

Background: Mutations in the cAMP-binding domains of the regulatory subunit of PKA (PRKAR1A) can cause either acrodysostosis or Carney complex.
Results: All mutations impaired cAMP binding, but those resulting in Carney complex had additional effects that increased PKA activity.
Conclusion: A single mechanism explains acrodysostosis, whereas diverse mechanisms cause Carney complex.
Significance: This is the first functional characterization of a series of PRKAR1A missense mutations causing acrodysostosis.
Keywords: bioluminescence resonance energy transfer (BRET), cell signaling, functional genomics, protein kinase A (PKA), site-directed mutagenesis, Acrodysostosis, CRE-luciferase, Carney complex, PRKAR1A
Abstract
The main target of cAMP is PKA, the main regulatory subunit of which (PRKAR1A) presents mutations in two genetic disorders: acrodysostosis and Carney complex. In addition to the initial recurrent mutation (R368X) of the PRKAR1A gene, several missense and nonsense mutations have been observed recently in acrodysostosis with hormonal resistance. These mutations are located in one of the two cAMP-binding domains of the protein, and their functional characterization is presented here. Expression of each of the PRKAR1A mutants results in a reduction of forskolin-induced PKA activation (measured by a reporter assay) and an impaired ability of cAMP to dissociate PRKAR1A from the catalytic PKA subunits by BRET assay. Modeling studies and sensitivity to cAMP analogs specific for domain A (8-piperidinoadenosine 3′,5′-cyclic monophosphate) or domain B (8-(6-aminohexyl)aminoadenosine-3′,5′-cyclic monophosphate) indicate that the mutations impair cAMP binding locally in the domain containing the mutation. Interestingly, two of these mutations affect amino acids for which alternative amino acid substitutions have been reported to cause the Carney complex phenotype. To decipher the molecular mechanism through which homologous substitutions can produce such strikingly different clinical phenotypes, we studied these mutations using the same approaches. Interestingly, the Carney mutants also demonstrated resistance to cAMP, but they expressed additional functional defects, including accelerated PRKAR1A protein degradation. These data demonstrate that a cAMP binding defect is the common molecular mechanism for resistance of PKA activation in acrodysosotosis and that several distinct mechanisms lead to constitutive PKA activation in Carney complex.
Introduction
Many hormones and factors act on cells via specific G-protein-coupled receptors to activate Gs proteins and adenylyl cyclases and result in the production of cAMP (1–3). The most studied target of cAMP is the cAMP-dependent protein kinase (PKA) (2). When in the inactive state, this serine/threonine kinase is composed of two catalytic subunits associated with two regulatory subunits. Binding of cAMP to the regulatory subunits results in the dissociation of catalytic subunits from the tetrameric complex and their activation (4). PRKAR1A is the most abundant and ubiquitous of the four known regulatory subunits (5). This 381-amino acid protein encompasses four identified domains, ordered as follows from the N terminus: the dimerization domain, an “inhibitor sequence” that interacts with the catalytic subunit, cAMP-binding domain A, and cAMP-binding domain B (6).
Inactivating mutations of PRKAR1A have long been recognized as a genetic cause of Carney complex, a multiple endocrine neoplasia syndrome associating pigmented micronodular adrenal hyperplasia, cardiac myxoma, lentiginosis, pituitary tumors, and other abnormalities (7, 8). Most of the mutations are nonsense or insertion/deletion mutations resulting in nonsense-mediated decay of the mRNA and the absence of protein translation (9). The haploinsufficiency results in constitutive activation of PKA as the consequence of increased concentrations of free catalytic subunits. In a very limited number of cases, Carney complex is associated with a missense mutation in PRKAR1A, some occurring in the cAMP-binding domains. Studies evaluating the functional consequences of these missense mutations indicated that, besides haploinsufficiency, altered PRKAR1A function can elevate PKA activity (10).
Recently, we identified a unique mutation in the gene coding PRKAR1A in three unrelated patients with acrodysostosis, a heterogeneous group of rare skeletal dysplasia (11). The patients also presented resistance to several hormones, including PTH3 and TSH, and were defined as having acrodysostosis type 1. The mutation was a nonsense mutation of the last exon of PRKAR1A, producing a truncated protein with a partial deletion of cAMP-binding domain B. Functional characterization of this mutant protein demonstrated that it associated normally with the catalytic subunits, but an alteration of the dissociation of the subunits by cAMP was observed as the consequence of a reduced affinity of the PRKAR1A mutant for cAMP binding (11). Subsequently, other mutations of PRKAR1A have been identified in patients with acrodysostosis, including both missense and nonsense mutations, all of which were localized in one of the two cAMP-binding domains of the protein (12–16). All reported patients with PRKAR1A mutations presented hormonal resistance, in contrast to patients affected with acrodysostosis type 2 (caused by PDE4D mutations) (17). Assessment of the functional consequences of recently described PRKAR1A mutations has not been reported. Interestingly, two of these mutations involve amino acids (Ala-213 and Gly-289) for which alternative amino acid substitutions have been reported to cause the Carney complex phenotype (10). The reason why these alternative substitutions at the same site produce such strikingly different phenotypes has also not been previously explored.
To address this issue, we have evaluated the functional characteristics of PRKAR1A regulatory subunits carrying eight different mutations identified in patients with acrodysostosis and compared the results with those obtained for the two alternative mutations involved in the Carney complex. Using a CRE-luciferase expression system, we have assessed the ability of cAMP to induce PKA-dependent protein expression. Using BRET techniques, we quantified the ability of regulatory subunits to bind to catalytic subunits in the absence of cAMP and to dissociate from the catalytic subunits following exposure to cAMP analogs. For selected mutants, we evaluated degradation rates using luciferase-PRKAR1A fusion proteins. Finally, we sought correlations between the functional defects observed and selected clinical characteristics of the patients.
Materials and Methods
PRKAR1A Mutations and Patients
Eight PRKAR1A mutations causing acrodysostosis with hormonal resistance and two causing the Carney complex were studied. Six acrodysostosis PRKAR1A mutations, all localized in cAMP-binding domain B, were previously identified and reported by us together with the patients' clinical, biochemical, and skeletal phenotype (11, 13) (Q285R, G289E, A328V, R335L, the recurrent R368X mutation, and Q372X). The mutation A213T, localized in cAMP-binding domain A, was reported by Muhn et al. (15). The mutation Y175C (c.524A>G), also localized in the cAMP-binding domain A, has not been reported previously. The affected patient was referred at the age of 9 years for evaluation of brachydactyly. She presented with bilateral brachymetacarpy and metatarsy affecting the IV and V metacarpal bones and the II, III, IV, and V metatarsal bones. Cone-shaped epiphyses and chunky bones were noted, but no widening of the lumbar interpediculate distance or ectopic ossification was observed. Her height was 1 m 30 cm (+1 S.D. relative to the general population), and her weight was 30 kg. Biochemical and endocrine studies revealed increased serum PTH (106 pg/ml; normal range, 10–65 pg/ml), increased serum TSH (8.1 μIU/ml; normal, <5 μIU), normal T4 (14 pmol/liter; normal range, 10–18 pmol/liter), normocalcemia (2.33 mmol/liter), slightly increased phosphatemia (1.6 mmol/liter; normal range, 1–1.5 mmol/liter), and urinary cAMP at the upper limit of the normal range (0.95 μmol/mmol creatinine; normal range, 0.3–0.99 μmol/mmol). The parents showed neither brachydactyly or abnormal endocrine functions. Informed written consent was obtained from the patient's parents. Analysis of the GNAS locus, including sequencing of GNAS exons 1–13 and evaluation of methylation at the four differentially methylated regions, gave normal results (data not shown), ruling out abnormalities in the expression or function of the Gαs protein coded by this locus, which can produce similar developmental and skeletal features (pseudohypoparathyroidism type 1a). Exons and intron-exon junctions for the PRKAR1A gene (NM_002734) (12 exons) were amplified using intronic primers, and PCR products were analyzed by direct nucleotide sequence analysis as described previously (11). This mutation was not seen in the mother (the father's DNA was not available) or in 200 controls and has not been reported in available databases (Single Nucleotide Polymorphism Performance Database/National Center for Biotechnology Information, 1000 genomes, exon variant servers, Human Genome Mutation Database, PRKAR1A mutation database). Correlations between clinical and molecular phenotype were performed using the clinical data reported previously (11, 13) for the Q285R, G289E, A328V, R335L, Q372X, and R368X mutations, described above for the Y175C mutation, and reported by Muhn et al. (15) for the A213T mutation. The two homologous mutations that result in the Carney complex, G289W and A213D, were reported by Greene et al. (10).
Constructions and Cell Culture
As described previously (11), the entire cDNA coding sequences of the human PRKAR1A and the mouse PRKACA genes were obtained by RT-PCR and subcloned in either pCDNA3.1 (Invitrogen) or pRLucN2(h) (Biosignal, PerkinElmer Life Sciences) plasmids for PRKAR1A and pYFP-C3 (Clontech/BD Bioscience) for PRKACA. Site-directed mutagenesis introducing the eight mutations associated with acrodysostosis and the two mutations associated with Carney complex was performed using the QuikChange site-directed mutagenesis kit (Stratagene) on both pCDNA-PRKAR1A and pR-PRKAR1A-Luc plasmids. The list of oligonucleotides used for PCR and site-directed mutagenesis is available upon request. All of the constructions were verified by sequencing of the entire coding sequence and plasmid promoter.
These plasmids were transfected alone or in combination in HEK293 cells (ATCC) using Effectene according to the manufacturer's instructions (Qiagen). Two days later, the transfected cells were either lysed using passive lysis buffer (Promega) and frozen or used to perform the experiments described below.
Western Blotting
Western blotting was performed using cell lysates obtained from HEK293 cells transfected with the empty plasmid or with the different PRKAR1A mutants subcloned in pCDNA3.1. Proteins were solubilized in MOSLB buffer (50 mm sodium pyrophosphate, 50 mm NaF, 50 mm NaCl, 5 mm EDTA, 5 mm EGTA, 100 mm NaVO4, 10 mm Hepes, pH 7.4, and 0.1% Triton X-100) with Complete miniprotease inhibitor mixture tablets (catalog no. 11836153001, Roche Applied Science). 10–40 μg of protein in Laemmli buffer were electrophoresed into a 10% acrylamide gel and then transferred onto a 0.2-μm nitrocellulose membrane (catalog no. BA83, GE Healthcare), incubated with anti-PKA(R1) monoclonal antibody (1:500; catalog no. 610166, Transduction Laboratories) or GAPDH polyclonal antibody (1:1000; catalog no. Ab37168, Abcam) in Tris-buffered saline/TweenR 20 (TBST: 137 mm NaCl, 20 mm Tris, 0.1% Tween 20, pH 7.6) + 5% nonfat milk. Secondary anti-mouse (1:5000) and anti-rabbit (1:10,000) antibodies were incubated also in TBST + 5% nonfat milk, and bands were revealed using an ECL kit (Super Signal, West Pico, chemiluminescence substrate, Thermo Scientific, catalog no. 34080).
PKA-induced Transcriptional Activity
Transcriptional PKAactivity was measured as described previously (11). Briefly, HEK293 cells were co-transfected with 225 ng of a pCDNA3.1 construct encoding WT PRKAR1A or the various mutants; 250 ng of the pCRE-Luc plasmid (Stratagene), containing a firefly luciferase reporter gene under control of a cAMP-responsive element (CRE); and 25 ng of the pRL-TK plasmid (Promega), containing a constitutively expressed Renilla luciferase reporter gene, which served as an internal control for transfection efficiency. Cells were either untreated (basal PKA-induced transcriptional activity) or treated with 0.3, 1, 5, or 10 μm forskolin (Sigma-Aldrich) in the presence of 1 mm 3-isobutyl-1-methylxanthine (Sigma-Aldrich). Luciferase activity was determined 6 h after the addition of forskolin as described previously (11), using a Centro LB 960 luminometer (Berthold). For each culture, the ratio of firefly luciferase activity to Renilla luciferase activity was calculated to correct for transfection efficiency. Results were expressed as the -fold increase in CRE-luciferase activity relative to 3-isobutyl-1-methylxanthine-treated cultures not stimulated with forskolin. To evaluate the EC50 for forskolin stimulation, pooled data for each mutant relating the forskolin dose and the increase in CRE-luciferase activity were fitted to a sigmoid curve with variable slope.
Interactions of PRKAR1A Mutants with Catalytic Subunits Using BRET Technique
The apparent affinity of wild type and the different mutant PRKAR1A subunits for the PRKACA subunit was evaluated by BRET. In each experiment, a fixed amount of PRKAR1A-luciferase plasmid (which varied from 1 to 5 ng in different experiments and for the different mutants) was transfected in HEK293 cells (6-well plates) in association or not with increasing amounts of the YFP-PRKACA plasmid (1–100 ng) as described (11, 18). For each transfection point, the luciferase, YFP, and BRET signals were measured using a Mithras LB 940 multimode reader (Berthold). The results are expressed as the percentage of maximal BRET signal for a given ratio of YFP/luciferase. The specific interaction between the regulatory and catalytic PKA subunits is identified by a hyperbolic increase of the BRET signal up to a plateau corresponding to the saturation of all donor molecules. BRET50 corresponds to the binding of 50% of BRET donor molecules and represents an indirect quantitative measurement of the affinity between the two subunits (19).
Dissociation of PKA Interactions by cAMP Analogs Using BRET Technique
The capacity of cAMP analogs to dissociate the wild type or mutant PRKAR1A-PRKACA complex was also tested using BRET. In each experiment, a fixed amount of PRKAR1A-luciferase plasmid (which varied from 20 to 30 ng in different experiments and for the different mutants) was used to transfect cells in a 60% confluent T-75 flask with or without a saturating amount of YFP-PRKACA plasmid (100–250 ng). The BRET signal in the absence of YFP-PRKACA is considered 0 and 100% in the presence of YFP-PRKACA without cAMP analog. Increasing concentrations of cAMP analogs (10−10 to 10−5 m) reduce the maximal BRET signal, resulting in a sigmoid dose-response curve whose EC50 is proportional to the PRKAR1A affinity for this analog. The three tested analogs were 8-bromo-cAMP, known to bind with the same affinity to the A and B domains of PRKAR1A; 8-PIP-cAMP, binding preferentially to the A domain; and 8-AHA-cAMP, binding preferentially to the B domain (20, 21).
Degradation of PRKAR1A Mutants
Analysis of the PRKAR1A mutant degradation takes advantage of reliable and accurate measurement of the PRKAR1A-luciferase recombinant protein. Therefore, low amounts of this protein can be expressed, and its degradation can be analyzed, avoiding the artifacts of overexpression. The degradation rate of the untagged WT PRKAR1A evaluated by Western blotting and that of WT PRKAR1A-luciferase evaluated by luminescence gave concordant results (see Fig. 8C). For each mutant, 10–30 ng of plasmid was used to transfect HEK293 cells in a 60% confluent T-75 flask. Twenty-four hours after transfection, the cells were trypsinized and seeded in 6-well plates. Twenty-four hours later, cycloheximide (20 μg/ml) was added to half of the wells in order to block protein synthesis. Cells, treated or not with cycloheximide, were lysed 0, 3, 6, 9, and 20 h after the addition of cycloheximide, and the luciferase signal was measured in the presence of coelenterazine using a Mithras LB 940 multimode reader (Berthold). Each experiment was done in duplicate and repeated at least three times for each mutant. Degradation was calculated as the reduction of the luminescent signal in the presence of cycloheximide compared with the luminescent signal at t = 0.
FIGURE 8.

Effect of missense mutations in the same amino acids of PRKAR1A causing either Carney complex or acrodysostosis on the degradation of PRKAR1A. HEK293 cells were transfected with PRKAR1A-luciferase constructs, plated in duplicate wells, and 24 h later, cycloheximide was added to half of the wells. At the indicated times after the addition of cycloheximide, luciferase activity was measured by luminometry. Results, expressed as the percentage of the time 0 fluorescence, are the mean ± S.E. (error bars) for at least three independent experiments, each performed in duplicate. Shown are findings for WT PRKAR1A (solid red lines) and PRKAR1A carrying mutations causing Carney complex (solid lines) and acrodysostosis (dotted lines) for the indicated mutations affecting Ala-213 (A) and Gly-289 (B). C, comparison of WT PRKAR1A degradation using Western blot or luminometry. HEK293 cells were transfected with WT PRKAR1A or PRKAR1A-luciferase constructs and plated in duplicate wells, and 24 h later, cycloheximide was added. At the indicated times after the addition of cycloheximide, the PRKAR1A protein was analyzed by Western blot (blue curve), or luciferase activity was measured by luminometry (red curve). Results, expressed as the percentage of results at time 0, are the mean ± S.E. for 3–5 independent experiments (unpaired Student's t test; *, p < 0.05; **, p < 0.01; ***, p < 0.001).
Three-dimensional Modeling of PRKAR1A Mutants
Human PRKAR1A was modeled as described previously (11). Molecular dynamic simulations using the CHARMM29 force field were applied on human WT and mutant PRKAR1A three-dimensional models in VEGAZZ software (Drug Design Laboratory, Milano, Italy) and NAMD software (University of Illinois, Urbana, IL) for 1 ns to check general reasonability of the modeled structure (22). The low frequency normal modes of human WT and mutant PRKAR1A three-dimensional models were performed in elNémo software (Université de Nantes, Nantes, France). In order to establish the positioning of cAMP in the binding pocket of domain A or domain B of human PRKAR1A mutants, cAMP was docked into human WT PRKAR1A domain A or B according to the structural data from the x-ray structure of bovine PRKAR1A (23). The positioning of cAMP was then refined by calculation using Autodock version 4.0 software (The Scripps Research Institute, La Jolla, CA). Docked cAMP in domain A or B of WT and mutant PRKAR1A was submitted to energy minimization using the MMFF94 force field. The r.m.s. deviation (ligand) and binding pocket surface of the mutant PRKAR1As were estimated and compared with values obtained for WT PRKAR1A. Positioning of the side chains of substituted residues in single mutants was determined using Chimera software (University of California, San Francisco, CA) and the Dunbrack rotamer library. Analysis and representation of ligand conformation in the domain A and B binding pockets of PRKAR1A was performed using Chimera software.
Statistical Analysis
All results are expressed as mean ± S.D. unless otherwise indicated. For studies evaluating CRE-luciferase activity, the activity in cells expressing some PRKAR1A mutants was still increasing exponentially at 10 μm forskolin, the highest dose tested, and the EC50 could not be accurately determined. In these cases, the EC50 is reported as “>5 μm” and was evaluated using the Kruskal-Wallace test as implemented in R (version 3.1.2). Post-test comparisons, performed only if p was <0.05, were made using the nonparametric Wilcoxon matched pairs test. -Fold stimulation was evaluated by analysis of variance. Post-test comparisons, performed only if p was <0.05; were made by pairwise t test. Data from BRET analysis was evaluated by analysis of variance. Post-test comparisons, performed only if p was <0.05, were made using the paired t test with unequal variance. Clinical parameters in patient groups were compared using the Mann-Whitney test.
Results
The functional consequences of eight PRKAR1A mutations resulting in acrodysostosis, comprising six novel missense mutations, one novel nonsense mutation (12–16), and the initially described R368X mutation (11), were characterized on the cAMP/PKA signaling pathway.
Expression of the PRKAR1A Mutants
Following overexpression in HEK293 cells, all of the PRKAR1A mutants were easily identifiable by Western blotting (Fig. 1). Most of the mutants had an apparent molecular mass of 48 kDa, comparable with the WT PRKAR1A protein and with the expected size of the protein. The two nonsense mutants (R368X and Q372X) produced truncated proteins with a reduced mass. Interestingly, two of the missense mutants had either a slightly increased (G289E) or reduced (Q285R) apparent molecular mass, suggesting a change in the molecular structure that had been preserved during SDS-PAGE.
FIGURE 1.

Expression of mutant PRKAR1A subunits analyzed by Western blotting. Cell lysates were prepared from HEK293 cells transfected with plasmids, resulting in the expression of WT PRKAR1A or PRKAR1A carrying the indicated mutations (A, WT, R368X, G289E, A328V, R335L, Q372X, and Q285R; B, WT, D183Y, A213D, and G289W; C, WT, Y175C, and A213T). Soluble proteins were electrophoresed into 10% SDS-polyacrylamide gels and transferred to nitrocellulose membranes, and PRKAR1A (top gels) and GAPDH (bottom gels) were detected by chemiluminescence after sequential treatment of the membranes with specific primary antibodies, peroxidase-conjugated secondary antibodies, and ECL substrate. The arrows show the migration of proteins of the indicated molecular mass (kDa).
Acrodysostosis PRKAR1A Mutants Exhibit Reduced PKA Activity
We first analyzed the PKA-induced transcriptional activity in HEK293 cells overexpressing WT PRKAR1A or the eight mutant PKA subunits following forskolin stimulation using a CRE-luciferase reporter system. In the absence of forskolin, luciferase activity was low in transfected cells and not significantly different from that of cells transfected with an empty vector (data not shown).
The dose-response curves for cells expressing the mutant PRKAR1A subunits were all shifted to the right compared with that observed for cells expressing the WT PRKAR1A subunit (Fig. 2A). Following stimulation with low doses of forskolin (0.3 and 1 μm), the -fold increase in CRE-luciferase activity was reduced in cells transfected with the mutant PRKAR1A subunits compared with that of cells transfected with WT PRKAR1A (Fig. 2B). At higher doses of forskolin, CRE-luciferase activity for cells expressing many of the mutants increased to levels that approached that seen for cells overexpressing wild-type PRKAR1A. Only cells overexpressing the Q372X, R368X, and R335L mutants continued to manifest significant reductions in CRE-luciferase activity following stimulation with both 5 and 10 μm forskolin (data not shown). Taken together, these findings suggest that all of the mutant regulatory subunits are less sensitive to cAMP, resulting in an impaired activation of PKA. As observed previously by us and others, forskolin induced less stimulation of PKA in cells overexpressing wild-type PRKAR1A than in cells transfected with the empty vector, reflecting the inhibitory effect of PRKAR1A subunit overexpression on PKA activity (11, 24) (Fig. 2A).
FIGURE 2.

PRKAR1A mutations occurring in acrodysostosis impair PKA-induced transcriptional activity. A, HEK293 cells were co-transfected with plasmids encoding (i) WT PRKAR1A or the indicated mutants, (ii) firefly luciferase reporter gene under control of a cAMP-responsive element, and (iii) a constitutively expressed Renilla luciferase reporter gene. 48 h later, cells were either left untreated or exposed to the indicated doses of forskolin in the presence of 3-isobutyl-1-methylxanthine. Luciferase activity was determined 6 h after the addition of forskolin, as described previously (11), and expressed as -fold stimulation over basal. To evaluate the EC50 for forskolin stimulation, pooled data for each mutant relating the forskolin dose and the increase in CRE-luciferase activity were fitted to a sigmoid curve with variable slope (top panel). The median, range, and statistical analysis for data from the individual experiments are shown in the bottom panel. †, paired results for four experiments are required to perform the Wilcoxon matched pairs test; EC50 of these mutants was greater than EC50 of WT PRKAR1A in all experiments. B, histograms of the luciferase activity after stimulation with 0.3 and 1 μm forskolin. Results are presented as mean ± S.E. (error bars) for at least three independent experiments. Results were analyzed by analysis of variance; post-tests (performed only if p was <0.05) were made using the paired t test with unequal variance. *, p < 0.05; **, p < 0.01. Values for 5 μm forskolin were not significantly different by analysis of variance, and post-tests were not performed.
PKA Subunit Basal Interactions and Affinity Are Not Modified by Acrodysostosis Mutations
The interaction between the catalytic and regulatory subunits of PKA was analyzed using a BRET donor saturation assay, as described previously (11, 25).
As expected, the two WT PKA subunits interact specifically as shown by a hyperbolic curve (Fig. 3). Similar hyperbolic curves were observed for all mutants (Fig. 3), and the BRET50 values were not significantly different from that of WT (Table 1).
FIGURE 3.

Donor saturation assay analyzed by BRET for the WT and the seven mutated PRKAR1A variants associated with acrodysostosis. The specific interaction between the catalytic and regulatory subunits is identified by a hyberbolic increase of the BRET signal expressed as a percentage of maximal BRET (y) as a function of increases in the YFP/luciferase ratio (x).
TABLE 1.
Apparent affinity of wild type and the different mutant PRKAR1A subunits for the PRKACA subunit
n represents the number of experiments. p values are calculated by comparing the results of each mutant with the results for the wild type protein (unpaired Student's t test).
| BRET50 (mean ± S.E.) | n | p | |
|---|---|---|---|
| WT | 0.48 ± 0.08 | 5 | |
| Y175C | 0.40 ± 0.15 | 3 | 0.65 |
| A213T | 0.22 ± 0.11 | 3 | 0.11 |
| A213D | 0.40 ± 0.15 | 4 | 0.65 |
| Q285R | 0.48 ± 0.35 | 2 | 0.99 |
| G289E | 0.34 ± 0.17 | 4 | 0.45 |
| G289W | 0.51 ± 0.27 | 3 | 0.91 |
| A328V | 0.28 ± 0.18 | 2 | 0.30 |
| R335L | 0.53 ± 0.43 | 2 | 0.85 |
| R368X | 0.35 ± 0.14 | 5 | 0.46 |
| Q372X | 0.76 ± 0.19 | 2 | 0.17 |
Dissociation of PKA Subunits Induced by cAMP Analogs
Because the major role of cAMP is to dissociate the regulatory subunits from catalytic subunits of PKA, thereby liberating the enzymatically active subunits, it was interesting to analyze the dose-dependent reduction of the BRET signal using cAMP analogs. To refine the analysis of cAMP action, three different cAMP analogs were studied: 8-PIP-cAMP, 8-AHA-cAMP, and 8-bromo-cAMP, which interact more specifically with the PRKAR1A cAMP-binding domain A, domain B, and both domains, respectively (20, 21).
Using the nonspecific analog 8-bromo-cAMP, the dissociation of PRKAR1A subunits carrying mutations identified in acrodysostosis patients was significantly less efficient than that of WT PRKAR1A subunits (Fig. 4 and Table 2). The EC50 values for this analog were significantly increased for all of the mutant subunits. This result strongly suggests that the common molecular alteration of mutants in acrodysostosis is a reduction of cAMP binding, resulting in a reduced efficiency of cAMP-induced dissociation of the regulatory PKA subunits and activation of the catalytic PKA subunits.
FIGURE 4.
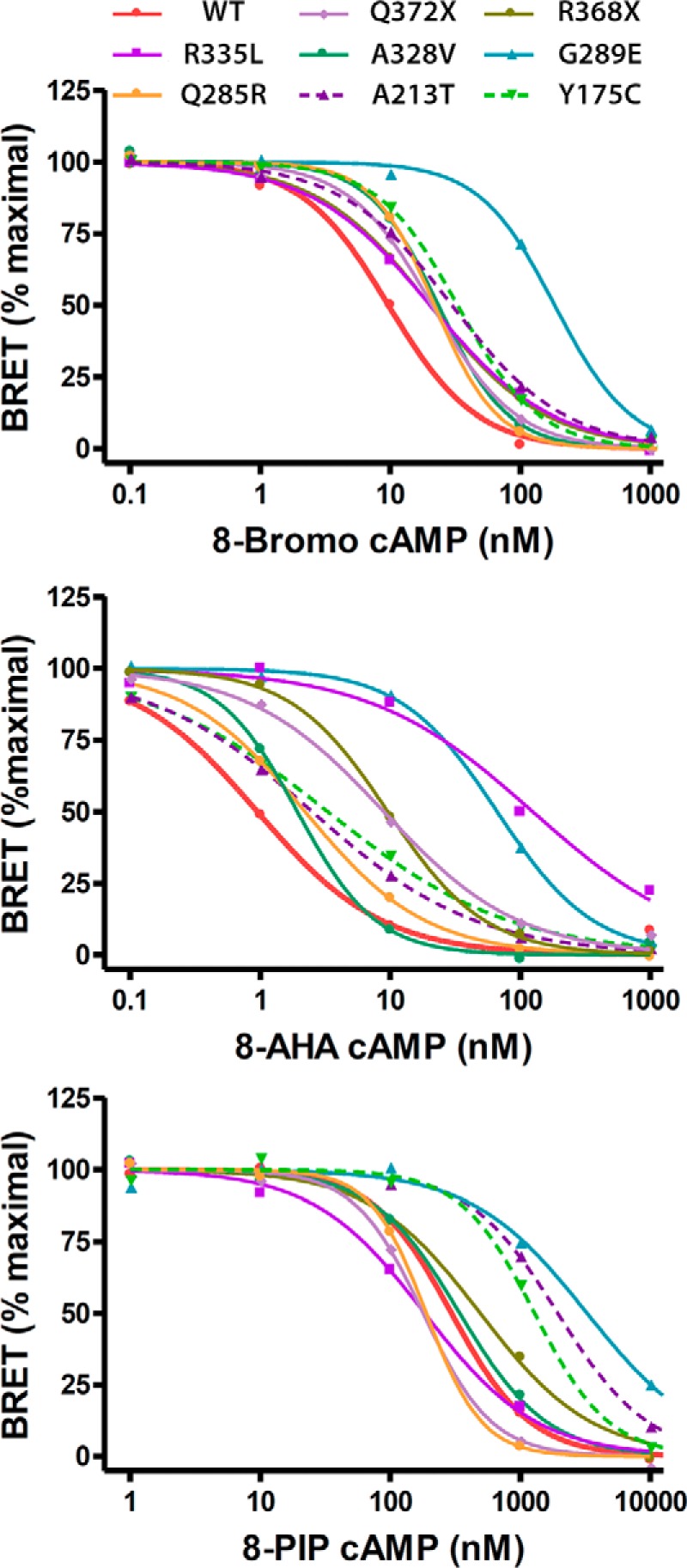
Effect of PRKAR1A mutations occurring in acrodysostosis on PKA regulatory and catalytic subunit dissociation. HEK293 cells were co-transfected with a fixed amount of plasmid, resulting in the expression of PRKAR1A-luciferase constructs carrying either the WT PRKAR1A sequence or the indicated mutations with or without a saturating amount of plasmid resulting in the expression of YFP-PRKACA. 48 h later, cells were lysed, and cell lysates were exposed or not to the indicated doses of 8-bromo-cAMP (top), 8-AHA-cAMP (middle), or 8-PIP-cAMP (bottom), and the BRET signal was measured as described under “Materials and Methods.” The signal in the absence of YFP-PRKACA was considered as 0%, and that in the presence of YFP-PRKACA without the cAMP analog was considered as 100%. To evaluate the EC50, pooled data for each mutant relating the cAMP analog concentration and the BRET signal were fitted to a sigmoid curve with variable slope. Values for variants with mutations in domain A are connected by dashed lines. The mean, S.D., and statistical analysis for data from the individual experiments are shown in Table 2.
TABLE 2.
Dissociation of PKA regulatory and catalytic subunits induced by cAMP analogs
| Mutant | 8-Bromo-cAMP |
8-AHA cAMP |
8-PIP cAMP |
||||||
|---|---|---|---|---|---|---|---|---|---|
| EC50 (mean ± S.D.) | n | p versus WT | EC50 (mean ± S.D.) | n | p versus WT | EC50 (mean ± S.D.) | n | p versus WT | |
| nm | nm | nm | |||||||
| WT | 9.0 ± 4.1 | 9 | 1.0 ± 0.4 | 7 | 300.3 ± 135.4 | 8 | |||
| Q372X | 21.2 ± 5.9 | 5 | 0.010 | 8.5 ± 3.2 | 5 | 0.010 | 176.5 ± 36.4 | 5 | 0.050 |
| R368X | 20.9 ± 5.5 | 4 | 0.023 | 12.1 ± 5.6 | 4 | 0.044 | 553.5 ± 182.3 | 3 | 0.177 |
| R335L | 21.1 ± 6.1 | 5 | 0.012 | 64.2 ± 21.9 | 4 | 0.015 | 732.2 ± 1242.0 | 5 | 0.526 |
| A328V | 23.6 ± 7.8 | 5 | 0.016 | 1.8 ± 0.4 | 4 | 0.017 | 374.5 ± 149.7 | 4 | 0.492 |
| G289E | 182.2 ± 41.3 | 6 | <0.001 | 67.7 ± 17.8 | 4 | 0.007 | 3415.1 ± 2091.4 | 7 | 0.011 |
| Q285R | 21.3 ± 5.7 | 5 | 0.008 | 2.4 ± 1.0 | 5 | 0.055 | 191.2 ± 80.6 | 5 | 0.122 |
| A213T | 31.5 ± 13.1 | 6 | 0.011 | 3.3 ± 2.5 | 5 | 0.136 | 1948.0 ± 387.7 | 4 | 0.004 |
| Y175C | 35.3 ± 11.7 | 6 | 0.003 | 2.8 ± 2.3 | 4 | 0.273 | 1303.8 ± 238.8 | 5 | <0.001 |
Except for one mutant (G289E), the results obtained using the cAMP analogs specific for cAMP-binding domain A or B indicated that mutations in a given cAMP-binding domain specifically affected the binding of cAMP to that domain. Thus, using 8-AHA-cAMP (B-domain analog), the PRKAR1A subunits carrying mutations in domain B, but not those with domain A mutations, showed significant (Q372X, R368X, R335L, A328V, G289E) or nearly significant (Q285R) increases in EC50 (Fig. 4 and Table 2). Conversely, using 8-PIP cAMP (A-domain analog), the two PRKAR1A subunits carrying mutations in domain A (Y175C and A213T) showed significant increases in EC50 using this analog (Fig. 4 and Table 2). Only the G289E mutation in domain B showed also a significant increase of 8-PIP-cAMP EC50.
Comparison of Molecular Phenotypes after Substitutions of Ala-213 and Gly-289 Causing either Acrodysostosis or Carney Complex
Among the few reported missense mutations in PRKAR1A causing the Carney complex, two (A213D and G289W) (10) affect amino acids for which alternative substitutions cause acrodysostosis (A213T and G289E). In an effort to understand how mutations of the same amino acid cause such strikingly different phenotypes, we compared the two sets of mutations using the techniques described above.
PKA-induced transcriptional activity was reduced in cells overexpressing the G289W Carney mutant, as was observed for cells overexpressing the G289E acrodysostosis mutant (Fig. 5). In contrast, the Carney-associated mutant A213D presented a specific pattern, with transcriptional activity comparable with the WT in cells treated with a low dose of forskolin (0.3 μm), but a profound decrease in PKA-induced transcriptional activity following stimulation with higher doses of forskolin (Fig. 5).
FIGURE 5.
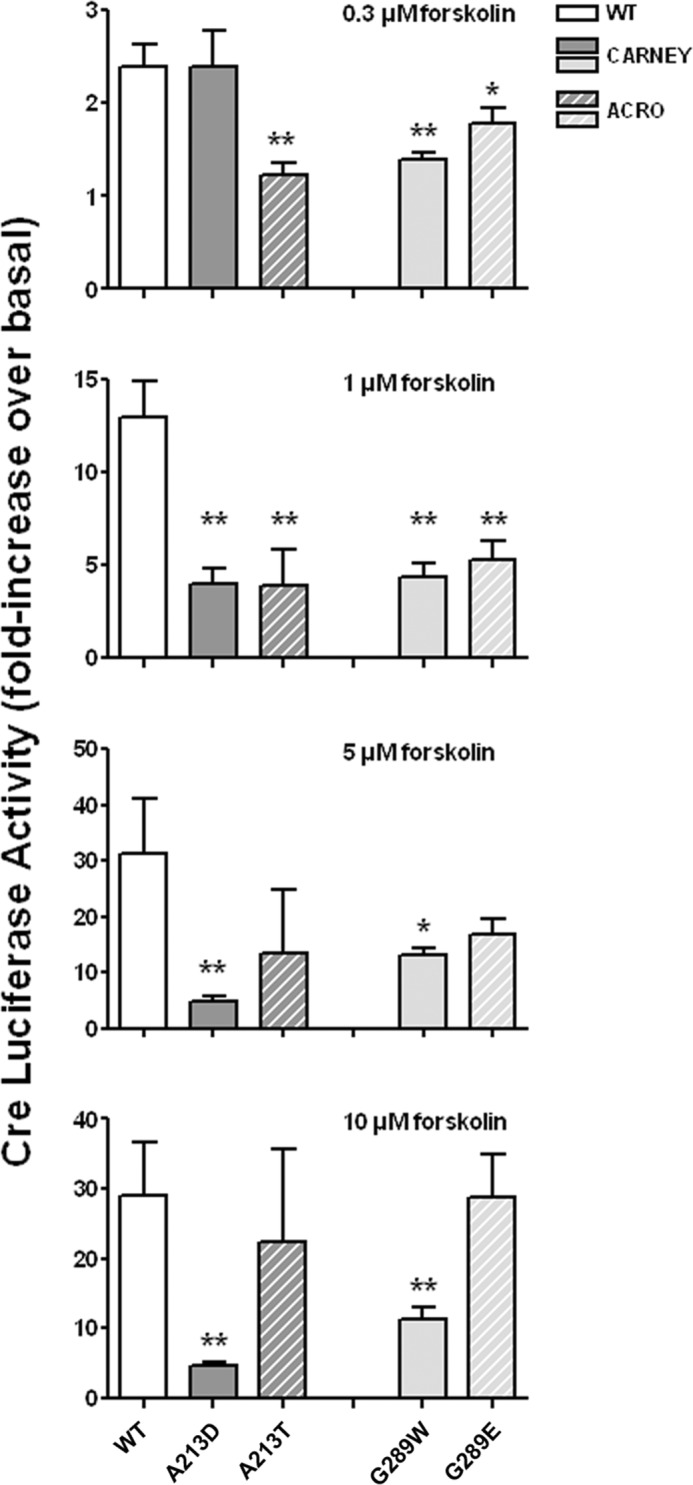
Comparison of cAMP-induced PKA transcriptional activity in cells expressing missense mutations of the same PRKAR1A amino acids causing either Carney complex or acrodysostosis. CRE-luciferase activity in HEK293 cells overexpressing WT or mutant PRKAR1A was measured as described in the legend of Fig. 2 and is expressed as the -fold increase over basal level observed following stimulation with the indicated doses of forskolin. Results (mean ± S.E. (error bars)) for mutations causing Carney complex (solid bars) and acrodysostosis (ACRO; hatched bars) are shown for mutations affecting Ala-213 (dark gray) and Gly-289 (light gray) and for WT PRKAR1A (solid white bars).
Using BRET, both Carney-associated mutants (A213D and G289W) demonstrated hyperbolic and saturable association curves with corresponding BRET50 values similar to those seen for WT PRKAR1A and A213T and G289E mutants causing acrodysostosis (Fig. 6 and Table 1).
FIGURE 6.

Donor saturation assay analyzed by BRET for the WT PRKAR1A and the variants carrying mutations at positions Ala-213 and Gly-289 associated with acrodysostosis and the Carney complex. The specific interaction between the catalytic and regulatory subunits is identified by a hyberbolic increase of the BRET expressed as a percentage of maximal BRET (y) as a function of increases in the YFP/luciferase ratio (x).
The dissociation of PKA subunits by cAMP analogs was also evaluated by BRET for the same four mutant regulatory subunits (Fig. 7). For the Carney-associated mutant G289W, the results show a significant impairment of subunit dissociation for all three cAMP analogs, similar to that observed for the acrodysostosis-associated mutant G289E, although the defect was somewhat less severe than for the G289E mutant. For the Carney-associated mutant A213D, a major defect in subunit dissociation was observed in response to high doses of 8-PIP-cAMP and 8-bromo-cAMP but not 8-AHA-cAMP, consistent with its localization within cAMP-binding domain A. The impairment in subunit dissociation seen at high doses of 8-PIP-cAMP and 8-bromo-cAMP was more severe than that observed for the acrodysosotosis-associated mutant A213T, similar to the greater impairment of PKA activation for A213D mutant compared with A213T for high doses of forskolin. Surprisingly, in cells treated with low doses (0.1–1 nm) of 8-bromo-cAMP, the dissociation of the A213D mutant seems to be greater than that of the WT PRKAR1A subunit. This observation raised the possibility that this mutant permits an increased, albeit low, level of dissociation of PRKAR1A from PRKACA in the presence of low concentrations of cAMP, but a greater extent of dissociation at higher cAMP concentrations is strongly impaired due to the effect of the mutation on the cAMP-binding domain A.
FIGURE 7.
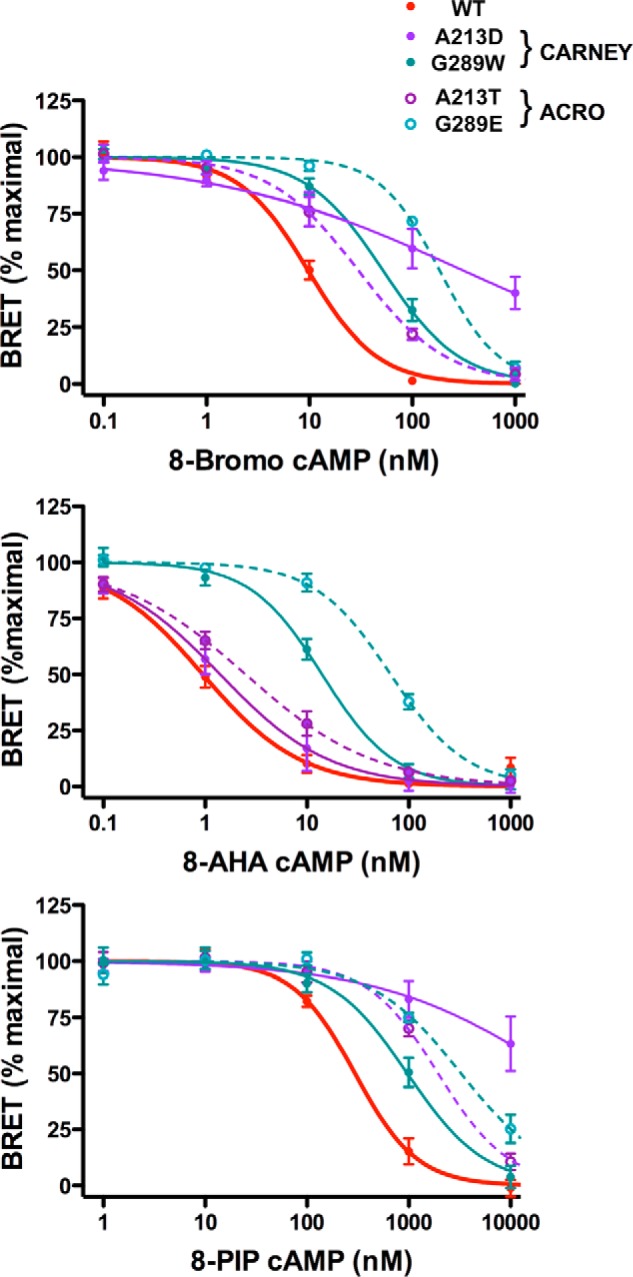
Comparison of the ability of cAMP analogs to dissociate PRKACA subunits from PRKAR1A subunits expressing missense mutations at the same amino acids causing either Carney complex or acrodysostosis. The dissociation of catalytic and regulatory subunits of PKA induced by the indicated doses of 8-bromo-cAMP (top), 8-AHA-cAMP (middle), and 8-PIP-cAMP (bottom) was evaluated by BRET as described in the legend of Fig. 3. Results are shown as the mean ± S.E. (error bars) for WT PRKAR1A (solid red lines) and PRKAR1A carrying mutations causing Carney complex (solid lines) and acrodysostosis (dotted lines) for the indicated mutations affecting Ala-213 (purple lines) and Gly-289 (blue lines).
Because most of the Carney-associated mutations are nonsense mutations resulting in mRNA degradation by nonsense-mediated decay (7, 8), we also investigated whether the two present mutations can result in accelerated protein degradation. As shown in Fig. 8, the Carney-associated mutant A213D presents a reduced degradation rate, similar to those of the two acrodysostosis-associated mutants G289E and A213T, as expected for regulatory subunits with impaired dissociation from the catalytic subunit (2, 11, 26). In contrast, the Carney-associated mutant G289W presents a significantly accelerated rate of degradation, offering a likely explanation for its association with the Carney complex.
We also analyzed PRKAR1A structural consequences of Ala-213 and Gly-289 mutations by three-dimensional modeling. As indicated by analyses of trajectories using 1-ns molecular dynamics simulations (r.m.s. deviation <0.2 Å), substitution of Ala-213 and Gly-289 by aspartate or threonine residues and tryptophan or glutamic acid residues, respectively, does not directly impact the structure of the PRKAR1A subunit.
The glycyl residue at position 289 is a quite flexible amino acid. A small increase of flexibility was observed for the Glu substitution causing acrodysostosis (r.m.s. deviation (WT) = 0.0087 versus r.m.s. deviation (G289E) = 0.0099), whereas a clear loss of flexibility was found for the Trp substitution causing Carney complex (r.m.s. deviation (WT) = 0.0087 versus r.m.s. deviation (G289W) = 0.0024), as calculated by the low frequency normal modes. These changes in flexibility may impact the cooperativity between cAMP A and B domains of the protein because the Gly-289 residue is located in the extended hydrophobic surface forming a contact between the two domains.
The Ala-213 is located in the site of affinity labeling with 8-N3-cAMP (sequence 211–213) of the WT PRKAR1A cAMP-binding domain A (27). Interestingly, Ala-213 is close to the Arg-211 residue, which had a crucial role in the interaction network involved in cAMP binding in domain A (Fig. 9A). The substitution of the alanine residue by an aspartate (A213D) introduces two new negative charges in the binding site of domain A (Fig. 9B), resulting in the formation of two hydrogen bonds between Arg-211 and Asp-213 (Fig. 9B) and disrupting ionic bonds between (i) Asn-173, Asp-172, and Arg-211 and (ii) Ala-213 and the cAMP molecule (27). The inability to dock the cAMP molecule into the molecular model containing this A213D mutation reinforces this hypothesis (data not shown). The acrodysostosis-associated A213T substitution also modified this interaction network (Fig. 9C), but modeling suggested that the affinity of this mutant for cAMP would be intermediate between that of WT PRKAR1A and the A213D mutant.
FIGURE 9.

Stick representations of models showing cAMP docking into the domain A binding pocket of PRKAR1A. Shown are models of the cAMP-binding domain A for WT PRKAR1A (A), mutant A213D (B), and mutant A213T (C). Dashed lines, ionic interactions. All images were generated with Chimera software.
Clinical and Molecular Phenotype Correlations in Acrodysostosis
Although all PRKAR1A mutations occurring in acrodysostosis impaired its function, heterogeneity in the level of the functional defects was observed. Thus, we sought to identify correlations between the intensity of the hormonal resistance/clinical phenotype and the PKA molecular phenotype of the patients.
To do so, we divided the patients into two groups based on forskolin-induced CRE-luciferase activity: those for whom the -fold stimulation over basal level was (Group A: Q372X, R368X, R335L) or was not (Group B: A328V, G289E, Q285R, A213T, Y175C) significantly reduced compared with that of wild-type PRKAR1A following stimulation with both 5 and 10 μm forskolin. Hormonal resistance to PTH, but not TSH, was significantly greater in Group A, as indicated by a greater increase in serum PTH levels (Fig. 10). No significant difference was observed between the two groups for serum calcium and phosphate (data not shown).
FIGURE 10.
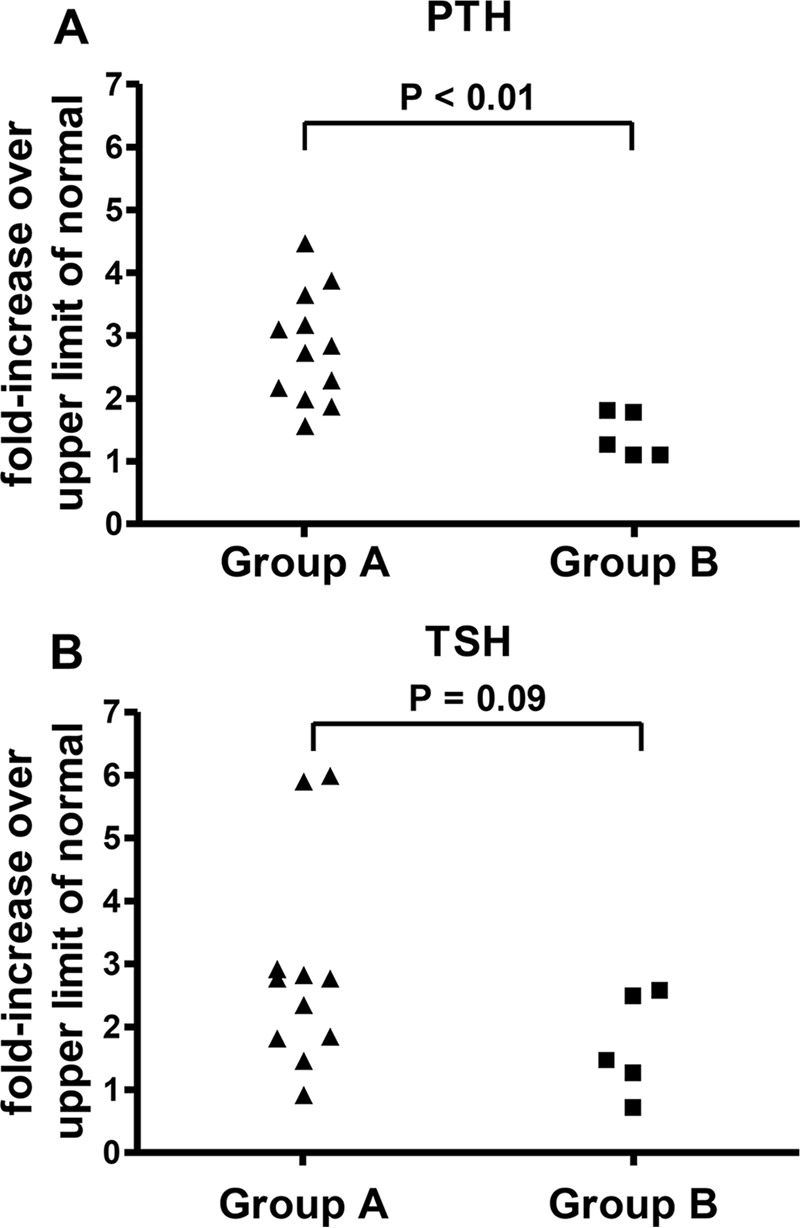
Hormonal resistance to PTH and TSH in patients with acrodysostosis as a function of the impact of the causative mutation on cAMP-induced PKA transcriptional activity. Acrodysostosis patients were divided into two groups based on forskolin-induced CRE-luciferase activity: those for whom the -fold stimulation over basal was (Group A: Q372X, R368X, R335L) or was not (Group B: A328V, G289E, Q285R, A213T, Y175C) significantly reduced compared with that of wild type at both 5 and 10 μm forskolin. Plasma PTH (A) and TSH (B) are expressed as the -fold increase over the upper limit of normal for the assay used. Groups were compared using the Mann-Whitney test.
Discussion
In this study, we have characterized the functional consequences of eight different mutations in the gene coding PRKAR1A identified in patients with acrodysostosis. Our results indicate that in general terms, the overall mechanism responsible for acrodysostosis is similar in patients expressing different mutations. In each case, the mutation impaired the ability of cAMP to dissociate the regulatory and catalytic subunits of PKA, thereby reducing the intensity of PKA signaling induced after stimulation of cells with low to moderate amounts of cAMP. None of the mutations appeared to influence the binding of PRKAR1A to the catalytic subunits in the absence of cAMP, regardless of whether the mutations were missense mutations or nonsense mutations resulting in truncated regulatory subunits. In addition, mutations occurring in cAMP-binding domains A and B cause defects in PKA signaling of similar magnitude.
Despite these overall similarities, several differences were observed while comparing these variants, providing new insights into the molecular pathogenesis of this clinical syndrome. Our results using BRET to evaluate the dissociation of regulatory subunits supported the idea that mutations occurring in the two cAMP-binding domains appeared to act locally, impairing cAMP binding specifically to the involved domain. Mutations affecting domain A resulted in significant increases in the EC50 for 8-PIP-cAMP, an analog that binds preferentially to domain A, but did not increase the EC50 for 8-AHA-cAMP, which binds preferentially to domain B. All mutations affecting domain B resulted in significant increases in the EC50 for 8-AHA cAMP. Interestingly, however, the G289E mutation, but not the other mutations affecting domain B, did increase the EC50 for 8-PIP cAMP. It is conceivable that the G289E mutation induced conformational changes that could influence the structure of both cAMP-binding domains not detected by the present modeling studies. Indeed, the cAMP-binding domain B is the “gatekeeper” for PKA activation, because binding of cAMP to this domain is required to induce conformational changes necessary to expose cAMP-binding domain A (27, 28). Thus, although 8-PIP-cAMP binds preferentially to domain A, sufficient binding to domain B is probably required to render the domain A accessible. Some mutations in domain B, such as G289E, may more strongly inhibit 8-PIP-cAMP binding to domain B and/or impair the subsequent rearrangement required to expose domain A, resulting in an increase in the EC50 for this analog. In this regard, the G289E mutation, located in a loop connecting the β2 and β3 strands and included in a hydrophobic region of domain B in contact with domain A, appears to alter the flexibility of the protein and induces changes that might alter the dynamic cooperativity of the two domains.
An important finding in our study was the observation that although all of the PRKAR1A mutations reduced PKA signaling in response to cAMP, the extent of the reduction could be different, depending on the mutations. These findings raise the possibility that the clinical characteristics observed in acrodysostosis may be, at least to some extent, mutation-dependent. Consistent with this idea, we found that patients carrying mutations that led to greater increases in the EC50 for forskolin-induced CRE-luciferase activity had, as a group, significantly higher plasma PTH levels. Further studies are needed to confirm this finding and evaluate the extent of the influence of functional differences in PRKAR1A activity on other clinical features of acrodysostosis, including skeletal dysplasia, growth, body weight, and resistance to other hormones.
Carney complex is due to PRKAR1A mutations causing inappropriate constitutive activation of the catalytic subunits of PKA. In the large majority of the cases, nonsense mutations or insertions/deletions trigger nonsense-mediated mRNA decay, resulting in protein haploinsufficiency of the regulatory subunit (7, 8). In the case of the two Carney missense mutations studied here, premature degradation of mRNA by nonsense-mediated decay cannot be invoked. Possible alternative explanations for constitutive activation of PKA include (i) an abnormally high affinity of PRKAR1A for cAMP, (ii) an impaired association of PRKAR1A and PRKACA under basal conditions, (iii) inappropriate activation of PKA in response to low physiological concentrations of cAMP, or (iv) an accelerated degradation of the protein.
A previous report analyzing the molecular defects of Carney-associated missense mutations identified abnormal binding between the two PKA subunits and with cAMP as the causes of increased PKA activity (10). Using different materials and methods, we investigated two of the mutants (A213D and G289W) because the same positions are also mutated in acrodysostosis. This comparison provides new insights, indicating various mutation-dependent mechanisms for PKA activation.
The Carney-associated mutant A213D is located in the cAMP-binding domain A and, like the A213T mutant occurring in acrodysostosis, resulted in a markedly impaired binding of 8-PIP-cAMP, but not 8-AHA cAMP, blunted PKA-induced transcriptional activity in response to high doses of forskolin, and a reduced protein degradation pattern. Several findings, however, supported the possibility that the Carney-associated A213D mutation, but not the acrodysostosis mutation A213T mutation, presents a peculiar phenotype. Domain A of this mutant has a markedly impaired ability to bind cAMP, as suggested both by BRET experiments and three-dimensional modeling. This alteration does not change the basal interaction between the regulatory and catalytic subunits but, in addition to impairing cAMP binding to domain A, may also facilitate enzyme activation in the absence of subunit dissociation or subunit dissociation to some extent in the presence of cAMP concentrations insufficient to activate wild-type PKA (29). Consistent with this scenario, we observed (i) no significant increase in basal PKA-induced transcriptional activity; (ii) preserved PKA-induced transcriptional activity in cells stimulated with a low dose, but not a high dose, of forskolin; and (iii) relative increased dissociation of regulatory and catalytic subunits measured by BRET in response to low dose 8-bromo-cAMP. In this regard, the similarities of our results for the A213D with those previously published by Greene et al. (10) are noteworthy. In their dose-response curves, PKA activity for lysates from cells expressing WT PRKAR1A plateaued at 0.1 μm cAMP, whereas for lysates from cells expressing A213D, activity continued to increase up to 5 μm cAMP, consistent with a higher EC50 for the A213D mutants, as also observed by us using the CRE-luciferase system. Conversely, following stimulation with low doses of cAMP (0.005–0.01 μm), the PKA activity of extracts from cells expressing the A213D mutant was consistently greater than that of cells expressing WT PRKAR1A, a finding compatible with inappropriate PKA activation occurring at low suprabasal levels of cAMP.
The Carney-associated G289W mutant exhibited a reduced affinity for cAMP without any change in the basal interaction between PRKAR1A and PRKACA, findings similar to what was observed with the acrodysostosis-associated G289E mutant. The accelerated degradation of this mutant is the most probable explanation for haploinsufficiency. The binding of PRKAR1A to the catalytic subunits is thought to prolong its half-life, and the G289W mutant was relatively resistant to cAMP-induced dissociation, suggesting that the accelerated degradation of the G289W mutant occurs at an early step in the process of synthesis, folding, and/or transport. This early and premature degradation of the Carney-associated G289W mutant probably prevails on the other acrodysostosis-like defects, explaining the clinical phenotype.
Altogether, our results and those reported by Greene et al. (10) indicate that Carney-associated mutations induce modest molecular alterations, which are difficult to visualize following overexpression in heterologous cells using the present available tools. Nevertheless, different mechanisms, including accelerated protein degradation and abnormal association of the two subunits in the presence of physiological concentrations of cAMP, appear to be involved. Further studies are required to directly demonstrate that the A213D mutant permits abnormally high PKA activity in the presence of suboptimal concentrations of cAMP, despite impairing cAMP binding to domain A.
In conclusion, the data presented here show for the first time and unambiguously that mutations of the PRKAR1A observed in acrodysosotosis all result in a reduced affinity for cAMP by this regulatory PKA subunit. Quantitative and qualitative differences in this defect are discernible for different mutations, and these differences may have clinical implications that require further evaluation. Mutations occurring at the same position in PRKAR1A as seen in acrodysostosis but resulting in the introduction of another amino acid can profoundly modify the observed phenotype, resulting in Carney complex.
Author Contributions
Y. R., C. L. S., W. A. K., C. A., A. C., and E. C. performed the experiments. J. B., A. L., C. S., and E. C. participated in the design of the project and analyzed the data. A. C. did the modeling analysis. C. S. and E. C. wrote the manuscript. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We are very grateful to Allan Hance for tremendous help with data analysis and writing of the manuscript and for interesting and friendly scientific discussions. We also thank Dr. M. O. North, M. Nigou, and V. Duchaussoy for the identification of the new mutations of PRKAR1A in acrodysosotosis and S. Marullo for valuable discussion of BRET results. We thank Pierre Bougnères for continuing support.
This work was supported by recurrent funding from INSERM and Assistance Publique, Hôpitaux de Paris.
- PTH
- parathyroid hormone
- TSH
- thyrotropin
- 8-bromo-cAMP
- 8-bromo-cyclic AMP
- 8-PIP-cAMP
- 8-piperidinoadenosine 3′,5′-cyclic monophosphate
- 8-AHA-cAMP
- 8-(6-aminohexyl)aminoadenosine-3′,5′-cyclic monophosphate
- 8-N3-cAMP
- 8-azidoadenosine-3′,5′-cyclic monophosphate
- CRE
- cAMP-responsive element
- BRET
- bioluminescence resonance energy transfer
- r.m.s.
- root mean square.
References
- 1.Parnot C., Miserey-Lenkei S., Bardin S., Corvol P., and Clauser E. (2002) Lessons from constitutively active mutants of G protein-coupled receptors. Trends Endocrinol. Metab. 13, 336–343 [DOI] [PubMed] [Google Scholar]
- 2.Spaulding S. W. (1993) The ways in which hormones change cyclic adenosine 3′,5′-monophosphate-dependent protein kinase subunits, and how such changes affect cell behavior. Endocr. Rev. 14, 632–650 [DOI] [PubMed] [Google Scholar]
- 3.Spiegel A. M., and Weinstein L. S. (2004) Inherited diseases involving G proteins and G protein-coupled receptors. Annu. Rev. Med. 55, 27–39 [DOI] [PubMed] [Google Scholar]
- 4.Taylor S. S., Buechler J. A., and Yonemoto W. (1990) cAMP-dependent protein kinase: framework for a diverse family of regulatory enzymes. Annu. Rev. Biochem. 59, 971–1005 [DOI] [PubMed] [Google Scholar]
- 5.Uhler M. D., Carmichael D. F., Lee D. C., Chrivia J. C., Krebs E. G., and McKnight G. S. (1986) Isolation of cDNA clones coding for the catalytic subunit of mouse cAMP-dependent protein kinase. Proc. Natl. Acad. Sci. U.S.A. 83, 1300–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Solberg R., Sandberg M., Natarajan V., Torjesen P. A., Hansson V., Jahnsen T., and Taskén K. (1997) The human gene for the regulatory subunit RI α of cyclic adenosine 3′,5′-monophosphate-dependent protein kinase: two distinct promoters provide differential regulation of alternately spliced messenger ribonucleic acids. Endocrinology 138, 169–181 [DOI] [PubMed] [Google Scholar]
- 7.Bertherat J., Horvath A., Groussin L., Grabar S., Boikos S., Cazabat L., Libe R., René-Corail F., Stergiopoulos S., Bourdeau I., Bei T., Clauser E., Calender A., Kirschner L. S., Bertagna X., Carney J. A., and Stratakis C. A. (2009) Mutations in regulatory subunit type 1A of cyclic adenosine 5′-monophosphate-dependent protein kinase (PRKAR1A): phenotype analysis in 353 patients and 80 different genotypes. J. Clin. Endocrinol. Metab. 94, 2085–2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bossis I., and Stratakis C. A. (2004) Minireview: PRKAR1A: normal and abnormal functions. Endocrinology 145, 5452–5458 [DOI] [PubMed] [Google Scholar]
- 9.Kirschner L. S., Sandrini F., Monbo J., Lin J. P., Carney J. A., and Stratakis C. A. (2000) Genetic heterogeneity and spectrum of mutations of the PRKAR1A gene in patients with the Carney complex. Hum. Mol. Genet. 9, 3037–3046 [DOI] [PubMed] [Google Scholar]
- 10.Greene E. L., Horvath A. D., Nesterova M., Giatzakis C., Bossis I., and Stratakis C. A. (2008) In vitro functional studies of naturally occurring pathogenic PRKAR1A mutations that are not subject to nonsense mRNA decay. Hum. Mutat. 29, 633–639 [DOI] [PubMed] [Google Scholar]
- 11.Linglart A., Menguy C., Couvineau A., Auzan C., Gunes Y., Cancel M., Motte E., Pinto G., Chanson P., Bougnères P., Clauser E., and Silve C. (2011) Recurrent PRKAR1A mutation in acrodysostosis with hormone resistance. N. Engl. J. Med. 364, 2218–2226 [DOI] [PubMed] [Google Scholar]
- 12.Lee H., Graham J. M. Jr., Rimoin D. L., Lachman R. S., Krejci P., Tompson S. W., Nelson S. F., Krakow D., and Cohn D. H. (2012) Exome sequencing identifies PDE4D mutations in acrodysostosis. Am. J. Hum. Genet. 90, 746–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Linglart A., Fryssira H., Hiort O., Holterhus P. M., Perez de Nanclares G., Argente J., Heinrichs C., Kuechler A., Mantovani G., Leheup B., Wicart P., Chassot V., Schmidt D., Rubio-Cabezas Ó., Richter-Unruh A., Berrade S., Pereda A., Boros E., Muñoz-Calvo M. T., Castori M., Gunes Y., Bertrand G., Bougnères P., Clauser E., and Silve C. (2012) PRKAR1A and PDE4D mutations cause acrodysostosis but two distinct syndromes with or without GPCR-signaling hormone resistance. J. Clin. Endocrinol. Metab. 97, E2328–E2338 [DOI] [PubMed] [Google Scholar]
- 14.Michot C., Le Goff C., Goldenberg A., Abhyankar A., Klein C., Kinning E., Guerrot A. M., Flahaut P., Duncombe A., Baujat G., Lyonnet S., Thalassinos C., Nitschke P., Casanova J. L., Le Merrer M., Munnich A., and Cormier-Daire V. (2012) Exome sequencing identifies PDE4D mutations as another cause of acrodysostosis. Am. J. Hum. Genet. 90, 740–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muhn F., Klopocki E., Graul-Neumann L., Uhrig S., Colley A., Castori M., Lankes E., Henn W., Gruber-Sedlmayr U., Seifert W., and Horn D. (2013) Novel mutations of the PRKAR1A gene in patients with acrodysostosis. Clin. Genet. 84, 531–538 [DOI] [PubMed] [Google Scholar]
- 16.Nagasaki K., Iida T., Sato H., Ogawa Y., Kikuchi T., Saitoh A., Ogata T., and Fukami M. (2012) PRKAR1A mutation affecting cAMP-mediated G protein-coupled receptor signaling in a patient with acrodysostosis and hormone resistance. J. Clin. Endocrinol. Metab. 97, E1808–E1813 [DOI] [PubMed] [Google Scholar]
- 17.Silve C., Le-Stunff C., Motte E., Gunes Y., Linglart A., and Clauser E. (2012) Acrodysostosis syndromes. Bonekey Rep. 1, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prinz A., Diskar M., Erlbruch A., and Herberg F. W. (2006) Novel, isotype-specific sensors for protein kinase A subunit interaction based on bioluminescence resonance energy transfer (BRET). Cell. Signal. 18, 1616–1625 [DOI] [PubMed] [Google Scholar]
- 19.Achour L., Kamal M., Jockers R., and Marullo S. (2011) Using quantitative BRET to assess G protein-coupled receptor homo- and heterodimerization. Methods Mol. Biol. 756, 183–200 [DOI] [PubMed] [Google Scholar]
- 20.Schwede F., Christensen A., Liauw S., Hippe T., Kopperud R., Jastorff B., and Døskeland S. O. (2000) 8-Substituted cAMP analogues reveal marked differences in adaptability, hydrogen bonding, and charge accommodation between homologous binding sites (AI/AII and BI/BII) in cAMP kinase I and II. Biochemistry 39, 8803–8812 [DOI] [PubMed] [Google Scholar]
- 21.Skålhegg B. S., Landmark B. F., Døskeland S. O., Hansson V., Lea T., and Jahnsen T. (1992) Cyclic AMP-dependent protein kinase type I mediates the inhibitory effects of 3′,5′-cyclic adenosine monophosphate on cell replication in human T lymphocytes. J. Biol. Chem. 267, 15707–15714 [PubMed] [Google Scholar]
- 22.Bae E., and Phillips G. N. Jr. (2005) Identifying and engineering ion pairs in adenylate kinases: insights from molecular dynamics simulations of thermophilic and mesophilic homologues. J. Biol. Chem. 280, 30943–30948 [DOI] [PubMed] [Google Scholar]
- 23.Su Y., Dostmann W. R., Herberg F. W., Durick K., Xuong N. H., Ten Eyck L., Taylor S. S., and Varughese K. I. (1995) Regulatory subunit of protein kinase A: structure of deletion mutant with cAMP binding domains. Science 269, 807–813 [DOI] [PubMed] [Google Scholar]
- 24.Kopperud R., Krakstad C., Selheim F., and Døskeland S. O. (2003) cAMP effector mechanisms. Novel twists for an “old” signaling system. FEBS Lett. 546, 121–126 [DOI] [PubMed] [Google Scholar]
- 25.Guillaud Bataille M., Rhayem Y., Sousa S. B., Libé R., Dambrun M., Chevalier C., Nigou M., Auzan C., North M. O., Sa J., Gomes L., Salpea P., Horvath A., Stratakis C. A., Hamzaoui N., Bertherat J., and Clauser E. (2014) Systematic screening for PRKAR1A gene rearrangement in Carney complex: identification and functional characterization of a new in-frame deletion. Eur. J. Endocrinol. 170, 151–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Daalen Wetters T., and Coffino P. (1983) Reversion of an S49 cell cyclic AMP-dependent protein kinase structural gene mutant occurs primarily by functional elimination of mutant gene expression. Mol. Cell. Biol. 3, 250–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim C., Cheng C. Y., Saldanha S. A., and Taylor S. S. (2007) PKA-I holoenzyme structure reveals a mechanism for cAMP-dependent activation. Cell 130, 1032–1043 [DOI] [PubMed] [Google Scholar]
- 28.Taylor S. S., Keshwani M. M., Steichen J. M., and Kornev A. P. (2012) Evolution of the eukaryotic protein kinases as dynamic molecular switches. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367, 2517–2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lefkimmiatis K., and Zaccolo M. (2014) cAMP signaling in subcellular compartments. Pharmacol. Ther. 143, 295–304 [DOI] [PMC free article] [PubMed] [Google Scholar]