey TANK (Q4R5I0); mouse TANK (Q8R383); and EMCV HB10 (JQ864080.1).
Results
TANK Is a Novel Target of EMCV 3C Protease
Previous studies demonstrated that viral proteases encoded by several picornaviruses could cleave the key molecules in type I interferon (IFN) and NF-κB signaling pathways, which resulted in the inhibition of IFN production and NF-κB activation (26, 30–32). To investigate which host protein related to IFN and NF-κB signaling pathways is cleaved by EMCV 3C, HEK293T cells were transfected with a plasmid ectopically expressing FLAG-tagged RIG-I, MDA5, MAVS, TANK, TBK1, IKKϵ, NEMO, IKKα, TRAF3, TRAF6, or IRF3, in combination with empty vector or a plasmid expressing HA-EMCV 3C. As shown in Fig. 1A, RIG-I and TANK, but none of the other proteins chosen in this study, were cleaved by EMCV 3C. We observed that the reduced abundance of full-length TANK (55 kDa) was accompanied by the appearance of two N-terminal fragments of TANK with apparent molecular masses of about 35 and 25 kDa (designated TANK-N1 and TANK-N2), suggesting that EMCV 3C caused at least two scissions within human TANK.
FIGURE 1.

TANK is a novel substrate of EMCV 3C. A, EMCV 3C cleaves TANK. HEK293T cells were transfected with 2 μg of a plasmid expressing FLAG-tagged RIG-I, MDA5, MAVS, TANK, TBK1, IKKϵ, NEMO, IKKα, TRAF6, TRAF3, or IRF3, along with 2 μg of empty vector or 2 μg of a plasmid encoding HA-EMCV 3C. At 36 hpt, the cell lysates were analyzed by Western blotting (WB) with the indicated antibodies. B, HEK293T cells were transfected with a plasmid expressing FLAG-TANK alone or along with increasing doses (0, 0.5, 1, or 2 μg) of plasmid encoding HA-EMCV 3C. The cell lysates were analyzed with the indicated antibodies. C, recombinant GST-TANK was incubated with purified EMCV 3C (His6-EMCV 3C). The reaction products were analyzed with anti-GST and anti-TANK antibodies. D, EMCV 3C interacts with TANK. HEK293T cells were transfected with a plasmid encoding FLAG-TANK, in combination with a plasmid encoding HA-EMCV 3C or HA-EMCV 3D as indicated. The cell lysates were immunoprecipitated (IP) with anti-FLAG antibody. The immunoprecipitants and aliquots of the whole cell lysates were subjected to Western blotting analysis with anti-HA, anti-FLAG, or anti-GAPDH antibodies.
To confirm whether TANK is cleaved by EMCV 3C in a dose-dependent manner, HEK293T cells were cotransfected with a plasmid encoding FLAG-TANK and increasing amounts of a plasmid expressing EMCV 3C. The result showed that the intensity of FLAG-TANK diminished when the protein levels of EMCV 3C were increased, and two N-terminal fragments began to appear (Fig. 1B, lanes 2–4), although there exists at least one additional C-terminal fragment, but it is degraded via the proteasome pathway (refer to Fig. 5). To further confirm that EMCV 3C cleaves TANK in vitro, purified EMCV 3C (His6-EMCV 3C) and GST-TANK were incubated, and the reaction products were detected with anti-EMCV 3C and anti-TANK antibodies. As shown in Fig. 1C, we found that purified EMCV 3C cleaved GST-TANK and produced two N-terminal fragments in vitro (60 and 45 kDa).
FIGURE 5.

Stability of the cleaved TANK fragments. A and B, HEK293T cells were transfected with a plasmid encoding FLAG-TANK or its cleaved fragments in the presence or absence of MG132 (10 μm). The fixed cells were stained with anti-FLAG antibody, and the localization of TANK and its mutants was visualized by confocal microscopy (A). The cell lysates were analyzed by Western blotting (WB) (B). C and D, HEK293T cells were cotransfected with a plasmid expressing FLAG-TANK-291N (C) or TANK(197C) (D), in combination with a plasmid expressing HA-EMCV 3C or its mutant, in the presence or absence of the proteasome inhibitor MG132 (10 μm). The cell lysates were analyzed with antibodies against FLAG, HA, and GAPDH. E, HEK293T cells were transfected with a plasmid expressing TANK(197C) or TANK(197–291) and then treated with or without MG132; the protein levels of TANK(197C) and TANK(197–291) were analyzed with anti-FLAG antibody. F, schematic of human TANK showing the positions of EMCV 3C cleavage sites (*) at Gln-197 and Gln-291 and the cleaved fragments.
To test whether TANK interacts with EMCV 3C, EMCV 3C and FLAG-TANK were coexpressed in HEK293T cells, and co-immunoprecipitation assay was performed. Our data illustrated that EMCV 3C was co-immunoprecipitated with FLAG-TANK and its cleaved fragments, despite the fact that TANK had been cleaved into two N-terminal fragments (Fig. 1D), suggesting that TANK and its cleaved N-terminal fragments specifically interacted with EMCV 3C.
EMCV 3C Protease Activity Is Required for TANK Cleavage
To test whether other EMCV proteins or EMCV 3C-processing intermediates are capable of cleaving TANK, these EMCV proteins were coexpressed with FLAG-TANK in HEK293T cells. As expected, only EMCV 3C and its processing intermediates (3ABCD, 3ABC, and 3CD) possess the ability to cleave TANK (Fig. 2, A and B). These results were consistent with the fact that only EMCV 3C and its processing intermediates have protease activity.
FIGURE 2.

EMCV 3C protease activity is required for TANK cleavage. A and B, HEK293T cells were transfected with a plasmid expressing FLAG-TANK alone or along with a plasmid expressing individual HA-EMCV protein (A) or EMCV 3C-processing intermediates (B). At 36 hpt, the cell lysates were analyzed by Western blotting (WB) with antibodies against HA, FLAG, and GAPDH. C, schematic of EMCV 3C constructs showing the positions of EMCV 3C protease activity sites (*) at H46A and C159A. D, HEK293T cells were transfected with a plasmid encoding FLAG-TANK alone or in combination with a plasmid expressing HA-EMCV 3C or its mutants. The cell lysates were prepared and analyzed by Western blotting.
To determine whether TANK cleavage by EMCV 3C is dependent on its protease activity, we constructed three plasmids expressing different mutant versions of EMCV 3C (H46A, C159A, and H46A/C159A) (Fig. 2C) and utilized them to analyze TANK cleavage. As shown in Fig. 2D, EMCV 3C cleaved TANK and produced two N-terminal fragments, whereas the three mutants of EMCV 3C failed to cleave TANK.
Apoptotic Caspases Are Not Required for TANK Cleavage
To investigate whether EMCV 3C-mediated TANK cleavage is dependent on apoptosis-related cysteine caspase activity, proteasome or lysosome signaling, HEK293T cells expressing FLAG-TANK and HA-EMCV 3C were treated with a pan-caspase inhibitor (Z-VAD-FMK), proteasome inhibitor (MG132), or lysosome inhibitor (NH4Cl), respectively. We found that none of the three inhibitors prevented EMCV 3C-mediated TANK cleavage (Fig. 3A, lanes 3–6). Consistent with these results, we found that TANK could not be cleaved in puromycin-induced apoptotic cells (Fig. 3B), although pro-caspase-3 and its substrate PARP1 were cleaved, which demonstrated that pro-caspase-3 was activated.
FIGURE 3.
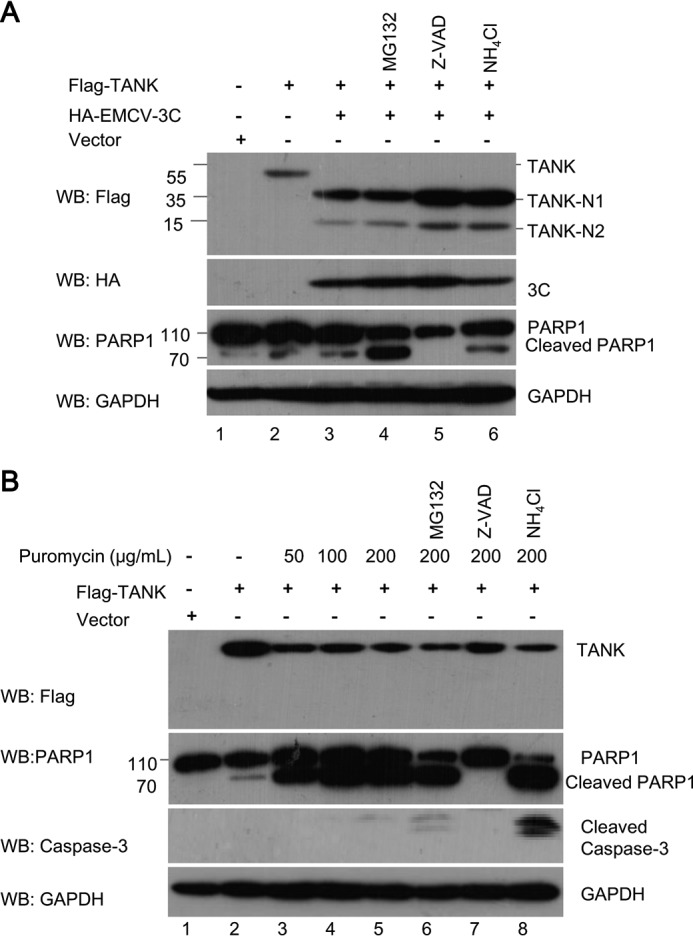
Apoptotic caspases are not required for TANK cleavage. A, HEK293T cells were transfected with a plasmid expressing FLAG-TANK, along with a plasmid expressing HA-EMCV 3C or empty vector. At 24 hpt, the cells were pretreated with MG132 (10 μm), Z-VAD-FMK (50 μm), or NH4Cl (10 mm) for another 4–8 h, and the cell lysates were analyzed by Western blotting (WB). B, HEK293T cells were transfected with a plasmid expressing FLAG-TANK. At 24 hpt, the cells were pretreated with MG132, Z-VAD-FMK, or NH4Cl as in A, and the cells were treated with different doses of puromycin as indicated. The cell lysates were analyzed by Western blotting.
TANK Is Cleaved at Gln-197 and Gln-291 by EMCV 3C
Our results indicated that EMCV 3C cleaved TANK and yielded two N-terminal fragments, TANK-N1 and TANK-N2 (Fig. 2, A–C). It has been demonstrated that EMCV 3C preferentially cleaves Gln-Ala, Gln-Ser, and Gln-Cys amino acid pairs (33–35). According to these data, we examined the amino acid sequence of TANK for potential EMCV 3C cleavage sites and inferred that at least two potential EMCV 3C cleavage sites may exist between amino acids 180–210 and 260–310 within human TANK. As shown in Fig. 4A, the two regions bear several glutamines (Gln) that resemble the signature Gln-Gly and Gln-Ala sequences of proteolytic sites for EMCV 3C (Fig. 4F).
FIGURE 4.

Mapping the specific cleavage sites within TANK. A, sequence of amino acids 180–307 within human TANK. In this region, glutamine residues (Q) (labeled in red) at 182, 190, 197, 266, 291, or 301 were substituted with arginine residue (R) or alanine residue (A). B and C, HEK293T cells were cotransfected with a plasmid expressing FLAG-TANK or its mutants alone or in combination with a plasmid expressing HA-EMCV 3C. At 24 hpt, the cell lysates were analyzed by Western blotting (WB). D, schematic of human TANK showing the positions of EMCV 3C cleavage sites (*) at Gln-197 and Gln-291 and its deletion mutant constructs. E, EMCV 3C cleaves TANK proteins from different species. HEK293T cells were transfected with a plasmid encoding FLAG-tagged human, mouse, monkey, or swine TANK, in combination with a plasmid expressing HA-EMCV 3C. At 36 hpt, cell lysates were analyzed with anti-FLAG, HA, and GAPDH antibodies as indicated. F, alignment of the junction sequences of viral proteins of EMCV (UniProtKB: P03304) and putative cleavage sites within TANK from different species. +1 means amino acids residues cleavage sites of EMCV 3C.
To define the putative cleavage site, a series of TANK substitution mutants were constructed in which the invariant glutamine at each potential P1 position was replaced with arginine or alanine (Fig. 4A). TANK or its mutants were coexpressed with EMCV 3C in HEK293T cells. As shown in Fig. 4B (lanes 8 and 12), TANKQ197R and TANKQ291A were partially resistant to EMCV 3C. In contrast to the Q197R and Q291A mutants, mutations at other glutamine residues (Q182A, Q190A, Q266A, and Q301A) did not prevent EMCV 3C cleavage. To further test whether Gln-197 and Gln-291 are indeed the cleavage sites, we generated a Q197R/Q291A double mutant (TANK-DM) and coexpressed it with EMCV 3C. As shown in Fig. 4C, we found that TANK-DM was completely resistant to EMCV 3C. Taken together, these findings suggest that TANK is cleaved by EMCV 3C at the 197 and 291 glutamine residues, producing at least four fragments (Fig. 4D). Interestingly, we also found that EMCV 3C cleaved mouse and monkey TANK and yielded two N-terminal fragments (Fig. 4E, 4th and 6th lanes), whereas EMCV 3C cleaved swine TANK to produce only one fragment (Fig. 4E, 8th lane). Compared with human TANK and mouse TANK, Glu-285 in swine TANK is not conserved, which may not be recognized and cleaved by EMCV 3C. These data suggest that TANK proteins from different species are able to be cleaved by EMCV 3C, which is consistent with the fact that EMCV has a broad range of susceptible hosts.
C-terminal Fragment of TANK Is Unstable
To test the stability of TANK and its cleaved fragments, HEK293T cells were transfected with a plasmid expressing TANK or its deletion mutants corresponding to the cleaved fragments in the presence or absence of the proteasome inhibitor MG132. We found that TANK-291C was an unstable fragment, which degraded via the ubiquitin-proteasome system because the proteasome inhibitor MG132 could block its degradation (Fig. 5, A and B). To test whether the TANK-291N and TANK(197C) fragments are cleaved by EMCV 3C and dependent on EMCV 3C protease activity, the two fragments were coexpressed with EMCV 3C or EMCV 3C-DM in the presence or absence of MG132. As shown in Fig. 5, C and D, TANK-291N and TANK(197C) were cleaved by EMCV 3C, but not EMCV 3C-DM. In addition, we also found that the TANK(197C) was an unstable fragment, which could be cleaved by EMCV 3C, yielding two unstable fragments (Fig. 5, E and F).
TANK Is Cleaved at Gln-291 in EMCV-infected Cells
To test whether TANK is cleaved during EMCV infection, HEK293T cells were either mock-infected or infected with EMCV at different multiplicities of infection, and the integrity of TANK was investigated. As shown in Fig. 6A, upon infection with increasing doses of EMCV, the levels of endogenous TANK decreased in a dose-dependent manner, and only a 35-kDa cleavage product (designated TANK-N1) was recognized by anti-TANK antibody against N terminus of TANK. Consistent with this result, a 35-kDa fragment was observed when HEK293T cells expressing FLAG-TANK were infected with the same dose of EMCV (Fig. 6B, lanes 3–5). As EMCV infection can induce apoptosis of target cells, to test whether EMCV-induced TANK cleavage is associated with apoptosis, TANK cleavage following EMCV infection was analyzed in the presence of Z-VAD-FMK, MG132, or NH4Cl. As shown in Fig. 6B (lanes 5, 6, and 8), PARP1 was cleaved when cells were infected with EMCV at an m.o.i. of 1.0. However, TANK cleavage in EMCV-infected cells could not be blocked by Z-VAD-FMK, MG132, or NH4Cl (Fig. 6B, lanes 6–8). These results demonstrate that TANK could not be cleaved by apoptotically related caspases and proteasome or lysosome signaling did not participate in TANK cleavage.
FIGURE 6.

TANK was cleaved at the 291 glutamine in EMCV-infected cells. A, HEK293T cells were either mock-infected or infected with EMCV HB10 at an m.o.i. of 0.01, 0.1, or 1.0, respectively. After 12 h, the whole cell lysates were analyzed by Western blotting. B, HEK293T cells were transfected with a plasmid expressing FLAG-TANK and treated with or without MG132 (10 μm), Z-VAD-FMK (50 μm), or NH4Cl (10 mm) for another 4–8 h. The cells were then infected with EMCV for another 12 h, and the cell lysates were analyzed by Western blotting (WB). C, HEK293T cells were transfected with a plasmid expressing FLAG-TANK or its mutants (Q197R or Q291A) as indicated and then infected with EMCV (m.o.i. 0.1) for another 12 h. The cell lysates were analyzed by Western blotting. D, HEK293T cells expressing FLAG-TANK-HA were treated with or without MG132 and then mock-infected or infected with EMCV at an m.o.i. of 0.01, 0.1, or 1.0, respectively. After 12 h, the cells were analyzed by Western blotting. E, schematic of human TANK showing the positions of EMCV 3C cleavage sites at Gln-291 and its cleaved fragments of human TANK.
Based on these results, we proposed that TANK might be preferentially cleaved at the Gln-291 but not at Gln-197 during EMCV infection. To test this hypothesis, HEK293T cells expressing TANK or its mutants were infected with EMCV as indicated. As shown in Fig. 6C (lanes 5–8), TANK-Q291A and TANK-DM were completely resistant to cleaving during EMCV infection, but not TANK and TANK-Q197R. To further confirm these results, a plasmid expressing dual-tagged TANK (FLAG-TANK-HA containing a FLAG tag at the N-terminal end and an HA tag at the C-terminal end of TANK) was transfected into HEK293T cells, which were then infected with EMCV in the presence or absence of MG132. Proteolysis of TANK was examined with anti-FLAG and anti-HA antibodies. Consistent with our previous results, a 35-kDa N-terminal fragment was detected by anti-FLAG antibody (Fig. 6D, lanes 1–4). However, the C-terminal fragment(s) could not be detected with anti-HA antibody in the absence of MG132. In addition, we noticed that a 15-kDa C-terminal fragment was detected by Western blotting with anti-HA antibody in the presence of MG132. These data indicate that the C-terminal fragment of TANK produced by EMCV 3C is unstable, which is degraded via the ubiquitin-proteasome system (Fig. 6D, lanes 7 and 8).
Taken together, our findings demonstrate that TANK is specifically cleaved at Gln-291 during EMCV infection, yielding two fragments (TANK-291N and TANK (291C)), and TANK (291C) is an unstable fragment that is degraded via the proteasome signaling pathway (Fig. 6E).
Cleavage of TANK Is Involved in NF-κB Activation during EMCV Infection
To detect the effect of EMCV infection in the NF-κB signaling pathway, HEK293T cells were infected with EMCV at the different times. As shown in Fig. 7, A and B, we found that TANK cleavage was accompanied by the expression of EMCV 3C, and the mRNA level of TNFα was significantly increased along with the EMCV titer. In addition, we observed that IκB was degraded, and phosphorylation of p65 was increased, whereas the EMCV titer is increased (Fig. 7C). These results suggest that EMCV infection activates NF-κB signaling in HEK293T cells. To investigate whether TANK affects EMCV-induced NF-κB activation, HEK293T cells were transfected with control shRNA (shNC) or shRNA targeting TANK (shTANK), along with the NF-κB-Luc reporter gene. The cells were then infected with EMCV. We found that knockdown of TANK expression significantly increased NF-κB promoter activity in the mock-infected cells (Fig. 7D, columns 2 and 3). However, knockdown of TANK expression did not significantly reduce NF-κB activation when the cells were infected with EMCV (Fig. 7D, columns 4 and 6). To test whether TANK cleavage affects NF-κB activation during EMCV infection, HEK293T cells were transfected with plasmids expressing NF-κB-Luc, TANK, or TANK-DM and then infected with EMCV at an m.o.i. of 1.0. As shown in Fig. 7E, compared with TANK, TANK-DM inhibited EMCV-mediated NF-κB-Luc promoter activity. These results demonstrate that cleavage of TANK is involved in EMCV infection-mediated NF-κB activation.
FIGURE 7.

Cleavage of TANK by EMCV 3C promotes NF-κB activation. A–C, HEK293T cells were infected with EMCV at an m.o.i. of 10 for 12 h, and the cells were collected at indicated time points to detect EMCV titer by TCID50 assay (A). The mRNA levels of TNFα were analyzed by quantitative RT-PCR (B). The expression of p65, p65-p, IκB, TANK, 3C, and GAPDH were analyzed by Western blotting (WB) with the indicated antibodies (C). Data are representative of three independent experiments performed in triplicate. D, HEK293T cells were transfected with either control shRNA (shNC) or shRNA targeting TANK (shTANK) in combination with an NF-κB-Luc reporter gene and pRL-SV40 plasmid. At 48 hpt, the cells were either mock-infected or infected with EMCV at an m.o.i. of 1.0 for 12 h. Luciferase assay was performed, and the expression of TNAK, 3C, and GAPDH was detected by Western blotting. E, HEK293T cells were transfected with a plasmid expressing wild-type TANK (TANK-wt) or TANK double mutant Q197R/Q291A (TANK-DM) in combination with an NF-κB-Luc reporter gene and pRL-SV40. At 24 hpt, the cells were either mock-infected or infected with EMCV at an m.o.i. of 1.0 for 12 h. Luciferase assay was performed, and the expressions of TNAK, 3C, and GAPDH were detected by Western blotting. Data are representative of the independent experiments performed in triplicate. *, 0.01 < p < 0.05; **, p < 0.01; ***, p < 0.001; NS, not statistically significant.
To detect which protein encoded by EMCV plays major roles in activating NF-κB signaling, a plasmid expressing individual EMCV protein was transfected into HEK293T cells along with NF-κB-Luc. As a result, ectopic expression of EMCV 2B and 3C significantly enhanced the activation of the NF-κB promoter, whereas the structural proteins (VP1–VP4) and 2A, 3AB, and 3D had little impact on NF-κB activation (data not shown). Recently, the effect of 2B on the NF-κB signaling pathway has been reported (3). However, the molecular mechanism of EMCV 3C-mediated NF-κB activation is not fully understood.
EMCV 3C Enhances TRAF6-mediated NF-κB Signaling by Targeting TANK
It has been demonstrated that TRAF6 is required for NF-κB activity in the IL-1R signaling pathway. TANK associates with TRAF6 and inhibits TRAF6-mediated NF-κB activity (12, 36, 37). In this study, we found that TANK is cleaved by EMCV 3C during EMCV infection. To test whether cleavage of TANK by EMCV 3C impairs its effects on TRAF6-mediated NF-κB activation, TANK or its mutants were coexpressed with EMCV 3C and TRAF6 in HEK293T cells. As shown in Fig. 8A, TANK and its mutants inhibited TRAF6-mediated NF-κB promoter activation. Interestingly, the inhibitory effect of TANK on TRAF6 was relieved by EMCV 3C. The result suggests that the cleavage of TANK by EMCV 3C is required to relieve TANK inhibitory effect on NF-κB signaling. To further test the effects of TANK cleavage fragments on TRAF6-mediated NF-κB signaling, HEK293T cells were transfected with plasmids expressing TANK or its cleaved fragments, along with the NF-κB-Luc reporter gene. As shown in Fig. 8, B and C, TANK, but not its cleaved N-terminal fragments, inhibited TRAF6-mediated NF-κB promoter activation, although the full-length TANK and N-terminal fragments of TANK (TANK-197N and TANK-291N) still bound TRAF6. Taken together, our data show that TANK cleavage by EMCV 3C relieves its inhibitory effect on TRAF6-mediated NF-κB signaling, indicating that intact TANK is required for the inhibition of TRAF6-mediated NF-κB-Luc activation. These findings are consistent with previous observations that TANK negatively regulates TRAF6 function (37).
FIGURE 8.

Cleavage of TANK enhances TRAF6-mediated NF-κB activation. A, HEK293T cells were transfected with plasmids expressing TRAF6, TANK-WT, or TANK-DM, in combination with a plasmid expressing HA-EMCV 3C, along with NF-κB-Luc and pRL-SV40. At 24 hpt, luciferase activity was measured. The expression of TRAF6, TANK, EMCV 3C, and GAPDH were analyzed by Western blotting (WB) with indicated antibodies. B, HEK293T cells were transfected with plasmids expressing TRAF6 and TANK or its cleaved fragments, in combination with NF-κB-Luc and pRL-SV40. At 24 hpt, luciferase assay was performed. The expressions of TRAF6, TANK, and GAPDH were analyzed by Western blotting with the indicated antibodies. C, HEK293T cells were transfected with plasmids expressing TRAF6 and TANK or its cleaved fragments. The cell lysates were immunoprecipitated (IP) with anti-FLAG antibody, and immunoprecipitants and aliquots of whole cell lysates (WCL) were analyzed by Western blotting. D, HEK293T cells were transfected with plasmids expressing FLAG-TRAF6 and HA-ubiquitin-Lys-63, along with a plasmid encoding Myc-TANK or its double mutant, in combination with a plasmid expressing EMCV 3C. FLAG-TRAF6 was immunoprecipitated with anti-FLAG antibody. The polyubiquitination of TRAF6 was detected with anti-HA antibody. E and F, HEK293T cells were transfected with a plasmid expressing GFP-p65 alone or in combination with a plasmid expressing TRAF6, along with a plasmid expressing EMCV-3C or its double mutant. The cells that GFP-p65 localized in the nucleus were numbered, and the expression of GFP-p65, TRAF6, TANK, 3C, and GAPDH were detected by Western blotting (E). GFP-p65 localization was visualized by confocal microscope (F). **, p < 0.01; ***, p < 0.001.
TRAF6 is an E3 ubiquitin ligase that serves as a platform to recruit the kinase TAK1 and the IKK complex, which subsequently activates NF-κB-dependent target genes in the IL-1R signaling pathway. Following stimulation, TRAF6 begins to oligomerize and activates its ubiquitin ligase activity, which leads to lysine 63 (Lys-63)-linked polyubiquitination of target proteins such as NEMO and TRAF6 itself (38). It has been demonstrated that TANK could inhibit TRAF6 polyubiquitination (12). To investigate whether EMCV 3C affects TRAF6 polyubiquitination, HEK293T cells were transfected with plasmids expressing TRAF6 and HA-Ub-K63, along with plasmids expressing TANK or TANK-DM and EMCV 3C. As shown in Fig. 8D, we found that both TANK and TANK-DM inhibited TRAF6 polyubiquitination (Fig. 8D, lanes 4 and 5). However, EMCV 3C relieved the TANK inhibitory effect on TRAF6 polyubiquitination but not TANK-DM (Fig. 8D, lanes 7 and 8). In addition, we found that TRAF6 overexpression promoted the nuclear translocation of the NF-κB subunit (p65), which was inhibited by TANK (Fig. 8, E and F). Interestingly, we also found that EMCV 3C, but not EMCV 3C-DM, relieved the inhibitory effect of TANK on the TRAF6-mediated nuclear translocation of p65 (Fig. 8, E and F).
TANK Is Cleaved by FMDV 3C Protease
To test whether TANK is cleaved by proteases encoded by other members of picornaviridae, the BHK-21 cells expressing FLAG-TANK were infected with FMDV, EV71, or CVB3 (Table. 1), respectively. The cells were harvested when the cytopathic effects were visible and the cell lysates were analyzed with the antibodies as indicated. As shown in Fig. 9, A, D, and F, TANK was cleaved in FMDV-infected cells, but not in EV71- and CVB3-infected cells. The FMDV genome encodes two proteases, FMDV L and FMDV 3C. Therefore, we continued to test which protease could cleave TANK. As shown in Fig. 9, B and C, TANK was cleaved by FMDV 3C and produced a 15-kDa N-terminal fragment. The abundance of TANK was reduced when FMDV L was coexpressed with TANK, but no cleaved fragment was detected (Fig. 9B, lanes 2 and 3). In addition, we found TANK cleavage by FMDV 3C is dependent on its protease activity because the protease with activity site mutant (H46Y), designated as FMDV 3C-M, did not cleave TANK (Fig. 9C). We also noticed that EV71 3C protease (EV71 3C) did not cleave TANK (Fig. 9E).
FIGURE 9.

FMDV 3C cleaves TANK. A, BHK-21 cells were transfected with a plasmid expressing FLAG-TANK. At 36 hpt, the cells were either mock-infected or infected with different doses of FMDV at an m.o.i. of 0, 0.001, 0.01, 0.1, or 5, respectively. At 8 hpi, the cell lysates were analyzed by Western blotting (WB) with anti-FLAG, anti-FMDV VP2, or anti-GAPDH antibodies. B and C, HEK293T cells were cotransfected with plasmids expressing FLAG-TANK and FMDV L protease (FMDV L) or FMDV 3C (B) or FMDV 3C or its double mutant (FMDV 3C-DM) (C). The cell lysates were analyzed by Western blotting with anti-FLAG or anti-HA antibody. D and E, HEK293T cells were transfected with a plasmid expressing FLAG-TANK and then infected with different doses of EV71 (D) or FLAG-TANK and EV71 3C or its mutants were coexpressed in HEK293T cells for 24 h (E). The cell lysates were analyzed by Western blotting. F, HEK293T cells were transfected with a plasmid expressing FLAG-TANK and then infected with different doses of CVB3 for 12 h as indicated. The cell lysates were analyzed by Western blotting. Anti-eIF4G antibody was used as a positive control.
TANK Is Cleaved by Other Viral Proteases
The viral genomic RNAs of positive RNA viruses are translated into one or two polyproteins. The polyprotein(s) can be processed into mature functional proteins by viral proteases (1, 39, 40). It has been proposed that these viral proteases could cleave host proteins to evade host innate immune response (39–41). To test whether TANK is cleaved by these viral proteases, TANK was coexpressed with these major proteases encoded by several positive RNA viruses belonging to the Arteriviridae, Coronaviridae, and Flaviviridae (Table 1). As shown in Fig. 10, we found that TANK was cleaved by PRRSV nonstructural protein 4 (Nsp4) and EAV Nsp4, but not NS3 proteases encoded by SARS NS3, TGEV NS3, JEV NS3, and CSFV NS3. These results demonstrate that TANK is a novel common target of several positive RNA viral proteases. Further investigation of the cleavage sites and effects of the cleavage products on the host antiviral response and viral replication are warranted.
FIGURE 10.

TANK is cleaved by other viral proteases. HEK293T cells were transfected with a plasmid encoding FLAG-TANK-Myc, in combination with a plasmid expressing individual viral proteases encoded by various positive RNA viruses as indicated (Table 1). The cell lysates were subjected to Western blotting (WB) analysis with antibodies against HA, FLAG, or Myc.
Discussion
EMCV 3C Cleaves TANK
Many viruses have evolved different strategies to evade host innate immune responses. For example, some viral proteases can inhibit type I IFN production and NF-κB signaling through the cleavage of receptors, adaptors, and regulators involved in these two pathways (30–32). Zaragoza et al. (42) reported that coxsackievirus 3C protease cleaves the inhibitor of NF-κBα (IκBα), and the proteolytic fragment forms a stable complex with NF-κB. The complex is translocated to the nucleus where it inhibits NF-κB transcriptional activation, which consequently increases cell apoptosis and decreases viral replication (42). Recently, NEMO, an adaptor that bridges the NF-κB and type I IFN signaling (43), is identified as a substrate of FMDV 3C and PRRSV Nsp4 (32, 44). Cleavage of NEMO impairs its ability to induce downstream IFN production and act as a signaling adaptor in the RIG-I/MDA5 pathway (32, 44).
EMCV 3C is the only cysteine protease encoded by the EMCV genome. Once EMCV 3C is translated, it is immediately activated and starts to cleave the two viral polyproteins and host proteins in cis or in trans with a high degree of substrate specificity (1). Through analysis of these known cleavage sites within the EMCV polyprotein, we found that EMCV 3C preferentially cleaves between Gln or Glu and Gly, Ser, or Ala residues. Recently, several cellular substrates of EMCV 3C were reported. RIG-I is found to be cleaved by EMCV 3C, which may explain why RIG-I is not the functioning sensor for EMCV RNA (45). Poly(A)-binding protein is also found as a substrate of EMCV 3C. EMCV 3C cleaves poly(A)-binding protein at a specific site, which inhibits EMCV replication (46).
In this study, we found that TANK is a novel substrate of EMCV 3C, which is cleaved at Gln-291 to yield an N-terminal fragment in EMCV-infected cells. However, TANK is cleaved at Gln-197 and Gln-291 by EMCV 3C in vitro, yielding at least five cleaved fragments (Figs. 4 and 5). Interestingly, TANK(197–291) and TANK(291C) fragments are unstable and rapidly degraded via the ubiquitin-proteasome system (Fig. 5, C and D). Why EMCV 3C does not cleave TANK at Gln-197 during EMCV infection remains unclear. We speculated that EMCV 3C might interact with other viral proteins or host proteins during EMCV replication, which blocks the TANK Gln-197 site to expose to EMCV 3C through steric hindrance. However, there are also some other possibilities. EMCV 3C might preferentially cleave TANK at the Gln-Gly rather than Gln-Ala or a much lower amount of EMCV 3C is produced during EMCV infection compared with EMCV 3C overexpression in vitro.
Cleavage of TANK Enhances TRAF6-mediated NF-κB Activation
It has been demonstrated that mice lacking the p50 subunit of NF-κB exhibit multifocal defects in immune responses involving B lymphocytes and nonspecific responses to infection. Additionally, mice lacking p50 are more resistant to infection with murine EMCV (47). The survival rate of p50−/− mice is higher than that of p50+/+ mice after EMCV infection, which is tightly linked to the animals' ability to clear the virus from the heart in vivo (48). Kawagoe et al. (37) also demonstrated that macrophages and B cells isolated from tank−/− mice have a greater potential to activate canonical NF-κB signaling in response to the stimulation of TLRs and B cell receptors. Recently, Ito et al. (3) reported that EMCV 2B is sufficient to induce the NLRP3 inflammasome activation and the IL-1β secretion. As a result, secreted IL-1β can bind to IL-1 receptors and activate NF-κB. These data suggest that NF-κB signaling is important for the EMCV-mediated pathogenicity.
TANK is identified as a TRAF2- and TRAF6-interacting protein (11, 49). It has been demonstrated that TANK contains a positive regulatory domain at its N terminus (N-TANK, 1–190 amino acids) and a negative regulatory domain at its C terminus (C-TANK) (11). N-TANK can activate NF-κB synergistically with full-length TRAF2, whereas C-TANK has a potent inhibitory effect. The synergistic activation of NF-κB by TRAF2 and N-TANK is completely abolished when equal amounts of N-TANK and C-TANK plasmids are cotransfected with TRAF2 (11). Interestingly, TANK inhibits the activation of NF-κB by TNFα, IL-1, and CD40 through inhibition of TRAF6-mediated NF-κB activation (11, 12, 37, 50). Consistent with these results, our results reveal that TANK inhibits TRAF6-mediated NF-κB activity (Fig. 8, A and B). Furthermore, we found that N-terminal fragments of TANK produced by EMCV 3C lose their abilities to inhibit TRAF6-mediated NF-κB activation, although the cleavage products TANK-197N and TANK-291N could still interact with TRAF6 (Fig. 8C). Our findings reveal that the inhibitory TANK (291C) is degraded, which provides one clue why the inhibitory effect of TANK on TRAF6-mediated NF-κB activation is relieved by EMCV 3C in EMCV-infected cells.
It has been demonstrated that TANK suppresses TLR signaling by down-regulating TRAF6 auto-ubiquitination (12) and that TLR-induced polyubiquitination of TRAF6 is up-regulated in tank−/− macrophages (37). In agreement with these results, we found that TANK inhibits the polyubiquitination of TRAF6 (Fig. 8D, lane 4), and the cleavage of TANK by EMCV 3C enhances the polyubiquitination of TRAF6 (Fig. 8D, lane 7). Our data suggest that cleavage of TANK is required for the activation of NF-κB in EMCV-infected cells. In summary, the cleavage of TANK by EMCV 3C relieves the inhibitory effect of TANK on TRAF6-mediated NF-κB activation by enhancing TRAF6 polyubiquitination, which may partially explain why NF-κB is activated in EMCV-infected cells.
Cleavage of TANK Is a Novel Strategy for Modulating NF-κB Signaling by Several Viruses
Post-translational modification of TANK is important for its functions (8, 51–53). TNFα stimulation promotes TANK recruitment to the IKK complex, where TANK is phosphorylated by IKKβ, and the interaction between TANK and NEMO is reduced (51). Recently, Shi et al. (53) demonstrated that the mitochondrial outer membrane protein MARCH5 interacts with TANK and catalyzes its Lys-63-linked polyubiquitination, which impairs its ability to inhibit TRAF6-mediated NF-κB signal transduction. To our knowledge, this is the first report that EMCV 3C specifically cleaves TANK in vitro. Interestingly, we found that EMCV 3C is not the only protease in picornaviruses that targets TANK. FMDV 3C also cleaves TANK, although the cleavage sites are different (Fig. 9, A–C). Additionally, we found that TANK is cleaved by PRRSV Nsp4 and EAV Nsp4, which are the main proteases required for viral polyprotein cleavage (39–41). However, four other viral proteases encoded by different members of coronaviridae and flaviviridae are unable to cleave TANK (Fig. 10). Therefore, we proposed that TANK is a novel target of some certain single-stranded positive-sense RNA viruses, which is involved in viral infection-mediated NF-κB activation.
Previous reports demonstrated that TRAF6 participates in the NF-κB signaling pathway (12, 54, 55). The absence of TRAF6 results in enhancement of viral replication and a significant reduction in the production of IL-6 and type I IFN after viral infection. Activation of NF-κB and IRF7 is also significantly impaired during RIG-I-like helicase signaling in the absence of TRAF6 (14). As TANK is a known binding partner of TRAF6, the cleavage of TANK by different viral proteases may represent a common mechanism to regulate host innate immune and inflammatory responses. However, the importance of viral protease-mediated cleavage of TANK and the functions of the cleaved fragments under normal physiological conditions remain unknown.
In this study, we are the first to demonstrate that the cleavage of TANK by EMCV 3C impairs the ability of TANK to inhibit TRAF6-mediated NF-κB signaling (Fig. 8). Cleavage of TANK is a novel common mechanism for some positive RNA viruses to evade the host innate immune responses. Our findings provide a novel insight on the prevention and treatment of virus-related diseases.
Author Contributions
C. J. W. conceived and coordinated the study and wrote the paper. L. H., Q. F. L., L. J. Z., X.H. C., and Q. Z. designed, performed, and analyzed the experiments shown in Figs. 1–5. L. I. H., C. Y. L., S. N. W., J. N. L. designed, performed, and analyzed the experiments shown in Figs. 6–10. S. J. C., T. X., H. B. Y., and L. Y. prepared EMCV and FMDV. X. J. W. prepared EAV. Y. A. W., G. H. D., and Z. H. Z. designed, performed, and analyzed the experiments shown in Figs. 9 and 10. X. S. X. and Y. F. Z. made the monoclonal antibodies.
Acknowledgments
We are grateful to Dr. Zhijun Tian for providing us with the PRRSV-HuN4 strain. We thank Dr. Li Feng and Dr. Ronghong Hua for providing the TGEV and JEV stains. We thank Dr. Wenhai Feng and Dr. Hong Tang for critical review of the manuscript.
This work was supported by the National Natural Science Foundation of China Grants 31172333 and 31300139 (to C. J. W. and L. H.) and State Key Laboratory of Veterinary Biotechnological Foundation Grant SKLVBP201432 (to L. H.). The authors declare that they have no conflict of interest with the contents of this article.
- EMCV
- encephalomyocarditis virus
- TANK
- TRAF family member-associated NF-κB activator
- FMDV
- foot and mouth diseases virus
- PRRSV
- porcine reproductive and respiratory syndrome virus
- EAV
- equine arteritis virus
- hpt
- hours post-transfection
- TRITC
- tetramethylrhodamine isothiocyanate
- Z
- benzyloxycarbonyl
- FMK
- fluoromethyl ketone
- m.o.i.
- multiplicity of infection
- IKK
- IκB kinase
- TLR
- toll-like receptor.
References
- 1.Carocci M., and Bakkali-Kassimi L. (2012) The encephalomyocarditis virus. Virulence 3, 351–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Loughran G., Firth A. E., and Atkins J. F. (2011) Ribosomal frameshifting into an overlapping gene in the 2B-encoding region of the cardiovirus genome. Proc. Natl. Acad. Sci. U.S.A. 108, E1111–E1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ito M., Yanagi Y., and Ichinohe T. (2012) Encephalomyocarditis virus viroporin 2B activates NLRP3 inflammasome. PLoS Pathog. 8, e1002857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ng C. S., Jogi M., Yoo J. S., Onomoto K., Koike S., Iwasaki T., Yoneyama M., Kato H., and Fujita T. (2013) Encephalomyocarditis virus disrupts stress granules, the critical platform for triggering antiviral innate immune responses. J. Virol. 87, 9511–9522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Napetschnig J., and Wu H. (2013) Molecular basis of NF-κB signaling. Annu. Rev. Biophys. 42, 443–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kawai T., and Akira S. (2007) Signaling to NF-κB by Toll-like receptors. Trends Mol. Med. 13, 460–469 [DOI] [PubMed] [Google Scholar]
- 7.Israël A. (2010) The IKK complex, a central regulator of NF-κB activation. Cold Spring Harb. Perspect. Biol. 2, a000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Häcker H., and Karin M. (2006) Regulation and function of IKK and IKK-related kinases. Science's STKE 2006, re13. [DOI] [PubMed] [Google Scholar]
- 9.Sun S. C. (2012) The noncanonical NF-κB pathway. Immunol. Rev. 246, 125–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiu Y. H., Zhao M., and Chen Z. J. (2009) Ubiquitin in NF-κB signaling. Chem. Rev. 109, 1549–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng G., and Baltimore D. (1996) TANK, a co-inducer with TRAF2 of TNF- and CD 40L-mediated NF-κB activation. Genes Dev. 10, 963–973 [DOI] [PubMed] [Google Scholar]
- 12.Maruyama K., Kawagoe T., Kondo T., Akira S., and Takeuchi O. (2012) TRAF family member-associated NF-κB activator (TANK) is a negative regulator of osteoclastogenesis and bone formation. J. Biol. Chem. 287, 29114–29124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lomaga M. A., Yeh W. C., Sarosi I., Duncan G. S., Furlonger C., Ho A., Morony S., Capparelli C., Van G., Kaufman S., van der Heiden A., Itie A., Wakeham A., Khoo W., Sasaki T., et al. (1999) TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 13, 1015–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Konno H., Yamamoto T., Yamazaki K., Gohda J., Akiyama T., Semba K., Goto H., Kato A., Yujiri T., Imai T., Kawaguchi Y., Su B., Takeuchi O., Akira S., Tsunetsugu-Yokota Y., and Inoue J. (2009) TRAF6 establishes innate immune responses by activating NF-κB and IRF7 upon sensing cytosolic viral RNA and DNA. PLoS One 4, e5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu H., and Arron J. R. (2003) TRAF6, a molecular bridge spanning adaptive immunity, innate immunity and osteoimmunology. BioEssays 25, 1096–1105 [DOI] [PubMed] [Google Scholar]
- 16.Matsumura T., Degawa T., Takii T., Hayashi H., Okamoto T., Inoue J., and Onozaki K. (2003) TRAF6-NF-κB pathway is essential for interleukin-1-induced TLR2 expression and its functional response to TLR2 ligand in murine hepatocytes. Immunology 109, 127–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loiarro M., Ruggiero V., and Sette C.. Targeting TLR/IL-1R signalling in human diseases. Mediators Inflamm. 2010, 674363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loiarro M., Ruggiero V., and Sette C. (2013) Targeting the Toll-like receptor/interleukin 1 receptor pathway in human diseases: rational design of MyD88 inhibitors. Clin. Lymphoma Myeloma Leuk. 13, 222–226 [DOI] [PubMed] [Google Scholar]
- 19.O'Neill L. A. (2008) The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol. Rev. 226, 10–18 [DOI] [PubMed] [Google Scholar]
- 20.Oeckinghaus A., Hayden M. S., and Ghosh S. (2011) Crosstalk in NF-κB signaling pathways. Nat. Immunol. 12, 695–708 [DOI] [PubMed] [Google Scholar]
- 21.Wang Z., Liu Y., Lin W., and Cui S. (2012) A real-time PCR to detect and analyze virulent EMCV loads in sows and piglets. Mol. Biol. Rep. 39, 10013–10017 [DOI] [PubMed] [Google Scholar]
- 22.Xue M., Wang H., Li W., Zhou G., Tu Y., and Yu L. (2012) Effects of amino acid substitutions in the VP2 B-C loop on antigenicity and pathogenicity of serotype Asia1 foot-and-mouth disease virus. Virol. J. 9, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y., Li Z., Ge X., Guo X., and Yang H. (2011) Autophagy promotes the replication of encephalomyocarditis virus in host cells. Autophagy 7, 613–628 [DOI] [PubMed] [Google Scholar]
- 24.Jiang J., and Tang H. (2010) Mechanism of inhibiting type I interferon induction by hepatitis B virus X protein. Protein Cell 1, 1106–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lei X., Liu X., Ma Y., Sun Z., Yang Y., Jin Q., He B., and Wang J. (2010) The 3C protein of enterovirus 71 inhibits retinoid acid-inducible gene I-mediated interferon regulatory factor 3 activation and type I interferon responses. J. Virol. 84, 8051–8061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lei X., Sun Z., Liu X., Jin Q., He B., and Wang J. (2011) Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by Toll-like receptor 3. J. Virol. 85, 8811–8818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang B., Xi X., Lei X., Zhang X., Cui S., Wang J., Jin Q., and Zhao Z. (2013) Enterovirus 71 protease 2Apro targets MAVS to inhibit anti-viral type I interferon responses. PLoS Pathog. 9, e1003231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weng C., Li Y., Xu D., Shi Y., and Tang H. (2005) Specific cleavage of Mcl-1 by caspase-3 in tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in Jurkat leukemia T cells. J. Biol. Chem. 280, 10491–10500 [DOI] [PubMed] [Google Scholar]
- 29.Li D., Li S., Sun Y., Dong H., Li Y., Zhao B., Guo D., Weng C., and Qiu H. J. (2013) Poly(C)-binding protein 1, a novel N(pro)-interacting protein involved in classical swine fever virus growth. J. Virol. 87, 2072–2080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lei X., Xiao X., Xue Q., Jin Q., He B., and Wang J. (2013) Cleavage of interferon regulatory factor 7 by enterovirus 71 3C suppresses cellular responses. J. Virol. 87, 1690–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mukherjee A., Morosky S. A., Delorme-Axford E., Dybdahl-Sissoko N., Oberste M. S., Wang T., and Coyne C. B. (2011) The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog. 7, e1001311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang D., Fang L., Li K., Zhong H., Fan J., Ouyang C., Zhang H., Duan E., Luo R., Zhang Z., Liu X., Chen H., and Xiao S. (2012) Foot-and-mouth disease virus 3C protease cleaves NEMO to impair innate immune signaling. J. Virol. 86, 9311–9322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li X. D., Sun L., Seth R. B., Pineda G., and Chen Z. J. (2005) Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. U.S.A. 102, 17717–17722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hall D. J., and Palmenberg A. C. (1996) Cleavage site mutations in the encephalomyocarditis virus P3 region lethally abrogate the normal processing cascade. J. Virol. 70, 5954–5961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun J. Y., Liu M. Q., Xu Y. L., Xu Z. R., Pan L., and Gao H. (2005) Improvement of the thermostability and catalytic activity of a mesophilic family 11 xylanase by N-terminus replacement. Protein Expr. Purif. 42, 122–130 [DOI] [PubMed] [Google Scholar]
- 36.Jakus P. B., Kalman N., Antus C., Radnai B., Tucsek Z., Gallyas F. Jr., Sumegi B., and Veres B. (2013) TRAF6 is functional in inhibition of TLR4-mediated NF-κB activation by resveratrol. J. Nutr. Biochem. 24, 819–823 [DOI] [PubMed] [Google Scholar]
- 37.Kawagoe T., Takeuchi O., Takabatake Y., Kato H., Isaka Y., Tsujimura T., and Akira S. (2009) TANK is a negative regulator of Toll-like receptor signaling and is critical for the prevention of autoimmune nephritis. Nat. Immunol. 10, 965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen Z. J. (2012) Ubiquitination in signaling to and activation of IKK. Immunol. Rev. 246, 95–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fang Y., and Snijder E. J. (2010) The PRRSV replicase: exploring the multifunctionality of an intriguing set of nonstructural proteins. Virus Res. 154, 61–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Snijder E. J., Kikkert M., and Fang Y. (2013) Arterivirus molecular biology and pathogenesis. J. Gen. Virol. 94, 2141–2163 [DOI] [PubMed] [Google Scholar]
- 41.Tian X., Lu G., Gao F., Peng H., Feng Y., Ma G., Bartlam M., Tian K., Yan J., Hilgenfeld R., and Gao G. F. (2009) Structure and cleavage specificity of the chymotrypsin-like serine protease (3CLSP/Nsp4) of porcine reproductive and respiratory syndrome virus (PRRSV). J. Mol. Biol. 392, 977–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zaragoza C., Saura M., Padalko E. Y., Lopez-Rivera E., Lizarbe T. R., Lamas S., and Lowenstein C. J. (2006) Viral protease cleavage of inhibitor of κBα triggers host cell apoptosis. Proc. Natl. Acad. Sci. U.S.A. 103, 19051–19056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao T., Yang L., Sun Q., Arguello M., Ballard D. W., Hiscott J., and Lin R. (2007) The NEMO adaptor bridges the nuclear factor-κB and interferon regulatory factor signaling pathways. Nat. Immunol. 8, 592–600 [DOI] [PubMed] [Google Scholar]
- 44.Huang C., Zhang Q., Guo X. K., Yu Z. B., Xu A. T., Tang J., and Feng W. H. (2014) Porcine reproductive and respiratory syndrome virus nonstructural protein 4 antagonizes beta interferon expression by targeting the NF-κB essential modulator. J. Virol. 88, 10934–10945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barral P. M., Sarkar D., Fisher P. B., and Racaniello V. R. (2009) RIG-I is cleaved during picornavirus infection. Virology 391, 171–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kobayashi M., Arias C., Garabedian A., Palmenberg A. C., and Mohr I. (2012) Site-specific cleavage of the host poly(A) binding protein by the encephalomyocarditis virus 3C proteinase stimulates viral replication. J. Virol. 86, 10686–10694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sha W. C., Liou H. C., Tuomanen E. I., and Baltimore D. (1995) Targeted disruption of the p50 subunit of NF-κB leads to multifocal defects in immune responses. Cell 80, 321–330 [DOI] [PubMed] [Google Scholar]
- 48.Schwarz E. M., Badorff C., Hiura T. S., Wessely R., Badorff A., Verma I. M., and Knowlton K. U. (1998) NF-κB-mediated inhibition of apoptosis is required for encephalomyocarditis virus virulence: a mechanism of resistance in p50 knockout mice. J. Virol. 72, 5654–5660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rothe M., Xiong J., Shu H. B., Williamson K., Goddard A., and Goeddel D. V. (1996) I-TRAF is a novel TRAF-interacting protein that regulates TRAF-mediated signal transduction. Proc. Natl. Acad. Sci. U.S.A. 93, 8241–8246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pomerantz J. L., and Baltimore D. (1999) NF-κB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. 18, 6694–6704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bonif M., Meuwis M. A., Close P., Benoit V., Heyninck K., Chapelle J. P., Bours V., Merville M. P., Piette J., Beyaert R., and Chariot A. (2006) TNFα- and IKKβ-mediated TANK/I-TRAF phosphorylation: implications for interaction with NEMO/IKKγ and NF-κB activation. Biochem. J. 394, 593–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gatot J. S., Gioia R., Chau T. L., Patrascu F., Warnier M., Close P., Chapelle J. P., Muraille E., Brown K., Siebenlist U., Piette J., Dejardin E., and Chariot A. (2007) Lipopolysaccharide-mediated interferon regulatory factor activation involves TBK1-IKKϵ-dependent Lys(63)-linked polyubiquitination and phosphorylation of TANK/I-TRAF. J. Biol. Chem. 282, 31131–31146 [DOI] [PubMed] [Google Scholar]
- 53.Shi H. X., Liu X., Wang Q., Tang P. P., Liu X. Y., Shan Y. F., and Wang C. (2011) Mitochondrial ubiquitin ligase MARCH5 promotes TLR7 signaling by attenuating TANK action. PLoS Pathog. 7, e1002057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Walsh M. C., Kim G. K., Maurizio P. L., Molnar E. E., and Choi Y. (2008) TRAF6 autoubiquitination-independent activation of the NFkappaB and MAPK pathways in response to IL-1 and RANKL. PLoS One 3, e4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Muroi M., and Tanamoto K. (2008) TRAF6 distinctively mediates MyD88- and IRAK-1-induced activation of NF-κB. J. Leukocyte Biol. 83, 702–707 [DOI] [PubMed] [Google Scholar]