

Background: Ca2+ transport by Ca2+-ATPase includes phosphoenzyme isomerization with luminal Ca2+ release.
Results: Mutation of Leu119, Tyr122, or Ile179 in an interdomain hydrophobic cluster retards release relative to isomerization.
Conclusion: There is a transient Ca2+-bound state and affinity reduction during release governed by cluster assembly.
Significance: Ca2+ release is a multistep process directed by head domain gathering on transmembrane helix M2.
Keywords: calcium ATPase, calcium transport, enzyme kinetics, enzyme structure, site-directed mutagenesis
Abstract
The mechanism whereby events in and around the catalytic site/head of Ca2+-ATPase effect Ca2+ release to the lumen from the transmembrane helices remains elusive. We developed a method to determine deoccluded bound Ca2+ by taking advantage of its rapid occlusion upon formation of E1PCa2 and of stabilization afforded by a high concentration of Ca2+. The assay is applicable to minute amounts of Ca2+-ATPase expressed in COS-1 cells. It was validated by measuring the Ca2+ binding properties of unphosphorylated Ca2+-ATPase. The method was then applied to the isomerization of the phosphorylated intermediate associated with the Ca2+ release process E1PCa2 → E2PCa2 → E2P + 2Ca2+. In the wild type, Ca2+ release occurs concomitantly with EP isomerization fitting with rate-limiting isomerization (E1PCa2 → E2PCa2) followed by very rapid Ca2+ release. In contrast, with alanine mutants of Leu119 and Tyr122 on the cytoplasmic part of the second transmembrane helix (M2) and Ile179 on the A domain, Ca2+ release in 10 μm Ca2+ lags EP isomerization, indicating the presence of a transient E2P state with bound Ca2+. The results suggest that these residues function in Ca2+ affinity reduction in E2P, likely via a structural rearrangement at the cytoplasmic part of M2 and a resulting association with the A and P domains, therefore leading to Ca2+ release.
Introduction
Sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA1a),2 a representative member of P-type ion-transporting ATPases, catalyzes Ca2+ transport coupled with ATP hydrolysis (Fig. 1) (for recent reviews, see Refs. 1–3). The enzyme is activated by the binding of two cytoplasmic Ca2+ ions at the high-affinity transport sites (E2 to E1Ca2, steps 1 and 2) and autophosphorylated at Asp351 with MgATP to form an ADP-sensitive phosphoenzyme (E1P, step 3), which reacts with ADP to regenerate ATP in the reverse reaction. Upon E1P formation, the two bound Ca2+ are occluded in the transport sites (E1PCa2). The subsequent isomeric transition to the ADP-insensitive E2P form results in rearrangements of the Ca2+ binding sites to deocclude Ca2+, open the release path, and reduce the affinity, therefore releasing Ca2+ into the lumen (steps 4 and 5). Finally, the Asp351-acylphosphate in E2P is hydrolyzed to form a Ca2+-unbound inactive E2 state (step 6).
FIGURE 1.
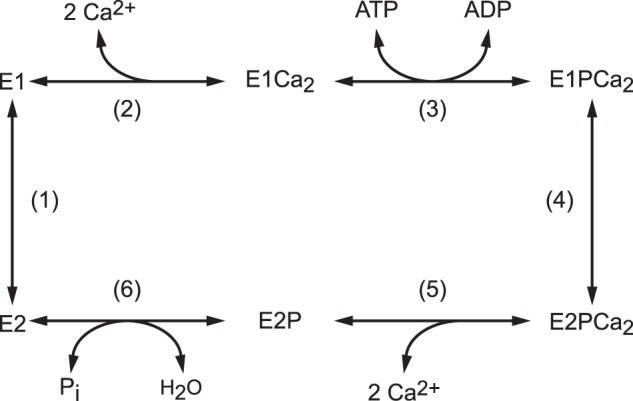
Reaction schematic of Ca2+-ATPase.
The EP isomerization associated with luminal Ca2+ release is a key rate-limiting process and involves a large rotation of the A domain, its association with the P domain and the cytoplasmic part of M2, and an inclination of associated A and P domains and the connected helices M2/M1 and M4/M5 via a steric effect of a M1/M2 V-shaped body (4–20). These motions are coupled to a rearrangement of the transport sites. We have found previously that the hydrophobic association of Leu119/Tyr122 on the cytoplasmic part of M2 with the A and P domains (Tyr122 hydrophobic cluster) is critical for formation of the Ca2+-released E2P ground state structure, with hydrolytic ability at the catalytic site and a properly opened luminal Ca2+ release path with reduced affinity at the transport sites (16–18). The Tyr122 hydrophobic cluster is formed with residues on the cytoplasmic part of M2 (Tyr122/Leu119), A domain (Ile179/Leu180), P domain (Val705/Val726), and A/M3 linker (Ile232), thereby producing a most compactly organized head in E2P. The postulated E2PCa2 transient intermediate was successfully trapped in the Ca2+-occluded state (before deocclusion) by elongation of the A/M1′ linker with a two- or four-glycine insertion. Consistently in this state, the Tyr122 hydrophobic cluster was shown to not yet be fully formed. The finding demonstrates the critical role of the strain in this linker, probably to effect inclination of the A and P domains and connected helices required for deocclusion and Ca2+ release (19, 20).
Further understanding of Ca2+ binding/release processes in the transport cycle and roles of residues involved, especially the dynamic key process E1PCa2 → E2PCa2 → E2P + 2Ca2+, is hampered by an inability to determine and detect the bound but deoccluded Ca2+ in minute amounts of expressed wild type and mutants obtained from cultured cells. In this study, we developed a method to determine the bound Ca2+ in the non-phosphorylated state as well as in the phosphorylated state of expressed enzymes. We took advantage of the rapid occlusion in E1PCa2 of bound Ca2+, from either E1Ca2 or E2P species (E2P and, possibly, the transient E2P state with bound Ca2+) by addition of a high concentration (10 mm) of Ca2+ (plus ATP for E1Ca2) (21–25), probably trapping the bound Ca2+ before possible exchange with the added Ca2+. The E1PCa2 thus formed is very stable, probably because of Ca2+ substitution of Mg2+ bound at the catalytic Mg2+ subsite, as found previously (23, 26–29), therefore withstanding membrane filtration and extensive washing. We then applied this new method to the EP isomerization and Ca2+ release kinetic processes in alanine substitution mutants of the Tyr122 hydrophobic cluster because they are critical for the Ca2+-released E2P ground state structure (16–18). The results indicate the presence of a transient E2P state with bound but deoccluded Ca2+ and show that Leu119 and Tyr122 on the cytoplasmic part of M2 and Ile179 on the A domain function via their association to reduce Ca2+ affinity during E2P processing and, thereby, to accelerate Ca2+ release into the lumen. The detailed analyses suggest that a possibly stepwise assembly of the residues into the Tyr122 hydrophobic cluster takes place for proper Ca2+ handling (namely, deocclusion, affinity reduction, and release) and, therefore, Ca2+ transport coupled with EP processing.
Experimental Procedures
Mutagenesis and Expression
QuikChangeTM site-directed mutagenesis (Stratagene) was utilized for the substitution of residues in rabbit SERCA1a cDNA. The ApaI-KpnI or KpnI-SalI restriction fragment was ligated back into the corresponding region in the full-length SERCA1a cDNA in the pMT2 expression vector (30). The pMT2 DNA was transfected into COS-1 cells with Lipofectamine and PlusTM reagent (Invitrogen), and the microsomes were prepared from COS-1 cells as described previously (18, 31).
Determination of EP
Microsomes expressing wild-type or mutant SERCA1a prepared from the COS-1 cells were phosphorylated with [γ-32P]ATP or 32Pi under the conditions described in the figure legends. The total amount of EP was determined following the addition of trichloroacetic acid. The amount of E2P was determined by adding an equal volume of a solution containing 2 mm ADP and 5 mm EGTA, followed by the trichloroacetic acid addition 1 s after the ADP addition. The amount of EP was quantified with digital autoradiography after separation by 5% SDS-PAGE at pH 6.0 according to Weber and Osborn (32) and as described previously (33). The amount of EP in expressed SERCA1a was obtained by subtracting the background radioactivity determined in the absence of Ca2+. In all mutants and the wild type, the background level was less than 1% of the total amount of EP.
Determination of Bound Ca2+
Microsomes were incubated with 0–10 μm 45Ca2+ at 4 °C in the absence or presence of 1 μm thapsigargin (TG), a highly specific and subnanomolar affinity inhibitor of SERCA that fixes the enzyme in the Ca2+-unbound E2 state (34). 50 μl of reaction mixture was spotted on a membrane filter (Millipore, 0.45-μm mixed cellulose membrane HAWP) and washed for 3 s with 1 ml of Ca2+ binding assay medium containing 50 mm HEPES/Tris (pH 8.0), 0.1 m KCl, 10 mm CaCl2, 7 mm MgCl2, and 0.1 mm ATP. Other experimental conditions are described in detail in the figure legends. The 45Ca2+ remaining on the filter was quantified by digital autoradiography. The amount of Ca2+ specifically bound to the Ca2+-ATPase (trapped as occluded in E1PCa2) was obtained by subtracting the amount of nonspecific Ca2+-binding background determined in the presence of TG. We confirmed that all mutants retained TG sensitivity by observing that TG (1 μm used in this study) completely inhibits EP formation from [γ-32P]ATP in the presence of Ca2+, showing its validity for the background determination. The background level of nonspecific Ca2+ binding (in the presence of 10 μm Ca2+) was ∼100 pmol/mg of microsomal protein, which is 30–50% of total Ca2+ binding (see Fig. 4, A and B).
FIGURE 4.
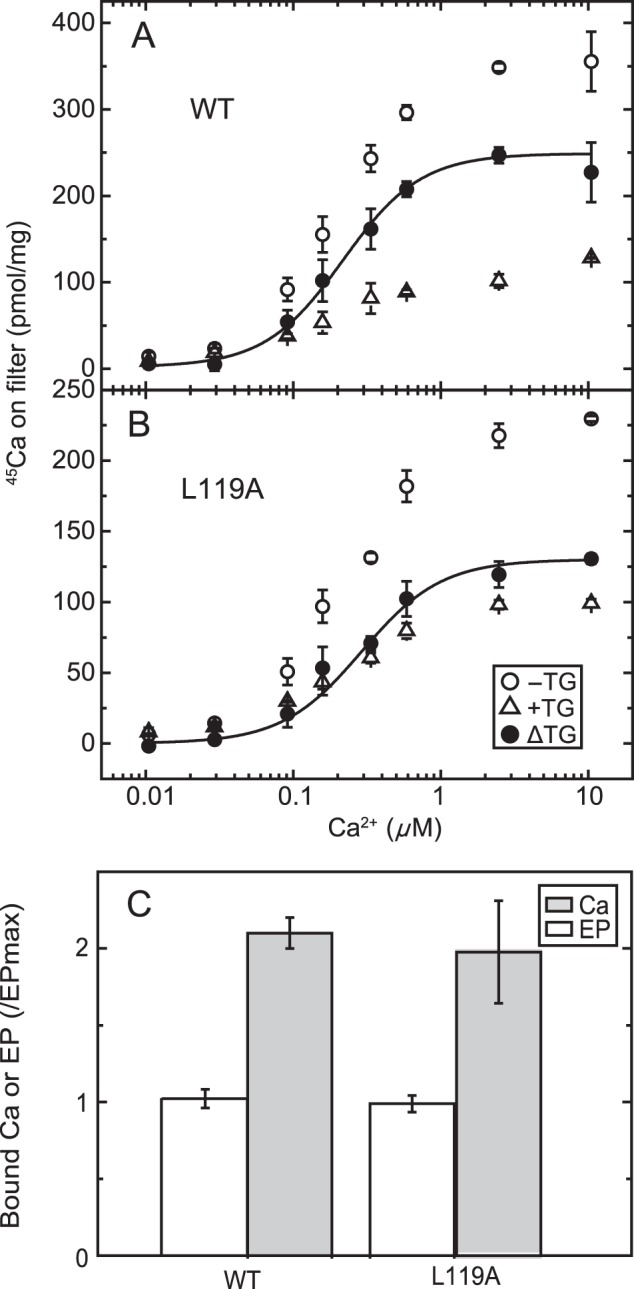
Ca2+ concentration dependence of Ca2+ binding in non-phosphorylated Ca2+-ATPase from COS-1 cells. A and B, microsomes expressing the wild type (A) or mutant L119A (B) were incubated at 4 °C for 20 s with various concentrations of 45Ca2+ in a mixture containing 20 μg/ml microsomal protein, 50 mm MOPS/Tris (pH 7.3), 0.1 m KCl, 7 mm MgCl2, 3 μm A23187, and 10 μm 45CaCl2 with 0–0.09 mm EGTA to give the indicated free Ca2+ concentration in the presence (open triangles) or absence (open circles) of 1 μm TG. Then, as described under “Experimental Procedures,” 50 μl of the mixture was spotted on a membrane filter and washed immediately for 3 s with 1 ml of a Ca2+ binding assay medium containing 10 mm CaCl2 and 0.1 mm ATP in 50 mm HEPES/Tris (pH 8.0), 0.1 m KCl, and 7 mm MgCl2. The values presented are the mean ± S.D. (n = 3–4). The amount of Ca2+ specifically bound to the Ca2+-ATPase (trapped as occluded Ca2+ in E1PCa2, closed circles) was obtained by subtracting the amount of nonspecific Ca2+ binding determined in the presence of TG. The solid lines show the least squares fit to the Hill equation with the fitting parameters Kd and Hill coefficient of 0.22 μm and 1.6 for the wild type and 0.29 μm and 1.6 for the mutant L119A. C, the maximum amount of specific 45Ca2+ binding in 10 μm 45Ca2+ was determined as above (Ca) and compared with the maximum amount of EP (EP) determined in the presence of 10 mm Ca2+ as in Fig. 2A (i.e. essentially under the same conditions as described for the determination of the catalytic site content in the preparation (45)). It should be mentioned here that, for the comparison, one preparation of microsomes was used throughout for the wild type and the mutant. Actual values of the amount of bound Ca2+ and that of EP (pmol/mg of microsomal protein (n = 4)) for the wild type were 287 ± 14 and 136 ± 8, respectively, giving a stoichiometry of Ca/EP = 2.11. Those for the L119A mutant were 95.6 ± 16.2 and 48.6 ± 2.6, respectively, providing a stoichiometry of Ca/EP = 1.97. Note that these maximum Ca2+ binding values are slightly different from those in A and B, in which different preparations were used.
Miscellaneous
Protein concentrations were determined according to Lowry et al. (35). Data were analyzed by nonlinear regression using the program Origin (Microcal Software, Inc.). Free Ca2+ concentrations were calculated by the Calcon program. Three-dimensional models of SERCA1a were produced by the program VMD (36).
Results
Rapid E1PCa2 Formation and Its Stabilization
To fix the bound Ca2+ in E1Ca2 in an occluded form in E1PCa2, it is necessary to rapidly phosphorylate E1Ca2 to E1PCa2 before Ca2+release and to stabilize E1PCa2 and prevent its decay during thorough washing. In analysis of the EP isomerization/Ca2+ release process E1PCa2 → E2PCa2 → E2P + 2Ca2+, the E2P species need to be rapidly converted to a stabilized E1PCa2 state. The crucial feature for the forward reaction is to add a very high concentration (10 mm) of Ca2+ together with ATP. E1PCa2 is extremely stable in a high concentration of Ca2+, probably because of Ca2+ replacing catalytic subsite Mg2+, as shown previously (23, 26–29). This stabilization prevents possible Ca2+ exchange at the transport sites in the reverse conversion in E1PCa2 ↔ E2P + 2Ca2+. Actually, the Mg2+ at the catalytic subsite is not “occluded” in E1PCa2 and exchanges rapidly with Ca2+ at such a high concentration (26–28). Also, addition of a high concentration of Ca2+ rapidly converts E2P to the stable E1PCa2 in the reverse reaction (18, 22).
In Fig. 2A, wild-type SERCA1a expressed in microsomes of COS-1 cells was incubated with 10 μm Ca2+ to form the E1Ca2 state in 7 mm Mg2+, and then [γ-32P]ATP plus an excess of EGTA or [γ-32P]ATP plus 10 mm Ca2+ was added in 0.1 m KCl, which accelerates E2P hydrolysis, causing E1PCa2 accumulation (37). When ATP is added with EGTA, E1PCa2 forms very rapidly despite the removal of free Ca2+ and then decays slowly via the rate-limiting EP isomerization, followed by rapid hydrolysis. The maximum EP level immediately after ATP/EGTA addition, estimated by an extrapolation of the single exponential decay to zero time, is very close to that obtained with ATP plus 10 mm Ca2+. E1PCa2 formation in 10 mm Ca2+ is also rapid and reaches a maximum within ∼3 s, albeit slightly slower than that without free Ca2+ (i.e. with the substrate MgATP, consistent with previous kinetic studies (21–25)). Importantly, during this 3 s period, ∼90% of E1PCa2 remains when free Ca2+ is removed by EGTA. Therefore, E1PCa2 formation and resulting Ca2+ occlusion by the simultaneous addition of ATP and 10 mm Ca2+ is rapid enough to trap almost all 45Ca2+ originally bound in E1Ca2 as an occluded E1PCa2 species before Ca2+ release and possible 45Ca2+-Ca2+ exchange (as demonstrated in Fig. 4).
FIGURE 2.
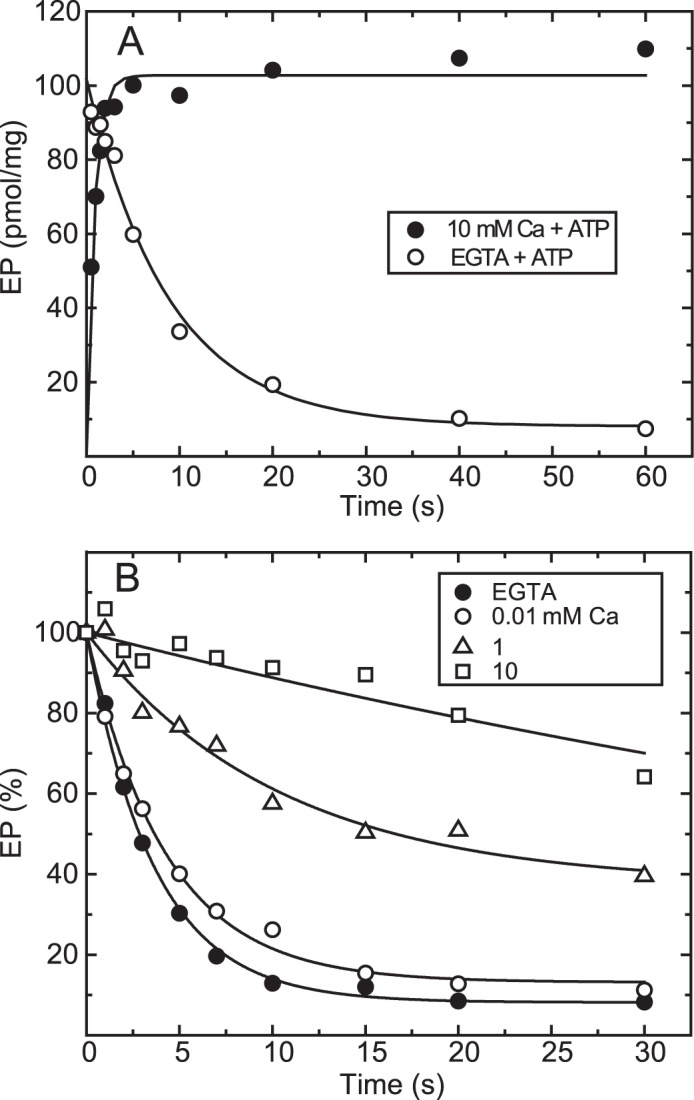
Time course of EP formation and its decay in the wild type. A, microsomes expressing wild-type Ca2+-ATPase prepared from COS-1 cells were incubated with 10 μm Ca2+ in a mixture containing 20 μg/ml microsomal protein, 20 mm MOPS/Tris (pH 7.3), 0.1 m KCl, 7 mm MgCl2, 10 μm CaCl2, and 3 μm A23187 at 4 °C. Then EP formation was initiated at zero time by mixing with an equal volume of a solution containing 20 μm [γ-32P]ATP, 0.1 m HEPES/Tris (pH 8.0), 0.1 m KCl, and 7 mm MgCl2 with 10 mm CaCl2 (closed circles) or 2 mm EGTA (open circles). B, EP was formed in 20 mm MOPS/Tris (pH 7.3), 0.1 m KCl, 7 mm MgCl2, 10 μm CaCl2, 3 μm A23187, and 10 μm [γ-32P]ATP at 4 °C for 10 s. Then the reaction was chased at zero time by mixing with an equal volume of a solution containing non-radioactive 0.2 mm ATP and various concentrations of CaCl2 to give the indicated final Ca2+ concentrations (0.01, 1, and 10 mm) or 2 mm EGTA to remove free Ca2+ in 0.1 m HEPES/Tris (pH 8.0), 0.1 m KCl, 7 mm MgCl2, and 3 μm A23187. The amount of EP was normalized to the value at zero time of the chase.
In Fig. 2B, the decay of E1PCa2 formed with [γ-32P]ATP in 10 μm Ca2+ and 7 mm Mg2+ was initiated by a cold (non-radioactive) ATP chase in various concentrations of Ca2+. The decay is slowed markedly with increasing Ca2+. Actually, E1PCa2 in 10 mm Ca2+ remains almost completely stable during the initial 3 s, a characteristic exploited in the Ca2+ binding assays in Figs. 4–10 as a period for membrane filter washing with 10 mm Ca2+.
FIGURE 5.
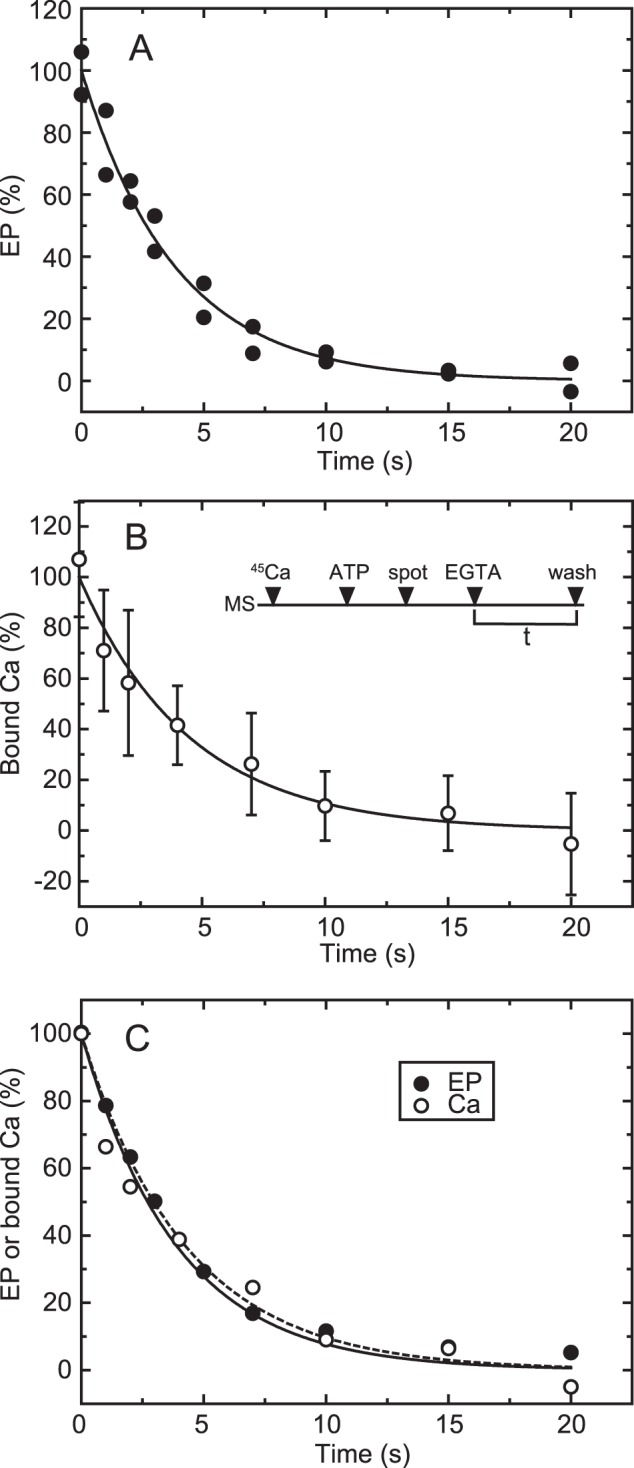
Time courses of EP isomerization and Ca2+ release in the wild type. A, for the determination of EP, wild-type Ca2+-ATPase was phosphorylated in a mixture containing 20 μg/ml microsomal protein, 20 μm [γ-32P]ATP, 10 μm CaCl2, 50 mm MOPS/Tris (pH 7.3), 0.1 m KCl, 7 mm MgCl2, and 3 μm A23187. Then the phosphorylation was chased at zero time by mixing with an equal volume of EGTA chase solution containing 1 mm EGTA, 50 mm MOPS/Tris (pH 7.3), 0.1 m KCl, 7 mm MgCl2, and 3 μm A23187. The reaction was quenched by acid at the indicated times. Here it should be noted that nearly all the EP was E1P in the presence of KCl (actually more than 95%), as we observed under essentially the same conditions (cf. Fig. 2A in Ref. 42). Therefore, EP decay represents EP isomerization (which is followed by rapid E2P hydrolysis). B, for the Ca2+ binding assay, wild-type Ca2+-ATPase in microsomes (MS) was incubated for 5 s with 10 μm 45Ca2+ in 50 mm MOPS/Tris (pH 7.3), 0.1 m KCl, 7 mm MgCl2, and 3 μm A23187 and phosphorylated by addition of a small volume of ATP to give 10 μm (ATP) for 20 s. Immediately, the mixture (50 μl) was spotted on the membrane filter (spot), and, at zero time (EGTA), free Ca2+ was removed to initiate EP decay by continuous rinsing with the above EGTA chase solution. After various periods (t), the amount of 45Ca2+ specifically bound to Ca2+-ATPase was determined by washing the filter with 1 ml of the Ca2+ binding assay medium as in Fig. 4 (wash). The values presented are the mean ± S.D. (n = 3–6). C, the time courses of EP decay (closed circles) and decrease in bound 45Ca2+ (open circles) determined in A and B are replotted in the same panel. The plots show the mean values in A and B. The solid and broken lines show a single exponential fit in which the rates are 0.23 s−1 for EP decay and 0.22 s−1 for the decrease in bound 45Ca2+, respectively.
FIGURE 6.
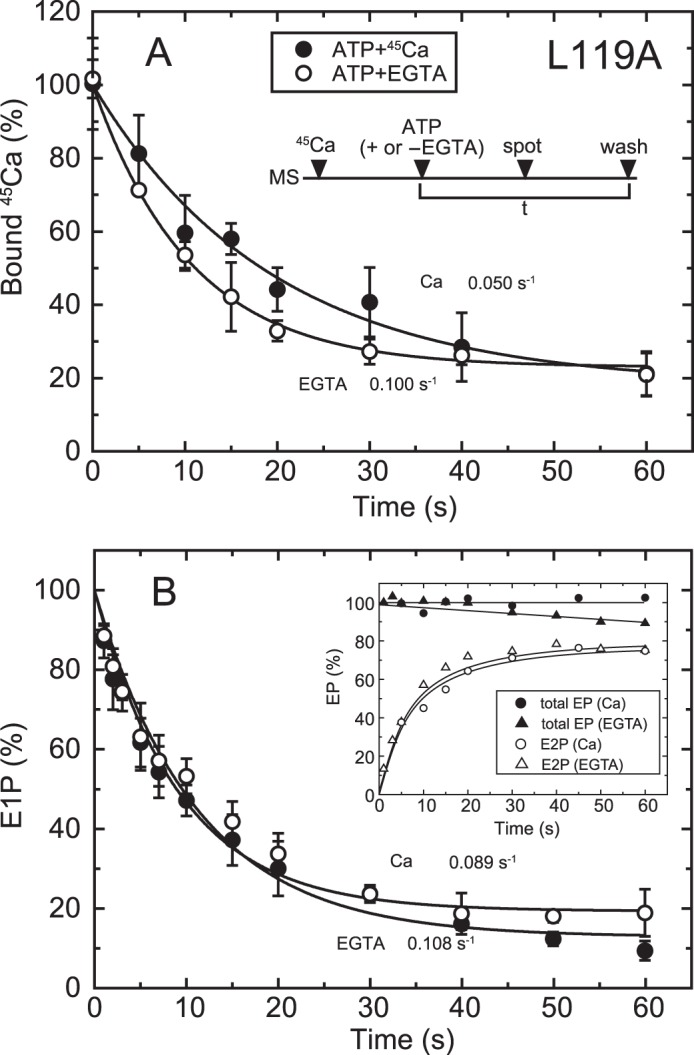
Time courses of Ca2+ release and EP isomerization in mutant L119A. A, microsomes (MS) expressing mutant L119A (20 μg/ml microsomal protein) were incubated for 20 s with 10 μm 45Ca2+ in 50 mm MOPS/Tris (pH 7.3), 0.1 m KCl, 7 mm MgCl2, and 3 μm A23187. Then, at zero time (ATP + or −EGTA), phosphorylation was initiated by addition of 10 μm ATP with (open circles) or without 1 mm EGTA (closed circles), the mixture was spotted on the filter, and the amount of 45Ca2+ specifically bound to the Ca2+-ATPase was determined as in Fig. 5B. The values presented are the mean ± S.D. (n = 3–5). The solid lines show a single exponential fit, and the rates of the 45Ca2+ release thus determined are 0.10 s−1 in an excess of EGTA (open circles) and 0.05 s−1 in the presence of 10 μm 45Ca2+ (closed circles). B, inset, microsomes expressing the mutant L119A (40 μg/ml microsomal protein) in 10 μm Ca2+ were phosphorylated at zero time by mixing with an equal volume of a solution containing 20 μm [γ-32P]ATP and 10 μm CaCl2 (circles) or 2 mm EGTA (triangles), and the total amount of EP (closed symbols) and the amount of E2P (open symbols) were determined at the indicated times as in Fig. 5A. Main panel, the fraction of E1P in the total amount of EP was calculated at each time point by subtracting the amount E2P from the total amount of EP (E1P plus E2P). The solid lines show a single exponential fit, and the rates of the decrease in E1P fraction, i.e. the E1P to E2P isomerization, thus determined are 0.108 s−1 in an excess of EGTA (open circles) and 0.089 s−1 in the presence of 10 μm Ca2+ (closed circles).
FIGURE 7.
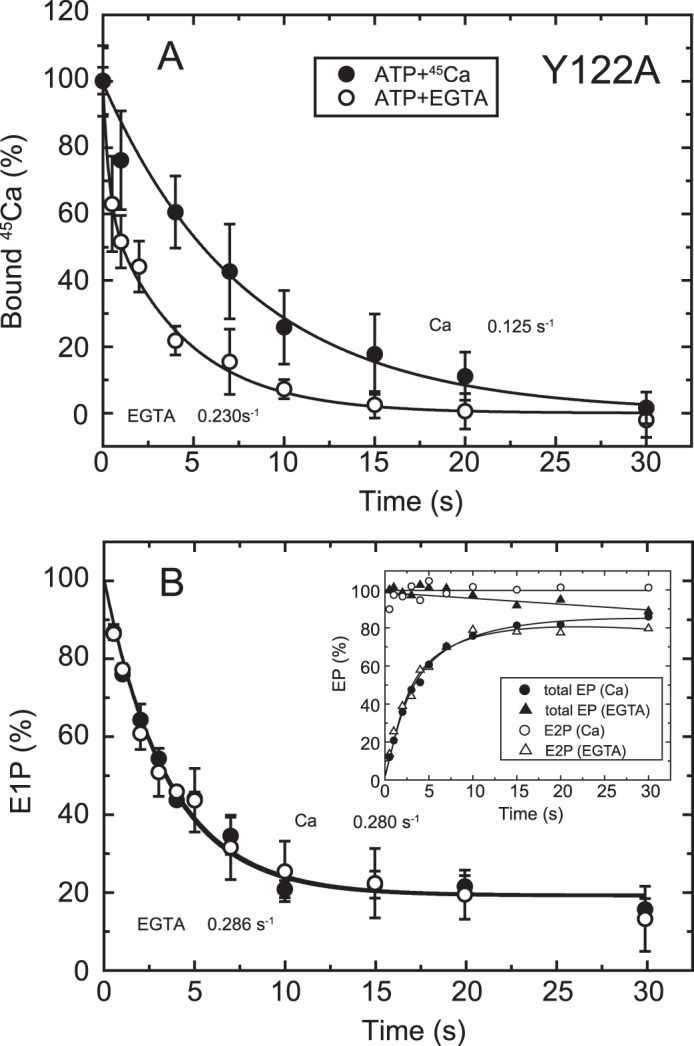
Time courses of Ca2+ release and EP isomerization in mutant Y122A. A, Ca2+ release from EP in the mutant Y122A was determined as in Fig. 6A. The values presented are the mean ± S.D. (n = 3–5). The solid lines show a single exponential fit, and the rates of the 45Ca2+ release thus determined are 0.23 s−1 in an excess of EGTA (open circles) and 0.125 s−1 in the presence of 10 μm 45Ca2+ (closed circles). B, inset, the total amount of EP (closed symbols) and the amount of E2P (open symbols) were determined at the indicated times with the mutant Y122A as described in Fig. 6B. Main panel, the fraction of E1P in the total amount of EP was calculated at each time point by subtracting the amount E2P from the total amount of EP (E1P plus E2P). The solid lines show a single exponential fit, and the rates of the decrease in E1P fraction, i.e. the E1P to E2P isomerization, thus determined are 0.286 s−1 in an excess EGTA (open circles) and 0.280 s−1 in the presence of 10 μm Ca2+ (closed circles).
FIGURE 8.
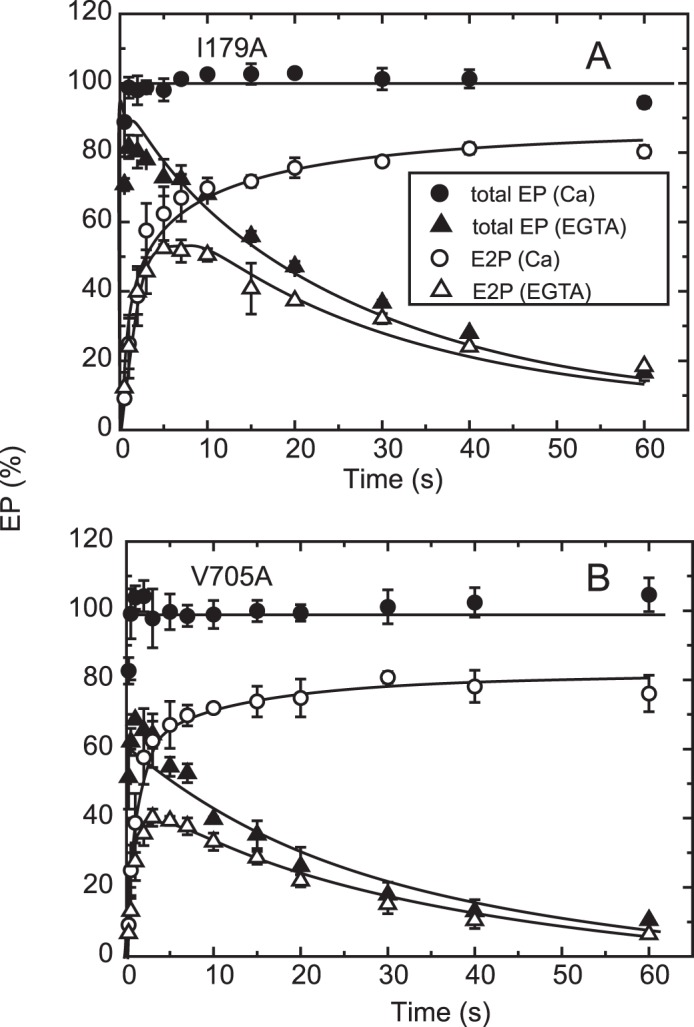
Time courses of EP isomerization in mutants I179A and V705A. The mutants I179A (A) and V705A (B) were phosphorylated with ATP, and the total amount of EP (closed symbols) and the amount of E2P (open symbols) were determined at the indicated time in an excess of EGTA (triangles) or in 10 μm Ca2+ (circles) as described in the inset in Fig. 6B. The fraction of E1P in the total amount of EP in the presence of 10 μm Ca2+ was calculated at each time point by subtracting the amount E2P from the total amount of EP (E1P plus E2P) and is depicted in Fig. 9B.
FIGURE 9.
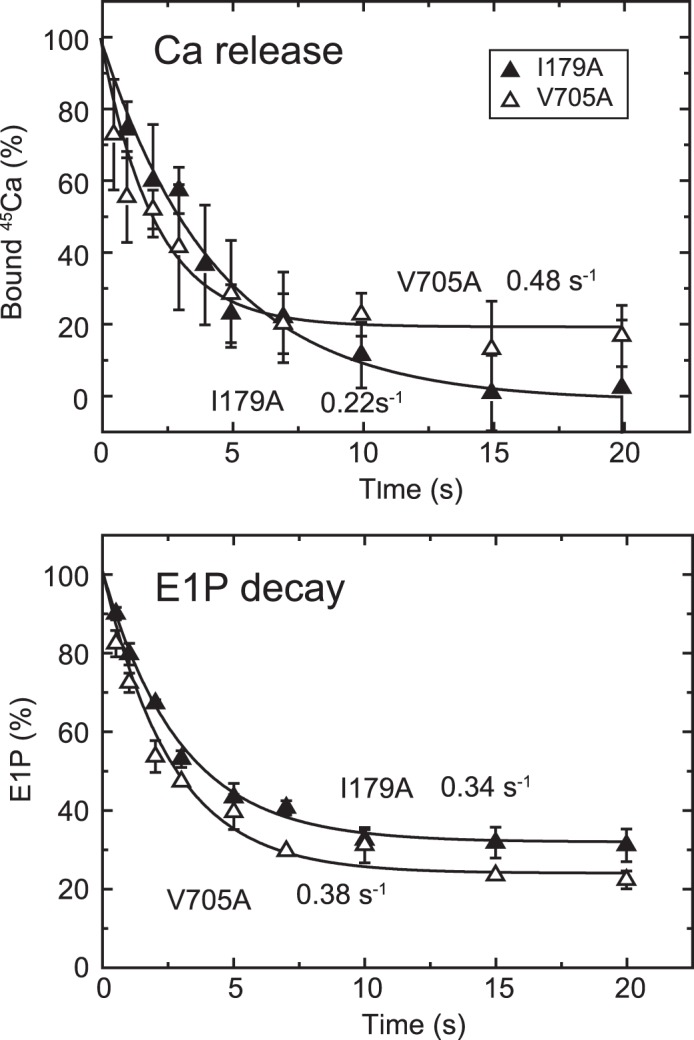
Time courses of Ca2+ release and EP isomerization in the presence of 10 μm Ca2+ in mutants I179A and V705A. A, Ca2+ release from EP in the mutants I179A and V705A was determined in the presence of 10 μm 45Ca2+ as described in Fig. 6A. The values presented are the mean ± S.D. (n = 3–5). The solid lines show a single exponential fit, and the rates of the 45Ca2+ release thus determined are 0.22 s−1 in I179A (closed triangles) and 0.48 s−1 in V705A (open triangles). B, the fraction of E1P in the total amount of EP was calculated at each time point in the presence of 10 μm Ca2+ in Fig. 8 by subtracting the amount E2P from the total amount of EP (E1P plus E2P). The values presented are the mean ± S.D. (n = 3). The solid lines show a single exponential fit, and the rates of the decrease in E1P fraction, i.e. the E1P to E2P isomerization, thus determined are 0.34 s−1 in I179A (closed triangles) and 0.38 s−1 in V705A (open triangles).
FIGURE 10.
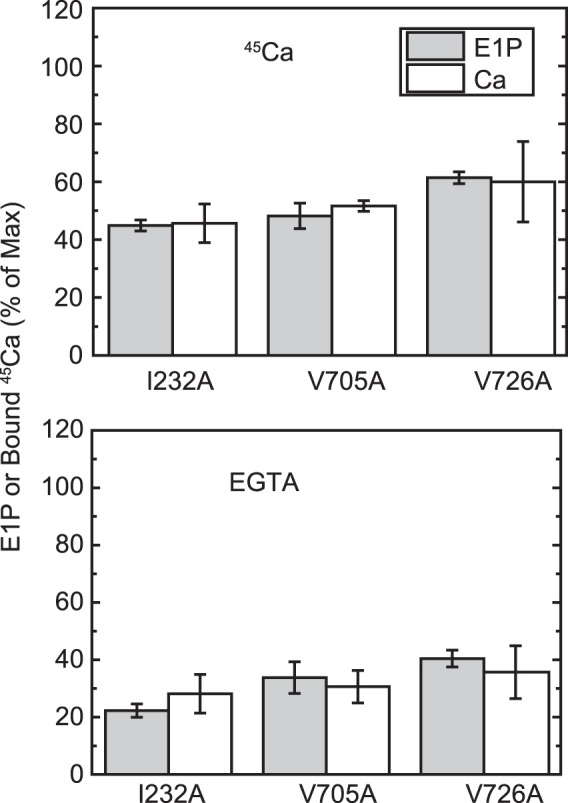
E1P fraction and bound Ca2+. The amount of bound Ca2+ and the E1P fraction in total amount of EP (E1P plus E2P) were determined repeatedly in the presence of an excess 1 mm EGTA (bottom panel) or 10 μm Ca2+ (top panel) or at one selected time point during the EP isomerization and Ca2+ release time courses (i.e. 2 s after the start of reaction) for the mutants I232A and V726A in comparison with the mutant V705A, as described in Fig. 6. The E1P fraction in the total amount of EP and the amount of bound 45Ca2+ relative to the maximum 45Ca2+ binding determined at zero time are shown as indicated (E1P and bound 45Ca, respectively). The values presented are the mean ± S.D. (n = 3–4). It should be noted that the Ca2+-ATPase is dephosphorylated to the E2 state upon Ca2+ removal by an excess of EGTA (bottom panel) and is in all phosphorylated states in the presence of 10 μm Ca2+ (top panel, see Figs. 6–8). Therefore, there is no Ca2+ bound to non-phosphorylated enzyme under our experimental conditions.
Rapid Luminal Ca2+-induced Reverse Conversion E2P + 2Ca2+ → E2PCa2 → E1PCa2 and Stabilization of E1PCa2
In Fig. 3, E2P was first formed with Pi and Mg2+ in the reverse reaction of hydrolysis, and then, at zero time, its decay was initiated by a large dilution with or without 10 mm Ca2+ in the presence of the Ca2+ ionophore A23187. When 10 mm Ca2+ is added, all E2P becomes stable E1P (as judged from its ADP sensitivity) because of Ca2+ binding to the luminally oriented, low-affinity Ca2+ transport sites (18, 22), in contrast to the rapid E2P hydrolysis without Ca2+ addition. The results show that E2P is very rapidly converted to E1PCa2 upon addition of 10 mm Ca2+, and E1PCa2 thus formed is stabilized in 10 mm Ca2+, in agreement with previous reports (23, 26–29). The results also suggest that E2P with bound Ca2+ can be fixed in the stable E1PCa2 by reverse conversion.
FIGURE 3.
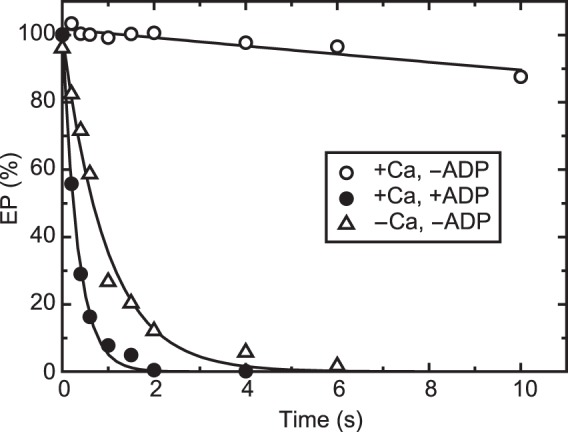
Stabilization of E1PCa2 formed by reverse conversion from E2P. Wild-type Ca2+-ATPase in microsomes was phosphorylated at 25 °C with 0.1 mm 32Pi in a mixture containing 200 μg/ml microsomal protein, 50 mm MOPS/Tris (pH 7.3), 7 mm MgCl2, 1 mm EGTA, 30 μm A23187, 7 mm MgCl2, and 20% (v/v) Me2SO that strongly favors E2P formation. The mixture was chilled at 4 °C and then, at zero time, diluted with a 19-fold volume of a solution containing 50 mm HEPES/Tris (pH 8.0), 0.105 m KCl, 7 mm MgCl2, and 10.5 mm CaCl2 (open and closed circles) or 1 mm EGTA (open triangles) in the absence (open symbols) or presence (closed circles) of 10.5 mm ADP, and the amount of EP was determined at the indicated times.
Ca2+ Binding to the Non-phosphorylated Wild Type
Considering all of these advantages of rapid formation and stabilization of E1PCa2 by the simultaneous addition of high concentrations of Ca2+ (10 mm) and ATP, we first determined the bound but non-occluded Ca2+ in wild type E1Ca2 by membrane filtration, as shown in Fig. 4, top panel. Microsomes containing expressed Ca2+-ATPase were incubated with various concentrations of 45Ca2+, spotted on a membrane filter, and exposed to continuous washing with Ca2+ binding assay medium containing 0.1 mm ATP and 10 mm non-radioactive Ca2+ for 3 s, during which E1PCa2 is rapidly formed and stabilized and free 45Ca2+ is washed out. The amount of 45Ca2+ specifically bound to the Ca2+-ATPase was obtained by subtracting the background determined in the presence of TG from that in its absence. The 45Ca2+ concentration dependence shows a saturation curve with a Kd value and Hill coefficient of 0.22 μm and 1.64, respectively, and a stoichiometry of maximum binding of ∼2 Ca2+ per catalytic site (maximum amount of EP, Fig. 4C), in complete agreement with the properties of the high-affinity transport sites established with sarcoplasmic reticulum vesicle Ca2+-ATPase (38) (whose values actually observed by this method are a Kd of 0.25 μm, a Hill coefficient of 1.61, and a stoichiometry of ∼2). Therefore, this method clearly fixes the 45Ca2+ originally bound in E1Ca2 of expressed SERCA1a in an occluded form as E1PCa2 before its release and exchange with the non-radioactive Ca2+ at 10 mm in the Ca2+ binding assay medium. This validates the method for measuring Ca2+ binding for small quantities of expressed SERCA1a.
The results also imply that there is virtually no exchange of the bound 45Ca2+ in E1PCa2 with the subsequently added 10 mm non-radioactive Ca2+. As noted above, this is probably due to stabilization of E1PCa2 by Ca2+ substituting for Mg2+ at the catalytic Mg2+ subsite (23, 26–29) rather than the Ca2+-induced reverse conversion in the equilibrium E1PCa2 ↔ E2P + 2Ca2+, which would result in Ca2+ exchange at the transport sites. Notably, also, previous analyses of Ca2+ binding/release in non-phosphorylated enzyme E1Ca2 ↔ E2 + 2Ca2+ shows (39–41) that the Ca2+ bound at transport site II is exposed to the medium and exchangeable with medium Ca2+, whereas Ca2+ bound at the deeply located site I is not released (unless site II becomes empty). ATP-induced phosphorylation to E1PCa2 requires both Ca2+ to be bound. Our results indicate that E1PCa2 formation upon addition of ATP and 10 mm Ca2+ is sufficiently fast to avoid such a Ca2+ exchange at site II, thereby trapping both of the 45Ca2+ originally bound in the E1Ca2 state as E1PCa2.
Time Course of Ca2+ Release and EP Isomerization in the Wild Type
The Ca2+ binding assay was applied to analyze the EP isomerization and Ca2+ release process in the wild type (Fig. 5). The experiments were performed in the presence of 0.1 m KCl, in which E2P does not accumulate because of its rapid hydrolysis (37), and, therefore, the EP decay reflects rate-limiting E1PCa2 isomerization to E2P (as shown in our previous report, cf. Fig. 2A in Ref. 42). In Fig. 5B, the expressed wild type was incubated with 10 μm 45Ca2+ and phosphorylated with ATP to form E1PCa2, and then an aliquot of the sample was placed on the membrane filter. At zero time, E1PCa2 decay was initiated by removal of 45Ca2+ by excess EGTA, and, after the indicated time periods, the sample was washed for 3 s with Ca2+ binding assay medium containing 10 mm non-radioactive Ca2+. For the determination of EP, [γ-32P]ATP and non-radioactive Ca2+ were used. The decrease in EP (representing EP isomerization) and that in bound Ca2+ coincide perfectly with the established mechanism in the wild type. The rate-limiting E1PCa2 to E2PCa2 isomerization is followed by the very rapid Ca2+ release E2PCa2 → E2P + 2Ca2+. The results obtained by this method are also in complete agreement with those established with sarcoplasmic reticulum vesicles, which contain a large proportion of Ca2+-ATPase, by the usual direct Ca2+ binding assay (i.e. without washing of the membrane filter) (42). Therefore, the Ca2+ release kinetics associated with EP isomerization can be followed by this assay using even small quantities of enzyme expressed in COS-1 cell microsomes.
Ca2+ Release in 10 μm Ca2+ Is Slower than EP Isomerization in the L119A Mutant
Detailed kinetic measurements of EP isomerization and Ca2+ release were made, first with the mutant L119A. We focused on residue Leu119 first because proteinase K-specific cleavage occurs at Leu119 in E2PCa2 (E2P with occluded Ca2+ trapped by elongation of the A/M1′ linker) and its analog E2·BeF3−·Ca2, but not in the Ca2+-released E2P ground state and its analog E2·BeF3− (7, 19, 20), and Leu119 is critical in the E2P ground state with reduced Ca2+ affinity (18). We hypothesized that this residue on the cytoplasmic part of M2, possibly its gathering into the Tyr122 hydrophobic cluster, may be involved in Ca2+ release during E2P processing and Ca2+ release (E2PCa2 → E2P + 2Ca2+). First, we found, as shown in Fig. 4B, that the properties of the Ca2+ binding sites of non-phosphorylated L119A (expression level about half of that of the wild type) are a Kd of 0.29 μm, a Hill coefficient of 1.61, and the stoichiometry 2 per one catalytic site (Fig. 4C), essentially the same as those of the wild type (cf. Fig. 4A). After establishing this stoichiometry of Ca2+ binding, we compared, in Fig. 6, the rate of Ca2+ release with that of EP isomerization.
In Fig. 6A, E1Ca2 was formed with saturating 10 μm 45Ca2+ and ionophore A23187, and phosphorylated at zero time to form E1PCa2 by the addition of 10 μm ATP with and without excess EGTA, and then the sample was spotted on the membrane filter, otherwise as in Fig. 5B. E1PCa2 is rapidly formed, and, as seen in Fig. 6B, there is almost no change in the total amount of EP during the time period because of the nearly complete block of E2P hydrolysis by the Leu119 mutation, whereas the E2P fraction increases gradually upon EP isomerization. When free 45Ca2+ is removed by EGTA, 45Ca2+ release takes place at the same rate as that of E2P formation. On the other hand, in the presence of 10 μm 45Ca2+, 45Ca2+ release is slower than E2P formation by ∼2-fold as a result of EP isomerization. The results suggest that, in 10 μm Ca2+, there may be a transient intermediate E2P state with bound Ca2+ at the transport sites with still a rather high affinity. Therefore, in this L119A mutant, Ca2+ affinity reduction at the transport sites likely occurs after EP isomerization (E1PCa2 → E2PCa2 at the catalytic site) and release gate opening, causing Ca2+ release to be retarded. Note that, for the wild type, this type of kinetic experiment in the presence of 10 μm Ca2+ is not feasible because, after Ca2+ release, E1Ca2 is quickly regenerated upon rapid E2P hydrolysis to E2, followed by immediate Ca2+ rebinding. The results with the L119A mutant nevertheless suggest that some structural rearrangement takes place at the Leu119 region on the cytoplasmic part of M2, possibly formation of the Tyr122 hydrophobic cluster, to effect Ca2+ affinity reduction and release.
EP Isomerization and Ca2+ Release in other Tyr122 Hydrophobic Cluster Mutants
To further explore possible roles in the Ca2+ release process of other residues in the Tyr122 hydrophobic cluster, i.e. the hydrophobic interactions of the cytoplasmic part of M2 with the A and P domains and A/M3 linker (Leu119/Tyr122 (M2), Ile179/Leu180 (A), Val705/Val726 (P), and Ile232 (A/M3 linker), see Fig. 11), we performed the same set of detailed kinetic measurements with mutant Y122A on the cytoplasmic part of M2 and I179A and V705A as representative for A and P domain interactions, respectively. It should be mentioned here that the stoichiometry of the maximum 45Ca2+ binding in Y122A, I179A, and V705A (Ca/EP determined as in Fig. 4C) are 2.03, 1.72, and 2.18, respectively, very similar to those in L119A and the wild type (1.97 and 2.11, respectively, see the legend for Fig. 4). Furthermore, the Kd (μm) and Hill coefficient of the Ca2+ binding sites determined with the Ca2+ concentration dependence of E1PCa2 formation from E1Ca2 are also in essential agreement in these mutants and the wild type, showing respective values 0.31 and 1.93 (Y122A), 0.24 and 1.98 (I179A), 0.35 and 1.90 (L119A), 0.61 and 1.51 (V705A), and 0.29 and 1.91 (wild type) (see Table 1 in Ref. 17).
FIGURE 11.

Structures at Leu119/Tyr122 region on the cytoplasmic part of M2 and Tyr122 hydrophobic cluster formation in E2·BeF3− and E2·BeF3−(TG). The structures in E2·BeF3− (PDB codes 2ZBE (Ref. 12) and 3B9B (Ref. 13)) and in E2·BeF3−(TG) (PDB code 2ZBF (Ref. 12)) are shown as a schematic. The cytoplasmic domains A, P, and N and the cytoplasmic part of M2 are colored yellow, cyan, pink, and purple, respectively. The residues involved in the formation of Tyr122 hydrophobic cluster (Leu119/Tyr122 on the cytoplasmic part of M2, Ile179/Leu180 on the A domain, Val705/Val726 on the P domain, and Ile232 on the A/M3 linker) are shown with van der Waals spheres and are colored green (Leu119/Tyr122), brown (Ile179/Leu180), and orange (Val705/Val726/Ile232).
In Y122A, as in L119A, E2P hydrolysis was nearly completely blocked (17), and the single exponential E2P accumulation in EP isomerization was observed with the same rates in the presence and absence of 10 μm Ca2+ (Fig. 7B). The 45Ca2+ release in 10 μm 45Ca2+ occurs in a single exponential manner (Fig. 7A) and is slower than E2P formation by more than 2-fold. Therefore, in Y122A, as in L119A, Ca2+ release after E2P formation is retarded in 10 μm Ca2+.
The 45Ca2+ release in Y122A upon 45Ca2+ removal by EGTA exhibits interesting kinetics. Immediately after removal, some portion of the bound 45Ca2+ (∼35% of the total, Fig, 7A, open circles) disappeared rapidly before formation of E2P (Fig. 7B, inset, closed triangles). The remaining part of the bound Ca2+ is released in a single exponential manner at the same rate as that of E2P formation in EP isomerization. This result suggests that the Ca2+-occluded structure in E1PCa2 is destabilized in this mutant so that a fraction of the Ca2+ of E1PCa2 is released rapidly (without affinity reduction because such a rapid release does not occur in 10 μm Ca2+). Similarly, we previously observed Ca2+ release (escape) from E1PCa2 without affinity reduction in the wild type in the absence of K+ and indicated that the stabilization of E1PCa2, in this case by the K+ binding on its specific site on the P domain, is crucial for stabilizing the Ca2+-occluded structure of E1PCa2 (42). Therefore, similar to the bound K+, Tyr122 is likely important for the structural stabilization of E1PCa2 with occluded Ca2+.
In the alanine mutants of Ile179 and Val705 that form the Tyr122 hydrophobic cluster, EP decay occurs fairly fast because of the faster E2P hydrolysis, as found previously (16, 17) (Fig. 8, see the decay after the Ca2+ removal (triangles), although it is still much slower than the wild type (cf. Fig. 5)). Nevertheless, within the initial time period of ∼20 s in the presence of 10 μm Ca2+, during which the amount of original EP species remains significant, we were able to compare E2P formation and Ca2+ release. We found, with V705A (Fig. 9), that the Ca2+ release almost coincides with E2P formation, therefore the Ca2+ release from E2P is not retarded by this P domain mutation. For the mutants V726A on the P domain and I232A on the A/M3 linker, we repeatedly determined the amount of E2P and that of bound Ca2+ at a time point during the EP isomerization when the amount of E2P fraction reaches ∼40–60% (Fig. 10). In both mutants, the bound Ca2+ and the E2P fraction are nearly the same, therefore there is no indication of retardation of Ca2+ release from E2P in these mutants as in V705A.
On the other hand, with mutant I179A on the A domain, Ca2+ release was obviously slower than the EP isomerization (Fig. 9); therefore, Ca2+ release is retarded. A curious observation with this mutant was that apparently all of the 45Ca2+ is released, although ∼30% of the total amount of EP remains as E1P. This again suggests that the Ca2+-occluded structure of E1PCa2 is partly perturbed in the mutant I179A, in this case causing a slow Ca2+ release. The L180A mutant does not accumulate E2P significantly at steady state, as found previously (18). Therefore, it was not possible to compare the kinetics of E2P formation and Ca2+ release.
Discussion
We have been able to show that several mutations at an interdomain junction at the cytoplasmic part of transmembrane helix M2 allow a separation of steps during luminal Ca2+ release and phosphoenzyme isomerization, steps that, in the wild type, are too fast to distinguish. This has been possible through the development of an assay that specifically measures bound, not occluded, Ca2+ during the release process. The difficulties in determining bound Ca2+ are to fix it to the protein during washing and to obtain a sufficiently high specific signal over the background, both hurdles especially acute with the small quantity of Ca2+-ATPase expressed in COS-1 cell microsomes (∼1% of the total protein). Our assay overcomes such problems by almost instantaneously converting the bound Ca2+ to a stable occluded form.
EP Isomerization with Delayed Ca2+ Release
In the mutants L119A and Y122A on the cytoplasmic part of M2, Ca2+ release in the presence of 10 μm 45Ca2+ is slower than EP isomerization, whereas they coincide in the absence of Ca2+. Retarded release in 10 μm 45Ca2+ is also observed with the mutant I179A on the A domain. Such results indicate an E2P species with partially exchangeable Ca2+ that binds luminal Ca2+ with relatively high affinity and slowly proceeds to the Ca2+-released form of E2P with affinity reduction. These attributes fit with our previous findings in which we analyzed luminal Ca2+-induced reverse conversion of E2P (formed by Pi without Ca2+) to E1PCa2 (18) and found that the E2P of these mutants have luminally facing transport sites with a Kd 120–250 μm for Ca2+, which is ∼10 times lower than that of the wild type (Kd 1.48 mm). The kinetic analysis of the reverse conversion further indicated that the Ca2+ release path is not fully opened in these mutants compared with the wild type (18). It seems that, in the transient E2P with bound but deoccluded Ca2+ in the mutant L119A, Y122A, or I179A, the affinity reduction is less (Kd change from low micromolar to 120–250 μm), allowing Ca2+ release at moderate free Ca2+ concentrations.
These findings can be described by assuming a transient state *E2P that possesses a high affinity Ca2+ site(s) facing the lumen (therefore, Ca2+ release and rebinding takes place in the presence of luminal Ca2+ as low as 10 μm) and proceeds to the E2P state with affinity reduction, as E1PCa2 ↔ *E2PCa2 ↔ E2PCa2 ↔ E2P + 2Ca2+. In the wild type, *E2PCa2 ↔ E2PCa2 and subsequent Ca2+ (45Ca2+) release are very rapid; therefore, the *E2P species before the affinity reduction with bound 45Ca2+ is not trapped by the added non-radioactive 40Ca2+. The trapping of bound 45Ca2+ in the *E2P species in the mutants L119A, Y122A, and I179A in the presence of 10 μm 45Ca2+ upon addition of non-radioactive 10 mm 40Ca2+ points to a possible sequential Ca2+ release/rebinding as E1PCa2 ↔ *E2PCa2 ↔ *E2PCa + Ca2+ ↔ *E2P + 2Ca2+ ↔ E2P + 2Ca2+, in which at least one bound 45Ca2+ is trapped (as *E2P45Ca40Ca by 45Ca2+-40Ca2+ exchange in *E2P) in the reverse conversion to E1PCa2 by the added 10 mm 40Ca2+. Such a sequential mechanism fits with the Ca2+ release path in E2P being not fully opened in these mutants. However, it is also possible that nonspecific Ca2+ binding to luminal polar and negatively charged (gating) residues (43) prevents full 45Ca2+ release or that Ca2+ substitution for Mg2+ at the catalytic Mg2+ subsite stabilizes *E2P with bound Ca2+, as happens in E1PCa2 (23, 26–29). Whatever the mechanism, we do not mean to imply that all bound 45Ca2+ in E2P is fixed in the conversion to the occluded E1PCa2 state but that at least some is.
Structural Rearrangements for Luminal Ca2+ Release during E2P Processing
In the intermediate state E2P with occluded Ca2+ (E2PCa2) and its structural E2·BeF3−·Ca2 analog trapped by elongation of the A/M1′ linker, the Leu119-specific site is cleaved by proteinase K and, therefore, sterically exposed, which is in contrast to the complete resistance in the Ca2+-released E2P ground state and its analog E2·BeF3−, formed from E2 in the absence of Ca2+ and in its TG-bound state E2·BeF3−(TG) (7, 19, 20). Evidently, some structural rearrangement takes place at the flexible Leu119 and Tyr122 region on the cytoplasmic part of M2 and Ile179 region on the A domain to effect Ca2+ deocclusion and release. Tyr122/Leu119 on the cytoplasmic part of M2 forms a hydrophobic interaction network (Tyr122 hydrophobic cluster) with five other residues on the A and P domains and the A/M3 linker (Ile179/Leu180, Val705/Val726, and Ile232) in E2P (Fig. 11), and Ile179 is positioned most closely to Leu119/Tyr122 on the cytoplasmic part of M2 (Fig. 11, 2ZBE and 3B9B). This cluster formation has been found previously (17, 18) to be critical for the E2P ground state structure with potential hydrolytic activity at the catalytic site and luminally opened transport sites with reduced Ca2+ affinity. Therefore, the assembly of Tyr122/Leu119 on the cytoplasmic part of M2 with the other residues and formation of the Tyr122 hydrophobic cluster is almost certainly accomplished during E2P processing to the E2P ground state (E2PCa2 → E2P + 2Ca2+) (Fig. 12, schematic).
FIGURE 12.

Schematic for E2P processing and Ca2+ handling. Top panel, structures of E1Ca2·AlF4−·ADP (a structural analog for the transition state in phosphorylation, E1PCa2·ADP‡, PDB code 1T5T (Ref. 10)), E2·BeF3− (PDB code 2ZBE (Ref. 12)), E2·BeF3−(TG) (PDB code 2ZBF (Ref. 12)), and E2·AlF4−(TG) ((a structural analog for the transition state in hydrolysis, E2P‡, PDB code 1XP5 (Ref. 11)) are shown as a schematic. In these structures, the residues involved in the formation of the Tyr122 hydrophobic cluster (Y122-HC) are depicted with van der Waals spheres and are colored as in Fig. 10 (see the residue numbers in Fig. 10). The cytoplasmic part of M2 and the TGES184 loop are colored in purple and blue, respectively, and the A, P, and N domains are colored in yellow, cyan, and pink, respectively. The open or closed state of the Ca2+ path (luminal gate) and the assembling state of the Tyr122 hydrophobic cluster are indicated above the structures. Bottom panel, schematic of the structural changes for Tyr122 hydrophobic cluster formation and for the property of the Ca2+ transport sites and release gate during E2P processing with Ca2+ release (E2PCa2 to E2P). In this model, the main body of Ca2+-ATPase is shown in red, orange, and yellow to indicate the open high-affinity, open low-affinity, and closed states, respectively, for the property of the Ca2+ transport sites and release gate. The green semicircle indicates the part of Tyr122 hydrophobic cluster formed in E2PCa2 upon EP isomerization (E1PCa2 → E2PCa2) and composed of Ile179/Leu180 on the A domain, Val705/Val726 on the P domain, and Ile232 on the A/M3 linker. The pink structure indicates the cytoplasmic part of M2, including Leu119 and Tyr122. During E2P processing, Tyr122/Leu119 gathers to Ile179 and then to the assembled five residues, completing the Tyr122-hydrophobic cluster, which is coupled with affinity reduction. Mutation of Leu119, Tyr122, or Ile179 probably destabilizes the hydrophobic cluster, thereby retarding affinity reduction. Also note that our previous detailed kinetic study has revealed (18) that the E2P ground state structure has a closed luminal gate and that the Tyr122 hydrophobic cluster is tightly fixed (as described under “Discussion”), which is depicted here with a yellow body.
However, in the crystal structure E2·BeF3−, Leu119/Tyr122 are not yet associated with the other five clustered residues despite being very close to Ile179, which is already gathered with the other four hydrophobic residues (Leu180, Val705/Val726, and Ile232) (12, 13), whereas all seven residues are assembled in the thapsigargin-fixed structure E2·BeF3−(TG) (Fig. 11) as well as in E2·AlF4−(TG), E2·MgF42−(TG), and E2(TG) and E2 (6, 9, 12, 13, 15).
Therefore, at first glance, the presently available E2·BeF3− crystal structures seem to not fit with the clustering having a critical function in the E2P ground state. However, it is possible that interaction of the cytoplasmic part of M2 Leu119/Tyr122 with the A domain Ile179 is the final process in assembling the Tyr122 hydrophobic cluster, and, for some reason, it is not seen in the analog crystal structures. The cytoplasmic part of M2 in the E2P ground state appears rather flexible, judging from the lack of interactions here in the crystal structures, and the absence of Ca2+ at the transport sites may have something to do with this. We can hypothesize that even mild perturbations, such as detergent solubilization, as well as mutations here could keep the residues apart. In fact, TG binding rearranges the helices in this state to produce a tightly closed gate.
It is also of interest to note that, in our detailed analyses of the Ca2+ dependences of the luminal Ca2+-induced reverse conversion kinetics E2P + 2Ca2+ → E1PCa2 and its analog E2·BeF3− + 2Ca2+ → E1Ca2·BeF3−, the K0.5 of luminal Ca2+ was found to be 1.5 mm for E2P (18) and 0.4 mm for E2·BeF3− (14). Therefore, if this is not a kinetic effect, then the affinity in E2·BeF3− is somewhat higher than that for E2P even though E2·BeF3− seemingly possesses all of the characteristics of the E2P ground state; i.e. a hydrophobic catalytic site, the same cytoplasmic domain organization, luminally open low-affinity transport sites, and the same intrinsic tryptophan fluorescence level that reflects arrangement of the transmembrane helices. It is definitely an E2P ground state structural analog (7), and yet there may be subtle differences that may arise from a slight difference in the active site between the covalently bound phosphate at the catalytic aspartate (Asp351) and the E2·BeF3− directly ligated with this aspartate. Alternatively, the crystallization process in detergent may impose a difference, possibly by selecting one of the putative flexible/fluctuating structures, somewhat between an “open high-affinity” state and an “open low-affinity” state. Again, we assert that the E2P ground state does, in fact, have a fully assembled hydrophobic cluster that completes the Ca2+ release process, otherwise it is difficult to explain the mutational effects.
We summarized our findings in a schematic in Fig. 12, which depicts that, after EP isomerization (E1PCa2 → E2PCa2 and loss of ADP sensitivity), the transport sites proceed from a state of open high affinity to one of open low affinity for Ca2+ release. The rearrangement on the cytoplasmic part of M2 (Tyr122/Leu119) and its assembly into a hydrophobic cluster are accomplished in this affinity reduction process by gathering of Tyr122/Leu119 on the cytoplasmic part of M2 with the Ile179/Leu180 region on the A domain that is already associated with the P domain during the EP isomerization E1PCa2 → E2PCa2. In the mutants L119A/Y122A and I179A, the Tyr122 hydrophobic cluster is destabilized, and formation of the open low-affinity state is retarded.
It is not clear why the mutations of residues Val705/Val726 on the P domain and Ile232 on the A/M3 linker in the hydrophobic cluster do not retard Ca2+ release, but it is probably because there are several interactions between the A and P domains, and a single conservative substitution here is not enough to disrupt A-P domain association. Also, these two domains are already associated in E2PCa2, and mild perturbations may be without effect on the later affinity reduction and release. There are large changes during EP isomerization. The A domain rotates and docks on the P domain, allowing Ile179/Leu180 on the A domain to cluster with Val705/Val726 on the P domain and Ile232 on the A/M3 linker associated with the P domain. In E2PCa2, the interactions at the Val200 loop on the A domain with polar residues on the P domain (19, 44) and those at the TGES184 loop on the A domain with the catalytic site on the P domain (which causes the loss of ADP sensitivity) are already produced. To effect affinity reduction and Ca2+ release, the associated A and P domains move together, allowing the cytoplasmic part of M2-A domain interaction at Tyr122/Leu119 and Ile179 to form and complete the critical Tyr122 hydrophobic cluster.
Regarding a possible “closed” state in the E2P ground state (Fig. 12), we have found previously, with detailed kinetics in the wild type (18), that the rate of luminal Ca2+-induced reverse conversion E2P + 2Ca2+ → E1PCa2 increases linearly with an increase in the luminal Ca2+ concentration and is not saturated even at 3 mm. This suggests that luminal Ca2+ access to the open low-affinity transport sites in E2P is rate-limiting, and, therefore, the gate in E2P may actually be closed, and the rate-limiting Ca2+ access may reflect opening of the closed state to an open low-affinity state. We have also found that this reverse conversion is retarded by alanine mutation of each of the seven residues in the Tyr122 hydrophobic cluster, and the extent of the retardation was almost the same in all seven mutants (Figs. 8 and 9 and supplemental Fig. 3 in Ref. 18). Therefore, all residues in the cluster seem important for reverse opening.
This finding (18) and the mutation-induced retardation of the forward Ca2+ release process found here can be accounted for by a destabilization of the open low-affinity state and transition states in the forward and reverse processes by the cluster mutations. Furthermore, the fact that all seven residues in the cluster influence the closed state implies that the change from the open low-affinity state to a tightly fixed closed state requires further rearrangements, which then permits advance to the transition state for hydrolysis (E2-P‡ mimicked by E2·AlF4−), in which the gate becomes tightly closed (7).
P-type ATPases possess a common molecular structure with N, P, and A domains connected to transmembrane helices, including the long M2 helix critical for gating (1–3), and almost certainly all of them utilize a common mechanism for ion pumping. We predict that ion release to the trans side of the membrane will entail rearrangements at the cytoplasmic part of M2 and engagement of the A and P domains. A stepwise affinity reduction and release on this trans side during phosphoenzyme isomerization may be discernable even in some wild-type pumps of other P-type ATPases.
Author Contributions
K. Y. and H. S. conceived and coordinated the study and wrote the paper. K. Y. designed, performed, and analyzed the experiments. T. D. provided critical discussions and technical advice. S. D. provided technical assistance and contributed to the preparation of the figures and manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. David H. MacLennan (University of Toronto), for SERCA1a cDNA and Dr. Randal J. Kaufman (Genetics Institute, Cambridge, MA) for the expression vector pMT2. We also thank Dr. David B. McIntosh for reviewing and improving the manuscript.
This work was supported by Grants-in-Aid for Scientific Research (C) (to K. Y.) and (B) (to H. S.) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan. The authors declare that they have no conflicts of interest with the contents of this article.
- SERCA
- sarcoplasmic reticulum Ca2+-ATPase
- TG
- thapsigargin.
References
- 1.Toyoshima C. (2008) Structural aspects of ion pumping by Ca2+-ATPase of sarcoplasmic reticulum. Arch. Biochem. Biophys. 476, 3–11 [DOI] [PubMed] [Google Scholar]
- 2.Toyoshima C. (2009) How Ca2+-ATPase pumps ions across the sarcoplasmic reticulum membrane. Biochim. Biophys. Acta 1793, 941–946 [DOI] [PubMed] [Google Scholar]
- 3.Møller J. V., Olesen C., Winther A.-M. L., and Nissen P. (2010) The sarcoplasmic Ca2+-ATPase: design of a perfect chemi-osmotic pump. Q. Rev. Biophys. 43, 501–566 [DOI] [PubMed] [Google Scholar]
- 4.Toyoshima C., Nakasako M., Nomura H., and Ogawa H. (2000) Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 Å resolution. Nature 405, 647–655 [DOI] [PubMed] [Google Scholar]
- 5.Danko S., Yamasaki K., Daiho T., Suzuki H., and Toyoshima C. (2001) Organization of cytoplasmic domains of sarcoplasmic reticulum Ca2+-ATPase in E1P and E1ATP states: a limited proteolysis study. FEBS Lett. 505, 129–135 [DOI] [PubMed] [Google Scholar]
- 6.Toyoshima C., and Nomura H. (2002) Structural changes in the calcium pump accompanying the dissociation of calcium. Nature 418, 605–611 [DOI] [PubMed] [Google Scholar]
- 7.Danko S., Yamasaki K., Daiho T., and Suzuki H. (2004) Distinct natures of beryllium fluoride-bound, aluminum fluoride-bound, and magnesium fluoride-bound stable analogues of an ADP-insensitive phosphoenzyme intermediate of sarcoplasmic reticulum Ca2+-ATPase. J. Biol. Chem. 279, 14991–14998 [DOI] [PubMed] [Google Scholar]
- 8.Toyoshima C., and Mizutani T. (2004) Crystal structure of the calcium pump with a bound ATP analogue. Nature 430, 529–535 [DOI] [PubMed] [Google Scholar]
- 9.Toyoshima C., Nomura H., and Tsuda T. (2004) Lumenal gating mechanism revealed in calcium pump crystal structures with phosphate analogues. Nature 432, 361–368 [DOI] [PubMed] [Google Scholar]
- 10.Sørensen T. L., Møller J. V., and Nissen P. (2004) Phosphoryl transfer and calcium ion occlusion in the calcium pump. Science 304, 1672–1675 [DOI] [PubMed] [Google Scholar]
- 11.Olesen C., Sørensen T. L., Nielsen R. C., Møller J. V., and Nissen P. (2004) Dephosphorylation of the calcium pump coupled to counterion occlusion. Science 306, 2251–2255 [DOI] [PubMed] [Google Scholar]
- 12.Toyoshima C., Norimatsu Y., Iwasawa S., Tsuda T., and Ogawa H. (2007) How processing of aspartylphosphate is coupled to lumenal gating of the ion pathway in the calcium pump. Proc. Natl. Acad. Sci. U.S.A. 104, 19831–19836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olesen C., Picard M., Winther A. M., Gyrup C., Morth J. P., Oxvig C., Møller J. V., and Nissen P. (2007) The structural basis of calcium transport by the calcium pump. Nature 450, 1036–1042 [DOI] [PubMed] [Google Scholar]
- 14.Danko S., Daiho T., Yamasaki K., Liu X., and Suzuki H. (2009) Formation of the stable structural analog of ADP-sensitive phosphoenzyme of Ca2+-ATPase with occluded Ca2+ by beryllium fluoride. J. Biol. Chem. 284, 22722–22735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Toyoshima C., Iwasawa S., Ogawa H., Hirata A., Tsueda J., and Inesi G. (2013) Crystal structures of the calcium pump and sarcolipin in the Mg2+-bound E1 state. Nature 495, 260–264 [DOI] [PubMed] [Google Scholar]
- 16.Yamasaki K., Daiho T., Danko S., and Suzuki H. (2004) Multiple and Distinct effects of mutations of Tyr122, Glu123, Arg324, and Arg334 involved in interactions between the top part of second and fourth transmembrane helices in sarcoplasmic reticulum Ca2+-ATPase. J. Biol. Chem. 279, 2202–2210 [DOI] [PubMed] [Google Scholar]
- 17.Wang G., Yamasaki K., Daiho T., and Suzuki H. (2005) Critical hydrophobic interactions between phosphorylation and actuator domains of Ca2+-ATPase for hydrolysis of phosphorylated intermediate. J. Biol. Chem. 280, 26508–26516 [DOI] [PubMed] [Google Scholar]
- 18.Yamasaki K., Wang G., Daiho T., Danko S., and Suzuki H. (2008) Roles of Tyr122-hydrophobic cluster and K+ binding in Ca2+-releasing process of ADP-insensitive phosphoenzyme of sarcoplasmic reticulum Ca2+-ATPase. J. Biol. Chem. 283, 29144–29155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Daiho T., Yamasaki K., Danko S., and Suzuki H. (2007) Critical role of Glu40-Ser48 loop linking actuator domain and first transmembrane helix of Ca2+-ATPase in Ca2+ deocclusion and release from ADP-insensitive phosphoenzyme. J. Biol. Chem. 282, 34429–34447 [DOI] [PubMed] [Google Scholar]
- 20.Daiho T., Danko S., Yamasaki K., and Suzuki H. (2010) Stable structural analog of Ca2+-ATPase ADP-insensitive phosphoenzyme with occluded Ca2+ formed by elongation of A-domain/M1′-linker and beryllium fluoride binding. J. Biol. Chem. 285, 24538–24547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamada S., and Ikemoto N. (1980) Reaction mechanism of calcium-ATPase of sarcoplasmic reticulum. Substrates for phosphorylation reaction and back reaction, and further resolution of phosphorylated intermediates. J. Biol. Chem. 255, 3108–3119 [PubMed] [Google Scholar]
- 22.de Meis L., and Inesi G. (1982) ATP synthesis by sarcoplasmic reticulum ATPase following Ca2+, PH, temperature, and water activity jumps. J. Biol. Chem. 257, 1289–1294 [PubMed] [Google Scholar]
- 23.Shigekawa M., Wakabayashi S., and Nakamura H. (1983) Reaction mechanism of Ca2+-dependent adenosine triphosphatase of sarcoplasmic reticulum. ATP hydrolysis with CaATP as a substrate and role of divalent cation. J. Biol. Chem. 258, 8698–8707 [PubMed] [Google Scholar]
- 24.Petithory J. R., and Jencks W. P. (1986) Phosphorylation of the calcium adenosinetriphosphatase of sarcoplasmic reticulum: rate-limiting conformational change followed by rapid phosphoryl transfer. Biochemistry 25, 4493–4497 [DOI] [PubMed] [Google Scholar]
- 25.Lacapere J. J., and Guillain F. (1990) Reaction mechanism of Ca2+ ATPase of sarcoplasmic reticulum: equilibrium and transient study of phosphorylation with Ca·ATP as substrate. J. Biol. Chem. 265, 8583–8589 [PubMed] [Google Scholar]
- 26.Shigekawa M., Wakabayashi S., and Nakamura H. (1983) Effect of divalent cation bound to the ATPase of sarcoplasmic reticulum: activation of phosphoenzyme hydrolysis by Mg2+. J. Biol. Chem. 258, 14157–14161 [PubMed] [Google Scholar]
- 27.Wakabayashi S., and Shigekawa M. (1984) Role of divalent cation bound to phosphoenzyme intermediate of sarcoplasmic reticulum ATPase. J. Biol. Chem. 259, 4427–4436 [PubMed] [Google Scholar]
- 28.Wakabayashi S., and Shigekawa M. (1987) Effect of metal bound to the substrate site on calcium release from the phosphoenzyme intermediate of sarcoplasmic reticulum ATPase. J. Biol. Chem. 262, 11524–11531 [PubMed] [Google Scholar]
- 29.Picard M., Jensen A. M., Sørensen T. L., Champeil P., Møller J. V., and Nissen P. (2007) Ca2+ versus Mg2+ coordination at the nucleotide-binding site of the sarcoplasmic reticulum Ca2+-ATPase. J. Mol. Biol. 368, 1–7 [DOI] [PubMed] [Google Scholar]
- 30.Kaufman R. J., Davies M. V., Pathak V. K., and Hershey J. W. (1989) The phosphorylation state of eucaryotic initiation factor 2 alters translational efficiency of specific mRNAs. Mol. Cell. Biol. 9, 946–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maruyama K., and MacLennan D. H. (1988) Mutation of aspartic acid-351, lysine-352, and lysine-515 alters the Ca2+ transport activity of the Ca2+-ATPase expressed in COS-1 cells. Proc. Natl. Acad. Sci. U.S.A. 85, 3314–3318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weber K., and Osborn M. (1969) The reliability of molecular weight determinations by dodecyl sulfate-polyacrylamide gel electrophoresis. J. Biol. Chem. 244, 4406–4412 [PubMed] [Google Scholar]
- 33.Daiho T., Suzuki H., Yamasaki K., Saino T., and Kanazawa T. (1999) Mutations of Arg198 in sarcoplasmic reticulum Ca2+-ATPase cause inhibition of hydrolysis of the phosphoenzyme intermediate formed from inorganic phosphate. FEBS Lett. 444, 54–58 [DOI] [PubMed] [Google Scholar]
- 34.Sagara Y., and Inesi G. (1991) Inhibition of the sarcoplasmic reticulum Ca2+ transport ATPase by thapsigargin at subnanomolar concentrations. J. Biol. Chem. 266, 13503–13506 [PubMed] [Google Scholar]
- 35.Lowry O. H., Rosebrough N. J., Farr A. L., and Randall R. J. (1951) Protein measurement with the folin phenol reagent. J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
- 36.Humphrey W., Dalke A., and Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 37.Shigekawa M., and Dougherty J. P. (1978) Reaction mechanism of Ca2+-dependent ATP hydrolysis by skeletal muscle sarcoplasmic reticulum in the absence of added alkali metal salts: II: kinetic properties of the phosphoenzyme formed at the steady state in high Mg2+ and low Ca2+ concentrations. J. Biol. Chem. 253, 1451–1457 [PubMed] [Google Scholar]
- 38.Inesi G., Kurzmack M., Coan C., and Lewis D. E. (1980) Cooperative calcium binding and ATPase activation in sarcoplasmic reticulum vesicles. J. Biol. Chem. 255, 3025–3031 [PubMed] [Google Scholar]
- 39.Inesi G. (1987) Sequential mechanism of calcium binding and translocation in sarcoplasmic reticulum adenosine triphosphatase. J. Biol. Chem. 262, 16338–16342 [PubMed] [Google Scholar]
- 40.Petithory J. R., and Jencks W. P. (1988) Binding of Ca2+ to the calcium adenosinetriphosphatase of sarcoplasmic reticulum. Biochemistry 27, 8626–8635 [DOI] [PubMed] [Google Scholar]
- 41.Orlowski S., and Champeil P. (1991) Kinetics of calcium dissociation from its high-affinity transport sites on sarcoplasmic reticulum ATPase. Biochemistry 30, 352–361 [DOI] [PubMed] [Google Scholar]
- 42.Yamasaki K, Daiho T, Danko S, and Suzuki H. (2010) Ca2+ release to lumen from ADP-sensitive phosphoenzyme E1PCa2 without bound K+ of sarcoplasmic reticulum Ca2+-ATPase. J. Biol. Chem. 285, 38674–38683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Webb R. J., Khan Y. M., East J. M., and Lee A. G. (2000) The importance of carboxyl groups on the lumenal side of the membrane for the function of the Ca2+-ATPase of sarcoplasmic reticulum. J. Biol. Chem. 275, 977–982 [DOI] [PubMed] [Google Scholar]
- 44.Kato S., Kamidochi M., Daiho T., Yamasaki K., Gouli W., and Suzuki H. (2003) Val200 residue in Lys189-Lys205 outermost loop on the A domain of sarcoplasmic reticulum Ca2+-ATPase is critical for rapid processing of phosphoenzyme intermediate after loss of ADP sensitivity. J. Biol. Chem. 278, 9624–9629 [DOI] [PubMed] [Google Scholar]
- 45.Barrabin H., Scofano H. M., and Inesi G. (1984) Adenosinetriphosphatase site stoichiometry in sarcoplasmic reticulum vesicles and purified enzyme. Biochemistry 23, 1542–1548 [DOI] [PubMed] [Google Scholar]