

Background: Mutations in retinal degeneration slow (RDS) lead to rod and cone-dominant retinal degeneration by mechanisms that remain unknown.
Results: Knockin mice carrying unglycosylated RDS have reduced RDS levels and functional defects in cones.
Conclusion: RDS glycosylation is important for cone but not rod function.
Significance: Differential reliance on glycosylation may help explain why some RDS disease mutations target cones and others target rods.
Keywords: glycosylation, oligomerization, photoreceptor, retina, tetraspanin, N229S, peripherin-2
Abstract
The photoreceptor-specific glycoprotein retinal degeneration slow (RDS, also called PRPH2) is necessary for the formation of rod and cone outer segments. Mutations in RDS cause rod and cone-dominant retinal disease, and it is well established that both cell types have different requirements for RDS. However, the molecular mechanisms for this difference remain unclear. Although RDS glycosylation is highly conserved, previous studies have revealed no apparent function for the glycan in rods. In light of the highly conserved nature of RDS glycosylation, we hypothesized that it is important for RDS function in cones and could underlie part of the differential requirement for RDS in the two photoreceptor subtypes. We generated a knockin mouse expressing RDS without the N-glycosylation site (N229S). Normal levels of RDS and the unglycosylated RDS binding partner rod outer segment membrane protein 1 (ROM-1) were found in N229S retinas. However, cone electroretinogram responses were decreased by 40% at 6 months of age. Because cones make up only 3–5% of photoreceptors in the wild-type background, N229S mice were crossed into the nrl−/− background (in which all rods are converted to cone-like cells) for biochemical analysis. In N229S/nrl−/− retinas, RDS and ROM-1 levels were decreased by ∼60% each. These data suggest that glycosylation of RDS is required for RDS function or stability in cones, a difference that may be due to extracellular versus intradiscal localization of the RDS glycan in cones versus rods.
Introduction
The RDS/PRPH2 gene codes for a photoreceptor-specific glycoprotein called retinal degeneration slow (RDS2 or peripherin-2), which is a member of the tetraspanin family of proteins and is critical for the proper formation of both rod and cone photoreceptor outer segments (OSs) (1–8). The OS is an organelle comprising a series of flattened opsin-packed discs and is specialized to detect incoming light and initiate phototransduction (9–11). Given the central importance of this organelle to normal vision, changes to the RDS/PRPH2 gene can have a negative effect on vision. Over 80 individual mutations of the RDS/PRPH2 gene in humans have been associated with significant diseases of the retina that can range from the rod-dominant retinitis pigmentosa to the cone-dominant diseases cone-rod dystrophy and macular degeneration (12). Additionally, extensive studies in mouse models have clearly demonstrated the importance of the RDS protein to retinal structure and function (2, 13–18). In mice heterozygous for the Rds gene (rds+/−), the OSs become highly disorganized, and the discs adopt a bizarre whorl-like morphology. These shorter and abnormally formed OSs are not able to function as well as normal OSs, resulting in severe early-onset defects in rod function and later-onset defects in cone function (4, 13). In the complete absence of RDS (rds−/−), OSs completely fail to form, with rod photoreceptors only able to build a connecting cilium, whereas cones build a sack-like OS that lacks all flattened cone lamellae (3, 19, 20). Both rds+/− and rds−/− mice also exhibit significant photoreceptor degeneration (4, 21). Although this cell death occurs through apoptosis, the mechanism that links OS disruption to the activation of the apoptosis pathway remains unclear with many competing theories (22).
The RDS protein itself is a small (∼35-kD) glycosylated four-pass transmembrane protein that localizes to the rim region of rod discs and the equivalent cone lamellae (8, 23). Its N- and C-terminal domains are cytosolic, whereas its two extracellular loop domains (D1 and D2) project into the enclosed discs in rods and the extracellular space between lamellae in cones (24). The C-terminal domain is known to direct the targeting of RDS complexes to the OS, engage in protein-protein interactions, and form an amphipathic helix that is thought to play a role in maintaining the unique membrane topology of the OS itself by potentially regulating membrane fusion or curvature (25–29).
In addition to the C terminus, the D2 loop is another region of functional significance. It is glycosylated and has a large mushroom shape that protrudes into the extracellular space. The D2 loop has been mapped as the region responsible for the assembly of RDS complexes, which are absolutely essential for the formation of OSs (14, 18, 30–32). RDS assembles into homo- and heterotetramers with its unglycosylated homologue rod outer segment membrane protein 1 (ROM-1) that are held together primarily through D2 loop-mediated non-covalent interactions (30, 33). These tetramers are subsequently linked into larger intermediate- and higher-order oligomers that are assembled via covalent disulfide bonds (33, 34). Critically, despite the mix of both RDS and ROM-1 in tetramers and intermediate disulfide-linked complexes, only RDS homotetramers are able to engage in the formation of the higher-order complexes of RDS (35). The D2 loop contains seven cysteines, six of which form intramolecular disulfide bonds that stabilize the complex three-dimensional structure of the D2 loop and promote proper folding (36). The seventh cysteine at position 150 is responsible for the intermolecular disulfide bonds that mediate the formation of larger oligomers (18, 31, 33, 35–37).
Despite our knowledge of the RDS protein and its importance to OS development and maintenance, we currently do not know how RDS is able to fulfil its cellular role inside the cell, nor do we understand why rods and cones appear to respond to the loss of RDS differently and why RDS mutations cause such widely varying disease phenotypes (12). One of the features of RDS that has stood out as a potential modulator of RDS function in rods versus cones is the N-linked glycosylation at asparagine 229 in the D2 loop. This posttranslational modification is conserved between all examined RDS genes and is markedly absent from ROM-1 (8). The glycosylation site on RDS is located within the critical oligomer-forming domain of RDS, suggesting that it could play a role in modulating the ability of RDS to form important complexes in rods or cones. Despite this, the RDS N-glycan has no identified function. To help study the role of RDS glycosylation, previous work utilized a transgenic mouse line expressing unglycosylated RDS in rod photoreceptors, but there was no observed loss of function or phenotype resulting from the lack of glycosylation in rods (16).
Because of the high conservation of the glycosylation site within a critical oligomer-forming domain for RDS and the known differential requirements for RDS in rods versus cones, we hypothesized that the N-linked glycan was important in cone photoreceptors. To test this, we generated and examined a knockin mouse model that expresses unglycosylated RDS (N229S) in the native RDS genetic locus. Our data show that the RDS glycan plays a role in modulating the interaction of RDS with its homologue ROM-1 and that it is critical for proper cone function. These data suggest that the RDS glycan is important for fine-tuning the cone OS and further our understanding of the RDS molecule and its regulation in rods versus cones.
Experimental Procedures
Animals
The University of Oklahoma Institutional Animal Care and Use Committee approved all experimental procedures, maintenance, and handling of mice. All animal work also followed the guidelines set forth by the Association for Research in Vision and Ophthalmology. N229S-RDS knockin mice were generated by inGenious Targeting Laboratory, Inc. (Ronkonkoma, NY). A positively identified bacterial artificial chromosome was used to isolate an 8.21-kbp region of genomic DNA that was used to construct the targeting vector. The long homology arm consisted of an ∼5.34-kbp region of DNA 5′ from the site of the mutation in exon 2, and the short homology arm extended 2.02 kbp 3′ of the end of the Neo cassette. A LoxP/FRT-Neomycin selection cassette was inserted in the RDS second intron 710 bp downstream of exon 2. Two point mutations were added to exon 2 of the rds gene. One was a silent GCG>GCA to eliminate a Hha1 restriction site to aid in genotyping, whereas the other was the AAC>AGC missense mutation to produce N229S. The bacterial artificial chromosome was subcloned into a 2.45-kbp pSP72 (Promega, Madison, WI) backbone vector, linearized with Not1, and electroporated into ES cells. The final targeting construct was 12.4 kbp. Five positive ES clones were found and injected into C57BL/6 blastocysts and implanted. Germline transmission was confirmed, and the resulting mice were bred to FLPeR-expressing mice (stock no. 003946, The Jackson Labs, Bar Harbor, ME) to remove the Neo cassette. PCR genotyping was used to confirm the absence of the rd8 mutation. The rds−/− mice were bred from founders originally provided by Dr. Neeraj Agarwal (University of North Texas Health Science Center, Fort Worth, TX), and rom1−/− mice were bred from founders originally provided by Dr. Roderick McInnes (McGill University, Montreal, QC, Canada). All animals were reared under cyclic light conditions (12-h light/dark, 30 lux), and animals of both sexes were used in all studies. In this study, all animals used were postnatal day (P) 30 or P180 as indicated.
Antibodies
The antibodies that were used in this study were as follows: RDS-CT (rabbit polyclonal, generated in-house, 1:1000) for immunofluorescence (IF) (18, 31), RDS-2B7 (mouse monoclonal, generated by Precision Antibodies, 1:1000) for Western blot (WB) (31, 38), ROM1-CT (rabbit polyclonal, generated in-house, 1:1000) for WB (14), ROM1–2H5 (mouse monoclonal, generated in-house,1:10) for WB (32), Na+K+ATPase (mouse monoclonal, Developmental Studies Hybridoma Bank, developed under the auspices of the NICHD/National Institutes of Health and maintained by the University of Iowa Department of Biology (Iowa City, IA), 1:500) for IF, Rhodopsin-1D4 (mouse monoclonal, provided by Dr. Robert Molday, University of British Columbia, 1:1000) for IF and WB, S-opsin (goat polyclonal, N-20, Santa Cruz Biotechnology (Santa Cruz, CA), 1:500) for IF, and β-actin (HRP-conjugated mAb, Sigma-Aldrich (St. Louis, MO) 1:10,000) for WB.
Immunofluorescence
For IF analysis, eyes were enucleated following euthanasia and fixed in 4% paraformaldehyde prior to dissection and cryoprotection as described previously (31). Eye cups were sectioned at 10 μm and subjected to IF staining with procedures described previously (39). The primary antibodies used are described above. Alexa Fluor 488, Alexa Fluor 555, and Alexa Fluor 647 (Life Technologies) secondary antibodies specific to goat, rabbit, or mouse were used. Images were captured with a Hamamatsu C-4742 camera through UPlanSApo objectives (Olympus, Tokyo, Japan) on an Olympus BX62 upright microscope equipped with a spinning disc confocal unit. Slidebook v5 software (Intelligent Imaging Innovations, Denver, CO) was used to analyze the resulting images. All experiments shown were repeated at least three times using independent animal samples, and images presented are single planes from a confocal stack.
Transmission Electron Microscopy and Immunogold Labeling
Samples at P30 or P180 were processed for EM and immunogold labeling as described previously (18, 19, 31). Plastic-embedded thin sections (600–800 Å) were used for EM/immunogold labeling, and thick sections (0.75 μm) were used for light microscopy. For nuclei counts, 200 μm2 areas were centered at the indicated distances from the optic nerve, and all nuclei in the outer nuclear layer (ONL) within these areas were counted manually. Immunogold experiments used rabbit polyclonal S-opsin antibodies shared by Dr. Cheryl Craft (University of Southern California, Los Angeles, CA). Secondary antibodies (AuroProbe 10-nm gold-conjugated goat anti-rabbit IgG or goat anti-mouse IgG, GE/Amersham Biosciences) were used at 1:50 dilution. Images were collected using a JEOL 100CX electron microscope at an accelerating voltage of 60 kV. Morphometry was performed as described previously (17) on 3–5 eyes/genotype.
Electroretinography
The electroretinography (ERG) procedures used have been described previously (14, 18, 19). Briefly, after an overnight period of dark adaptation, mice were anesthetized via intramuscular injection of 85 mg/kg ketamine and 14 mg/kg xylazine (Butler Schein Animal Health, Dublin, OH) in the dark, and their eyes were dilated using 1% cyclogyl (Pharmaceutical Systems, Inc., Tulsa, OK). Full-field ERG was recorded using a UTAS system (LKC, Gaithersburg, MD) with platinum wire loop electrodes that were placed to be in contact with the cornea though a thin layer of methylcellulose (Pharmaceutical Systems, Inc.). A single flash stimulus of 157 candelas/s/m2 was used to generate the scotopic response in dark-adapted animals. The resulting A-wave is generated primarily by the rod photoreceptors with minor contributions from the cone populations. Following scotopic analysis, a 5-min light adaptation using 29.03 candelas/m2 background light was performed to bleach the rod photoreceptor response prior to 25 flashes at 157 candelas/s/m2 to measure the photopic (cone) response. Statistical analysis was performed using one-way ANOVA with Bonferroni multicomparison post-test.
Protein Extraction and Analysis
All retinas were flash-frozen immediately after collection and stored at −80 °C before processing. For all protein extractions, the procedures used have been described in detail previously (17). In brief, crude retinal extracts were prepared by solubilization in extraction buffer (PBS (pH 7.0) containing 1% Triton X-100, 5 mm EDTA, 5 mg/ml N-ethylmaleimide, and a standard protease inhibitor mixture) with a short sonication to homogenize the tissue. Following extraction for 1 h at 4 °C, insoluble material was spun down, and the resulting protein concentration was measured using a colorimetric protein assay (Bradford reagent, Bio-Rad). Samples were kept on ice and used immediately to avoid any nonspecific aggregation. Western blots, SDS-PAGE, and immunoprecipitation were performed as described previously (18, 34, 40). The 5–20% sucrose gradient velocity sedimentation has been described previously (18, 34, 40). In multiplex blotting experiments, secondary antibodies were Alexa Fluor 647 anti-rabbit (pseudocolored red) and Alexa Fluor 488 anti-mouse (pseudocolored green). All experiments and analyses were repeated a minimum of three times. Blots were imaged using ChemiDocTM MP imaging system (Bio-Rad), and densitometric analysis was performed using Image Lab software v4.1 (Bio-Rad). Plot profiles were generated in National Institutes of Health ImageJ, background-subtracted, and normalized to the maximum intensity.
RNA Preparation and Analysis
Retinas were used to isolate total RNA as described previously (41) with TRIzol reagent (Life Technologies) and treated with RNase-free DNase I (Promega) using the procedures recommended by the manufacturer. An oligo(dT) primer was used along with Superscript III reverse transcriptase (Life Technologies) to generate cDNA. A C1000 thermal cycler (Bio-Rad) was used for quantitative RT-PCR as described previously (19). All resulting values were normalized to the HPRT housekeeping gene. Primer sequences used for this analysis have been published previously (17) For Northern blot analysis, 20 μg of total RNA was run on a 1% agarose gel containing 18% (v/v) formaldehyde. Prior to blotting onto Ambian® Brightstar®-Plus (Life Technologies), 18S and 28S ribosomal RNAs were imaged using a ChemiDocTM MP imaging system (Bio-Rad) for size control. The Amersham Biosciences (GE Healthcare) random priming kit was used on a linearized pcDNA3.1 vector containing wild-type RDS cDNA to generate 32P-labeled oligonucleotides (7). Kodak BioMax MS film with a BioMax Transcreen-LE intensifying screen (Carestream Health, Rochester, NY) was used to visualize the resulting bands.
Results
The N229S Knockin Mutation Leads to Unglycosylated RDS That Traffics Properly to the OS
Changes in the expression of the RDS protein can lead to significant defects in OS structure and function, including haploinsufficiency-associated retinitis pigmentosa phenotypes (12, 13, 15, 42). Therefore, to avoid these level of expression issues that could complicate our work on the role of glycosylation in RDS function, we used a knockin strategy to introduce an Ala-to-Gly missense mutation resulting in the conversion of asparagine 229 to serine (N229S) in exon 2 of the native RDS gene (Fig. 1A). For clarity, the N229S mutant Rds allele is abbreviated as N, with rdsN/+ and rdsN/N used to refer to the heterozygous and homozygous genetic backgrounds, respectively. To confirm that our genetic knockin approach yielded proper expression of the Rds gene, we used quantitative RT-PCR to assess the levels of Rds transcript in the rdsN/+ and rdsN/N relative to the WT and found no significant differences (Fig. 1B). Additionally we showed, by Northern blot analysis, that the genetic mutation did not alter the normal splicing or transcript size of the mouse Rds gene (Fig. 1C).
FIGURE 1.

Knockin of the N229S mutation removes RDS glycosylation while preserving normal RDS message and trafficking. A, the N229S mutation was introduced to exon 2 of the Rds gene to drive expression of unglycosylated RDS in the native regulatory environment. B, at P30, total RNA was isolated from murine retinas of the indicated genotypes, analyzed by quantitative RT-PCR for RDS, and normalized to the housekeeping gene HPRT. N.S., not significant. C, Northern blotting with radiolabeled random primed RDS cDNA as a probe was performed on equivalent RNAs in B. 28S and 18S ribosomal RNAs were used as a size reference, and 28S is shown in the bottom panel. D, retinal extracts from WT and rdsN/N were subjected to enzymatic deglycosylation by PNGase F. Actin reactivity was used as a loading control. IB, immunoblot. E, retinal cross-sections at P30 from WT and rdsN/N were immunolabeled with antibodies for S-opsin (green), Na+K+ATPase (blue), and either RDS (red, left), ROM-1 (red, center), or rhodopsin (red, right). Nuclei were counterstained with DAPI (gray). IS, inner segment; OPL, outer plexiform layer. Scale bar = 20 μm.
Next we wanted to confirm that the N229S mutation resulted in the expression of an unglycosylated form of RDS. We digested retinal extracts from WT and rdsN/N with PNGase F enzyme, which cleaves all N-linked glycans (Fig. 1D). RDS from the WT retina shows a marked downshift when subjected to PNGase F digestion, whereas the RDS from the rdsN/N retina does not, and the undigested form also runs at a lower molecular size, confirming that N229S RDS is not N-glycosylated (Fig. 1D). Changes in the D2 loop of RDS have been shown previously to alter trafficking of the RDS protein (37). To determine whether the N229S mutation affected RDS localization, we immunofluorescently labeled frozen retinal sections with antibodies against OS markers as well as Na+K+ATPase (blue), which is a marker for the inner segment and cell bodies of photoreceptors. Neither RDS nor ROM-1 show any signs of mislocalization outside the OS (Fig. 1E, left and center). Because sometimes disruptions in RDS lead to defects in the localization of other OS constituents, such as the rod and cone opsins (18, 20, 34), we also assessed their localization in the rdsN/N and WT. However, both rhodopsin (Fig. 1E, right, red) and S-opsin (Fig. 1E, green) were properly localized to the OS without any signs that subcellular trafficking was affected by the loss of RDS glycosylation.
The Loss of RDS Glycosylation Results in Less High-order RDS Oligomer Formation and an Increase in RDS·ROM-1 Dimerization in Intermediate Complexes
We next analyzed the levels of N229S-RDS protein. Whole retinal extracts were prepared, and Western blot analysis using antibodies specific for RDS and ROM-1 was used to analyze the levels of RDS and ROM-1 protein. Levels of RDS and ROM-1 in rdsN/+ and rdsN/N retinas were not significantly different from the WT (Fig. 2A). The formation of RDS·ROM-1 oligomers is critical for RDS function, and they play a key role in maintaining proper OS structure. To explore whether altering RDS glycosylation ablated RDS·ROM-1 binding, we subjected WT and rdsN/N retinal extracts to reciprocal co-immunoprecipitation with antibodies against RDS (Fig. 2B) and ROM-1 (Fig. 2C). As in the WT, we find that RDS pulls down ROM-1 and vice versa in the rdsN/N (Fig. 2, B and C; In, input; B, bound; Ft, flow-through), suggesting that deglycosylation does not eliminate the ability of RDS to bind ROM-1.
FIGURE 2.

Lack of RDS glycosylation does not change steady-state levels of RDS protein and reveals RDS·ROM-1 heterodimerization. A, total retinal extracts were prepared from the indicated genotypes at P30 and analyzed by reducing SDS-PAGE/Western blotting. Membranes were probed with the indicated antibodies. Band densities were analyzed and normalized to actin and set relative to the average WT values (n = 4 retinas/genotype). Values are mean ± S.E. IB, immunoblot. B and C, reciprocal co-immunoprecipitation was performed on WT and rdsN/N retinal extracts with antibodies specific to either RDS (B) or ROM-1 (C), followed by reducing SDS-PAGE/Western blotting with the indicated antibodies. In, input; B, bound; Ft, flow-through. D, retinal extracts from the indicated genotypes were analyzed by non-reducing SDS-PAGE/Western blot and probed with antibodies to RDS (left panel) or ROM-1 (right panel). Arrows highlight size shift of RDS monomers and dimers and ROM-1 dimers because of loss of the glycosylation of RDS.
During the process of assembling into large- and intermediate-sized oligomers, RDS and ROM-1 both form covalent disulfide-linked complexes, mediated by Cys-150 (in RDS) and Cys-153 (in ROM-1). Initial studies have suggested that these disulfide bonds are homomeric, i.e. that RDS·RDS and ROM-1·ROM-1 can form covalent interactions but that RDS·ROM-1 interactions are non-covalent (16, 33). However, later work has suggested that RDS·ROM-1 could form heteromeric disulfide linkages (35). We assessed intermolecular disulfide bonds in the absence of glycosylation by evaluating non-reducing SDS-PAGE/Western blots. On non-reducing gels, RDS and ROM-1 in covalently linked complexes run as a dimer-sized band of ∼75 kDa, whereas RDS·ROM-1 in non-covalent complexes run as a monomer at ∼35 kDa (Fig. 2D). As expected, the loss of RDS glycosylation causes a downshift of both the monomer and dimer of RDS (Fig. 2D, left panel, arrows). The ROM-1 monomer does not show any appreciable size shift in the WT versus rdsN/N as expected. However, in mice expressing the unglycosylated form of RDS, the entire ROM-1 dimer band is shifted to a smaller size (Fig. 2D, right panel, arrows). Given that the ROM-1 gene/protein is unaltered in rdsN/N, the only explanation for this result is that ROM-1 dimerizes with RDS when RDS is unglycosylated. In addition, the fact that the entire visible ROM-1 dimer band shifts down suggests that ROM-1 almost exclusively forms heterodimers with RDS as opposed to homodimers in the rdsN/N. However, previous results were conflicting regarding whether or not RDS and ROM-1 form heteromeric covalent linkages in the WT background, so it was unclear whether the RDS·ROM-1 covalent linkages visible because of the size shift in rdsN/N were normal or were an outcome of the mutation.
Therefore, we repeated our non-reducing SDS-PAGE/Western blot experiment but included rds−/− retinal extracts (in which ROM-1 is present but not RDS) and rom1−/− retinal extracts (in which RDS is present but not ROM-1). Blots were probed with antibodies against RDS and ROM-1, and fluorescent secondary antibodies were used to simultaneously image both proteins (Fig. 3, A–C). The left panels in Fig. 3 show the blots, whereas the right panels show profile analyses (averages from three experiments) of the region of the blot corresponding to the RDS (red) and ROM-1 (green) dimer band (the region in the plot profile is indicated by the bracket on the side of the blot). The profile plots show percent of maximum band intensity as a function of apparent molecular weight and enable the detection of small shifts. To facilitate visualization, the blots and profiles are broken down by channel, with RDS alone shown in Fig. 3B (red) and ROM-1 alone shown in Fig. 3C (green). Although RDS and ROM-1 are quite close in size, ROM-1 typically runs at a slightly lower apparent molecular weight than WT RDS (but larger than unglycosylated RDS). Interestingly, both RDS and ROM-1 monomers (indicated by M in Fig. 3A) often appear as a doublet, but the cause of this is unknown. However, although the intensity of the various bands in the doublet can vary, the sizes of the two bands are quite consistent, making size comparison possible. The slightly smaller apparent molecular weight of ROM-1 can be easily seen by comparing the size of the RDS (red) monomer bands in rom1−/− with the ROM-1 (green) monomer doublet bands in rds−/− in Fig. 3A, left panel. This subtle difference in size (RDS > ROM-1 > unglycosylated RDS) enables us to assess the composition (i.e. homo- versus heteromeric) of RDS and ROM-1 dimers.
FIGURE 3.
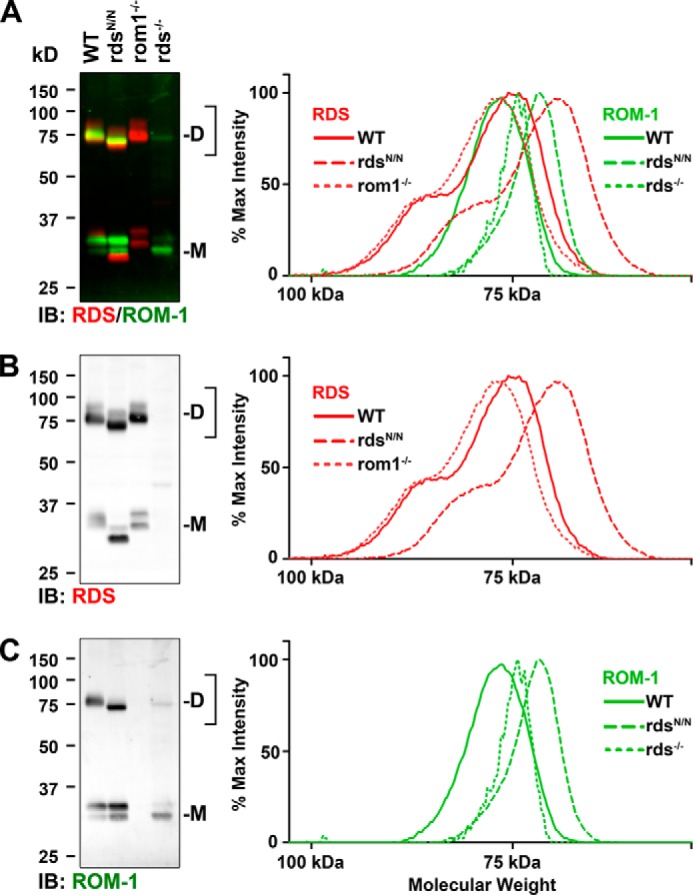
WT and N229S RDS form covalent linkages with ROM-1. Non-reducing SDS-PAGE/Western blots were performed on P30 retinal extracts of the indicated genotypes (WT, rdsN/N, and rom1−/−, 10 μg/lane; rds−/−, 40 μg/lane). Blots were probed simultaneously with antibodies against RDS and ROM-1 and imaged using multiplexed fluorescent secondary antibodies. D, dimer; M, monomer; IB, immunoblot. The bracketed region was subjected to profile analysis using ImageJ, and background-subtracted plot profiles are presented in the right panels. A, RDS·ROM-1 channels (red/green, respectively) combined. B and C, the same data are separated to more easily visualize the RDS and ROM-1 channels separately.
In the rom1−/− retina, the absence of ROM-1 means that all covalently linked (i.e. dimeric) RDS is comprised of RDS/RDS homomeric linkages (Fig. 3B, dotted line). The slight right shift (i.e. toward a smaller molecular weight) in the WT plot profile (Fig. 3B, solid line) compared with the rom1−/− plot profile (Fig. 3B, dotted line) is consistent with the formation of RDS·ROM-1 heteromeric dimers that are slightly smaller than RDS·RDS homodimers because of the presence of the smaller ROM-1. As expected, in rdsN/N, the RDS dimer plot profile (Fig. 3B, dashed line) is significantly right-shifted (i.e. toward a smaller molecular weight) compared with the WT because RDS in rdsN/N is unglycosylated.
The dotted line in Fig. 3C, right panel, represents ROM-1 homodimers (i.e. the location of the ROM-1 dimer band in rds−/− retinal extracts). When WT RDS is present, the ROM-1 dimer peak shifts to the left (Fig. 3C, solid line), consistent with the formation of covalent linkages between ROM-1 and RDS. Most importantly, in rdsN/N, the ROM-1 dimer peak shifts significantly to the right (i.e. smaller) (Fig. 3C, dashed line). Because the loss of RDS glycosylation does not affect ROM-1 monomer size, this result is most consistent with the formation of covalent linkages between ROM-1 and unglycosylated RDS. These data confirm that both WT and unglycosylated RDS can form covalent linkages with ROM-1 in vivo.
Because evaluation by SDS-PAGE results in the interruption of non-covalent complexes of RDS·ROM-1, we next examined how the loss of RDS glycosylation alters the assembly of RDS·ROM-1 complexes under native conditions using a 5–20% non-reducing sucrose gradient (Fig. 4). We have observed previously that higher-order RDS homo-oligomers appear in the heaviest gradient fractions, 1–3, whereas intermediate oligomers appear in fractions 4 and 5 and tetramers appear in fractions 6–9. After separating complexes on the gradients, the percent of total RDS or ROM-1 in each fraction was examined by reducing SDS-PAGE/Western blot (Fig. 4, A and B). This revealed a decrease in the relative percent of high-order RDS oligomers in rdsN/N compared with the WT (Fig. 4A, arrow) without an overall change in the ROM-1 oligomers (Fig. 4B). Analysis of the same non-reducing gradient fractions under non-reducing SDS-PAGE allows us to determine the relative contribution of monomers and dimers in each complex type (Fig. 4, C and D). This more detailed analysis demonstrates that the decrease in the percent of higher-order oligomers in rdsN/N (Fig. 4, A and C, arrows) is associated with an increase in the amount of RDS dimers in intermediate fractions (Fig. 4C, arrowhead). In addition, we also see an increase in the relative amount of ROM-1 dimers (as opposed to monomers) in intermediate gradient fractions (Fig. 4D, arrowhead). Together, these data suggest a subtle role for RDS glycosylation in regulating RDS·ROM-1 complex assembly.
FIGURE 4.

The loss of RDS glycosylation alters the assembly of RDS·ROM-1 complexes. A and B, retinal extracts from WT (blue) and rdsN/N (red) were separated on a 5–20% sucrose gradient under non-reducing conditions. Fractions were prepared by drip collection (fraction 1, 20% to fraction 12, 5%) and analyzed by reducing SDS-PAGE/Western blot probed with antibodies specific to RDS (A) and ROM-1 (B). IB, immunoblot. C and D, retinal extracts were fractionated along non-reducing 5–20% sucrose gradients as in A and B but were then analyzed by non-reducing SDS-PAGE/Western blot and probed with antibodies to RDS (C) and ROM-1 (D). In the quantification (top panels, C and D), monomers are depicted in black/green, and dimers are depicted in blue/red. A reduction in the higher-order oligomers (arrow) and an increase in intermediate dimers (arrowheads) is observed. Each quantification graph plots RDS or ROM-1 band intensities in each gradient fraction as a percent of the total RDS or ROM-1. Presented are means (n = 4/genotype) ± S.E.
Unglycosylated RDS Leads to a Late-onset Functional Defect in Cones
Given the defects in complex assembly we observed in rdsN/N retinas, we used full-field ERG to ask whether there were functional defects associated with expression of unglycosylated RDS and whether the N229S mutant allele could rescue the severe functional phenotype associated with RDS haploinsufficiency (i.e. in rds+/− (13)). Scotopic recordings were performed at P30 and P180, and representative traces are shown in Fig. 5, A and B. Although these recording conditions can generate a signal from both rods and cones, cone-generated a-waves under these conditions are undetectable, so the A-wave is a useful measure of rod ERG response. Rod function is severely decreased in rds+/− as early as P30 (13), and we here find that the N229S allele (in rdsN/+) can completely rescue rod function to WT levels (Fig. 5A). This is consistent with what has been reported previously (16), and, importantly, this rescue is long-lasting. rdsN/+ scotopic ERG amplitudes are significantly higher than rds+/− and not significantly different from the WT even at P180 (Fig. 5B; **, p < 0.01 by one-way ANOVA). We also found that rod function (analyzed under scotopic conditions) is not changed between WT and rdsN/N at P30 (Fig. 5, A and B). At P180, although mean scotopic A-wave amplitudes are lower in rdsN/N than in the WT, the difference is not statistically significant. To determine whether this difference was magnified at later ages, a small cohort of rdsN/N and WT animals were aged to P360, but no significant differences in scotopic ERG amplitudes were observed at that age. Consistent with this lack of rod functional decrease, we found that overall retinal structure and lamination were not altered in rdsN/N versus WT, and there was no detectable degeneration at P30 (Fig. 5C). At P180, overall retinal structure is still normal, as is the thickness of the OS layer (Fig. 5, D and E). However, measurements from across the retina show that there is modest thinning of the ONL at P180 in rdsN/N versus WT (Fig. 5F; *, p < 0.05; **, p < 0.01; ***, p < 0.001 by two-way ANOVA). To determine whether this thinning was due to cell loss or to other changes in the retina, we counted nuclei in the ONL. We observed that the mean number of ONL nuclei in rdsN/N was slightly lower than that in the WT, but the difference did not achieve statistical significance (Fig. 5G). These data suggest that mild retinal degeneration may be ongoing, although it is not yet detectable by ERG. Consistent with the lack of significant defects in OS length and scotopic ERG, rod OS ultrastructure is also grossly normal at P180 in rdsN/N (Fig. 5H shows WT OS ultrastructure, whereas Fig. 5I shows several examples of OS ultrastructure in rdsN/N).
FIGURE 5.

Moderate retinal degeneration is observed by P180 in mice expressing unglycosylated RDS. A and B, rod photoreceptor function was analyzed using the mixed full-field scotopic ERG at P30 (A) and P180 (B). Under these conditions, the initial negative displacement A-wave is generated primarily by the large number of rod photoreceptors. Representative traces are shown in the left panels, with means ± S.E. shown in the right panels. For ERG, n = 14–16 mice/genotype, statistical analysis was done by one-way ANOVA. **, p < 0.01. C and D, plastic-embedded retinal sections were imaged in the central retina at P30 (C) and P180 (D). IS, inner segment; INL, inner nuclear layer; IPL, inner plexiform layer; ONH, optic nerve head. E, the thickness of the OS layer was measured in 5–10 eyes/genotype at P180. Plotted are means ± S.E. F and G, images were taken at increasing distances from the optic nerve head, and outer nuclear layer thickness (F) and outer nuclear layer nucleus counts (G) were measured in 3–5 eyes/genotype. Plotted are means ± S.E. Statistical analysis was done by two-way ANOVA. *, p < 0.05; **, p < 0.01; ***, p < 0.001. H and I, TEM images at P180 from WT and rdsN/N. OSs from multiple different rdsN/N eyes are shown in the first, second, and third panels in I. Scale bars = C and D, 50 μm; H and I, 10 μm (left) and 500 nm (right).
When photopic ERG was used to analyze cone function at early ages (P30) no significant differences were observed in rdsN/+ or rdsN/N compared with the WT (Fig. 6A). Interestingly, by P180, maximum photopic ERG amplitudes were decreased significantly by 22% (**, p < 0.01) in rdsN/N compared with the WT (Fig. 6B), suggesting that RDS glycosylation is critical for long-term cone function. When a small cohort of WT and rdsN/N animals was aged to P360, we found that WT signals had dropped gradually because of age, but rdsN/N values plateaued post-P180, suggesting that the ability of cells to respond to the aging process may be reduced in the rdsN/N retina.
FIGURE 6.

Cone function is decreased at later time points in mice that lack RDS glycosylation without an associated loss of cone photoreceptor cells. A and B, cone photoreceptor function was analyzed using full-field photopic ERG at P30 (A) and P180 (B). **, p < 0.05 by one-way ANOVA with Bonferroni post hoc comparison. Shown are mean values from n = 4–16 mice/genotype. C, frozen retinal sections along the superior-inferior axis (S-I) were prepared from P180 WT and rdsN/N mice, and cones were labeled with a mixture of antibodies specific to M-opsin and S-opsin (red), with nuclei counterstained with DAPI. INL, inner nuclear layer; IPL, inner plexiform layer. D, starting at the optic nerve, total cones were counted in 200-μm2 areas at the indicated distances from the optic nerve. Shown are means (n = 3/genotype) ± S.E. E and F, retinal sections at P30 or P180 were immunogold-labeled with antibodies against S-opsin to identify cone OSs. Arrows indicate cone OSs in low-magnification EM images. The right panels show higher-magnification images of cone OSs. Most cone OSs in rdsN/N exhibit normal morphology, but abnormal cone OSs are also seen (far right panel, F). Scale bars = C, 50 μm; E and F, 2 μm.
A loss of cone function could be due to a decrease in the ability of cones to generate a robust signal or because of cone degeneration. To determine whether cone degeneration was occurring, we labeled central retinal cross-sections with a mix of antibodies to S- and M-opsin (representative images are shown in Fig. 6C, red) and counted total cones in 200-μm2 areas at increasing distances from the optic nerve head. Despite the significant drop in cone photoreceptor function, we did not observe any concomitant drop in cone photoreceptors anywhere within the retina (Fig. 6D) in rdsN/N compared with the WT. To ensure that the loss of cone function was not due to unexpected localized loss of cone cells, we also counted cones on retinal flat mounts and observed a similar outcome. These data suggest that the loss of cone function is a direct result of a decrease in the function of the cone photoreceptor itself instead of a decrease in cone cell number. Finally, we asked whether cone OS ultrastructure was altered in rdsN/N at P180. Because cones are rare in the murine retina (∼3–5% of photoreceptors), we immunogold-labeled with antibodies against S-opsin to identify them prior to performing EM. The left panels of Fig. 6, E and F, show low-magnification EM of cone outer segments (arrows), whereas the right panels of Fig. 6, E and F, show higher-magnification images of cone OSs. At P180, the majority of cone OSs in rdsN/N appeared normal (Fig. 6F, center panels), but we also observed cone OSs that exhibited abnormal disc stacking and alignment (Fig. 6F, right panel).
RDS and ROM-1 Levels Are Decreased Significantly in rdsN/N Animals on the Cone-dominant nrl−/− Background
Our functional analysis suggests a cone-specific role for RDS glycosylation. However, the mouse retina is rod-dominant, and, as a result, analysis of cone-specific biochemical or cell biological defects is difficult. To overcome this obstacle, we crossed the rdsN/N mouse onto the nrl−/− background. In the absence of the transcription factor Nrl, rod photoreceptors are converted to cone-like cells that express cone proteins (43, 44). To determine whether nrl−/− would be an effective background to study the effects of unglycosylated RDS in cones, we first asked whether rdsN/N/nrl−/− modeled the ERG phenotype seen in rdsN/N. Evaluation was limited to P30 because the ongoing degeneration in the nrl−/− can complicate the interpretation of the effects because of other factors. Photopic ERG showed that there is a significant (32%) decrease in maximum photopic A-wave amplitude (Fig. 7A, left and center panels) in rdsN/N/nrl−/− versus the nrl−/− as early as P30. Photopic B-wave amplitudes are normal at P30 in rdsN/N/nrl−/−, similar to the case in the WT (Fig. 7A, right panel). In the WT background, cone A-waves are too small to measure, so B-waves are used as a proxy for cone function. These data show that unglycosylated RDS-associated defects in cone function are replicated in nrl−/− and confirm that RDS glycosylation is needed for cone function.
FIGURE 7.

Cone function and steady-state levels of RDS and ROM-1 are decreased in the cone-dominant nrl−/− retina in the absence of RDS glycosylation. A, photopic ERG of nrl−/−, rdsN/+/nrl−/−, and rdsN/N/nrl−/− mice was used to assess cone function at P30. Representative traces are shown in the left panel. Maximum photopic A-wave amplitudes (center panel) represent the signal generated by cone photoreceptors, whereas photopic B amplitudes (right panel) measure inner retinal responses to photoreceptor signaling. Shown are means (n = 4–16 mice/genotype) ± S.E. **, p < 0.01 by one-way ANOVA with Bonferroni post-hoc comparison. B–D, total retinal extracts were prepared from the indicated genotypes and analyzed by reducing SDS-PAGE/Western blot. Immunoblots (IB)were probed with antibodies specific for RDS (B), ROM-1 (C), S-opsin (D), and actin (B–D). Band densities were analyzed, normalized to actin, and set relative to the average nrl−/− values. Shown are means ± S.E. from 5–18 retinas/genotype. **, p < 0.01 by one-way ANOVA with Bonferroni post-hoc comparison (rdsN/N/nrl−/− versus nrl−/−).
To determine whether cones have a different biochemical response to the loss of RDS glycosylation than rods, we assessed the levels of RDS (Fig. 7B) and ROM-1 (Fig. 7C) protein in the cone-dominant nrl−/− retina. Although RDS and ROM-1 protein levels are normal in rdsN/+/nrl−/−, we observed a significant 40% drop in both RDS and ROM-1 levels in rdsN/N/nrl−/− compared with nrl−/− (Fig. 7, B and C; **, p < 0.01). However, when we examined the levels of cone S-opsin, we found no difference in rdsN/+/nrl−/− or rdsN/N/nrl−/− compared with nrl−/− (Fig. 7D). Together, these data support the hypothesis that RDS glycosylation may be required for the stability of RDS and ROM-1 in cones.
Given the interesting phenotype observed in cones in the absence of RDS glycosylation, we decided to compare RDS·ROM-1 oligomer formation between the nrl−/− and rdsN/N/nrl−/− retina. We have found previously that some mutations in RDS can affect its ability to bind ROM-1 in cones but not rods (18), so we conducted reciprocal co-immunoprecipitation for RDS and ROM-1 from nrl−/− and rdsN/N/nrl−/−. Our results confirmed that unglycosylated RDS retains the ability to interact with ROM-1 in cones (Fig. 8). We next utilized 5–20% sucrose gradient fractionation to analyze the distribution of RDS and ROM-1 in various complex types in the rdsN/N/nrl−/− and nrl−/−. Fig. 9, A and B, shows fractions from non-reducing sucrose gradients separated on reducing SDS-PAGE. As we have observed previously (31, 32), cones in nrl−/− have a higher total percent of RDS in the large oligomer fractions 1–3 than rods (e.g. Fig. 9A). However, we did not observe any differences in the distribution of RDS or ROM-1 between nrl−/− and rdsN/N/nrl−/−. When we separate gradient fractions on non-reducing gels to analyze the amount of RDS·ROM-1 involved in covalent linkages in each gradient fraction, we find that there is a small but replicable increase in monomeric (i.e. non-covalently linked) RDS in tetramer fractions in the rdsN/N/nrl−/− compared with the nrl−/− retina (Fig. 9C, arrow), whereas the distribution of covalent versus non-covalently linked ROM-1 is not altered significantly altered (Fig. 9D). These results suggest that the effect of unglycosylated RDS on RDS·ROM-1 complex formation in the nrl−/− background is minor, although RDS and ROM-1 protein levels are decreased significantly decreased in rdsN/N/nrl−/− and that phenotypic outcomes may be due to haploinsufficiency in cones but not rods.
FIGURE 8.
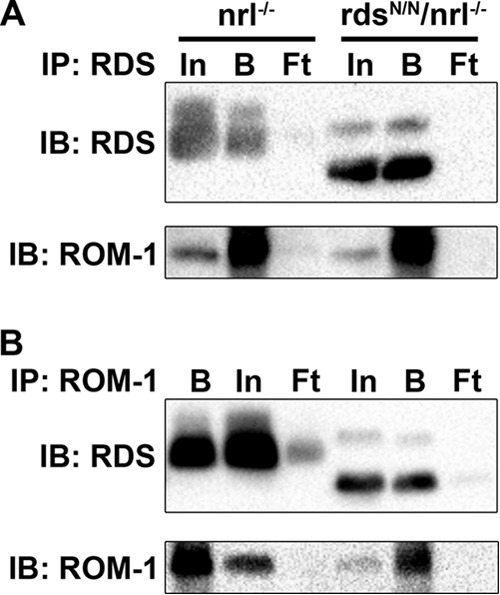
Deglycosylation of RDS does not affect the ability of RDS to bind ROM-1 in cones. Reciprocal co-immunoprecipitation (IP) was performed on P30 nrl−/− and rdsN/N/nrl−/− retinal extracts using antibodies against RDS (A) or ROM-1 (B). Reducing SDS-PAGE/Western blots were probed with the indicated antibodies. In, input; B, bound; Ft, flow-through; IB, immunoblot.
FIGURE 9.

The loss of rds glycosylation induces subtle alterations in RDS·ROM-1 complex formation cones. Retinal extracts from the nrl−/− (red) and rdsN/N/nrl−/− (blue) were separated on non-reducing 5–20% sucrose gradients, and gradient fractions were subsequently separated by reducing SDS-PAGE/Western blot (A and B) or non-reducing SDS-PAGE/Western blot (C and D). Blots were probed with RDS (A and C) or ROM-1 (B and D). In the quantifications in A and B, nrl−/− values are plotted in blue, and rdsN/N/nrl−/− values are plotted in red. In the quantifications in C and D, nrl−/− and rdsN/N/nrl−/− monomers are plotted in black/green, respectively, whereas dimers are plotted in blue/red, respectively. Shown are means (n = 4) ± S.E. IB, immunoblot.
Discussion
RDS N-glycosylation is a highly conserved posttranslational modification and is one of the key differences between RDS and its homologue ROM-1. Despite this conservation, no known function for RDS glycosylation has yet been identified. This work expands our understanding of how RDS functions differentially in rods versus cones, a critical but still unexplained phenomenon that contributes to widely varying RDS-associated disease pathology, by examining the role of RDS glycosylation in all aspects of RDS function. By utilizing a knockin approach, we are able to detect subtle changes associated with the loss of RDS glycosylation that might have been hidden in the “noise” associated with level of expression challenges in transgenics. We found that, in rod photoreceptor cells, RDS and ROM-1 protein levels were not affected by the lack of RDS glycosylation but that RDS complexes shifted away from higher-order oligomers toward intermediate oligomers without inducing severe structural or functional phenotypes in rods. In contrast, cones complexes were only subtly affected by loss of glycosylation. However, RDS and ROM-1 protein levels were decreased significantly, leading to a reduction in cone function.
RDS·ROM-1 complex formation is critical for proper OS formation, and we have observed previously that rods and cones have differences in RDS·ROM-1 complex assembly and distribution. Specifically, cones have a larger percentage of total RDS present in high molecular weight homomeric complexes compared with intermediate and tetrameric heteromers (18, 31, 32). In addition, results from C150S transgenic mice (in which RDS cannot form intermolecular disulfide bonds) suggest that, in cones, covalent intermolecular disulfide bonds in RDS·ROM-1 complexes are formed earlier in the RDS·ROM-1 biosynthetic pathway than in rods (18, 31). Rods and cones also tend to respond differently to different disease mutations. In general, rods tend to be more sensitive to the total quantity of RDS present and more resilient in the face of slightly abnormal complexes (e.g. in rds+/−, C214S, and rod-specific-C150S models (13, 15, 18)). Therefore, it makes sense that, in rods where unglycosylated RDS is just as stable as in the WT (i.e. RDS protein levels are normal in the rdsN/N), structural and functional phenotypes are mild (consistent with results reported previously (16)) even though RDS·ROM-1 complex assembly is slightly altered.
In contrast, cones are much more sensitive to slight aberrations in RDS·ROM-1 complex assembly (e.g. R172W and cone-specific C150S (14, 18, 31, 32)) rather than RDS levels. However, haploinsufficiency does cause later-onset functional defects in cones (13). Interestingly, in the nrl−/− background, this time course is accelerated, and modest reductions in cone function because of RDS haploinsufficiency (i.e. in rds+/−/nrl−/−) are detected as early as P30 (16). These observations are entirely consistent with our findings here. Reduced levels of RDS in rdsN/N/nrl−/− suggest that unglycosylated RDS is less stable in cones than WT RDS, leading to reduced cone ERG phenotypes consistent with haploinsufficiency.
However, this brings up the question of why levels of unglycosylated RDS are lower than WT RDS in cones but not rods. Two potential explanations exist. The first is that the unglycosylated RDS may be trafficked or packaged into complexes less efficiently in cones, leading to degradation in the inner segment. This would be consistent with our prior observation that complex assembly/trafficking and RDS·ROM-1 interactions are different in rods versus cones (18). However, the molecular mechanism by which RDS glycosylation would affect trafficking is not known. The second possibility is that the unique architecture of cone photoreceptor OSs may be a factor. In rods, the OS discs are isolated from the plasma membrane, whereas in cones, many discs are contiguous with the plasma membrane. In addition, it is thought that disc-plasma membrane fusion is ongoing and reversible in cones and not limited to the base of the OSs as in rods. One of the outcomes of this difference between rods and cones is that the D2 loop of RDS (where the N-glycan occurs) is extracellular in cones but intradiscal in rods. Therefore, the more dynamic nature of the cone OS coupled with the composition of the extracellular space may confer a cone-specific requirement for RDS glycosylation and lead to RDS·ROM-1 degradation, reflected here by a decrease in their levels in the rdsN/N/nrl−/−.
The role of RDS glycosylation has remained a mystery for many years, and here we show that RDS glycosylation plays a role in fine-tuning RDS·ROM-1 oligomer formation and promotes the stability of RDS in cones. Furthermore, we were able to show that RDS glycosylation plays an important role in the maintenance of cone function. Taken together, this work expands our understanding of the cellular mechanisms that regulate OS formation by providing a cell-specific role for RDS glycosylation and suggests that this highly conserved posttranslational modification contributes to the differences in RDS function in rods and cones.
Author Contributions
M. I. N. conceived the study. M. I. N., M. W. S., and S. M. C. designed the study, conducted the study, analyzed the data, and wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Barb Nagel (St. Louis University Department of Pathology) and Layne Rodden for technical assistance.
This work was supported by NEI/National Institutes of Health Grants R01EY010609-MIN and T32EY023202-MWS, by the Oklahoma Center for the Advancement of Science and Technology (to S. M. C.), and by the Knights Templar Eye Foundation (to S. M. C.).
- RDS
- retinal degeneration slow
- OS
- outer segment
- P
- postnatal day
- IF
- immunofluorescence
- WB
- Western blot
- ONL
- outer nuclear layer
- ERG
- electroretinography
- ANOVA
- analysis of variance.
References
- 1.Démant P., Iványi D., and van Nie R. (1979) The map position of the rds gene on the 17th chromosome of the mouse. Tissue Antigens 13, 53–55 [DOI] [PubMed] [Google Scholar]
- 2.Sanyal S., De Ruiter A., and Hawkins R. K. (1980) Development and degeneration of retina in rds mutant mice: light microscopy. J. Comp. Neurol. 194, 193–207 [DOI] [PubMed] [Google Scholar]
- 3.Sanyal S., and Jansen H. G. (1981) Absence of receptor outer segments in the retina of rds mutant mice. Neurosci. Lett. 21, 23–26 [DOI] [PubMed] [Google Scholar]
- 4.Hawkins R. K., Jansen H. G., and Sanyal S. (1985) Development and degeneration of retina in rds mutant mice: photoreceptor abnormalities in the heterozygotes. Exp. Eye Res. 41, 701–720 [DOI] [PubMed] [Google Scholar]
- 5.Molday R. S., Hicks D., and Molday L. (1987) Peripherin: a rim-specific membrane protein of rod outer segment discs. Invest. Ophthalmol. Vis. Sci. 28, 50–61 [PubMed] [Google Scholar]
- 6.Connell G., Bascom R., Molday L., Reid D., McInnes R. R., and Molday R. S. (1991) Photoreceptor peripherin is the normal product of the gene responsible for retinal degeneration in the rds mouse. Proc. Natl. Acad. Sci. U.S.A. 88, 723–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng T., al Ubaidi M. R., and Naash M. I. (1997) Structural and developmental analysis of the mouse peripherin/rds gene. Somat. Cell Mol. Genet. 23, 165–183 [DOI] [PubMed] [Google Scholar]
- 8.Conley S. M., Stuck M. W., and Naash M. I. (2012) Structural and functional relationships between photoreceptor tetraspanins and other superfamily members. Cell. Mol. Life Sci. 69, 1035–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steinberg R. H., Fisher S. K., and Anderson D. H. (1980) Disc morphogenesis in vertebrate photoreceptors. J. Comp. Neurol. 190, 501–508 [DOI] [PubMed] [Google Scholar]
- 10.Mustafi D., Engel A. H., and Palczewski K. (2009) Structure of cone photoreceptors. Prog. Retin. Eye Res. 28, 289–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sung C. H., and Chuang J. Z. (2010) The cell biology of vision. J. Cell Biol. 190, 953–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boon C. J., den Hollander A. I., Hoyng C. B., Cremers F. P., Klevering B. J., and Keunen J. E. (2008) The spectrum of retinal dystrophies caused by mutations in the peripherin/RDS gene. Prog. Retin. Eye Res. 27, 213–235 [DOI] [PubMed] [Google Scholar]
- 13.Cheng T., Peachey N. S., Li S., Goto Y., Cao Y., and Naash M. I. (1997) The effect of peripherin/rds haploinsufficiency on rod and cone photoreceptors. J. Neurosci. 17, 8118–8128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ding X. Q., Nour M., Ritter L. M., Goldberg A. F., Fliesler S. J., and Naash M. I. (2004) The R172W mutation in peripherin/rds causes a cone-rod dystrophy in transgenic mice. Hum. Mol. Genet. 13, 2075–2087 [DOI] [PubMed] [Google Scholar]
- 15.Stricker H. M., Ding X. Q., Quiambao A., Fliesler S. J., and Naash M. I. (2005) The Cys214 → Ser mutation in peripherin/rds causes a loss-of-function phenotype in transgenic mice. Biochem. J. 388, 605–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kedzierski W., Bok D., and Travis G. H. (1999) Transgenic analysis of rds/peripherin N-glycosylation: effect on dimerization, interaction with rom1, and rescue of the rds null phenotype. J. Neurochem. 72, 430–438 [DOI] [PubMed] [Google Scholar]
- 17.Stuck M. W., Conley S. M., and Naash M. I. (2014) The Y141C knockin mutation in RDS leads to complex phenotypes in the mouse. Hum. Mol. Genet. 23, 6260–6274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chakraborty D., Ding X. Q., Conley S. M., Fliesler S. J., and Naash M. I. (2009) Differential requirements for retinal degeneration slow intermolecular disulfide-linked oligomerization in rods versus cones. Hum. Mol. Genet. 18, 797–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farjo R., Skaggs J. S., Nagel B. A., Quiambao A. B., Nash Z. A., Fliesler S. J., and Naash M. I. (2006) Retention of function without normal disc morphogenesis occurs in cone but not rod photoreceptors. J. Cell Biol. 173, 59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chakraborty D., Conley S. M., Al-Ubaidi M. R., and Naash M. I. (2014) Initiation of rod outer segment disc formation requires RDS. PLoS ONE 9, e98939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jansen H. G., and Sanyal S. (1984) Development and degeneration of retina in rds mutant mice: electron microscopy. J. Comp. Neurol. 224, 71–84 [DOI] [PubMed] [Google Scholar]
- 22.Chang G. Q., Hao Y., and Wong F. (1993) Apoptosis: final common pathway of photoreceptor death in rd, rds, and rhodopsin mutant mice. Neuron 11, 595–605 [DOI] [PubMed] [Google Scholar]
- 23.Goldberg A. F. (2006) Role of peripherin/rds in vertebrate photoreceptor architecture and inherited retinal degenerations. Int. Rev. Cytol. 253, 131–175 [DOI] [PubMed] [Google Scholar]
- 24.Wrigley J. D., Nevett C. L., and Findlay J. B. (2002) Topological analysis of peripherin/rds and abnormal glycosylation of the pathogenic Pro216 → Leu mutation. Biochem. J. 368, 649–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boesze-Battaglia K., Song H., Sokolov M., Lillo C., Pankoski-Walker L., Gretzula C., Gallagher B., Rachel R. A., Jenkins N. A., Copeland N. G., Morris F., Jacob J., Yeagle P., Williams D. S., and Damek-Poprawa M. (2007) The tetraspanin protein peripherin-2 forms a complex with melanoregulin, a putative membrane fusion regulator. Biochemistry 46, 1256–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edrington T. C. 5th, Yeagle P. L., Gretzula C. L., and Boesze-Battaglia K. (2007) Calcium-dependent association of calmodulin with the C-terminal domain of the tetraspanin protein peripherin/rds. Biochemistry 46, 3862–3871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tam B. M., Moritz O. L., and Papermaster D. S. (2004) The C terminus of peripherin/rds participates in rod outer segment targeting and alignment of disk incisures. Mol. Biol. Cell 15, 2027–2037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khattree N., Ritter L. M., and Goldberg A. F. (2013) Membrane curvature generation by a C-terminal amphipathic helix in peripherin-2/rds, a tetraspanin required for photoreceptor sensory cilium morphogenesis. J. Cell Sci. 126, 4659–4670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boesze-Battaglia K., Lamba O. P., Napoli A. A. Jr., Sinha S., and Guo Y. (1998) Fusion between retinal rod outer segment membranes and model membranes: a role for photoreceptor peripherin/rds. Biochemistry 37, 9477–9487 [DOI] [PubMed] [Google Scholar]
- 30.Ding X. Q., Stricker H. M., and Naash M. I. (2005) Role of the second intradiscal loop of peripherin/rds in homo and hetero associations. Biochemistry 44, 4897–4904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chakraborty D., Conley S. M., Stuck M. W., and Naash M. I. (2010) Differences in RDS trafficking, assembly and function in cones versus rods: insights from studies of C150S-RDS. Hum. Mol. Genet. 19, 4799–4812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Conley S. M., Stuck M. W., Burnett J. L., Chakraborty D., Azadi S., Fliesler S. J., and Naash M. I. (2014) Insights into the mechanisms of macular degeneration associated with the R172W mutation in RDS. Hum. Mol. Genet. 23, 3102–3114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goldberg A. F., and Molday R. S. (1996) Subunit composition of the peripherin/rds-rom-1 disk rim complex from rod photoreceptors: hydrodynamic evidence for a tetrameric quaternary structure. Biochemistry 35, 6144–6149 [DOI] [PubMed] [Google Scholar]
- 34.Chakraborty D., Ding X. Q., Fliesler S. J., and Naash M. I. (2008) Outer segment oligomerization of Rds: evidence from mouse models and subcellular fractionation. Biochemistry 47, 1144–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loewen C. J., and Molday R. S. (2000) Disulfide-mediated oligomerization of Peripherin/Rds and Rom-1 in photoreceptor disk membranes. Implications for photoreceptor outer segment morphogenesis and degeneration. J. Biol. Chem. 275, 5370–5378 [DOI] [PubMed] [Google Scholar]
- 36.Goldberg A. F., Loewen C. J., and Molday R. S. (1998) Cysteine residues of photoreceptor peripherin/rds: role in subunit assembly and autosomal dominant retinitis pigmentosa. Biochemistry 37, 680–685 [DOI] [PubMed] [Google Scholar]
- 37.Loewen C. J., Moritz O. L., Tam B. M., Papermaster D. S., and Molday R. S. (2003) The role of subunit assembly in peripherin-2 targeting to rod photoreceptor disk membranes and retinitis pigmentosa. Mol. Biol. Cell 14, 3400–3413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Conley S. M., Stricker H. M., and Naash M. I. (2010) Biochemical analysis of phenotypic diversity associated with mutations in codon 244 of the retinal degeneration slow gene. Biochemistry 49, 905–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stuck M. W., Conley S. M., and Naash M. I. (2012) Defects in the outer limiting membrane are associated with rosette development in the Nrl−/− retina. PLoS ONE 7, e32484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Conley S. M., Stuck M. W., Burnett J. L., Chakraborty D., Azadi S., Fliesler S. J., and Naash M. I. (2014) Insights into the mechanisms of macular degeneration associated with the R172W mutation in RDS. Hum. Mol. Genet. 23, 3102–3114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cai X., Nash Z., Conley S. M., Fliesler S. J., Cooper M. J., and Naash M. I. (2009) A partial structural and functional rescue of a retinitis pigmentosa model with compacted DNA nanoparticles. PLoS ONE 4, e5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saga M., Mashima Y., Akeo K., Oguchi Y., Kudoh J., and Shimizu N. (1993) A novel Cys-214-Ser mutation in the peripherin/RDS gene in a Japanese family with autosomal dominant retinitis pigmentosa. Hum. Genet. 92, 519–521 [DOI] [PubMed] [Google Scholar]
- 43.Daniele L. L., Lillo C., Lyubarsky A. L., Nikonov S. S., Philp N., Mears A. J., Swaroop A., Williams D. S., and Pugh E. N. Jr. (2005) Cone-like morphological, molecular, and electrophysiological features of the photoreceptors of the Nrl knockout mouse. Invest. Ophthalmol. Vis. Sci. 46, 2156–2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mears A. J., Kondo M., Swain P. K., Takada Y., Bush R. A., Saunders T. L., Sieving P. A., and Swaroop A. (2001) Nrl is required for rod photoreceptor development. Nat. Genet. 29, 447–452 [DOI] [PubMed] [Google Scholar]