

Background: IRF4 is a master transcription regulator critical in immune cell development and thermogenic gene expression.
Results: We report the crystal structure of IRF4-IAD and SAXS studies of full-length IRF4.
Conclusion: IRF4 has a flexible autoinhibitory region and a compact semistructured linker.
Significance: These studies identified new structural features that provide insights into the function and regulation of IRF4.
Keywords: gene regulation, interferon regulatory factor (IRF), small angle x-ray scattering (SAXS), structural biology, transcription factor, x-ray crystallography
Abstract
IRF4 is a unique member of the interferon regulatory factor (IRF) family playing critical regulatory roles in immune cell development, regulation of obesity-induced inflammation, and control of thermogenic gene expression. The ability of IRF4 to control diverse transcriptional programs arises from its proficiency to interact with numerous transcriptional partners. In this study, we present the structural characterization of full-length IRF4. Using a combination of x-ray and small angle x-ray scattering studies, we reveal unique features of the interferon activation domain, including a set of β-sheets and loops that serve as the binding site for PU.1, and also show that unlike other IRF members, IRF4 has a flexible autoinhibitory region. In addition, we have determined the small angle x-ray scattering solution structure of full-length IRF4, which, together with circular dichroism studies, suggests that the linker region is not extended but folds into a domain structure.
Introduction
Interferon regulatory factors (IRFs)2 are versatile transcription regulators that mediate homeostatic mechanisms of host defense against pathogens (1–4). Moreover, they also participate in cell growth regulation, differentiation of hematopoietic cells, and apoptosis (5). Promoters of genes regulated by IRFs have copies of the consensus IRF recognition sequence 5′-AANNGAAA-3′ found in cis-acting elements called interferon-stimulated response elements (ISREs) (6–9). Members of the IRF family (IRF1 through -9 in mice and humans) have two conserved functional domains: an N-terminal helix-turn-helix DNA-binding domain (DBD) with a signature five conserved tryptophan residues and a C-terminal interferon activation domain (IAD) critical in mediating protein-protein interactions (1, 9–12). IRF3 is the best studied member of the family and has provided the overall mechanism of IRF activation mediated by viral infection (13, 14). In this view, IRF3, which is ubiquitously and constitutively expressed, is localized in the cytoplasm as an inactive monomer. Dimerization and nuclear translocation is induced upon viral infection via IKKϵ/TBK1-mediated phosphorylation of specific serine-threonine clusters present in the C-terminal autoinhibitory region (AR) of the IAD domain (13–15). Phosphorylation of corresponding residues in IRF7 and IRF5 has also been shown to be important for their activation in the regulation of IFN α/β genes (16, 17). Thus, virus-induced phosphorylation/dimerization appears to be the mechanism by which a subset of IRF proteins (IRF3, IRF7, and IRF5) regulate gene expression. However, other IRF members regulate genes through different activation mechanisms. IRF4 (also called Pip (PU.1-interacting protein), ICSAT (interferon consensus sequence-binding protein for activated T-cells), and LSIRF (lymphocyte-specific IRF)) is the only IRF factor that is not regulated by interferons (IFNs) and is quite different from other members in multiple ways (18–20). Originally, IRF4 expression was thought to be restricted to cells of the immune system, but recently, it has been detected in the heart, kidney, liver, and brain (21–23). In contrast to other IRF proteins, IRF4 binds DNA with low affinity and requires interaction with different binding partners to bind DNA. These include other IRF family members, the leucine zipper heterodimer BATF-JunB, STAT6, PU.1, and PGC-1α among others (24–29). The low DNA binding affinity of IRF4 has been attributed to the presence of an AR residing in the last 30 residues of the IAD. It has been suggested that this region physically interacts with the DBD and maintains the protein in an autoinhibited state (30–32). Upon interaction with a binding partner, the inhibitory mechanism is relieved, allowing IRF4 to bind its recognition DNA sequence (32). However, how this inhibition is relieved when IRF4 binds to ISRE sites as a homodimer is not known. Thus, there are multiple ways by which IRF4 can be recruited to DNA. For instance, during activation of genes containing Ets-IRF composite elements (EICEs), IRF4 interacts with PU.1, phosphorylated at serine 148, through residues located in the IAD, particularly Lys-399 and Arg-398 (32). On the other hand, binding of IRF4 to genes regulated by AP1-IRF-consensus elements requires cooperative interaction with BATF-JunB heterodimer through residues Glu-77, Lys-63, and His-55 of BATF (26). Moreover, at high local concentrations, IRF4 regulates genes containing ISRE sites presumably by IRF4 dimerization (33). The diversity of mechanisms by which IRF4 can be recruited to DNA sites suggests that is capable of providing binding sites to accommodate the diverse number of interacting partners and that the autoinhibitory mechanism can be released in multiple ways. To delineate the structural and molecular details unique to IRF4, we determined the x-ray structure of an IAD construct lacking the last 30 residues (IADΔC). The structure reveals several features that are exclusive to IRF4, such as an open binding pocket that serves as the PU.1 binding site. Small angle x-ray scattering (SAXS) studies of the complete IAD show a flexible AR, which is not folded into the IAD. Furthermore, we show that the full-length protein is an elongated molecule with the putative linker region being most likely folded into a domain. Taken together, our crystallography and solution scattering data reveal key differences between the IADs of IRF4 and other members of the IRF family and provide a low resolution structure of a full-length IRF protein.
Experimental Procedures
Protein Purification
Murine IRF4 IADΔC construct 238–420 was subcloned into pET-15b TEV vector (pet15TEV_NESG (EvNO00338203) from the DNASU plasmid repository; the vector has a tobacco etch virus (TEV) protease-cleavable site following the His6 site). Positive clones were sequenced and subsequently transformed into BL21-pLysS* Escherichia coli expression cells were used to start an overnight preculture with 100 μg/ml ampicillin and 25 μg/ml chloramphenicol antibiotics at 37 °C. The next day, cells were grown at 37 °C, induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside at optical density ∼0.8, and harvested after 4 h. Protein was eluted from an Ni-NTA column with 25 mm Tris-base, pH 8.0, 500 mm NaCl, 2 mm TCEP, 300 mm imidazole, 10% glycerol. Next, the His tag was removed using the TEV protease (34). The cleaved protein was further purified on an Ni-NTA column to remove the His tag as well as the TEV protease, followed by gel filtration on a Superdex 75 column in 25 mm Tris-base, pH 8.0, 300 mm NaCl, 2 mm TCEP, and 5% glycerol. Purified protein was concentrated to 16 mg/ml (0.753 mm) and stored at −80 °C in 60-μl aliquots.
Murine IRF4 construct 238–450 (IAD) was subcloned into pET-15b, and positive clones were grown at 37 °C until the optical density reached ∼0.8, induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside, and harvested after 4 h. Following Ni-NTA-based affinity purification, the His affinity tag was removed using thrombin (1 unit of thrombin/mg of protein). Thrombin was precipitated with p-aminobenzamidine-agarose beads, and finally the protein was purified on a Superdex 75 column in 25 mm Tris-base, pH 8.0, 500 mm NaCl, 5 mm β-mercaptoethanol, 10% glycerol, after which the protein was concentrated to 1 mg/ml (0.04 mm) and stored in 100-μl aliquots.
The full-length IRF4 construct 1–450 (IRF4FL) was subcloned into pET-15b TEV using the appropriate primers. Positive clones of IRF4WT were transformed into BL21-pLysS* E. coli expression cells, and an overnight preculture was grown using the appropriate antibiotics. IRF4FL was grown at 37 °C until the optical density reached ∼0.6, induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside, and harvested after 6 h. Protein was eluted from an Ni-NTA column with 300 mm imidazole, and the His tag was removed using TEV protease. The cleaved protein was further purified on an Ni-NTA column to remove the His tag as well as the TEV protease. This was followed by additional purification on a phenyl-Sepharose column with 25 mm Tris-base, pH 8.0, 2 m NaCl, 1 mm TCEP, 1 mm EDTA eluted with low salt. The final step of purification was done using a gel filtration on a Superdex 75 column in 25 mm Tris-base, pH 8.0, 300 mm NaCl, 2 mm TCEP, and 5% glycerol. The protein was concentrated to ∼10 mg/ml (0.196 mm) and stored at −80 °C in 100-μl aliquots. IRF4ΔNC was cloned using the appropriate primers and expressed and purified using a similar protocol described here for IRF4FL.
Crystallization
Crystals of IRF4 IADΔC (amino acids 238–420) were obtained in 2.5 m NaCl, 0.1 m imidazole, pH 8.0, at 20 °C. Optimization led to separate, single crystals growing in 1.5–1.7 m KCl, 0.1 m imidazole, pH 8.0, at 4 °C. The crystals were flash-frozen in liquid nitrogen after soaking in a solution of 75% mother liquor and 25% glycerol for 15–20 s.
Analytical Ultracentrifugation Studies
Analytical ultracentrifugation experiments were carried out in a XL-I analytical centrifuge (Beckman Coulter) using 12-mm Epon double sector cells with sapphire windows and loaded into an An-60 Ti 4-hole rotor. All protein constructs were buffer-exchanged into 25 mm Tris·HCl, 300 mm NaCl, 1 mm EDTA, 3 mm TCEP (pH 7.9). Sedimentation velocity experiments were performed at different loading concentrations (between 0.5 and 2 mg/ml) at 20 °C. Density and viscosity of solutions and partial specific volumes were calculated using the program SEDNTRP (35, 36). Samples were centrifuged at 35,000 rpm, and data were collected with both absorbance and interference detectors. The data were fit using the continuous distribution c(S) model in SEDFIT (37).
SAXS
SAXS data were collected at the undulator-based beamline X9 at the National Synchrotron Light Source part of the Brookhaven National Laboratory using an MAR165 charge-coupled device area detector located at a distance around 3.5 meters from the sample and an x-ray beam energy of ∼2 keV with an exposure time of 60 s each. Merging, trimming, and scaling were performed using PRIMUS, a part of the ATSAS suite (38). Buffer subtraction was carried out using beamline-specific software. Radii of gyration (Rg) were evaluated using the Guinier approximation sRg < 1.3. Distance distribution functions and maximum diameters (Dmax) were calculated using the program GNOM (39).SAXS molecular envelopes were generated using GASBOR and DAMMIN (40, 41).
Circular Dichroism Experiments
CD experiments were carried out at the University of Richmond Biochemistry Department. Spectra of IRF4 protein constructs were obtained using a JASCO J-720 spectropolarimeter. CD measurements (190–260 nm) were collected in quartz cells of 0.1-cm path length at 20 °C with a bandwidth of 0.1 nm. IRF4ΔNC and IADΔC were buffer-exchanged into a CD buffer, 50 mm sodium phosphate buffer (pH 8.0), whereas the DBD was in same buffer but at pH 7.0. Protein concentrations were calculated using a NanoDrop spectrophotometer using the calculated extinction coefficient for each of the proteins. The spectra were analyzed by BestSel (42).
Results
Structure Determination
IRF4 IADΔC (amino acids 238–420) was crystallized in 1.5–1.7 m KCl, 0.1 m imidazole, pH 8.0, at 4 °C. Diffraction data were collected from multiple single thin plate crystals at beamlines X4A and X25 at the National Synchrotron Light Source. The crystals belong to the P21221 space group with unit cell dimensions a = 45.5 Å, b = 84.9 Å, c = 149.9 Å, and α = β = γ = 90°. The structure was solved by molecular replacement using the structure of IRF5 IAD (Protein Data Bank entry 3DSH) as a search model using the program Phaser in CCP4 (43). Model building was carried out using COOT (44). The structure was refined to an R-factor of 25.5% and a final Rfree of 19.3%. The data collection and refinement statistics are presented in Table 1. The crystals contain two polypeptide chains per asymmetric unit, namely subunit A (residues 239–420) and subunit B (240–420), and 115 solvent molecules. Overall, we obtained an excellent Ramachandran plot with ∼98% of residues in the most favored region, the remaining 2% of residues in the generally allowed region, and no residues in the disallowed region.
TABLE 1.
Data collection and refinement statistics
| Parameters | Values |
|---|---|
| Data collection | |
| Space group | P21221 |
| Cell dimensions | |
| a, b, c (Å) | 45.5, 84.9, 149.9 |
| α, β, γ (degrees) | 90, 90, 90 |
| Wavelength (Å) | 1.1 |
| Resolution (Å) | 30–2.36 (2.4–2.36) |
| No. of measured reflections | 92,367 |
| No. of unique reflections | 23,107 |
| Data coverage (%)a | 93 (83) |
| Rmerge(%)a,b | 0.153 (0.662) |
| I/σa | 8.9 (2.1) |
| Refinement | |
| Resolution range | 32.4–2.6 |
| Reflections | 17,124 |
| Rcryst (%)c | 19.3 |
| Rfree (%)d | 25.5 |
| Non-hydrogen atoms | 3039 |
| Water | 115 |
| Average B-factors (Å2) | 49.8 |
| Protein | 36 |
| Water | 16 |
| r.m.s. deviations | |
| Bonds (Å) | 0.009 |
| Angles (Å) | 1.145 |
| Ramachandran plot quality | |
| Most favored (%) | 97.5 |
| Additional allowed (%) | 2.5 |
| Generously allowed (%) | 0.0 |
| Disallowed (%) | 0 |
aValues for the outmost shells are given in parentheses.
b Rmerge = Σ|I − 〈I〉|/ΣI, where I is the integrated intensity of a given reflection.
cRcryst = Σ [‖Fo| − |Fc‖Σ|Fo|.
dFor R-free calculations, 5% of data was excluded from refinement.
Overall Structure of IRF4 IADΔC
The IRF4 IADΔC structure contains the modified MH2 fold of the Smad family of proteins previously seen in IRF3 and IRF5 structures (11, 12, 45). The domain has a sickle-like shape with four α-helices (labeled α1–α4) surrounding a β-barrel (β1–β11) (Fig. 1A). Helices α1, α3, and α4 are at one end of the molecule, forming a helix bundle where the N- and C-terminal ends of the domain are also located. The remaining helix, α2, sits at the opposite end of the domain and loosely packs against one of the β-sheets (Fig. 1A). Five long loops connect different secondary structure elements protruding from the main core of the domain. The two molecules in the asymmetric unit form a dimer through interaction with α3 of the helix bundle burying a surface area of ∼1400 Å2 (Fig. 1B). However, this interface appears to be an effect of crystallization because the protein is monomeric in solution (Fig. 6A). The two molecules are almost identical, superimposing with an r.m.s. deviation of 0.98 Å for 175 Cα. However, helix α1 is in a different conformation with an r.m.s. deviation of 3.7 Å (Fig. 1C). This difference suggests that α1 is loosely packed against helices α3–α4 and has a dynamic character. To determine structural differences with other IRF IAD structures, we aligned the IRF4 to equivalent regions of IRF3 (amino acids 189–382) and IRF5 (amino acids 222–422), resulting in an r.m.s. deviation of 1.35 and 1 Å, respectively (Fig. 2). The analysis reveals several features that are unique to IRF4, in particular the structure of loops L3 and L5. In IRF3, loop L3 hovers on top of the β barrel covering strands 7, 10, and 11 (Fig. 2A). In contrast, the loop in IRF4 projects out of the core of the protein in a conformation similar to IRF5 (Fig. 2B). In IRF3 and IRF5, the loop L5 has a short α-helix that is absent in IRF4. Moreover, the overall direction of the loop is opposite to IRF4 with an r.m.s. deviation of 9.7 Å for IRF3 and 7.5 Å for IRF5 (Fig. 2). The conformation of both loops produces IADs with different accessible surface areas; in particular, the accessible surface area in IRF3 is ∼1500 Å2 smaller than IRF4 (Fig. 2C). Structure and sequence alignment of the three IADs reveal features that lead to the different loop L3 conformations. In IRF3, β7 and β10 are longer, causing L3 to “tilt” in the direction of strand β10 (Fig. 3A). In contrast, β7 in IRF4 and IRF5 is shorter by three residues and is followed by Gly-Pro. This combination breaks the β-strand and causes a turn in the polypeptide chain away from the core of the protein (Fig. 3B). The other IRF member that may have similar loop L3 conformation is IRF6, which is highly similar to IRF5 in sequence (Fig. 5B).
FIGURE 1.

Overall structure of IRF4 IADΔC. A, schematic representation showing the domain arrangement of IRF4 and the IRF4 constructs used in this study. Green, DBD; blue, IAD. B, ribbon diagram representation of the IRF4 IADΔC structure. β-Barrel strands are labeled 1–11, and α-helices are labeled 1–4. Five loops connect several secondary structure elements. C, ribbon representation of the dimer in the asymmetric unit. Contacts are through α-helix 3. D, superposition of the two molecules in the asymmetric unit showing the different conformation of α-helix 1.
FIGURE 6.

SAXS studies of IRF4 IAD. A, sedimentation coefficient distribution profiles of IAD domain (black) and the IADΔC (red). B, Kratky plots for IAD (black) and IADΔC (red). C, left, the experimental scattering profile (black) for IADΔC agrees very well with the theoretical scattering profile (red) generated from the crystal structure of IADΔC (65, 66); right, the experimental scattering profile (black) for IAD agrees well with the theoretical scattering profile (red) generated from the model of IAD. D, SAXS molecular envelopes of IADΔC (left) and IAD (right) with overall dimensions. E, BILBOMD IRF4 IAD model showing one representative of the AR conformations.
FIGURE 2.
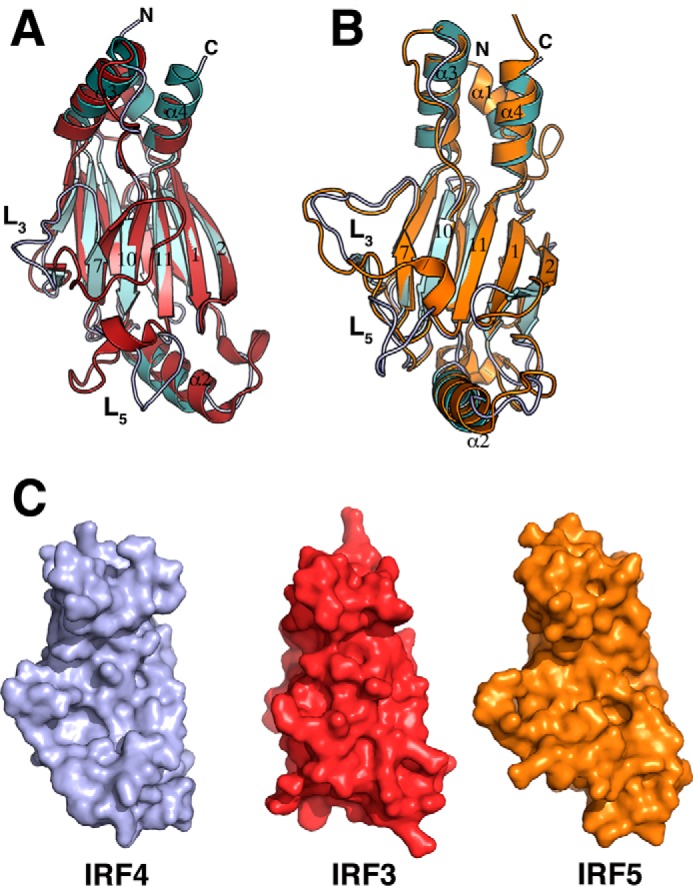
Comparison of IRF IAD structures. Shown are ribbon representations of superposition of IRF4 (cyan) and IRF3 (red) (A) and superposition of IRF4 (cyan) and IRF5 (orange) (B). Conformations of loops L3 between IRF4 and IRF3 are drastically different. Loops L5 in IRF3 and IRF5 contain an additional α-helix. C, accessible surface area representations of IRF4 (light purple), IRF3 (red), and IRF5 (orange).
FIGURE 3.
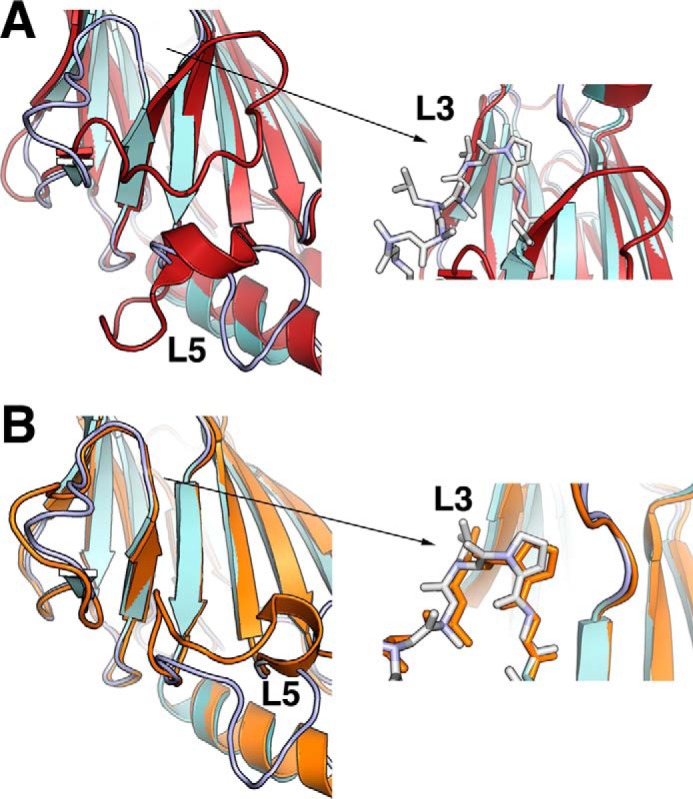
IRF IAD conformation of loops L3 and L5. A, details of the differences in loops L3 and L5 between IRF4 (cyan) and IRF3 (red); the right panel shows the shorter β-strand 7 and the presence of Gly-Pro in IRF4, which shifts the direction of the loop L3. B, details of differences in loop L5 between IRF4 (cyan) and IRF5 (orange); the right panel shows a similar conformation of loop L3 and the presence of the Gly-Pro sequence in both structures.
FIGURE 5.

Helix bundle differences between IRFs. A, hydrophobicity differences in the helix bundle between IRF4 (left) and IRF3 (right). Coloring indicates regions of high hydrophobicity (red) to low hydrophobicity (white). B, sequence alignment of IRF IADs showing the different secondary structure elements as blue squares (helices) and red squares (β-sheets). Residues in green are serine residues located in the AR. C, superposition of IRF4 (light purple), IRF3 (red), and IRF5 (orange), showing the different conformations of helix α1.
Structural Basis of the PU.1 Binding Site
IRF4 requires interaction with phosphorylated PU.1 to bind to EICE sites found in many promoters and enhancers (46). Previous studies, including alanine-scanning mutational analysis, identified several IRF4 residues that are critical for the interaction with phosphorylated PU.1, in particular Lys-399 and Arg-398 (Fig. 4, A and B) (32). Our structure shows that the mutations affecting complex formation with PU.1 and transcription of EICE sites are distributed along loop L5 and strand β11 and part of a binding pocket for phosphorylated PU.1 (Fig. 4B). Analysis of the structure using CASTp (Computed Atlas of Surface Topography of Proteins) was carried out to identify binding pockets in the IAD. CASTp can identify and measure protein pockets using precise computational geometry methods that include α shape and Delauney triangulation (47). The main IAD pocket includes, in addition to β11 and loop L5, residues from β7, β8, and loop L3 spanning 69 Å2 of accessible area, as shown in Fig. 4C. Residue Lys-399 points straight up into the solvent and is found in a highly positive patch region (Fig. 4D). This binding pocket appears to be specific for IRF4 only due to the structural architecture of the loops L3 and L5 described previously. In IRF3, loop L3 acts as a lid blocking any access to residues residing in β11 and loop L5 (Fig. 3A). In IRF5, there is a similar size pocket localized between β11 and β12; however, the presence of several bulky hydrophobic residues in β10, such as Trp-393 and Phe-386, restricts access to the loop L5 residues. In addition, the Lys-399 equivalent residue (Lys-401) is located underneath the pocket in IRF5 and is not accessible for interactions (data not shown). Thus, in IRF4, the open structure of the IAD generates a large surface that can be used for interaction with different transcriptional partners.
FIGURE 4.

PU.1 binding pocket. A, schematic representation of IRF4 and position of mutations that affect PU.1 complex formation. B, mutations that affect complex formation with PU.1 mapped on the IRF4 surface. C, binding pocket formed by residues from β7, β8, and loop L3 in addition to β11 and loop L5. Residue Lys-399 points into the solvent and is located in a highly localized positive patch region. D, electrostatic surface representation of the pocket. Residues Arg-398 and Lys-399 of loop L5 are shown. E, Leu-400 and Ile-401 are shown here as part of a hydrophobic core interacting with residues from α-helix 2.
IRF4 IAD Helical Bundle
The x-ray structure of IRF3 IAD showed that the helical bundle located at one end of the IAD keeps the C-terminal AR in a conformation that maintains IRF3 in a monomeric state (11, 12). The AR structural elements pack against a mostly hydrophobic surface generated by α1, α3, and α4 (11, 12). In IRF4, the hydrophobicity of this region is significantly less than in IRF3 (Fig. 5). Sequence and structural alignment reveal that key hydrophobic residues found in IRF3 are missing in IRF4 (Fig. 5C). For example, residues corresponding to Val-391, Leu-393, and Ile-395 in helix α5 of IRF3 are not found in IRF4. Hence, it is likely that the conformation of the AR in IRF4 will be different from the one seen in IRF3. Moreover, IRF4 lacks the serine residues found in IRF3 and IRF5, whose phosphorylation release the autoinhibitory conformation inducing dimerization. In addition, the IRF4 structure shows that although the conformation of helices α3 and α4 does not change much among the three IRF proteins (r.m.s. deviation of 1.04 Å), their respective helices α1 are in different configurations (Fig. 5B). For instance, superposition of IRF3 and IRF4 gives an r.m.s. deviation of 3.22 Å for α1. Hence, α1 in IRF4 has a dynamic character, which can be seen in the two different conformations of the two molecules in the asymmetric unit (Fig. 1D). Interestingly, several studies have shown that the integrity of the helical bundle has a direct effect on the transcriptional ability of IRF proteins. For instance, mutation of Leu-368 to proline results in an IRF4 molecule that is unable to form a ternary complex with phosphorylated PU.1 (48). The corresponding residues in IRF8 and IRF9 were also shown to be important in IFN signaling as well (49). Leu-368 is part of a hydrophobic core in IRF4 that includes Leu-246 and Glu-243 in α1; Phe-364, Leu-365, and Phe-371 in α3; and Leu-409, Leu-413, and Tyr-414 in α4.
SAXS Studies Reveal That the IRF4 Autoinhibitory Region Is Flexible and Does Not Dock into the IAD Domain
To obtain structural information about the IRF4 autoinhibitory region (residues 420–450), we performed solution studies using sedimentation velocity and SAXS on the complete IAD domain and the IADΔC constructs. Sedimentation velocity studies show that both constructs sediment around 2 S and are monomeric (Fig. 6A and Table 2). SAXS-calculated parameters for IAD show larger Rg and Dmax values than IADΔC, suggesting that the AR is probably a flexible tail unlike IRF3 (Table 2). This is supported by the Kratky plots showing a departure from a bell shape in IAD but not for IADΔC (Fig. 6B). We generated ab initio models using the programs DAMMIN and GASBOR with merged data sets from different concentrations. The generated SAXS molecular envelopes have overall shapes that resemble the IADΔC x-ray structure (Fig. 6D). However, the envelope for IAD is larger, with additional density at one end of the envelope that most likely corresponds to the autoinhibitory region. To obtain a more detailed description of the conformation of this region, we made a model of the complete IAD (residues 238–450) and used BILBOMD to generate minimum ensemble search models consistent with the SAXS data (50). The initial IAD model had the last 30 residues in an extended conformation, and several BILBOMD models showed that the AR does not fold into the core of the helical bundle like IRF3 (Fig. 6E). The χ of the fit between the experimental and the model scattering curves was 0.21. Interestingly, if we generate an IRF4 model with the AR region acquiring an IRF3-like conformation, the χ of the fit is the worst of all of the models (data not shown). Incorporation of 2–3 alternative conformations marginally improves the fitting to a χ of 0.19. Taken together, our data show a flexible unstructured IRF4 autoinhibitory region that does not dock into the helical bundle, as seen in IRF3.
TABLE 2.
Hydrodynamic parameters
| Protein | Sa | Rgb | Dmaxc | χ |
|---|---|---|---|---|
| nm | nm | |||
| IAD | 2.2 | 28 | 76.2 | 0.239d |
| IADΔC | 2.16 | 20 | 62.8 | 0.04d |
| IRF4FL | 3.6 | 39 | 135 | 0.78e |
| IRF4ΔNC | 3.4 | 34 | 123 | 0.9e |
aExperimentally determined by sedimentation velocity.
bExperimentally determined by Guinier analysis.
cExperimentally determined by P(r) analysis using GNOM.
dGoodness of fit atomic models to scattering data determined by FoXS.
eGoodness of fit rigid body model to scattering data determined by BUNCH.
SAXS Studies Show That IRF4FL Has an Elongated Shape with Flexible N- and C-terminal Inhibitory Regions
It has been hypothesized that IRF4 is in an autoinhibited “closed” conformation in which the C-terminal AR of the IAD interacts with the DBD domain, preventing it from binding DNA (51). To obtain structural information about the arrangement of the IRF4 domains and the conformations of N- and C-terminal autoinhibitory regions in the context of the full-length protein, we performed sedimentation velocity and SAXS-based structural characterization of IRF4FL (residues 1–450). We obtained a sedimentation coefficient of 3.4 s20,w for IRF4FL, which is significantly smaller than expected for a globular protein with the same molecular weight (4.9 S), suggesting that IRF4FL has an elongated shape. Removal of the N- and C-terminal autoinhibitory regions produces a protein construct, IRF4ΔNC (residues 20–420), that sediments at 3.2 s20,w, implying that the elongated shape is not due to the N- and C-terminal ends but due to the DBD-linker-IAD domain arrangement (Fig. 7A). This conclusion is supported by the SAXS parameters showing that the Rg and Dmax have similar values in both constructs (Table 2). Comparison of the Kratky plot shows that removal of the N- and C-terminal regions (IRF4ΔNC) results in a flat region in the high q regions, whereas the IRF4FL data shows an upward behavior that is characteristic of folded proteins with flexible tails (Fig. 7B) (52). This conclusion is supported by the Porod-Debye plot with a loss of the plateau region in the IRF4FL but not in IRF4ΔNC (Fig. 7C). Furthermore, the Kratky-Debye plot also supports a model of IRF4FL containing flexible regions (Fig. 7, D and E) (53). SAXS data were next used to calculate ab initio envelopes using GASBOR, resulting in models with elongated shapes (Fig. 7F). The dimensions of the averaged envelopes are consistent with the experimentally determined Rg and Dmax values (Table 2).
FIGURE 7.

SAXS studies on full-length IRF4. A, sedimentation coefficient distribution profiles of IRF4FL (black) and IRF4ΔNC (red). B, Kratky plots for IRF4FL (black) and IRF4ΔNC (red). C, Porod-Debye plots of IRF4FL (black) and IRF4ΔNC (red). D, Kratky-Debye plot for IRF4FL. E, Kratky-Debye plot for IRF4ΔNC. The plot shows that whereas IRF4FL plateaus, the plot for IRF4ΔNC has a downward behavior. F, GASBOR generated envelopes for IRF4FL (gray) and IRF4ΔNC (red).
SAXS Rigid Body Modeling of Full-length IRF4 Shows That the Linker Region Folds into a Compact Domain-like Structure
Docking of the DBD and IAD into the IRF4ΔNC SAXS envelope was carried out using the chimera-fitting volume function and SUPCOMB from the ATSAS package to obtain a rough estimate of the relative position of the two functional domains (38, 54). The docking leaves a small volume to be occupied by the linker region. However, because the linker region contains ∼104 residues, it is most likely to fold into a compact domain structure. We should point out that the docking of the domains represents just one of the many configurations that can fit into the SAXS envelope, and we have used it to illustrate that the linker is not in an extended conformation. To determine whether the linker region has secondary structure elements, we performed CD experiments on DBD, IADΔC, and IRF4ΔNC. Secondary structure content was calculated from the individual CD spectra using the program BestSel (42). The CD spectra calculated for each sample are shown in Fig. 8B. The calculated percentages of helix, strand, turn, and disorder are shown in Table 3. The data show an increase in the overall secondary structure character in IRF4ΔNC compared with the calculated content from the individual domains. Moreover, secondary structure prediction of the linker region using the program Jpred calculates a short β-strand region between residues 214 and 221 (42, 55). Rigid body modeling was performed using IRF4ΔNC with BUNCH, using IRF4 DBD residues 20–134 and IAD residues 238–420 (56, 57). The resulting model has a linker region that is highly condensed and is located between the DBD and IAD domains (Fig. 8C). The IRF4ΔNC rigid model fits well with the experimental data with a χ value of 0.9 (Fig. 8B). A similar model was obtained using the program CORAL (56). Next, we used BUNCH to generate an IRF4FL model, resulting in a structure with a comparable arrangement of DBD and IAD domains and a highly folded linker region (Fig. 8C). The IRF4FL and IRF4ΔNC models superimpose with an r.m.s. deviation of ∼6 Å, and although there are differences in the rotational position of the domains, the distance with respect to each other is the same. The difference in the positions may reflect conformational changes occurring upon removal of the N- and C-terminal regions but may just be a result of the low resolution of the data. Nevertheless, the IRF4FL model shows that the AR is extended, thus supporting the results found for the IAD.
FIGURE 8.

SAXS rigid body modeling of IRF4. A, docking of IRF4 DBDΔN and IADΔC onto SAXS envelope of IRF4ΔNC. B, CD spectra of IRF4 DBDΔN, IADΔC, and IRF4ΔNC. C, BUNCH-generated IRF4ΔNC rigid body model showing the DBD and IAD on either end of the envelope with a compact region for the linker. The experimental scattering profile superimposes well with the one generated using the model. D, BUNCH-generated IRF4FL rigid body model and fit to the experimental data.
TABLE 3.
Characterization of IRF4 secondary structure by CD
| Total helix | Total strand | Turns | Disordered | |
|---|---|---|---|---|
| % | % | % | % | |
| DBDΔN | 24.5 | 28.0 | 10.2 | 37.4 |
| IADΔC | 8.5 | 31 | 10.2 | 50.4 |
| IRF4ΔNC (predicted) | 11.39 | 22.76 | 7.80 | 34.5 |
| IRF4ΔNC (experimental) | 5.3 | 37.2 | 12.7 | 44.8 |
The conformation of the linker shown in the two models in Fig. 8 can be interpreted only as an indicator of the high compactness that it must have to fit the SAXS data and not as an accurate representation of its three-dimensional structure. Thus, IRF4 has an elongated structure with the DBD and IAD domains at either end of the molecule and separated by a compact linker domain.
Discussion
Our studies provide a structural view of a full-length IRF protein. The crystal structure of IRF4 IADΔC shows some of the structural features that make it unique among IRF family members. The structure displays an open binding pocket on one side of the β-barrel that exposes residues that are critical for the interaction with phosphorylated PU.1 and probably other transcriptional partners. The PU.1 binding site is located on one face of the β-barrel, where the conformation of loops L3 and L5 generate an open surface that is accessible for binding. In other members of the IRF family, the conformation of loop L3 acts as a lid that prevents access to the binding pocket. In addition, loop L5 has a small α-helix that partially blocks the binding pocket. Mutations in IRF4 that affect complex formation with PU.1 have been determined previously, and they validate our structure (32). Not surprisingly, they are all located in loop L5 and include critical residues Lys-399 and Arg-398 that interact directly with PU.1 phosphoserine 148 (32). Mutations in residues that weakly affect complex formation, such as Glu-389, Glu-390, Phe-391, and Pro-392, are all located in the N-terminal half of loop L5 and may affect its conformation, preventing access to the binding site. However, mutations of Leu-400 and Ile-401 also affect complex formation, and both are located in the buried face of L5 and β11, where they are part of a hydrophobic pocket interacting with helix α2 that stabilizes the overall structure (Fig. 4E). Our SAXS studies show that the AR in IRF4 is flexible and does not fold into the helical bundle as in IRF3. This difference in the AR reflects the diversity of the sequences found in this region among IRF family members (Fig. 5B). In particular, the IRF4 AR is significantly shorter than IRF3 or IRF5 and does not have many of the hydrophobic residues that participate in the folding and docking of the AR into the helical bundle (Fig. 5B). This parallels the properties of the different helical bundles. In IRF3 and IRF5, helices 1, 3, and 5 generate a large hydrophobic region, whereas in IRF4, this is not the case. Interestingly, Leu-368 in helix α3 seems to be critical in stabilizing the helix bundle in IRF4 by making hydrophobic interactions with Leu-413 in helix α4. A mutation of Leu-368 results in a protein incapable of forming the ternary complex with PU.1 and DNA (48). Thus, the stability of the helical bundle has further repercussions on the overall function of IRF4.
Our SAXS studies show the overall domain architecture of a full-length IRF protein. It shows that the linker region most likely adopts a folded conformation, where it may interact with the DBD and IAD domains located at either end of the molecule. This finding is supported by our CD studies and a report suggesting that the linker in IRF3 is not unfolded but may also adopt a folded conformation (58). Thus, our SAXS IRF4 envelope may represent the general domain architecture for all IRF proteins and suggest that the linker domain may play a role in the regulation of IRF function. Indeed, a study by Mamane et al. (59) showed that FKBP52, a peptidyl-prolyl isomerase, inhibited the transactivation activity of IRF4 and binding to EICE DNA sites. They mapped the site of interaction to the linker region and proposed a mechanism of posttranslational modification of IRF4 activity. In another study, Wang et al. (60) showed that phosphorylation of residues in the linker region of IRF3, which has 15% serine content, negatively regulates its transactivation activity. Moreover, it was shown that ubiquitination of IRF8 enhances its activity to regulate expression of IL-12p40, and the linker is the site of interaction for the E3 ligase Ro52 (61). Thus, it appears that the linker region can be used to regulate IRF activity through different mechanisms.
Based on biochemical data and insights from the structures of IRF3 and IRF5, it has been proposed that upon phosphorylation, the AR undergoes a large conformational change promoting dimerization and binding to DNA. The fact that the ARs of IRF proteins are diverse in terms of their sequence homology and their length suggests the possibility of alternative mechanisms that could induce IRF dimerization. IRF4 in particular has evolved two mechanisms in order to bind DNA: 1) its ability to interact with other transcription factors and binding to composite sites and 2) formation of homodimers at high concentrations to bind ISREs. Thus, the critical event in the activation of IRF4 in both cases is the formation of homo- or heterodimers that leads to an increase in DNA affinity. The fact that the C-terminal autoinhibitory region is flexible supports the notion that IRF4 homodimerization may not require a trigger, such as phosphorylation, but is prompted by an increase in protein concentration, as suggested by the fact that the IRF4 binds to ISRE sites as a homodimer at high concentrations, and this property is critical for B-cell differentiation into plasma cells (33, 62–64). Further studies are required to determine the structural determinants of IRF4 oligomerization.
Author Contributions
S. G. R. cloned, expressed, and purified the proteins; collected and determined the x-ray structure and the SAXS studies; performed initial analytical ultracentrifugation experiments; analyzed CD data; and wrote the original draft of the paper. V. S. performed analytical centrifugation and CD experiments and protein preparation for SAXS studies and analyzed CD data. C. R. E. conceived and analyzed data, wrote the manuscript, and secured funding.
Acknowledgments
We thank members of the Brookhaven National Synchrotron Light Source beamlines for help in data collection: Vivian Stojanoff, Jean Jakoncic, and Edwin Lazo (X6a); Annie Heroux and Howard Robinson (X25); Randy Abramowitz and John Schwanof (X4); and Lin Yang and Vito Graziano (X9). We also thank Kevin Dyer, Jane Tanamachi, and Michal Hammel (ALS SIBYLS beamline). We thank Dr. Jonathan Dattelbaum and Dr. J. Ellis Bell (Biochemistry Department, University of Richmond) for generous help and access to the circular dichroism spectrophotometer. We also thank laboratory members Francisco Zarate-Perez, Faik Musayev, and John Burgner.
This work is supported by National Institutes of Health Grants R01-GM092854 and R21-CA179008 and a Virginia Commonwealth University PRQF award. The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 5BVI) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- IRF
- interferon regulatory factor
- SAXS
- small angle x-ray scattering
- ISRE
- interferon-stimulated response element
- DBD
- DNA-binding domain
- AR
- autoinhibitory region
- EICE
- Ets-IRF composite element
- TEV
- tobacco etch virus
- r.m.s.
- root mean square
- IAD
- interferon activation domain
- Ni-NTA
- nickel-nitrilotriacetic acid
- TCEP
- tris(2-carboxyethyl)phosphine.
References
- 1.Taniguchi T., Ogasawara K., Takaoka A., and Tanaka N. (2001) IRF family of transcription factors as regulators of host defense. Annu. Rev. Immunol. 19, 623–655 [DOI] [PubMed] [Google Scholar]
- 2.Takaoka A., and Yanai H. (2006) Interferon signalling network in innate defence. Cell Microbiol. 8, 907–922 [DOI] [PubMed] [Google Scholar]
- 3.Ozato K., Tailor P., and Kubota T. (2007) The interferon regulatory factor family in host defense: mechanism of action. J. Biol. Chem. 282, 20065–20069 [DOI] [PubMed] [Google Scholar]
- 4.Paun A., and Pitha P. M. (2007) The IRF family, revisited. Biochimie 89, 744–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tamura T., Yanai H., Savitsky D., and Taniguchi T. (2008) The IRF family transcription factors in immunity and oncogenesis. Annu. Rev. Immunol. 26, 535–584 [DOI] [PubMed] [Google Scholar]
- 6.Fujita T., Shibuya H., Hotta H., Yamanishi K., and Taniguchi T. (1987) Interferon-β gene regulation: tandemly repeated sequences of a synthetic 6 bp oligomer function as a virus-inducible enhancer. Cell 49, 357–367 [DOI] [PubMed] [Google Scholar]
- 7.Goodbourn S., and Maniatis T. (1988) Overlapping positive and negative regulatory domains of the human β-interferon gene. Proc. Natl. Acad. Sci. U.S.A. 85, 1447–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Escalante C. R., Yie J., Thanos D., and Aggarwal A. K. (1998) Structure of IRF-1 with bound DNA reveals determinants of interferon regulation. Nature 391, 103–106 [DOI] [PubMed] [Google Scholar]
- 9.Fujii Y., Shimizu T., Kusumoto M., Kyogoku Y., Taniguchi T., and Hakoshima T. (1999) Crystal structure of an IRF-DNA complex reveals novel DNA recognition and cooperative binding to a tandem repeat of core sequences. EMBO J. 18, 5028–5041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mamane Y., Heylbroeck C., Génin P., Algarté M., Servant M. J., LePage C., DeLuca C., Kwon H., Lin R., and Hiscott J. (1999) Interferon regulatory factors: the next generation. Gene 237, 1–14 [DOI] [PubMed] [Google Scholar]
- 11.Qin B. Y., Liu C., Lam S.S., Srinath H., Delston R., Correia J.J., Derynck R., and Lin K. (2003) Crystal structure of IRF-3 reveals mechanism of autoinhibition and virus-induced phosphoactivation. Nat. Struct. Biol. 10, 913–921 [DOI] [PubMed] [Google Scholar]
- 12.Takahasi K., Suzuki N. N., Horiuchi M., Mori M., Suhara W., Okabe Y., Fukuhara Y., Terasawa H., Akira S., Fujita T., and Inagaki F. (2003) X-ray crystal structure of IRF-3 and its functional implications. Nat. Struct. Biol. 10, 922–927 [DOI] [PubMed] [Google Scholar]
- 13.Yoneyama M., Suhara W., Fukuhara Y., Fukuda M., Nishida E., and Fujita T. (1998) Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 17, 1087–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin R., Mamane Y., and Hiscott J. (1999) Structural and functional analysis of interferon regulatory factor 3: localization of the transactivation and autoinhibitory domains. Mol. Cell Biol. 19, 2465–2474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Servant M. J., ten Oever B., LePage C., Conti L., Gessani S., Julkunen I., Lin R., and Hiscott J. (2001) Identification of distinct signaling pathways leading to the phosphorylation of interferon regulatory factor 3. J. Biol. Chem. 276, 355–363 [DOI] [PubMed] [Google Scholar]
- 16.Marié I., Smith E., Prakash A., and Levy D. E. (2000) Phosphorylation-induced dimerization of interferon regulatory factor 7 unmasks DNA binding and a bipartite transactivation domain. Mol. Cell Biol. 20, 8803–8814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barnes B. J., Kellum M. J., Field A. E., and Pitha P. M. (2002) Multiple regulatory domains of IRF-5 control activation, cellular localization, and induction of chemokines that mediate recruitment of T lymphocytes. Mol. Cell Biol. 22, 5721–5740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eisenbeis C. F., Singh H., and Storb U. (1995) Pip, a novel IRF family member, is a lymphoid-specific PU.1-dependent transcriptional activator. Genes Dev. 9, 1377–1387 [DOI] [PubMed] [Google Scholar]
- 19.Matsuyama T., Grossman A., Mittrücker H. W., Siderovski D. P., Kiefer F., Kawakami T., Richardson C. D., Taniguchi T., Yoshinaga S. K., and Mak T. W. (1995) Molecular cloning of LSIRF, a lymphoid-specific member of the interferon regulatory factor family that binds the interferon-stimulated response element (ISRE). Nucleic Acids Res. 23, 2127–2136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamagata T., Nishida J., Tanaka S., Sakai R., Mitani K., Yoshida M., Taniguchi T., Yazaki Y., and Hirai H. (1996) A novel interferon regulatory family transcription factor, ICSAT/Pip/LSIRF, that negatively regulates the activity of interferon-regulated genes. Mol. Cell Biol. 16, 1283–1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eguchi J., Wang X., Yu S., Kershaw E. E., Chiu P. C., Dushay J., Estall J. L., Klein U., Maratos-Flier E., and Rosen E. D. (2011) Transcriptional control of adipose lipid handling by IRF4. Cell Metab. 13, 249–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang D. S., Bian Z. Y., Zhang Y., Zhang S. M., Liu Y., Zhang R., Chen Y., Yang Q., Zhang X. D., Fan G. C., and Li H. (2013) Role of interferon regulatory factor 4 in the regulation of pathological cardiac hypertrophy. Hypertension 61, 1193–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo S., Li Z. Z., Jiang D. S., Lu Y. Y., Liu Y., Gao L., Zhang S. M., Lei H., Zhu L. H., Zhang X. D., Liu D. P., and Li H. (2014) IRF4 is a novel mediator for neuronal survival in ischaemic stroke. Cell Death Differ. 21, 888–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li P., Spolski R., Liao W., Wang L., Murphy T. L., Murphy K. M., and Leonard W. J. (2012) BATF-JUN is critical for IRF4-mediated transcription in T cells. Nature 490, 543–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glasmacher E., Agrawal S., Chang A. B., Murphy T. L., Zeng W., Vander Lugt B., Khan A. A., Ciofani M., Spooner C. J., Rutz S., Hackney J., Nurieva R., Escalante C. R., Ouyang W., Littman D. R., Murphy K. M., and Singh H. (2012) A genomic regulatory element that directs assembly and function of immune-specific AP-1-IRF complexes. Science 338, 975–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tussiwand R., Lee W. L., Murphy T. L., Mashayekhi M., KC W., Albring J. C., Satpathy A. T., Rotondo J. A., Edelson B. T., Kretzer N. M., Wu X., Weiss L. A., Glasmacher E., Li P., Liao W., Behnke M., Lam S. S., Aurthur C. T., Leonard W. J., Singh H., Stallings C. L., Sibley L. D., Schreiber R. D., and Murphy K. M. (2012) Compensatory dendritic cell development mediated by BATF-IRF interactions. Nature 490, 502–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gupta S., Jiang M., Anthony A., and Pernis A. B. (1999) Lineage-spacific modulation of interleukin 4 signaling by interferon regulatory factor 4. J. Exp. Med. 190, 1837–1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pongubala J. M., Nagulapalli S., Klemsz M. J., McKercher S. R., Maki R. A., and Atchison M. L. (1992) PU.1 recruits a second nuclear factor to a site important for immunoglobulin κ 3′ enhancer activity. Mol. Cell Biol. 12, 368–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kong X., Banks A., Liu T., Kazak L., Rao R. R., Cohen P., Wang X., Yu S., Lo J. C., Tseng Y. H., Cypess A. M., Xue R., Kleiner S., Kang S., Spiegelman B. M., and Rosen E. D. (2014) IRF4 is a key thermogenic transcriptional partner of PGC-1α. Cell 158, 69–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perkel J. M., and Atchison M. L. (1998) A two-step mechanism for recruitment of Pip by PU.1. J. Immunol. 160, 241–252 [PubMed] [Google Scholar]
- 31.Yee A. A., Yin P., Siderovski D. P., Mak T. W., Litchfield D. W., and Arrowsmith C. H. (1998) Cooperative interaction between the DNA-binding domains of PU.1 and IRF4. J. Mol. Biol. 279, 1075–1083 [DOI] [PubMed] [Google Scholar]
- 32.Brass A. L., Zhu A. Q., and Singh H. (1999) Assembly requirements of PU.1-Pip (IRF-4) activator complexes: inhibiting function in vivo using fused dimers. EMBO J. 18, 977–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ochiai K., Maienschein-Cline M., Simonetti G., Chen J., Rosenthal R., Brink R., Chong A. S., Klein U., Dinner A. R., Singh H., and Sciammas R. (2013) Transcriptional regulation of germinal center B and plasma cell fates by dynamical control of IRF4. Immunity 38, 918–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bazan J. F., and Fletterick R. J. (1988) Viral cysteine proteases are homologous to the trypsin-like family of serine proteases: structural and functional implications. Proc. Natl. Acad. Sci. U.S.A. 85, 7872–7876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hayes D., Laue T., and Philo J. (1995) Program Sednterp: Sedimentation Interpretation Program, University of New Hampshire, Durham, NH [Google Scholar]
- 36.Laue T. M., Shah B. D., Ridgeway T. M., and Pelletier S. L. (1992) in Analytical Ultracentrifugation in Biochemistry and Polymer Science (Harding S. E., Rowe A. J., and Horton J. C., eds) pp. 90–125, Royal Society of Chemistry, Cambridge, UK [Google Scholar]
- 37.Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Konarev P. V., Petoukhov M. V., Volkov V. V., and Svergun D. I. (2006) ATSAS 2.1, a program package for small-angle scattering data analysis. J. Appl. Crystallogr. 39, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Svergun D. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 40.Svergun D. I. (1999) Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76, 2879–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Svergun D. I., Petoukhov M. V., and Koch M. H. (2001) Determination of domain structure of proteins from x-ray solution scattering. Biophys. J. 80, 2946–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Micsonai A., Wien F., Kernya L., Lee Y. H., Goto Y., Réfrégiers M., and Kardos J. (2015) Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 112, E3095–3103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen W., Lam S. S., Srinath H., Jiang Z., Correia J. J., Schiffer C. A., Fitzgerald K. A., Lin K., and Royer W. E. Jr. (2008) Insights into interferon regulatory factor activation from the crystal structure of dimeric IRF5. Nat. Struct. Mol. Biol. 15, 1213–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marecki S., and Fenton M. J. (2002) The role of IRF-4 in transcriptional regulation. J. Interferon Cytokine Res. 22, 121–133 [DOI] [PubMed] [Google Scholar]
- 47.Dundas J., Ouyang Z., Tseng J., Binkowski A., Turpaz Y., and Liang J. (2006) CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 34, W116–W118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meraro D., Hashmueli S., Koren B., Azriel A., Oumard A., Kirchhoff S., Hauser H., Nagulapalli S., Atchison M. L., and Levi B. Z. (1999) Protein-protein and DNA-protein interactions affect the activity of lymphoid-specific IFN regulatory factors. J. Immunol. 163, 6468–6478 [PubMed] [Google Scholar]
- 49.Levi B. Z., Hashmueli S., Gleit-Kielmanowicz M., Azriel A., and Meraro D. (2002) ICSBP/IRF-8 transactivation: a tale of protein-protein interaction. J. Interferon Cytokine Res. 22, 153–160 [DOI] [PubMed] [Google Scholar]
- 50.Pelikan M., Hura G. L., and Hammel M. (2009) Structure and flexibility within proteins as identified through small angle x-ray scattering. Gen. Physiol. Biophys. 28, 174–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brass A. L., Kehrli E., Eisenbeis C. F., Storb U., and Singh H. (1996) Pip, a lymphoid-restricted IRF, contains a regulatory domain that is important for autoinhibition and ternary complex formation with Ets factor PU.1. Genes Dev. 10, 2335–2347 [DOI] [PubMed] [Google Scholar]
- 52.Hammel M. (2012) Validation of macromolecular flexibility in solution by small-angle x-ray scattering (SAXS). Eur. Biophys. J. 41, 789–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rambo R. P., and Tainer J. A. (2011) Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers 95, 559–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 55.Cuff J. A., and Barton G. J. (2000) Application of multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins 40, 502–511 [DOI] [PubMed] [Google Scholar]
- 56.Petoukhov M. V., and Svergun D. I. (2005) Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 89, 1237–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikhney A. G., Gajda M., Gorba C., Mertens H. D. T., Konarev P. V., and Svergun D. I. (2012) New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 45, 342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shukla H., Vaitiekunas P., Majumdar A. K., Dragan A. I., Dimitriadis E. K., Kotova S., Crane-Robinson C., and Privalov P. L. (2012) The linker of the interferon response factor 3 transcription factor is not unfolded. Biochemistry 51, 6320–6327 [DOI] [PubMed] [Google Scholar]
- 59.Mamane Y., Sharma S., Petropoulos L., Lin R., and Hiscott J. (2000) Posttranslational regulation of IRF-4 activity by the immunophilin FKBP52. Immunity 12, 129–140 [DOI] [PubMed] [Google Scholar]
- 60.Wang J. T., Chang L. S., Chen C. J., Doong S. L., Chang C. W., and Chen M. R. (2014) Glycogen synthase kinase 3 negatively regulates IFN regulatory factor 3 transactivation through phosphorylation at its linker region. Innate Immun. 20, 78–87 [DOI] [PubMed] [Google Scholar]
- 61.Kong H. J., Anderson D. E., Lee C. H., Jang M. K., Tamura T., Tailor P., Cho H. K., Cheong J., Xiong H., Morse H. C. 3rd, and Ozato K. (2007) Cutting edge: autoantigen Ro52 is an interferon inducible E3 ligase that ubiquitinates IRF-8 and enhances cytokine expression in macrophages. J. Immunol. 179, 26–30 [DOI] [PubMed] [Google Scholar]
- 62.Sciammas R., Li Y., Warmflash A., Song Y., Dinner A. R., and Singh H. (2011) An incoherent regulatory network architecture that orchestrates B cell diversification in response to antigen signaling. Mol. Syst. Biol. 7, 495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sciammas R., Shaffer A. L., Schatz J. H., Zhao H., Staudt L. M., and Singh H. (2006) Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity 25, 225–236 [DOI] [PubMed] [Google Scholar]
- 64.Klein U., Casola S., Cattoretti G., Shen Q., Lia M., Mo T., Ludwig T., Rajewsky K., and Dalla-Favera R. (2006) Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat. Immunol. 7, 773–782 [DOI] [PubMed] [Google Scholar]
- 65.Schneidman-Duhovny D., Hammel M., Tainer J. A., and Sali A. (2013) Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophys. J. 105, 962–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schneidman-Duhovny D., Hammel M., and Sali A. (2010) FoXS: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res. 38, W540–W544 [DOI] [PMC free article] [PubMed] [Google Scholar]