

Background: The impact of DISC1, a key protein driving the endophenotypes of major mental disorders, on GABAergic inhibition is unknown.
Results: DISC1 knockdown or overexpression altered GABAAR-mediated synaptic transmission by regulating kinesin motor/microtubule-based GABAAR trafficking.
Conclusion: DISC1 exerts an important effect on GABAAR trafficking and synaptic inhibition.
Significance: Our observations may be relevant for the role of DISC1 in mental disorders associated with altered GABAergic inhibition.
Keywords: electrophysiology, GABA receptor, intracellular trafficking, schizophrenia, synapse
Abstract
Association studies have suggested that Disrupted-in-Schizophrenia 1 (DISC1) confers a genetic risk at the level of endophenotypes that underlies many major mental disorders. Despite the progress in understanding the significance of DISC1 at neural development, the mechanisms underlying DISC1 regulation of synaptic functions remain elusive. Because alterations in the cortical GABA system have been strongly linked to the pathophysiology of schizophrenia, one potential target of DISC1 that is critically involved in the regulation of cognition and emotion is the GABAA receptor (GABAAR). We found that cellular knockdown of DISC1 significantly reduced GABAAR-mediated synaptic and whole-cell current, whereas overexpression of wild-type DISC1, but not the C-terminal-truncated DISC1 (a schizophrenia-related mutant), significantly increased GABAAR currents in pyramidal neurons of the prefrontal cortex. These effects were accompanied by DISC1-induced changes in surface GABAAR expression. Moreover, the regulation of GABAARs by DISC1 knockdown or overexpression depends on the microtubule motor protein kinesin 1 (KIF5). Our results suggest that DISC1 exerts an important effect on GABAergic inhibitory transmission by regulating KIF5/microtubule-based GABAAR trafficking in the cortex. The knowledge gained from this study would shed light on how DISC1 and the GABA system are linked mechanistically and how their interactions are critical for maintaining a normal mental state.
Introduction
The Disrupted-in-schizophrenia 1 (DISC1) gene was originally identified in a unique Scottish pedigree in which all members with a major mental illness carried the inherited chromosomal translocation that interrupts the coding sequence of DISC1, resulting in the loss of DISC1 expression (1–4). Subsequent association studies have reported a link between genetic variations of DISC1 and specific endophenotypes that are commonly associated with schizophrenia, depression, and bipolar disorder (5–9). DISC1 is not only highly expressed in the developing brain but also in various areas of the adult brain, including the cortex and hippocampus, and in both pyramidal neurons and interneurons (10). During early development, DISC1 regulates progenitor cell proliferation via glycogen synthase kinase 3β-regulated WNT-β-catenin activity and postmitotic neuronal migration via interacting with proteins in the dynein motor complexes that are associated with microtubules and the centrosome (11–13). In the adult brain, DISC1 regulates neurogenesis and orchestrates the tempo of neuronal integration (14, 15).
DISC1 is enriched in the postsynaptic density of excitatory synapses (16), where it regulates spine size, spine number, and AMPA receptor synaptic trafficking via a kalirin 7-Rac1-PAK pathway (17). DISC1 also regulates synapse maintenance through interacting with TRAF2 and NCK-interacting protein kinase (TNIK) (18) and exerts an important effect on NMDA receptor expression and function through a mechanism depending on the transcription factor cAMP response element-binding protein (19). DISC1 has also been found at inhibitory synapses (16). However, the role of DISC1 in regulating inhibitory synaptic transmission is largely unknown.
A convergent finding in individuals with schizophrenia is the reduced perisomatic inhibition of pyramidal neurons in the dorsolateral prefrontal cortex (PFC),3 which leads to the diminished synchronized neuronal activity required for cognitive functions (20, 21). The GABAA receptor is the major mediator of fast synaptic inhibition in the mammalian brain. The dynamic regulation of GABAA receptor composition, trafficking, and function plays a key role in controlling network and cellular activity in normal and disease conditions (22, 23). Selective alterations in GABAA receptors, GABA content, and GABAergic local circuit neurons have been discovered in the PFC of schizophrenia and depression patients (21, 24). In this study, we examined the effect of DISC1 on GABAA receptors and GABAergic transmission in cortical pyramidal neurons, which may help our understanding of the role of DISC1 in regulating synaptic inhibition.
Experimental Procedures
Primary Neuronal Culture
Rat PFC cultures were prepared by modification of methods described previously (25). Briefly, the PFC was dissected from 18-day-old rat embryos, and cells were dissociated using trypsin and titrated through a Pasteur pipette. The neurons were plated on coverslips coated with poly-l-lysine in DMEM with 10% fetal calf serum at a density of 1 × 105 cells/cm2. When neurons attached to the coverslip within 24 h, the medium was changed to Neurobasal with a B27 supplement (1.0% penicillin/streptomycin, 2% B27, and 0.5 mm Glutamine). Two to three days after plating, 5 μm cytosine arabinoside was added to inhibit glial growth. Subsequently, half of the medium was changed to a conditional medium once a week. Neurons were maintained for 2–3 weeks before being used for recordings.
Small Interfering RNA and Dominant Negative Constructs
To suppress the expression of DISC1 in cultured neurons, we used shRNA, a potent agent for sequence-specific gene silencing (26). DISC1 shRNA (5′-GGCAAACACTGTGAAGTGC-3′) or scramble shRNA (control shRNA) (5′-GGAGCAGACGCTGAATTAC-3′) was inserted into the vector pSuper-Venus and driven by the H1-RNA polymerase III promoter. The construct also carried CMV promoter-driven enhanced GFP (12, 17, 19). The DISC1 shRNA-encoding plasmid (DISC1 shRNA) has been shown to induce a strong suppression of DISC1 expression (12). Cultured PFC neurons (DIV 15–16) were transfected with control shRNA or DISC1 shRNA using the Lipofectamine 2000 method. Full-length DISC1 (17) plasmid was used to overexpress DISC1. To suppress total KIF5 activity or overexpress KIF5, we co-transfected KLC1 siRNA (Santa Cruz Biotechnology, Santa Cruz, CA), dominant negative KIF5C, or the full-length heavy chains of KIF5C (a gift from Dr. Josef T. Kittler, University College London) (27) with control shRNA or DISC1 shRNA. Neurons were used in experiments 2–3 days after transfection.
Whole-cell Recordings of Ionic Currents
Recordings of whole cell GABAAR-mediated currents used standard voltage clamp techniques (28). The internal solution consisted of 180 mm N-methyl-d-glucamine, 40 mm HEPES, 4 mm MgCl2, 0.5 mm 1,2-bis(2-aminohenoxy) ethane N,N,N_,N_-tetraacetic acid, 12 mm phosphocreatine, 2 mm Na2ATP, 0.2 mm Na3GTP, and 0.1 mm leupeptin (pH 7.2–7.3, 265–270 mosmol/liter). The external solution consisted of 135 mm NaCl, 20 mm CsCl, 1 mm MgCl2, 10 mm HEPES, 5 mm BaCl2, 10 mm glucose, and 0.001 mm tetrodotoxin (pH 7.3–7.4, 300–305 mosmol/liter). Recordings were obtained using an Axopatch 200B amplifier controlled and monitored with a computer running pClamp 8 with a DigiData 1320 series interface. Electrode resistances were typically 2–4 MΩ in the bath. After seal rupture, series resistance (4–10 MΩ) was compensated (70–90%) and monitored periodically. The cell membrane potential was held at 0 mV. GABA (50 μm) was applied for 2 s every 30 s to minimize the desensitization-induced decrease of current amplitude. Drugs were applied using a gravity-fed “sewer pipe” system. The array of application capillaries (∼150-μm inner diameter) was positioned a few hundred micrometers from the cell being recorded. Solution changes were affected by the SF-77B fast-step solution stimulus delivery device (Warner Instruments, Hamden, CT). Data analyses were performed with Clampfit (Axon Instruments, Sunnyvale, CA) and Kaleidagraph (Albeck Software, Reading, PA). Current density was calculated by taking the average peak amplitude (picoampere) of a single recording and dividing that value by the capacitance (picofarad).
Electrophysiological Recording of Synaptic Currents
Recording of miniature inhibitory postsynaptic currents (mIPSC) in cultured PFC neurons (DIV 14–16) used the whole-cell patch technique. Electrodes (3–5 MΩ) were filled with the following internal solution: 100 mm CsCl, 30 mm N-methyl-d-glucamine, 10 mm HEPES, 4 mm NaCl, 1 mm MgCl2 5 mm EGTA, 5 mm MgATP, 0.5 mm Na2GTP, 12 mm phosphocreatine, 0.2 mm leupeptin, and 2 mm QX-314 (pH 7.2–7.3, 265–270 mosmol/liter). Oxygenated artificial cerebral spinal fluid (130 mm NaCl, 3 mm KCl, 5 mm MgCl2, 1 mm CaCl2, 26 mm NaHCO3, 1.25 mm NaH2PO4, and 10 mm glucose (pH 7.4, 300 mosmol/liter) was used as the external solution. Tetrodotoxin (0.5 μm), D-AP5 (50 μm), and 6,7-dinitroquinoxaline-2,3-dione (10 μm) were added to cultures to block action potentials N-methyl-d-aspartic acid and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid/kainate receptors, respectively. The cell membrane potential was held at −70 mV. A mini analysis program (Synaptosoft, Leonia, NJ) was used to analyze the spontaneous synaptic events. For each different condition, mIPSC recordings of 8 min were used for analysis.
Immunocytochemical Staining
For the detection of surface GABAA receptors, the primary cultures (2.5 × 104 cells/cm2, DIV 17–18) were fixed, blocked, and incubated with a monoclonal antibody against the GABAAR β2/3 subunits (1:250, Millipore, catalog no. MAB341 or 1:250, NeuroMab, catalog no. 75-363) overnight at 4 °C. After washing, neurons were permeabilized and incubated with a polyclonal antibody against MAP2 (1:250; Santa Cruz Biotechnology, catalog no. sc-20172) or a polyclonal anti-VIAAT (1:1000, a gift from Dr. Stephen Moss, Tufts University) for 2 h at room temperature. After washing, the neurons were incubated with Alexa Fluor 568-conjugated secondary antibody (1:250, Invitrogen) and Alexa Fluor 647-conjugated secondary antibody (1:250, Invitrogen) for 2 h at room temperature. After washing in PBS three times, coverslips were mounted on slides with Vectashield mounting medium (Vector Laboratories, Burlingame, CA).
For the detection of GABAAR at synapses, neurons (2.5 × 104 cells/cm2, DIV 17–18) were fixed, permeabilized, blocked, and incubated with a monoclonal anti-GABAAR β2/3 antibody (1:250, Millipore, catalog no. MAB341, Ref. 29) and a polyclonal anti-VIAAT antibody (1:1000) overnight at 4 °C. After washing, the neurons were incubated with Alexa Fluor 568-conjugated secondary antibody (1:250, Invitrogen) and Alexa Fluor 647-conjugated secondary antibody (1:250, Invitrogen) for 2 h at room temperature. After washing, the coverslips were mounted on slides with Vectashield mounting medium.
Fluorescence images were obtained using a ×100 oil immersion objective with a cooled charge-coupled device camera mounted on a Nikon fluorescence microscope (Nikon Eclipse E600). Images were captured digitally using SPOT basic image capture software. All specimens were imaged under identical conditions and analyzed with identical parameters using ImageJ software. To define dendritic clusters, a single threshold was chosen manually so that clusters corresponded to puncta with the mean intensity equal to ∼2-fold of the diffuse fluorescence mean intensity on the dendritic shaft. When GABAAR β2/3 and VIAAT clusters were identified, the co-localized clusters were highlighted as white points by an analysis tool in ImageJ. On each coverslip, the cluster density, cluster size, and cluster fluorescence intensity of three to five neurons (two to three dendritic segments 50 μm in length per neuron) were measured. Quantitative analyses were conducted blindly (without knowledge of experimental treatment).
Statistics
Student's t test or analysis of variance with post hoc Dunnett or Turkey tests (Kaleidagraph) were performed to detect statistical significance among groups with different treatments.
Results
DISC1 Knockdown or Overexpression Alters GABAAR-mediated Currents
To examine the effect of DISC1 knockdown on GABAergic signaling, we measured mIPSC, a synaptic response to the quantal release of single GABA vesicles, in cultured PFC pyramidal neurons. Neurons (DIV 14–16) were transfected with a scrambled shRNA (control shRNA) or DISC1 shRNA, and recordings were performed 2 days after transfection. As shown in Fig. 1, A–C, DISC1 shRNA-transfected neurons had a significant reduction in mIPSC amplitude (control shRNA, 31.8 ± 1.8 pA, n = 19; DISC1 shRNA, 24.4 ± 1.3 pA, n = 23; p < 0.01), whereas there was no significant change in mIPSC frequency (control shRNA, 4.73 ± 0.74 Hz, n = 19; DISC1 shRNA, 3.93 ± 0.66 Hz, n = 23; p > 0.05). Transfecting the full-length shRNA-resistant DISC1 (DISC1-FLR) rescued mIPSC amplitude (29.6 ± 1.4 pA, n = 17, p > 0.05), confirming that the effect of DISC1 shRNA on mIPSC is mediated by the specific loss of DISC1.
FIGURE 1.

DISC1 knockdown reduces mIPSC amplitude and GABAAR current density in cultured cortical pyramidal neurons. A, cumulative plots of mIPSC amplitudes in neurons transfected with control shRNA, DISC1 shRNA, or DISC1 shRNA plus an shRNA-resistant full-length DISC1 (DISC1R). Note that DISC1 knockdown caused a leftward shift in the distribution of mIPSC amplitudes, indicative of a reduction of mIPSC sizes. B, representative mIPSC traces taken from neurons in A. C, summary (mean ± S.E.) of mIPSC amplitudes and frequencies in neurons with different transfections. **, p < 0.01; Dunnett test. con, control. D, representative whole-cell GABAAR current traces in neurons transfected with control or DISC1 shRNA. E, summary (mean ± S.E.) of GABAAR current density in neurons with different transfections. *, p < 0.05, Student's t test.
To test whether the DISC1 shRNA-induced reduction in mIPSC amplitude was due to alteration of receptor density and/or activity, we measured the GABA-elicited whole-cell current mediated by both synaptic and extrasynaptic GABAA receptors. Application of GABA (50 μm) evoked a partially desensitizing outward current in cultured PFC pyramidal neurons (held at 0 mV) that could be blocked by the GABAA receptor antagonist bicuculline (30 μm). As shown in Fig. 1, D and E, neurons transfected with DISC1 shRNA had a significantly smaller GABAAR current density (control shRNA, 85.3 ± 7.3 pA/pF, n = 7; DISC1 shRNA, 62.7 ± 5.5 pA/pF, n = 7; p < 0.05).
In addition to DISC1 knockdown, we also examined the impact of overexpressing full-length DISC1 (FL-DISC1) on GABAergic signaling. As shown in Fig. 2, A and B, transfecting GFP-tagged FL-DISC1 caused a significant increase in mIPSC amplitude in cultured PFC pyramidal neurons (GFP, 32.7 ± 1.4 pA, n = 14; FL-DISC1, 40.3 ± 2.8 pA, n = 16; p < 0.05). Whole-cell GABAAR current density (picoampere/picofarad) was also increased significantly by full-length DISC1 (Fig. 2, C and D; GFP, 79.5 ± 5.8 pA/pF, n = 14; FL-DISC1, 104.9 ± 7.2 pA/pF, n = 14; p < 0.05). Together, these results indicate that DISC1 knockdown or overexpression leads to a decrease or increase in GABAAR currents, respectively.
FIGURE 2.

DISC1 overexpression increases mIPSC amplitude and GABAAR current density in cultured cortical pyramidal neurons. A, cumulative plots of mIPSC amplitudes in neurons transfected with GFP or FL-DISC1. Note that DISC1 overexpression caused a rightward shift in the distribution of mIPSC amplitudes, indicative of an increase in the sizes of mIPSCs. Inset, representative mIPSC traces. B, summary (mean ± S.E.) of mIPSC amplitudes and frequencies in neurons with different transfections. *, p < 0.05; Student's t test. C, representative whole-cell GABAAR current traces in neurons transfected with GFP or FL-DISC1. D, summary (mean ± S.E.) of GABAAR current density in neurons with different transfections. *, p < 0.05; Student's t test.
DISC1 Knockdown or Overexpression Alters GABAAR Surface and Synaptic Distribution
Emerging evidence suggests that the trafficking of GABAARs underlies dynamic changes in synaptic receptor numbers and inhibitory postsynaptic current amplitudes, providing an effective mechanism for regulating the strength and plasticity of synaptic inhibition (23, 30). Therefore, we examined whether the change in GABA responses by DISC1 knockdown or overexpression was associated with altered membrane trafficking of GABAARs by using a quantitative surface immunostaining assay. Surface GABAAR clusters were compared in PFC cultures transfected with control shRNA, DISC1 shRNA, or full-length DISC1. An antibody against the N-terminal extracellular domain of GABAAR β2/3 subunits was used to detect surface GABAARs, and immunostaining was performed under nonpermeabilized conditions. As shown in Fig. 3, A and B, DISC1 knockdown significantly decreased surface GABAAR cluster density (number of clusters/30 μm dendrite; control shRNA, 21.7 ± 1.2, n = 23; DISC1 shRNA, 14.1 ± 0.8, n = 19; p < 0.01) and size (square micrometer; control shRNA, 0.19 ± 0.03, n = 23; DISC1 shRNA, 0.11 ± 0.01, n = 19; p < 0.05). In contrast, overexpression of FL-DISC1 significantly increased surface GABAAR cluster density and size (Fig. 3, C and D) (cluster density: GFP, 19.3 ± 1.1, n = 53; FL-DISC1, 28.6 ± 1.2, n = 51; p < 0.001; cluster size: GFP, 0.27 ± 0.03, n = 53; FL-DISC1, 0.46 ± 0.03, n = 51; p < 0.001) without altering the expression of the inhibitory synapse marker VIAAT (27, 31) (Fig. 3, C and D) (cluster density: GFP, 14.3 ± 0.8, n = 26; FL-DISC1, 14.5 ± 1.5, n = 26; p > 0.05; cluster size: GFP, 0.64 ± 0.05, n = 26; FL-DISC1, 0.63 ± 0.05, n = 26; p > 0.05). These results suggest that the changes in surface GABAAR expression are not due to the formation of new inhibitory synapses.
FIGURE 3.
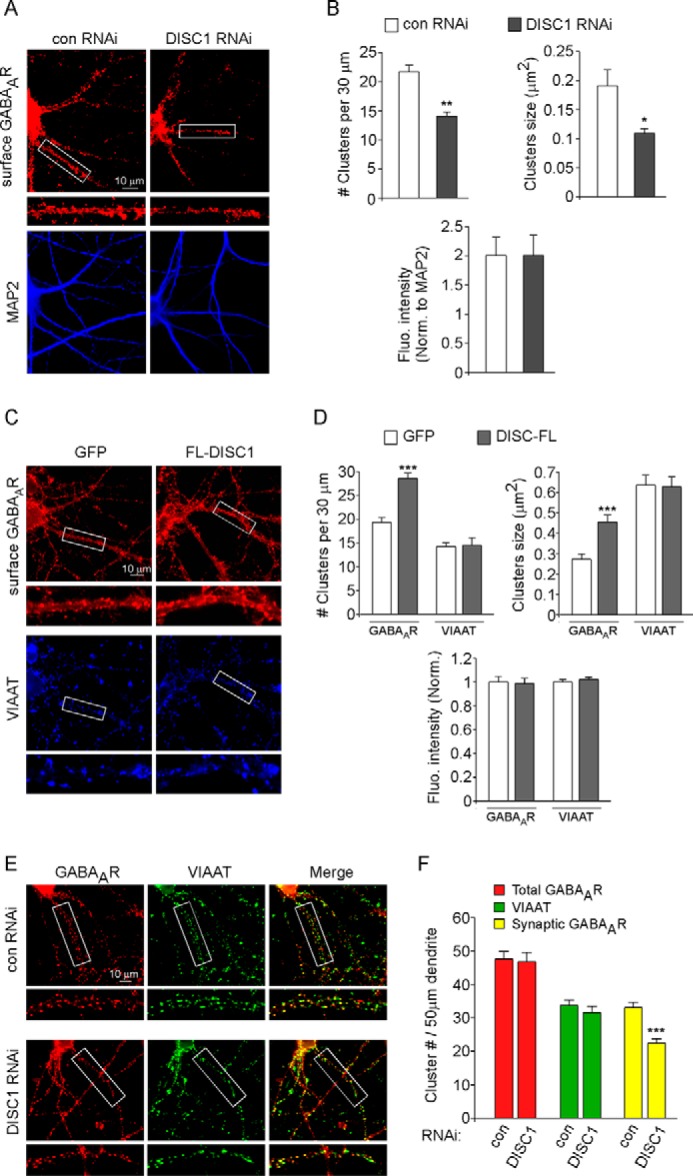
DISC1 knockdown reduces surface and synaptic GABAAR clusters, whereas DISC1 overexpression increases surface GABAAR clusters. A and C, immunocytochemical images of surface GABAAR β2/3 subunits in cortical cultures transfected with a control (con) versus DISC1 shRNA (A) or GFP versus FL-DISC1 (C). Following surface GABAAR β2/3 labeling, neurons were permeabilized and co-stained with MAP2 (A) or VIAAT (C). Enlarged versions of the boxed regions of dendrites are also shown. B and D, quantitative analysis of surface GABAAR β2/3 or VIAAT clusters in cortical cultures with different transfections. ***, p < 0.001; **, p < 0.01; *, p < 0.05; Student's t test. Fluo, fluorescence; Norm, normalized. E, immunocytochemical images of control shRNA- versus DISC1 shRNA-transfected cortical neurons co-stained with GABAAR β2/3 and VIAAT. Enlarged versions of the boxed regions of dendrites are also shown. F, quantitative analysis of the densities of total GABAAR clusters, VIAAT clusters, and synaptic GABAAR clusters (co-localized with VIAAT). ***, p < 0.001, Student's t test.
To examine synaptic GABAARs, we co-stained the GABAAR subunit with the inhibitory synapse marker VIAAT. GABAARs opposed to VIAAT-labeled inhibitory synaptic terminals were considered postsynaptic GABAARs. As shown in Fig. 3, E and F, DISC1 knockdown did not significantly change the density of total GABAAR clusters (control shRNA, 47.6 ± 2.2, n = 27; DISC1 shRNA, 46.8 ± 2.8, n = 28; p > 0.05) or VIAAT clusters (control shRNA, 33.7 ± 1.6, n = 27; DISC1 shRNA, 31.6 ± 1.8, n = 28; p > 0.05) but significantly reduced synaptic GABAAR cluster density (control shRNA, 33.0 ± 1.6, n = 27; DISC1 shRNA, 22.4 ± 1.4, n = 28; p < 0.001). These results suggest that DISC1 regulates GABAergic transmission mainly by changing the number of GABAARs in the synapse.
DISC1 Regulation of GABAARs Depends on the Microtubule Motor Protein KIF5
Next we investigated the potential mechanism underlying DISC1 regulation of GABAARs at the cell surface. It has been shown that DISC1 can directly interact with KIF5, a microtubule motor protein (32). Our previous studies have found that GABAARs are trafficked to synapses along microtubules by KIF5 (27). Using co-immunoprecipitation assays, we have demonstrated that immunoprecipitated GABAARs could readily bring down tubulin and KIF5 light chain (33), suggesting that GABAAR-KIF5-tubulin form a complex in vivo. Therefore, it is possible that DISC1 may affect the interaction between GABAARs and KIF motor proteins, influencing GABAAR transport along dendritic microtubules and inhibitory synaptic transmission.
To test the role of KIF5 in GABAAR trafficking and function, we transfected neurons with the siRNA against KIF5 light chain 1 (KLC1), which caused an effective suppression of KLC1 expression (34). KLCs are essential for the proper function or localization of KIF5 heavy chains (35). As shown in Fig. 4, A and D, KLC1 knockdown resulted in a significant reduction in mIPSC amplitude (control shRNA, 32.9 ± 1.4 pA, n = 14; KLC1 siRNA, 27.4 ± 1.4 pA, n = 16; p < 0.01) but not mIPSC frequency (control shRNA, 1.91 ± 0.6 Hz, n = 17; KLC1 siRNA, 0.96 ± 0.3 Hz, n = 14; p > 0.05). These results suggest that GABAARs rely upon KIF5 for trafficking to the postsynaptic membrane.
FIGURE 4.

DISC1 regulation of synaptic inhibition requires KIF5. A, cumulative plots of mIPSC amplitudes in cultured cortical pyramidal neurons transfected with control or KLC1 siRNA. Note that KLC1 knockdown caused a leftward shift in the distribution of mIPSC amplitudes, indicative of a reduction of mIPSC sizes. B and C, cumulative plots of mIPSC amplitudes in neurons transfected with control (con) or DISC1 shRNA in the presence of KLC1 siRNA (B) or dominant-negative (DN) KIF5C (C). E, cumulative plots of mIPSC amplitudes in neurons with or without FL-DISC1 transfection in the presence of DN-KIF5C. Insets (A–C and E), representative mIPSC traces. D and F, summary (mean ± S.E.) of mIPSC amplitudes in neurons with different transfections. *, p < 0.05; **, p < 0.01; NS, not significant; analysis of variance.
To examine the role of KIF5 in DISC1 regulation of GABAARs, we blocked the function of KIF by either KLC1 siRNA or a dominant-negative KIF5C construct (DN-KIF5C) that lacks the motor domain (27) and examined the effect of DISC1 knockdown or overexpression on mIPSC. As shown in Fig. 4, B and D, knockdown of KLC resulted in the occlusion of DISC1 shRNA effects on mIPSC amplitude (control shRNA + KLC1 siRNA, 27.6 ± 1.9 pA, n = 6; DISC1 shRNA + KLC1 siRNA, 27.1 ± 2.0 pA, n = 4; p > 0.05). Such an occlusion was also observed in the presence of DN-KIF5C (Fig. 4, C and D, control shRNA + DN-KIF5C, 26.5 ± 1.4 pA, n = 9; DISC1 shRNA + DN-KIF5C, 25.0 ± 2.1 pA, n = 8; p > 0.05). Blocking the function of KIF17, another kinesin family motor protein involved in NMDA receptor transport (36), failed to alter the reducing effect of DISC1 shRNA effects on mIPSC amplitude (Fig. 4D) (control shRNA + KIF17 antisense, 33.0 ± 1.9 pA, n = 23; DISC1 shRNA + KIF17 antisense, 25.5 ± 1.2 pA, n = 13; p < 0.01). In addition, the enhancing effect of FL-DISC1 on mIPSC amplitude was blocked in the presence of DN-KIF5C (Fig. 4, E and F) (DN-KIF5, 25.8 ± 0.8 pA, n = 13; FL-DISC1 + DN-KIF5, 25.1 ± 1.2 pA, n = 20; p > 0.05).
Finally, we examined whether overexpressing KIF5 could block the effect of DISC1 knockdown on mIPSC. As shown in Fig. 5, A–D, the reducing effect of DISC1 shRNA on mIPSC amplitude (control shRNA, 33.3 ± 2.3 pA, n = 15; DISC1 shRNA, 26.4 ± 1.0 pA, n = 15; p < 0.05) was prevented in the presence of full-length KIF5C (control shRNA + KIF5C, 30.7 ± 1.6 pA, n = 15; DISC1 shRNA + KIF5C, 30.7 ± 1.7 pA, n = 13; p > 0.05). There was no significant change in mIPSC frequency with any of these treatments (control shRNA, 5.35 ± 1.53 Hz, n = 15; DISC1 shRNA, 3.92 ± 0.95 Hz, n = 15; control shRNA + KIF5C, 3.96 ± 0.57 Hz, n = 15; DISC1 shRNA + KIF5C, 4.04 ± 0.79 Hz, n = 13; p > 0.05). Together, these results suggest that the kinesin motor protein KIF5 is necessary and sufficient for DISC1 regulation of GABAARs.
FIGURE 5.

Overexpression of KIF blocks the reducing effect of DISC1 knockdown on synaptic inhibition. A, cumulative plots of mIPSC amplitudes in cultured cortical pyramidal neurons transfected with control (con) or DISC1 shRNA in the absence or presence of full-length KIF5C. B, representative mIPSC traces taken from neurons in A. C and D, summary (mean ± S.E.) of mIPSC amplitudes (C) or frequencies (D) in neurons with different transfections. *, p < 0.05; analysis of variance.
Discussion
DISC1, a key protein driving the endophenotypes of major mental disorders (9), is located at both excitatory and inhibitory synapses (16). From the network of protein-protein interactions around DISC1, DISC1 is implicated in processes of cytoskeletal stability, intracellular transport, and synaptic activity (37). It has been found that DISC1 interacts with multiple proteins, such as the second messenger PDE4B, which hydrolyzes cAMP and regulates the PKA signaling pathway (4), the transcription factor ATF4/CREB2, which controls gene expression (38), and the motor protein kinesin 1, which mediates the microtubule-based transport of neuronal cargos (32). The role of DISC1 in regulating glutamate receptors and spine synapses has been documented (17–19). In this study, we revealed the role of DISC1 in regulating GABAA receptors and synaptic inhibition. DISC1 knockdown led to a significant decrease of GABAAR-mediated synaptic and whole-cell current (Fig. 1), which was accompanied by reduced surface and synaptic GABAAR clusters (Fig. 3), whereas DISC1 overexpression significantly increased GABAAR currents (Fig. 2) and surface expression (Fig. 3). These data suggest that DISC1 facilitates the delivery of GABAARs to the synaptic membrane.
The trafficking of GABAARs begins in the endoplasmic reticulum, where they are assembled and then forward-trafficked to the Golgi network. This is followed by long-range transport along microtubules on dendrites and insertion into extrasynaptic sites, where they can diffuse laterally into synapses (23). The anterograde transport of neuronal cargos along microtubules relies on the kinesin superfamily of motor proteins, which typically consists of two identical heavy chains and two identical light chains (39–41). Kinesin heavy chain has a conserved N-terminal motor domain that is responsible for microtubule binding and a diverse C-terminal non-motor domain containing kinesin light chain binding domain and cargo binding domain. Previous studies by us and others have found that kinesin motor proteins are important for the microtubule-based transport of ionotropic glutamate receptors and the efficacy of excitatory synaptic transmission (34, 36, 42–45). The role of KIF5 motors in regulating GABAAR trafficking and the strength of inhibitory synaptic transmission was later revealed by our studies (27, 33).
Because DISC1 can interact directly with KIF5 (32), we hypothesized that KIF5 may be involved in DISC1 regulation of GABAA receptor trafficking. Consistently, the reducing effect of DISC1 knockdown on GABAergic transmission was occluded by suppressing KIF5 (Fig. 4) and blocked by overexpressing KIF5 (Fig. 5), suggesting that KIF5 is necessary and sufficient for DISC1 regulation of synaptic inhibition.
Deficits in microtubule/KIF-based transport have been suggested to underlie the pathogenesis of a number of neurodegenerative diseases (46). Our results suggest that the loss of DISC1 function in mental disorder conditions may interfere with the interaction between GABAARs and KIF motor proteins, leading to the disruption of GABAAR transport along dendritic microtubules and impairment of inhibitory synaptic transmission. The diminished synaptic inhibition could contribute to the reduced capability to generate γ frequency-synchronized neuronal activity, leading to the impaired working memory functions shared by various mental illnesses (21).
Author Contributions
J. W., N. M. G., and Z. G. performed the experiments and analyzed data. Z. Y. designed the experiments and wrote the paper.
Acknowledgments
We thank Xiaoqing Chen for technical support and Dr. Akira Sawa (Johns Hopkins University School of Medicine) for pSuper-Venus-DISC1/scramble shRNA constructs. We also thank Dr. Josef T. Kittler (University College London) for DN-KIF5C and full-length KIF5C constructs and Dr. Stephen Moss (Tufts University) for the VIAAT antibody.
This work was supported by National Institutes of Health Grant MH101690 (to Z. Y.). The authors declare that they have no conflicts of interest with the contents of this article.
- PFC
- prefrontal cortex
- DIV
- days in vitro
- GABAAR
- GABAA receptor
- mIPSC
- miniature inhibitory postsynaptic current
- VIAAT
- vesicular inhibitory amino acid transporter.
References
- 1.St Clair D., Blackwood D., Muir W., Carothers A., Walker M., Spowart G., Gosden C., and Evans H. J. (1990) Association within a family of a balanced autosomal translocation with major mental illness. Lancet 336, 13–16 [DOI] [PubMed] [Google Scholar]
- 2.Blackwood D. H., Fordyce A., Walker M. T., St Clair D. M., Porteous D. J., and Muir W. J. (2001) Schizophrenia and affective disorders-cosegregation with a translocation at chromosome 1q42 that directly disrupts brain-expressed genes: clinical and P300 findings in a family. Am. J. Hum. Genet. 69, 428–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Millar J. K., Wilson-Annan J. C., Anderson S., Christie S., Taylor M. S., Semple C. A., Devon R. S., St Clair D. M., Muir W. J., Blackwood D. H., and Porteous D. J. (2000) Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum. Mol. Genet. 9, 1415–1423 [DOI] [PubMed] [Google Scholar]
- 4.Millar J. K., Pickard B. S., Mackie S., James R., Christie S., Buchanan S. R., Malloy M. P., Chubb J. E., Huston E., Baillie G. S., Thomson P. A., Hill E. V., Brandon N. J., Rain J. C., Camargo L. M., Whiting P. J., Houslay M. D., Blackwood D. H., Muir W. J., and Porteous D. J. (2005) DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signaling. Science 310, 1187–1191 [DOI] [PubMed] [Google Scholar]
- 5.Callicott J. H., Straub R. E., Pezawas L., Egan M. F., Mattay V. S., Hariri A. R., Verchinski B. A., Meyer-Lindenberg A., Balkissoon R., Kolachana B., Goldberg T. E., and Weinberger D. R. (2005) Variation in DISC1 affects hippocampal structure and function and increases risk for schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 102, 8627–8632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cannon T. D., Hennah W., van Erp T. G., Thompson P. M., Lonnqvist J., Huttunen M., Gasperoni T., Tuulio-Henriksson A., Pirkola T., Toga A. W., Kaprio J., Mazziotta J., and Peltonen L. (2005) Association of DISC1/TRAX haplotypes with schizophrenia, reduced prefrontal gray matter, and impaired short- and long-term memory. Arch. Gen. Psychiatry 62, 1205–1213 [DOI] [PubMed] [Google Scholar]
- 7.Prata D. P., Mechelli A., Fu C. H., Picchioni M., Kane F., Kalidindi S., McDonald C., Kravariti E., Toulopoulou T., Miorelli A., Murray R., Collier D. A., and McGuire P. K. (2008) Effect of disrupted-in-schizophrenia-1 on pre-frontal cortical function. Mol. Psychiatry 13, 915–917, 909 [DOI] [PubMed] [Google Scholar]
- 8.Carless M. A., Glahn D. C., Johnson M. P., Curran J. E., Bozaoglu K., Dyer T. D., Winkler A. M., Cole S. A., Almasy L., MacCluer J. W., Duggirala R., Moses E. K., Göring H. H., and Blangero J. (2011) Impact of DISC1 variation on neuroanatomical and neurocognitive phenotypes. Mol. Psychiatry 16, 1096–1104, 1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brandon N. J., and Sawa A. (2011) Linking neurodevelopmental and synaptic theories of mental illness through DISC1. Nat. Rev. Neurosci. 12, 707–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schurov I. L., Handford E. J., Brandon N. J., and Whiting P. J. (2004) Expression of disrupted in schizophrenia 1 (DISC1) protein in the adult and developing mouse brain indicates its role in neurodevelopment. Mol. Psychiatry 9, 1100–1110 [DOI] [PubMed] [Google Scholar]
- 11.Mao Y., Ge X., Frank C. L., Madison J. M., Koehler A. N., Doud M. K., Tassa C., Berry E. M., Soda T., Singh K. K., Biechele T., Petryshen T. L., Moon R. T., Haggarty S. J., and Tsai L. H. (2009) Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3β/β-catenin signaling. Cell 136, 1017–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kamiya A., Kubo K., Tomoda T., Takaki M., Youn R., Ozeki Y., Sawamura N., Park U., Kudo C., Okawa M., Ross C. A., Hatten M. E., Nakajima K., and Sawa A. (2005) A schizophrenia-associated mutation of DISC1 perturbs cerebral cortex development. Nat. Cell Biol. 7, 1167–1178 [DOI] [PubMed] [Google Scholar]
- 13.Ishizuka K., Kamiya A., Oh E. C., Kanki H., Seshadri S., Robinson J. F., Murdoch H., Dunlop A. J., Kubo K., Furukori K., Huang B., Zeledon M., Hayashi-Takagi A., Okano H., Nakajima K., Houslay M. D., Katsanis N., and Sawa A. (2011) DISC1-dependent switch from progenitor proliferation to migration in the developing cortex. Nature 473, 92–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duan X., Chang J. H., Ge S., Faulkner R. L., Kim J. Y., Kitabatake Y., Liu X. B., Yang C. H., Jordan J. D., Ma D. K., Liu C. Y., Ganesan S., Cheng H. J., Ming G. L., Lu B., and Song H. (2007) Disrupted-in-schizophrenia 1 regulates integration of newly generated neurons in the adult brain. Cell 130, 1146–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim J. Y., Liu C. Y., Zhang F., Duan X., Wen Z., Song J., Feighery E., Lu B., Rujescu D., St Clair D., Christian K., Callicott J. H., Weinberger D. R., Song H., and Ming G. L. (2012) Interplay between DISC1 and GABA Signaling regulates neurogenesis in mice and risk for schizophrenia. Cell 148, 1051–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kirkpatrick B., Xu L., Cascella N., Ozeki Y., Sawa A., and Roberts R. C. (2006) DISC1 immunoreactivity at the light and ultrastructural level in the human neocortex. J. Comp. Neurol. 497, 436–450 [DOI] [PubMed] [Google Scholar]
- 17.Hayashi-Takagi A., Takaki M., Graziane N., Seshadri S., Murdoch H., Dunlop A. J., Makino Y., Seshadri A. J., Ishizuka K., Srivastava D. P., Xie Z., Baraban J. M., Houslay M. D., Tomoda T., Brandon N. J., Kamiya A., Yan Z., Penzes P., and Sawa A. (2010) Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat. Neurosci. 13, 327–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Q., Charych E. I., Pulito V. L., Lee J. B., Graziane N. M., Crozier R. A., Revilla-Sanchez R., Kelly M. P., Dunlop A. J., Murdoch H., Taylor N., Xie Y., Pausch M., Hayashi-Takagi A., Ishizuka K., Seshadri S., Bates B., Kariya K., Sawa A., Weinberg R. J., Moss S. J., Houslay M. D., Yan Z., and Brandon N. J. (2011) The psychiatric disease risk factors DISC1 and TNIK interact to regulate synapse composition and function. Mol. Psychiatry 16, 1006–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei J., Graziane N. M., Wang H., Zhong P., Wang Q., Liu W., Hayashi-Takagi A., Korth C., Sawa A., Brandon N. J., and Yan Z. (2014) Regulation of N-methyl-d-aspartate receptors by disrupted-in-schizophrenia-1. Biol. Psychiatry 75, 414–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Volk D. W., Pierri J. N., Fritschy J. M., Auh S., Sampson A. R., and Lewis D. A. (2002) Reciprocal alterations in pre- and postsynaptic inhibitory markers at chandelier cell inputs to pyramidal neurons in schizophrenia. Cereb. Cortex 12, 1063–1070 [DOI] [PubMed] [Google Scholar]
- 21.Lewis D. A., Hashimoto T., and Volk D. W. (2005) Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 6, 312–324 [DOI] [PubMed] [Google Scholar]
- 22.Moss S. J., and Smart T. G. (2001) Constructing inhibitory synapses. Nat. Rev. Neurosci. 2, 240–250 [DOI] [PubMed] [Google Scholar]
- 23.Jacob T. C., Moss S. J., and Jurd R. (2008) GABAA receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat. Rev. Neurosci. 9, 331–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luscher B., Shen Q., and Sahir N. (2011) The GABAergic deficit hypothesis of major depressive disorder. Mol. Psychiatry 16, 383–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X., Zhong P., Gu Z., and Yan Z. (2003) Regulation of NMDA receptors by dopamine D4 signaling in prefrontal cortex. J. Neurosci. 23, 9852–9861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McManus M. T., and Sharp P. A. (2002) Gene silencing in mammals by small interfering RNAs. Nat. Rev. Genet. 3, 737–747 [DOI] [PubMed] [Google Scholar]
- 27.Twelvetrees A. E., Yuen E. Y., Arancibia-Carcamo I. L., MacAskill A. F., Rostaing P., Lumb M. J., Humbert S., Triller A., Saudou F., Yan Z., and Kittler J. T. (2010) Delivery of GABAARs to synapses is mediated by HAP1-KIF5 and disrupted by mutant huntingtin. Neuron 65, 53–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang X., Zhong P., and Yan Z. (2002) Dopamine D4 receptors modulate GABAergic signaling in pyramidal neurons of prefrontal cortex. J. Neurosci. 22, 9185–9193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCann C. M., Bracamontes J., Steinbach J. H., and Sanes J. R. (2006) The cholinergic antagonist α-bungarotoxin also binds and blocks a subset of GABA receptors. Proc. Natl. Acad. Sci. U.S.A. 103, 5149–5154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomas P., Mortensen M., Hosie A. M., and Smart T. G. (2005) Dynamic mobility of functional GABAA receptors at inhibitory synapses. Nat. Neurosci. 8, 889–897 [DOI] [PubMed] [Google Scholar]
- 31.Dumoulin A., Rostaing P., Bedet C., Lévi S., Isambert M. F., Henry J. P., Triller A., and Gasnier B. (1999) Presence of the vesicular inhibitory amino acid transporter in GABAergic and glycinergic synaptic terminal boutons. J. Cell Sci. 112, 811–823 [DOI] [PubMed] [Google Scholar]
- 32.Taya S., Shinoda T., Tsuboi D., Asaki J., Nagai K., Hikita T., Kuroda S., Kuroda K., Shimizu M., Hirotsune S., Iwamatsu A., and Kaibuchi K. (2007) DISC1 regulates the transport of the NUDEL/LIS1/14-3-3ϵ complex through kinesin-1. J. Neurosci. 27, 15–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuen E. Y., Wei J., Zhong P., and Yan Z. (2012) Disrupted GABAAR trafficking and synaptic inhibition in a mouse model of Huntington's disease. Neurobiol. Dis. 46, 497–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mandal M., Wei J., Zhong P., Cheng J., Duffney L. J., Liu W., Yuen E. Y., Twelvetrees A. E., Li S., Li X. J., Kittler J. T., and Yan Z. (2011) Impaired AMPA receptor trafficking and function by mutant huntingtin. J. Biol. Chem. 286, 33719–33728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rahman A., Kamal A., Roberts E. A., and Goldstein L. S. (1999) Defective kinesin heavy chain behavior in mouse kinesin light chain mutants. J. Cell Biol. 146, 1277–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Setou M., Nakagawa T., Seog D. H., and Hirokawa N. (2000) Kinesin superfamily motor protein KIF17 and mLin 10 in NMDA receptor-containing vesicle transport. Science 288, 1796–1802 [DOI] [PubMed] [Google Scholar]
- 37.Camargo L. M., Collura V., Rain J. C., Mizuguchi K., Hermjakob H., Kerrien S., Bonnert T. P., Whiting P. J., and Brandon N. J. (2007) Disrupted in schizophrenia 1 interactome: evidence for the close connectivity of risk genes and a potential synaptic basis for schizophrenia. Mol. Psychiatry 12, 74–86 [DOI] [PubMed] [Google Scholar]
- 38.Sawamura N., Ando T., Maruyama Y., Fujimuro M., Mochizuki H., Honjo K., Shimoda M., Toda H., Sawamura-Yamamoto T., Makuch L. A., Hayashi A., Ishizuka K., Cascella N. G., Kamiya A., Ishida N., Tomoda T., Hai T., Furukubo-Tokunaga K., and Sawa A. (2008) Nuclear DISC1 regulates CRE-mediated gene transcription and sleep homeostasis in the fruit fly. Mol. Psychiatry 13, 1138–1148, 1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldstein L. S., and Yang Z. (2000) Microtubule-based transport systems in neurons: the roles of kinesins and dyneins. Annu. Rev. Neurosci. 23, 39–71 [DOI] [PubMed] [Google Scholar]
- 40.Hirokawa N., and Takemura R. (2005) Molecular motors and mechanisms of directional transport in neurons. Nat. Rev. Neurosci. 6, 201–214 [DOI] [PubMed] [Google Scholar]
- 41.Hirokawa N., and Noda Y. (2008) Intracellular transport and kinesin superfamily proteins, KIFs: structure, function, and dynamics. Physiol. Rev. 88, 1089–1118 [DOI] [PubMed] [Google Scholar]
- 42.Setou M., Seog D. H., Tanaka Y., Kanai Y., Takei Y., Kawagishi M., and Hirokawa N. (2002) Glutamate -receptor-interacting protein GRIP1 directly steers kinesin to dendrites. Nature 417, 83–87 [DOI] [PubMed] [Google Scholar]
- 43.Guillaud L., Setou M., and Hirokawa N. (2003) KIF17 dynamics and regulation of NR2B trafficking in hippocampal neurons. J. Neurosci. 23, 131–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yuen E. Y., Jiang Q., Chen P., Gu Z., Feng J., and Yan Z. (2005) Serotonin 5-HT1A receptors regulate NMDA receptor channels through a microtubule-dependent mechanism. J. Neurosci. 25, 5488–5501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yuen E. Y., Jiang Q., Feng J., and Yan Z. (2005b) Microtubule regulation of N-methyl-d-aspartate receptor channels in neurons. J. Biol. Chem. 280, 29420–29427 [DOI] [PubMed] [Google Scholar]
- 46.Goldstein L. S. (2003) Do disorders of movement cause movement disorders and dementia? Neuron 40, 415–425 [DOI] [PubMed] [Google Scholar]