

Background: Syk associates with proteins that are found in stress granules.
Results: Stress granules contain Syk, Grb7, and phosphotyrosine and persist in cells treated with inhibitors of autophagy.
Conclusion: Syk is recruited to stress granules and promotes their clearance through autophagy.
Significance: The regulation of both stress granule clearance and autophagy is important for the survival of cells exposed to external stress.
Keywords: autophagy, cancer biology, signal transduction, stress granule, tyrosine-protein kinase (tyrosine kinase), Syk, Grb7
Abstract
Syk is a cytoplasmic kinase that serves multiple functions within the immune system to couple receptors for antigens and antigen-antibody complexes to adaptive and innate immune responses. Recent studies have identified additional roles for the kinase in cancer cells, where its expression can either promote or suppress tumor cell growth, depending on the context. Proteomic analyses of Syk-binding proteins identified several interacting partners also found to be recruited to stress granules. We show here that the treatment of cells with inducers of stress granule formation leads to the recruitment of Syk to these protein-RNA complexes. This recruitment requires the phosphorylation of Syk on tyrosine and results in the phosphorylation of proteins at or near the stress granule. Grb7 is identified as a Syk-binding protein involved in the recruitment of Syk to the stress granule. This recruitment promotes the formation of autophagosomes and the clearance of stress granules from the cell once the stress is relieved, enhancing the ability of cells to survive the stress stimulus.
Introduction
The Syk protein-tyrosine kinase is best known for its many roles in the adaptive and innate immune systems as a component of the molecular machinery that couples immune recognition receptors to intracellular signaling pathways in multiple hematopoietic cell types, including B cells, mast cells, platelets, NK cells, macrophages, and neutrophils (1, 2). In these cells, receptor engagement leads to the phosphorylation of a pair of tyrosines present within immunoreceptor tyrosine-based activation motifs on receptor components, which leads to the recruitment of Syk to the receptor and its subsequent activation. The participation of Syk in pro-inflammatory signaling events has led to considerable interest in the use of small molecule Syk inhibitors for the treatment of diseases such as allergic asthma, rheumatoid arthritis, and systemic lupus erythematosus (3). The identification of additional roles for Syk in tumorigenesis has increased interest in the use of Syk inhibitors as chemotherapeutic agents for the treatment of disorders in which the expression of Syk serves as a pro-survival factor (4). The inhibition of or knockdown in expression of Syk promotes apoptosis in multiple cancer cell types, including many hematopoietic malignancies as well as K-Ras-dependent lung and pancreatic cancer (5), small cell lung cancer (6), ovarian cancer (7), and retinoblastoma (8). For many cancer cells, the expression of Syk can protect them from cell death due to genotoxic or oxidative stress (9).
Eukaryotic cells respond to external stresses in a variety of ways, including the reprogramming of translational processes (10, 11). Insults due to heat shock, viral infection, oxidative stress, exposure to arsenite, or glucose deprivation activate kinases that phosphorylate eIF2α3 to stall translation of mRNAs bound to initiation complexes and promote polysome disassembly. Messenger RNAs are rerouted along with their associated proteins into stress granules (SGs), large ribonucleoprotein (RNP) complexes scaffolded by RNA binding proteins with glycine-rich self-associating domains. Cytoplasmic poly(A) mRNAs stalled in the translational initiation phase are sequestered in SGs and potentially protected from degradation until stress is released and normal protein synthesis can resume. SGs also can sequester signaling molecules important for apoptosis and contribute in this fashion to the protection of cells from stress (12, 13).
The formation of RNP aggregates, however, also can be detrimental to cell survival. This is particularly apparent within the nervous system, where mutations in RNPs that promote self-assembly can be major drivers of neurodegenerative diseases (14–17). Similarly, defective clearance of SGs leads to the pathological accumulation of RNP particles (18, 19). Genetic studies of regulators of SG dynamics in yeast identified granulophagy, a form of macroautophagy (referred to here simply as autophagy), functioning in conjunction with Cdc48/valosin-containing protein (VCP), as critical for SG clearance (20). Mutations in VCP are known to cause amyotrophic lateral sclerosis and frontotemporal dementia, consistent with a role for defects in SG clearance in disease pathology (18, 19). VCP also is up-regulated in many human cancers (21–23), and tumor cells use autophagy as a survival strategy when encountering external stresses due to increased metabolic demands, a challenging microenvironment, or exposure to therapeutic agents (24–27).
To better understand the pro-survival pathways in which Syk functions, we conducted a series of proteomic analyses to identify substrates and interacting partners of the kinase (28–30). Among the binding partners we identified were several with functions in mRNA metabolism that are known to associate with SGs. This finding raised the interesting possibility that Syk also associates with SGs and plays a role in SG function or dynamics. In this study, we demonstrate that Syk is, in fact, recruited to SGs in response to the treatment of cells with either sodium arsenite or a proteasome inhibitor, both of which are known inducers of SG formation (31). This recruitment requires the phosphorylation of a pair of tyrosines on Syk located in a region where the kinase interacts with the SG component, Grb7. In SGs, Syk is active and catalyzes the phosphorylation of proteins on tyrosine. Interestingly, the recruitment of Syk to SGs promotes their clearance from cells through autophagy and enhances the survival of cells in which SGs are induced. This previously unknown function of Syk may underlie many aspects of its ability to promote the survival of cancer cells in the presence of metabolic or therapeutic stress.
Experimental Procedures
DNA Constructs and Cell Lines
Lentiviral vectors for the expression of enhanced green fluorescent protein (EGFP)-tagged Syk (Syk-EGFP) and various mutant forms of Syk-EGFP (Syk-EGFP(K396R), Syk-EGFP(Y342F), Syk-EGFP(Y346F), and Syk-EGFP(Y342F/Y346F)) were described previously (9). The cDNA for Syk-EGFP(R428Q/M429L/M442A) (Syk-AQL-EGFP)was amplified by PCR from the corresponding EGFP-N2 (Clontech) construct (32) and cloned into the Tet-inducible lentiviral vector pLVX-Tight-Puro (Clontech). Lentiviral particles were packaged in HEK293T cells. MCF7 cells obtained from ATCC and MCF7 cells lacking endogenous Syk (MCF7-BD) (33) were cultured in Dulbecco's modified Eagle's medium containing 10% FCS, 100 units/ml penicillin, and 100 μg/ml streptomycin. MCF7-BD cells were infected with lentiviruses, selected in medium containing 1 μg/ml puromycin, expanded, and screened for kinase expression by fluorescence microscopy and Western blotting. Syk-deficient DT40 chicken B cells transfected to stably express Syk-EGFP were described previously (34). DG75 human B lymphoma cells were obtained from ATCC. DG75 cells with reduced levels of Syk due to expression of shRNA directed against the Syk mRNA were described previously (9). Mouse Grb7 cDNA was obtained from the DNASU plasmid repository and cloned into the VQ Ad5CMV (ViraQuest) shuttle vector containing the mCherry cDNA sequence. A vector coding for Grb7-mCherry(R461M) was generated by site-directed mutagenesis using the transformer mutagenesis kit (Clontech). MCF7-BD cells stably expressing Syk-EGFP were transiently transfected with expression plasmids encoding Grb7-mCherry or Grb7-mCherry(R461M) using Lipofectamine 2000 (Invitrogen). A GST-SH2 domain construct of human Grb7 cloned into the pGEX6P1 vector was obtained from Addgene (plasmid 46444 (35)).
MCF7 cells were transfected with Silencer® predesigned siRNA against ATG7 or scrambled siRNA (Life Technologies) using Lipofectamine RNAiMAX and used 72 h after transfection. For the knockout of ATG3, three sets of lentiviral all-in-one guide RNA plus Cas9 (pCLIP-ALL hCMV-BLAST) expression vectors were purchased from Transomic Technologies. Lentiviral particles were packaged in HEK293T and used to infect MCF7 cells. The ATG7 and ATG3 antibodies were purchased from Cell Signaling Technology (D12B11) and Abcam (ab10825), respectively.
Stress Granule Formation and Clearance
To induce the formation of SGs, cells were treated with either sodium arsenite (500 μm or as indicated) or MG132 (Sigma-Aldrich) (100 μm) for 2 or 3 h, respectively, unless indicated otherwise. For some experiments, cells were pretreated with the Src inhibitor PP1 (20 nm) (Cayman Chemical) for 15 min prior to the induction of SG formation. For SG clearance assays, cells were treated with sodium arsenite (250 μm) for 2 h or MG132 (100 μm) for 3 h and then washed and allowed to recover in fresh medium in the absence of drug for an additional 4 h. Endogenous Syk was inhibited during recovery by the addition of R406 (5 μm) where indicated. Syk-AQL-EGFP was selectively inhibited during SG formation or clearance by the addition of either 1-NM-PP1 (Cayman Chemical) or 3-MB-PP1 (Calbiochem) at a concentration of 5 μm. In some experiments, inhibitors of autophagy (5 mm 3-methyladenine or 10 μm N2,N4-dibenzylquinazoline-2,4-diamine) were added during SG clearance. The ability of cells to recover following the removal of stress was measured using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay.
To visualize SGs, cells were fixed with 10% cold methanol for 10 min, permeabilized with 1% Triton X-100 in PBS, and blocked with PBS containing 1 mg/ml BSA, 0.05% Tween 20, and 10% goat serum. Cells were stained with antibodies against G3BP (BD Biosciences (611126)), TIAR (Cell Signaling Technology (D26E4)), phosphotyrosine (Millipore (4G10)), Grb7 (Santa Cruz (A-12)), and/or LC3A/B (Cell Signaling Technology (D3U4C)). Bound primary antibodies were detected using AlexaFluor 594-conjugated goat anti-mouse IgG and/or AlexaFluor 405-conjugated goat anti-rabbit IgG secondary antibodies (Invitrogen). Slides were examined using either an Olympus BH2-RFCA fluorescence microscope equipped with a Sony DXC-950 3CCD color camera, an EVOS FL imaging system, or a Zeiss LSM 710 confocal microscope. The amount of Syk-EGFP versus G3BP was compared by measuring the fluorescence intensity of each protein in each individual punctum using ImageJ.
Cellular Fractionation and Co-immunoprecipitation Assays
For the preparation of cell fractions based on detergent solubility, MCF7-BD cells stably expressing Syk-EGFP were lysed in buffer A (50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1% Nonidet P-40, 0.025% sodium deoxycholate, 1 mm EGTA, 10% glycerol, and protease inhibitor mixture (Abcam (65621)). The detergent-soluble fraction was separated from the insoluble fraction by centrifugation for 1 min at 18,000 × g. The insoluble fraction was resuspended in buffer A containing 1% SDS. Proteins in the supernatant of each fraction were separated by SDS-PAGE and analyzed by Western blotting with antibodies against Syk (Santa Cruz Biotechnology (N-19)), GAPDH (Ambion (AM4300)), eIF2α (Santa Cruz Biotechnology (FL-315)), phospho-eIF2α (AbCam (32157)), Grb7 (Santa Cruz Biotechnology (A-12)), or phosphotyrosine (Millipore (4G10)).
For co-immunoprecipitation assays, MCF7-BD cells stably expressing Syk-EGFP were lysed in buffer A containing 2 mm Na3VO4. EGFP-tagged proteins were immunoprecipitated using GFP-Trap agarose beads (ChromoTek). Bound immune complexes were washed with buffer A, separated by SDS-PAGE, and analyzed by Western blotting with the indicated antibodies. For SG component immunoprecipitation assays, cells were lysed in 100 mm KCl, 5 mm MgCl2, 10 mm HEPES (pH 7.4), 0.5% Triton X-100, 100 units/ml RNase inhibitor, 1 mm dithiothreitol, 1 mm phenylmethylsulfonyl fluoride, and protease inhibitor mixture. The supernatants from the lysate were precleared for 30 min by agitation with a mixture of protein A and G magnetic beads. Cleared lysates were incubated with the indicated antibodies bound to magnetic beads. Immune complexes were washed five times with buffer A containing 100 units/ml RNase inhibitor and analyzed by Western blotting.
Protein Interaction Screens
The yeast two-hybrid screen for Syk-binding proteins expressed from a human mammary gland cDNA library was performed as described previously (33). For additional interaction assays, AH109 cells were transformed simultaneously with two plasmids expressing Grb7 and wild-type Syk, Syk(K396R), or truncated versions of Syk (Syk tandem SH2 domains, Syk catalytic domain, or Syk catalytic domain plus linker B) and then tested for growth on selection medium.
The GST-SH2 domain of Grb7 was expressed in and isolated from Escherichia coli by adsorption onto glutathione-agarose. Immobilized GST or GST-SH2 (10 μg) was incubated with lysates of MCF7-BD cells expressing either Syk-EGFP or Syk-EGFP(Y342F/Y346F) and then washed with 50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 10% glycerol, and 1% Nonidet P-40. Bound proteins were eluted in SDS-sample buffer and detected by Western blotting.
Results
Syk Is Recruited to SGs
Previous screens from our laboratory for Syk substrates and interaction partners identified multiple proteins mapped to specific complexes or pathways involved in mRNA dynamics (28–30). Specifically, we identified as Syk-interacting proteins an extensive set of SG components, including G3BP, a known scaffolding protein necessary for SG formation (10, 29, 36–37). To investigate a possible direct association of Syk with SGs, we asked whether Syk was recruited to SGs under conditions that promoted their formation. For this, we used a line of MCF7 breast cancer cells that lacks endogenous Syk (MCF7-BD) but stably expresses Syk-EGFP (33). Cells were treated with either the proteasome inhibitor MG132 for 3 h or sodium arsenite for 2 h, fixed and stained with an antibody against G3BP, and examined by fluorescence microscopy. Both treatments resulted in the formation of cytoplasmic puncta containing G3BP consistent with the formation of SGs (Fig. 1A). A fraction of Syk-EGFP co-localized with G3BP-containing bodies in cells treated with either stimulus. Syk-EGFP also co-localized with another SG marker, TIAR, as shown in Fig. 1B for cells treated with MG132. Similar findings were observed in other cell types, as shown for Syk-EGFP recruited to G3BP-containing puncta in Syk-EGFP-expressing DT40 lymphoma cells treated with MG132 (Fig. 1C) and endogenous Syk co-localized with TIAR in human DG75 B lymphoma cells treated with sodium arsenite (Fig. 1D).
FIGURE 1.
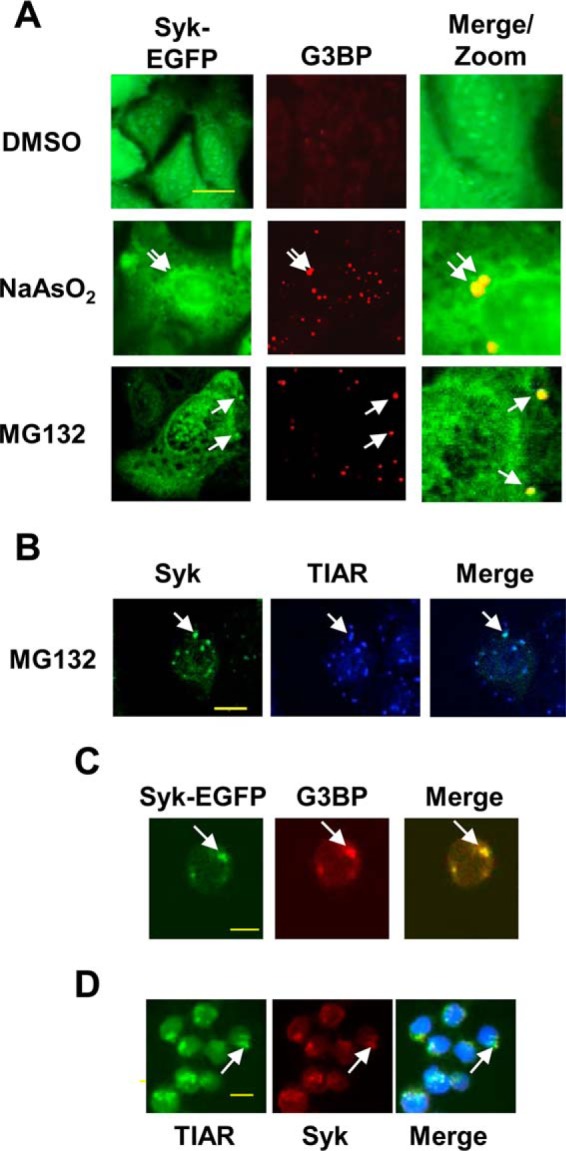
Syk associates with SGs in multiple cell types. A, MCF7-BD cells stably expressing Syk-EGFP were treated with sodium arsenite for 2 h or either MG132 or DMSO (solvent carrier for MG132) for 3 h. Cells were fixed, stained using antibodies against G3BP (red), and examined by fluorescence microscopy. B, Syk-EGFP-expressing MCF7-BD cells were treated with MG132 for 3 h, fixed, stained for TIAR, and examined by confocal microscopy. C, Syk-deficient DT40 B cells ectopically expressing Syk-EGFP were treated with MG132 for 3 h, fixed, and stained with antibodies against G3BP (red). D, DG75 B lymphoma cells were treated with sodium arsenite for 2 h, fixed, and stained with antibodies against TIAR (green) and endogenous Syk (red). Staining of the nucleus with DAPI is indicated in blue. Examples of SGs are indicated by the arrows. Bars, 10 μm.
A redistribution of Syk within the cell following the induction of SGs also was examined biochemically. MCF7-BD cells stably expressing Syk-EGFP were treated with MG132 to induce SG formation. At timed intervals, cells were lysed and separated into detergent-soluble and -insoluble fractions (SG proteins appear in the insoluble fraction). Western blot analyses demonstrated an increased level of Syk-EGFP in the detergent-insoluble fraction following the addition of MG132 (Fig. 2A). The accumulation of Syk-EGFP in the insoluble fraction increased at each time point over the 4-h treatment period. Increased levels of eIF2α, another SG component, also appeared in the insoluble fraction as a function of treatment time. The level of Syk-EGFP in MG132-induced cytoplasmic puncta continued to increase over an extended 12-h time period, as detected by fluorescence microscopy (Fig. 2B).
FIGURE 2.
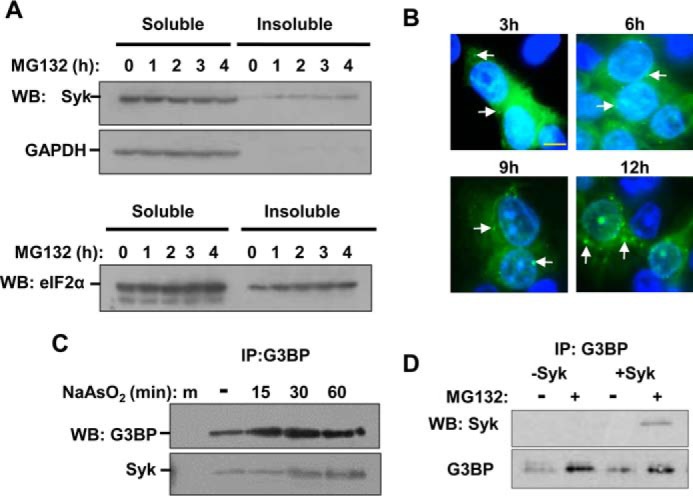
Syk associates with SG markers. A, MCF7-BD cells expressing Syk-EGFP were treated with MG132 for the indicated times. Lysates were separated into detergent-soluble and -insoluble fractions, which were examined by Western blotting (WB) for Syk-EGFP (Syk) or glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (top panels) or eIF2α (bottom panel). B, MCF7-BD cells expressing Syk-EGFP were treated with MG132 for the indicated times, fixed, stained with DAPI, and examined under the fluorescence microscope. Nuclei were stained with DAPI (blue). Examples of SGs are indicated by the arrows. Bar, 10 μm. C, G3BP was immunoprecipitated (IP) from lysates of Syk-EGFP-expressing MCF7-BD cells pretreated with sodium arsenite for the indicated times. Immune complexes were separated by SDS-PAGE and examined by Western blotting for the presence of G3BP and Syk-EGFP (Syk). D, G3BP was immunoprecipitated from lysates of MCF7-BD cells lacking (−) or expressing Syk-EGFP (+) and pretreated with MG132 for 3 h. Immune complexes were separated by SDS-PAGE and examined by Western blotting for the presence of G3BP and Syk-EGFP (Syk).
To further confirm a recruitment of Syk to SGs, we immunoprecipitated G3BP from lysates of Syk-EGFP-expressing MCF7-BD cells that had been treated with sodium arsenite to induce SG formation. The anti-G3BP immune complexes were collected on magnetic beads and then separated by SDS-PAGE and examined for the presence of Syk-EGFP by Western blotting. We observed a time-dependent association of Syk with G3BP in arsenite-treated cells (Fig. 2C), consistent with the two proteins being present in the same complex. Similarly, we found Syk-EGFP in anti-G3BP immune complexes isolated from lysates of cells treated with MG132 (Fig. 2D). Thus, evidence from both biochemical and fluorescence microscopy approaches indicated that Syk was actively recruited to SGs. Syk was particularly prevalent in SGs formed during prolonged exposure to stress-inducing agents.
Syk Is Active in SGs
To determine whether Syk was active when recruited to SGs, we monitored by Western blotting Syk-dependent protein phosphorylation in cells induced to form SGs by treatment with either sodium arsenite or MG132. For these studies, we used a line of MCF7-BD cells in which the expression of Syk-EGFP could be induced with tetracycline (9). Cells either treated or not treated with tetracycline were exposed to sodium arsenite for increasing lengths of time. Western blotting analyses of whole cell lysates with antibodies against phosphotyrosine revealed a pronounced increase in the level of tyrosine-phosphorylated proteins selectively in cells induced to express Syk-EGFP (Fig. 3A). An examination of Syk-EGFP immunoprecipitated from cells and examined by Western blotting with phosphotyrosine antibodies confirmed that Syk itself was phosphorylated on tyrosine in cells treated with sodium arsenite (Fig. 3B). An increase in the tyrosine phosphorylation of Syk also was observed in cells treated with MG132 (Fig. 3C).
FIGURE 3.

The formation of SGs leads to the activation of Syk. A, Tet-responsive MCF7-BD cells were not induced (−tet) or induced (+tet) to express Syk-EGFP and treated for the indicated times with sodium arsenite. Cell lysates were examined by Western blotting with antibodies against phosphotyrosine (pTyr), Syk, or GAPDH. The migration positions of molecular weight markers (kDa) are indicated. B, MCF7-BD cells induced to express Syk-EGFP were treated with sodium arsenite for the indicated times. Syk-EGFP was immunoprecipitated with GFP-Trap beads. Bound proteins were separated by SDS-PAGE and examined by Western blotting with antibodies against Syk and phosphotyrosine. C, MCF7-BD cells stably expressing Syk-EGFP were treated with sodium arsenite (NA) for 2 h or MG132 (MG) for 3 h. Cell lysates and anti-GFP immune complexes were separated by SDS-PAGE and examined by Western blotting (WB) with antibodies against phosphotyrosine, Syk, and GAPDH. IP, immunoprecipitation.
We next asked whether the phosphorylation on tyrosine of proteins within SGs was enhanced in cells expressing Syk. MCF7-BD cells lacking Syk or stably expressing Syk-EGFP were left untreated or were treated with either MG132 or sodium arsenite, fixed, stained using antibodies against phosphotyrosine and TIAR, and examined by confocal microscopy. There was a significant increase in a punctate pattern of phosphotyrosine staining in cells stably expressing Syk-EGFP when treated with either MG132 (Fig. 4A) or sodium arsenite (Fig. 4B). These puncta co-localized with structures containing both TIAR and Syk-EGFP, confirming the identification of these as SGs. This enhanced tyrosine phosphorylation was largely dependent on the expression of Syk-EGFP (Fig. 4A).
FIGURE 4.
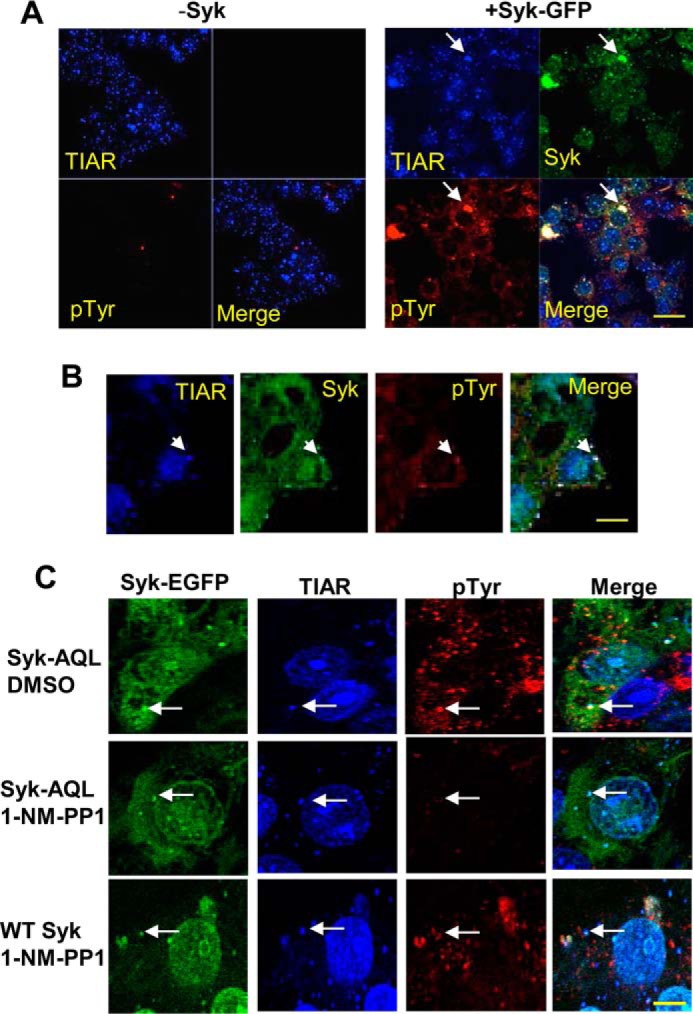
Syk catalyzes the phosphorylation of proteins on tyrosine within SGs. A, MCF7-BD cells lacking Syk (−Syk) or expressing Syk-EGFP were treated with MG132 for 3 h, fixed, stained with antibodies against TIAR (blue) or phosphotyrosine (pTyr) (red), and examined by confocal microscopy. B, MCF7-BD cells expressing Syk-EGFP were treated with sodium arsenite for 2 h, fixed, stained with antibodies against TIAR (blue) or phosphotyrosine (red), and examined by confocal microscopy. C, MCF7-BD cells stably expressing Syk-EGFP (WT Syk) or Syk-AQL-EGFP (Syk-AQL) were treated with MG132 in the presence of 1-NM-PP1 or DMSO solvent carrier for 3 h. Cells were fixed, stained with antibodies against TIAR (blue) or phosphotyrosine (red) and examined by confocal microscopy. Examples of SGs are indicated by the arrows. Bars, 10 μm.
To confirm that this phosphorylation within SGs was specifically dependent on Syk and its catalytic activity, we compared MCF7-BD cells stably expressing Syk-EGFP with cells expressing an analog-sensitive mutant of Syk-EGFP (Syk-AQL-EGFP (32)). Syk-AQL is an engineered form of Syk with mutations within the ATP-binding site that make it uniquely sensitive to inhibition by analogs of PP1 that have bulky side chains that preclude their binding to the wild-type kinase (38). Both sets of cells were pretreated with 1-NM-PP1, a selective orthogonal inhibitor of Syk-AQL, and then treated with MG132 for 3 h to induce SG formation. Cells were fixed and stained using antibodies against phosphotyrosine and TIAR (Fig. 4C). The increase in tyrosine phosphorylation stimulated by MG132 was largely inhibited by 1-NM-PP1 in cells expressing Syk-AQL-EGFP but not in cells expressing Syk-EGFP, which is not sensitive to the inhibitor. Thus, the enhanced phosphorylation of proteins within SGs in cells treated with MG132 was largely dependent on the catalytic activity of Syk.
Recruitment of Syk to SGs Requires Its Phosphorylation
Despite the low level of SG-associated phosphotyrosine observed in cells expressing Syk-AQL-EGFP and pretreated with 1-NM-PP1, the inhibited kinase was still localized to SGs (Fig. 4C). This suggested that the recruitment of Syk to SGs or its retention there might not be dependent on its catalytic activity. To assess this, we compared MCF7-BD cells stably expressing wild-type Syk-EGFP with those expressing a catalytically inactive mutant, Syk-EGFP(K396R). Both active and inactive forms of Syk-EGFP co-localized with G3BP in SGs when cells were treated with MG132 (Fig. 5). Thus, the kinase activity of Syk was not required for its association with SG components.
FIGURE 5.
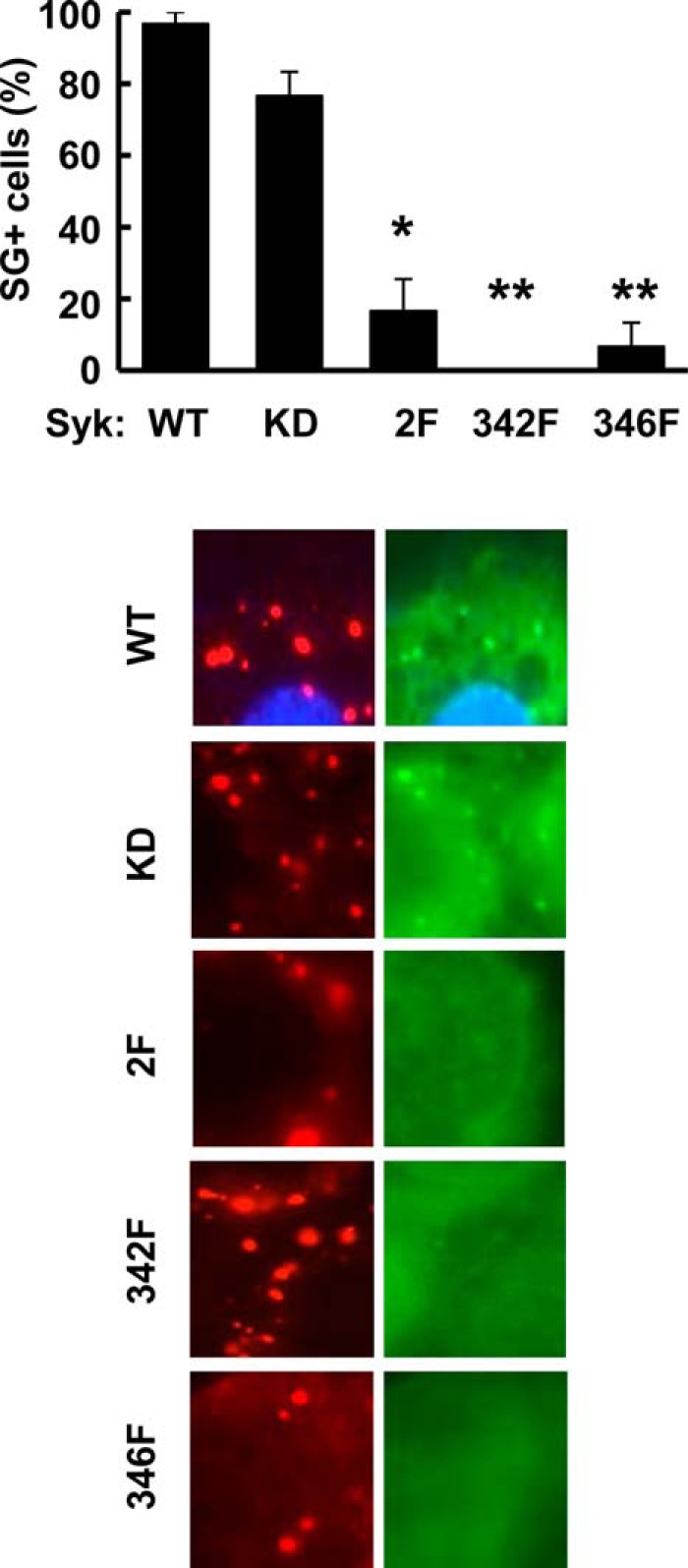
The phosphorylation of Syk on tyrosines 342 and 346 promotes its binding to SGs. MCF7-BD cells stably expressing Syk-EGFP (WT), Syk-EGFP(K396R) (KD), Syk-EGFP(Y342F/Y346F) (2F), Syk-EGFP(Y342F) (342F), or Syk-EGFP(Y346F) (346F) were treated with MG132 for 3 h, fixed, stained with antibodies against G3BP (red), and examined by fluorescence microscopy. Regions of cells containing stress granules visualized by the presence of G3BP-containing puncta were examined for co-localization with puncta containing Syk-EGFP. Cells containing three or more puncta containing both G3BP and Syk-EGFP were scored as SG+. Results represent the means and S.E. (error bars) of three separate experiments. *, p = 0.006; **, p = 0.0002.
When activated, Syk becomes phosphorylated on multiple residues, including tyrosines 342 and 346 (based on the murine Syk numbering system), which are found in the linker B region that separates the tandem pair of SH2 domains from the catalytic domain (1). These residues, when phosphorylated, serve as multifunctional docking sites that mediate interactions with several proteins that contain SH2 domains (1, 2). To assess whether these tyrosines played a role in the association of Syk with SGs, we generated MCF7-BD cells expressing forms of Syk-EGFP in which one or both tyrosines were replaced by phenylalanine. Cell lines were generated that stably expressed Syk-EGFP(Y342F/Y346F), Syk-EGFP(Y342F), or Syk-EGFP(Y346F). These were treated with MG132 for 3 h and then fixed and stained for G3BP to mark SGs. Cells were examined for G3BP-containing puncta that co-localized with puncta containing EGFP-tagged wild-type or mutant Syk. Unlike Syk-EGFP, Syk-EGFP(Y342F/Y346F) largely failed to localize to SGs following treatment with MG132 (Fig. 5). Similarly, the co-localization with G3BP in SGs of both single point mutants of the kinase was defective. Thus, the phosphorylation of both linker B tyrosines 342 and 346 was important for the recruitment of Syk to SGs.
Because the linker B tyrosines on Syk are phosphorylated in B cells either by autophosphorylation or by Src family kinases (39), we examined the effects of an Src family kinase inhibitor on the recruitment of Syk to SGs. MCF7-BD cells stably expressing Syk-EGFP were incubated with the Src kinase inhibitor PP1 for 15 min prior to the induction of SG formation by the addition of MG132. We quantified the fluorescence intensity of Syk-EGFP and G3BP in the puncta that contained both proteins, comparing the control and PP1-treated groups. The fluorescence intensity of Syk within SGs decreased significantly with PP1 treatment, whereas the fluorescence intensity of G3BP within these same puncta did not change (Fig. 6, A and B). These results are consistent with a contribution of Src family kinases to the recruitment of Syk to SGs, probably through the phosphorylation of Syk on Tyr-342 and Tyr-346.
FIGURE 6.
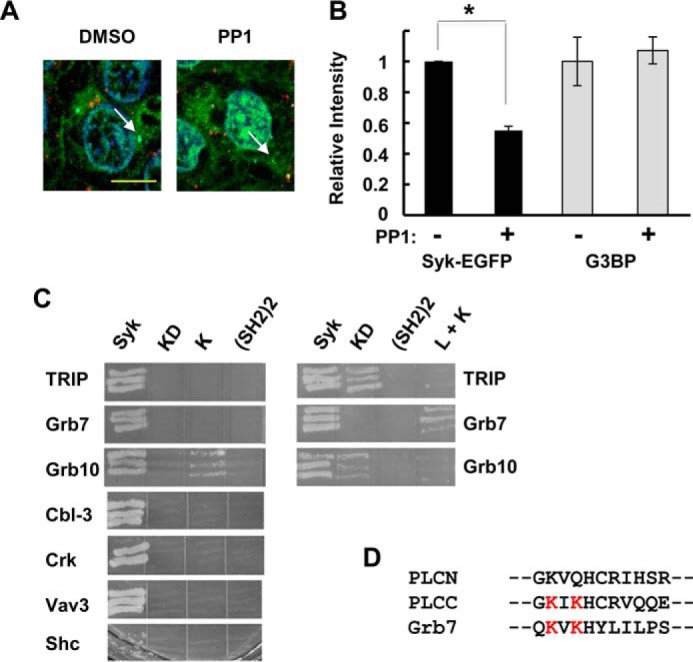
Syk interacts with Grb7. A, MCF7-BD cells stably expressing Syk-EGFP were treated with MG132 in the presence of PP1 or DMSO carrier for 3 h, fixed, and stained for G3BP. Examples of SGs are indicated by the arrows in the illustrated merged images. Bar, 10 μm. B, the relative pixel intensities of SGs from the experiment in A were quantified by ImageJ for content of G3BP and Syk-EGFP. Data represent means ± S.E. (error bars) for triplicate experiments analyzing a minimum of 15 SGs in each. *, p = 0.0001 (Student's t test). C, Gal4 DNA binding domain fusion proteins containing full-length Syk (Syk), Syk(K396R) (KD), the kinase domain only (K), or the tandem SH2 domains ((SH2)2) were examined in a yeast two-hybrid assay for binding to the indicated proteins. D, amino acid sequence of the Tyr(P)-346-binding region of the C-terminal SH2 domain of PLC-γ (PLCC) compared with the comparable regions from the N-terminal SH2 domain of PLC-γ (PLCN), which does not bind, and the SH2 domain of Grb7. Conserved lysines involved in direct interactions with the phosphotyrosine are indicated in red.
Syk Is Recruited to SGs by Grb7
The requirement for two linker B tyrosines for the recruitment of Syk to SGs suggested a role for an adaptor protein that could bind to Syk when it is phosphorylated on both Tyr-342 and Tyr-346. To identify potential adapters, we performed a yeast two-hybrid screen using as prey a human mammary gland cDNA library and as bait both an active and a catalytically inactive form of Syk. When expressed as an Syk-GAL4 DNA-binding domain fusion protein, wild-type Syk becomes tyrosine-phosphorylated in yeast, whereas the catalytically inactive mutant does not (33). Several proteins were identified that bound selectively to active Syk but not to the catalytically inactive mutant (Fig. 6C). These included Grb7, Grb10, Cbl-3, Vav3, Crk, and Shc (Fig. 6C). All of these contain an SH2 domain and/or a structurally related tyrosine kinase binding domain that binds phosphotyrosine, consistent with their selective binding to active, tyrosine-phosphorylated Syk. Of these, only Grb7 had been identified previously as a component of SGs (40). In the yeast two-hybrid screen, Grb7 bound to full-length Syk and failed to bind to the catalytic domain of Syk or to the tandem pair of SH2 domains but was capable of binding to a construct containing both the linker B region and the catalytic domain, consistent with an interaction mediated by residues within linker B (Fig. 6C). A comparison of the sequence of the Grb7 SH2 domain with that of the C-terminal SH2 domain of PLC-γ revealed in common a pair of lysine residues known to be important in the PLC-γ SH2 domain for the formation of a second phosphotyrosine binding pocket that allows it to bind to phosphopeptides containing two phosphotyrosines (41) (Fig. 6D). To explore this interaction further, we performed an in vitro pull-down assay in which the immobilized GST-SH2 domain of Grb7 was incubated with lysates of MCF7-BD cells expressing either wild-type Syk-EGFP or Syk-EGFP(Y342F/Y346F). As shown in Fig. 7A, the SH2 domain of Grb7 preferentially interacted with the wild-type kinase, supporting a role for Tyr-342 and Tyr-346 in the association of Syk with Grb7. Fractions of both Syk-EGFP and Grb7 relocated into the detergent-insoluble fraction of MCF7-BD cells following their treatment with MG132 (Fig. 7B), consistent with both proteins localizing to SGs. In addition, endogenous Grb7 could be co-immunoprecipitated with G3BP selectively under conditions that promoted the formation of SGs, and this interaction did not require the presence of Syk (Fig. 7C). In contrast, an association of Syk with Grb7 did not require SG formation (Fig. 7D).
FIGURE 7.

Grb7 associates with Syk and with SGs. A, lysates from MCF7-BD cells expressing Syk-EGFP (WT) or Syk-EGFP(Y342F/Y346F) (2F) were adsorbed to glutathione-Sepharose beads containing bound GST or GST-Grb7 SH2 domain. Syk-EGFP was detected in cell lysates or in the bead-bound fraction by Western blotting (WB) with antibodies against Syk. GST and GST-Grb7 SH2 domain were detected using antibodies against GST. B, MCF7-BD cells expressing Syk-EGFP were treated with MG132 for the indicated times, lysed, and separated into detergent-soluble and -insoluble fractions. The presence of Syk-EFGP (Syk) and endogenous Grb7 was detected by Western blotting. C, G3BP was immunoprecipitated (IP) from MCF7-BD cells pretreated without (−) or with (+) MG132 for 3 h. The immune complexes were separated by SDS-PAGE, and the presence of endogenous G3BP and Grb7 was detected by Western blotting. A mock (m) immunoprecipitation carried out in the absence of anti-G3BP was included as a control for nonspecific binding. D, Grb7 was immunoprecipitated from MCF7-BD cells expressing or lacking Syk-EGFP that were pretreated with or without MG132 for 3 h. Syk-EGFP, Grb7, and GAPDH were detected in cell lysates and immune complexes by Western blotting with antibodies against Syk or Grb7.
To confirm the localization of Grb7 to SGs, we treated MCF7-BD cells expressing Syk-EGFP with MG132 and then stained cells with antibodies against TIAR and endogenous Grb7. Both Syk and Grb7 proteins localized to cytoplasmic puncta that also contained TIAR (Fig. 8A). To investigate further whether the SH2 domain of Grb7 was involved in the recruitment of Syk to SGs, we inactivated it by mutating the essential arginine in the canonical phosphotyrosine binding site to a methionine. Wild-type Grb7 with an mCherry tag at the C terminus (Grb7-mCherry) and Grb7-mCherry(R451M) were transiently expressed in MCF7-BD cells expressing Syk-EGFP, which were then treated with MG132 to induce SG formation. Whereas wild-type Grb7-mCherry localized to SGs along with Syk-EGFP and TIAR (Fig. 8B), neither Grb7-mCherry(R451M) nor Syk-EGFP localized to TIAR-containing SGs in cells expressing the mutant form of Grb7 (Fig. 8C). Thus, a functional SH2 domain of Grb7 promotes the recruitment of both Syk and Grb7 to SGs.
FIGURE 8.

Grb7 promotes the binding of Syk to SGs. A, MCF7-BD cells expressing Syk-EGFP were treated with MG132 for 3 h, fixed, stained with antibodies against endogenous TIAR (blue) and Grb7 (red), and examined by confocal microscopy. B and C, MCF7-BD cells expressing Syk-EGFP were transiently transfected with expression plasmids for Grb7-mCherry (B) or Grb7-mCherry(R461M) (Grb7-R461M) (C) (red). Cells were treated with MG132 for 3 h, fixed, stained with antibodies against TIAR (blue), and examined by confocal microscopy. Examples of SGs are indicated by the arrows. Bars, 10 μm.
Syk Promotes the Clearance of SGs
We compared the number of SGs formed in MCF7-BD cells expressing or lacking Syk-EGFP following a 3-h treatment with MG132. The median number of SGs formed per cell was higher in Syk-deficient cells than in cells stably expressing Syk-EGFP (Fig. 9A). This suggested a role for Syk in modulating SG dynamics. To explore this further, we examined the effect of Syk on the ability of cells to clear SGs following their release from the stress stimulus. MCF7-BD cells expressing Syk-EGFP or lacking Syk were treated with MG132 for 3 h to induce SGs. The cells were washed and allowed to recover in drug-free medium for 4 h. Cells were then fixed and immunostained with an antibody against G3BP to detect SGs. The number of cells containing more than three SGs were counted as SG-positive (SG+), whereas those with three or fewer SGs were counted as SG-negative. The presence or absence of Syk in MCF7-BD cells had little influence on the number of SG+ cells appearing during the initial 3-h incubation. However, far fewer Syk-EGFP-expressing cells retained SGs following the 4-h recovery period as compared with cells lacking Syk (Fig. 9B). To confirm this observation, we performed a biochemical assay to assess changes in the level of phospho-eIF2α by Western blot analysis. In MCF7-BD cells, the induction of SGs with MG132 resulted in an increase in the extent of phosphorylation of eIF2α in both Syk-expressing and Syk-deficient cells (Fig. 9C). However, the level of phospho-eIF2α decreased to a greater extent following the 4-h recovery phase in cells expressing Syk-EGFP than in cells lacking the kinase, consistent with the presence of fewer SGs.
FIGURE 9.

Syk promotes the clearance of SGs. A, MCF7-BD cells lacking Syk (−Syk) or expressing Syk-EGFP (+Syk) were treated with MG132 for 3 h. Cells were fixed and stained with antibodies against G3BP. The number of SGs present per cell was counted. B, MCF7-BD cells lacking Syk (Syk(−)) or expressing Syk-EGFP (Syk(+)) were treated with MG132 for 3 h and then washed and placed in fresh medium lacking MG132 for 4 h. Cells were fixed and stained with DAPI and antibodies against G3BP. The number of cells containing three or more SGs was counted as SG+ prior to (Formation) or following (Clearance) removal of MG132. Data represent means ± S.E. (error bars) of triplicate experiments in which at least 30 cells were examined in each. *, p < 0.005 comparing clearance in Syk-expressing cells with that in Syk-deficient cells (Student's t test). C, lysates from MCF7-BD cells lacking Syk (−) or expressing Syk-EGFP (+) that were pretreated with MG132 for 3 h and then washed and placed in fresh medium lacking MG132 for 4 h were separated by SDS-PAGE and analyzed by Western blotting (WB) with antibodies against phospho-eIF2α, eIF2α, Syk-EGFP (Syk), or GAPDH. Replicate samples are illustrated for formation and clearance.
To assess whether the catalytic activity of Syk was necessary for SG clearance, we compared Syk-deficient MCF7-BD cells with those stably expressing either Syk-AQL-EGFP or Syk-EGFP. Cells were treated with MG132 for 3 h to induce SG formation and were then washed and allowed to recover in fresh medium for 4 h. Where indicated, an orthogonal inhibitor of Syk-AQL, 3-MB-PP1 (42), was added either immediately following the removal of MG132 or 1 h later (Fig. 10, A and B). Cells were fixed, stained using a G3BP antibody, and examined for the presence of SGs. As observed previously, most cells lacking Syk retained SGs after the 4-h recovery period. In contrast, most cells expressing either Syk-EGFP or Syk-AQL-EGFP were largely free of visible SGs 4 h after removal of the MG132. The addition of 3-MB-PP1 effectively blocked the clearance of SGs from cells expressing Syk-AQL-EGFP but had no effect on the reduction in SGs in the Syk-EGFP-expressing cells. When 3-MB-PP1 was added 1 h after the removal of MG132, the number of Syk-AQL-EGFP-expressing cells that were SG+ was reduced by ∼50%. Thus, many cells were able to clear most SGs in the first hour following removal of MG132, but further clearance could be blocked by the Syk kinase inhibitor.
FIGURE 10.
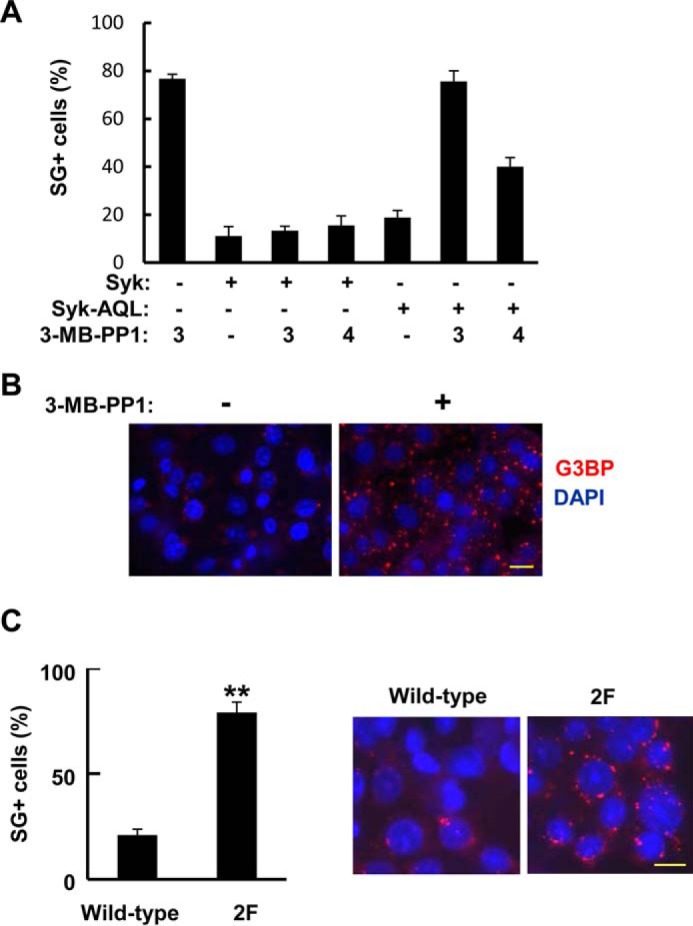
SG clearance requires Syk activity and its association with SGs. A, MCF7-BD cells lacking Syk or expressing either Syk-EGFP (Syk) or Syk-AQL-EGFP (Syk-AQL) were treated with MG132 for 3 h and then washed and placed in fresh medium for 4 h. Cells were treated with 3-MB-PP1 (5 μm) at the 3 h time point (when MG132 was removed) (3) or 1 h later (4). Cells were fixed and stained with DAPI and antibodies against G3BP. The number of cells containing three or more SGs was counted as SG+. Data represent means ± S.E. (error bars) for triplicate experiments in which SGs were counted in at least 30 cells. B, example of stained and fixed cells from the experiment in A. Cells had been pretreated for 3 h with MG132, washed, and placed in fresh medium in the absence or presence of 3-MB-PP1 for 4 h and then fixed and stained with DAPI (blue) and with antibodies against G3BP (red) to mark SGs. C, MCF7-BD cells expressing Syk-EGFP (Wild-type) or Syk-EGFP(Y342F/Y346F) (2F) were treated with MG132 for 3 h and then washed and placed in fresh medium lacking MG132 for 4 h. Cells were fixed and stained with DAPI (blue) and antibodies against G3BP (red). The number of cells containing three or more SGs was counted as SG+. Data represent means ± S.E. for triplicate experiments in which SGs were counted in at least 30 cells. **, p = 0.0002. Bars, 10 μm.
To determine whether the association of Syk with SGs also was required for their clearance, we compared MCF7-BD cells stably expressing Syk-EGFP with those expressing Syk-EGFP(Y342F/Y346F), which is catalytically active but fails to bind to SGs. Cells again were treated with MG132 for 3 h and then washed and allowed to recover for 4 h. The number of SG+ cells was much higher for cells expressing Syk-EGFP(Y342F/Y346F) as compared with those expressing wild-type Syk-EGFP (Fig. 10C). Thus, both the catalytic activity of Syk and its association with SGs were required for the clearance of SGs from cells after removal of the stress stimulus.
To determine whether endogenously expressed Syk also regulated SG clearance, we compared MCF7 cells purchased from ATCC (MCF7-ATCC), which express endogenous Syk, with the Syk-deficient MCF7-BD cell line (Fig. 11A). Cells from both lines formed SGs to similar extents when treated with sodium arsenite (Fig. 11, A and B). However, MCF7-ATCC cells were much more effective at clearing SGs following removal of the stress-inducing agent than were MCF7-BD cells. This enhanced ability of MCF7-ATCC cells to clear SGs was blocked by the addition of the Syk inhibitor R406 (Fig. 11C). Similarly, whereas shRNA-mediated knockdown of Syk in DG75 B cells had no measurable effect on SG formation, the reduction in Syk attenuated SG clearance (Fig. 11D). These data indicate that Syk, when expressed at normal endogenous levels, enhances SG clearance.
FIGURE 11.
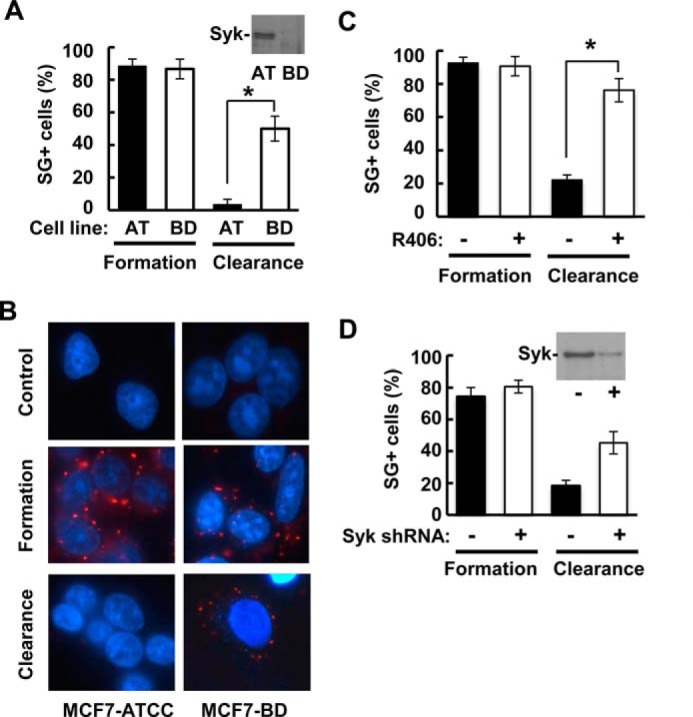
Syk expressed at endogenous levels regulates SG clearance. A, MCF7-ATTC cells (AT), which contain endogenous Syk as confirmed by Western blotting (inset), and Syk-deficient MCF7-BD cells (BD) were treated with sodium arsenite (250 μm) for 2 h and then washed and placed in fresh medium for 4 h. The number of cells containing three or more SGs was counted as SG+. Data represent means ± S.E. (error bars) for triplicate experiments in which SGs were counted in at least 30 cells. *, p = 0.01. B, MCF7-ATCC and MCF7-BD cells were untreated (control) or treated as described in A, fixed, stained with DAPI (blue) and antibodies against G3BP (red), and examined by fluorescence microscopy. C, MCF7-ATCC cells were treated with sodium arsenite (250 μm) for 2 h in the presence of either DMSO carrier alone (−) or 5 μm R406 (+) and then washed and placed in medium containing DMSO or R406 but lacking sodium arsenite for 4 h. The number of cells containing three or more SGs (as determined by the presence of G3BP-containing puncta) was counted as SG+. Data represent means ± S.E. for triplicate experiments in which SGs were counted in at least 30 cells. *, p = 0.002. D, DG75 B lymphoma cells lacking (−) or expressing an shRNA against the Syk message (+) were treated with sodium arsenite (250 μm) for 2 h and then washed and placed in fresh medium for 4 h. The number of cells containing three or more SGs (as determined by the presence of G3BP-containing puncta) was counted as SG+. Data represent means ± S.E. for triplicate experiments in which SGs were counted in at least 30 cells. *, p = 0.008. A Western blot showing the relative levels of expression of endogenous Syk is shown in the inset.
Syk Promotes SG Clearance through Autophagy
Recent data indicate that SGs can be cleared from cells through the process of autophagy (20). To determine whether autophagy was involved in the Syk-dependent clearance of SGs, we treated MCF7-BD cells stably expressing Syk-EGFP with MG132 for 3 h to induce SG formation. Cells were washed and allowed to recover in the absence of MG132 but in the presence of one of two known inhibitors of autophagy: N2,N4-dibenzylquinazoline-2,4-diamine or 3-methyladenine. Cells were then fixed and stained with a G3BP antibody to detect SGs. Both autophagy inhibitors blocked the ability of Syk-EGFP to promote SG clearance in cells recovering from MG132-induced stress (Fig. 12A). Similarly, knockout of ATG3 using CRISPR/Cas9 (Fig. 12B) or knockdown of ATG7 using siRNA (Fig. 12C) significantly reduced their clearance following stress removal (Fig. 13A).
FIGURE 12.
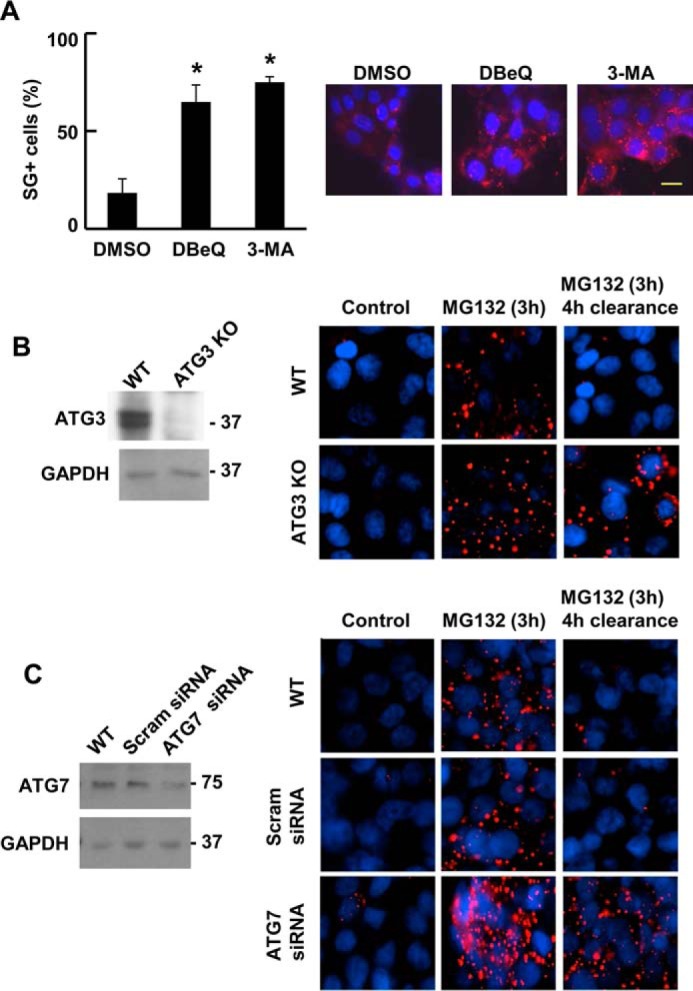
Syk promotes the clearance of SGs through autophagy. A, MCF7-BD cells expressing Syk-EGFP were treated with MG132 for 3 h and then washed and placed in fresh medium lacking MG132 but containing DeBQ, 5-MA, or DMSO carrier for 4 h. Cells were fixed and stained with DAPI (blue) and antibodies against G3BP (red). The number of cells containing three or more SGs was counted as SG+. Data represent means ± S.E. (error bars) for triplicate experiments in which SGs were counted in at least 30 cells. *, p < 0.01 (drug-treated as compared with DMSO only control). B, ATG3 was knocked out in MCF7 ATCC cells using CRISPR/Cas9-mediated gene editing. Levels of ATG3 in wild-type cells (WT) and ATG3 knock-out cells (ATG3KO) were measured by Western blotting. Wild-type cells or cells lacking ATG3 were treated without (Control) or with MG132 for 3 h and then washed and placed in fresh medium lacking MG132 for 4 h. Cells were fixed and stained with DAPI (blue) and antibodies against G3BP (red) and examined by fluorescence microscopy. C, MCF7 ATCC cells were transfected with scrambled siRNA (Scram) or siRNA against ATG7. Levels of ATG7 were measured by Western blotting. Wild-type cells or cells transfected with scrambled siRNA or ATG7 siRNA were treated with MG132 for 3 h and then washed and placed in fresh medium lacking MG132 for 4 h. Cells were fixed and stained with DAPI (blue) and antibodies against G3BP (red) and examined by fluorescence microscopy.
FIGURE 13.
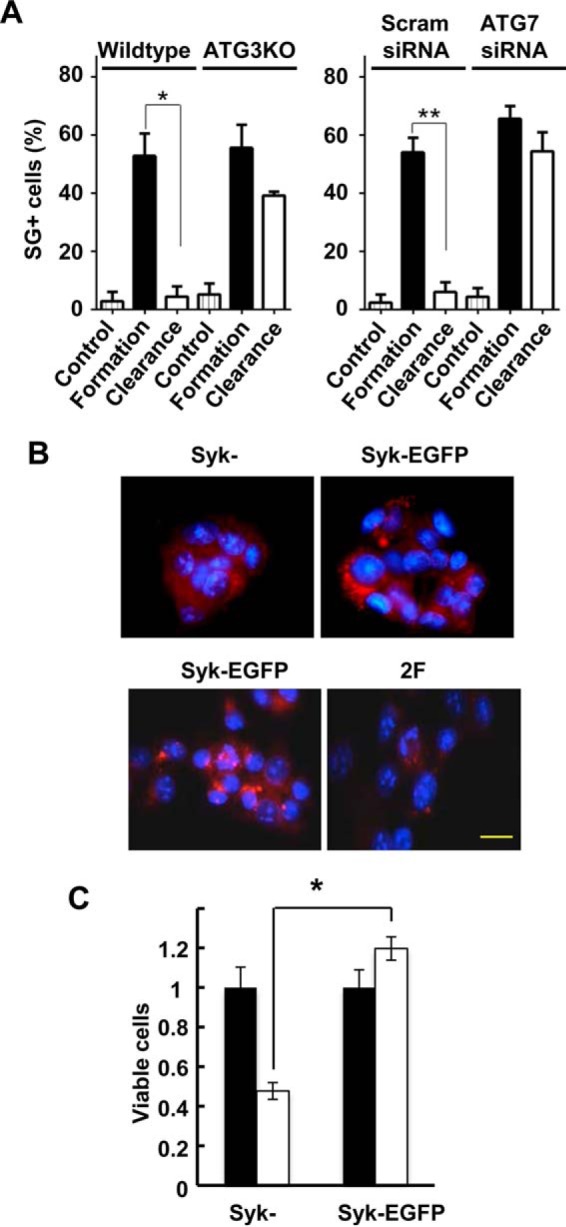
Syk promotes the appearance of LC3-containing vesicles and cell survival. A, the number of cells containing three or more SGs (as determined by the presence of G3BP-containing puncta) from experiments in Fig. 12, B and C, were counted as SG+. Data represent means ± S.E. (error bars) for counts from 10 random frames of cells containing at least 25 cells each from three replicates. *, p = 0.001; **, p = 0.01. B, MCF7-BD cells lacking Syk (Syk−) or expressing either Syk-EGFP or Syk-EGFP(Y342F/Y346F) (2F) were treated with MG132 for 3 h and then washed and placed in fresh medium lacking MG132 for 4 h. Cells were fixed and stained with DAPI (blue) and antibodies against LC3A/B (red). C, MCF7-BD cells lacking Syk (Syk−) or expressing Syk-EGFP were treated without (closed bars) or with (open bars) sodium arsenite (1 μm) for 3 h. Cells were washed and placed in fresh medium for 24 h. Cell viability was measured using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Data represent means ± S.E. for triplicate experiments. *, p = 0.05. Bars, 10 μm.
Because cells expressing Syk-EGFP effectively cleared SGs, whereas cells lacking Syk did not, we compared MCF7-BD cells expressing or lacking the kinase for the formation of autophagosomes during recovery from MG132-induced stress. Cells were treated with MG132 for 3 h, washed, and allowed to recover for 4 h in the absence of the proteasome inhibitor. Cells were fixed and stained for LC3A/B, which is processed and incorporated into maturing autophagosomal membranes and thus serves as a useful marker for these vesicles (43). Abundant LC3-positive vesicles were observed in cells expressing Syk-EGFP as opposed to cells lacking Syk, which exhibited few autophagosomes (Fig. 12C). To determine whether the association of Syk with SGs was important for autophagosome formation, we compared MCF7-BD cells expressing Syk-EGFP with those expressing Syk-EGFP(Y342F/Y346F). Cells were treated with MG132 and allowed to recover as described above. LC3-positive vesicles were much more plentiful in the Syk-EGFP-expressing cells as compared with cells expressing Syk-EGFP(Y342F/Y346F) (Fig. 13A). These results indicate that the recruitment of Syk to SGs promotes their clearance through the recruitment or formation of autophagosomes.
SG Clearance Promotes Cell Survival
Because Syk functions as a pro-survival factor in numerous cancer cell types (4), we asked whether the expression of the kinase might alter the capacity of cells to survive an insult that results in the formation of SGs. MCF7-BD cells expressing Syk-EGFP or lacking Syk were treated with a low concentration of sodium arsenite for 3 h, conditions under which cells form numerous small SGs. Cells were washed and then allowed to recover and proliferate in fresh medium for an additional 24 h. Viable cells were then measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Whereas cells expressing Syk grew normally following removal of the stress stimulus, the growth of cells lacking Syk was greatly attenuated (Fig. 13B).
Discussion
Our study supports a model in which Syk is recruited to SGs that are induced in response to external agents that cause oxidative stress and disrupt protein folding or interfere with the ubiquitin-proteasome system. This recruitment requires the phosphorylation of Syk on a pair of tyrosines in the linker B region, which creates a docking site for the SG component, Grb7. Once at the SG, Syk is active and catalyzes the phosphorylation of proteins on tyrosine that promote the clearance from the cell of SGs through autophagy, thus enabling the cells to recover from the initial stress stimulus.
The recruitment of Syk to SGs requires its phosphorylation on the two tyrosines found within the sequence, YESPYADP, in the linker B region of the protein. Multiple proteins have been described that contain SH2 domains capable of binding within this region, and many of these SH2 domains recognize Syk preferentially when both tyrosines (Tyr-342 and Tyr-346) are phosphorylated (41, 44). This was first demonstrated for the C-terminal SH2 domain of phospholipase C-γ (41). In this case, phosphotyrosine 342 binds in the canonical phosphotyrosine binding pocket found within all SH2 domains, whereas phosphotyrosine 346 binds at a second site found only in a subset of SH2 domains. Here the phosphate group interacts with two critical lysines. Interestingly, these two lysines are conserved in the sequence of the Grb7, suggesting that the Grb7 SH2 domain too can recognize doubly phosphorylated peptides. The expression of a form of Grb7 with an SH2 domain disabled by a point mutation in what is typically the most high affinity binding pocket (41) blocks the recruitment of Syk to the SG consistent with an SH2 domain-mediated interaction between Syk and Grb7. Unexpectedly, the association of Grb7 with the SG also is inhibited by this mutation. This suggests that the Grb7 SH2 domain is important for both its interaction with Syk and with SGs. This may reflect the fact that both Grb7 and its isolated SH2 domain are dimers, capable of binding simultaneously more than one ligand (45, 46). The site(s) on the SG to which the Grb7 SH2 domain binds is not known; nor is it known whether or not this site contains phosphotyrosine because the Grb7 SH2 domain is capable also of recognizing amino acid sequences that lack a phosphoamino acid (47, 48).
The ability of a catalytically inactive form of Syk to associate with SGs suggests that the phosphorylation of Tyr-342 and Tyr-346 can be catalyzed by kinases other than Syk itself. This is consistent with previous phosphopeptide mapping experiments demonstrating the phosphorylation of catalytically inactive Syk on Tyr-342 and Tyr-346 in B cells activated through the antigen receptor in a manner dependent on the expression of Lyn, a Src family kinase (39). Consistent with this idea, the inclusion of a Src kinase inhibitor reduced the content of Syk in SGs. Interestingly, Src kinases also are known to phosphorylate at least one other SG component, Sam68/KHDR1 (10, 49).
We find that the presence of Syk in MCF7 cells is not required for the formation of SGs. In fact, the number of SGs present in Syk-expressing cells treated for 3 h with MG132 is reduced compared with the number in cells lacking the kinase. The presence or absence of Syk, however, does have a dramatic effect on the disappearance of SGs from cells once the stress is removed. This clearance of SGs requires the association of Syk with the SG because Syk-EGFP(Y342F/Y346F) fails to promote SG clearance. The addition of an inhibitor orthogonal to Syk-AQL-EGFP to cells expressing the analog-sensitive kinase, but not to cells expressing Syk-EGFP, also blocks SG clearance. This indicates an essential and specific role for Syk-catalyzed tyrosine phosphorylation in this process. Visualization of protein tyrosine phosphorylation by immunofluorescence clearly demonstrates a substantial increase in the phosphorylation of proteins resident in or near SGs. This phosphorylation is largely Syk-dependent because it is selectively blocked in cells expressing Syk-AQL-EGFP upon treatment with the Syk-AQL-specific inhibitor. However, the exact protein(s) that Syk phosphorylates to promote SG clearance remains to be determined.
SGs are thought to disassemble following the removal of stress, allowing sequestered mRNAs to return to ribosomes for translation (11). A recent genetic screen in yeast, however, also clearly indicates a critical role for components of the autophagy pathway in modulating the dynamics of SG removal (20). Consistent with a role for autophagy in SG dynamics, the addition of inhibitors of autophagy, the knockout of ATG3, and a reduction in the level of ATG7 in MCF7 cells all attenuate the Syk-dependent clearance of SGs following removal of the stress stimulus. Also consistent with this mechanism, a substantial increase in LC3-positive vesicles is seen in cells recovering from stress when cells contain a catalytically active form of Syk that also is capable of binding to SGs. These observations implicate Syk as an important factor in regulating the removal from cells of SGs through autophagy. Grb7, which promotes SG formation or enhances SG stability, has been reported also to recruit to SGs focal adhesion kinase, which phosphorylates Grb7 to promote SG disassembly (40). It is not known whether autophagy contributes to the loss of SGs that is seen following the recruitment of FAK and phosphorylation of Grb7.
The consequences of the presence or absence of Syk in tumor cells are manifested in different ways, depending on the tumor cell type and its stage of development (4). In many tumors, the expression of Syk enhances cell survival, especially in the face of external or internal factors, including oxidative stress, exposure to chemotherapeutic agents, expression of oncogenes like activated K-Ras, or loss of Rb (4, 5, 8, 9). Multiple mechanisms have been described to account for many of the protective effects of Syk on cells, including activation of the PI3K/Akt pro-survival pathway leading to the stabilization of the anti-apoptotic proteins Mcl-1 and/or XIAP, activation of mTOR, activation of STAT3 or STAT5, enhanced transcription of NF-κB-regulated genes, and stabilization of the mRNA for Bcl-xL through an association of Syk with nucleolin (8, 9, 50–56). The contributions of autophagy to cancer cell survival also are context-dependent. Although a lack of autophagy can contribute to tumorigenesis in some cancer types, in more established tumors, autophagy instead confers a survival advantage that is particularly important for the cancer cell resistance to external stresses, including hypoxia and exposure to radiation or chemotherapeutic agents (24–27). Thus, many established tumors rely on autophagy as a mechanism to survive elevated metabolic or therapeutic stress, and inhibitors of autophagy can sensitize some tumors to the actions of chemotherapeutic agents (26). MCF7 cells are relatively insensitive to the inhibition of autophagy for basal cell growth (57). However, we find that MCF7 cells expressing Syk are better able to recover from exposure to sodium arsenite, which induces SG formation, than are cells lacking the kinase. This suggests that the increased autophagy promoted by the presence of Syk facilitates cell survival in response to stress. Thus, Syk-dependent enhancement of autophagy provides an additional mechanism by which the kinase can enable cancer cells to evade stress-induced cell death. This suggests that Syk inhibitors would be useful in sensitizing a subset of Syk-expressing cancer cells to the actions of chemotherapeutic agents.
Author Contributions
R. L. G conceived of the project; M. O. K., R. L. H., S. G., J. S. T., and W.-H. W. performed the cell and molecular biology experiments; Q. Z. performed the yeast two-hybrid screen; and R. L. G., M. O. K., and R. L. H. wrote the manuscript.
This work was supported by Public Health Service Grants R01AI098132 and R01CA115465 (to R. L. G.) from the National Institutes of Health. The DNA sequencing facility was supported by National Institutes of Health, NCI, Grant CCSG CA23168 to the Purdue University Center for Cancer Research. The authors declare that they have no conflicts of interest with the contents of this article.
- eIF2α
- eukaryotic initiation factor 2α
- SG
- stress granule
- EGFP
- enhanced green fluorescent protein
- G3BP
- Ras-GTPase activating protein-binding protein
- LC3
- microtubule-associated protein light chain 3
- RNP
- ribonucleoprotein
- TIAR
- T-cell-restricted intracellular antigen-1 related protein
- VCP
- valosin-containing protein
- SH2
- Src homology 2.
References
- 1.Geahlen R. L. (2009) Syk and pTyr'd: Signaling through the B cell antigen receptor. Biochim. Biophys. Acta 1793, 1115–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mócsai A., Ruland J., and Tybulewicz V. L. J. (2010) The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat. Rev. Immunol. 10, 387–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geahlen R. L. (2014) Getting Syk: spleen tyrosine kinase as a therapeutic target. Trends Pharmacol. Sci. 35, 414–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krisenko M. O., and Geahlen R. L. (2015) Calling in SYK: SYK's dual role as a tumor promoter and tumor suppressor in cancer. Biochim. Biophys. Acta 1853, 254–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singh A., Greninger P., Rhodes D., Koopman L., Violette S., Bardeesy N., and Settleman J. (2009) A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell 15, 489–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Udyavar A. R., Hoeksema M. D., Clark J. E., Zou Y., Tang Z., Li Z., Li M., Chen H., Statnikov A., Shyr Y., Liebler D. C., Field J., Eisenberg R., Estrada L., Massion P. P., and Quaranta V. (2013) Co-expression network analysis identifies spleen tyrosine kinase (SYK) as a candidate oncogenic driver in a subset of small-cell lung cancer. BMC Syst. Biol. 7, Suppl. 5, S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prinos P., Garneau D., Lucier J.-F., Gendron D., Couture S., Boivin M., Brosseau J.-P., Lapointe E., Thibault P., Durand M., Tremblay K., Gervais-Bird J., Nwilati H., Klinck R., Chabot B., Perreault J.-P., Wellinger R. J., and Elela S. A. (2011) Alternative splicing of SYK regulates mitosis and cell survival. Nat. Struct. Mol. Biol. 18, 673–679 [DOI] [PubMed] [Google Scholar]
- 8.Zhang J., Benavente C. A., McEvoy J., Flores-Otero J., Ding L., Chen X., Ulyanov A., Wu G., Wilson M., Wang J., Brennan R., Rusch M., Manning A. L., Ma J., Easton J., Shurtleff S., Mullighan C., Pounds S., Mukatira S., Gupta P., Neale G., Zhao D., Lu C., Fulton R. S., Fulton L. L., Hong X., Dooling D. J., Ochoa K., Naeve C., Dyson N. J., Mardis E. R., Bahrami A., Ellison D., Wilson R. K., Downing J. R., and Dyer M. A. (2012) A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature 481, 329–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang W.-H., Childress M. O., and Geahlen R. L. (2014) Syk interacts with and phosphorylates nucleolin to stabilize Bcl-xL mRNA and promote cell survival. Mol. Cell. Biol. 34, 3788–3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buchan J. R., and Parker R. (2009) Eukaryotic stress granules: the ins and outs of translation. Mol. Cell 36, 932–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anderson P., and Kedersha N. (2008) Stress granules: the Tao of RNA triage. Trends Biochem. Sci. 33, 141–150 [DOI] [PubMed] [Google Scholar]
- 12.Kim W. J., Back S. H., Kim V., Ryu I., and Jang S. K. (2005) Sequestration of TRAF2 into stress granules interrupts tumor necrosis factor signaling under stress conditions. Mol. Cell. Biol. 25, 2450–2462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li W., Simarro M., Kedersha N., and Anderson P. (2004) FAST is a survival protein that senses mitochondrial stress and modulates TIA-1-regulated changes in protein expression. Mol. Cell. Biol. 24, 10718–10732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ash P. E. A., Vanderweyde T. E., Youmans K. L., Apicco D. J., and Wolozin B. (2014) Pathological stress granules in Alzheimer's disease. Brain Res. 1584, 52–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dormann D., and Haass C. (2011) TDP-43 and FUS: a nuclear affair. Trends Neurosci. 34, 339–348 [DOI] [PubMed] [Google Scholar]
- 16.Kim H. J., Kim N. C., Wang Y.-D., Scarborough E. A., Moore J., Diaz Z., MacLea K.S., Freibaum B., Li S., Molliex A., Kanagaraj A. P., Carter R., Boylan K. B., Wojtas A. M., Rademakers R., Pinkus J. L., Greenberg S. A., Trojanowski J. Q., Traynor B. J., Smith B. N., Topp S., Gkazi A.-S., Miller J., Shaw C. E., Kottlors M., Kirschner J., Pestronk A., Li Y. R., Ford A. F., Gitler A. D., Benatar M., King O. D., Kimonis V. E., Ross E. D., Weihl C. C., Shorter J., and Taylor J. P. (2013) Mutations in prion- like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495, 467–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.King O. D., Gitler A. D., and Shorter J. (2012) The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 1462, 61–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Watts G. D. J., Wymer J., Kovach M. J., Mehta S. G., Mumm S., Darvish D., Pestronk A., Whyte M. P., and Kimonis V.E. (2004) Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 36, 377–381 [DOI] [PubMed] [Google Scholar]
- 19.Johnson J. O., Mandrioli J., Benatar M., Abramzon Y., Van Deerlin V. M., Trojanowski J. Q., Gibbs J. R., Brunetti M., Gronka S., Wuu J., Ding J., McCluskey L., Martinez-Lage M., Falcone D., Hernandez D. G., Arepalli S., Chong S., Schymick J. C., Rothstein J., Landi F., Wang Y.-D., Calvo A., Mora G., Sabatelli M., Monsurrò M. R., Battistini S., Salvi F., Spataro R., Sola P., Borghero G., The ITALSGEN Consortium, Galassi G., Scholz S. W., Taylor J. P., Restagno G., Chiò A., and Traynor B. J. (2010) Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68, 857–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buchan J. R., Kolaitis R.-M., Taylor J. P., and Parker R. (2013) Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 153, 1461–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamamoto S., Tomita Y., Hoshida Y., Iizuka N., Monden M., Yamamoto S., Iuchi K., and Aozasa K. (2004) Expression level of valosin-containing protein (p97) is correlated with progression and prognosis of non-small-cell lung carcinoma. Ann. Surg. Oncol. 11, 697–704 [DOI] [PubMed] [Google Scholar]
- 22.Tsujimoto Y., Tomita Y., Hoshida Y., Kono T., Oka T., Yamamoto S., Nonomura N., Okuyama A., and Aozasa K. (2004) Elevated expression of valosin-containing protein (p97) is associated with poor prognosis of prostate cancer. Clin. Cancer Res. 10, 3007–3012 [DOI] [PubMed] [Google Scholar]
- 23.Laguë M. N., Romieu-Mourez R., Bonneil É., Boyer A., Pouletty N., Mes-Masson A.-M., Thibault P., Nadeau M.-È., and Boerboom D. (2012). Proteomic profiling of a mouse model for ovarian granulosa cell tumor identifies VCP as a highly sensitive serum tumor marker in several human cancers. PLoS One 7, e42470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi K. S. (2012) Autophagy and cancer. Exp. Mol. Med. 44, 109–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kimmelman A. C. (2011) The dynamic nature of autophagy in cancer. Gene Dev. 25, 1999–2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen S., Rehman S. K., Zhang W., Wen A., Yao L., and Zhang J. (2010) Autophagy is a therapeutic target in anticancer drug resistance. Biochim. Biophys. Acta 1806, 220–229 [DOI] [PubMed] [Google Scholar]
- 27.White E. (2012) Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 12, 401–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iliuk A. B., Martin V. A., Alicie B. M., Geahlen R. L., and Tao W.A. (2010) In-depth analyses of kinase-dependent tyrosine phosphoproteomes based on metal ion-functionalized soluble nanopolymers. Mol. Cell. Proteomics 9, 2162–2172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Galan J. A., Paris L. L., Zhang H.-J., Adler J., Geahlen R. L., and Tao W. A. (2011) Identification of Syk-interacting proteins using a novel amine-specific isotope tag and GFP nanotrap. J. Am. Soc. Mass Spectrom. 22, 319–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xue L., Wang W.-H., Iliuk A., Hu L., Galan J. A., Yu S., Hans M., Geahlen R. L., and Tao W. A. (2012) Sensitive kinase assay linked with phosphoproteomics for identifying direct kinase substrates. Proc. Natl. Acad. Sci. U.S.A. 109, 5615–5620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mazroui R., Di Marco S., Kaufman R. J., and Gallouzi I. E. (2007) Inhibition of the ubiquitin-proteasome system induces stress granule formation. Mol. Biol. Cell 18, 2603–2618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oh H., Ozkirimli E., Shah K., Harrison M. L., and Geahlen R. L. (2007) Generation of an analog-sensitive Syk tyrosine kinase for the study of signaling dynamics from the B cell antigen receptor. J. Biol. Chem. 282, 33760–33768 [DOI] [PubMed] [Google Scholar]
- 33.Zhou Q., and Geahlen R. L. (2009) Interaction of the protein tyrosine kinase Syk and TRAF-interacting protein (TRIP) in the tumor necrosis factor (TNF)-mediated survival pathway in breast epithelial cells. Oncogene 28, 1348–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou F., Hu J., Ma H., Harrison M. L., and Geahlen R. L. (2006) Nucleocytoplasmic trafficking of the Syk protein-tyrosine kinase. Mol. Cell. Biol. 26, 3478–3491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Machida K., Thompson C. M., Dierck K., Jablonowski K., Kärkkäinen S., Liu B., Zhang H., Nash P. D., Newman D. K., Nollau P., Pawson T., Renkema G. H., Saksela K., Schiller M. R., Shin D. G., and Mayer B. J. (2007) High-throughput phosphotyrosine profiling using SH2 domains. Mol. Cell 26, 899–915 [DOI] [PubMed] [Google Scholar]
- 36.Kedersha N., Cho M. R., Li W., Yacono P. W., Chen S., Gilks N., Golan D. E., and Anderson P. (2000) Dynamic shuttling of Tia-1 accompanies the recruitment of mRNA to mammalian stress granules. J. Cell Biol. 151, 1257–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kwon S., Zhang Y., and Matthias P. (2007) The deacetylase HDAC6 is a novel critical component of stress granules involved in the stress response. Genes Dev. 21, 3381–3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bishop A. C., Ubersax J. A., Petsch D. T., Matheos D. P., Gray N. S., Blethrow J., Shimizu E., Tsien J. Z., Schultz P. G., Rose M. D., Wood J. L., Morgan D. O., and Shokat K. M. (2000) A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 407, 395–401 [DOI] [PubMed] [Google Scholar]
- 39.Keshvara L. M., Isaacson C. C., Yankee T. M., Sarac R., Harrison M. L., and Geahlen R. L. (1998) Syk- and Lyn-dependent phosphorylation of Syk on multiple tyrosines following B-cell activation includes a site that negatively regulates signaling. J. Immunol. 161, 5276–5283 [PubMed] [Google Scholar]
- 40.Tsai N.-P., Ho P.-C., and Wei L.-N. (2008) Regulation of stress granule dynamics by Grb7 and FAK signalling pathway. EMBO J. 27, 715–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Groesch T. D., Zhou F., Mattila S., Geahlen R. L., and Post C. B. (2006) Structural basis for the requirement of two phosphotyrosine residues in signaling mediated by Syk tyrosine kinase. J. Mol. Biol. 356, 1222–1236 [DOI] [PubMed] [Google Scholar]
- 42.Levin S. E., Zhang C., Kadlecek T. A., Shokat K. M., and Weiss A. (2008) Inhibition of ZAP-70 kinase activity via an analog-sensitive allele blocks T cell receptor and CD28 superagonist signaling. J. Biol. Chem. 283, 15419–15430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., and Yoshimori T. (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen C.-H., Martin V. A., Gorenstein N. M., Geahlen R. L., and Post C. B. (2011) Two closely-spaced tyrosines regulate NFAT signaling in B cells via Syk association with Vav. Mol. Cell. Biol. 31, 2984–2996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Porter C. J., Matthews J. M., Mackay J. P., Pursglove S. E., Schmidberger J. W., Leedman P. J., Pero S. C., Krag D. N., Wilce M. C. J., and Wilce J. A. (2007) Grb7 SH2 domain structure and interactions with a cyclic peptide inhibtor of cancer cell migration and proliferation. BMC Struct. Biol. 7, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Porter C. J., Wilce M. C. J., Mackay J. P., Leedman P., and Wilce J. A. (2005) Grb7-SH2 domain dimerisation is affected by a single point mutation. Eur. Biophys. J. 34, 454–460 [DOI] [PubMed] [Google Scholar]
- 47.Oligino L., Lung F. D., Sastry L., Bigelow J., Cao T., Curran M., Burke T. R. Jr., Wang S., Krag D., Roller P. P., and King C. R. (1997) Nonphosphorylated peptide ligands for the Grb2 Src homology 2 domain. J. Biol. Chem. 272, 29046–29052 [DOI] [PubMed] [Google Scholar]
- 48.Hart C. P., Martin J. E., Reed M. A., Keval A. A., Pustelnik M. J., Northrop J. P., Patel D. V., and Grove J. R. (1999) Potent inhibitory ligands of the GRB2 SH2 domain from recombinant peptide libraries. Cell. Signal. 11, 453–464 [DOI] [PubMed] [Google Scholar]
- 49.Fumagalli S., Totty N. F., Hsuan J. J., and Courtneidge S. A. (1994) A target for Src in mitosis. Nature 368, 871–874 [DOI] [PubMed] [Google Scholar]
- 50.Baudot A. D., Jeandel P. Y., Mouska X., Maurer U., Tartare-Deckert S., Raynaud S. D., Cassuto J. P., Ticchioni M., and Deckert M. (2009) The tyrosine kinase Syk regulates the survival of chronic lymphocytic leukemia B cells through PKCδ and proteasome-dependent regulation of Mcl-1 expression. Oncogene 28, 3261–3273 [DOI] [PubMed] [Google Scholar]
- 51.Chen L., Monti S., Juszczynski P., Ouyang J., Chapuy B., Neuberg D., Doench J. G., Bogusz A. M., Habermann T. M., Dogan A., Witzig T. E., Kutok J. L., Rodig S. J., Golub T., and Shipp M. A. (2013) SYK inhibition modulates distinct PI3K/AKT-dependent survival pathways and cholesterol biosynthesis in diffuse large B cell lymphomas. Cancer Cell 23, 826–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Caldwell R. G., Wilson J. B., Anderson S. J., and Longnecker R. (1998) Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity 9, 405–411 [DOI] [PubMed] [Google Scholar]
- 53.Carnevale J., Ross L., Puissant A., Banerji V., Stone R. M., DeAngelo D. J., Ross K. N., and Stegmaier K. (2013) SYK regulates mTOR signaling in AML. Leukemia 27, 2118–2128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oellerich T., Oellerich M. F., Engelke M., Münch S., Mohr S., Nimz M., Hsiao H. H., Corso J., Zhang J., Bohnenberger H., Berg T., Rieger M. A., Wienands J., Bug G., Brandts C., Urlaub H., and Serve H. (2013) β2 integrin-derived signals induce cell survival and proliferation of AML blasts by activating a Syk/STAT signaling axis. Blood 121, 3889–3899, S1–S66 [DOI] [PubMed] [Google Scholar]
- 55.Kanie T., Abe A., Matsuda T., Kuno Y., Towatari M., Yamamoto T., Saito H., Emi N., and Naoe T. (2004) TEL-Syk fusion constitutively activates PI3-K/Akt, MAPK and JAK2-independent STAT5 signal pathways. Leukemia 18, 548–555 [DOI] [PubMed] [Google Scholar]
- 56.Uckun F. M., Qazi S., Ma H., Tuel-Ahlgren L., and Ozer Z. (2010) STAT3 is a substrate of SYK tyrosine kinase in B-lineage leukemia/lymphoma cells exposed to oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 107, 2902–2907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maycotte P., Gearheart C. M., Barnard R., Aryal S., Mulcahy Levy J. M., Fosmire S. P., Hansen R. J., Morgan M. J., Porter C. C., Gustafson D. L., and Thorburn A. (2014) STAT3-mediated autophagy dependence identifies subtypes of breast cancer where autophagy inhibition can be efficacious. Cancer Res. 74, 2579–2590 [DOI] [PMC free article] [PubMed] [Google Scholar]