

Background: R146G and R21C mutations in cardiac TnI are associated with hypertrophic cardiomyopathy.
Results: Both mutations blunt PKA-mediated effects on weakening cTnI-cTnC interaction and accelerating myofibril relaxation.
Conclusion: Both mutations result in hypercontraction and impaired relaxation, which may contribute to increased risk to traumatic heart failure.
Significance: This study increases mechanistic understanding of how single amino acid mutations result in cardiac contractile dysfunction.
Keywords: cardiomyopathy, kinetics, molecular modeling, mutant, troponin, β-adrenergic, C-I interaction, contraction, myofibril, relaxation
Abstract
Two hypertrophic cardiomyopathy-associated cardiac troponin I (cTnI) mutations, R146G and R21C, are located in different regions of cTnI, the inhibitory peptide and the cardiac-specific N terminus. We recently reported that these regions may interact when Ser-23/Ser-24 are phosphorylated, weakening the interaction of cTnI with cardiac TnC. Little is known about how these mutations influence the affinity of cardiac TnC for cTnI (KC-I) or contractile kinetics during β-adrenergic stimulation. Here, we tested how cTnIR146G or cTnIR21C influences contractile activation and relaxation and their response to protein kinase A (PKA). Both mutations significantly increased Ca2+ binding affinity to cTn (KCa) and KC-I. PKA phosphorylation resulted in a similar reduction of KCa for all complexes, but KC-I was reduced only with cTnIWT. cTnIWT, cTnIR146G, and cTnIR21C were complexed into cardiac troponin and exchanged into rat ventricular myofibrils, and contraction/relaxation kinetics were measured ± PKA phosphorylation. Maximal tension (Tmax) was maintained for cTnIR146G- and cTnIR21C-exchanged myofibrils, and Ca2+ sensitivity of tension (pCa50) was increased. PKA phosphorylation decreased pCa50 for cTnIWT-exchanged myofibrils but not for either mutation. PKA phosphorylation accelerated the early slow phase relaxation for cTnIWT myofibrils, especially at Ca2+ levels that the heart operates in vivo. Importantly, this effect was blunted for cTnIR146G- and cTnIR21C-exchanged myofibrils. Molecular dynamics simulations suggest both mutations inhibit formation of intra-subunit contacts between the N terminus and the inhibitory peptide of cTnI that is normally seen with WT-cTn upon PKA phosphorylation. Together, our results suggest that cTnIR146G and cTnIR21C blunt PKA modulation of activation and relaxation kinetics by prohibiting cardiac-specific N-terminal interaction with the cTnI inhibitory peptide.
Introduction
Familial hypertrophic cardiomyopathy (HCM)2 has been identified as a major autosomal dominant disease and is highly correlated with mutations detected in myofilament contractile proteins (1). Although the majority of mutations are found in myosin and cardiac myosin-binding protein C (cMyBP-C), mutations have also been identified in thin filament regulatory proteins such as cardiac troponin I (cTnI), which is a subunit of the cardiac troponin (cTn) complex that has a critical role in the activation and relaxation of cardiac muscle (2). At the beginning of systole, with the rise of intracellular Ca2+ in cardiomyocytes, Ca2+ binding to cardiac troponin C (cTnC) initiates a chain of events involving dynamic and structural changes in troponin that result in the activation of the thin filament (3). In the absence of Ca2+ (diastole), cTnC exists in its “closed” conformation, and cTnI binds actin tightly (and only weakly with cTnC), inhibiting actin-myosin interaction (3, 4). In systole, Ca2+ binding to site II of cTnC induces an “open” conformation that increases interaction between the N terminus of cTnC (NcTnC) and the cTnI switch peptide, resulting in decreased binding of the cTnI inhibitory peptide with actin (3, 5). Consequently, this permits increased tropomyosin mobility, myosin interaction with actin to form cross-bridges, resulting in force generation (3).
β-Adrenergic stimulation serves as an essential physiological mechanism to meet increases in circulatory demand, acting through positive inotropic-lusitropic effects (6). During β-adrenergic stimulation, cTnI is phosphorylated by protein kinase A (PKA) at sites Ser-23 and Ser-24 (Ser(P)-23/Ser(P)-24) that reside in the cardiac-specific N terminus of cTnI (NcTnI) (6). We (7, 8) and others (6, 9–11) have demonstrated that phosphorylation of these sites reduces the affinity of cTnC for cTnI (KC-I), reduces Ca2+ sensitivity (pCa50) of tension production, increases cross-bridge cycling kinetics, and accelerates cardiac muscle relaxation. We have also reported that PKA phosphorylation of cTnI or bis-phosphomimic substitutions of cTnI (cTnIS23D/S24D) accelerates and shortens the initial slow phase of cardiac myofibril relaxation, particularly during contraction with physiological (sub-maximal) Ca2+ conditions, and thus it increases the overall speed of relaxation (7).
HCM-associated cTnI mutations were first reported by Kimura et al. in 1997 (12), including R145G/R145Q, R162W, G203S, and K206Q. Among them, the cTnIR145G mutation (cTnIR146G in rodent), which is located in the inhibitory peptide of cTnI, has received prominent attention (13–25). Most previous studies investigating this mutation have focused on the Ca2+ sensitivity of tension, and ATPase activity in cardiomyocytes, demembranated cardiac muscle, and transgenic mice. It is well established that cTnI-R146G mutation increases Ca2+ sensitivity of myofibrillar ATPase activity and force (18–20), reduces inhibition of actin-tropomyosin-activated myosin ATPase (14, 18, 19), and may have no direct effect on the cross-bridge cycle (20). It has also been reported that the cTnI-R145G mutation has a significant effect on energy cost and has been associated with diastolic dysfunction (20). Another mutation, cTnIR21C, is the only identified HCM-associated mutation located at the cardiac-specific N terminus of cTnI (26–29). In transgenic mice, the cTnIR21C mutation has been reported to prevent PKA-mediated phosphorylation in vivo (27, 28). It has also been reported that isolated cardiac myocytes from R21C mice older than 12 months of age have significantly delayed Ca2+ transient decay and relaxation (28). However, the mechanism for these effects and how these mutations affect the contraction and relaxation kinetics of cardiac muscle have not been studied.
Previous studies have proposed the formation of an intra-molecular interaction between the N terminus and the inhibitory peptide region of cTnI upon PKA phosphorylation of Ser-23/Ser-24 of cTnI (11, 30–32). Recently, our computational modeling results demonstrated that introduction of the S23D/S24D substitutions (bis-phosphomimic substitutions) on cTnI (cTnIS23D/S24D) led to the formation of an intra-subunit interaction between the N terminus and the inhibitory peptide of cTnI (8). We hypothesized that this interaction may be the structural correlate for shortening the duration and increasing the rate of the early phase of relaxation by destabilizing cTnI switch peptide interaction with NcTnC (8). Therefore, we hypothesized that introduction of an HCM mutation located in either the N terminus or the inhibitory peptide of cTnI may disrupt the formation of this intra-subunit interaction and blunt the effects of Ser-23/Ser-24 phosphorylation by PKA during β-adrenergic stimulation. In this work, we tested this hypothesis by studying the two HCM mutations cTnIR146G and cTnIR21C (see Fig. 1 for the location) that are located in the inhibitory peptide and the N terminus of cTnI (respectively) using combined protein biochemistry, myofibril mechanics, and computational (molecular dynamics) simulation studies. Our studies indicate that both of these cTnI mutants increase Ca2+ binding of cTn (KCa) and KC-I in solution, increase the Ca2+ sensitivity of myofibril tension development, and also prolong the early slow phase of relaxation. Importantly, both mutants blunt the ability of PKA to reduce KC-I and the Ca2+ sensitivity of tension (pCa50) and speed relaxation of myofibrils. Our computational modeling of cTn suggests that introduction of either mutation inhibits the formation of the intra-subunit interaction between the N terminus and the inhibitory peptide of cTnI normally seen for cTn with phosphorylation (or bis-phosphomimic substitutions) of Ser-23/Ser-24. Thus, in addition to being hyper-contractile during systole, hearts with these mutations may have impaired initiation of diastole during β-adrenergic stimulation.
FIGURE 1.

Human cTnI sequence (A) and ternary structure (B) with HCM-related mutation sites (R21C and R145G) and PKA phosphorylation sites (Ser-23/Ser-24). In the ternary structure, cTnC(1–161) is shown in blue; cTnI(1–172) is in red, and cTnT(236–285) is in yellow. The asterisks indicate the key positions in cTnI.
Experimental Procedures
Proteins, cTnC Labeling, cTnI Phosphorylation, and cTn Complex Reconstitution
WT rat cTnC, cTnI, and cTnT in the pET24 vectors were constructed and expressed as described previously (33). cTnCC35S, cTnIS23D/S24D, cTnIR146G, cTnIR21C, and cTnIR21C/S23D/S24D were constructed from WT cTnC and cTnI plasmids, respectively, using a site-directed mutagenesis kit (Stratagene, La Jolla, CA). We (Fig. 2A) and others (27, 28) demonstrated that cTnIR21C disrupts the PKA phosphorylation on Ser-23/Ser-24 of cTnI. Thus, the bis-phosphomimic substitutions (S23D/S24D) were introduced on sites Ser-23/Ser-24 of cTnIR21C (cTnIR21C/S23D/S24D) to mimic the effects of PKA-mediated phosphorylation of these sites. All WT and mutant proteins were expressed with a pET-24 (Novagen, Madison, WI.) vector containing the T7 promoter, lac operator, and a kanamycin-resistant gene, and the expressed constructs were finally confirmed by the DNA sequence analysis.
FIGURE 2.

A, phosphorylation (phos) profile of Ser-23/Ser-24 for purified cTnIWT, cTnIR146G, and cTnIR21C from the cTnC affinity column. Raw (B) and normalized (C) PKA phosphorylation profiles of cTnI for rat LV cardiac myofibrils exchanged with cTn-containing cTnIWT or cTnIR146G before and after PKA treatment are shown.
The cTnCC35S substitution was labeled with a fluorescent probe {N-[2-(iodoacetoxy)ethyl]-N-methyl}-nitrobenz-2-oxa-1,3-diazole (IANBD, Mr = 406.14, Life Technologies, Inc., catalog no. I-9) at Cys-84 (cTnCIANBDC35S) in the dark overnight at 4 °C to monitor the Ca2+-cTn (KCa) and cTnC-cTnI (KC-I) binding affinities, as described previously (7, 34–36). The labeling efficiency was determined by measuring the IANBD fluorophore to protein molar concentration ratio (7, 36). The concentration of protein was determined using Bio-Rad protein assay (based on Bradford method), and the IANBD concentration in the labeled protein was determined by dividing the absorbance of the labeled protein at the maximal absorbance for the fluorophore by the extinction coefficient of IANBD (21000 m−1cm−1) at a wavelength of 481 nm. The final labeling efficiency was then determined to be ∼90%.
Purified cTnIWT, cTnIR146G, and cTnIR21C were phosphorylated using a cTnC affinity column by adding 500 units of the catalytic subunit of PKA (Sigma, catalog no. P2645). The reaction was initiate by adding 0.5 mm ATP and 6 mg/ml DTT to the column, and the column was incubated in a pre-warmed water bath at 30 °C for 30 min (37). The phosphorylation profile of cTnI was determined by calculating the percentage of phosphorylated and the total amount of cTnI from Western blot (7).
Whole cTn complexes were formed using rat cTnC (WT or cTnCIANBDC35S), rat cTnI (WT, WT-Ser(P)-23/Ser(P)-24, WT-S23D/S24D, R146G, R146G/Ser(P)-23/Ser(P)-24, R21C, or R21C/S23D/S24D), and rat cTnT (WT) at a 1:1:1 molar ratio and then dialyzed through a series of buffers with gradually decreased KCl concentration at 4 °C (without stirring) as described previously (38, 39). Here, cTn complexes with cTnCIANBDC35S, cTnISer(P)-23/Ser(P)-24 (or cTnIR146G/Ser(P)-23/Ser(P)-24)were used for Ca2+-cTn binding measurement.
Steady-state Fluorescence Measurements
All steady-state fluorescence experiments were measured using an LS50B luminescence spectrometer (PerkinElmer Life Sciences) at 15 °C as described previously (7, 36, 40). Solution composition for this fluorescence measurement was as follows (in mm): 150 KCl, 20 MOPS, 3 MgCl2, 2 EGTA, and 1 DTT, pH 7.0. Fluorescence signal of 2 ml of cTnIANBDC35S or cTnCIANBDC35S (0.6 μm) was monitored during the titration of microliter amounts of Ca2+ or cTnI variants (WT, Ser(P)-23/Ser(P)-24, S23D/S24D, R146G, R146G/Ser(P)-23/Ser(P)-24, R21C or R21C/S23D/S24D) in the presence of Ca2+ (100 μm) at ∼530 nm with an excitation wavelength of 490 nm. The concentration of free Ca2+ was computed using Maxchelator (41). The Ca2+ sensitivity (measured as pCa50, the pCa (pCa = −log [Ca2+]) value at half-maximal fluorescence signal change) was collected by fitting the binding curve with the sigmoid Hill equation as described previously (42). The reported values are the means ± S.E. of three to six successive titrations.
Ethical Approval and Tissue Preparation
Animal procedures were conducted in accordance with the National Institutes of Health Policy on Humane Care and Use of Laboratory Animals and were approved by the University of Washington Institutional Animal Care and Use Committee (IACUC). Rats were housed in the Department of Comparative Medicine, University of Washington, and cared for according to IACUC procedures. Male Sprague-Dawley rats (3 months old, 150–250 g) were anesthetized with an intraperitoneal injection of pentobarbital (50 mg/kg) after initial exposure to isoflurane (3–5% in oxygen). When the rat had no reflexive response, its heart was rapidly excised and dissected in oxygenated physiological saline solution containing the following (in mm): 100 NaCl, 24 NaHCO3, 2.5 KCl, 1 MgSO4·7H2O, 1 Na2HPO4, and 1 CaCl2 (43). After this, both ventricles were cut open, and the whole heart was demembranated in skinning solution containing (in mm): 100 KCl, 9 MgCl2, 4 Na2ATP, 5 K2EGTA, 10 MOPS, 1% Triton X-100, pH 7.0, 50% v/v glycerol, and 1:100 dilution “protease inhibitor mixture” (Sigma, catalog no. P8340) overnight at 4 °C (44, 45). The heart was then washed three times in the same solution without Triton X-100 and stored at −20 °C for using up to 1 week. Myofibrils from the left ventricles (LV) were used for the mechanical measurements described below.
Solutions
Composition of solution used for mechanical measurements was determined by an iterative algorithm that computes the equilibrium concentration of ions and ligands based on published affinity constants (46). Composition of relaxing solution was as follows (in mm): 80 MOPS, 1 Mg2+, 5 MgATP, 83 K+, 52 Na+, 15 EGTA, and 15 creatine phosphate, pH 7.0, at 15 °C. The solution ionic strength was 170 mm, and the inorganic Pi concentration that was determined by NMR measurement was 0.5 mm (47). All mechanical measurements were performed at a constant temperature of 15 °C. The Ca2+ levels (expressed as pCa = −log [Ca2+]) for activation solutions were adjusted by adding CaCl2. To study the effects of PKA, isolated myofibrils were incubated with 200 μl of relaxing solution containing 100 units of PKA and 6 mm DTT for 45 min at 20 °C.
Exchange of Recombinant cTn Complexes into Myofibrils
Muscle bundles obtained from the rat LV were rinsed twice in Rigor solution containing the following (in mm): 100 KCl, 50 Tris, 2 MgCl2, 1 EGTA, 1 DTT and 1:100 dilution of protease inhibitor mixture before being homogenized for 2 pulses of 30 s on ice at high speed. cTn complexes containing cTnIWT, cTnIS23D/S24D, cTnIR146G, cTnIR21C, or cTnIR21C/S23D/S24D at a final concentration of ∼1 mg/ml were passively exchanged into isolated rat LV myofibrils in a buffer containing the following (in mm): 200 KCl, 20 MOPS, 5 MgCl2, 2 EGTA, 1 DTT, 4 ATP, and 1:100 dilution of protease inhibitor mixture on a slow rocker overnight at 4 °C (7). Following exchange, myofibrils were washed with relaxing solution containing 1 mg/ml bovine serum albumin (BSA) twice for 30 min to remove any non-specifically bound exogenous cTn.
Myofibril Mechanical/Kinetics Measurement
Myofibril mechanical/kinetics measurements were performed on a custom-built setup as described previously (48). Briefly, single or small bundles (∼2–4) of cardiac myofibrils were attached between two glass micro-tools forged from borosilicate glass capillary tubes (outer diameter 1.0 mm and inner diameter 0.5 mm, Sutter Instruments, Novato, CA), with the initial sarcomere length set as ∼2.3 μm, and perfused with solutions that can be rapidly switched. One of the needles acted as a force transducer, which deflected in a predictable manner upon application of force (48). Needle stiffness was determined by first deflecting the needle with a known amount of force using a galvanometer. Needle deflections were measured under a ×40 lens, and this yielded stiffness in nN μm−1. The stiffness of needles used for the experiments ranged between 5 and 11 nN μm−1. This force transducer needle was positioned over a dual diode system, which records needle displacement and correlates displacement to force development. A second straight needle was attached to the other end of the myofibril and was applied to rapidly shorten and re-stretch the myofibril through a computer interface and a Piezo-controller motor (PZT Servo controller, LVPZT amplifier, Physik Instrumente, Irvine, CA). At the end of each experiment, a calibration curve was performed in which the force transducer needle was moved in 1-μm steps over the range of the diodes using micromanipulators (MP-285, Sutter Instruments, Novato, CA).
A double-barreled borosilicate θ glass pipette (capillary glass tubing outer diameter 2.0 mm and inner diameter 1.4 mm, SEP 0.2 mm, modified in-house to outer diameter of 0.55 mm, Warner Instruments, Hamden, CT) was used to stream low (10−9 m, pCa 9.0) and high (10−4 m, pCa 4.0) Ca2+-containing solutions to the mounted preparation, and stepping for solution switch over the preparation was controlled by a computerized motor (SF-77B Perfusion Fast Step, Warner Instruments). The solution change was complete in ∼10 ms (48, 49).
Activation and relaxation data were collected at 15 °C and fit as described previously (48–50). The kinetics of contractile activation (kact; with rapid increase in Ca2+) was obtained from a single-exponential rise to a maximum. A rapid release-restretch protocol (a sudden 20% decrease in optimal length followed by rapid stretching back to the original length after 25 ms of unloaded shortening) was applied to measure the rate of force redevelopment (ktr). A slow phase relaxation rate (krel, slow) was reported as the slope of a regression line fit to the tension trace and normalized to the tension amplitude, and the slow phase duration (trel, slow) was measured from the onset of solution change at the myofibril to the shoulder marking the beginning of fast phase. Transition from slow to rapid phase was determined through multiple factors. An apparent change in the slope of the data or a change in the signal-to-noise ratio was often apparent at the transition. The fast phase relaxation rate (krel, fast) was measured from a single exponential decay fitted to the data. A t½ estimation was made in cases where the decay was not well described by a single exponential, and this was converted to a rate τ = ln(2)/t½. Myofibrils that contracted >10% of their optimal length were excluded from the analysis as non-isometric.
Protein Phosphorylation Profile
The cTnI phosphorylation profile was quantified using Western blot by calculating the amount of phosphorylated cTnI relative to the total amount of cTnI (7). The phosphorylated cTnI was detected using a rabbit polyclonal to cTnI (phospho-Ser-22 + Ser-23, from Abcam, catalog no. ab58545) as primary antibody (1:1000) and goat anti-rabbit IgG-HRP (Santa Cruz Biotechnology, sc-2004) as secondary antibody (1:5000). The total cTnI was quantified using antibodies rabbit polyclonal IgG to troponin I (H170, from Santa Cruz Biotechnology, sc-15368) (1:1000), and goat anti-rabbit IgG-HRP (1:5000).
Statistics
Comparisons between groups of data were performed using paired or unpaired Student's t test as appropriate. All reported data are expressed as mean ± S.E, and “n” represents the number of experimental samples in each group. Results with p < 0.05 were considered statistically significant. In this study, the R146G, R21C, WT + PKA, and S23D/S24D data were compared with the WT sets; the R146G + PKA data were compared with WT + PKA sets, and the R21C/S23D/S24D results were compared with S23D/S24D sets.
Computational Modeling
The initial structure of the cTn complex was built up based on the core crystal structure of Takeda et al. (51) with the addition of the N terminus of cTnI from the NMR structure provided by Howarth et al. (30). To mimic phosphorylation, a bis-phosphomimics model (cTnIS23D/S24D) was constructed by mutating Ser-23/Ser-24 of cTnI to aspartic acid (Asp). Two systems of human cTn were prepared for simulations as follows: a cTnIR21C Ca2+-bound cTnC(1–161)-cTnI(1–172)-cTnT(236–285) (cTnIR21C cTn model), and a cTnIR21C/S23D/S24D Ca2+-bound cTnC(1–161)-cTnI(1–172)-cTnT(236–285) (cTnIR21C/S23D/S24D cTn model). The cTnI mutations were performed using the Mutate Residue module in VMD (52). The build-up models were immersed with TIP3P water molecules in a truncated rectangular box, which extended minimally 14 Å away from any solute atoms (53). Then, K+ and Cl− ions were added to neutralize the systems and brought to 150 mm ionic strength. The fully solvated systems contained 112,758 (cTnIR21C cTn model), and 112,759 (cTnIR21C/S23D/S24D cTn model) atoms, respectively. Prior to the MD simulations, we performed three steps of minimizations. Next, 150-ns MD simulations were performed under the NPT ensemble and 300 K using NAMD 2.9 (54) and the CHARMM27 force field (55). The SHAKE procedure was applied on the bonds involving hydrogen atoms, and the time step was set to 2.0 fs (56). During the sampling process, the coordinates were saved every 10 ps. The stability between site II Ca2+ and its coordinating residues (Asp-65, Asp-67, Ser-69, Thr-71, Asp-73, and Glu-76) of cTnC was monitored by calculating the following distances for each 150-ns simulation as described previously (8, 36). Simulations were run in triplicate.
The residue-residue contacts between cTnC and key regions of cTnI (N terminus, inhibitory peptide and switch peptide regions) were monitored over the course of the entire 450-ns simulations. Contacts between two residues were defined as described previously (36), with a carbon-carbon distance of ≤5.4 Å and a distance between any other non-carbon atoms of ≤4.6 Å being a contact. Contacts between NcTnC-switch peptide of cTnI, and cTnC-inhibitory peptide of cTnI were monitored. The intra-subunit interaction between the N terminus and the inhibitory peptide region of cTnI were also recorded. For each residue contact pair, the fraction of the simulation time that these residues were in contact was calculated for both simulation systems.
Results
Purified cTnI Phosphorylation Level from cTnC Column
Both WT, R146G, and R21C cTnI were phosphorylated by PKA using the cTnC affinity column, and the extent of cTnI phosphorylation was determined by computing the percentage of Ser-23/Ser-24 phosphorylated versus the total amount of cTnI, using Western blot analysis (7). As shown in Fig. 2A, our results suggested that our phosphorylation protocol was very efficient, with >85% phosphorylation for both WT and R146G cTnI. Consistent with previous reports (27, 28), the phosphorylation of cTnIR21C was <5%, suggesting cTnIR21C may disrupt the PKA phosphorylation process at Ser-23/Ser-24 of cTnI, and thus resulted in “blunted” β-adrenergic stimulation effects. This may be the actual physiological/pathogenic mechanism of cTnIR21C. Thus, to determine whether it is the cTnIR21C mutation per se that is altering function or just the inability to get Ser-23/Ser-24 phosphorylated, we introduced the bis-phosphomimic substitutions S23D/S24D into cTnIR21C (cTnIR21C/S23D/S24D) to mimic the effect of PKA phosphorylation. We (7, 8, 25, 57) and others (58–61) previously demonstrated that cTnIS23D/S24D can mimic the PKA phosphorylation effects on Ser-23/Ser-24 of cTnI (cTnI Ser(P)-23/Ser(P)-24) both structurally and functionally.
Steady-state Fluorescence Measurements of KC-I and KCa
The effects of R146G or R21C mutation ± PKA phosphorylation (or bis-phosphomimic substitutions) on cTn KCa and the binding affinity of cTnC for cTnI (KC-I) were determined by steady-state fluorescence measurements using a fluoroprobe IANBD, as described previously (7, 36). IANBD, a sulfhydryl-reactive and environment-sensitive extrinsic fluorophore, has been widely used to study the intra-molecular interactions of proteins, and labeling at Cys-84 of cTnCC35S reflects conformational and environmental changes of NcTnC that arise from Ca2+ binding and/or interaction with cTnI (7, 35, 36, 40). We first measured the conformational changes with Ca2+ binding to cTn containing either cTnIR146G or cTnIR146G/Ser(P)-23/Ser(P)-24compared with WT cTnI. As shown in the Ca2+ titration curves in Fig. 3A, cTnIR146G increased (left shift) Ca2+ binding affinity (KCa) compared with cTn containing cTnIWT, in agreement with previous studies (18–20). The Ca2+ sensitivity of the fluorescence intensity (reported as pCa50) was shifted by 0.24 pCa units, from 7.07 ± 0.03 (cTn with cTnIWT) to 7.31 ± 0.03 (cTn with cTnIR146G). Consistent with our previous finding (7), phosphorylation of cTnIWT at Ser-23 and −24 (cTnISer(P)-23/Ser(P)-24) also reduced KCa, resulting in a 0.31 pCa unit decrease (right shift). Similarly, PKA phosphorylation of cTnIR146G (cTnIR146G/Ser(P)-23/Ser(P)-24)reduced the Ca2+ sensitivity (pCa50 = 7.03 ± 0.03), resulting in a 0.28 pCa unit decrease (right shift).
FIGURE 3.

Changes in the IANBD fluorescence emission intensity of cTnCC35S in complex with cTnTWT and cTnI variants with titration of Ca2+ (A and C) and of cTnCC35S alone with titration of cTnI variants in the presence of 100 μm Ca2+ (B and D).
In view of the “gatekeeper” role of cTnC-cTnI interaction in translating the Ca2+ signal to myofilament proteins to initiate cardiac muscle contraction, we also tested how the cTnIR146G mutation ± PKA phosphorylation affected KC-I. The KC-I was measured by titrating cTnIR146G or cTnIR146G/Ser(P)-23/Ser(P)-24 into cTnCIANBDC35S in the presence of 100 μm Ca2+. Fig. 3B shows the IANBD fluorescence signal change as the concentration of cTnI was increased up to 0.8 μm in solutions containing 0.6 μm cTnCIANBDC35S. There was no further change in the fluorescence signal beyond 0.6 μm cTnI, suggesting strong binding of cTnI to cTnC such that 1:1 binding was achieved. Similar to KCa, cTnIR146G left-shifted KC-I compared with cTnIWT. As we reported previously (7), phosphorylation of cTnIWT reduced KC-I. However, this effect was completely eliminated (blunted) for the cTnIR146G mutant.
Consistent with our previous finding (7), with respect to the cTnIWT, cTnIS23D/S24D substitutions also reduced (right-shifted) both Ca2+ sensitivity (pCa50 = 6.78 ± 0.03) and KC-I, similar to phosphorylation of cTnIWT at Ser-23 and -24. As with cTnIR146G, cTnIR21C also increased both KCa (pCa50 = 7.29 ± 0.02) and KC-I compared with cTnIWT (Fig. 3, C and D), and upon introduction of the bis-phosphomimic substitutions at Ser-23/Ser-24 of cTnIR21C (cTnIR21C/S23D/S24D), KCa also decreased (pCa50 = 7.03 ± 0.03), and the effect on KC-I was blunted. Interestingly, this suggests that both mutations, in different regions of cTnI, behave similarly in solution.
Recombinant Troponin (cTn) Complex Exchange Profiles
The native cTn in isolated myofibrils was passively exchanged with recombinant rat cTn containing either cTnIWT, cTnIR146G, or cTnIR21C. The extent of exchange (exchange efficiency) for this procedure was periodically determined by exchanging cTn containing a cTnT-labeled at the N terminus with a c-Myc tag, to compare the c-Myc tag band versus the native cTnT band in gels and with Western blot analysis (7, 57). Using this approach, we consistently see >80% endogenous cTn replacement by cTn containing the c-Myc tagged cTnT in myofibrils (7). This suggests the exchange protocol is very efficient and changes in contractile parameters should be attributed to the exchanged cTn containing either cTnIR146G or cTnIR21C.
To study the effects of PKA phosphorylation, myofibrils exchanged with cTn containing either cTnIWT or cTnIR146G were incubated with relaxing solution containing 100 units of PKA and DTT for 45 min. For cTnIR21C, PKA effects were studied by exchanging cTn containing cTnIR21C/S23D/S24D and were compared with the cTn containing cTnIS23D/S24D. The phosphorylation profile for cMyBP-C and titin (also phosphorylated by PKA incubation) was not measured; however, they should be similar for each group as paired comparisons of myofibrils containing cTnIR146G or cTnIR21C versus cTnIWT were made from each heart. The extent of cTnI phosphorylation in exchanged myofibrils (prior to PKA treatment) was inversely related to the exchange efficiency. The phosphorylation profile is plotted in Fig. 2, B and C. A very small amount of residual phosphorylated cTnI was likely present in every exchange preparation because the exchange efficiency was not 100%. It is clear that the cTnI phosphorylation level in exchanged myofibrils was quite low, as recombinant cTnI was not phosphorylated, further confirming high exchange efficiency. PKA treatment significantly increased the cTnI phosphorylation level, resulting in over 90% of cTnI phosphorylated, which is consistent with our previous observation (7).
cTnI-R146G and cTnI-R21C Mutations Effects on Myofibril Contraction
The effects of mutations on tension development and relaxation kinetics (at 15 °C) were determined from isolated myofibrils from rat LV cardiac muscle exchanged with cTn containing either cTnIR146G or cTnIR21C and compared with the WT-cTn complex. Myofibrils were exposed to continually flowing solutions that were rapidly switched to provide step increases and decreases in bathing [Ca2+], from relaxing solution (pCa 9.0) to either maximal (pCa 4.0) or submaximal (pCa 5.4, pCa 5.6, and pCa 5.8) [Ca2+] and then back to 9.0. Representative example tension traces for cTnIWT, cTnIR146G, and cTnIR21C exchanged myofibrils during the submaximal [Ca2+] (pCa 5.4) activation-relaxation protocol are presented in Fig. 4. A summary of tension magnitude and kinetic parameters for rat LV myofibrils exchanged with cTn containing cTnIWT, cTnIR146G, or cTnIR21C is presented in Table 1 and Figs. 5 and 6.
FIGURE 4.
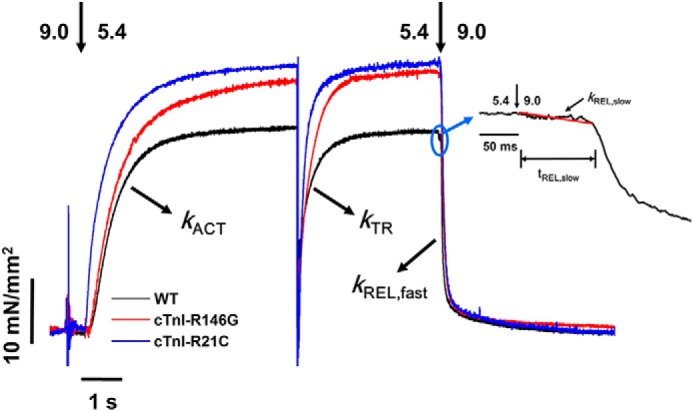
Representative tension trace (at pCa 5.4) for isolated rat LV cardiac myofibril after exchanging with recombinant cTn complexes containing cTnIWT, cTnIR146G, and cTnIR21C. The inset is a close-up of slow phase of relaxation demonstrating how krel, slow and trel, slow are measured.
TABLE 1.
Tension generation and relaxation parameters after recombinant rat WT cTn or rat cTn contained either cTnI-R146G or cTnI-R21C exchange into rat ventricular myofibrils at 15 °C
Values given are mean ± S.E. Number in parentheses indicates the number of myofibrils. a, p < 0.05 versus WT; b, p < 0.01 versus WT; and a,d, p < 0.05 versus WT + PKA; b,d, p < 0.01 versus WT + PKA; c,d, p < 0.001 versus WT + PKA; and a,e, p < 0.05 versus S23D/S24D; b,e, p < 0.01 versus. S23D/S24D; c,e p < 0.01 versus S23D/S24D.
| Myofibril Batches | pCa | Tension generation |
ktr | Relaxation |
|||
|---|---|---|---|---|---|---|---|
| Tension | kact | trel, slow | krel slow | krel fast | |||
| mN/mm2 | s−1 | s−1 | ms | s−1 | s−1) | ||
| WT | 4.0 | 73 ± 4 (30) | 3.2 ± 0.3 (30) | 5.4 ± 0.5 (30) | 79 ± 6 (28) | 1.1 ± 0.1 (28) | 18 ± 2 (29) |
| WT + PKA | 4.0 | 69 ± 7 (29) | 2.3 ± 0.2a (29) | 5.6 ± 0.9 (27) | 66 ± 3a (26) | 1.8 ± 0.2b (26) | 21 ± 2 (28) |
| S23D/S24D | 4.0 | 66 ± 5 (23) | 2.6 ± 0.2a (23) | 6.1 ± 0.6 (23) | 65 ± 5a (20) | 2.1 ± 0.4a (20) | 22 ± 3 (20) |
| R146G | 4.0 | 69 ± 7 (27) | 2.7 ± 0.2 (27) | 4.6 ± 0.5 (27) | 105 ± 7b (25) | 1.3 ± 0.2 (25) | 18 ± 2 (27) |
| R146G + PKA | 4.0 | 69 ± 10 (23) | 2.8 ± 0.3 (23) | 4.9 ± 0.6 (22) | 91 ± 5c,d (21) | 1.3 ± 0.2a,d (21) | 18 ± 2 (22) |
| R21C | 4.0 | 74 ± 6 (23) | 3.0 ± 0.3 (23) | 5.6 ± 0.3 (23) | 100 ± 6b (22) | 1.1 ± 0.1 (22) | 19 ± 2 (23) |
| R21C/S23D/S24D | 4.0 | 70 ± 5 (24) | 2.7 ± 0.2 (24) | 5.3 ± 0.4 (23) | 89 ± 7b,e (23) | 1.2 ± 0.2a,e (23) | 18 ± 2 (23) |
| WT | 5.4 | 31 ± 3 (23) | 1.6 ± 0.2 (23) | 3.6 ± 0.5 (23) | 84 ± 5 (21) | 2.0 ± 0.3 (21) | 18 ± 2 (23) |
| WT + PKA | 5.4 | 18 ± 3a (16) | 1.4 ± 0.2 (16) | 3.0 ± 0.4 (15) | 67 ± 3a (14) | 5.9 ± 1.4a (14) | 19 ± 3 (15) |
| S23D/S24D | 5.4 | 17 ± 3a (13) | 1.9 ± 0.3 (13) | 4.2 ± 0.5 (13) | 58 ± 6a (13) | 6.1 ± 1.5a (13) | 20 ± 3 (13) |
| R146G | 5.4 | 39 ± 4a (17) | 2.0 ± 0.2 (17) | 3.4 ± 0.3 (16) | 103 ± 10a (16) | 2.1 ± 0.3 (16) | 18 ± 3 (16) |
| R146G + PKA | 5.4 | 36 ± 6a,d (15) | 1.7 ± 0.2 (15) | 3.6 ± 0.4 (15) | 87 ± 6c,d (15) | 2.0 ± 0.3a,d (15) | 21 ± 2 (15) |
| R21C | 5.4 | 40 ± 4a (16) | 1.7 ± 0.2 (16) | 3.9 ± 0.3 (16) | 90 ± 5 (15) | 1.8 ± 0.2 (15) | 18 ± 2 (16) |
| R21C/S23D/S24D | 5.4 | 37 ± 3b,e (15) | 1.9 ± 0.2 (15) | 4.0 ± 0.4 (14) | 88 ± 5c,e (14) | 2.1 ± 0.2a,e (14) | 18 ± 3 (14) |
FIGURE 5.

Tension (A) and pCa-tension relationship (B) for cTnIWTversus cTnIR146G-exchanged myofibrils prior to and after PKA treatment. Tension (C) and pCa-tension relationship (D) for cTnIWT versus cTnIR21C-exchanged myofibrils prior to PKA treatment and after introduction of the bis-phosphomimic mutations. The kact/ktr ratio for cTnIWT-versus cTnIR146G-(E) and cTnIWT- versus cTnIR21C- (F) exchanged myofibrils prior to and after PKA treatment (or introduction of the bis-phosphomimic mutations). *, p < 0.05; #, p < 0.01.
FIGURE 6.

Slow phase relaxation at sub-maximal Ca2+ level (pCa 5.4) for WT-cTn (A), cTnIR146G cTn- (B) and cTnIR21C cTn (C)-exchanged rat LV cardiac myofibrils before (black) and after (red) PKA treatment. The kinetics (krel, slow, D) and duration (trel, slow, E) of slow phase relaxation for cTnIWT- versus cTnIR146G-exchanged myofibrils prior to and after PKA treatment are shown. The kinetics (krel, slow, F) and duration (trel, slow, G) of slow phase relaxation for cTnIWT- versus cTnIR21C-exchanged myofibrils prior to PKA treatment and after introduction of the bis-phosphomimic mutations are shown. *, p < 0.05; #, p < 0.01; **, p < 0.005.
Maximal tension (Tmax) did not differ for myofibrils exchanged with cTnIR146G (69 ± 7 mN/mm2) compared with cTnIWT myofibrils (73 ± 4 mN/mm2, Fig. 5A). Tension was also measured at multiple submaximal Ca2+ levels, and the pCa50 was left-shifted 0.13 pCa units (Fig. 5B), from 5.32 ± 0.02 (cTnIWT myofibrils) to 5.45 ± 0.03 (cTnIR146G myofibrils), demonstrating an increase in the Ca2+ sensitivity of tension, as reported previously by others for the cTnIR146G mutation (18–20). Similarly, Tmax was maintained for the cTnIR21C exchanged myofibrils (74 ± 6 mN/mm2, Fig. 5C), and the Ca2+ sensitivity of tension (pCa50 = 5.42 ± 0.02) also increased, displaying a 0.10 pCa unit left shift of the curve (Fig. 5D). The above results are clearly demonstrated in the example traces in Fig. 4, which were collected at pCa 5.4, showing that both cTnIR146G and cTnIR21C myofibrils have significantly higher tension development compared with the cTnIWT myofibrils.
The rate of contractile activation (kact) by rapid switching of solution [Ca2+] from pCa 9.0 to 4.0 (or submaximal Ca2+ levels) includes the kinetic processes of Ca2+-dependent thin filament activation, myosin cross-bridge binding, and the subsequent tension development. Compared with the cTnIWT-exchanged samples (3.2 ± 0.3 s−1), kact did not differ for either the cTnIR146G- (2.7 ± 0.2 s−1) or cTnIR21C (3.0 ± 0.3 s−1)-exchanged myofibrils at pCa 4.0 or any sub-maximal Ca2+ level tested (Table 1). For all myofibrils, kact was significantly slower at sub-maximal Ca2+ levels than during maximal Ca2+ activations, as reported previously for rodent cardiac myofibrils (7, 47), suggesting that the Ca2+ sensitivity of cardiac contraction kinetics is maintained upon introduction of HCM-associated mutations. Once the activation was completed (i.e. tension was in steady state), a rapid release-restretch protocol was applied on myofibrils to measure the rate of tension redevelopment (ktr). The ktr protocol is designed to measure the rate of myosin cross-bridge attachment and subsequent tension generation (45) when Ca2+ binding to troponin is in near steady state (e.g. the thin filament is already activated). This measurement can help to differentiate the contribution of Ca2+-mediated thin filament activation versus the cross-bridge cycling kinetics to kact. At all measured conditions (cTnIR146G- or cTnIR21C-exchanged myofibrils), ktr was faster than kact at both maximal and submaximal Ca2+ levels, as reported previously for cTnIWT-exchanged myofibrils (7), suggesting thin filament activation is rate-limiting for rat cardiac myofibril tension generation from rest (diastole) at 15 °C. Comparing the kact/ktr ratio can give an indication of whether thin filament activation kinetics is more rate-limiting to tension development in the R146G or R21C exchanged myofibrils as compared with the WT myofibrils. Fig. 5, E and F, demonstrates that this ratio did not change upon introduction of either mutation, suggesting the activation process is maintained.
cTnI-R146G and cTnI-R21C Mutations Affect Myofibril Relaxation
Rapid deactivation of myofibrils by switching from a maximal or sub-maximal [Ca2+] solution to relaxing solution (pCa 9.0) induced a biphasic relaxation, an initial linear tension decay followed by a more rapid (fast) exponential decay back to the baseline tension (see example trace in the inset of Fig. 4). The rate of the slow phase relaxation (krel, slow) is thought to be predominantly reflective of the cross-bridge detachment rate (47, 50, 62–65), whereas the duration of slow phase relaxation (trel, slow) may be influenced by the time for the troponin to move back to a “blocked” state (66). For the maximal activations, krel, slow was unchanged for cTnIR146G-treated myofibrils (1.3 ± 0.2 s−1, Fig. 6D) compared with cTnIWT exchanged myofibrils (1.1 ± 0.1 s−1), whereas trel, slow of the cTnIR146G-exchanged myofibrils (105 ± 7 ms, Fig. 6E) was significantly prolonged compared with those of the cTnIWT-treated myofibrils (79 ± 6 ms). Similarly, at pCa 4.0, cTnIR21C-exchanged myofibrils also prolonged trel, slow (100 ± 6 ms, Fig. 6G) and did not affect krel, slow (1.1 ± 0.1 s−1, Fig. 6F). By analyzing the contribution of slow phase on whole relaxation, we found the contributions for the cTnIWT-, cTnIR146G-, and cTnIR21C-exchanged myofibrils at pCa 4.0 were 6, 7, and 7% of the total amplitude, respectively, suggesting cross-bridge detachment was not affected by the mutations. In contrast to the slow phase of relaxation, the much larger, rapid phase of relaxation (krel, fast) was determined by several sarcomeric properties as well as uneven relaxation kinetics between sarcomeres in series (63–65). There was no difference in krel, fast for either cTnIR146G (18 ± 2 s−1) or cTnIR21C (19 ± 2 s−1) versus cTnIWT-(18 ± 2 s−1) exchanged myofibrils. At a more physiological level of Ca2+, pCa 5.4, krel, slow was twice as fast compared with maximal activation (pCa 4.0) for all myofibrils (Table 1), suggesting faster cross-bridge detachment. Akin to pCa 4.0, at sub-maximal Ca2+ levels there was no difference in krel, slow or krel, fast between cTnIWT-, cTnIR146G-, and cTnIR21C-exchanged myofibrils. However, trel, slow was also prolonged for the cTnIR146G-exchanged myofibrils (Tables 1 and Fig. 6E). We calculated the times to reach 50% (RT50) and 90% (RT90) relaxation time (RT) and found that only cTnIR146G significantly prolonged the RT50 with respect to the cTnIWT exchanged myofibrils.
cTnI-R146G and cTnI-R21C Mutations Blunt the PKA Effects on Myofibril Contraction and Relaxation
We next studied the effects of PKA (or introduction of bis-phosphomimic substitutions at Ser-23/Ser-24 of cTnI) on myofibril contraction and relaxation. Consistent with previous studies (7), we found that after treating the cTnIWT-exchanged myofibrils with PKA, Tmax (69 ± 7 mN/mm2) was maintained (Fig. 5A), and pCa50 was right-shifted 0.2 pCa units to 5.12 ± 0.03, demonstrating reduced Ca2+ sensitivity of tension development (Fig. 5B). Akin to the PKA-treated WT-exchanged myofibrils, Tmax (66 ± 5 mN/mm2) was also maintained (Fig. 5C), and pCa50 was also decreased for the S23D/S24D-exchanged myofibrils (Fig. 5D), as we previously reported (7). PKA treatment of cTnIWT myofibrils (or S23D/S24D-exchanged myofibrils) also slowed kact (2.3 ± 0.3 s−1) during maximal Ca2+ activation, although the ktr (5.6 ± 0.8 s−1) was unchanged, suggesting PKA phosphorylation affects the kinetics of thin filament activation prior to cross-bridge binding and tension development. This can be clearly seen by calculating the kact/ktr ratio (Fig. 5, E and F) for PKA-treated WT-exchanged myofibril (or S23D/S24D-exchanged myofibrils), which is significantly decreased with respect to WT-exchanged myofibrils prior to PKA treatment. Following PKA treatment (or introduction of bis-phosphomimic substitutions), maximal tension was also maintained for cTnIR146G-(69 ± 10 mN/mm2) and cTnIR21C/S23D/S24D-(70 ± 5 mN/mm2) exchanged myofibrils. In contrast to cTnIWT, the Ca2+ sensitivity of tension development following PKA phosphorylation was not reduced (blunted) for either cTnIR146G or cTnIR21C (Fig. 5, B and D). Additionally, maximal kact (pCa 4.0) did not change for myofibrils exchanged with cTnIR146G following PKA phosphorylation or cTnIR21C/S23D/S24D, which can be clearly observed in the plots of kact/ktr ratio from Fig. 5, E and F, suggesting the slowing of thin filament activation at maximal Ca2+ was also blunted.
One of the main effects of β-adrenergic stimulation on cardiac function is an increase in heart rate, so faster relaxation is crucial to ensure maintained or increased diastolic ventricular filling. Thus, we measured how relaxation rates were affected following PKA treatment of cTn-exchanged myofibrils. As we observed previously (7), PKA treatment of cTnIWT-exchanged myofibrils (or cTnIS23D/S24D-exchanged myofibrils) significantly increased krel, slow (1.8 ± 0.2 s−1) and decreased trel, slow (66 ± 3 ms) during maximal Ca2+ activation, speeding up the overall relaxation. Moreover, these effects were greater at sub-maximal Ca2+ levels where the heart operates. Interestingly, for all PKA-phosphorylated cTnIR146G myofibrils or cTnIR21C/S23D/S24D-exchanged myofibrils, there were no changes in either the rate (1.3 ± 0.2 s−1 versus 1.2 ± 0.2 s−1) or the duration (91 ± 5 ms versus 89 ± 7 ms) of slow phase relaxation, suggesting the effects of PKA to speed relaxation were blunted for both mutations. Fig. 6, A–C, is a set of example tension traces demonstrating these findings. Additionally, no changes were detected in krel, fast with PKA phosphorylation. We also saw that upon PKA phosphorylation (or introduction of the bis-phosphomimic substitutions) to cTnIWT, both RT50 and RT90 were significantly decreased. Importantly, these effects were blunted with the introduction of either cTnIR146G or cTnIR21C mutations.
Molecular Dynamics Simulations
We recently reported on differences in cTn dynamics between molecular dynamics models containing the WT versus R146G cTnI (8, 25). Here, we studied the dynamics of WT versus R21C cTnI in our cTn model for comparison with the R146G model. Triplicate 150-ns MD simulations were compared. The root-mean-square fluctuations (RMSF) versus the protein residue numbers of each subunit were calculated, and the average (± S.D.) RMSF of the cTnC and cTnI subunits for both cTnIWT and cTnIR21C cTn systems is presented in Fig. 7, A and B. In Fig. 7, we highlight site I (pink) and site II (the Ca2+-binding loop, blue) of cTnC, and the inhibitory peptide (green) and switch peptide regions (orange) of cTnI. Similar to the cTnIR146G model (25), fluctuations were comparable for the cTnIR21C cTn model with respect to the WT model throughout most of the residues (average RMSFs of cTnC are 2.8 and 2.9 Å, respectively; and the average RMSFs of cTnI are 2.8 and 2.9 Å, respectively). Most of the regions had no changes larger than the standard deviations. The most pronounced differences to cTnC were seen in site I (cTnC residues 28–38, 2.2 Å for cTnIWT versus 2.6 Å for cTnIR21C cTn model) and the linker loop (cTnC residues 80–100, 2.5 Å for cTnIWT versus 2.3 Å for cTnIR21C cTn model) connecting the N- and C-terminal lobe of cTnC. The most fluctuating region detected in cTnI for both complexes was the N terminus (cTnI residues 1–41), although the helical bundle identified as the I-T arm (cTnI residues 42–137) was the most stable region in the cTnI subunits, again suggesting its structural rather than regulatory function.
FIGURE 7.

A–D, comparison of average (± S. D.) RMSF values of cTnC and cTnI for WT and cTnIR21C and cTnIR21C/S23D/S24D cTn systems in triplicate rounds of MD simulations. Here, site I and site II (the Ca2+ binding loop) of cTnC are highlighted in blue and pink, respectively, and the inhibitory peptide and switch peptide regions of cTnI are highlighted in green and orange, respectively. E–G, superposition of snapshots in schematic representation extracted every 10 ns during 150-ns MD simulations for three complexes. The cTnC is shown in blue; cTnI is in red; cTnT is in yellow, and all the key regions are highlighted with dashed circles.
We next investigated how introduction of bis-phosphomimic substitutions to Ser-23/Ser-24 of cTnI affected the dynamics of cTn containing the cTnIR21C mutation. Fig. 7, C and D, shows the average (± S.D.) RMSF of the cTnC and cTnI subunits for cTnIR21C and cTnIR21C/S23D/S24D cTn systems, respectively. Fluctuations were increased slightly in the cTnIR21C/S23D/S24D system (average RMSF 3.1 Å) with respect to the cTnIR21C system (average RMSF 2.8 Å), akin to what we previously reported for WT and cTnIS23D/S24D simulations (8), as well as the cTnIR146G and cTnIR146G/S23D/S24D simulations (25). Previously, we observed a significant change (p < 0.001) in NcTnI upon introduction of bis-phosphomimic substitutions to the WT model (8). Interestingly, introduction of bis-phosphomimic substitutions to the cTnIR21C model had very little impact on the stability of the N terminus of cTnI, similar to what we recently observed for cTnIR146G model (25). To better visualize how introduction of the R21C and/or bis-phosphomimic substitutions influences the subunit interactions among the cTn complexes, 15 snapshots taken every 10 ns during the entire MD simulations were superimposed (Fig. 7, E–G). For clarity, cTnC is shown in blue, cTnI in red, and cTnT in yellow. In contrast to the greater flexibility exhibited for the NcTnI in the cTnIS23D/S24D cTn model with respect to the WT model (8), the introduction of S23D/S24D to the cTnIR21C model had very little impact on the overall structures. As we discuss below, we speculate that this difference between the WT and R21C model upon introduction of the bis-phosphomimic substitutions may result in interference of the interaction of NcTnI with other regions of the cTn complex, and thus blunt the effects of PKA normally seen for the WT system.
We next measured the time evolution of distances between the bound Ca2+ ion and the six coordinating residues of site II (Asp-65, Asp-67, Ser-69, Thr-71, Asp-73, and Glu-76) over the course of each 150-ns simulation. Among these six coordinating residues, four (Asp-65, Asp-67, Asp-73, and Glu-76) exhibited no significant difference in fluctuations in any of the simulations, and these residues were always coordinated with Ca2+ (results not shown). The distances for the other two coordinating residues, Ser-69 and Thr-71, fluctuated much more and varied for each run in all the four systems. Thr-71 generally did not coordinate, in agreement with the structural data from x-ray crystallography (results not shown) (51). Fig. 8 shows the distances between Ca2+ and Ser-69. It is clear that Ser-69 had the most pronounced difference for Ca2+ coordination, in agreement with our previous observations (8, 25, 27, 40). The percentage of contact time for Ser-69 varies among different systems. Compared with the WT system (10%), the coordinating time of Ser-69 was increased to 23% in the cTnIR21C system. Although this was not statistically significant (p = 0.3981), it suggests a stronger interaction that could provide stabilization. This may interpret the increased Ca2+ binding affinity of cTnIR21C with respect to the WT observed from the steady-state fluorescence measurements. Interestingly, the contact time was decreased to 17% upon introduction of the phosphomimic substitutions to R21C, in agreement with the reduction of Ca2+ binding affinity (KCa) observed from the steady-state fluorescence measurements. Interestingly, we previously (25) found that compared with the WT system, the coordinating time of Ser-69 in the cTnIR146G system was increased to 29%. Upon introduction of the bis-phosphomimic substitutions to cTnIR146G, the contact time was decreased to 6%. Thus, the results obtained for cTnIR146G were similar to cTnIR21C (prolongation of the coordinating time). However, upon introduction of the bis-phosphomimic substitutions, cTnIR146G displayed a greater reduction of the coordinating time compared with cTnIR21C.
FIGURE 8.
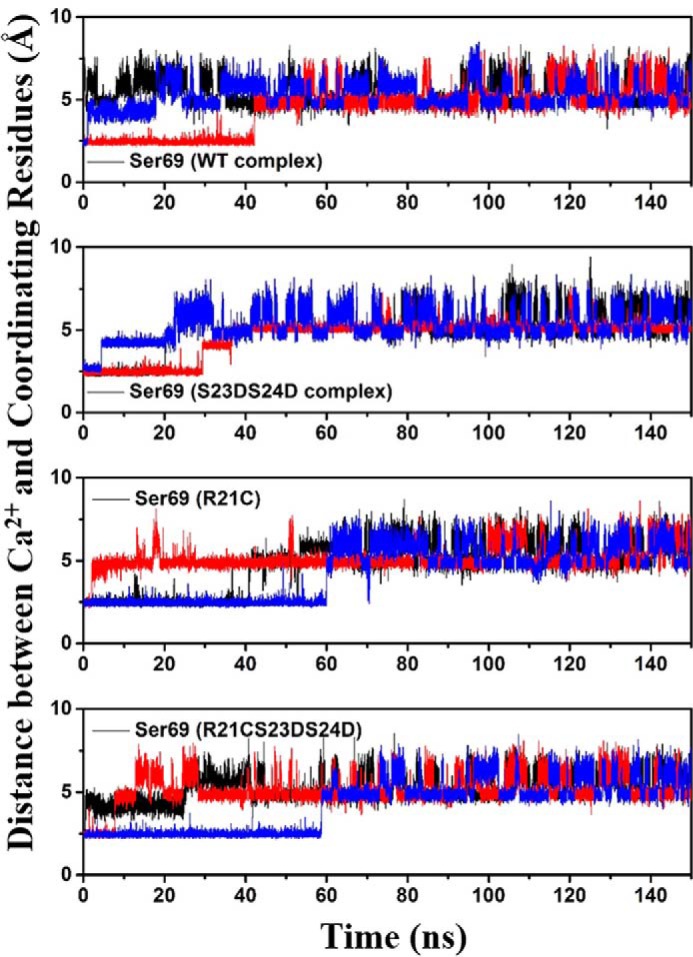
Distances between Ca2+ and its coordinating residue Ser-69 of cTnC site II over the course of each MD simulation for four complexes. Here, the 1st run result is shown in black; the 2nd run result is in red, and the 3rd run result is in blue.
Next, the residue-residue contacts of key regions were monitored over the time course of the entire 450-ns simulations. Fig. 9 shows the corresponding average contact map of residue-residue pairs (left) and the representative binding pattern (right) between the N terminus (cTnI residues 1–41, shown in red) and inhibitory peptide region (cTnI residues 138–147, shown in blue) of cTnI for the WT (A), cTnI-S23D/S24D (B), cTnI-R21C (C) and cTnI-R21C/S23D/S24D (D) cTn models. As we reported previously (8), there were no interactions between the N terminus and the inhibitory peptide region of cTnI (Fig. 9A) for the WT model. A dramatic change was observed upon introduction of the bis-phosphomimic substitutions to Ser-23/Ser-24 of the WT complex (cTnI-S23D/S24D) (8), with residues 9–14 interacting with residues 140–142 of the inhibitory peptide for ∼50% of the entire simulation (Fig. 9B). No intra-subunit interactions of cTnI were observed in the cTnI-R21C system (Fig. 9C), similar to the WT system, which is not surprising because bis-phosphomimic substitutions were not present. However, when the bis-phosphomimic residues were introduced to the cTnI-R21C system (cTnI-R21C/S23D/S24D, Fig. 9D), the intra-subunit interaction still did not form, as we observed previously for the cTnI-R146G system (25). We have suggested that this intra-subunit interaction may destabilize contacts between the cTnI switch peptide and hydrophobic residues in the NcTnC that occur following Ca2+ binding to activate contraction.
FIGURE 9.

Average contact map of residue-residue pairs (left) and the representative binding pattern (right) between the N terminus (shown in red) and inhibitory peptide region (shown in blue) of cTnI for the WT (A), cTnI-S23D/S24D (B), cTnI-R21C (C), and cTnI-R21C/S23D/S24D (D) cTn models. The blue end of the spectrum (value 0) reflects no contact between residue-residue pair, and the red end of the spectrum (value 1) represents 100% contact between residue-residue pair.
To examine this region of direct interaction between cTnC and cTnI following Ca2+ binding, we examined the contact stability of the cTnI switch peptide with hydrophobic NcTnC residues. Fig. 10 shows the different contact maps of residue-residue pairs between 14 hydrophobic residues of NcTnC (from left to right: they are Phe-20, Ala-23, Phe-24, Ile-26, Phe-27, Ile-36, Leu-41, Val-44, Leu-48, Leu-57, Met-60, Phe-77, Met-80, and Met-81) and the cTnI switch peptide (cTnI residues 148–164) for the different systems. Because Ca2+ and the cTnI switch peptide were present at the start of the MD model simulations, and we did not remove Ca2+ during simulations, we did not expect to see dramatic structural changes in this region. However, a change in the fluctuation of contacts can be considered as an indicator of stability of the cTnC-cTnI interaction associated with activation. As compared with the WT complex, there was little change in contact time upon introduction of the R21C mutant (Fig. 10A). A more dramatic change was seen upon introduction of the bis-phosphomimic residues to Ser-23/Ser-24 of WT complex (cTnI-S23D/S24D), suggesting decreased interaction between NcTnC hydrophobic residues and cTnI switch peptide upon phosphorylation (Fig. 10B). However, with introduction of the bis-phosphomimic to cTnI-R21C system (Fig. 10C), there was little change in fluctuation for the contacts compared with the cTnI-R21C or WT systems. Together with the contact information for the cTnI intra-subunit interaction (Fig. 9), our MD simulations suggest that phosphorylation of Ser-23/Ser-24 cTnI results in intra-subunit interaction of the cTnI N terminus with the inhibitory peptide, which reduces stability of cTnI switch peptide contacts with the cTnC hydrophobic patch, and that both the R21C and R146G cTnI mutations abrogate this action. In turn, this suggests a potential structure-based mechanism of how these mutations impair PKA regulation of contraction and relaxation.
FIGURE 10.
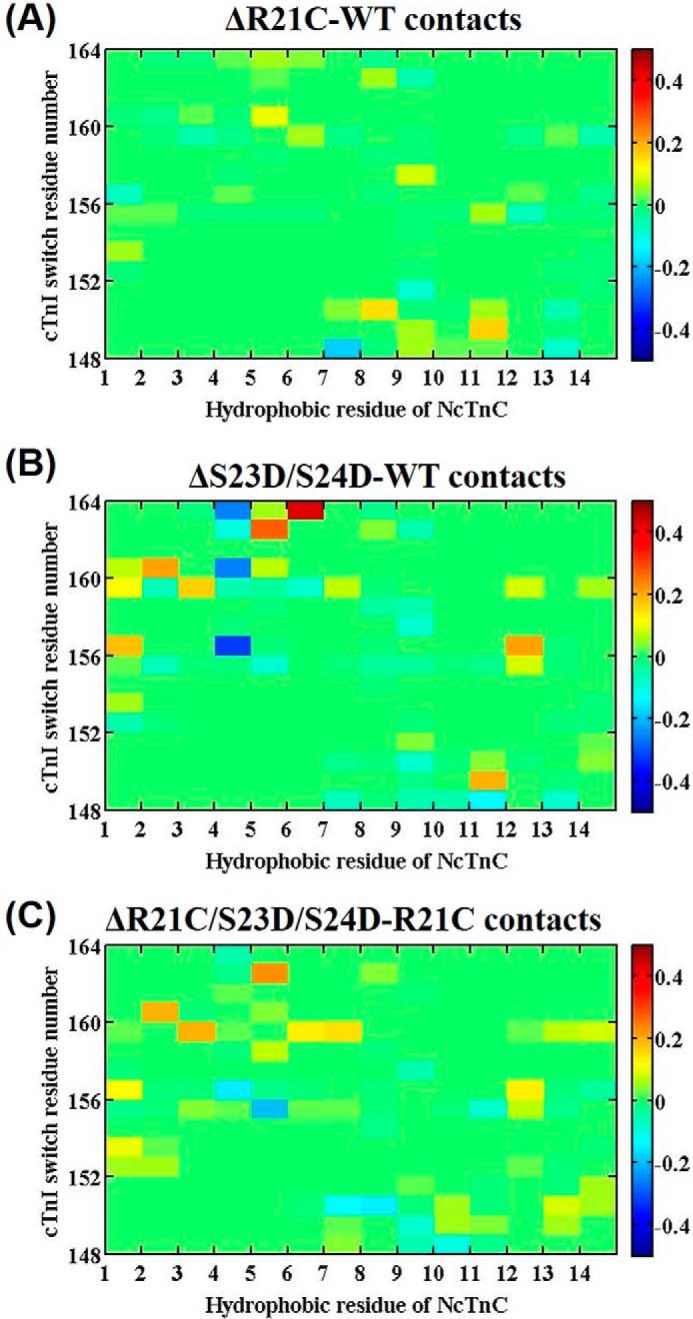
A–C, difference contact map of residue-residue pairs between 14 hydrophobic NcTnC residues and the switch peptide of cTnI mostly affected upon introduction of mutation or the phosphomimic mutations. The 14 hydrophobic residues of NcTnC are (from left to right): Phe-20, Ala-23, Phe-24, Ile-26, Phe-27, Ile-36, Leu-41, Val-44, Leu-48, Leu-57, Met-60, Phe-77, Met-80, and Met-81. Color green (value 0) reflects no difference between the two systems; the red end of the spectrum (values above 0) reflects more contacts in the R21C, S23D/S24D, or R21C/S23D/S24D cTn system, and the blue of the spectrum (values below 0) indicates more contacts in WT, WT, or R21C model.
Discussion
In this study, we tested whether HCM-associated mutations located in either the N terminus or inhibitory peptide of cTnI may disrupt the formation of an interaction between these regions that occurs with Ser-23/Ser-24 phosphorylation by PKA, thus blunting the regulatory effects on cTn that normally occur during β-adrenergic stimulation. We report here the effects of cTnIR146G or cTnIR21C ± PKA phosphorylation (or bis-phosphomimic substitutions) on KC-I, the contractile properties of isolated rat LV cardiac myofibrils, and the whole troponin structure and dynamics changes. The most significant findings of the current study were as follows: 1) Both mutations significantly increased KCa and KC-I compared with the cTnIWT. However, although PKA phosphorylation of cTnI resulted in a similar reduction of KCa for WT and both mutant-containing cTn complexes, the reduction in KC-I seen for cTnIWT was eliminated for both mutations. 2) Tmax was maintained for both cTnIR146G- and cTnIR21C-exchanged myofibrils, and the Ca2+ sensitivity of tension (pCa50) was left-shifted. However, although PKA phosphorylation (or bis-phosphomimic substitutions) decreased pCa50 (0.2 pCa units) for WT myofibrils, this effect was blunted for both mutations. 3) PKA phosphorylation of WT myofibrils accelerated the early slow phase relaxation, especially during the sub-maximal Ca2+ levels that heart operates in vivo, but most importantly, this effect was blunted for both cTnIR146G- and cTnIR21C-exchanged myofibrils. 4) MD simulations suggest the mechanism by which cTnIR146G and cTnIR21C blunt PKA-mediated reduction of KC-I, Ca2+ sensitivity of tension, and the early phase of relaxation is inhibition of the formation of an intra-subunit interaction between the N terminus and the inhibitory peptide of cTnI. This is consistently seen for WT-cTn with introduction of the bis-phosphomimic substitutions of cTnI (S23D/S24D).
Effects of Mutations on Contractile Activation and Relaxation Kinetics
Previous studies to determine the effects of cTnIR146G on the maximal tension production and the Ca2+ sensitivity of tension generation in the cardiac muscle have produced complex and sometimes contradictory results. For example, Takahashi-Yanaga et al. (19) reported that the cTnIR145G resulted in an increase in the Ca2+ sensitivity of force generation and myofibrillar ATPase activity in skinned muscle fibers. In contrast, using the reconstituted actin-tropomyosin-activated myosin ATPase assay, Lang et al. (14), Takahashi-Yanaga et al. (19), and Elliot et al. (18) reported that cTnIR145G decreases the maximal ATPase in the presence of Ca2+ and reduces inhibition of actomyosin ATPase activity in the absence of Ca2+. Using the human cTnIR145G-exchanged into murine myofibrils, Kruger et al. (21) reported no change in the Ca2+ sensitivity of tension development; however, a slightly decreased Ca2+ sensitivity was detected in the myofibrils from transgenic cTnIR146G mice. In this study, compared with WT cTn-exchanged myofibrils, Tmax was maintained for cTnIR146G- and cTnIR21C-exchanged myofibrils, and the Ca2+ sensitivity of tension (pCa50) was left-shifted by 0.13 and 0.10 pCa units, respectively. This agrees with the steady-state fluorescence measurements that showed both mutations increased KCa. To understand the structural basis of this, we monitored the time evolution of distances between the Ca2+ ion (site II) and its six coordinating residues over the course of multiple 150-ns MD simulations and found that Ser-69 coordination with Ca2+ was increased with the cTnIR21C mutation (Fig. 7), in agreement with our previous report for simulations with cTn containing cTnIR146G (25). This may explain how the Ca2+ sensitivity of contractile activation is left-shifted for both cTnIR146G and cTnIR21C, in accordance with previous studies on cTnC mutations where Ca2+ binding at site II was stabilized (36).
To better determine how both mutations affect thin filament activation and cross-bridge kinetics, we compared the rapid release-restretch protocol (ktr) with the Ca2+-activation protocol (kact) during the same activation trial. Here, ktr was faster than kact for cTnIWT-, cTnIR146G-, and cTnIR21C-exchanged rat cardiac myofibrils at all Ca2+ levels we tested, indicating that thin filament activation was rate-limiting for tension generation of the rat myofibrils at 15 °C. This finding is consistent with our previous work in rodent hearts (7, 47) but different from several other references that reported no difference between kact and ktr (62, 66). As we have previously discussed (47), the presence of 0.5 mm Pi in our solutions (a level of Pi that is close to the physiological level of Pi present in the heart) can explain this difference, and when a phosphate “mop” is used in activation solutions, the difference is eliminated. The presence of Pi influences the cross-bridge tension generating isomerization specifically, without affecting thin filament activation kinetics (47). Our current results confirm those previous findings in rodents and extend them by demonstrating that a similar effect occurs at both maximal and sub-maximal Ca2+ levels with introduction of either of these putative HCM-associated cTnI mutations.
Rapid, complete Ca2+ removal from cardiac myofibrils (Fig. 3) by a rapid solution switching protocol induces two distinct relaxation phases (biphasic), starting with an initial early slow phase of relaxation and followed by a more rapid (fast) relaxation phase. During the slow phase, isometric conditions are maintained in sarcomeres and the force decays with a linear constant rate, indicating that krel, slow primarily reflects the turnover kinetics of cross-bridge cycling, dominated by the detachment rate (62, 66). The duration of the initial slow phase of relaxation (trel, slow) depends on the Ca2+ activation levels and can be influenced by the Ca2+ binding and, likely, the cTnC-cTnI interaction properties of cTn. Here, compared with WT myofibrils, krel, slow was maintained, whereas trel, slow was prolonged upon introduction of either cTnIR146G or cTnIR21C mutations at both maximal and submaximal Ca2+ levels. Consistent with previous observations (7), krel, slow also accelerated at sub-maximal Ca2+ level for all conditions. During submaximal Ca2+ activations, there is less Ca2+ binding to thin filaments (troponin) at any given time, so that it is easier for the thin filament to become deactivated when myosins detach. Moreover, the presence of 0.5 mm Pi in our study should exacerbate this effect, as it results in a reduction of the tension-bearing cross-bridges. This may accelerate the detachment of myosin cross-bridges from the thin filament, thus contributing to an increase in the slow phase of relaxation at submaximal Ca2+ levels.
Effects of PKA Phosphorylation on Contractile Activation and Relaxation Kinetics
β-Adrenergic stimulation is a major physiological mechanism to meet the increase in circulatory demand, acting through positive inotropic and lusitropic effects (6). For cTnI, β-adrenergic stimulation results in the phosphorylation of Ser-23/Ser-24 of cTnI by PKA (6). Considering its key role in the heart performance, we studied how both mutations influence the PKA responsiveness. With PKA phosphorylation (or bis-phosphomimic substitutions), Tmax did not differ for cTnIWT-, cTnIR146G-, and cTnIR21C-exchanged myofibrils. However, although PKA phosphorylation (or with bis-phosphomimic substitutions) right-shifted pCa50 (∼0.2 pCa units) for WT myofibrils, this effects was blunted for both mutations (Fig. 4, B and D). In addition, during the maximal Ca2+ activation, the ratio of kact/ktr was decreased from 0.67 ± 0.03 to 0.48 ± 0.05 for WT myofibril upon PKA treatment (or to 0.45 ± 0.05 with introduction of the bis-phosphomimic substitutions), suggesting a slowed thin filament activation process with PKA phosphorylation. Interestingly, kact/ktr did not differ with PKA phosphorylation (or bis-phosphomimic substitutions) in the presence of either cTnIR146G or cTnIR21C, suggesting the slowing of thin filament activation by PKA-mediated cTnI phosphorylation during maximal Ca2+ conditions is blunted. Consistent with previous work in our laboratory (7) and by others (6, 9), we demonstrated that PKA treatment (or with bis-phosphomimic substitutions) increased the speed of the slow phase of relaxation for WT myofibrils, especially at the submaximal Ca2+ levels that heart operates during a cardiac twitch. Most importantly, we found that this effect of PKA on slow phase relaxation was eliminated (blunted) for both mutations.
It is important to point out that the conditions for PKA phosphorylation and introduction of bis-phosphomimic substitutions (S23D/S24D) are different in vivo, because cMyBP-C and titin are also targets for PKA phosphorylation during β-adrenergic stimulation (67, 68). So, to determine the specific effect on cTnI phosphorylation, we exchanged recombinant cTn containing cTnIS23D/S24D into cardiac myofibrils. In steady-state fluorescence studies, we found that cTnIS23D/S24D and PKA-mediated phosphorylation of cTnI (cTnISer(P)-23/Ser(P)-24) resulted in an almost identical effect, a right shift of the KC-I and KCa curves (Fig. 3) compared with cTnIWT. The pCa50 shift was −0.31 and −0.29 pCa units for the cTn complex containing cTnISer(P)-23/Ser(P)-24 versus cTnIS23D/S24D, respectively. We also saw very similar results on the modulation of thin filament activation and myofibril relaxation for PKA-phosphorylated cTnIWT-exchanged myofibrils and cTnIS23D/S24D-exchanged myofibrils. After treating the cTnIWT-exchanged myofibrils with PKA, Tmax was maintained and pCa50 was right-shifted 0.2 pCa units. Similarly, Tmax was maintained, and pCa50 was also decreased (0.21 pCa units) for the cTnIS23D/S24D-exchanged myofibrils. For relaxation, both PKA treatment of cTnIWT-exchanged myofibrils and cTnIS23D/S24D-exchanged myofibrils significantly increased the rate and decreased the duration of early slow phase relaxation, especially at sub-maximal Ca2+ levels where the heart operates. All the above findings suggest similar functional effects of PKA phosphorylation and cTnIS23D/S24D substitutions in our systems. In this study, the PKA-phosphorylated cTnIR146G data were compared with PKA-phosphorylated cTnIWT sets, and the cTnIR21C/S23D/S24D results were compared with cTnIS23D/S24D sets. Considering the same (similar) functional effects of PKA phosphorylation and cTnIS23D/S24D substitutions, we decided it was fair to compare the PKA-phosphorylated cTnIR146G with the cTnIR21C/S23D/S24D data.
We and others (27, 28) have demonstrated that cTnIR21C disrupts PKA phosphorylation of Ser-23/Ser-24 on cTnI, and this abrogates the effect of β-adrenergic stimulation on cTnI regulation of contraction and relaxation. This may be the actual physiological/pathogenic mechanism of cTnIR21C, where normally PKA-mediated phosphorylation of Ser-23/Ser-24 would speed relaxation of the contractile apparatus. To investigate whether “forced phosphorylation” could overcome this, we studied bis-phosphomimic substitutions S23D/S24D of cTnIR21C. Our results indicated that even with bis-phosphomimic substitutions, the phosphorylation-mediated effects on KC-I and myofibril relaxation were still blunted, suggesting that the cTnIR21C mutation per se results in the cardiac dysfunction of modulation by phosphorylation, similar to our results for the cTnIR146G mutation. Considering the similar results for cTnIR146G and cTnIR21C (with/without PKA phosphorylation or bis-phosphomimic substitutions) in both solution biochemistry and myofibril kinetics/mechanics measurements, we wanted to compare their phenotypes in humans, but unfortunately the data are rather sparse. An affected individual with the R145G mutation had ventricular hypertrophy characteristic of severe HCM (12). The missense mutation R21C has been identified in at least two families (69). In one family, a patient had apical hypertrophy after presenting with atrial fibrillation. The patient's father, three siblings, and an 18-year-old daughter all succumbed to sudden cardiac death. The clinical evaluations of three surviving mutation carriers from this family revealed that one had asymmetrical septal hypertrophy; another had isolated left atrial enlargement, and the third one had normal cardiac dimensions despite an abnormal electrocardiogram. Another family was recently also identified with the R21C mutation. This family has four members with subaortic asymmetrical hypertrophy and one mutation carrier with normal cardiac dimensions who had to be resuscitated from sudden death. Thus the sparse amount of data available suggest human phenotypes for both mutations may be similar, but more information is needed to conclude this.
To understand the structural molecular level basis of how phosphorylation of cTnI residues Ser-23/Ser-24 changes the behavior of whole cTn complex and thus results in the changes in cardiac function, we performed paralleled MD simulations on WT, cTnIR146G, and cTnIR21C containing cTn and found that introduction of the bis-phosphomimic substitutions significantly altered the cTnC-cTnI interactions, particularly in the inhibitory-switch peptide regions (8). The most significant finding is that there were no intra-subunit interactions in the WT model in the absence of phosphorylation, but introduction of the bis-phosphomimic substitutions (S23D/S24D) for the WT model led to the formation of an intra-subunit interaction between the N terminus and the inhibitory peptide regions of cTnI (Fig. 9B) (8). This intra-subunit interaction has been suggested by Solaro and co-workers (11, 30, 32) based on solution biochemical and spectroscopic studies (31). In addition, we also compared our simulation results of bis-phosphomimic cTnI with biochemical studies from other laboratories (70–72). Dong et al. (70) found that bis-phosphorylation resulted in a reduction of the axial ratio of cTnI and the formation of a more compact structure upon phosphorylation using fluorescence studies. Heller et al. (71) reported that bis-phosphorylation induced a dramatic bending of the rod-like cTnI at the N-terminal extension that binds with cTnC, resulting in a significant decrease in the axial ratio of cTnI and the cTn complex overall. Reiffert et al. (72) used surface plasmon resonance to determine that the shape of cTnI changed from an asymmetrical structure to a more symmetrical one upon phosphorylation, which is consistent with the bending that results in a shorter and broader structure. Our simulations also suggest a bending at the N-terminal extension of cTnI and a more compact cTn structure upon phosphorylation that is consistent with these previous biochemical studies. We further speculate that this intra-subunit interaction may subsequently weaken interactions of the cTnI switch peptide with NcTnC, as demonstrated by increased fluctuation of contacts in MD simulations (Fig. 10B). This would allow stronger interaction between the inhibitory peptide of cTnI and actin and move the equilibrium toward thin filament deactivation. Interestingly, this intra-subunit interaction no longer formed in simulations with introduction of the cTnIR146G or cTnIR21C to the cTn complex, demonstrating that both mutations blunted the ability of cTnI phosphorylation to reposition the N-terminal extension to interact the inhibitory peptide region. These findings suggest a structural mechanism that can explain the loss of PKA-mediated modulation of thin filament activation and relaxation of myofibrils that need to occur with increasing heart rate during β-adrenergic stimulation and increased physical activity.
An important caution of using our simulations to explain our experimental data is that recombinant cTn subunit proteins were made from rat sequences, although the MD simulations were based on the human sequence. For cTnC, there is only one amino acid difference (Ile-119 in human and Met-119 in rat), which is a conservative substitution. For cTnT, there is also only one amino acid difference (Phe-251 in human and His-251 in rat) in the portion included in our computational model (residues 236–285), and this is also a conservative substitution. For cTnI, there are total 13 variants in the portion used for our computational model (residues 1–172). Four of those are located in the N terminus of cTnI (Gly-4, Arg-10, Arg-13, and Ile-19 in human and Glu-4, Gly-10, Gln-13, and Val-19 in rat). One variant is located in the switch peptide of cTnI (Ala-161 in human and Thr-162 in rat). All the other variants are resided in the I-T arm region of cTnI (Leu-53, Leu-61, Ala-75, Glu-84, Ala-86, Ala-91, Ile-114, and Phe-133 in human and Met-54, Met-62, Leu-76, Val-85, Asp-87, Glu-92, Val-115,and Tyr-134 in rat). Among them, G4E, R10G, E84V, A86D, and A91E substitutions change the electrostatic charge properties and size of the amino acid; R13Q changes the charge property of the amino acid; A75V changes the size of the amino acid; and, I19V, L53M, L61M, I114V, F133Y, and A161T are conservative substitutions. There is no evidence to suggest these variants alter structure-function of the cTn complex, but this has not been studied in any detail. None of these positions has been reported to be associated with disease, which supports the idea that these sequence variants among the two species have little or no effects on the function. Additionally, most results of ATPase assays and the Ca2+-force relationship show consistent results between rodents and human. Some minor differences have been attributed to the different myosin isoforms between the two species. For these reasons, we think it is fair to compare the in vitro findings with the computational results. In the future, it would be interesting to perform the in vitro study based on the human sequence (and/or also conduct the computational modeling based on the rodent sequence) and to compare those results with our current findings to further investigate the disease-unrelated variants among species.
Author Contributions
M. R., J. A. M., and A. D. M conceived this study. Y. C. and V. R. designed the experiment; Y. C., V. R., A. T., D. W., and L. O. performed the experiment and analyzed the data; Y. C. and S. L. built up the computational model and performed the computational study; Y. C. and M. R. wrote the initial draft of the paper. All authors reviewed, edited, and approved the manuscript.
Acknowledgments
We thank Dr. Charles Luo and Luping Xie for preparations of cTnI mutant proteins and protein isolation and Drs. Maria Razumova and Galina Flint for the assistance with rat tissue and solution preparation. We appreciate Drs. John Solaro and Paul Rosevear for structural information of the cTnI N-terminal extension. We appreciate Prof. Rommie Amaro and Dr. Peter Kekenes-Huskey for the support of this work, and Dr. Jordan Klaiman for helpful suggestions on writing the article. We are indebted to Martha Mathiason for the development of data acquisition and analysis software.
This work was supported in part by National Institutes of Health Grants R01 HL-111197 (to M. R.) and P41 GM103426 (to NBCR), by American Heart Association Grants 15POST25080292 (to Y. C.), 11POST7400069 (to V. R.), and 12POST11570005 (to S. L.), and by grants from National Science Foundation, National Institutes of Health, Howard Hughes Medical Institution, and the National Science Foundation Supercomputer Centers (to the J. A. M. group). The authors declare that they have no conflicts of interest with the contents of this article.
- HCM
- hypertrophic cardiomyopathy
- cTnC
- cardiac TnC
- cTnI
- cardiac troponin I
- NcTnC
- N terminus of TnC
- NcTnC
- N terminus of cTnC
- cTn
- cardiac troponin
- MD
- molecular dynamics
- IANBD
- {N-[2-(iodoacetoxy)ethyl]-N-methyl}-nitrobenz-2-oxa-1,3-diazole
- NcTnI
- N terminus of cTnI
- cTnT
- cardiac troponin T
- N
- newton
- RMSF
- root-mean-square fluctuation
- cMyBP-C
- cardiac myosin-binding protein C
- LV
- left ventricle.
References
- 1.Seidman C. E., and Seidman J. G. (2011) Identifying sarcomere gene mutations in hypertrophic cardiomyopathy, a personal history. Circ. Res. 108, 743–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Willott R. H., Gomes A. V., Chang A. N., Parvatiyar M. S., Pinto J. R., and Potter J. D. (2010) Mutations in troponin that cause HCM, DCM, and RCM: what can we learn about thin filament function? J. Mol. Cell. Cardiol. 48, 882–892 [DOI] [PubMed] [Google Scholar]
- 3.Gordon A. M., Homsher E., and Regnier M. (2000) Regulation of contraction in striated muscle. Physiol. Rev. 80, 853–924 [DOI] [PubMed] [Google Scholar]
- 4.Sia S. K., Li M. X., Spyracopoulos L., Gagné S. M., Liu W., Putkey J. A., and Sykes B. D. (1997) Structure of cardiac muscle troponin C unexpectedly reveals a closed regulatory domain. J. Biol. Chem. 272, 18216–18221 [DOI] [PubMed] [Google Scholar]
- 5.Spyracopoulos L., Li M. X., Sia S. K., Gagné S. M., Chandra M., Solaro R. J., and Sykes B. D. (1997) Calcium-induced structural transition in the regulatory domain of human cardiac troponin C. Biochemistry 36, 12138–12146 [DOI] [PubMed] [Google Scholar]
- 6.Zhang R., Zhao J., Mandveno A., and Potter J. D. (1995) Cardiac troponin I phosphorylation increases the rate of cardiac muscle relaxation. Circ. Res. 76, 1028–1035 [DOI] [PubMed] [Google Scholar]
- 7.Rao V., Cheng Y., Lindert S., Wang D., Oxenford L., McCulloch A. D., McCammon J. A., and Regnier M. (2014) PKA phosphorylation of cardiac troponin I modulates activation and relaxation kinetics of ventricular myofibrils. Biophys. J. 107, 1196–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng Y., Lindert S., Kekenes-Huskey P., Rao V. S., Solaro R. J., Rosevear P. R., Amaro R., McCulloch A. D., McCammon J. A., and Regnier M. (2014) Computational studies of the effect of the S23D/S24D troponin I mutation on cardiac troponin structural dynamics. Biophys. J. 107, 1675–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kentish J. C., McCloskey D. T., Layland J., Palmer S., Leiden J. M., Martin A. F., and Solaro R. J. (2001) Phosphorylation of troponin I by protein kinase A accelerates relaxation and cross-bridge cycle kinetics in mouse ventricular muscle. Circ. Res. 88, 1059–1065 [DOI] [PubMed] [Google Scholar]
- 10.Chandra M., Dong W.-J., Pan B.-S., Cheung H. C., and Solaro R. J. (1997) Effects of protein kinase A phosphorylation on signaling between cardiac troponin I and the N-terminal domain of cardiac troponin C. Biochemistry 36, 13305–13311 [DOI] [PubMed] [Google Scholar]
- 11.Solaro R. J., Rosevear P., and Kobayashi T. (2008) The unique functions of cardiac troponin I in the control of cardiac muscle contraction and relaxation. Biochem. Biophys. Res. Commun. 369, 82–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kimura A., Harada H., Park J. E., Nishi H., Satoh M., Takahashi M., Hiroi S., Sasaoka T., Ohbuchi N., Nakamura T., Koyanagi T., Hwang T. H., Choo J. A., Chung K. S., Hasegawa A., et al. (1997) Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat. Genet. 16, 379–382 [DOI] [PubMed] [Google Scholar]
- 13.Deng Y., Schmidtmann A., Redlich A., Westerdorf B., Jaquet K., and Thieleczek R. (2001) Effects of phosphorylation and mutation R145G on human cardiac troponin I function. Biochemistry 40, 14593–14602 [DOI] [PubMed] [Google Scholar]
- 14.Lang R., Gomes A. V., Zhao J., Housmans P. R., Miller T., and Potter J. D. (2002) Functional analysis of a troponin I (R145G) mutation associated with familial hypertrophic cardiomyopathy. J. Biol. Chem. 277, 11670–11678 [DOI] [PubMed] [Google Scholar]
- 15.Lindhout D. A., Li M. X., Schieve D., and Sykes B. D. (2002) Effects of T142 phosphorylation and mutation R145G on the interaction of the inhibitory region of human cardiac troponin I with the C-domain of human cardiac troponin C. Biochemistry 41, 7267–7274 [DOI] [PubMed] [Google Scholar]
- 16.Westfall M. V., Borton A. R., Albayya F. P., and Metzger J. M. (2002) Myofilament calcium sensitivity and cardiac disease–insights from troponin I isoforms and mutants. Circ. Res. 91, 525–531 [DOI] [PubMed] [Google Scholar]
- 17.Reis S., Littwitz C., Preilowski S., Mügge A., Stienen G. J., Pott L., and Jaquet K. (2008) Expression of cTnI-R145G affects shortening properties of adult rat cardiomyocytes. Pflügers Arch. 457, 17–24 [DOI] [PubMed] [Google Scholar]
- 18.Elliott K., Watkins H., and Redwood C. S. (2000) Altered regulatory properties of human cardiac troponin I mutants that cause hypertrophic cardiomyopathy. J. Biol. Chem. 275, 22069–22074 [DOI] [PubMed] [Google Scholar]
- 19.Takahashi-Yanaga F., Morimoto S., Harada K., Minakami R., Shiraishi F., Ohta M., Lu Q. W., Sasaguri T., and Ohtsuki I. (2001) Functional consequences of the mutations in human cardiac troponin I gene found in familial hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 33, 2095–2107 [DOI] [PubMed] [Google Scholar]
- 20.Wen Y., Pinto J. R., Gomes A. V., Xu Y., Wang Y., Wang Y., Potter J. D., and Kerrick W. G. (2008) Functional consequences of the human cardiac troponin I hypertrophic cardiomyopathy mutation R145G in transgenic mice. J. Biol. Chem. 283, 20484–20494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kruger M., Zittrich S., Redwood C., Blaudeck N., James J., Robbins J., Pfitzer G., and Stehle R. (2005) Effects of the mutation R145G in human cardiac troponin I on the kinetics of the contraction-relaxation cycle in isolated cardiac myofibrils. J. Physiol. 564, 347–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kobayashi T., and Solaro R. J. (2006) Increased Ca2+ affinity of cardiac thin filaments reconstituted with cardiomyopathy-related mutant cardiac troponin I. J. Biol. Chem. 281, 13471–13477 [DOI] [PubMed] [Google Scholar]
- 23.Zhou Z., Rieck D., Li K. L., Ouyang Y., and Dong W. J. (2013) Structural and kinetic effects of hypertrophic cardiomyopathy related mutations R146G/Q and R163W on the regulatory switching activity of rat cardiac troponin I. Arch. Biochem. Biophys. 535, 56–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brunet N. M., Chase P. B., Mihajlovic G., and Schoffstall B. (2014) Ca2+-regulatory function of the inhibitory peptide region of cardiac troponin I is aided by the C terminus of cardiac troponin T: effects of familial hypertrophic cardiomyopathy mutations cTnI R145G and cTnT R278C, alone and in combination, on filament sliding. Arch. Biochem. Biophys. 552, 11–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lindert S., Cheng Y., Kekenes-Huskey P., Regnier M., and McCammon J. A. (2015) Effects of HCM cTnI mutation R145G on troponin structure and modulation by PKA phosphorylation elucidated by molecular dynamics simulations. Biophys. J. 108, 395–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gomes A. V., Harada K., and Potter J. D. (2005) A mutation in the N terminus of troponin I that is associated with hypertrophic cardiomyopathy affects the Ca2+ sensitivity, phosphorylation kinetics, and proteolytic susceptibility of troponin. J. Mol. Cell. Cardiol. 39, 754–765 [DOI] [PubMed] [Google Scholar]
- 27.Wang Y., Pinto J. R., Solis R. S., Dweck D., Liang J., Diaz-Perez Z., Ge Y., Walker J. W., and Potter J. D. (2012) Generation and functional characterization of knock-in mice harboring the cardiac troponin I-R21C mutation associated with hypertrophic cardiomyopathy. J. Biol. Chem. 287, 2156–2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dweck D., Sanchez-Gonzalez M. A., Chang A. N., Dulce R. A., Badger C. D., Koutnik A. P., Ruiz E. L., Griffin B., Liang J., Kabbaj M., Fincham F. D., Hare J. M., Overton J. M., and Pinto J. R. (2014) Long term ablation of protein kinase A (PKA)-mediated cardiac troponin I phosphorylation leads to excitation-contraction uncoupling and diastolic dysfunction in a knock-in mouse model of hypertrophic cardiomyopathy. J. Biol. Chem. 289, 23097–23111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liang J., Kazmierczak K., Rojas A. I., Wang Y., and Szczesna-Cordary D. (2015) The R21C mutation in cardiac troponin I imposes differences in contractile force generation between the left and right ventricles of knock-in mice. Biomed. Res. Int. 2015, 742536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Howarth J. W., Meller J., Solaro R. J., Trewhella J., and Rosevear P. R. (2007) Phosphorylation-dependent conformational transition of the cardiac specific N-extension of troponin I in cardiac troponin. J. Mol. Biol. 373, 706–722 [DOI] [PubMed] [Google Scholar]
- 31.Sadayappan S., Finley N., Howarth J. W., Osinska H., Klevitsky R., Lorenz J. N., Rosevear P. R., and Robbins J. (2008) Role of the acidic N′ region of cardiac troponin I in regulating myocardial function. FASEB J. 22, 1246–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Warren C. M., Kobayashi T., and Solaro R. J. (2009) Sites of intra- and intermolecular cross-linking of the N-terminal extension of troponin I in human cardiac whole troponin complex. J. Biol. Chem. 284, 14258–14266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Köhler J., Chen Y., Brenner B., Gordon A. M., Kraft T., Martyn D. A., Regnier M., Rivera A. J., Wang C.-K., and Chase P. B. (2003) Familial hypertrophic cardiomyopathy mutations in troponin I (K183Δ, G203S, K206Q) enhance filament sliding. Physiol. Genomics 14, 117–128 [DOI] [PubMed] [Google Scholar]
- 34.Gordon A. M., Qian Y., Luo Z., Wang C. K., Mondares R. L., and Martyn D. A. (1997) Characterization of troponin-C interactions in skinned barnacle muscle: comparison with troponin-C from rabbit striated muscle. J. Muscle Res. Cell Motil. 18, 643–653 [DOI] [PubMed] [Google Scholar]
- 35.Martyn D. A., Regnier M., Xu D., and Gordon A. M. (2001) Ca2+- and cross-bridge-dependent changes in N- and C-terminal structure of troponin C in rat cardiac muscle. Biophys. J. 80, 360–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang D., Robertson I. M., Li M. X., McCully M. E., Crane M. L., Luo Z., Tu A.-Y., Daggett V., Sykes B. D., and Regnier M. (2012) Structural and functional consequences of the cardiac troponin C L48Q Ca2+-sensitizing mutation. Biochemistry 51, 4473–4487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dong W.-J., Xing J., Chandra M., Solaro J., and Cheung H. C. (2000) Structural mapping of single cysteine mutants of cardiac troponin I. Proteins. 41, 438–447 [PubMed] [Google Scholar]
- 38.Potter J. D. (1982) Preparation of troponin and its subunits. Methods Enzymol. 85, 241–263 [DOI] [PubMed] [Google Scholar]
- 39.Dong W.-J., Robinson J. M., Stagg S., Xing J., and Cheung H. C. (2003) Ca2+-induced conformational transition in the inhibitory and regulatory regions of cardiac troponin I. J. Biol. Chem. 278, 8686–8692 [DOI] [PubMed] [Google Scholar]
- 40.Wang D., McCully M. E., Luo Z., McMichael J., Tu A.-Y., Daggett V., and Regnier M. (2013) Structural and functional consequences of cardiac troponin C L57Q and I61Q Ca2+-desensitizing variants. Arch. Biochem. Biophys. 535, 68–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patton C., Thompson S., and Epel D. (2004) Some precautions in using chelators to buffer metals in biological solutions. Cell Calcium 35, 427–431 [DOI] [PubMed] [Google Scholar]
- 42.George S. E., Su Z., Fan D., Wang S., and Johnson J. D. (1996) The fourth EF-hand of calmodulin and its helix-loop-helix components: impact on calcium binding and enzyme activation. Biochemistry 35, 8307–8313 [DOI] [PubMed] [Google Scholar]
- 43.Adhikari B. B., Regnier M., Rivera A. J., Kreutziger K. L., and Martyn D. A. (2004) Cardiac length dependence of force and force redevelopment kinetics with altered cross-bridge cycling. Biophys. J. 87, 1784–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Regnier M., Rivera A. J., Chen Y., and Chase P. B. (2000) 2-Deoxy-ATP enhances contractility of rat cardiac muscle. Circ. Res. 86, 1211–1217 [DOI] [PubMed] [Google Scholar]
- 45.Brenner B., and Eisenberg E. (1986) Rate of force generation in muscle: correlation with actomyosin ATPase activity in solution. Proc. Natl. Acad. Sci. U.S.A. 83, 3542–3546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fabiato A. (1988) Computer programs for calculating total from specified free or free from specified total ionic concentrations in aqueous solutions containing multiple metals and ligands. Methods Enzymol. 157, 378–417 [DOI] [PubMed] [Google Scholar]
- 47.Kreutziger K. L., Piroddi N., McMichael J. T., Tesi C., Poggesi C., and Regnier M. (2011) Calcium binding kinetics of troponin C strongly modulate cooperative activation and tension kinetics in cardiac muscle. J. Mol. Cell. Cardiol. 50, 165–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Colomo F., Piroddi N., Poggesi C., te Kronnie G., and Tesi C. (1997) Active and passive forces of isolated myofibrils from cardiac and fast skeletal muscle of the frog. J. Physiol. 500, 535–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tesi C., Colomo F., Nencini S., Piroddi N., and Poggesi C. (2000) The effect of inorganic phosphate on force generation in single myofibrils from rabbit skeletal muscle. Biophys. J. 78, 3081–3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kreutziger K. L., Piroddi N., Scellini B., Tesi C., Poggesi C., and Regnier M. (2008) Thin filament Ca2+ binding properties and regulatory unit interactions alter kinetics of tension development and relaxation in rabbit skeletal muscle. J. Physiol. 586, 3683–3700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takeda S., Yamashita A., Maeda K., and Maéda Y. (2003) Structure of the core domain of human cardiac troponin in the Ca2+-saturated form. Nature 424, 35–41 [DOI] [PubMed] [Google Scholar]
- 52.Humphrey W., Dalke A., and Schulten K. (1996) VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 53.Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., and Klein M. L. (1983) Comparison of simple potential functions for simulation liquid water. J. Chem. Phys. 79, 926–935 [Google Scholar]
- 54.Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., and Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.MacKerell A. D. Jr., Banavali N., and Foloppe N. (2000) Development and current status of the CHARMM force field for nucleic acids. Biopolymers 56, 257–265 [DOI] [PubMed] [Google Scholar]
- 56.Ryckaert J.-P., Ciccotti G., and Berendsen H. J. C. (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 [Google Scholar]
- 57.Rao V. S., Korte F. S., Razumova M. V., Feest E. R., Hsu H., Irving T. C., Regnier M., and Martyn D. A. (2013) N-terminal phosphorylation of cardiac troponin-I reduces length-dependent calcium sensitivity of contraction in cardiac muscle. J. Physiol. 591, 475–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Finley N., Abbott M. B., Abusamhadneh E., Gaponenko V., Dong W., Gasmi-Seabrook G., Howarth J. W., Rance M., Solaro R. J., Cheung H. C., and Rosevear P. R. (1999) NMR analysis of cardiac troponin C-troponin I complexes: effects of phosphorylation. FEBS Lett. 453, 107–112 [DOI] [PubMed] [Google Scholar]
- 59.Sakthivel S., Finley N. L., Rosevear P. R., Lorenz J. N., Gulick J., Kim S., VanBuren P., Martin L. A., and Robbins J. (2005) In vivo and in vitro analysis of cardiac troponin I phosphorylation. J. Biol. Chem. 280, 703–714 [DOI] [PubMed] [Google Scholar]
- 60.Dohet C., al-Hillawi E., Trayer I. P., and Rüegg J. C. (1995) Reconstitution of skinned cardiac fibres with human recombinant cardiac troponin-I mutants and troponin-C. FEBS Lett. 377, 131–134 [DOI] [PubMed] [Google Scholar]
- 61.Takimoto E., Soergel D. G., Janssen P. M., Stull L. B., Kass D. A., and Murphy A. M. (2004) Frequency- and afterload-dependent cardiac modulation in vivo by troponin I with constitutively active protein kinase a phosphorylation sites. Circ. Res. 94, 496–504 [DOI] [PubMed] [Google Scholar]
- 62.Poggesi C., Tesi C., and Stehle R. (2005) Sarcomeric determinants of striated muscle relaxation kinetics. Pflugers Arch. 449, 505–517 [DOI] [PubMed] [Google Scholar]
- 63.Kress M., Huxley H. E., Faruqi A. R., and Hendrix J. (1986) Structural changes during activation of frog muscle studied by time-resolved x-ray diffraction. J. Mol. Biol. 188, 325–342 [DOI] [PubMed] [Google Scholar]
- 64.Huxley A. F., and Simmons R. M. (1970) Rapid 'give' and the tension 'shoulder' in the relaxation of frog muscle fibres. J. Physiol. 210, 32P-33P [PubMed] [Google Scholar]
- 65.Luo Y., Davis J., Tikunova S. B., Smillie L. B., and Rall J. A. (2003) Myofibrillar determinants of rate of relaxation in skinned skeletal muscle fibers. Adv. Exp. Med. Biol. 538, 573–582 [DOI] [PubMed] [Google Scholar]
- 66.Tesi C., Piroddi N., Colomo F., and Poggesi C. (2002) Relaxation kinetics following sudden Ca2+ reduction in single myofibrils from skeletal muscle. Biophys. J. 83, 2142–2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Colson B. A., Bekyarova T., Locher M. R., Fitzsimons D. P., Irving T. C., and Moss R. L. (2008) Protein kinase A-mediated phosphorylation of cMyBP-C increases proximity of myosin heads to actin in resting myocardium. Circ. Res. 103, 244–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yamasaki R., Wu Y., McNabb M., Greaser M., Labeit S., and Granzier H. (2002) Protein kinase A phosphorylates Titin's cardiac-specific N2B domain and reduces passive tension in rat cardiac myocytes. Circ. Res. 90, 1181–1188 [DOI] [PubMed] [Google Scholar]
- 69.Arad M., Penas-Lado M., Monserrat L., Maron B. J., Sherrid M., Ho C. Y., Barr S., Karim A., Olson T. M., Kamisago M., Seidman J. G., and Seidman C. E. (2005) Gene mutations in apical hypertrophic cardiomyopathy. Circulation 112, 2805–2811 [DOI] [PubMed] [Google Scholar]
- 70.Dong W. J., Chandra M., Xing J., She M., Solaro R. J., and Cheung H. C. (1997) Phosphorylation-induced distance change in a cardiac muscle troponin I mutant. Biochemistry 36, 6754–6761 [DOI] [PubMed] [Google Scholar]
- 71.Heller W. T., Finley N. L., Dong W. J., Timmins P., Cheung H. C., Rosevear P. R., and Trewhella J. (2003) Small-angle neutron scattering with contrast variation reveals spatial relationships between the three subunits in the ternary cardiac troponin complex and the effects of troponin I phosphorylation. Biochemistry 42, 7790–7800 [DOI] [PubMed] [Google Scholar]
- 72.Reiffert S. U., Jaquet K., Heilmeyer L. M. Jr., and Herberg F. W. (1998) Stepwise subunit interaction changes by mono- and bisphosphorylation of cardiac troponin I. Biochemistry 37, 13516–13525 [DOI] [PubMed] [Google Scholar]