

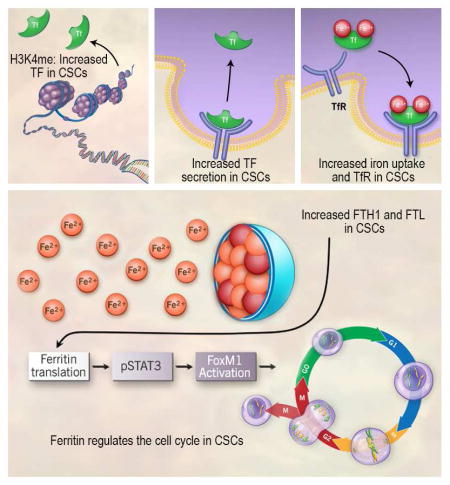
Summary
Glioblastomas display hierarchies with self-renewing cancer stem-like cells (CSCs). RNA sequencing and enhancer mapping revealed regulatory programs unique to CSCs causing upregulation of the iron transporter transferrin, the top differentially expressed gene compared to tissue-specific progenitors. Direct interrogation of iron uptake demonstrated CSCs potently extract iron from the microenvironment more effectively than other tumor cells. Systematic interrogation of iron flux determined that CSCs preferentially require transferrin receptor and ferritin - two core iron regulators - to propagate and form tumors in vivo. Depleting ferritin disrupted CSC mitotic progression, through the STAT3-FoxM1 regulatory axis, revealing an iron-regulated CSC pathway. Iron is a unique, primordial metal fundamental for earliest life forms, and on which CSCs have an epigenetically programmed, targetable dependence.
Graphical Abstract
Introduction
Glioblastoma (GBM, World Health Organization grade IV glioma) is the most lethal and prevalent primary malignant brain tumor in adults (Stupp et al., 2009). Even with the use of highly aggressive therapies, recurrence is inevitable and prognosis is dismal with a median survival of 14–15 months (Stupp et al., 2009). More effective therapeutic strategies are therefore urgent. We and others have reported that GBMs contain cellular hierarchies with cells featuring stem-like properties at the apex, governed by the microenvironment (Eyler et al., 2011; Li et al., 2009; Singh et al., 2004). The significance of cancer stem-like cells (CSCs) is underscored by their resistance to conventional therapy (Bao et al., 2006a; Chen et al., 2012) and promotion of tumor angiogenesis (Bao et al., 2006b). We and others have also demonstrated that CSCs can be defined functionally by their capacity to self-renew with an increased capacity to form tumorspheres, express stem cell markers, differentiate into multiple lineages, and phenocopy the original tumor in vivo (Eyler et al., 2011; Flavahan et al., 2013; Lathia et al., 2010; Li et al., 2009; Singh et al., 2004). CSCs endure and even thrive in stressful tumor conditions including hypoxia, oxidative stress, inflammation, acidic stress and low glucose (Flavahan et al., 2013; Hjelmeland et al., 2011; Li et al., 2009; Venere et al., 2014; Ye et al., 2012); all of these niches mediate effects in part through iron metabolism (Huang et al., 2013; Peyssonnaux et al., 2007; Torti and Torti, 2013). This coalescence suggests that exploiting aberrant iron regulation in tumors may uncover a direct driver of CSC tumorigenicity and therapeutic resistance.
Iron has specific functions in a non-cancerous cell; required for proteins or enzymes that regulate respiratory complexes, DNA and heme synthesis, and mitosis and epigenetic modifications, all of which are dysregulated in cancer (Lane et al., 2014; Torti and Torti, 2013). However, most anticancer therapies aimed at removing iron via chelation are not likely to be cancer tissue-specific, especially in the iron-dependent brain. Therefore, targeting iron regulation within tumor-specific and/or hyperactive pathways represents a potential approach to crippling a key cancer dependency.
Regulated iron scavenging is fundamental and ubiquitous throughout nature. We hypothesized that in GBM, CSCs scavenge iron by co-opting regulatory programs, typically reserved for the liver and specialized regions in the brain such as the choroid plexus (Leitner and Connor, 2012), to potentially secrete and uptake TF. To identify crucial downstream iron-mediated CSC pathways, it is necessary to perform “iron-tracing” experiments to measure iron uptake along with expression patterns of proteins needed for iron transport and storage. Intracellular iron entry typically requires TF binding ferric iron before complexing with transferrin receptor (TfR) followed by endocytosis and eventual iron release into the cytoplasm (Torti and Torti, 2013). TfR is highly expressed in many cancers including breast, lung, bladder, leukemia, lymphoma, and glioma; suggesting that tumor cells exhibit increased iron demands (Daniels et al., 2006a). Numerous oncology studies have attempted to harness this intracellular delivery system. Whether targeting TfR directly with antibodies, conjugating chemotherapeutics, such as doxorubicin, cisplatin, and chlorambucil, to TF and/or attaching genetic vectors to TF, it is evident that TfR activity is involved in tumor growth (Daniels et al., 2006a, 2006b). Yet, it is unclear how iron is driving tumor-specific pathways and/or if iron is preferentially utilized within a heterogeneous tumor population, as may be the case in GBM.
As free iron acts as a catalyst for producing free radicals via the Fenton reaction (Fe2+ + H2O2 → Fe3+ + ·OH + OH−), excess iron is stored in ferritin, a 24-subunit protein that can store up to 4,500 iron atoms. The clinical importance of ferritin in tumor growth is demonstrated in other cancers, contributing to tumorigenesis by acting as an autocrine growth factor, restoring tumor-dependent vessel growth, as well as being associated with invasion (Coffman et al., 2009; Holtkamp et al., 2005; Kikyo et al., 1994). Ferritin targeting also sensitizes glioma cells to chemotherapy (Liu et al., 2011). We hypothesize that altering iron availability in CSCs ablates drivers of the stem cell phenotype and targeting ferritin represents a point of fragility of iron addicted CSCs.
Results
CSCs utilize a hepatocyte and tumor specific epigenetic program to upregulate and secrete transferrin (TF)
To discover therapeutically viable gene and pathway targets in CSCs we performed next generation RNA sequencing on a validated GBM CSC model as well as normal brain progenitor cell populations (Figure 1A). We sought to discover targets highly expressed in the CSCs, but not in normal neuronal and glial progenitors to avoid targets likely to introduce CNS toxicity. TF was the top differentially expressed gene compared to oligodendrocyte progenitor cells, the most abundant CNS progenitor cell in the adult brain, as well as in the top 6 differentially regulated genes compared to both astroglial and neural progenitor cells (Figure 1B). We validated that TF protein was highly upregulated in GBM compared to normal brain (Figure S1A, S1B), with expression present in mitotic cells and perivascular niches in GBM (Figure S1C, S1D). While not statistically significant, likely due to limited sample size, there was a trend for worse survival in GBM patients with high TF expression (Figure S1E). In the normal brain, only the choroid plexus secretes TF (Leitner and Connor, 2012), potentially due to alternative splicing variants of TF in TF-producing cells, such oligodendrocytes (de Arriba Zerpa et al., 2000). However, we determined that CSCs (CD133+ cells) secrete high levels of TF, whereas TF secretion was barely detectable in non-CSCs (CD133− cells) (Figure 1C). To understand how TF was being regulated in CSCs, we performed epigenomic profiling of the same CSC model as in Figure 1A using Chromatin Immunoprecipitation (ChIP)-sequencing and compared this profile to published datasets. We focused our analysis on putative enhancer elements, marked by H3K4 mono-methylation (H3K4me1). Enhancers are tissue specific regulatory elements, and gained enhancers in cancer have been shown to mark cancer dependency genes (Akhtar-Zaidi et al., 2012). We used the algorithm PreSTIGE (Corradin et al., 2014) to call enhancers predicted to regulate TF in CSCs, as well as in other somatic and neoplastic cell types. Only CSCs and HepG2, a well-differentiated liver carcinoma cell line often used to study hepatocyte function, had enhancers predicted to target TF (Figure 1D). Both CSCs and HepG2 cells had multiple, overlapping enhancers targeting TF; some of which are lost upon CSC differentiation (Figure 1E). Together, these data suggest that CSCs gain a hepatocyte specific epigenetic regulatory program that allows them to upregulate TF expression.
Figure 1.

Upregulation of TF through hepatocyte specific epigenetic program and increased iron uptake via TF in CSCs. (A) Heat map of fold change of gene expression (FPKM from RNA-seq) in CSCs versus normal neural progenitors. Red represents increased expression in CSCs compared to normal neural progenitor cells, while blue represents a decrease in expression in CSCs compared to normal controls. Samples compared are IN528 CSCs versus human Embryonic Stem (hES)-cell derived OPCs, Glial Progenitors (GP) derived from acute dissociated epilepsy resection, and hES-cell derived neuronal precursor cell (NPC). OPC and GP data was generated from this study while NPC data is from (Sauvageau et al., 2013). (B) Top and bottom 10 differentially expressed genes from cell types above (CSC FPKM/OPC FPKM). (C) TF secretion was measured in CSCs and non-CSCs using 1 μg protein from each xenograft specimen added to a transferrin ELISA (Abcam). ***, p < 0.001. (D) PreSTIGE analysis (Corradin et al., 2014) was conducted on H3K4me1 ChIP-seq data from CSCs and 13 ENCODE cell lines to determine enhancers predicted to target TF. (E) UCSC Browser image depicting H3K4me1 peaks at the TF locus. Red background designates acutely dissociated (p0) CSCs or in vitro (MGG) CSCs, black designates in vitro serum differentiated CSCs. Enhancers predicted by PreSTIGE to target TF in p0 CSCs. (F) Diagram of the experimental procedure for ex vivo imaging of TF uptake in brain slices. (G) Reconstructions of representative fields showing TF (green) in CSCs (red) and non-CSCs (blue) and quantification of fluorescent-Tf as measured on the surface of the three-dimensional reconstruction. ***, p = 4.73 × 10−166. Scale bar = 10 μm. (H) Radiolabeled iron (100 μM 55Fe bound to TF) uptake after 3 hr in CSCs compared to non-CSCs, **, p < 0.01; ***, p < 0.001. (I) Scavenge of iron (100 μM 55Fe) by CSCs and matched non-CSCs, ***, p < 0.001. Error bars represent ± SEM. See also Figure S1 and Table S1.
Iron uptake is enhanced in CSCs
To demonstrate preferential iron scavenging ability in GBM CSCs, we developed an ex vivo transplant model in which non-neoplastic brain slice cultures were implanted with fluorescently-labeled CSCs and non-CSCs followed by TF loading and subsequent imaging (Figure 1F). We isolated matched CSC and non-CSC populations based on sorting for CD133 (Table S1), a protein epitope we have repeatedly validated as a marker of CSC self-renewal (Figure S1F) and tumor formation. While the use of this marker is controversial as it has not been universally informative across all tumors and no perfect CSC marker has been identified, our lab has repeatedly shown that CD133 informs a fraction of GBM cells that enrich for a functional CSC hierarchy of tumorigenic and non-tumorigenic cells (Figure S1F) (Bao et al., 2006a; Eyler et al., 2011; Flavahan et al., 2013; Hjelmeland et al., 2011; Lathia et al., 2010; Li et al., 2009; Venere et al., 2014; Yan et al., 2014). Furthermore, CSCs from our models express putative neural stem cell markers (i.e. Sox2, Olig2, A2B5, Tuj1, and Oct4), consistent with the xenografts used in the current studies (Yan et al., 2014).
Multiphoton imaging of our ex-vivo transplant model and 3-D reconstruction of TF co-localization with the tumor cell surface demonstrated that the amount of bound TF was over 20-fold higher in CSCs compared to non-CSCs under the same conditions (Figure 1G). As a complimentary approach to verify this difference, we quantified uptake of radiolabeled iron (55Fe). CSCs and non-CSCs grown in identical culture conditions were treated with TF-conjugated iron for 3 hr, resulting in up to 3-fold greater uptake in CSCs (Figure 1H). We hypothesized that if CSCs actively produce and secrete TF, unbound iron uptake should be elevated even in the absence of experimentally complexed iron with TF. This route of iron uptake was at least 2-fold higher in CSCs compared to uptake in non-CSCs (Figure 1I), even exceeding the uptake of activated human macrophages (data not shown), which commonly serve as a positive control (Kuhn et al., 1999).
TfR expression is indicative of increased tumorigenicity
We compared ChIP-sequencing of H3K27 acetylation at the TfR loci (encoded by TFRC) in normal brain, GBM, CSCs and non-CSCs (Figure S2A) as well immunohistochemically in non-neoplastic brains and in gliomas (WHO grades II, III, and IV) (Figure S2B). Consistent with in silico analysis (Figure 2A, B), neuropathological scoring of TfR immunohistochemical (IHC) intensity from a tumor microarray demonstrated a grade dependence with nearly 2-fold higher TfR in grade IV gliomas compared to lower grade tumors (Figure 2C). The Repository for Molecular Brain Neoplasia Database (REMBRANDT) and The Cancer Genome Atlas (TCGA) datasets along with our IHC analysis each revealed a negative correlation between TfR and survival (Figure 2D–F; Figure S2C).
Figure 2.

Elevated TfR expression in GBM indicates poor prognosis. (A, B) Analysis of brain tumor data sets from Oncomine for the mRNA level of TFRC in GBM compared to non-neoplastic brain (A; Sun dataset) and in high-grade glioma compared to lower grade glioma (B; Phillips dataset). (C) IHC scores of TfR in different grades of glioma in the Horbinski dataset. (D, E) Analysis of correlation between TFRC mRNA expression and glioma patient survival (D; REMBRANDT dataset and E; TCGA dataset). High expression represents 2-fold elevation vs. intermediate expression with log rank analysis. Low expression represents < 2-fold reduction vs. intermediate expression with log rank analysis. (F) Analysis of TfR protein expression score with patient survival; Horbinski dataset (WHO grades II–IV). (G) Immunofluorescence (IF) of TfR (red), Sox2 (green), and DAPI (blue) in GBM xenograft tissue. Scale bar = 50 μm. (H) Western blot of TfR and β-actin in CSCs and non-CSCs from 3 different GBM xenografts. (I) Membrane and cytosol fractions of TfR were determined in CSCs and non-CSCs. Error bars represent ± SEM. See also Figure S2.
Similar to patient GBM tissue, TfR was expressed in multiple GBM xenografts and co-localized with Sox2, a transcription factor recognized as a putative CSC marker (Figure 2G) and also increased in CSCs compared to non-CSCs (Figure 2H). Recycling of the TfR complex occurs following the release of iron from the endosome. We used a biotinylation assay to segregate and measure cell surface (membrane) and cytosolic protein fractions of TfR (Figure 2I). Cells were grown to equal confluency in identical media conditions to mitigate any potential differences in receptor expression that could be attributed to cell density. ERK1/2 served as a separation control, representing a molecule not normally expressed on the membrane surface. While cytosolic TfR was indistinguishable between cell types, the membrane fraction was noticeably elevated in CSCs, suggesting that the surface level expression accounts for the overall increase of TfR in CSCs, likely due to increased recycling or upregulation of TfR (Figure 2I).
TfR comprised an average of 55% of the total tumor population of freshly dissociated patient-derived xenografts as analyzed by flow cytometry (Figure S3A). Indeed, TfR expression is not restricted to CSCs nor is it likely a CSC marker, yet TfR/Sox2 co-localization prompted us to investigate if TfR was essential for CSC maintenance, including formation of tumorspheres from a single cell in vitro. Bulk GBM cells positive or negative for TfR were plated by limiting dilution and evaluated for tumorsphere formation 10 days later. TfR-positive cells consistently formed tumorspheres at a greater frequency than TfR-negative cells (Figure 3A). Likewise, plating TfR-positive cells increased the percentage of wells containing tumorspheres by 25-fold (plating 1 cell/well) and 10-fold (plating 10 cells/well) (Figure 3B). In vivo xenografting with limiting dilution based on high or low TfR expression showed that mice bearing TfR-high cells developed tumors with a shorter latency and increased tumor incidence with decreased survival compared to mice bearing TfR-low cells (Figure 3C–F; Figure S3B, C). These results demonstrate the importance of iron acquisition for CSC self-renewal and tumor initiation. This was further demonstrated by performing limited dilution assays in the context of CSCs that were either positive or negative for TfR, which revealed that TfR expression increased the stem cell frequency of CSCs by nearly 10-fold (Figure S3D, E).
Figure 3.

TfR drives sphere formation in vitro and tumor formation in vivo. (A) In vitro limiting dilution assays plating decreasing numbers of FACS TfR+ or TfR− tumor cells from freshly dissociated GBM xenografts calculated with extreme limiting dilution assay (ELDA) analysis (Top). Stem cell frequencies from TfR+ and TfR− cells were calculated as the ratio 1/x, where 1 = stem cell and x = all cells (Bottom). (B) Percentage of wells that formed tumorspheres from either TfR+ or TfR− GBM cells based on plating 1 cell/well (Left) or 10 cells/well (Right). Error bars represent ± SEM. (C, D) Survival curves of mice orthotopically implanted with TfR-high or TfR-low expressing GBM cells at dilutions of 1,000 cells (C) or 100 cells (D); n = 5 for all groups. (E, F) Median survival (E, in days post injection) and tumor incidence (F, in days) in mice bearing GBM cells. See also Figure S3.
Ferritin is a negative GBM prognostic factor and is increased in CSCs
Once shuttled out of the endosome, iron is either used immediately or stored in ferritin, which is made up of two subunits with non-overlapping functions – heavy chain (H-ferritin; FTH1) and light chain (L-ferritin; FTL). To circumvent the challenge of detecting free iron, we used FTH1 and FTL as indicators of iron flux into CSCs. Intracellular free iron leads to ferritin mRNA translation resulting in increased iron storage. Treating CSCs with iron for 3 hr increased FTH1 and FTL protein levels, which continued to increase for 24 hr (Figure 4A). These data suggest that CSCs scavenge iron to the extent that storage machinery needs to be increased to buffer any potential negative consequences of free iron and to regulate proper intracellular iron distribution.
Figure 4.

Ferritin is preferentially expressed in CSCs. (A) Western blot of FTH1 and FTL following treatment with 100 μM Fe. (B) In silico analysis of brain tumor datasets from Oncomine showing FTH1 and FTL expression in GBM compared to normal brain tissue. Middle box line represents median, top box line represents upper quartile, bottom box line represents lower quartile, top error bar represents 90th percent and bottom error bar represents 10th percent. ***, p < 0.001. (C) Quantification of FTH1 and FTL IHC intensity with glioma grade. Error bars represent ± SEM. (D) Correlation between overall survival of glioma patients and FTH1 and FTL IHC score of their tumor from the Horbinski dataset (WHO grades II–IV). (E) Correlations between FTH1 or FTL expression and patient survival in the TCGA dataset. (F) Ferritin IHC of control brain, low-grade and high-grade glioma tissue. Scale bar = 50 μm. (G) IF labeling of GBM xenografts with FTH1 (Top, red) or FTL (Bottom, red), Sox2 (green), and DAPI (blue). Scale bar = 50 μm. (H) Western blot of FTH1, FTL and β-actin in CSCs and non-CSCs from 6 different GBM xenografts.
Based on in silico analysis of published gene expression databases, FTH1 and FTL are expressed higher in GBM compared to non-neoplastic brain with a strong correlation with FTH1 and FTL gene expression patterns (Figure 4B). In a separate patient cohort, we performed IHC scoring for FTH1 and FTL expression, which increased with glioma grade (Figure 4C), correlated with worse survival (Figure 4D), and was further validated by comparing survival curves of GBM patients from the TCGA dataset (Figure 4E). Ferritin expression increased with tumor grade (Figure 4F) yet appeared confined to subsets of cells and tumor niches, suggesting that not every tumor cell is equally iron-dependent. In GBM-derived xenografts, ferritin co-localized with Sox2 (Figure 4G), prompting us to analyze ferritin protein expression in CSCs and non-CSCs. In all GBM specimens, FTH1 and FTL protein were elevated in CSCs compared to matched non-CSCs cultured identically (Figure 4H). These data indicate that iron storage is elevated in CSCs suggesting greater iron dependence compared to non-CSCs and the non-neoplastic brain.
Targeting ferritin expression decreases CSCs growth in vitro and in vivo
To determine if ferritin maintains CSC tumorigenicity, short hairpin RNA interference (shRNA) was transduced into CSCs to ablate ferritin activity. Validated constructs (Figure S4A–D) with unique sequences at non-overlapping regions exhibiting the best knockdown were chosen (referred to hereafter as FTH1 shRNA or FTL shRNA) along with a control, non-targeting shRNA sequence that does not target a mammalian sequence (referred to as NT shRNA). FTH1 and FTL shRNA depleted ferritin mRNA and protein levels in CSCs across multiple specimens (Figures 5A and 5B). As hypothesized, ferritin knockdown decreased TfR expression, likely due to released intracellular free iron causing a negative feedback loop. Both ferritin chains are necessary for proper storage function, yet it is unknown if targeting either subunit separately affects the overall tumorigenicity of CSCs. We hypothesized that preventing the ability to properly store and control iron would halt CSC proliferation, severely impacting their growth. This became evident in examining tumorsphere formation, which was robustly diminished by ferritin knockdown (Figure 5C, D). FTH1 or FTL knockdown affected cellular fitness in CSCs by stunting growth (Figure 5E). Proliferation, indicated by thymidine incorporation, decreased by more than sevenfold in CSCs transduced with either FTH1 or FTL shRNA (Figure 5F). Elevated ferritin expression in CSCs suggests an increased iron demand and/or dependence; therefore, we expected that targeting ferritin would be more detrimental to CSCs compared to non-CSCs or non-neoplastic brain cells. In this separate study, growth of non-neoplastic adult brain cells was least affected by ferritin knockdown (Figure S4E), suggesting a potentially high therapeutic index for ferritin targeting in GBM.
Figure 5.

Targeting ferritin decreases CSC growth in vitro and tumor formation in vivo. (A–B) qRT-PCR of FTH1 and FTL mRNA (A) and immunoblot of proteins (B) in CSCs following transduction with NT, FTH1, or FTL shRNA. (C) CSC-derived tumorsphere formation following ferritin knockdown. Scale bar = 200 μm. (D) In vitro limiting dilution assays of CSCs transduced with NT, FTH1, or FTL shRNA calculated with ELDA analysis. (E) ATP-dependent growth in CSCs after ferritin knockdown. *, p < 0.05; ***, p < 0.001. (F) CSC proliferation following ferritin knockdown, measured by 3H-thymidine incorporation. ***, p < 0.001. (G) In vivo imagining 2 weeks post-orthotopic injection of CSCs expressing luciferase, transduced with NT, FTH1, or FTL shRNA. (H) Kaplan-Meier curves indicating survival of mice bearing intracranial injections of 10,000 CSCs with ferritin knockdown. Error bars represent ± SEM. See also Figure S4.
Extending these studies to in vivo orthotopic models, 10,000 viable, luciferase-expressing CSCs acutely transduced with NT, FTH1 or FTL shRNA were intracranially implanted into mice followed by in vivo monitoring. While mice with NT shRNA CSCs began to develop tumor symptoms within two weeks, there were no signs of tumors in mice with FTH1 or FTL shRNA CSCs at this time point (Figure 5G). Validation with CSCs derived from an additional human specimen produced nearly identical results (data not shown) and tumor formation was virtually absent in mice that received CSCs from either specimen with ferritin knockdown (Figure 5H). These results demonstrate that iron storage is critical for tumor formation. Without ferritin, an abundance of uncontrolled iron likely blocks the necessary steps necessary for CSCs to propagate and create an environment enriched for forming tumors.
Molecular Mediators of Ferritin
The loss of proper iron storage severely decreases CSC survival and tumor forming capabilities, so we set out to identify molecular nodes downstream of ferritin that are vital to CSC biology, amenable to therapeutic targeting, and whose disruption may explain our observed results. As loss of either FTH1 or FTL is vital for CSC biology, we interrogated the TCGA database to confirm relative homologous gene correlations and ferritin expression across GBM subtypes (Figure 6A, B). Next, CSCs isolated from five different GBM patients were transduced with NT, FTH1, or FTL shRNA, with verification of ferritin knockdown followed by analysis with Affymetrix HuGene 2.1 chips for whole transcriptome profiling. Of the total 40,716 RefSeq transcripts profiled, genes were analyzed by computing the robust-multi array average (RMA) across all samples followed by selecting only genes that were increased or decreased by at least 2-fold following FTH1 and FTL knockdown across all five specimens (Table S2). Our initial studies focused on the 116 down-regulated genes (Figure 6C). Classifying these genes based on ontology, we discovered the largest subset of genes were primarily involved with cell cycle and mitosis (84 out of 116 or 71.8%) followed by DNA damage response (29 out of 116 or 24.8%) and Rho GTP signaling (7 out of 117 or 5.98%), keeping in mind that the role of some genes overlap across multiple pathways (Figure 6C). Consistent with these data were geneset enrichment plots verifying that loss FTH1 and FTL impairs expression of cell cycle associated genes (Figure 6D). To confirm that ferritin inhibition dysregulates the CSC cell cycle, we performed flow cytometry-based cell cycle profiling; FTH1 or FTL shRNA reduced time spent in S-phase (Figure 6E, Figure S5A, B). FTH1 knockdown increased time spent in G0/G1 while FTL knockdown caused G2/M arrest. Collectively, these results suggest that ferritin function critically regulates cell cycle progression in CSCs.
Figure 6.

Gene expression profiling reveals ferritin dependence for cell cycle progression. (A) Genome-wide analysis of TCGA GBM dataset that demonstrates genes (points on graph) displaying correlation coefficients with FTH1 (x-axis) and correlation coefficients with FTL (y-axis). (B) FTH1 and FTL expression in TCGA GBM dataset based on subtype. (C) Affymetrix HuGene 2.1 microarray analysis of all genes decreased ≥ 2-fold from 5 different GBM specimens following ferritin shRNA compared to NT shRNAs. (D) Geneset enrichment analysis using the C2 canonical pathways Broad MsigDB database on gene expression data compared non-targeting control against FTH1 (left) or FTL (right) knockdown. (E) Cell cycle analysis of two GBM specimens following transduction with NT, FTH1, or FTL shRNA. See also Figure S5 and Table S2.
STAT3 and FoxM1 regulation are impacted by ferritin knockdown
We analyzed our dataset for genes most correlated with ferritin expression and discovered that signal transducer and activator of transcription 3 (STAT3) had the highest correlation value (Figure 7A), providing a potential clue for downstream pathways of ferritin signaling. Ingenuity’s upstream regulator analysis identified transcriptional regulators that most likely explain the observed gene expression data changes within our dataset. From this analysis, the STAT3-regulated transcription factor FoxM1 was found to have the highest activation z-score in both FTH1 shRNA and FTL shRNA samples (Figure 7B). FoxM1 is a transcription factor known to play a pivotal role in cell cycle signaling many cancers, including being overexpressed in GBM (Hodgson et al., 2009; Liu et al., 2006; Zhang et al., 2011) and has recently been identified to interact with β-catenin (Zhang et al., 2011) and the mitotic kinase MELK (Joshi et al., 2013) in CSCs, regulating their self-renewal and proliferation. Based on our gene expression analysis, FOXM1 was decreased by twofold in CSCs depleted of FTH1 or FTL (Figure S6A). Ferritin knockdown in CSCs reduced FoxM1, phosphorylated and, to a lesser extent, total STAT3 protein levels as well as FoxM1 promoter activity (Figure 7C, D), suggesting a ferritin-STAT3-FoxM1 feedback loop. Further confirming the importance of ferritin for FoxM1 function, mRNA of FoxM1 downstream targets were all decreased following ferritin knockdown (Figure 7E), consistent with reduced FoxM1 occupancy as measured by chromatin immunoprecitation (Figure 7F). FoxM1 rescue experiments were performed in which CSCs were first infected with NT control sequence, FTH1, or FTL shRNA followed by transient transfection with either a FoxM1 overexpression construct or empty vector control. After 48 hr, FoxM1 overexpression restored and even improved CSC growth (Figure 7G, Figure S6B). These results support FoxM1 as an essential downstream mediator of ferritin in CSCs.
Figure 7.

FoxM1 signaling requires intact ferritin. (A) Correlation of STAT3 with total ferritin following ferritin knockdown. Colors represent individual xenograft specimens. (B) Ingenuity upstream analysis of pathways down-regulated by ferritin shRNA-derived gene expression data. (C) Immunoblot of FoxM1, pSTAT3 and total STAT3 following ferritin knockdown in CSCs. (D) Ferritin knockdown effect on FoxM1 promoter activity in 08-387 CSCs, measured with a luciferase reporter construct; ***, p < 0.001. (E) Relative mRNA levels of FoxM1 downstream targets derived from gene expression data (averages taken from specimens 4121, 3832, 3691, 08-387 and GBM10); *, p < 0.05; **, p < 0.01; ***, p < 0.001. (F) ChIP PCR of FoxM1 targets performed on 08-387 CSCs transduced with NT, FTH1, or FTL shRNA. (G) Transient FoxM1 overexpression in 08-387 CSCs 48h following ferritin knockdown; *, p < 0.05; ***, p < 0.001. Error bars represent ± SEM. See also Figure S6.
Discussion
Absolutely essential for life, iron regulation is prominently expressed in the CSC transcriptome, with iron mediating interplay with the tumor microenvironment, epigenetic modifications, genetic mutations, and altered metabolism; processes increasingly connected in driving CSC maintenance and therapeutic resistance. Given the unique ability to donate and accept electrons, iron homeostasis must always be maintained; yet in GBM, iron flux is asymmetrical with CSCs emulating a bacteria-siderophore-like system where TF is epigenetically regulated and secreted to potentially ensure that CSCs are able to scavenge sufficient iron. This program results in a cascade of events in which TfR recycling and ferritin translation increases. While iron regulators have long been recognized as involved in cancer biology, specifically how and why subsets of cancer cells use iron to sustain tumor growth has been unclear. We now demonstrate a targetable molecular fingerprint of iron in CSCs involving a series of tumor-specific feedback loops that are likely amplified and/or dysfunctional depending on cues derived from stem cell niches.
TfR and ferritin are intricately regulated by iron but expression may be induced by microenvironmental influences, including those we and others previously identified as promoters of the CSC phenotype, such as nitric oxide (NO) (Eyler et al., 2011; Recalcati et al., 1998) and the cytokines NF-κB (Hjelmeland et al., 2010; Kwak et al., 1995; Pham et al., 2004) and TGF-β (Koorts et al., 2011; Schober and Fuchs, 2011; Shinojima et al., 2013). In turn, ferritin can act as a proinflammatory cytokine, potentially independent of iron status (Ruddell et al., 2009). Likewise, the angiogenic and hypoxic CSC niches also likely rely on a bi-directional relationship with iron given that ferritin controls angiogenesis by binding to and inhibiting the anti-angiogenic properties of cleaved high molecular weight kininogen (HKa) (Coffman et al., 2009). Activation of HIF prolyl hydroxylases requires an iron chaperone, poly (rC) binding protein 1 (PCBP1), which delivers iron to ferritin (Nandal et al., 2011). Currently under investigation is whether these interactions exist in GBM, predominantly affecting CSCs and how ferritin preferentially directs iron towards being used in CSC-specific pathways, providing a new strategy for designing GBM therapies.
TF is a secreted protein, making it a potentially difficult part of the iron pathway to design therapeutic agents against and TfR is important even for the normal brain; therefore, we chose to focus on understanding the downstream effects of targeting ferritin – consisting of a set of proteins with cell-specific distribution patterns (Connor et al., 1994). We identified STAT3 phosphorylation and FoxM1 signaling as the most notable pathways affected by ferritin depletion. Out of all genes in our microarray, STAT3 correlated the most with ferritin expression. STAT3 is a critical CSC signaling node (Cao et al., 2010; Kim et al., 2013) upstream of FoxM1 (Mencalha et al., 2012) that also leads to promoter binding of Myc (Zhao et al., 2014), an oncogenic ferritin repressor (Wu et al., 1999), and Sox2 (Zhao et al., 2014), a stem core cell transcription factor that we found co-localized in CSCs expressing ferritin. The bidirectional FoxM1-STAT3 signaling axis has recently been fortified in GBM, whereas FoxM1 is also required for the STAT3 activation required to promote CSC self-renewal and tumorigenicity (Gong et al., 2015). We demonstrate that at a minimum, this pathway is sensitive to changes in iron homeostasis. We postulate another scenario; STAT3 and FoxM1 signaling are iron-dependent and/or require direct ferritin binding. Excessive free iron, not contained by ferritin (which we hypothesis to occur in our model using ferritin shRNAs), induces the formation of free radicals and reactive oxygen species, triggering the activation of antioxidant-responsive elements (AREs) located in FTH1 and FTL (Iwasaki et al., 2007; Wasserman and Fahl, 1997). ARE binding represses ferritin function; however, this is counteracted by the protein inhibitor of activated STAT3 (PIAS3) (Iwasaki et al., 2007). PIAS3 stabilization of ferritin may represent part of the feedback loop in which STAT3 and downstream signaling targets such as FoxM1 hinge on ferritin activity. Further supporting an iron-STAT3 regulatory axis is STAT3-mediated induction of hepcidin; an enzyme produced in the liver that prevents iron export (Wrighting and Andrews, 2006). Similar to the effects of CSCs upregulating TfR, hepcidin activity results in increased intracellular iron and ferritin translation. We hypothesize that the increased ferritin in CSCs directly interacts with STAT3 and/or potentiates STAT3 phosphorylation. Therefore, without ferritin intact, this autocrine system is inhibited and might explain the inability for CSCs to develop tumors when FTH1 and FTL were depleted. CSC dependence on ample iron trafficking through TfR and ferritin was further evident when we segregated acutely dissociated GBM cells solely off of TfR expression, which was enough to dictate tumor formation in vivo – occurring in the absence of mutations or growth factor stimuli.
CSCs represent a small tumor cell subset, ranging from 0.1% to 10% of the overall GBM cytoarchitecture (Bao et al., 2006a; Yan et al., 2014). Based on our microarray data, only ~1% of all genes in CSCs were decreased more than twofold (our threshold for significance) by ferritin knockdown, yet the functional consequences of losing these genes were significant. This suggests that the biological insult of altering iron flux through ferritin inhibition in CSCs is not a passive or random event. This needs to be taken into account when designing effective treatment strategies. Current iron-based treatment strategies are focused on iron chelation therapy, which removes intracellular iron from any cell, carrying significant side effects. Given that CSCs uptake more iron than non-CSCs, thereby providing a buffering capacity should there be a sudden loss of iron, it is unlikely that iron chelation will be effective in treating GBM or other CSC-driven tumors. Alternatively, we demonstrated that targeting the ability of a cell to control iron by inhibiting proper iron storage was the most detrimental to cells with the highest iron demands. Our studies demonstrate the importance of looking beyond considerations of iron as only a ubiquitous, un-targetable metal and instead indicate the feasibility of disrupting iron flux in CSCs – a cell type difficult to penetrate with current therapies. Moving forward, we envision broadening this understanding while integrating nanotechnology platforms and small molecule inhibitors against regulators of iron handling in designing effective therapies for GBM.
Experimental Procedures
For details of standard lab techniques see Supplemental Experimental Procedures.
Human GBM specimens and derivative CSCs
Glioblastoma (GBM) tissues were obtained from excess surgical materials from patients with informed consent at Duke University, Mayo Clinic, Cleveland Clinic, University of Kentucky and Odense University Hospital after review from a neuropathologist in accordance with an approved protocol by the Institutional Review Board (USA) and the Committee on Health Research Ethics (Denmark). Functionally validated GBM CSCs and/or non-CSCs were derived immediately after dissociation of xenograft (transiently) passaged in immunocompromised mice using prospective sorting and in some cases transiently cultured in vitro. For all experiments in which CSCs and non-CSCs were directly compared, Geltrex (Invitrogen) was used to attach cells to tissue-culture plates and a medium consisting of CSC medium (Neurobasal Medium (NBM; Invitrogen) + B27 (w/o vitamin A; Invitrogen) + 20ng/ml EGF/FGF) with 1% FBS was used for long-term studies (beyond 24 hr) whereas in short-term experiments (less than 6 h), a null medium consisting of only NBM was used. For further details see Supplemental Experimental Procedures.
hES-cell derived OPCs and isolation and culture of primary normal glial progenitors
Human OPCs were differentiated from the human embryonic stem cell line H7 over a 154 day protocol as previously demonstrated (Wang et al., 2013). Normal (NM), non-neoplastic cells were derived from patient tissue specimens of neurosurgical resection in accordance with a Cleveland Clinic Institutional Review Board-approved protocol. Cells that grew out were sorted for A2B5 and characterized by marker staining to be proliferating cells of the glial lineage. For details see Supplemental Experimental Procedures.
RNA-Sequencing
RNA was isolated using standard methods. Library was prepared with the Illumina TruSeq RNA Sample Preparation Kit and sequenced on the Illumina HiSeq 2500 platform at the Case Western Reserve University Genomics Core Facility. Reads were aligned to hg19 using Tophat and analyzed with Cufflinks. For details see Supplemental Experimental Procedures.
ChIP-sequencing
H3K4me1ChIP was performed from 5×106 crosslinked p0-CSCs and sequencing libraries were prepared as previously described (Corradin et al., 2014). For details, including predicting gene targets of enhancers with PreSTIGE, see Supplemental Experimental Procedures.
Tissue microarray immunohistochemistry and survival analysis
The glioma tissue microarray (TMA) and semiquantification of immunohistochemical data was performed as described previously (Gilbert et al., 2014). Briefly, three 2-mm diameter cores per deidentified tumor were obtained and semiquantified on a relative scale from 0 to 3, with 0 = negative and 3 = strongest. Results from all 3 cores were averaged together to produce a final score for a tumor. For details see Supplemental Experimental Procedures.
Cell cycle analysis
Cells were fixed, stained with PI- staining buffer (10 μg/ml propidium iodide, 10 μg/ml RNAse A, 0.5% Triton X-100 in PBS) for 30 min at 37°C, spun down and resuspended in PBS. Samples were acquired with FACScan (BD Biosciences) and analyzed using Modfit (Verity) and FlowJo (Tree Star) software.
In vitro limiting dilution assay and tumorsphere formation
Propidium iodide-negative cells (n ≥ 6) were flow sorted with decreasing numbers of cells per well (1, 5, 10, 20, 50, and 100) plated in 96-well plates. Extreme limiting dilution analysis was performed using software available at http://bioinf.wehi.edu.au/software/elda/. Tumorsphere formation assays were also performed similarly to those in our previous reports (Flavahan et al., 2013), with 1 cell per well and 10 cells per well plated in 96-well plates and the percentage of wells with tumorspheres were measured after 10 d.
Vectors and lentiviral constructs
Overexpression vectors (pcDNA 3.1 negative control and FoxM1-WT; a kind gift from Dr. Ichiro Nakano, Ohio State University) and FoxM1 promoter pGL3 luciferase vectors (a kind gift from Dr. Suyun Huang, MD Anderson Cancer Center) were transfected using Fugene HD (Promega) according to manufacturer’s instructions. Luciferase-based FoxM1 promoter activity was normalized to β-gal and renilla transfection levels. See Supplemental Experimental procedures for lentiviral production details, clone sequences for FTH1 and FTL shRNA and FoxM1 overexpression.
In vivo tumor initiation assay
All animal procedures were performed in accordance with Cleveland Clinic IACUC approved protocols. For in vivo limiting dilution studies, acutely dissociated GBM xenografts were sorted as TfR high (top 20%) and TfR low (bottom 20%) and recovered for 1 h in a 37°C incubator. Three dilutions (10,000, 1000, or 100 cells; n = 5 mice per group) were intracranially injected into mice 4–6 weeks of age. Brains were removed at the onset of neurological signs. For ferritin shRNA studies, luciferase-labeled CSCs were transduced with lentiviral vectors expressing NT, FTH1, or FTL shRNA. At 2 d post-transduction 10,000 viable cells were intracranially injected into mice as previously described (Flavahan et al., 2013; Yan et al., 2014). A Xenogen IVIS (Perkin Elmer) was used for in vivo imaging.
Retrospective analysis of TFRC, FTH1, FTL and FOXM1 gene expression in human gliomas
Correlations between glioma grade, patient survival and gene expression were determined through analysis of TCGA, Sun, Nutt, Freije, and Phillips brain data sets, which are available through Oncomine (Compendia Biosciences, http://www.oncomine.org/). For details see Supplemental Experimental Procedures
Statistical analysis
See Supplemental Experimental Procedures for all statistical analysis performed.
Supplementary Material
Significance.
Glioblastoma is the most lethal primary brain tumor due in part to chemoresistant and radioresistant cancer stem-like cells (CSCs). We used RNA and ChIP-sequencing to identify an epigenetic program used almost exclusively in the liver that allows CSCs to preferentially scavenge iron more effectively than other tumor cells, resembling the activity of iron-starved microbial pathogens. Systematic interrogation of iron flux determined that CSCs require transferrin receptor and ferritin – two core iron regulators – to propagate and form tumors in vivo. Disrupting iron metabolism via ferritin targeting prevented downstream signaling of critical STAT3 and FoxM1-mediated pathways. Our findings uncover a hyper-activated, epigenetically programmed CSC metabolic node amenable for therapeutic targeting.
Acknowledgments
We thank Cleveland Clinic Microscopy and Flow Cores, Monica Venere for her thoughtful insight, Dana Napier for her histologic expertise, and James Morrow for help with PreSTIGE Analysis. This work was supported by The American Brain Tumor Association (DLS), The Peter and Carmen Lucia Buck Training Program in Translational Clinical Oncology (CH), the University of Kentucky College of Medicine Physician Scientist Program (CH), James S. McDonnell Foundation (JNR) and NIH grants: T32 TRN508838 CWRU (DLS) F30 CA183510 (TEM) T32 GM007250 MSTP (TEM), K08 CA155764 (CH), 2P20 RR020171 COBRE pilot grant (CH), R01 CA154130 (JNR), R01 CA169117 (JNR), R01 CA171652 (JNR), R01 NS087913 (JNR), R01 NS089272 (JNR). The TMA construction was supported by the Biospecimen and Tissue Procurement Shared Resource Facility of the University of Kentucky Markey Cancer Center (P30CA177558).
Footnotes
Accession numbers
Super series accession number – GSE72204. Epigenetic profile – GSE72201, RNA-seq profile – GSE72202 and gene expression array after ferritin knockdown – GSE72203.
Conflict of Interest
The authors declare no conflict of interest.
Author Contribution List
David Schonberg: Conception and design, development of methodology, acquisition of data, analysis and interpretation of data, writing and/or revision of manuscript
Tyler Miller: Conception and design, development of methodology, acquisition of data, analysis and interpretation of data, writing and/or revision of manuscript
Quilian Wu: Acquisition of data, analysis and interpretation of data
William Flavahan: Acquisition of data, analysis and interpretation of data, writing and/or revision of manuscript
Nupur Das: Conception and design, development of methodology, acquisition of data
James Hale: Acquisition of data
Christopher Hubert: Conception and design, development of methodology, acquisition of data, analysis and interpretation of data
Stephen Mack: Acquisition of data
Awad Jarrar: Acquisition of data
Robert Karl: Acquisition of data
Ann Mari Rosager: Acquisition of data
Anne Dixon: Acquisition of data
Paul Tesar: Conception and design, study supervision and material support
Petra Hamerlik: Acquisition of data
Bjarne Kristensen: Acquisition of data
Craig Horbinski: Conception and design, acquisition of data, study supervision and material support
James Connor: Conception and design, analysis and interpretation of data, study supervision and material support
Paul Fox: Conception and design, analysis and interpretation of data, study supervision and material support
Justin Lathia: Conception and design, acquisition of data, analysis and interpretation of data, study supervision and material support
Jeremy Rich: Conception and design, development of methodology, analysis and interpretation of data, writing and/or revision of manuscript, study supervision and material support
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akhtar-Zaidi B, Cowper-Sal-lari R, Corradin O, Saiakhova A, Bartels CF, Balasubramanian D, Myeroff L, Lutterbaugh J, Jarrar A, Kalady MF, et al. Epigenomic enhancer profiling defines a signature of colon cancer. Science. 2012;336:736–739. doi: 10.1126/science.1217277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Arriba Zerpa GA, Saleh MC, Fernández PM, Guillou F, Espinosa de los Monteros A, de Vellis J, Zakin MM, Baron B. Alternative splicing prevents transferrin secretion during differentiation of a human oligodendrocyte cell line. J Neurosci Res. 2000;61:388–395. doi: 10.1002/1097-4547(20000815)61:4<388::AID-JNR5>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006a;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB, Shi Q, McLendon RE, Bigner DD, Rich JN. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006b;66:7843–7848. doi: 10.1158/0008-5472.CAN-06-1010. [DOI] [PubMed] [Google Scholar]
- Cao Y, Lathia JD, Eyler CE, Wu Q, Li Z, Wang H, McLendon RE, Hjelmeland AB, Rich JN. Erythropoietin Receptor Signaling Through STAT3 Is Required For Glioma Stem Cell Maintenance. Genes Cancer. 2010;1:50–61. doi: 10.1177/1947601909356352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, Parada LF. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–526. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffman LG, Parsonage D, D’Agostino R, Torti FM, Torti SV. Regulatory effects of ferritin on angiogenesis. Proc Natl Acad Sci U S A. 2009;106:570–575. doi: 10.1073/pnas.0812010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor JR, Boeshore KL, Benkovic SA, Menzies SL. Isoforms of ferritin have a specific cellular distribution in the brain. J Neurosci Res. 1994;37:461–465. doi: 10.1002/jnr.490370405. [DOI] [PubMed] [Google Scholar]
- Corradin O, Saiakhova A, Akhtar-Zaidi B, Myeroff L, Willis J, Cowper-Sal lari R, Lupien M, Markowitz S, Scacheri PC. Combinatorial effects of multiple enhancer variants in linkage disequilibrium dictate levels of gene expression to confer susceptibility to common traits. Genome Res. 2014;24:1–13. doi: 10.1101/gr.164079.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels TR, Delgado T, Rodriguez JA, Helguera G, Penichet ML. The transferrin receptor part I: Biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin Immunol. 2006a;121:144–158. doi: 10.1016/j.clim.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Daniels TR, Delgado T, Helguera G, Penichet ML. The transferrin receptor part II: targeted delivery of therapeutic agents into cancer cells. Clin Immunol Orlando Fla. 2006b;121:159–176. doi: 10.1016/j.clim.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Eyler CE, Wu Q, Yan K, MacSwords JM, Chandler-Militello D, Misuraca KL, Lathia JD, Forrester MT, Lee J, Stamler JS, et al. Glioma stem cell proliferation and tumor growth are promoted by nitric oxide synthase-2. Cell. 2011;146:53–66. doi: 10.1016/j.cell.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan WA, Wu Q, Hitomi M, Rahim N, Kim Y, Sloan AE, Weil RJ, Nakano I, Sarkaria JN, Stringer BW, et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat Neurosci. 2013;16:1373–1382. doi: 10.1038/nn.3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert MR, Liu Y, Neltner J, Pu H, Morris A, Sunkara M, Pittman T, Kyprianou N, Horbinski C. Autophagy and oxidative stress in gliomas with IDH1 mutations. Acta Neuropathol (Berl) 2014;127:221–233. doi: 10.1007/s00401-013-1194-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong A-H, Wei P, Zhang S, Yao J, Yuan Y, Zhou A-D, Lang FF, Heimberger AB, Rao G, Huang S. FoxM1 Drives a Feed-Forward STAT3-Activation Signaling Loop That Promotes the Self-Renewal and Tumorigenicity of Glioblastoma Stem-like Cells. Cancer Res. 2015 doi: 10.1158/0008-5472.CAN-14-2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjelmeland AB, Wu Q, Wickman S, Eyler C, Heddleston J, Shi Q, Lathia JD, Macswords J, Lee J, McLendon RE, et al. Targeting A20 decreases glioma stem cell survival and tumor growth. Plos Biol. 2010;8:e1000319. doi: 10.1371/journal.pbio.1000319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjelmeland AB, Wu Q, Heddleston JM, Choudhary GS, MacSwords J, Lathia JD, McLendon R, Lindner D, Sloan A, Rich JN. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ. 2011;18:829–840. doi: 10.1038/cdd.2010.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson JG, Yeh RF, Ray A, Wang NJ, Smirnov I, Yu M, Hariono S, Silber J, Feiler HS, Gray JW, et al. Comparative analyses of gene copy number and mRNA expression in glioblastoma multiforme tumors and xenografts. Neuro-Oncol. 2009;11:477–487. doi: 10.1215/15228517-2008-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtkamp N, Afanasieva A, Elstner A, van Landeghem FKH, Könneker M, Kuhn SA, Kettenmann H, von Deimling A. Brain slice invasion model reveals genes differentially regulated in glioma invasion. Biochem Biophys Res Commun. 2005;336:1227–1233. doi: 10.1016/j.bbrc.2005.08.253. [DOI] [PubMed] [Google Scholar]
- Huang J, Simcox J, Mitchell TC, Jones D, Cox J, Luo B, Cooksey RC, Boros LG, McClain DA. Iron regulates glucose homeostasis in liver and muscle via AMP-activated protein kinase in mice. Faseb J Off Publ Fed Am Soc Exp Biol. 2013;27:2845–2854. doi: 10.1096/fj.12-216929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki K, Hailemariam K, Tsuji Y. PIAS3 interacts with ATF1 and regulates the human ferritin H gene through an antioxidant-responsive element. J Biol Chem. 2007;282:22335–22343. doi: 10.1074/jbc.M701477200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi K, Banasavadi-Siddegowda Y, Mo X, Kim SH, Mao P, Kig C, Nardini D, Sobol RW, Chow LML, Kornblum HI, et al. MELK-dependent FOXM1 phosphorylation is essential for proliferation of glioma stem cells. Stem Cells Dayt Ohio. 2013;31:1051–1063. doi: 10.1002/stem.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikyo N, Hagiwara K, Fujisawa M, Kikyo N, Yazaki Y, Okabe T. Purification of a cell growth factor from a human lung cancer cell line: its relationship with ferritin. J Cell Physiol. 1994;161:106–110. doi: 10.1002/jcp.1041610113. [DOI] [PubMed] [Google Scholar]
- Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N, Oh YT, Kim H, Rheey J, Nakano I, et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell. 2013;23:839–852. doi: 10.1016/j.ccr.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koorts AM, Levay PF, Becker PJ, Viljoen M. Pro- and anti-inflammatory cytokines during immune stimulation: modulation of iron status and red blood cell profile. Mediators Inflamm. 2011;2011:716301. doi: 10.1155/2011/716301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn DE, Baker BD, Lafuse WP, Zwilling BS. Differential iron transport into phagosomes isolated from the RAW264.7 macrophage cell lines transfected with Nramp1Gly169 or Nramp1Asp169. J Leukoc Biol. 1999;66:113–119. doi: 10.1002/jlb.66.1.113. [DOI] [PubMed] [Google Scholar]
- Kwak EL, Larochelle DA, Beaumont C, Torti SV, Torti FM. Role for NF-kappa B in the regulation of ferritin H by tumor necrosis factor-alpha. J Biol Chem. 1995;270:15285–15293. doi: 10.1074/jbc.270.25.15285. [DOI] [PubMed] [Google Scholar]
- Lane DJR, Mills TM, Shafie NH, Merlot AM, Saleh Moussa R, Kalinowski DS, Kovacevic Z, Richardson DR. Expanding horizons in iron chelation and the treatment of cancer: role of iron in the regulation of ER stress and the epithelial-mesenchymal transition. Biochim Biophys Acta. 2014;1845:166–181. doi: 10.1016/j.bbcan.2014.01.005. [DOI] [PubMed] [Google Scholar]
- Lathia JD, Gallagher J, Heddleston JM, Wang J, Eyler CE, Macswords J, Wu Q, Vasanji A, McLendon RE, Hjelmeland AB, et al. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell. 2010;6:421–432. doi: 10.1016/j.stem.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner DF, Connor JR. Functional roles of transferrin in the brain. Biochim Biophys Acta Bba - Gen Subj. 2012;1820:393–402. doi: 10.1016/j.bbagen.2011.10.016. [DOI] [PubMed] [Google Scholar]
- Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, Shi Q, Cao Y, Lathia J, McLendon RE, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009;15:501–513. doi: 10.1016/j.ccr.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Dai B, Kang SH, Ban K, Huang FJ, Lang FF, Aldape KD, Xie T, Pelloski CE, Xie K, et al. FoxM1B is overexpressed in human glioblastomas and critically regulates the tumorigenicity of glioma cells. Cancer Res. 2006;66:3593–3602. doi: 10.1158/0008-5472.CAN-05-2912. [DOI] [PubMed] [Google Scholar]
- Liu X, Madhankumar AB, Slagle-Webb B, Sheehan JM, Surguladze N, Connor JR. Heavy chain ferritin siRNA delivered by cationic liposomes increases sensitivity of cancer cells to chemotherapeutic agents. Cancer Res. 2011;71:2240–2249. doi: 10.1158/0008-5472.CAN-10-1375. [DOI] [PubMed] [Google Scholar]
- Mencalha AL, Binato R, Ferreira GM, Du Rocher B, Abdelhay E. Forkhead box M1 (FoxM1) gene is a new STAT3 transcriptional factor target and is essential for proliferation, survival and DNA repair of K562 cell line. Plos One. 2012;7:e48160. doi: 10.1371/journal.pone.0048160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandal A, Ruiz JC, Subramanian P, Ghimire-Rijal S, Sinnamon RA, Stemmler TL, Bruick RK, Philpott CC. Activation of the HIF prolyl hydroxylase by the iron chaperones PCBP1 and PCBP2. Cell Metab. 2011;14:647–657. doi: 10.1016/j.cmet.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyssonnaux C, Zinkernagel AS, Schuepbach RA, Rankin E, Vaulont S, Haase VH, Nizet V, Johnson RS. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs) J Clin Invest. 2007;117:1926–1932. doi: 10.1172/JCI31370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham CG, Bubici C, Zazzeroni F, Papa S, Jones J, Alvarez K, Jayawardena S, De Smaele E, Cong R, Beaumont C, et al. Ferritin heavy chain upregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell. 2004;119:529–542. doi: 10.1016/j.cell.2004.10.017. [DOI] [PubMed] [Google Scholar]
- Recalcati S, Taramelli D, Conte D, Cairo G. Nitric oxide-mediated induction of ferritin synthesis in J774 macrophages by inflammatory cytokines: role of selective iron regulatory protein-2 downregulation. Blood. 1998;91:1059–1066. [PubMed] [Google Scholar]
- Ruddell RG, Hoang-Le D, Barwood JM, Rutherford PS, Piva TJ, Watters DJ, Santambrogio P, Arosio P, Ramm GA. Ferritin functions as a proinflammatory cytokine via iron-independent protein kinase C zeta/nuclear factor kappaB-regulated signaling in rat hepatic stellate cells. Hepatol Baltim Md. 2009;49:887–900. doi: 10.1002/hep.22716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvageau M, Goff LA, Lodato S, Bonev B, Groff AF, Gerhardinger C, Sanchez-Gomez DB, Hacisuleyman E, Li E, Spence M, et al. Multiple knockout mouse models reveal lincRNAs are required for life and brain development. Elife. 2013;2:e01749. doi: 10.7554/eLife.01749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober M, Fuchs E. Tumor-initiating stem cells of squamous cell carcinomas and their control by TGF-β and integrin/focal adhesion kinase (FAK) signaling. Proc Natl Acad Sci U S A. 2011;108:10544–10549. doi: 10.1073/pnas.1107807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinojima N, Hossain A, Takezaki T, Fueyo J, Gumin J, Gao F, Nwajei F, Marini FC, Andreeff M, Kuratsu JI, et al. TGF-β mediates homing of bone marrow-derived human mesenchymal stem cells to glioma stem cells. Cancer Res. 2013;73:2333–2344. doi: 10.1158/0008-5472.CAN-12-3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJB, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- Torti SV, Torti FM. Iron and cancer: more ore to be mined. Nat Rev Cancer. 2013;13:342–355. doi: 10.1038/nrc3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venere M, Hamerlik P, Wu Q, Rasmussen RD, Song LA, Vasanji A, Tenley N, Flavahan WA, Hjelmeland AB, Bartek J, et al. Therapeutic targeting of constitutive PARP activation compromises stem cell phenotype and survival of glioblastoma-initiating cells. Cell Death Differ. 2014;21:258–269. doi: 10.1038/cdd.2013.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Bates J, Li X, Schanz S, Chandler-Militello D, Levine C, Maherali N, Studer L, Hochedlinger K, Windrem M, et al. Human iPSC-derived oligodendrocyte progenitor cells can myelinate and rescue a mouse model of congenital hypomyelination. Cell Stem Cell. 2013;12:252–264. doi: 10.1016/j.stem.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman WW, Fahl WE. Functional antioxidant responsive elements. Proc Natl Acad Sci. 1997;94:5361–5366. doi: 10.1073/pnas.94.10.5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrighting DM, Andrews NC. Interleukin-6 induces hepcidin expression through STAT3. Blood. 2006;108:3204–3209. doi: 10.1182/blood-2006-06-027631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu KJ, Polack A, Dalla-Favera R. Coordinated Regulation of Iron-Controlling Genes, H-Ferritin and IRP2, by c-MYC. Science. 1999;283:676–679. doi: 10.1126/science.283.5402.676. [DOI] [PubMed] [Google Scholar]
- Yan K, Wu Q, Yan DH, Lee CH, Rahim N, Tritschler I, DeVecchio J, Kalady MF, Hjelmeland AB, Rich JN. Glioma cancer stem cells secrete Gremlin1 to promote their maintenance within the tumor hierarchy. Genes Dev. 2014;28:1085–1100. doi: 10.1101/gad.235515.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Xu S, Xin Y, Yu S, Ping Y, Chen L, Xiao H, Wang B, Yi L, Wang Q, et al. Tumor-Associated Microglia/Macrophages Enhance the Invasion of Glioma Stem-like Cells via TGF-β1 Signaling Pathway. J Immunol. 2012;189:444–453. doi: 10.4049/jimmunol.1103248. [DOI] [PubMed] [Google Scholar]
- Zhang N, Wei P, Gong A, Chiu WT, Lee HT, Colman H, Huang H, Xue J, Liu M, Wang Y, et al. FoxM1 promotes β-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell. 2011;20:427–442. doi: 10.1016/j.ccr.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D, Pan C, Sun J, Gilbert C, Drews-Elger K, Azzam DJ, Picon-Ruiz M, Kim M, Ullmer W, El-Ashry D, et al. VEGF drives cancer-initiating stem cells through VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene. 2014 doi: 10.1038/onc.2014.257. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.