

Synopsis
Huntington disease (HD) is caused by an expanded polyglutamine repeat in the huntingtin protein. GABAergic medium spiny neurons in the striatum are mostly affected in HD. However, mutant huntingtin (mhtt)-induced molecular changes in these neurons remain largely unknown. This study focuses on the effect of mhtt on the subcellular localization of glutamic acid decarboxylase (GAD), the enzyme responsible for synthesizing GABA. In this study, we report that the subcellular distribution of GAD is significantly altered in two neuronal cell lines that express either the N-terminus or full length mhtt. GAD65 is predominantly associated with Golgi membrane in cells expressing normal htt. However, it diffuses in the cytosol of cells expressing mhtt. As a result, vesicle-associated GAD65 trafficking is impaired. Since palmitoylation of GAD65 is required for GAD65 trafficking, we then demonstrate that palmitoylation of GAD65 is reduced in the HD model. Furthermore, overexpression of huntingtin-interacting protein 14, the enzyme responsible for palmitoylating GAD65 in vivo, could rescue GAD65 palmitoylation and vesicle-associated GAD65 trafficking. Taken together, our data support the idea that GAD65 palmitoylation is important for the delivery of GAD65 to inhibitory synapses and suggest that impairment of GAD65 palmitoylation by mhtt may lead to altered inhibitory neurotransmission in HD.
Keywords: huntingtin, protein palmitoylation, huntingtin-interacting protein 14, GAD65, axonal transport, protein aggregates
INTRODUCTION
Huntington disease (HD) is an inherited neurodegenerative disorder that is caused by an expanded polyglutamine repeat (polyQ) within the N-terminus of the huntingtin protein (htt). It is characterized by a progressive loss of GABAergic medium spiny neurons (MSNs) in the striatum. It is believed that the loss of their inhibitory inputs is the underlying cause of the uncontrolled movements characteristic of patients with HD [1]. Interestingly, it has also been reported that motor deficits develop without evidence of striatal atrophy in early-stage HD patients and transgenic mice [2, 3]. Therefore, it has been suggested that neuronal dysfunction, rather than cell death, is responsible for early neurological deficits observed in HD patients [4]. However, mhtt-induced neuronal dysfunction in MSNs remains largely unknown.
GABA, the inhibitory neurotransmitter used by MSNs, is synthesized by glutamic acid decarboxylase (GAD), which has two isoforms, GAD65 and GAD67. GAD65 is predominantly associated with synaptic vesicle membranes in nerve terminals and this association is crucial for GABAergic neurotransmission [5, 6]. There is evidence to suggest that GAD65, but not GAD67, forms a protein complex consisting of the heat shock protein 70, the vesicular GABA transporter and the cysteine string protein (CSP) [7]. This protein complex provides a structural basis for a functional coupling model between GABA synthesis and transport into synaptic vesicles [7]. It has been shown that palmitoylation of GAD65 at cysteines 30 and 45 is a critical post-translational modification of GAD65 that mediates the transport of GAD65 to synaptic terminals [8]. Palmitoylation dynamically regulates the partitioning of GAD65 between the ER-Golgi and the post-Golgi membrane [9], which further controls the trafficking of GAD65 from the Golgi membrane to presynaptic terminals [10, 11].
Palmitoylation of GAD65 is mediated by huntingtin-interacting protein 14 (HIP14), which was initially isolated as a huntingtin-interacting protein [12], and later proven to be a neuronal palmitoyl acyltransferase (PAT) due to the presence of a core Asp-His-His-Lys (DHHC) motif essential for PAT activity [13]. For this reason, HIP14 is also named ZDHHC-17 (Zinc Finger, DHHC-type containing 17). Increasing GAD65 palmitoylation by overexpression of HIP14 promotes the perinuclear accumulation of GAD65. On the other hand, disruption of HIP14 expression alters trafficking of GAD65 and significantly reduces axonal clustering of GAD65 [13]. HIP14 contributes to the neuronal dysfunction in HD by perturbing normal intracellular transport pathways in neurons [12]. Palmitoylation of htt by HIP14 at cysteine 214 is essential for htt trafficking and function [14]. HIP14 interacts with htt through its ankyrin domain and the interaction is inversely correlated to the polyQ length in htt [12]. In addition to htt and GAD65, HIP14 also shows remarkable substrate specificity for other neuronal proteins including synaptosomal-associated protein 25 (SNAP25), postsynaptic density protein 95 (PSD-95), synaptotagmin I and CSP [13, 15].
To gain insight into the mechanism underlying GABAergic neuronal dysfunction in HD, we examined the effect of mhtt on the subcellular localization of GAD65 in this study. We first demonstrate that the subcellular localization and trafficking of GAD65 is impaired in a neuronal cell line express the N-terminal fragment of mhtt. In addition, we also show that subcellular localization of GAD65 is altered in STHdhQ111 cells that express full length mhtt. This is possibly due to a reduced palmitoylation of GAD65 in the presence of mhtt. Furthermore, we demonstrate that overexpression of HIP14 could restore the palmitoylation of GAD65 and improve GAD65 trafficking. Thus, GAD65 palmitoylation is important for delivery of GAD65 to inhibitory synapses and disruption of this process by mhtt may lead to altered inhibitory neurotransmission in HD.
EXPERIMENTAL
Cell culture and differentiation
Neuro-2a cells (N2a, ATCC), STHdhQ7 and STHdhQ111 cells (obtained from HD Community BioRepository) were cultured in Eagle’s minimum essential medium (ATCC) supplemented with 10% fetal bovine serum (PAA laboratories). For N2a cell differentiation, cells were grown in the absence of serum for 48 hours.
Animals
Breeding pairs of R6/2 transgenic mice were purchased from the Jackson Laboratories, and the line was maintained by backcrossing to CBA3 C57BL/6 F1 in the animal facilities of Florida Atlantic University (FAU). The R6/2 mouse line, which expresses exon 1 of the human HD gene, contains 150 CAG repeats [16]. R6/2 mice display an aggressive phenotype, including deficits of motor co-ordination, altered locomotor activity, impaired cognitive performance and seizures, and therefore provide clear experimental endpoints [17]. The neuropathology of R6/2 mice is similar to human HD at the cellular level with development of nuclear huntingtin protein deposits before the onset of symptoms [18].
All animals were maintained under temperature- and light- controlled conditions (20–23°C, 12-hour-light/12-hour-dark cycle) with continuous access to food and water. Animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of FAU.
Plasmids and transfection
The N-terminal coding region (amino acids 1–68) of wild type human htt with 25 polyQ repeats (htt25Q, Addgene plasmid 1187) or 103 polyQ repeats (htt103Q, Addgene plasmid 1186) was attached to an enhanced green fluorescent protein (EGFP) and subcloned into a pcDNA3.1/HisB vector [19]. Human HIP14 cDNA in a pCMV6-Entry vector was purchased from Origene. Human GAD65 cDNA in pCR4-TOPO and GAD67 cDNA in pBluescriptR were obtained from Open Biosystems and subcloned into pcDNA3-mRFP (monomeric red fluorescent protein) which was obtained from Addgene (Addgene plasmid 13032). Truncated human GAD65 cDNA with the deletion of the amino acids 1–69 was obtained from human GAD65 cDNA by standard PCR and subcloned into pcDNA3-mRFP (forward primer: 5′-ggcgggatccaaaggccgcctgcgcctgcgac-3′; reverse primer: 5′-ggccctctagattaggcgccggtggagtg-3′). Truncated human GAD67 cDNA with the deletion of amino acids 1–90 was obtained from human GAD67 cDNA by standard PCR and subcloned into pcDNA3-mRFP (forward primer: 5′-ggcgggatccaacagagactgacttctctaatct -3′; reverse primer: 5′-ggccctctagattaggcgccggtggagtg-3′). Transfection was performed using the standard lipofectamine 2000 method according to the manufacturer’s instruction (Invitrogen). Unless stated elsewhere, cells were analyzed 48 hours after transfection.
Antibodies
The following antibodies were used: Affinity-purified rabbit polyclonal antibody against recombinant RFP (anti-RFP) was purchased from BioVision. Affinity-purified rabbit polyclonal antibody against the sequence surrounding Ala150 of human GAD65 isoform (anti-GAD65), monoclonal rabbit antibody against human protein disulfide isomerase (anti-PDI) and monoclonal rabbit antibody against a synthetic peptide corresponding to residues surrounding Gly190 of human Rab5 protein (anti-Rab5) were purchased from Cell Signaling Technology. Rabbit polyclonal antibody against amino acids 1–238 of full length GFP (anti-GFP) was purchased from Santa Cruz Biotechnology. Mouse monoclonal (ascites) antibody against GAD65 isoform (GAD6) was purchased from Developmental Studies Hybridoma Bank, University of Iowa. Purified mouse monoclonal antibody against recombinant GAD67 isoform (anti-GAD67) was obtained from Chemicon. Rabbit polyclonal antibody against C-terminus of human HIP14 (rabbit anti-HIP14), purified goat polyclonal antibodies against C-terminus of HIP14 (goat anti-HIP14), mouse monoclonal antibody against FLAG epitope (M2) were purchased from Sigma. Mouse monoclonal antibody against the Golgi matrix protein of 130 KDa (GM130) was obtained from BD Biosciences. Alexa Fluor 405, 488 or 594 goat antibodies against rabbit or mouse were purchased from Invitrogen.
Sample preparations
N2a cells were lysed in lysis buffer (50 mM Tris, pH=7.5, 150 mM NaCl, 0.5 mM EDTA, 1X complete protease inhibitor cocktail from Sigma, 1X phosphatase inhibitor cocktail from Pierce) containing 1% triton X-100 for 30 min at 4°C. For striatal sample preparation, striatum was first dissected from 11-week-old R6/2 mice and their control littermates and then lysed in lysis buffer containing 1% Triton X-100 at 4°C for 30 min. After lysis, samples were centrifuged at 13,000 × g for 20 min at 4°C to separate soluble and insoluble fractions. To dissolve insoluble fractions, the resulting pellets were further incubated with 100% formic acid at 37°C for 40 min as described [20]. After incubation, samples were dried by vacuum centrifugation, resuspended in 1x SDS-PAGE sample buffer and neutralized with 1 M Tris. All sample mixtures were boiled for 5 min before electrophoresis. Protein concentration was determined using the Bradford method.
Co-immunoprecipitation
Co-immunoprecipitation was performed using PureProteome™ protein G magnetic beads (Millipore). Cells were first lysed in lysis buffer containing 0.5% NP-40. About 500 μg of cell or striatal lysates at a concentration of 2 mg/ml was pre-cleared with 30 μl of protein G beads and then incubated with anti-RFP (1:50), anti-GAD65 (1:50) or goat anti-HIP14 (1:50) for 2 hours at 4°C with gentle shaking. At the end of the incubation, 30 μl of protein G beads were added to the antigen-antibody complex and further incubated with gentle mixing overnight at 4°C. The immobilized protein G–bound complexes were washed three times with cell lysis buffer containing 0.2% Triton X-100. For the detection of protein-protein interaction, 30 μl of 2X SDS sample buffer were added and boiled for 5 min to elute the protein complex for western blot analysis. For palmitoylation detection, the immobilized protein G-bound complexes were further processed as described below.
Detection of GAD palmitoylation
Palmitoylated GAD was detected using the “fatty acyl exchange labeling” method as described by Drisdel, et al [21, 22]. Free cysteines in GAD were first blocked with excessive N-ethylmaleimide (NEM). Cys-palmitoyl thioester linkage in palmitoylated cysteines was then cleaved by hydroxylamine (HA) which made the palmitoylated cysteine available for biotin labeling and detection by streptavidin-horseradish peroxidase (HRP). Briefly, immunopurified GAD65 attached to protein G beads was resuspended in 150 μl of lysis buffer containing 50 mM NEM and 0.2% Triton X-100 followed by incubation at 4°C overnight. After incubation, excess NEM was removed by washing the sample extensively with the lysis buffer. The complex was further incubated with 1 M hydroxylamine (pH=7.4) for 1 hour to cleave the Cys-palmitoyl thioester linkage. After removal of excess hydroxylamine, the complex was further incubated with 80 μM 1-biotinamido-4-[4-(maleimidomethyl) cyclohexane-carboxamido] butane (biotin-BMCC in lysis buffer, pH=6.2) for 2 hours at 4°C. At the end of incubation, the beads were washed extensively with lysis buffer and eluted with 2X SDS sample buffer. The palmitoylated GAD was then detected by western blot using streptavidin-HRP antibodies (Cell Signaling Technology). For densitometry analysis, the band intensity of total GAD65 and palmitoylated GAD65 were separately quantified by calculating the area under curve (AUC) of the specific signal using Image J. Palmitoylated GAD65 was then normalized with the total amount of GAD65. To compare the changes in palmitoylated GAD65 between different groups, we further normalized the palmitoylated GAD65 in cells expressing htt103Q to that of cells expressing htt25Q.
Western blot
Samples were separated by SDS-PAGE and transferred to a PVDF membrane. After the transfer, the PVDF membrane was blocked with TBS-T (20 mM Tris-HCl, pH=7.5, 150 mM NaCl, 0.1% Tween-20) containing 5% non-fat dry milk for 2 hours at room temperature. The membrane was then incubated with the following antibodies: anti-GFP (1:1000), GAD6 (1:500), anti-GAD67 (1:500), rabbit anti-HIP14 (1:500) or monoclonal anti-Flag M2 antibodies (1:1000) overnight at 4°C with gentle shaking. After extensive washing with TBS-T, the membrane was incubated with HRP-conjugated secondary antibodies. Signals were detected by enhanced chemiluminescence.
Immunofluorescence
N2a cells were grown on coverslips and transfected with the desired plasmids as indicated. Forty-eight hours after transfection, cells were gently washed with PBS, fixed in 4% paraformaldehyde for 30 min at room temperature and then permeabilized for 10 min with PBS containing 0.25% Triton X-100. Cells were then blocked in blocking buffer [10% normal goat serum (NGS), 1% BSA in PBS] for 1 hour at room temperature and incubated with GAD6 (1:1000 in PBS containing 1% NGS and 1% BSA), anti-GAD67 (1:1000), M2 antibody (1:500), anti-GM130 (1:500), Rab5 (1:500) or anti-PDI (1:500) for 1 hour at room temperature. Next, cells were incubated with the respective goat Alexa Fluor 405, 488 or 594 secondary IgG (Invitrogen, 1:2000 in PBS containing 1% NGS and 1% BSA) for 1 hour at room temperature with gentle shaking. Between steps, extensive washes (3 × 5 min) with PBS were performed to remove unbound reagents. The coverslips were then mounted in Prolong® Gold antifade reagent (Invitrogen). Immunofluorescence was detected using an immunofluorescent microscope (Zeiss Axio Imager D1) or a laser confocal microscope (Zeiss LSM700). The co-localization analysis of GAD65-RFP with Golgi marker GM130 or ER marker PDI was performed using Image J software. Background was first subtracted from each channel using Image J. Manders’ coefficient was calculated with the Intensity Correlation Analysis plug-in. The whole cell image was designated as the region of interest (ROI) during the analysis. A total of 10 cells from three independent transfection experiments were analyzed per condition.
Statistical analysis
Data were expressed as means + S.E.M. To establish significance, data were subjected to unpaired student’s t-tests using StatView 5.0 (SAS Institute Inc.) or GraphPad Prism 5.0 (GraphPad Software, Inc.). The criterion for significance was set at p ≤ 0.05.
RESULTS
The subcellular distribution of GAD65 and GAD67 is altered in the presence of mhtt
We initially examined whether mhtt expression would affect the subcellular distribution of GAD in a neuronal N2a cell line. For easier visualization of GAD expression, we attached mRFP to the C-terminus of GAD. In all cells that co-transfected with GAD65-mRFP and htt25Q-EGFP, confocal analysis revealed that GAD65-mRFP accumulated in vesicular structures in the perinuclear region of N2a cells, which is largely co-localized with Golgi marker, GM130 (Fig. 1A, 1E). This is consistent with the report that GAD65-GFP localizes predominantly to Golgi membrane in the cell body of neurons and COS-7 cells [9]. In contrast, GAD65-mRFP was detected in a diffuse pattern in the cytosol of N2a cells expressing htt103Q and correlated poorly with GM130 staining (Fig. 1B, 1E). We also noticed that, in about 50% of the cells that had htt103Q aggregate formation, part of GAD65-mRFP accumulated around htt103Q aggregates (Supplementary Fig. S1).
Fig. 1. Subcellular localization of GAD is altered in N2a cells expressing htt103Q and STHdhQ111 cells.
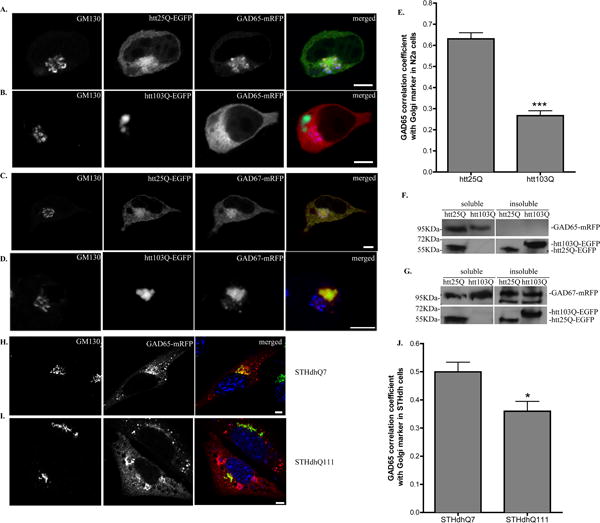
A–D Confocal analysis of N2a cells co-transfected with (A) GAD65-mRFP and htt25Q-EGFP, (B) GAD65-mRFP and htt103Q-EGFP, (C) GAD67-mRFP and htt25Q-EGFP or (D) GAD67-mRFP and htt103Q-EGFP. Co-transfected N2a cells were fixed 48 hours after transfection and co-stained with GM130 (blue in merged images). Scale bar represents 5 μm. E. Correlation coefficient analysis for co-localization of GAD65-mRFP with ER Golgi marker, GM130 in N2a cells. n=10 cells per group. *** p < 0.0001. F–G. Western blot analysis of the distribution of (H) GAD65 or (I) GAD67 in N2a cells expressing htt25Q or htt103Q. Cell lysates were separated into detergent-soluble and -insoluble fractions. Insoluble fraction was further dissolved with formic acid. The two fractions were subjected to western blot analysis for the presence of GAD or htt as indicated. H–I. Confocal analysis of STHdhQ7 (E) and STHdhQ111 (F) cells transfected with GAD65-mRFP. Cells were fixed 48 hours after transfection and co-stained with GM130 (green in merged images) and Hoechst 333432 (blue in merged images). Scale bar represents 5 μm. J. Correlation coefficient analysis for co-localization of GAD65-mRFP with GM130 in STHdh cells. n=8 cells for STHdhQ7 and n=10 cells for STHdhQ111 cells. * p < 0.05.
Since GAD exists in two isoforms, we also examined the subcellular distribution of GAD67 in N2a cells expressing htt25Q or htt103Q. Similar to GAD65-mRFP, GAD67-mRFP was also largely associated with Golgi membrane (Fig. 1C compared to Fig. 1A). However, more GAD67-mRFP was also present outside of the Golgi compartment (Manders’ coefficient with GM130 is 0.386 + 0.036, n=10). Unexpectedly, we found that, in all cells expressing htt103Q aggregates, GAD67-mRFP formed tight aggregates around htt103Q aggregates and no diffuse expression pattern of GAD67 in the cytosol was observed compared to that of GAD65-mRFP (Fig. 1D, compared to Fig. 1B). Furthermore, the aggregates did not co-localize with Golgi marker (Fig. 1D). It is necessary to mention that mhtt had no effect on the expression pattern of RFP alone (data not shown). To rule out the possibility that the attachment of mRFP may cause GAD to form aggregates or change its subcellular localization, we compared the expression pattern of GAD-mRFP to that of GAD without the attachment of mRFP in N2a cells. We did not find any significant difference in their expression patterns. Both GAD65 and GAD67 were predominantly distributed in the Golgi compartment regardless of the presence of mRFP (Supplementary Fig. S2A and S2B). This was also confirmed with endogenous GAD staining in primary cortical neurons (Supplementary Fig. S2C and S2D).
It was reported that mhtt forms detergent-insoluble aggregates which can be further dissolved using formic acid [20]. To investigate whether GAD forms insoluble aggregates with mhtt, we performed immunoblotting studies on the detergent-soluble and insoluble fractions in N2a cells expressing mhtt. As shown in the bottom panels of Fig. 1F and 1G, htt25Q was present in the soluble fraction while htt103Q was mainly present in the detergent-insoluble fraction due to the aggregate formation. We also detected the presence of htt25Q in the insoluble fraction. This was probably due to the insufficient wash of the insoluble pellet before dissolving with formic acid. GAD65-mRFP was present in the soluble fractions in both htt25Q- and htt103Q-expressing cells (top panel, Fig. 1F). On the contrary, GAD67-mRFP was found to be present in both soluble and insoluble fractions (top panel, Fig. 1G).
More importantly, we also investigated the subcellular localization of GAD in striatal cell lines that express full length murine htt with either 7 (STHdhQ7) and 111 (STHdhQ111) glutamines [23]. In STHdhQ7 cells, most GAD65-mRFP accumulated in the Golgi compartment, as suggested by GM130 staining (Fig. 1H and 1J). However, this pattern was changed in STHdhQ111 cells. GAD65-mRFP was found to be more diffusely expressed in STHdhQ111 cells (Fig. 1I and 1J), which is similar to what we observed in N2a cells expressing N-terminal fragment of mhtt (Fig. 1B). Since the N-terminal fragment of mhtt is more toxic, it is reasonable to expect that htt103Q has a more severe effect on GAD65 subcellular expression than full length mhtt (Fig. 1E compared to Fig. 1J). Interestingly, GAD67-mRFP also showed aggregate formation or diffuse pattern in STHdhQ111 cells (Supplementary Fig. S3). Taken together, these data suggest that mhtt expression could affect the subcellular localization of GAD65 and GAD67 in different manners.
GAD65 is largely re-located to ER membrane in the presence of mhtt in N2a cells
Since GAD65 appeared in a diffuse pattern in cells expressing mhtt, we next investigated whether GAD65 in htt103Q-expressing cells is associated with ER membrane with a specific ER marker, protein disulfide isomerase (PDI). Due to the heavy bleed-through of EGFP aggregate signal into the blue channel, we transfected N2a cells with htt25Q or htt103Q that contains no EGFP. In N2a cells expressing htt25Q, GAD65 was found to have little overlap with PDI staining (Fig. 2A), indicating that the majority of GAD65-mRFP is not present in ER. However, in the presence of mhtt, GAD65 was re-directed to ER membrane as shown in Fig. 2B. Colocalization coefficient analysis revealed that there is a significant degree of colocalization between GAD65 and PDI in cells expression htt103Q (Fig. 2C).
Fig. 2. GAD65 accumulates in ER membrane in the presence of mhtt in N2a cells.
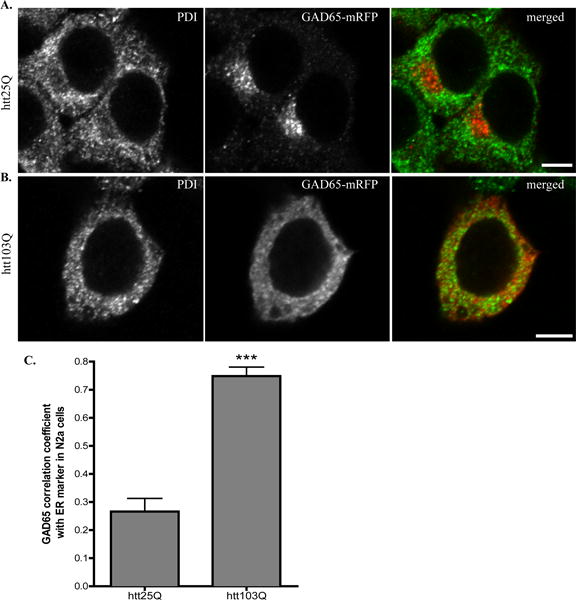
A–B Confocal analysis of N2a cells co-transfected with (A) GAD65-mRFP and htt25Q, (B) GAD65-mRFP and htt103Q. Co-transfected N2a cells were fixed 48 hours after transfection and co-stained with ER marker, PDI (green in merged images). Scale bar represents 5 μm. C. Correlation coefficient analysis for co-localization of GAD65-mRFP with PDI in N2a cells. n=8 cells per group. *** p < 0.0001.
N-terminus of GAD is responsible for the different subcellular distribution of GAD in the presence of mhtt
GAD65 and GAD67 showed different subcellular expression patterns in N2a cells expressing htt103Q (Fig. 1), suggesting that GAD65 and GAD67 respond differently to the presence of mhtt. Since GAD65 and GAD67 differ substantially at the N-terminus only [24, 25], we reasoned that the N-terminus of GAD may be responsible for the different responses to mhtt expression. To test this hypothesis, we generated a truncated GAD65 with the deletion of the first 69 amino acids, tGAD65-mRFP [26] and a truncated GAD67 with the deletion of the first 90 amino acids, tGAD67-mRFP [27]. Both truncated GADs were linked to mRFP at the C-terminus. In N2a cells that were co-transfected with htt25Q-EGFP and tGAD65-mRFP, tGAD65 was detected in a diffuse appearance (Fig. 3A). A similar pattern was found for tGAD67-mRFP in N2a cells expressing htt25Q (Fig. 3C and 3E). Interestingly, both tGAD65 and tGAD67 were present as aggregates in cells expressing htt103Q (Fig. 3B and 3D), indicating that the N-terminus of GAD65 may prevent the full length GAD65 from forming aggregates in cells expressing htt103Q (Fig. 1B). We further determined the subcellular localization of tGAD65 in N2a cells transfected with htt25Q. Compared to full length GAD65, removal of N-terminal fragment of GAD65 appeared to abolish the association of GAD65 with Golgi membrane as indicated by lack of co-staining with GM130 (Fig. 3E, Supplementary Fig. S4). Moreover, tGAD65 had a different staining pattern from PDI staining, indicating that tGAD65 mainly diffuses in the cytosol (Supplementary Fig. S4). This is consistent with the report that N-terminus of GAD65 is important for membrane association [28]. Similar findings were obtained for tGAD67 when co-stained with GM130 or PDI (data not shown).
Fig. 3. N-terminus deletion affects the subcellular localization of GAD in N2a cells expressing htt25Q or htt103Q.
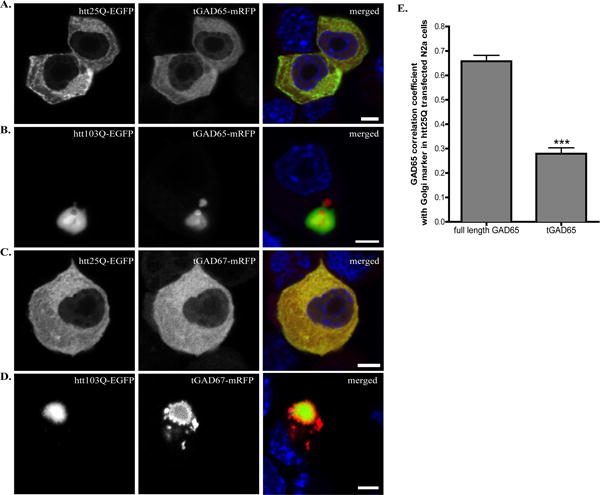
A–D N2a cells were co-transfected with (A) tGAD65-mRFP and htt25Q-EGFP, (B) tGAD65-mRFP and htt103Q-GFP, (C) tGAD67-mRFP and htt25Q-EGFP or (D) tGAD67-mRFP and htt103Q-GFP. Co-transfected N2a cells were fixed 48 hours after transfection and the nuclei were stained with Hoechst 333432 (blue in merged images). Scale bar represents 5 μm. E. Correlation coefficient analysis for co-localization of tGAD65-mRFP with GM130 in N2a cells expressing htt25Q. n=10 cells per group. *** p < 0.0001.
Palmitoylation of GAD65 is reduced in the presence of mhtt
Palmitoylation is an important post-translational modification of GAD65 that regulates its subcellular localization [9]. This prompted us to investigate whether GAD65 palmitoylation was affected in N2a cells expressing mhtt. The extent of palmitoylation was determined using the “fatty acyl exchange labeling” method [21]. We first determined whether GAD was palmitoylated in N2a cells that were transfected with GAD65-mRFP or GAD67-mRFP alone. We demonstrated that GAD65 but not GAD67 was palmitoylated in N2a cells (Fig. 4A). As expected, in the absence of HA treatment, no signal was detected since no free cysteine was available for biotin-BMCC labeling (Fig. 4A). We then investigated whether GAD65 palmitoylation was affected by mhtt expression. Palmitoylation of GAD65 in cells expressing htt103Q was greatly reduced compared to cells expressing htt25Q, while the total GAD65 input remained the same between groups (Fig. 4B). We estimated the percentage of GAD65 palmitoylation by densitometry analysis and found that GAD65 palmitoylation in cells expressing htt103Q was reduced to ~20% of the control group (Fig. 4B). Additionally, we also analyzed the palmitoylation of endogenous GAD65 in R6/2 mice. As shown in Fig. 4C, palmitoylation of GAD65 in the striatum of R6/2 mice was also significantly decreased. Palmitoylation of endogenous GAD65 was mediated by HIP14 [13]. Recently, it was reported that the enzymatic activity of HIP14 is regulated by palmitoylation and HIP14 palmitoylation is decreased in the YAC128 HD mouse model [29, 30]. We therefore also checked the palmitoylation of HIP14 in R6/2 mice. Compared to the control animals, HIP14 palmitoylation was decreased in R6/2 mice (Fig. 4D).
Fig. 4. GAD65 palmitoylation is reduced in the presence of mhtt.
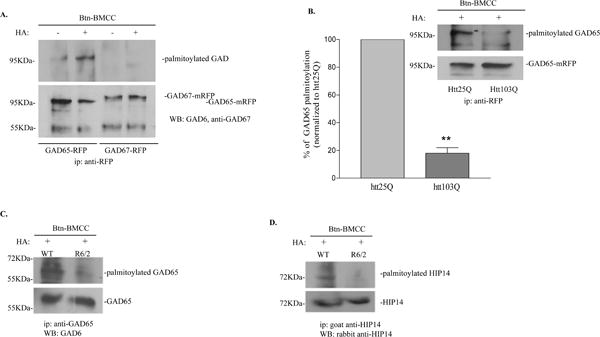
A. GAD65 but not GAD67 was palmitoylated in N2a cells with HA treatment (top panel). Immunoprecipitated GAD65 and GAD67 were detected using GAD6 and anti-GAD67, respectively (bottom panel). B. Palmitoylation of GAD65 was reduced in N2a cells expressing htt103Q. N2a cells were co-transfected with GAD65 and htt plasmids as indicated. Immunoprecipitated GAD65-mRFP was detected using GAD6. Top panel, A representative image from three independent experiments is shown. Bottom panel, densitometry analysis of the percentage of palmitoylated GAD65 in cells expressing htt103Q compared to cells expressing htt25Q. **p<0.01, n=3, unpaired student’s t-test. C. Palmitoylation of GAD65 was reduced in R6/2 mice. Top panel, palmitoylated GAD65. Bottom panel, total input of immunoprecipitated GAD65. D. Palmitoylation of HIP14 was reduced in R6/2 mice. Top panel, palmitoylated HIP14. Bottom panel, total input of immunoprecipitated HIP14.
mHtt affects the transport of GAD65 in differentiated N2a cells
Since palmitoylation is required for post-Golgi trafficking of GAD65 to synaptic nerve terminals [10], it is reasonable to suspect that the active trafficking of GAD65 along axons is impaired in HD. We tested this hypothesis in differentiated N2a cells which have neuronal-like morphology and express neuronal markers. As shown in Fig. 5, differentiated N2a cells had elongated processes. GAD65 was associated with punctuate vesicular structures in processes of cells transfected with GAD65-mRFP alone (Fig. 5A, arrowheads) or co-transfected with GAD65-mRFP and htt25Q (Fig. 5B, arrowheads), suggesting that GAD65 was actively transported along the process in vesicle-associated structures (also see Supplementary movie 1). It was reported that GAD65 colocalizes with early endosomal marker, Rab5, which is associated with axonal endosomal vesicles [10]. Consistently, we showed that GAD65-mRFP colocalized with Rab5 in processes of differentiated N2a cells expressing htt25Q, especially in regions close to cell bodies (Fig. 5C). However, in htt103Q-expressing cells, GAD65 passively diffused along the process (Fig. 5D, and supplementary movie 2) and did not colocalize with Rab5 (Fig. 5E).
Fig. 5. GAD65 trafficking is impaired in differentiated N2a cells expressing mhtt.
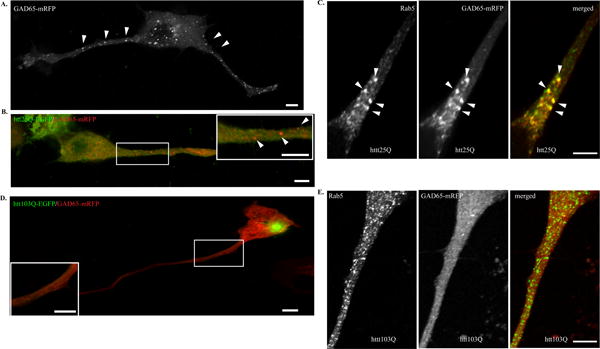
Cells were then allowed to differentiate after transfection in the absence of serum for 48 hours. A. N2a cells were transfected with GAD65-mRFP alone. Arrowheads indicate GAD65 is present in vesicle-like structures. B. N2a cells were co-transfected with GAD65-mRFP and htt25Q-EGFP. C. N2a cells were co-transfected with GAD65-mRFP and htt25Q. Cells were stained with anti-Rab5 (green in the merged image). D. N2a cells were co-transfected with GAD65-mRFP and htt103Q-EGFP. GAD65 is present as diffuse pattern in the process of differentiated N2a cells. E. N2a cells were co-transfected with GAD65-mRFP and htt103Q. Cells were stained with anti-Rab5 (green in the merged image). Scale bar in all images represents 5 μm.
Overexpression of HIP14 improves GAD65 trafficking in cells expressing htt103Q
We next investigated whether increasing GAD65 palmitoylation would rescue the defective trafficking of GAD65 in N2a cells expressing mhtt. Therefore, we performed triple-transfection in N2a cells using HIP14-Flag, GAD65-mRFP and htt plasmids. Overexpression of all three proteins was first confirmed by western blot (Fig. 6A). We noticed that GAD65 and htt103Q expression in the triple transfection was lower than those in the double transfection. This is probably due to lesser amounts of plasmids used in the triple transfection. We then investigated the effect of HIP14 overexpression on GAD65 subcellular localization by immunofluorescence. We consistently observed the diffused expression pattern of GAD65 in cells expressing htt103Q only (Fig. 6B). Interestingly, when HIP14 was overexpressed, GAD65-mRFP was re-associated with vesicular structures in N2a cells expressing htt103Q (Fig. 6C). Lastly, we tested whether overexpression of HIP14 increased GAD65 palmitoylation which caused the redistribution of GAD65. Indeed, we found that GAD65 palmitoylation was increased when HIP14 was overexpressed (Fig. 6D).
Fig. 6. Overexpression of HIP14 improves GAD65 trafficking and palmitoylation.
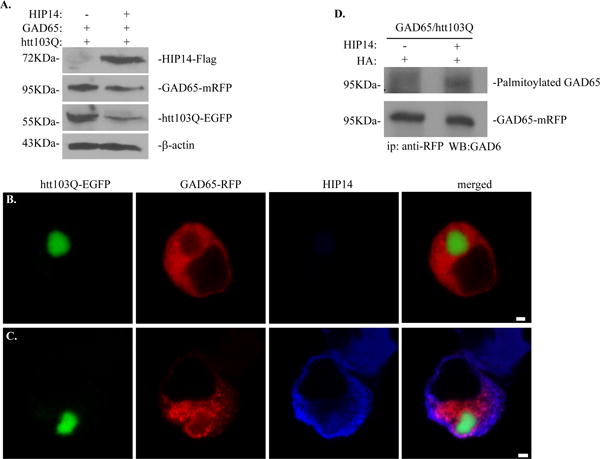
A. Western blot analysis of HIP14 overexpression in cells triple-transfected with GAD65-mRFP, htt103Q-EGFP and HIP14-Flag. HIP14-Flag was detected using M2 antibodies. GAD65-mRFP was detected using GAD6 antibodies and htt103Q-EGFP was detected using anti-GFP antibodies. B–C. Effect of HIP14 overexpression on the subcellular localization of GAD65. N2a cells were co-transfected with (B) htt103Q-EGFP and GAD65-mRFP or (C) triple-transfected with HIP14-Flag, htt103Q-EGFP and GAD65-mRFP. Forty-eight hours later, cells were fixed and stained with M2 antibody. Scale bar represents 2 μm. D. Overexpression of HIP14 increased GAD65 palmitoylation in N2a cells expressing htt103Q.
HIP14 does not interact with GAD65
Finally, we wanted to address how mhtt expression would impair GAD65 palmitoylation at the molecular level. Could mhtt down-regulate the expression of HIP14, which directly reduces its activity? We tested this hypothesis in R6/2 mice. As shown in Fig. 7A, HIP14 expression level was not significantly affected by mhtt expression in the striatum of R6/2 mice at 11 weeks old. In addition, the predominant localization of HIP14 in the Golgi compartment was not affected in R6/2 mice (Supplementary Fig. S5). Increasing evidence suggests that HIP14, as a PAT, interacts with its substrates such as htt [12] and PSD-95 [31]. This may also be true for other DHHC palmitoylating enzymes [31]. We therefore tested whether HIP14 also interacts with GAD65 and if so, whether mhtt expression disrupts the interaction. We first performed a co-immunoprecipitation assay using lysates of N2a cells that had over-expressed HIP14-Flag and GAD65-mRFP. As shown in Fig. 7B, we detected the immunoprecipitated GAD65 (bottom panel). However, we did not detect the presence of HIP14 in the immunoprecipitated complexes (top panel). To rule out the possibility that the tags we used in this study may interfere with the interaction between HIP14 and GAD65, we immunoprecipitated endogenous GAD65 from striatal lysates of 11-week-old wild type and R6/2 mice. As seen in Fig. 7C, HIP14 was not present in the immunoprecipitated complex from the striatal lysates of either wild type or R6/2 mice. Taken together, our data suggest that GAD65 does not interact with HIP14.
Fig. 7. GAD65 does not interact with HIP14.
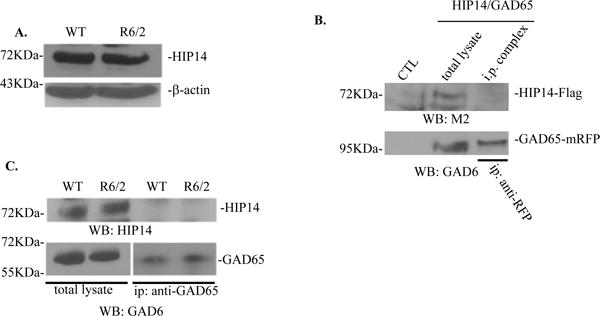
A. Western blot analysis of HIP14 protein expression in the striatum of WT and R6/2 mice at 11 weeks old. B. No interaction was found between overexpressed GAD65-mRFP and HIP14-Flag in N2a cells. GAD65 was immunoprecipitated from N2a cells co-transfected with HIP14 and GAD65 using anti-RFP antibodies. GAD65 and HIP14 were detected using GAD6 and anti-Flag antibodies respectively. Total lysate from N2a cells alone (CTL) and co-transfected N2a cells were used as negative and positive control, respectively. C. HIP14 did not co-immunoprecipitate with GAD65 in WT and R6/2 mice at 11 weeks old.
DISCUSSION
Palmitoylation plays an important role in regulating trafficking and function of neuronal proteins. The reversible nature of S-palmitoylation provides a potential mechanism for protein shuttling between different intracellular compartments [32]. GAD65 is predominantly associated with synaptic vesicles in nerve terminals [7] and regulates GABAergic neurotransmission [5, 6]. Palmitoylation is crucial for efficient targeting of GAD65 to presynaptic clusters. It has been shown that palmitoylation-deficient GAD65 mutants have a diffuse appearance in dendrites and proximal axons of primary hippocampal neurons [10]. However, whether GAD65 palmitoylation is affected under neuropathological conditions has not been reported. In this study, we demonstrate that palmitoylation of GAD65 is decreased in HD, which further impairs the axonal transport of GAD65 and its function.
GAD65 and GAD67 are the two isoforms of GAD in the brain with different subcellular localizations and functions. Our current data strongly support the notion that GAD65 and GAD67 are subjected to different regulatory mechanisms in neurons. GAD65 is palmitoylated at Cys30 and Cys45 [11]. Although GAD67 has similar cysteine-containing motifs in the N-terminus (Cys33 and C48), there have been no reports of GAD67 palmitoylation. Instead, evidence from our present study and others [33, 34] indicates that unlike GAD65, GAD67 is not palmitoylated in neurons. Therefore, the transport of GAD67 to synaptic nerve terminals may be mediated through distinct mechanisms that involve GAD65-dependent and –independent pathways [35]. Interestingly, we found that GAD65 and GAD67 show different subcellular expression patterns in neuronal cell lines expressing mhtt. GAD65 remains soluble and diffuse in the presence of mhtt while GAD67 becomes partially insoluble and forms aggregates with mhtt. What causes this difference remains to be investigated. Our data suggest that the N-terminus of GAD65 may play an essential role since the removal of the N-terminus in GAD65 led to the formation of GAD65 aggregates with mhtt.
In mammals, up to 23 different kinds of PATs have been reported using a systemic screening method [36]. They all share the conserved DHHC domain and exhibit distinct but overlapping substrate specificity [31]. For instance, DHHC-3 specifically enhances palmitoylation of GABAA receptor γ2 subunit in vivo [37]. It is reported that four PATs (DHHC-3, DHHC-8, DHHC-13 and HIP14) can enhance the palmitoylation of GAD65 in co-transfected COS cells [31]. This raises the possibility that other PATs may also palmitoylate GAD65. However, palmitoylation of endogenous GAD65 in neuronal culture is decreased by knocking down HIP14 but is not affected by DHHC-3 knockdown [31]. Consistently, it has been shown that overexpression of HIP14 enhances perinuclear accumulation of GAD65 in cultured neurons while HIP14 siRNA significantly reduces axonal clustering of GFP-tagged GAD65 [13]. Consequently, it is suggested that HIP14 is the major endogenous enzyme for GAD65 palmitoylation.
We have demonstrated that GAD65 palmitoylation is decreased in HD. In addition to GAD65, reduced palmitoylation of other neuronal proteins in HD have also been reported in several studies. Palmitoylation of htt by HIP14 is essential for its trafficking and function. In HD, expansion of polyQ in htt results in a significant reduction in htt palmitoylation, leading to an accelerated formation of inclusion bodies [14]. Another study has shown that palmitoylation and function of glial glutamate transporter-1 (GLT-1) is reduced in the YAC128 mouse model of HD [38]. The enzyme that palmitoylates GLT-1 has not been identified. On the other hand, not all palmitoylated proteins are affected in HD. For instance, it has been reported that palmitoylation of SNAP25 is not altered in HD. It is possible that other DHHC enzymes such as DHHC-3 can palmitoylate SNAP25 in vivo [31]. Another possibility is that different palmitate turnover rates exist among proteins. It has been shown that palmitate on PSD-95, CDC42 and two G-proteins (H-RAS and N-RAS) rapidly and continually turns over. On the contrary, palmitate on SNAP25 and synaptotagmin I is relatively stable since 2-bromopalmitate treatment, an inhibitor of protein palmitoylation, has little or no effect [39]. Conceivably, GAD65 may have a higher palmitoylation turnover rate; hence changes in GAD65 palmitoylation can be readily detected under pathological conditions.
The molecular basis for reduced palmitoylation of neuronal proteins in HD, except for htt, remains elusive. The decreased palmitoylation of htt in HD is believed to be due to a decreased direct interaction between mhtt and HIP14, which is mediated through the ankryin repeat domain in HIP14 [12, 14]. Furthermore, it has been reported that other DHHC palmitoylating enzymes can interact with their respective substrates in overexpressed cell lysates but not in brain extracts. For example, DHHC-8 interacts with paralemmin and DHHC-3 interacts with SNAP-25 [31]. Therefore, it is reasonable to hypothesize that HIP14 interacts with GAD65. Unexpectedly, we could not detect any interaction between GAD65 and HIP14 by co-immunoprecipitation in either overexpressed cell extracts or brain samples. While our data suggest that there is no interaction between HIP14 and GAD65, there is still possibility that GAD65 and HIP14 do not form a long-lived enzymatic reaction intermediate as others [31], which prevents the detection of the complex formation between GAD65 and HIP14 in the overexpressed cell lysates.
In the present study, we showed that HIP14 protein levels are not affected in HD. Therefore, if there is any change of HIP14 activity in HD, it is more likely related to some post-translational modifications of HIP14, such as palmitoylation. Recently, it is reported that palmitoylation of HIP14 is essential for the enzymatic activity of HIP14, which is modulated by wild type htt [29]. HIP14 palmitoylation is decreased in the brains of mice lacking one htt allele and further reduced in neuronal culture with htt knockdown [29]. It is also shown that exon 1 of htt, which does not interact with HIP14, can further comprise the ability of mhtt to modulate HIP14 activity [29]. Soon after, the same group showed that HIP14 activity is indeed reduced in a full length HD mouse model, YAC128 [30]. Consistently, we also showed that HIP14 palmitoylation was reduced in R6/2 mice. Therefore, it is likely that HIP14 activity is decreased in HD, which accounts for the reduced palmitoylation of GAD65.
Since protein palmitoylation is a reversible process, another possibility that cannot be excluded is that GAD65 de-palmitoylation is increased which results in the net decrease of palmitoylated GAD65. Protein de-palmitoylation is mediated by palmitoyl protein thioesterases (PPT). The identified PPTs are very limited and include acyl-protein thioesterase 1 (APT1), palmitoyl protein thioesterase 1 (PPT1) and PPT2. No specific PPT has been reported to de-palmitoylate GAD65. PPT1 is a promising candidate for neuronal proteins as it has been reported that PPT1 deficiency impairs synaptic vesicle recycling at nerve terminals [40]. It would be interesting to investigate whether PPT1 de-palmitoylates GAD65 and whether its activity is up-regulated in HD.
As to the functional consequences of reduced GAD65 palmitoylation in HD, we have shown that GAD65 trafficking is affected. It is well established that normal htt is involved in neuronal protein trafficking, such as brain derived neurotrophic factor (BDNF) [41] and GABAA receptor [42], by forming a complex with huntingtin associated protein-1 (HAP1), which is disrupted by mhtt [43, 44]. It is not clear whether huntingtin is directly involved in GAD65 trafficking. Our data indicate that it is less likely since the shorter fragment of htt (amino acid 1–68) used in this study may not interact with HAP1 and possesses the function of full length huntingtin in vesicular trafficking. Moreover, the N-terminal fragment of mhtt used in this study is missing the palmitoylation site C214, which is essential for htt trafficking and function upon palmitoylation [14]. Therefore we believe that disrupted GAD65 trafficking is more likely due to the decreased GAD65 palmitoylation rather than a direct involvement of htt. In summary, our results indicate that GAD65 palmitoylation is decreased in HD which leads to disrupted GAD65 trafficking and reduced synaptic clustering of GAD65 in nerve terminals. This contributes to the understanding of the altered synaptic inhibition and increased neuronal excitability observed in HD.
Supplementary Material
Acknowledgments
The authors wish to thank Barbara A. Nambu, PhD, for her editorial and writing assistance. This work was supported by the National Institute of Neurological Disorders and Stroke [grant number NS066339 (to J.W)].
Abbreviations
- APT1
acyl-protein thioesterase
- Biotin-BMCC
1-biotinamido-4-[4-(maleimidomethyl)cyclohexane-carboxamido]butane
- CSP
cysteine string protein
- EGFP
enhanced green fluorescent protein
- GAD
glutamic acid decarboxylase
- GLT-1
glial glutamate transporter-1
- HA
hydroxylamine
- HAP1
huntingtin associated protein 1
- HD
Huntington disease
- HIP14
huntingtin-interacting protein 14
- Htt25Q
N-terminal of huntingtin protein with 25 polyQ repeats
- Htt103Q
N-terminal of huntingtin protein with 103 polyQ repeats
- MSNs
medium spiny neurons
- N2a
neuro-2a cell line
- NEM
N-ethylmaleimide
- NGS
normal goat serum
- PAT
palmitoyl acyltransferase
- PDI
protein disulfide isomerase
- PolyQ
expanded polyglutamine repeats
- PPT
palmitoyl protein thioesterases
- PSD-95
postsynaptic density protein 95
- mRFP
monomeric red fluorescent protein
- SNAP25
synaptosomal-associated protein 25
- ZDHHC-17
Zinc Finger, DHHC-type containing 17
Footnotes
AUTHOR CONTRIBUTION
Daniel Rush conducted the experiments, contributed to data analysis and helped with the manuscript preparation. Rebecca Leon helped with HIP14 experiments and contributed to data discussion. Mark McCollum contributed to experiments and data discussion. Ryan Treu performed molecular cloning of truncated GADs. Jianning Wei designed the experiments, analyzed the data and wrote the paper.
References
- 1.Gil J, Rego A. Mechanisms of neurodegeneration in Huntington’s disease. Eur J Neurosci. 2008;27:2803–2820. doi: 10.1111/j.1460-9568.2008.06310.x. [DOI] [PubMed] [Google Scholar]
- 2.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies S. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 3.Vonsattel J, Myers R, Stevens T, Ferrante R, Bird E, Richardson EJ. Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Yuan J, Yankner B. Apoptosis in the nervous system. Nature. 2000;407:802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- 5.Asada H, Kawamura Y, Maruyama K, Kume H, Ding R, Ji F, Kanbara N, Kuzume H, Sanbo M, Yagi T. Mice Lacking the 65 kDa Isoform of Glutamic Acid Decarboxylase (GAD65) Maintain Normal Levels of GAD67 and GABA in Their Brains but Are Susceptible to Seizures. Biochem Biophys Res Commun. 1996;229:891–895. doi: 10.1006/bbrc.1996.1898. [DOI] [PubMed] [Google Scholar]
- 6.Tian N, Petersen C, Kash S, Baekkeskov S, Copenhagen D, Nicoll R. The role of the synthetic enzyme GAD65 in the control of neuronal-aminobutyric acid release. Proc Natl Acad Sci USA. 1999;96:12911–12916. doi: 10.1073/pnas.96.22.12911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jin H, Wu H, Osterhaus G, Wei J, Davis K, Sha D, Floor E, Hsu C, Kopke R, Wu J. Demonstration of functional coupling between-aminobutyric acid (GABA) synthesis and vesicular GABA transport into synaptic vesicles. Proc Natl Acad Sci USA. 2003;100:4293–4298. doi: 10.1073/pnas.0730698100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baekkeskov S, Kanaani J. Palmitoylation cycles and regulation of protein function (Review) Mol Membr Biol. 2009;26:42–54. doi: 10.1080/09687680802680108. [DOI] [PubMed] [Google Scholar]
- 9.Kanaani J, Patterson G, Schaufele F, Lippincott-Schwartz J, Baekkeskov S. A palmitoylation cycle dynamically regulates partitioning of the GABA-synthesizing enzyme GAD65 between ER-Golgi and post-Golgi membranes. J Cell Sci. 2008;121:437–439. doi: 10.1242/jcs.011916. [DOI] [PubMed] [Google Scholar]
- 10.Kanaani J, Diacovo M, El-Husseini A, Bredt D, Baekkeskov S. Palmitoylation controls trafficking of GAD65 from Golgi membranes to axon-specific endosomes and a Rab5a-dependent pathway to presynaptic clusters. J Cell Sci. 2004;117:2001–2013. doi: 10.1242/jcs.01030. [DOI] [PubMed] [Google Scholar]
- 11.Kanaani J, El-Husseini A, Aguilera-Moreno A, Diacovo J, Bredt D, Baekkeskov S. A combination of three distinct trafficking signals mediates axonal targeting and presynaptic clustering of GAD65. J Cell Biol. 2002;158:1229–1238. doi: 10.1083/jcb.200205053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singaraja R, Hadano S, Metzler M, Givan S, Wellington C, Warby S, Yanai A, Gutekunst C, Leavitt B, Yi H. HIP14, a novel ankyrin domain-containing protein, links huntingtin to intracellular trafficking and endocytosis. Hum Mol Genet. 2002;11:2815–2828. doi: 10.1093/hmg/11.23.2815. [DOI] [PubMed] [Google Scholar]
- 13.Huang K, Yanai A, Kang R, Arstikaitis P, Singaraja R, Metzler M, Mullard A, Haigh B, Gauthier-Campbell C, Gutekunst C. Huntingtin-interacting protein HIP14 is a palmitoyl transferase involved in palmitoylation and trafficking of multiple neuronal proteins. Neuron. 2004;44:977–986. doi: 10.1016/j.neuron.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 14.Yanai A, Huang K, Kang R, Singaraja R, Arstikaitis P, Gan L, Orban P, Mullard A, Cowan C, Raymond L. Palmitoylation of huntingtin by HIP14 is essential for its trafficking and function. Nat Neurosci. 2006;9:824–831. doi: 10.1038/nn1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohyama T, Verstreken P, Ly C, Rosenmund T, Rajan A, Tien A, Haueter C, Schulze K, Bellen H. Huntingtin-interacting protein 14, a palmitoyl transferase required for exocytosis and targeting of CSP to synaptic vesicles. J Cell Biol. 2007;179:1481–1496. doi: 10.1083/jcb.200710061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW. Exon I of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic Mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 17.Heng MY, Detloff PJ, Albin RL. Rodent genetic models of Huntington disease. Neurobiol Dis. 2008;32:1–9. doi: 10.1016/j.nbd.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 18.DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 19.Leon R, Bhagavatula N, Ulukpo O, McCollum M, Wei J. BimEL as a possible molecular link between proteasome dysfunction and cell death induced by mutant huntingtin. Eur J Neurosci. 2010;31:1915–1925. doi: 10.1111/j.1460-9568.2010.07215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hazeki N, Tukamoto T, Goto J, Kanazawa I. Formic acid dissolves aggregates of an N-terminal huntingtin fragment containing an expanded polyglutamine tract: applying to quantification of protein components of the aggregates. Biochem Biophys Res Commun. 2000;277:386–393. doi: 10.1006/bbrc.2000.3682. [DOI] [PubMed] [Google Scholar]
- 21.Drisdel R, Green W. Labeling and quantifying sites of protein palmitoylation. Biotechniques. 2004;36:276–285. doi: 10.2144/04362RR02. [DOI] [PubMed] [Google Scholar]
- 22.Drisdel R, Manzana E, Green W. The role of palmitoylation in functional expression of nicotinic {alpha} 7 receptors. J Neurosci. 2004;24:10502–10510. doi: 10.1523/JNEUROSCI.3315-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME. Dominant phenotypes produced by the HD mutation in STHdhQ111 striatal cells. Hum Mol Genet. 2000;9:2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- 24.Bu D, Erlander M, Hitz B, Tillakaratne N, Kaufman D, Wagner-McPherson C, Evans G, Tobin A. Two human glutamate decarboxylases, 65-kDa GAD and 67-kDa GAD, are each encoded by a single gene. Proc Natl Acad Sci USA. 1992;89:2115–2119. doi: 10.1073/pnas.89.6.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Erlander M, Tillakaratne N, Feldblum S, Patel N, Tobin A. Two genes encode distinct glutamate decarboxylases. Neuron. 1991;7:91–100. doi: 10.1016/0896-6273(91)90077-d. [DOI] [PubMed] [Google Scholar]
- 26.Wei J, Jin Y, Wu H, Sha D, Wu J. Identification and functional analysis of truncated human glutamic acid decarboxylase 65. J Biomed Sci. 2003;10:617–624. doi: 10.1159/000073527. [DOI] [PubMed] [Google Scholar]
- 27.Sha D, Wei J, Wu H, Jin Y, Wu J. Molecular cloning, expression, purification, and characterization of shorter forms of human glutamic decarboxylase 67 in an E. coli expression system. Mol Brain Res. 2005;136:255–261. doi: 10.1016/j.molbrainres.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 28.Shi Y, Veit B, Baekkeskov S. Amino acid residues 24–31 but not palmitoylation of cysteines 30 and 45 are required for membrane anchoring of glutamic acid decarboxylase, GAD65. J Cell Biol. 1994;124:927–934. doi: 10.1083/jcb.124.6.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang K, Sanders SS, Kang R, Carroll JB, Sutton L, Wan J, Singaraja R, Young FB, Liu L, El-Husseini A. Wild-type HTT modulates the enzymatic activity of the neuronal palmitoyl transferase HIP14. Hum Mol Genet. 2011;20:3356–3365. doi: 10.1093/hmg/ddr242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singaraja RR, Huang K, Sanders SS, Milnerwood AJ, Hines R, Lerch JP, Franciosi S, Drisdel RC, Vaid K, Young FB. Altered palmitoylation and neuropathological deficits in mice lacking HIP14. Hum Mol Genet. 2011;20:3899–3909. doi: 10.1093/hmg/ddr308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang K, Sanders S, Singaraja R, Orban P, Cijsouw T, Arstikaitis P, Yanai A, Hayden M, El-Husseini A. Neuronal palmitoyl acyl transferases exhibit distinct substrate specificity. FASEB J. 2009;23:2605–2615. doi: 10.1096/fj.08-127399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fukata Y, Fukata M. Protein palmitoylation in neuronal development and synaptic plasticity. Nat Rev Neurosci. 2010;11:161–175. doi: 10.1038/nrn2788. [DOI] [PubMed] [Google Scholar]
- 33.Kanaani J, Lissin D, Kash S, Baekkeskov S. The hydrophilic isoform of glutamate decarboxylase, GAD67, is targeted to membranes and nerve terminals independent of dimerization with the hydrophobic membrane-anchored isoform, GAD65. J Biol Chem. 1999;274:37200–37209. doi: 10.1074/jbc.274.52.37200. [DOI] [PubMed] [Google Scholar]
- 34.Solimena M, Aggujaro D, Muntzel C, Dirkx R, Butler M, De Camilli P, Hayday A. Association of GAD-65, but not of GAD-67, with the Golgi complex of transfected Chinese hamster ovary cells mediated by the N-terminal region. Proc Natl Acad Sci USA. 1993;90:3073–3077. doi: 10.1073/pnas.90.7.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kanaani J, Kolibachuk J, Martinez H, Baekkeskov S. Two distinct mechanisms target GAD67 to vesicular pathways and presynaptic clusters. J Cell Biol. 2010;190:911–925. doi: 10.1083/jcb.200912101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fukata M, Fukata Y, Adesnik H, Nicoll R, Bredt D. Identification of PSD-95 palmitoylating enzymes. Neuron. 2004;44:987–996. doi: 10.1016/j.neuron.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 37.Keller C, Yuan X, Panzanelli P, Martin M, Alldred M, Sassoe-Pognetto M, Luscher B. The {gamma} 2 subunit of GABAA receptors is a substrate for palmitoylation by GODZ. J Neurosci. 2004;24:5881–5891. doi: 10.1523/JNEUROSCI.1037-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang K, Kang M, Askew C, Kang R, Sanders S, Wan J, Davis N, Hayden M. Palmitoylation and function of Glial Glutamate Transporter-1 is reduced in the YAC128 mouse model of Huntington disease. Neurobiol Dis. 2010;40:207–215. doi: 10.1016/j.nbd.2010.05.027. [DOI] [PubMed] [Google Scholar]
- 39.Kang R, Wan J, Arstikaitis P, Takahashi H, Huang K, Bailey A, Thompson J, Roth A, Drisdel R, Mastro R. Neural palmitoyl-proteomics reveals dynamic synaptic palmitoylation. Nature. 2008;456:904–909. doi: 10.1038/nature07605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim S, Zhang Z, Sarkar C, Tsai P, Lee Y, Dye L, Mukherjee A. Palmitoyl protein thioesterase-1 deficiency impairs synaptic vesicle recycling at nerve terminals, contributing to neuropathology in humans and mice. J Clin Invest. 2008;118:3075–3086. doi: 10.1172/JCI33482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gauthier L, Charrin B, Borrell-Pages M, Dompierre J, Rangone H, Cordelieres F, De Mey J, MacDonald M, Lebmann V, Humbert S. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 42.Twelvetrees AE, Yuen EY, Arancibia-Carcamo IL, MacAskill AF, Rostaing P, Lumb MJ, Humbert S, Triller A, Saudou F, Yan Z. Delivery of GABAARs to synapses is mediated by HAP1-KIF5 and disrupted by mutant huntingtin. Neuron. 2010;65:53–65. doi: 10.1016/j.neuron.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Engelender S, Sharp A, Colomer V, Tokito M, Lanahan A, Worley P, Holzbaur E, Ross C. Huntingtin-associated protein 1 (HAP1) interacts with the p150Glued subunit of dynactin. Hum Mol Genet. 1997;6:2205–2212. doi: 10.1093/hmg/6.13.2205. [DOI] [PubMed] [Google Scholar]
- 44.Li XJ, Li SH, Sharp AH, Nucifora FC, Schilling G, Lanahan A, Worley P, Snyder SH, Ross CA. A huntingtin-associated protein enriched in brain with implications for pathology. Nature. 1995;378:398–402. doi: 10.1038/378398a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.