

Background: R2-like ligand-binding oxidases (R2lox) can assemble a Mn/Fe or diiron cofactor.
Results: The metal centers are structurally similar and activate oxygen, resulting in redox-coupled structural changes.
Conclusion: Oxygen activation likely proceeds via similar mechanisms at Mn/Fe and diiron clusters, while their redox state controls oxygen and substrate access.
Significance: R2lox proteins could provide novel catalysts for oxidative chemistry.
Keywords: ferritin, metalloprotein, small angle x-ray scattering (SAXS), X-ray absorption spectroscopy, X-ray crystallography
Abstract
Two recently discovered groups of prokaryotic di-metal carboxylate proteins harbor a heterodinuclear Mn/Fe cofactor. These are the class Ic ribonucleotide reductase R2 proteins and a group of oxidases that are found predominantly in pathogens and extremophiles, called R2-like ligand-binding oxidases (R2lox). We have recently shown that the Mn/Fe cofactor of R2lox self-assembles from MnII and FeII in vitro and catalyzes formation of a tyrosine-valine ether cross-link in the protein scaffold (Griese, J. J., Roos, K., Cox, N., Shafaat, H. S., Branca, R. M., Lehtiö, J., Gräslund, A., Lubitz, W., Siegbahn, P. E., and Högbom, M. (2013) Proc. Natl. Acad. Sci. U.S.A. 110, 17189–17194). Here, we present a detailed structural analysis of R2lox in the nonactivated, reduced, and oxidized resting Mn/Fe- and Fe/Fe-bound states, as well as the nonactivated Mn/Mn-bound state. X-ray crystallography and x-ray absorption spectroscopy demonstrate that the active site ligand configuration of R2lox is essentially the same regardless of cofactor composition. Both the Mn/Fe and the diiron cofactor activate oxygen and catalyze formation of the ether cross-link, whereas the dimanganese cluster does not. The structures delineate likely routes for gated oxygen and substrate access to the active site that are controlled by the redox state of the cofactor. These results suggest that oxygen activation proceeds via similar mechanisms at the Mn/Fe and Fe/Fe center and that R2lox proteins might utilize either cofactor in vivo based on metal availability.
Introduction
In the ferritin-like superfamily of proteins a four-helix bundle scaffold hosts a dinuclear metal cofactor (1, 2). Ferritins, bacterioferritins, and other related proteins assemble large protein cages from this scaffold that are used to store iron (3, 4). In these proteins, the metal-binding site in the four-helix bundle sequesters and oxidizes the iron to be stored in a mineralized form inside the cage (3, 4). Other ferritin-like proteins use dinuclear metal cofactors of different composition to perform a diverse array of catalytic functions. While the manganese catalases are more distantly related, bacterial multicomponent monooxygenases (BMMs),5 class I ribonucleotide reductases (RNRs), and fatty acid desaturases all share a common fold (1). In these proteins, the di-metal cofactor activates oxygen and catalyzes one- or two-electron redox chemistry from the high valent state of the metal ions (2, 5–7). In BMMs, a diiron cofactor is used to hydroxylate a large variety of both saturated and unsaturated hydrocarbons, including methane (6, 8). RNRs catalyze the reduction of ribonucleotides to deoxyribonucleotides via a radical-initiated mechanism. The three classes of RNRs differ in subunit composition and the way the catalytic radical is generated. Class I RNRs are composed of two types of subunits. The R1 subunit houses the catalytic site for ribonucleotide reduction and allosteric regulation sites, whereas the R2 subunit contains the di-metal center that generates the radical (5, 7, 9). Within class I RNRs, three subgroups can be distinguished that use different metal cofactors. A diiron cofactor is found in the prototypical class Ia R2 proteins (9), class Ib utilizes a dimanganese cluster (10–13), and the most recently identified class Ic contains a heterodinuclear Mn/Fe center (14–17). Whereas class Ia and Ib R2 proteins carry the stable radical on a tyrosine residue close to the metal cluster (10, 11, 13, 18, 19), this tyrosine residue is replaced by a redox inert phenylalanine in class Ic (20), and the radical equivalent is instead stored in form of the MnIV/FeIII state of the cofactor (14). Even more recently, another group of ferritin-like proteins utilizing a Mn/Fe cofactor was discovered in which the R2 scaffold is remodeled to house a hydrophobic ligand-binding channel (21–23). Although their physiological activity remains enigmatic, these proteins are clearly capable of an oxidase activity, as they catalyze formation of a tyrosine-valine ether cross-link in the protein scaffold close to the active site (21, 22). The two proteins from this group that have been structurally characterized to date were both found to contain a long-chain fatty acid ligand bound in the ligand-binding channel and coordinated to the metal cofactor (21, 22). These proteins were therefore called R2-like ligand-binding oxidases (R2lox). While their fold is most closely related to RNR R2 proteins, they appear to use the heterodinuclear cofactor for similar functions as BMMs or another recently discovered member of the ferritin superfamily, the cyanobacterial diiron aldehyde-deformylating oxygenase (24–28). The Mn/Fe cofactor is thus equally versatile as the diiron cofactor in that it can perform both one- and two-electron redox chemistry, but its full potential has yet to be uncovered.
We have recently shown that the R2lox protein itself can select for manganese in site 1 and iron in site 2, thereby assembling the heterodinuclear Mn/Fe cofactor (22), but it is not clear whether the coordination geometry in R2lox depends on metal identity. In our previous work, we presented the structures of an R2lox protein from the thermophilic bacterium Geobacillus kaustophilus in the metal-free, as well as the oxidized resting and nonactivated, reduced Mn/Fe-bound states (22). While in the oxidized state, metal-binding site 1 was almost exclusively populated by manganese, and site 2 contained mainly iron, the reduced state presented a roughly 1:1 mixture of Mn/Fe and Fe/Fe clusters. Potential differences between the Mn/Fe and Fe/Fe-bound states might be averaged out in this structure. To investigate how binding of manganese versus iron impacts coordination mode and larger conformational changes, we therefore determined the structures of R2lox also in the oxidized and reduced Fe/Fe-bound states, as well as the Mn/Mn-bound state, which does not activate oxygen. Likely routes for controlled oxygen and substrate access to the active site can be discerned in the structures. Small angle x-ray scattering (SAXS) and x-ray absorption spectroscopy (XAS) data corroborate the crystallographic results on the global and the local active site level. From these data, structural models for the reduced and oxidized homo- and heterodinuclear metal centers are derived that provide insight into the oxygen activation mechanism.
Experimental Procedures
Protein Production and Purification
An N-terminally His-tagged full-length construct of the R2lox protein I from G. kaustophilus (accession number yp_148624) was produced and purified as described previously (22). Briefly, protein was produced recombinantly in Escherichia coli BL21(DE3) (Novagen) grown in terrific broth (FormediumTM). To obtain metal-free protein, 0.5 mm EDTA was added to the cultures immediately before induction with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside. Apoprotein was purified via heat denaturation of contaminating proteins and nickel chelate affinity chromatography. Cells were disrupted by high pressure homogenization in 25 mm HEPES-Na, pH 7.0, 300 mm NaCl, 20 mm imidazole, 0.5 mm EDTA. The lysate was cleared by centrifugation, incubated at 60 °C for 10 min, and again cleared by centrifugation. The supernatant was applied to a nickel nitrilotriacetic acid-agarose (Qiagen) gravity flow column. The beads were washed with lysis buffer containing 40 mm imidazole, followed by the same buffer without EDTA. Protein was then eluted using lysis buffer containing 250 mm imidazole and without EDTA. The eluate was exchanged into 25 mm HEPES-Na, pH 7.0, 50 mm NaCl using a HiTrap desalting column (GE Healthcare), concentrated to ∼1 mm, aliquoted, flash-frozen in liquid nitrogen, and stored at −80 °C (22). Protein concentration was determined using an extinction coefficient at 280 nm of 47.76 and 50.56 mm−1 cm−1 for metal-free and metal-bound R2lox, respectively (29).
Mass Spectrometric Analysis of the Copurifying Ligand
Ligands were extracted from apoprotein with methanol. Samples (5 μl) were analyzed by LC-MS using a 6550 Agilent QTOF coupled to an Agilent 1290 LC system. Data were collected between m/z 70 and 1700 in positive/negative ion mode. The following ESI settings were used (Agilent Jetstream): gas temperature 300 °C; gas flow 8 liters/min; nebulizer pressure 40 p.s.i.; sheath gas temperature 350 °C; sheath gas flow 11 liters/min; Vcap 4000 V; fragmentor 100 V; Skimmer 145 V; Octapole RF Peak 750 V. All samples were separated using a reversed phase Kinetex C18 column (100 × 2.1 mm, 2.6-μm particle size, 100 Å pore size, Phenomenex). For elution, solvents used were 0.1% formic acid (FA) in water (solvent A) and 75:25% acetonitrile/isopropyl alcohol, 0.1% FA (solvent B). All solvents were of HPLC grade. Linear gradients were used for all separations and were devised as follows for reversed phase separation (0.5 ml/min): min 0, 5% B; min 8, 95% B; min 10, 95% B; min 10.2, 5% B; min 12, 5% B. Raw data were processed using Mass Hunter Qual (Agilent), with the “find by molecular feature” function, and generated CEF files were further aligned and statistically processed in Mass Profiler Professional (Agilent). Extracted protein samples were compared with blank extracted samples to identify potential ligands. Structural identification of ligands was done using accurate mass (<5 ppm mass error).
Crystallization and Data Collection
R2lox was crystallized by vapor diffusion at 22 °C in 12.5–27.5% (w/v) PEG 1500, 100 mm HEPES-Na, pH 7.0–7.4. To obtain full occupancy of the Mn/Fe cofactor, crystals of partially metal-bound protein were additionally soaked in mother liquor containing 5 mm each FeCl2 and MnCl2 for 30 min and then briefly washed in mother liquor supplemented with 20% (v/v) PEG 400 before flash-cooling in liquid nitrogen (22). To reconstitute the Mn/Fe cofactor in apoprotein, crystals of metal-free protein were soaked in mother liquor additionally containing 5 mm each MnCl2 and (NH4)2Fe(SO4)2 for 1–24 h under aerobic conditions and then briefly washed in 40% (w/v) PEG 1500, 100 mm HEPES-Na, pH 7.0. This procedure leads to the oxidized resting state of the Mn/Fe cofactor (with ∼1:0 Mn:Fe in metal site 1 and ∼1:2–1:3 Mn:Fe in site 2) (22). The oxidized Fe/Fe-bound state was obtained analogously by soaking with 5 mm (NH4)2Fe(SO4)2 only. Soaking with 5 mm MnCl2 only under the same conditions led to the nonactivated Mn/Mn-bound state. To obtain the nonactivated reduced Mn/Fe cofactor, apoprotein crystals were soaked in 1 ml of 40% (w/v) PEG 1500, 100 mm HEPES-Na, pH 7.0, 5 mm (NH4)2Fe(SO4)2, 5 mm MnCl2, 0.5% (w/v) sodium dithionite, 0.5 mm phenosafranin, and 0.05% (v/v) Tween 20 for 1 h and flash-cooled directly without washing (22). Using this procedure, a roughly equal mixture of nonactivated reduced Mn/Fe and Fe/Fe clusters was obtained (∼1:1 Mn:Fe in site 1 and ∼1:4 Mn:Fe in site 2) (22). The reduced Fe/Fe-bound state was reconstituted analogously by soaking with 5 mm (NH4)2Fe(SO4)2 only at pH 7.4 for 2 h. Soaking solutions were always freshly prepared immediately before use, using freshly dissolved (NH4)2Fe(SO4)2 and dithionite to ensure that the iron was ferrous and that oxygen was effectively removed from soaking solutions used to obtain reduced states, with phenosafranin serving as redox indicator. Data were collected at 100 K at beamlines PX14.1/BESSY (Helmholtz Center Berlin, Germany), ID23–2/ESRF (Grenoble, France), and X06SA/SLS (Villigen, Switzerland).
Structure Determination, Model Building, and Refinement
Data were processed with XDS (30). All structures of R2lox were solved using a previously determined structure in the same redox state (22) not containing any ligands as a starting model. Except for the reduced Fe/Fe-bound state, which was obtained in space group P21212 and was solved by molecular replacement using Phaser in Phenix (31, 32), all crystals were in space group I222, and structures were solved by Fourier synthesis. Refinement was carried out with phenix.refine (31, 33) and iterated with rebuilding in Coot (34). Refinement included individual atomic coordinate and isotropic B factor refinement, occupancy refinement for alternative conformations and metal ions bound on the protein surface (but generally not the active site metal ions), and bulk solvent corrections. Metal-ligand bond lengths were restrained. Solvent molecules were added with phenix.refine and manually. Hydrogens were added to the models in the later stages of refinement. In the structure of R2lox that had been isolated in (partially) metal-bound form and additionally soaked with manganese and iron under aerobic conditions (22), strong electron density connecting the phenolic oxygen of Tyr-162 and the Cβ of Val-72 was observed. This ether cross-link was restrained to an ideal distance of 1.45 ± 0.02 Å. Electron density for the cross-link was also observed in apoprotein crystals soaked with manganese and iron or iron only under aerobic conditions but was not strong enough to warrant restraining it. Structures were validated using MolProbity (35). Data and refinement statistics are given in Tables 1 and 2. All figures were prepared with PyMOL (version 1.6.0.0, Schrodinger, LLC). Electrostatic surface potentials were calculated with the Adaptive Poisson-Boltzmann Solver (APBS) in PyMOL (36, 37). A morph between the nonactivated Mn/Fe-bound state of R2lox before cross-link formation and the oxidized Mn/Fe-bound state containing the cross-link was generated by interpolating the two conformations using the corkscrew method without minimization with the UCSF Chimera package (38).
TABLE 1.
Crystallographic data statistics
Values in parentheses are for the highest resolution shell. Friedel pairs were merged.
| Soaking condition | Anoxic soaks |
Aerobic soaks |
||||||
|---|---|---|---|---|---|---|---|---|
| Mn + Fe (1 h)a | Fe (2 h)a | Mn + Feb | Mn + Fe (1 h )a | Mn + Fe (24 h)a | Fe (1 h)a | Fe (24 h)a | Mn (24 h)a | |
| PDB code | 4HR4 | 4XBV | 4HR0 | 5DCR | 5DCS | 5DCO | 4XB9 | 4XBW |
| Beamline | X06SA/SLS | X06SA/SLS | ID23–2/ESRF | X06SA/SLS | X06SA/SLS | PX14.1/BESSY | PX14.1/BESSY | PX14.1/BESSY |
| Wavelength (Å) | 1.00 | 1.00 | 0.87 | 1.00 | 1.00 | 0.92 | 0.92 | 0.92 |
| Resolution range (Å) | 50.00–1.90 (2.02–1.90) | 50.00–1.80 (1.91–1.80) | 50.00–1.90 (2.01–1.90) | 50.00–2.11 (2.24–2.11) | 50.00–2.01 (2.13–2.01) | 50.00–2.33 (2.47–2.33) | 50.00–1.80 (1.91–1.80) | 50.00–1.99 (2.11–1.99) |
| Space group | I222 | P21212 | I222 | I222 | I222 | I222 | I222 | I222 |
| Unit cell dimensions a, b, c (Å) | 55.93, 97.71, 128.13 | 55.99, 98.08, 128.59 | 56.15, 97.74, 130.85 | 56.19, 97.34, 129.76 | 56.06, 97.42, 129.32 | 55.63, 96.70, 128.34 | 56.04, 97.05, 129.74 | 56.37, 97.15, 128.07 |
| Unique reflections | 27,657 (4408) | 66,383 (10441) | 28,825 (4531) | 20,697 (3221) | 23,996 (3803) | 15,093 (2291) | 33,183 (5276) | 24,406 (3860) |
| Multiplicity | 3.7 | 6.4 | 4.1 | 3.7 | 3.7 | 4.1 | 4.0 | 4.1 |
| Completeness (%) | 98.5 (98.7) | 99.7 (98.5) | 99.3 (97.3) | 98.7 (97.2) | 99.3 (99.2) | 98.8 (95.3) | 99.6 (99.3) | 99.5 (98.7) |
| I/σ(I) | 10.75 (1.91) | 13.35 (1.75) | 15.73 (2.00) | 7.29 (1.76) | 5.35 (1.70) | 11.64 (1.90) | 18.94 (2.14) | 21.93 (1.92) |
| Rmerge | 6.1 (51.7) | 9.0 (95.6) | 5.4 (75.4) | 10.0 (44.6) | 15.5 (40.9) | 8.6 (70.4) | 3.8 (66.0) | 3.7 (73.2) |
| Rmeas | 7.2 (60.1) | 9.9 (104.1) | 6.3 (86.6) | 11.6 (51.9) | 18.1 (47.4) | 9.8 (80.8) | 4.4 (76.0) | 4.2 (83.9) |
| CC½c | 99.6 (79.2) | 99.7 (73.0) | 99.9 (70.6) | 99.0 (84.4) | 96.4 (84.1) | 99.8 (70.7) | 99.9 (73.5) | 100 (71.8) |
a The protein was produced and purified in metal-free form, and crystals were soaked with manganese and/or iron for the indicated durations under anoxic or aerobic conditions.
b The protein was isolated in partially metal-bound form, and the crystal was additionally soaked with manganese and iron under aerobic conditions for 30 min.
c Percentage of correlation between intensities from random half-datasets (107). The correlation is significant at the 0.1% level in all resolution shells in all datasets.
TABLE 2.
Refinement statistics
| Soaking condition | Anoxic soaks |
Aerobic soaks |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Mn + Fe (1 h)a |
Fe (2 h)a | Mn + Fec | Mn + Fe (1 h)a | Mn + Fe (24 h)a | Fe (1 h)a | Fe (24 h)a | Mn (24 h)a | ||
| Conf. Ab | Conf. Bb | ||||||||
| PDB code | 4HR4 | 4XBV | 4HR0 | 5DCR | 5DCS | 5DCO | 4XB9 | 4XBW | |
| Reflections used | 27,653 | 27,653 | 66,349 | 28,820 | 20,696 | 23,992 | 15,093 | 33,179 | 24,403 |
| Rwork/Rfree (%)d | 18.1/21.5 | 18.0/21.9 | 18.9/22.9 | 16.6/19.6 | 16.5/21.2 | 16.9/21.7 | 15.7/22.6 | 16.5/20.3 | 17.7/22.4 |
| Coordinate error (Å) | 0.20 | 0.22 | 0.28 | 0.18 | 0.19 | 0.23 | 0.29 | 0.19 | 0.24 |
| Non-H atoms | 2469 | 2467 | 4983 | 2486 | 2428 | 2468 | 2427 | 2482 | 2344 |
| Protein residuese | 286 (2–287) | 286 (2–287) | 571 (A: 2–286; B: 2–287) | 286 (2–287) | 285 (2–286) | 287 (2–288) | 285 (2–286) | 285 (2–286) | 274 (13–286) |
| Water molecules | 82 | 82 | 218 | 98 | 50 | 62 | 50 | 100 | 53 |
| Ligand molecules | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 1 |
| Metal ions | 3 | 3 | 6 | 4 | 3 | 3 | 2 | 2 | 3 |
| r.m.s.d. bonds (Å)f | 0.020 | 0.017 | 0.018 | 0.018 | 0.019 | 0.017 | 0.019 | 0.016 | 0.022 |
| r.m.s.d. angles (°)f | 1.331 | 1.316 | 1.286 | 1.244 | 1.332 | 1.216 | 1.394 | 1.264 | 1.313 |
| Ramachandran favored/allowed/outliers (%)g | 96.5/3.5/0.0 | 96.1/3.9/0.0 | 96.5/3.0/0.5 | 97.2/2.8/0.0 | 97.9/2.1/0.0 | 96.9/3.1/0.0 | 97.5/2.5/0.0 | 97.2/2.8/0.0 | 95.6/4.4/0.0 |
| Clashscoreg | 12.1 | 2.1 | 2.7 | 4.7 | 2.6 | 2.1 | 3.0 | 1.1 | 4.0 |
| Wilson B (Å2) | 45.5 | 37.4 | 29.1 | 39.8 | 40.4 | 37.8 | 39.6 | 30.2 | 38.1 |
| Isotropic B (Å2)h | |||||||||
| All atoms | 51.7 | 52.9 | 40.7 | 39.4 | 51.7 | 49.1 | 54.7 | 41.3 | 55.4 |
| Protein main and side chains | 51.8 | 53.0 | 40.7 | 39.3 | 51.7 | 49.2 | 54.9 | 41.3 | 55.5 |
| Metal ion in site 1 | 30.9 | 32.6 | 20.8 | 27.8 | 36.2 | 29.2 | 41.3 | 24.9 | 33.7 |
| Metal ion in site 2 | 33.8 | 35.7 | 24.8 | 30.8 | 37.3 | 32.0 | 40.9 | 26.3 | 41.9 |
| Other metal ions | 66.9 | 70.0 | 49.6 | 48.9 | 85.2 | 67.5 | - | - | 89.4 |
| Fatty acid | 59.5 | 55.7 | 45.5 | 42.4 | 58.6 | 57.6 | 49.8 | 46.4 | 61.1 |
| Water | 44.2 | 44.7 | 37.9 | 41.5 | 44.8 | 41.2 | 45.2 | 41.2 | 44.5 |
a The protein was produced and purified in metal-free form, and crystals were soaked with manganese and/or iron for the indicated durations under anoxic or aerobic conditions.
b Two alternative active site conformations were modeled, but conformation (conf.) A was judged to explain the data better than conformation B.
c The protein was isolated in partially metal-bound form, and the crystal was additionally soaked with manganese and iron under aerobic conditions for 30 min.
d Rfree is calculated from a randomly selected 5% subset of reflections excluded from refinement.
e Residues out of the 302-residue full-length protein included in the final model are given in parentheses.
f Root-mean-square deviation (r.m.s.d.) from ideal geometry.
g Geometry statistics were calculated with MolProbity (35).
h Average B factors were calculated with Baverage in the CCP4 suite (108).
Sample Preparation for X-ray Absorption Spectroscopy
To obtain the oxidized resting state of the Mn/Fe cofactor, 200 μm apoprotein was incubated with 2.4 eq of MnCl2 and 1.2 eq (per monomer) of (NH4)2Fe(SO4)2 in 100 mm HEPES-Na, pH 7.0, 50 mm NaCl for 1 h at room temperature under aerobic conditions. Excess metal ions were removed by passing the sample through a HiTrap desalting column (GE Healthcare) equilibrated in 25 mm HEPES-Na, pH 7.0, 50 mm NaCl. Following this reconstitution procedure, ∼50% of the protein contained Mn/Fe cofactors, the highest proportion that can be obtained in solution (22). (A much larger excess of metal ions, as used in the crystal soaking experiments, cannot be used for reconstitution in solution because it leads to protein precipitation.) The reconstituted protein was concentrated to 1–3 mm, and glycerol was added to a final concentration of 10% (v/v) before transfer into sample holders and flash-cooling in liquid nitrogen. The nonactivated reduced Mn/Fe cofactor was reconstituted in the same way in an anaerobic glove box with the addition of 1 mm sodium dithionite to all buffers. Iron-only loaded samples were prepared using a 4- or 5-fold molar excess of (NH4)2Fe(SO4)2 over polypeptide chains under aerobic or anaerobic conditions. No glycerol was added to reduced state samples.
TXRF
Metal contents of apoprotein preparations and XAS samples were quantified using total reflection x-ray fluorescence analysis on a Bruker PicoFox instrument (39). A gallium standard (Sigma) was added to the samples (v/v 1:1) prior to the measurements. TXRF spectra were analyzed using the routines provided with the spectrometer.
X-ray Absorption Spectroscopy
XAS experiments at the iron and manganese K-edges were carried out at beamline Samba of SOLEIL (Paris, France) using a standard setup for XAS (double-crystal Si[220] monochromator, liquid helium cryostat for holding samples at 20 K, 36 element energy-resolving germanium detector from Canberra for x-ray fluorescence monitoring) as described previously (40, 41). Dead-time corrected XAS spectra were averaged and normalized, and EXAFS oscillations were extracted as described previously (42). EXAFS data were processed, Fourier transforms (FTs) calculated, and spectral simulations carried out using in-house software (42). Phase functions were calculated with FEFF8.2 (43). FTs were calculated for k-values of 1.8–12.1 Å−1 using cos2 windows extending over 10% of both k-range ends.
Bond Valence Sum Calculation
BVS values were calculated using Equation 1 with a B value of 0.37 Å, values of R0i for Fe-O of 1.737 Å, Fe-N of 1.792 Å, Mn-O of 1.762 Å, and Mn-N of 1.843 Å, which represent the average values for metal(II) and metal(III) species (44), and using the coordination numbers (Ni) and mean metal-ligand distances (Ri) from the EXAFS analysis or the crystallographic models.
Mass Spectrometric Analysis of Cross-link Formation
100 μm apoprotein was incubated with 2 eq of MnCl2 and 1 eq (per monomer) of (NH4)2Fe(SO4)2 or 3 eq of (NH4)2Fe(SO4)2 only in 100 mm HEPES-Na, pH 7.0, 50 mm NaCl, 1 mm sodium dithionite under aerobic conditions for 4 h at room temperature. Excess metal ions were removed by passing the sample through a HiTrap desalting column (GE Healthcare) equilibrated in 25 mm HEPES-Na, pH 7.0, 50 mm NaCl. The reconstituted protein was concentrated to ∼1 mm. Six replicates of each sample were prepared. From each assay replicate, 80 μg of R2lox protein were subjected to proteolytic digestion by Glu-C (Promega, enzyme/substrate ratio 1:40) in phosphate buffer using the filter aided sample preparation method (45). In phosphate buffer (50 mm, pH 7.6), the enzyme Glu-C cleaves the protein at the C-terminal side of glutamic and aspartic residues. From each replicate, 300 μl of digested sample was collected and acidified by addition of 100 μl of 10% FA prior to LC-MS. The autosampler of an HPLC 1200 system (Agilent Technologies) injected 1 μl (∼200 ng of peptides) into a C18 guard desalting column (Zorbax 300SB-C18, 5 × 0.3 mm, 5 μm bead size, Agilent). Then a 15-cm-long C18 PicoFrit column (100 μm internal diameter, 5 μm bead size, Nikkyo Technos Co., Tokyo, Japan) installed onto the nano electrospray ionization (NSI) source was used. Solvent A was 97% water, 3% acetonitrile, 0.1% FA; and solvent B was 5% water, 95% acetonitrile, 0.1% FA. At a constant flow of 0.4 μl/min, a linear gradient went from 2% B up to 40% B in 45 min, followed by a steep increase to 100% B in 5 min, plateau at 100% B for 5 min, and subsequent re-equilibration with 2% B. On-line LC-MS was performed using an LTQ Orbitrap Velos Pro mass spectrometer (Thermo Scientific). FTMS master scans (AGC target of 1e6) were acquired with a resolution of 30,000 and were followed by data-dependent MS/MS (AGC target of 1e5) at a resolution of 7,500. In data-dependent MS/MS, the top two ions from the master scan were selected first for collision-induced dissociation (at 35% energy) and afterward for higher energy collision dissociation (at 30% energy). Precursors were isolated with a 2 m/z window. Dynamic exclusion was used with 60-s duration. Each sample was analyzed in technical triplicates amounting to a total of 36 LC-MS runs. The relative amount of ether cross-link in each sample was quantified using the Glu-C cleavage product cross-linked peptide AVIRAATVYNMIVE-AVTLD (where the underlined residues are the cross-linked Tyr and Val) as surrogate reporter (i.e. the 689.038 m/z ion). The area under the curve was calculated for the extracted ion chromatogram in the 689.028–689.048 m/z range in the retention time window 36–38 min using the Qual Browser in Xcalibur (Thermo Scientific). This area was then normalized to the total protein area of the respective LC-MS run using the precursor area quantification node of Proteome Discoverer 1.4 (Thermo Scientific).
Small Angle X-ray Scattering of Protein Solutions
To prepare samples suitable for SAXS measurements, metal-free and aerobically Mn/Fe-reconstituted protein was additionally purified via gel filtration on a Superdex 200 column (GE Healthcare) and was concentrated to yield samples in concentration ranges from 1 to 17 mg/ml in 25 mm HEPES-Na, pH 7.0, 150 mm NaCl. The flow-through of the concentration step was used as buffer reference for SAXS measurements. Samples were centrifuged immediately prior to measurement. SAXS data were collected at beamline I911-SAXS/Max II at an x-ray wavelength of 0.91 Å over an s range of 0.01–0.5 Å−1. The momentum transfer s is defined as s = 4π sinθ/λ, where 2θ is the scattering angle, and λ is the x-ray wavelength. Scattering profiles of lysozyme, bovine serum albumin, and alcohol dehydrogenase (Sigma) were measured as reference for molecular mass determination. The ATSAS package (46) was used to process and analyze data. The radius of gyration (Rg) was derived by the Guinier approximation (I(s) = I(0) exp(−s2Rg2/3) for s Rg <1.3). The molecular masses of the solutes were determined by extrapolating the scattering intensities to zero angle and using a standard curve obtained from I(0) values and known molecular masses of the reference proteins. Independent estimates of the molecular mass were obtained from the hydrated volume of the particles. Theoretical scattering profiles of atomic resolution models were calculated and fitted to measured profiles with CRYSOL (47). Ab initio models were reconstructed from the experimental data using the programs DAMMIF (48) and GASBOR (49), initially without imposing any symmetry or other restrictions on possible models. Because all models were clearly 2-fold symmetric, further models were calculated imposing 2-fold symmetry. Eight models that were independently reconstructed with GASBORi were aligned and averaged with SUPCOMB (50) and DAMAVER (51). The Situs package (52) was used to calculate an envelope representation and dock the atomic resolution model into it.
Results
A Mixture of Hydroxylated Long-chain Fatty Acids Copurifies with R2lox
A striking feature of R2lox proteins is the hydrophobic tunnel extending from the active site to the protein surface in which a long-chain fatty acid is bound that copurifies with the protein from the heterologous expression host (21, 22). To verify the nature of the ligand, a sample of R2lox produced in E. coli in metal-free form was subjected to mass spectrometry analysis. It was found to contain a mixture of different long-chain fatty acids, mainly hydroxylated octadecanoic (C18H34O3) and hexadecanoic acid (C16H32O3) (Fig. 1). In the crystal structures, the ligand was modeled as palmitic (hexadecanoic) acid. We have not attempted to assign the hydroxyl group in the crystal structures, as its location in the chain is not apparent either in the electron density or from the mass spectrometry data. Importantly, however, because the mass spectrometry data clearly indicates that the ligand is a fatty acid, it was modeled as such in all structures, although the density for the carboxyl group is not entirely clear in all of them (see below).
FIGURE 1.

Mass spectra of the copurifying ligand extracted from R2lox. It is a mixture of mainly C16 (top) and C18 (bottom) hydroxylated fatty acids. The position of the hydroxyl group could not be determined. The insets show the isotopic distributions for respective [M − H]−1.
Crystal Structures of R2lox in Different Metallation and Redox States
Structures of R2lox with a Mn/Fe cluster in the nonactivated, reduced, and oxidized resting state were previously determined (22). Here, we present a detailed structural analysis of differently metallated states of R2lox containing Mn/Fe, Fe/Fe, or Mn/Mn clusters. For simplicity, in the following we refer to structures in the oxidized resting state as “oxidized” and to nonactivated states as “reduced.” The assignment of redox states is based on the characteristic arrangement of the metal ligands in the structures. As detailed below, the metal ions are most likely divalent even in oxidized state structures due to x-ray photoreduction. To assess cofactor activity in crystals (see below), several oxidized Mn/Fe and Fe/Fe state crystals were analyzed (Tables 1 and 2), but because the structures were identical within error, we discuss only the respective highest resolution structures in detail (PDB codes 4HR0 and 4XB9 for the oxidized Mn/Fe and Fe/Fe cofactor, respectively).
The protein-derived metal ligands in R2lox consist of two histidines and four glutamates. In the reduced Mn/Fe-bound state, each metal ion is coordinated by one histidine and four carboxylates (Fig. 2A). One of the carboxylate groups is provided by the external fatty acid ligand, bridging the metal ions in bidentate coordination mode. Glu-102 provides the second bidentate bridge opposite the fatty acid. Each metal ion is further coordinated by one histidine and one monodentate glutamate (site 1, Glu-69 and His-105; site 2, Glu-167 and His-205). The last glutamate ligand, Glu-202, bridges the metal ions in monodentate coordination mode, while also coordinating the iron ion in site 2 with the second carboxyl oxygen (i.e. a μ-η1,η2 coordination). A water molecule is bound at the open coordination site of the manganese ion in site 1. Both metal ions are therefore coordinated in octahedral geometry, although the coordination sphere in site 2 is distorted.
FIGURE 2.

Stereo view of the active site of R2lox in the nonactivated reduced (A) and oxidized resting (B) Mn/Fe-bound states. Carbon atoms of first and selected second sphere amino acid residues are colored cyan and green, respectively, and those of the fatty acid ligand light blue (with oxygen colored red and nitrogen blue). Iron and manganese are shown as orange and purple spheres, respectively, and water/oxo/hydroxo ligands as smaller red spheres. Metal-ligand bonds are indicated by gray lines, hydrogen bonds by blue dashed lines, and bond lengths are given in Å.
Only Glu-202 changes its coordination mode upon oxidation. It shifts outward, leaving only the monodentate coordination to site 2, and a hydroxo ion, presumably derived from molecular oxygen, takes the place of the bridge (Fig. 2B). This bridging ligand was assigned as a hydroxo ion based on Mössbauer and EPR data, which also showed that the ligand to site 1 is a water molecule (22, 29). These changes lead to the coordination sphere in site 2 also becoming perfectly octahedral. No other major changes occur in the first coordination sphere, with the exception of the water ligand to site 1. Upon oxidation, this water molecule moves farther away from the metal ion and closer to its other coordinating ligand, the phenolic oxygen of Tyr-175 (Fig. 2). This movement appears to cause a concerted movement of Tyr-175 and a leucine residue in the second ligand sphere closer to site 1, Leu-198. Tyr-175 only rotates slightly to accommodate the hydrogen bond to the water ligand, but it would then clash with the reduced state conformation of Leu-198, which consequently adopts a different rotamer (Fig. 2).
With the fatty acid ligand coordinated to both metal ions in the reduced state, all coordination sites of both metal ions are occupied, and the cofactor cannot bind and reduce oxygen unless a ligand is released (Fig. 2). However, the cofactor clearly is activated under aerobic conditions. The electron density for the fatty acid is less well defined in the reduced than in the oxidized state, indicating that the ligand is mobile in the reduced state (Fig. 3, A and B). Because the reduced active site model described above (“conformation A”) does not appear to completely explain the crystallographic data, we attempted to model an alternate reduced active site conformation (“conformation B”) where the fatty acid carboxyl group is not coordinated to the metal ions, and Glu-167 instead coordinates the iron ion in bidentate mode (Fig. 3, B–F). Starting from this conformation, both Glu-202 and Glu-167 would have to rotate upon oxidation. However, conformation B does not explain the data better than conformation A (Fig. 3, E and F, and Table 2). The manganese ion in site 1 has an open coordination site, whereas Glu-167 coordinates the iron ion in site 2 with one rather short (1.8 Å) and one rather long (2.3 Å) bond. The positive difference density observed in conformation B cannot satisfactorily be explained by water molecules, as these would clash with surrounding atoms. Simultaneous refinement of both conformations led to worse geometry in the active site than separate refinements of either conformation, while not removing the positive difference density around Glu-167 (data not shown). Other active site models were also tested and found to be more unsatisfactory. We conclude that conformation A explains the available crystallographic data best, if not completely.
FIGURE 3.
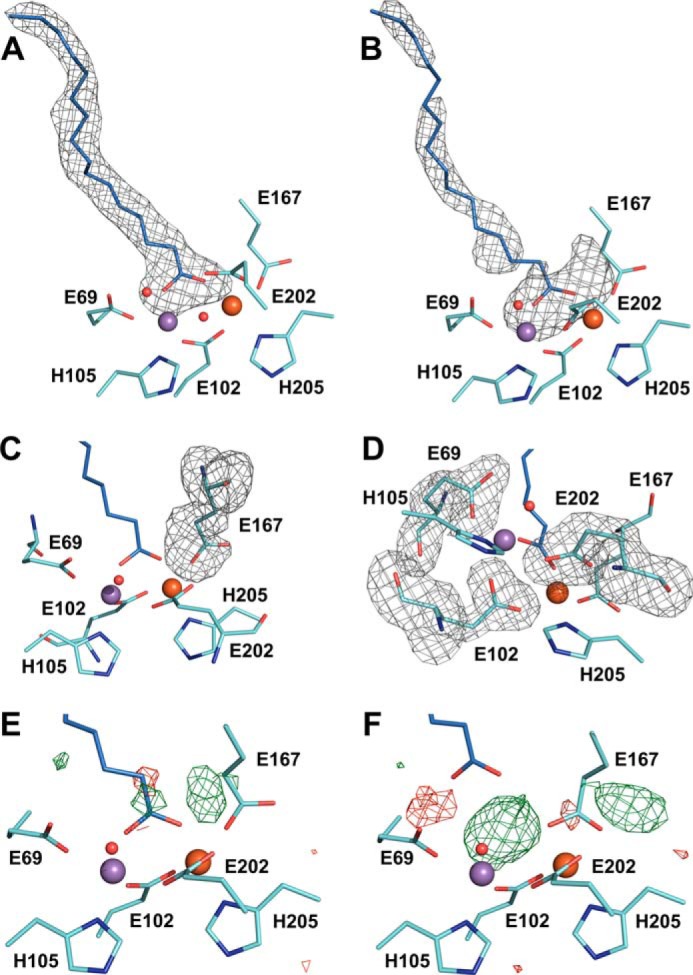
Structural heterogeneity in the reduced Mn/Fe-bound active site of R2lox. A and B, mFo − DFc omit electron density for the copurified ligand bound in the oxidized (A) and reduced (B) Mn/Fe-bound active site, contoured at 2 σ. The fatty acid is much more disordered in the reduced state, and its carboxyl head group may not be coordinated to the metal ions in all molecules. The omit electron density also suggests an alternative conformation of Glu-167 that is mutually exclusive with the position of the fatty acid headgroup as modeled. C and D, mFo − DFc omit electron density for Glu-167 (C) and the other three glutamate ligands (D) in the reduced Mn/Fe-bound active site, contoured at 3 σ. The other three glutamate ligands are much better defined in the electron density than Glu-167. A–D, omit electron density is shown as gray mesh. E and F, mFo − DFc difference electron density around the active site of R2lox in the reduced Mn/Fe-bound state contoured at +3 σ (green) and −3 σ (red). E, conformation A with Glu-167 as monodentate ligand to the iron ion in site 2 and the fatty acid coordinated to both metal ions as a bidentate bridge (PDB code 4HR4). F, conformation B with Glu-167 as bidentate ligand to the iron ion in site 2 and the fatty acid not coordinated to the metal ions.
No major structural differences are observed between the Mn/Fe- and Fe/Fe-bound structures in corresponding redox states. All structures superimpose with root-mean-square deviations between 0.11 and 0.36 Å, in the range of the coordinate error (Table 2). In particular, the active sites are virtually identical (Fig. 4, A and B). Generally, R2lox crystallizes in space group I222 with one molecule in the asymmetric unit, but crystals of the reduced Fe/Fe-bound state were obtained in P21212 with two molecules in the asymmetric unit. The two polypeptide chains do not display any significant differences, aside from chain B being slightly more disordered than chain A, as evidenced by its higher average B factor (39.1 and 42.3 Å2 for the whole chain A and B, respectively). Mobility of the active site, and especially the fatty acid ligand, is observed in both chains in the reduced Fe/Fe-bound state (data not shown). Oxygen activation therefore seems to proceed via similar mechanisms with the Mn/Fe and the Fe/Fe cofactor.
FIGURE 4.

A and B, the active site of R2lox in the reduced (A) and oxidized (B) Fe/Fe-bound state, superimposed with the Mn/Fe-bound structure in the corresponding redox state. C, the nonactivated Mn/Mn-bound active site, superimposed with the reduced Mn/Fe-bound state. The same color scheme is used as in Fig. 2, with the Mn/Fe structures shown in transparent gray.
In the aerobically reconstituted Mn/Mn-bound state, the metal ligands adopt a reduced state conformation (Fig. 4C). This is expected, as the dimanganese cluster cannot activate oxygen (19, 53). When R2lox is reconstituted with a low excess of only manganese in solution, manganese binds only to site 1 (22). In crystal soaking experiments, a far larger excess of manganese can be used (which would precipitate the protein in solution but does not harm the crystals). Under these conditions, manganese binds in both metal-binding sites of R2lox. However, the manganese ion in site 2 refines to a significantly higher B factor than the manganese ion in site 1, indicating that site 1 is more disordered and/or not fully occupied. Combined B factor and occupancy refinement yielded B factors and occupancies of 33.5 Å2/0.96 and 40.5 Å2/0.95 for site 1 and site 2, respectively. Because this is very near the B factors obtained with full occupancies, we chose to deposit the model with fully occupied metal sites (Table 2). In contrast, in mixed-metal and iron-only structures, both metal ions refine to full occupancy and very similar B factors (Table 2). The crystallographic data therefore indicate that although manganese can be forced into both metal sites when using a large excess of manganese over protein, it still does not bind equally well on its own as iron or manganese together with iron.
Refined Active Site Architectures Obtained by X-ray Absorption Spectroscopy
R2lox was reconstituted with manganese and iron or iron only under both anaerobic and aerobic conditions in solution to obtain reduced and oxidized Mn/Fe and Fe/Fe centers. The samples were subjected to XAS analysis to determine the metal oxidation states and interatomic distances in the metal centers from x-ray absorption near edge structure (XANES) and extended x-ray absorption fine structure (EXAFS) spectra, respectively. Metal contents in R2lox samples before and after reconstitution were determined by TXRF analysis (Table 3). The apoprotein preparation contained ∼10% diiron clusters, assuming that all iron was bound in dinuclear centers. The content of manganese and other transition metal ions (nickel, copper, and zinc) was negligible. The aerobically reconstituted iron-only sample contained near-stoichiometric amounts of iron per protein monomer, so that almost quantitative occupancy of dinuclear Fe/Fe centers was expected. Stoichiometric metal loading was likewise observed in the aerobically Mn/Fe-reconstituted sample. The manganese to iron ratio of about 1:3 indicates that approximately half of the cofactors were either of the Mn/Fe or Fe/Fe type. This assignment agrees with that previously made for EPR samples reconstituted in solution using the same procedure as for the XAS samples (22). In the anaerobically reconstituted samples, metal loading was sub-stoichiometric, suggesting the presence of large amounts of apoprotein or alternatively singly occupied centers besides dinuclear cofactors. However, the manganese to iron ratio in the reduced Mn/Fe sample was similar to the oxidized sample, indicating similar relative amounts of Mn/Fe and Fe/Fe clusters. The sub-stoichiometric loading of reduced state samples was probably caused by the desalting step used to remove excess metal ions after reconstitution, which might also remove weakly bound metal ions in the protein, as the metal ions only become “fixed” in the active site upon oxygen activation due to the formation of metal(III)-bridging μO(H) bonds (54).
TABLE 3.
Metal contents of R2lox samples before and after reconstitution with manganese and iron in solution
Ox is oxidized, aerobically prepared samples; red is reduced, anaerobically prepared samples.
| Sample | Concentrations |
Ratios |
Metal clusters |
|||||
|---|---|---|---|---|---|---|---|---|
| [Protein]a | [Mn] | [Fe] | Mn/protein | Fe/protein | Fe/Mn | Mn/Feb | Fe/Feb | |
| mm | mm | mm | % | % | ||||
| Apo | 1.18 | <0.01 | 0.23 | <0.01 | 0.19 | 23 | 0 | 9.5 |
| Fe/Fe-ox | 0.90 | <0.01 | 1.72 | <0.01 | 1.91 | >200 | 0 | 95 |
| Fe/Fe-red | 1.78 | <0.01 | 1.05 | <0.01 | 0.59 | >100 | 0 | 30 |
| Mn/Fe-ox | 1.00 | 0.49 | 1.57 | 0.49 | 1.57 | 3.2 | 49 | 51 |
| Mn/Fe-red | 2.23 | 0.22 | 0.50 | 0.10 | 0.22 | 2.3 | 10 | 6 |
a Concentrations refer to polypeptide chains.
b Percentages of metal centers are relative to polypeptide chains. The amounts of dinuclear cluster species were calculated assuming that all manganese was bound in Mn/Fe clusters.
For Fe/Fe-R2lox, the absolute energies of the iron K-edge spectra (∼7121.4 and ∼7124.8 eV) and the energy difference (∼3.4 eV) are indicative of mostly FeII in the reduced and FeIII in the oxidized sample (Fig. 5A) (55, 56). Accordingly, anaerobically reconstituted protein contains mainly FeII/FeII centers and aerobically reconstituted protein mainly FeIII/FeIII centers. The high edge energy of the oxidized sample and the relatively large energy difference, in comparison with related FeII and FeIII compounds (55, 57), suggest the presence of minor amounts (≤20%) of FeIV species in Fe/Fe-R2loxox. For Mn/Fe-R2lox, the manganese K-edge energies (∼6546.8 and ∼6549.2 eV) and the energy difference (∼2.4 eV) are indicative of almost quantitative amounts of MnII and MnIII in the reduced and oxidized sample, respectively (55, 57, 58). Thus the manganese-containing centers are mainly in the MnII/FeII and MnIII/FeIII states, respectively. Again, the relatively high manganese K-edge energy in the oxidized sample suggests a minor (≤20%) MnIV admixture. The predominant oxidation states observed in the XAS samples agree with the assignments made previously based on EPR and Mössbauer data (22, 29).
FIGURE 5.

XAS analysis of Fe/Fe and Mn/Fe centers in R2lox. Spectra at the iron (left) or manganese (right) K-edges of anaerobically reconstituted, reduced (red), aerobically reconstituted, x-ray photoreduced (xpr) and aerobically reconstituted, oxidized (ox) samples are shown (xpr denotes x-ray irradiation for ∼90 min, Fe/Fe center, or ∼50 min, Mn/Fe center, prior to the collection of the spectra). A, XANES spectra. Insets, iron or manganese K-edge energies (dots) for increasing x-ray irradiation periods determined from respective XANES spectra (data not shown) together with single exponential decay simulations (lines) with half-times of ∼33 min (iron) or ∼16 min (manganese). B, FTs of iron and manganese EXAFS oscillations in the insets (black lines, experimental data; colored lines, simulations with parameters in Table 4). Spectra are vertically displaced for comparison.
The rates of x-ray photoreduction (xpr) of iron and manganese in R2loxox were monitored by down-shifts of the K-edge energies in response to increasing periods of x-ray irradiation of samples held at 20 K (Fig. 5A). The iron K-edge shifted by ∼1.8 eV after ∼2 h of xpr, with a half-time of ∼33 min, suggesting that ∼50% of the initial FeIII was reduced to FeII in Fe/Fe-R2loxox and thus mostly mixed-valence FeIII/FeII centers were present after this exposure period. The manganese K-edge shifted by ∼1.8 eV after ∼1 h of xpr, with a half-time of ∼16 min, indicating the reduction of ∼75% of the initial MnIII to MnII in Mn/Fe-R2loxox, and therefore a majority of MnII-containing centers after this irradiation period. Similarly rapid xpr has been observed for typical R2 proteins (55, 56). The faster reduction of MnIII compared with FeIII can likely be explained by the about twice as large x-ray absorption cross-section of the protein sample at manganese K-edge energies compared with iron K-edge energies (56). We used a spot size of about 0.3 mm2 at ∼1011 photons s−1, i.e. a specific x-ray flux of ∼3 × 1011 photons mm−2 s−1 in the xpr experiments at the iron and manganese K-edges at 20 K. The specific flux at the used crystallography beamlines was in the range of 7 × 1013 to 4 × 1015 photons mm−2 s−1. The crystallographic measuring temperature was 100 K, considerably higher than for XAS, which may be expected to accelerate xpr by a factor of about 10 (56, 59). However, this factor may be compensated by the about 10 times lower absorption of the sample at the higher x-ray energies used in crystallography. Assuming a linear dose-rate relationship of xpr (56, 59), at least about 200–10,000 times faster reduction of metal(III) ions was therefore expected during crystallographic data collection of R2loxox. This means that the FeII and MnII levels likely were reached within less than 0.5–25 s of data collection, long before a complete dataset was obtained. Crystal structures of initially oxidized R2lox are thus expected to contain mainly divalent metal ions. Because crystallographic data collection proceeded at 100 K, movement of the amino acid side chains bound to the metal ions may be limited. Nevertheless, it is important to ascertain which structural changes accompany xpr in crystalline samples and how well aerobically reconstituted crystal structures therefore actually represent the oxidized resting state. In addition, the much higher precision of EXAFS for interatomic distance determination (∼0.02 Å) compared with protein crystallography yields more accurate metal-ligand bond lengths and metal-metal distances.
EXAFS spectra (FTs and EXAFS oscillations) of iron for Fe/Fe-R2lox and of manganese for Mn/Fe-R2lox samples are shown in Fig. 5B. Simulations of the spectra reveal the structural alterations at the metal sites in response to redox state changes of the cofactors (Table 4). Iron and manganese are most likely 6-coordinated both in the reduced and oxidized state.
TABLE 4.
EXAFS simulation parameters
The following abbreviations are used: N, coordination number; R, interatomic distance; 2σ2, Debye-Waller factor; RF, error sum calculated for reduced distances of 1–3.5 Å (42); red, reduced, anaerobically prepared samples; xpr, aerobically prepared, X-ray photoreduced samples; ox, oxidized, aerobically prepared samples.
| Fe/Fe |
N (per metal)/R (Å)/2σ2 × 103 (Å2) |
RF | ||||
|---|---|---|---|---|---|---|
| Fe–N/O | Fe–N/O | Fe–C/N/O | Fe–C/N/O | Fe–Fe | ||
| % | ||||||
| Red | 1.90a/1.98/5b | 4.10a/2.15/5b | 1.82/2.86/2c | 3.75/3.11/5c | 1c/3.64/12 | 15.2 |
| Xpr | 2.34a/1.97/8b | 3.66a/2.11/8b | 1.62/2.54/2c | 2.58/3.05/5c | 1c/3.46/9 | 12.5 |
| Ox | 2.30a/1.91/6b | 3.70a/2.05/6b | 1.48/2.52/2c | 2.66/2.98/5c | 1c/3.43/7 | 15.5 |
| Mn/Fe | Mn–N/O | Mn–N/O | Mn–C/N/O | Mn–C/N/O | Mn–Fe | |
|---|---|---|---|---|---|---|
| Red | 0.38a/1.97/16b | 5.62a/2.19/16b | 1.64/2.58/2c | 0.81/3.27/5c | 1c/3.65/23 | 13.1 |
| Xpr | 1.17a/1.92/13b | 4.83a/2.14/13b | 0.62/2.63/2c | 1.32/3.11/5c | 1c/3.47/14 | 13.2 |
| Ox | 2.20a/1.90/11b | 3.80a/2.13/12b | 0.95/2.67/2c | 1.92/3.02/5c | 1c/3.51/8 | 17.8 |
a First sphere coordination numbers were coupled to yield a sum of 6.
b Debye-Waller factors were coupled to yield equal values for the two Fe/Mn–N/O shells.
c Fixed parameters.
In the reduced Fe/Fe center, two resolved Fe-N/O bond lengths of close to 2.0 and 2.2 Å are likely attributable mostly to OHn (water species) ligands, as well as O(carboxylate) and N(histidine) ligands at FeII, respectively (Table 4). The coordination number of distances within about 2.9–3.1 Å, reflecting C/O/N atoms of amino acid side chains in the second coordination sphere (i.e. atoms not directly bound to the metal ions), was slightly increased compared with the oxidized sample, possibly due to the Fe-C and Fe-O distances from the bridging-chelating carboxylate (Glu-202) observed in the crystal structure of the reduced Fe/Fe center (Fig. 4A). An FeII–FeII distance of ∼3.65 Å, similar to the crystal structure, could be determined, but contributed only weakly to the EXAFS. The relatively small contribution of metal-metal distances to the EXAFS was likely due to the presence of considerable amounts of single-metal cofactor sites in the sub-stoichiometrically loaded reduced sample (see above, Table 3). Longer Fe-Fe distances around 4.2 Å, accounting for respective small FT peak features (Fig. 5B), could also be included in the EXAFS fit (data not shown) and may be due to Fe/Fe clusters lacking a bridging-chelating carboxylate.
In the oxidized Fe/Fe sample, metal-metal distances were relatively well discernable in the EXAFS data, in agreement with the expected near-quantitative presence of dinuclear cofactors (Table 3). The oxidized Fe/Fe centers revealed about 0.1 Å shorter FeIII–N/O bonds and an increased coordination number of the shorter bonds, in agreement with an additional Fe-μO(H)-Fe-bridging motif in the site (Table 4). The FeIII–FeIII distance was shortened by only ∼0.2 Å. Inclusion of a second Fe-Fe distance of ∼3.1 Å with a low coordination number slightly increased the fit quality (data not shown). Such a distance may reflect minor amounts of Fe/Fe centers with two μO(H) bridges (55–57, 60). Xpr of Fe/Fe-R2loxox led to an overall Fe–N/O bond elongation by ∼0.05 Å and to increased distance heterogeneity (larger Debye-Waller factor) in the mixed-valence cluster, and thus to structural parameters more similar to the reduced cofactor. The Fe-Fe distance after xpr, however, remained almost unchanged.
The reduced Mn/Fe cofactor showed rather homogeneous and typical MnII–N/O bond lengths close to 2.2 Å and second sphere Mn-C/N/O distances of about 2.6–3.3 Å (Table 4). The increased coordination number of Mn-C/N/O bonds of ∼2.6 Å compared with the oxidized center may be due to a bridging-chelating carboxylate (Glu-202), as in the case of the Fe/Fe center. A MnII–FeII distance of ∼3.65 Å was similar to the FeII–FeII distance and likewise contributed only weakly to the EXAFS, presumably due to the presence of large amounts of singly occupied cofactor sites. Longer Mn-Fe distances around 4.2 Å could also be included in the fit (data not shown), as in case of the Fe/Fe cluster.
For the oxidized Mn/Fe center, a considerably increased MnIII–N/O bond length spread was found, the shorter bonds (∼1.9 Å) presumably reflecting mostly Mn-OHn and Mn-μO(H)-Fe motifs, whereas the longer bonds (∼2.1 Å) correspond to the O(carboxylate) and N(histidine) ligands (Table 4). Like in the oxidized Fe/Fe sample, a small shortening of the MnIII–FeIII distance (by ∼0.15 Å) was observed, and metal-metal distances were likewise relatively well discernable in the EXAFS data. Inclusion of a second Mn-Fe distance at ∼3.0 Å with a low coordination number slightly increased the fit quality (data not shown). The xpr effects (after ∼50 min of irradiation) on the initially oxidized Mn/Fe center were similar to those on the Fe/Fe cluster. Overall, longer Mn-N/O bonds and increased distance heterogeneity more similar to the reduced cofactor were observed, but the Mn-Fe distance remained almost unchanged.
BVS calculations were carried out to derive the metal oxidation states on the basis of the bond lengths from EXAFS analysis (Table 5). The EXAFS data reveal mean Fe/Mn-N/O bond lengths decreasing by ∼0.1 Å in the R2lox sample in the order reduced > xpr > oxidized and an Fe/Mn-Fe distance shortening by ∼0.15 Å on the red → ox transition. The respective BVS increases from values close to 2 to values close to 3, in good agreement with the XANES data indicating mostly MII/FeII and MIII/FeIII (where M is manganese or iron) centers in the reduced and oxidized samples, respectively.
TABLE 5.
Metal-ligand bond lengths and BVS values from EXAFS and crystal structures
The first value in each line is from EXAFS, and the values in parentheses are from the crystal structures. The following abbreviations are used: Red, reduced, anaerobically prepared samples; xpr, aerobically prepared, x-ray photoreduced samples; ox, oxidized, aerobically prepared samples.
| Fe/Mn–Fe (Å) |
Fe/Mn–N/O mean (Å) |
BVS |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Red | xpr | Ox | Red | xpr | Ox | Red | xpr | Ox | |
| Fe/Fe | 3.64 (3.61a ) | 3.46 (-) | 3.42 (3.52) | 2.10 (2.06) | 2.06 (-) | 2.00 (2.06) | 2.4 (2.3b) | 2.6 (-) | 3.1 (2.3b) |
| Mn/Fe | 3.65 (3.61) | 3.47 (-) | 3.51 (3.48) | 2.18 (2.05) | 2.10 (-) | 2.05 (2.14) | 2.1 (2.5b) | 2.6 (-) | 3.0 (1.9b) |
a Average of chains A and B is shown.
b Unrealistically short Fe/Mn–O distances (≤1.72 Å) in the crystal structures were replaced by average bond lengths of the remaining Fe/Mn–N/O distances in the BVS calculations.
The bond length differences between reduced and oxidized states derived from EXAFS are so small that they fall within the range of the coordinate error of the crystal structures (Table 2). Although the mean Mn/Fe–N/O bond lengths in all crystal structures are similar to the bond lengths in the reduced state XAS samples, and the BVS values are consequently close to 2 (Table 5), we can therefore not safely conclude that the crystal structures reflect the predominant presence of divalent metal ions in both reduced and (initially) oxidized R2lox, although, as noted above, this is very likely the case. The global cofactor configurations, however, including the metal-metal distance, are not affected much by xpr, as revealed by the EXAFS data. The arrangement of amino acid ligands around the metal ions in the oxidized state crystal structures thus accurately represents the oxidized state. The EXAFS data also show, in agreement with the crystallographic data, that the overall structures of the Mn/Fe and Fe/Fe clusters are rather similar (Fig. 6). It appears that the presence of a manganese ion at site 1 changes the cofactor structure only insignificantly in comparison with a diiron center.
FIGURE 6.
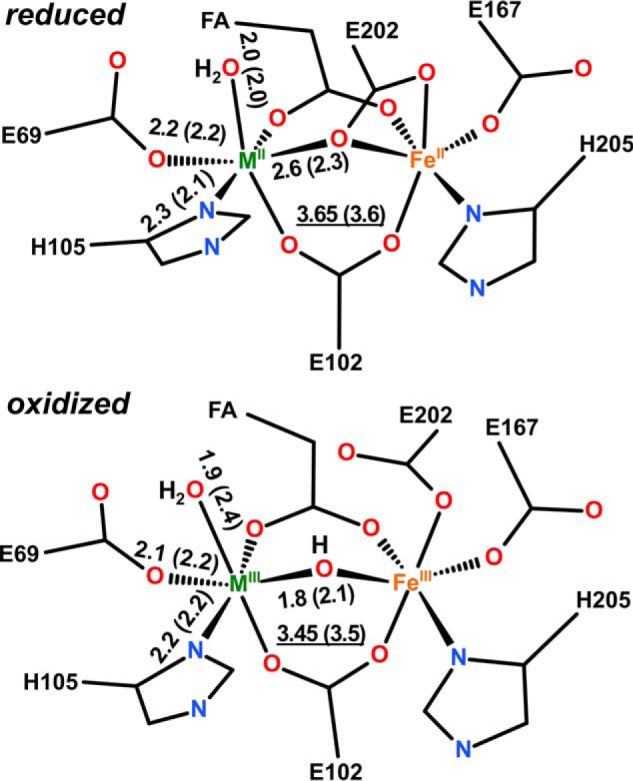
R2lox cofactor structures combining crystallographic and XAS results. Global structures of reduced (top) and oxidized (bottom) metal centers were based on crystallographic data (M, manganese or iron; FA, fatty acid ligand). Approximate metal-ligand and metal-metal (underlined) distances (in Å) are mean values for Fe/Fe and Mn/Fe centers from XAS or crystallography (in parentheses). The distance spread includes the Debye-Waller parameters (Table 4). Bond orders were neglected in the drawing for clarity. Protonation states of terminal and metal-bridging oxygen species are based on EPR assignments (22, 29).
There is also evidence for other (minor) cofactor structures in the XAS samples. These structures may be more similar to the ones observed in classical RNR R2 proteins. The XANES revealed small FeIV and MnIV contributions in the oxidized samples, which may remain due to incomplete reduction of MnIV/FeIV centers to MIII/FeIII centers. In reduced R2lox, potential metal-metal distances around ∼4.2 Å may correspond to cofactors that lack the fatty acid ligand, so that a center with only one bridging carboxylate (Glu-102) is formed, such as conformation B in Fig. 3F, as observed in many crystal structures of standard R2 proteins (see Refs. 55, 56 and references therein). This agrees with the structural heterogeneity observed in the crystal structures of reduced Mn/Fe and diiron states (Fig. 3). In oxidized R2lox, metal-metal distances of ∼3 Å may also correspond to (minor amounts of) R2-like centers formed in the absence of the fatty acid, which contain two metal-bridging μO(H) groups. (An ∼3-Å metal distance is typical for R2 proteins containing MIII2 clusters (55, 56).) An in-depth characterization of such minor cofactor species, however, would require samples in which they are quantitatively populated.
Tyrosine-Valine Ether Cross-link Formation in R2lox with Different Metal Cofactors
As described previously, upon oxygen activation the Mn/Fe cofactor of R2lox catalyzes formation of an ether cross-link between the phenolic oxygen of Tyr-162 and the Cβ of Val-72 (Figs. 2B and 7) (22). This cross-link is absent in the nonactivated reduced state, and instead the hydroxyl group of Tyr-162 is hydrogen-bonded to the carbonyl oxygen of Val-72 (Figs. 2A and 7). It should be noted that the absence of the cross-link in the reduced state is due to the fact that this structure was obtained by soaking apoprotein crystals with manganese and iron under anoxic conditions, i.e. the protein was never exposed to metal ions and oxygen at the same time. Cross-link formation is unlikely to be reversible (22).
FIGURE 7.

Tyrosine-valine ether cross-link formation catalyzed by different metal cofactors in R2lox. mFo − DFc omit electron density for residues Tyr-162 and Val-72 in apoprotein crystals soaked with manganese and/or iron in the absence or presence of oxygen for the indicated durations, and in protein isolated in metal-bound form, contoured at 3.0 σ. The cross-link is absent when the cofactor is reconstituted in the absence of oxygen, whereas in the presence of oxygen it is formed by both the Mn/Fe and the Fe/Fe cofactor, but not the Mn/Mn cluster. For clarity, the link is modeled here where the density shows it is present, but poor density fits and too long refined bond lengths indicate it is only partially formed in soaked apoprotein crystals. Quantification of the cross-link by mass spectrometry shows that it is formed equally efficiently by the Mn/Fe and the Fe/Fe cofactor (data not shown).
Because the dimanganese center cannot activate oxygen, it does not form the cross-link, as expected (Figs. 4C and 7). In the oxidized Fe/Fe state, electron density for the cross-link is present, but not strong enough to warrant modeling the link (Figs. 4B and 7). Cross-link formation during soaking of apoprotein crystals appears to be less efficient than in the cell, likely due to suboptimal reaction conditions. Electron density for the link is observed in apoprotein crystals that were soaked with iron or manganese and iron under aerobic conditions (Fig. 7), but when restrained to the optimal distance of 1.45 ± 0.02 Å, the cross-link does not fit the density well, and the bond refines to unrealistically long distances, indicating that the cross-link is not present in all protein molecules in the crystal. In contrast, with the same restraint a good density fit and refined bond distance of 1.52 Å were obtained for protein that was produced and purified in metal-bound form (Figs. 2B and 7) (22).
We used mass spectrometry to verify that the cross-link is indeed formed by both the Mn/Fe and the Fe/Fe cofactor. As shown previously (22), the precursor ion 689.0381 m/z, corresponding to the tyrosine-valine ether cross-linked peptideAVIRAATVYNMIVR-AVTLD (where the underlined residues are the cross-linked Tyr and Val) obtained by proteolytic digestion of R2lox with Glu-C, fragments into a series of b and y ions in MS2 spectra, including four ions containing the valinyl carbocation. These carbocations arise from the gas-phase cleavage of the ether bond between the tyrosine and valine side chains. The MS2 spectra were practically identical in Mn/Fe and iron-only samples, demonstrating that the cross-linked peptide was found in both conditions (data not shown). The amount of cross-linked peptide in R2lox varies somewhat from sample to sample, but is, within error, equal in Mn/Fe and iron-only samples, indicating that, at least under the conditions used in our assay, both cofactors are equally capable of catalyzing cross-link formation.
Likely Routes for Gated Oxygen and Substrate Access to the Active Site
In the reduced Mn/Fe and Fe/Fe state, a solvent-accessible hydrophobic channel is observed leading from the protein surface to metal site 2. This channel is occluded in the oxidized state (Fig. 8A). It may be the access channel for molecular oxygen, suggesting that oxygen first binds to the metal ion in site 2, in agreement with the structural mobility observed at this site in the reduced state. The channel appears to be gated by the oxidation state-dependent carboxylate shift of Glu-202 together with a rotamer shift in Ile-206. Although the “closed” conformation of Ile-206 is clearly defined in the oxidized state, in the reduced state the electron density is less clear, indicating that, like the site 2 metal ligand Glu-167, Ile-206 displays some structural mobility in the reduced state. Only the “open” conformation was modeled, but both may in fact be present. The crystal structures thus indicate that metal site 2 and the channel approaching it are structurally dynamic in the reduced state but not in the oxidized state.
FIGURE 8.

Likely routes for gated oxygen and substrate access to the active site. A, residues Glu-202 and Ile-206 of R2lox gate a hydrophobic channel leading from the protein surface to metal site 2. Rotamer changes open the channel in the reduced state (left) and close it in the oxidized state (right). B, residues Leu-61 and Phe-178 also adopt different rotamers in the reduced (left) and oxidized (right) state, creating a pocket branching off of the ligand-binding tunnel in the reduced state. C, two entrances to the ligand-binding pocket are gated by a flexible loop. They are open in the reduced Mn/Fe state (left) but occluded in the oxidized Mn/Fe state (right), whereas in the other metallation states they are found in various combinations of open and closed (data not shown). The tunnel entrances are labeled 1 and 2, and the pockets/channels shown in A and B are indicated. The same color scheme is used as in Fig. 2. Shown in gray is the molecular surface of R2lox, as calculated by CASTp (106) using a 1.4 Å probe radius.
Another apparently redox state-dependent rotamer switch occurs farther away from the active site, involving residues Leu-61 and Phe-178 that flank the ligand-binding tunnel at the upper end, at the backbone of Tyr-175 (Fig. 8B). This switch is likely to be coupled to the concerted water ligand–Tyr-175–Leu-198 movement. It does not appear to cause larger conformational changes, but it is tempting to speculate that it relays the redox state-dependent conformational changes of the metal ligands to the protein surface to control substrate binding.
The ligand-binding channel on the whole is somewhat remodeled in the different metallation and redox states (Fig. 8C). In the reduced Mn/Fe state, two entrances to the tunnel can be discerned, both of which appear to be gated by a flexible loop between helices G and H (Figs. 8C and 9, A and B). Small rotamer shifts in the loop residues open and close both entrances. Because of the very high B factors of this region in the crystal structures (Fig. 9A), its electron density is poorly defined, and the conformations of side chains in the loop cannot reliably be determined. We can therefore not ascertain whether the tunnel entrances are accessible or occluded depending on the redox state. However, as this loop is by far the most mobile region in the crystal structures, it appears likely that it controls substrate access.
FIGURE 9.
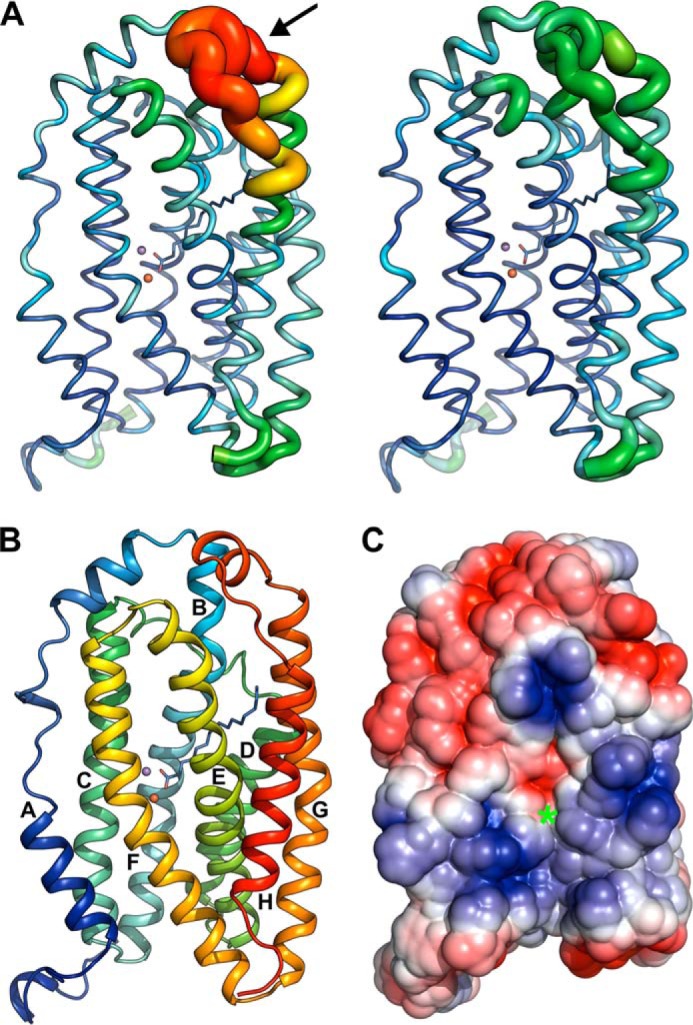
A, B factors of Cα-atoms in the reduced (left) and oxidized (right) Mn/Fe-bound state of R2lox. B factors are indicated both by the color range from dark blue, 17 Å2, to red, 127 Å2, and the thickness of the ribbon. The fatty acid ligand and metal ions are shown as stick model and spheres, respectively. The loop gating the ligand-binding tunnel entrances is indicated by an arrow. B, structure of the R2lox protomer in the oxidized Mn/Fe-bound state in rainbow coloring, with helices numbered according to R2 convention (note that the secondary structure assignment algorithm of PyMOL was used; this assignment differs slightly from the DSSP assignment). The 15 C-terminal residues are disordered. The dimer interface is formed by the N terminus and helices A–C (see Fig. 10C). The metal ions are depicted as spheres, and the fatty acid ligand as a stick model. C, electrostatic surface potential of the R2lox protomer in the oxidized Mn/Fe-bound state in the same orientation as in A and B. The entrance of the proposed oxygen channel is marked with a green asterisk. Positively charged regions are colored blue, negatively charged regions red, and neutral regions white.
Interestingly, in the dimanganese state Tyr-175, Leu-198, Leu-61, Phe-198, and Ile-206 adopt the same position as in the oxidized Mn/Fe and Fe/Fe states (Figs. 4C and 8, A and B). The proposed oxygen channel is thus closed, although Glu-202 is in a reduced state conformation. The fatty acid ligand, Glu-167 and Ile-206 also appear to be less mobile in the Mn/Mn structure than in the reduced Mn/Fe and Fe/Fe states. Because the Mn/Mn structure was obtained from an aerobically soaked crystal, closure of the oxygen channel and the concerted water ligand–Tyr-175–Leu-198–Leu-61–Phe-198 movement may therefore be coupled with oxygen binding rather than activation. (Unfortunately electron density for the water ligand, although it is most likely present, is not observed in the Mn/Mn structure.) Hence, the Mn/Mn state might represent an intermediate conformation in the oxygen binding and activation reaction.
Metal Binding Does Not Cause Larger Conformational Changes in R2lox
On a larger scale, the structural differences between the reduced and oxidized state crystal structures of R2lox are minor. A morph between the two states (supplemental Movie S1) illustrates that the helix bundle contracts slightly along the helical axis upon reduction. This compaction is mainly caused by the carboxylate shift of Glu-202 in helix F (see Fig. 9B for helix numbering). Helix B, containing the cross-link residue Val-72, is also more contracted in the absence of the cross-link. Once the cross-link is formed, its movement is likely to be more constrained during further redox cycles of the protein.
To assess whether metal binding and oxygen activation cause larger conformational changes in solution that might be prevented by packing interactions in crystals, we subjected R2lox, both in the metal-free and the Mn/Fe-bound state, to SAXS experiments (Fig. 10). No differences between the metal-free and metal-bound states can be observed in the scattering profiles (Fig. 10B). The SAXS data are thus in agreement with the relatively minor conformational changes accompanying metal binding and oxygen activation in crystals. The molecular mass of R2lox estimated from the SAXS data corresponds to that of a dimer. R2 proteins form dimers that interact with the dimeric R1 subunit for radical transfer to the active site in R1 (5, 61–63). The extensive dimer interface is conserved in R2lox proteins (∼2700 Å2 or 20% of the total surface area (64)), and the same dimeric assembly is found in crystal structures (21, 22, 65). A theoretical scattering profile calculated of the dimer observed in the crystal packing matches the observed scattering profile (Fig. 10B). Envelope reconstructions yield the typical heart shape of R2 proteins, and the crystallographic dimer fits well into the SAXS envelope (Fig. 10C). Hence, this is the dimeric assembly present in solution.
FIGURE 10.

Small angle x-ray scattering analysis of R2lox. A, quality assessment of SAXS data of Mn/Fe-R2lox at 9.1 mg/ml. Top, Guinier plot (ln I(s) versus s2) with linear regression (for points shown in red) used for determination of Rg (Rg = 2.95 ± 0.09 nm) and residuals (blue) of the linear regression. All samples in the concentration series yielded the same Rg value within error. Bottom, distance distribution function calculated with Dmax = 10.60 nm, used for solution envelope reconstructions. B, scattering profiles of the metal-free (blue) and Mn/Fe-bound (black) states of R2lox in solution in comparison with the profile calculated from the crystallographic model of the R2lox dimer (red, Chi2 = 1.05). C, solution envelope reconstruction of R2lox, superimposed with the crystallographic model of the R2lox dimer in the oxidized Mn/Fe-bound state. The SAXS envelope is shown as gray mesh, the atomic resolution model is depicted in schematic representation colored by chain. Metal ions are shown as spheres and the fatty acid ligand as stick model.
From the XAS and SAXS analysis together, we can therefore conclude that the main structural species of R2lox, both at the active site and the global level, are the same in solution and in crystals.
Discussion
R2lox could be described as a hybrid of BMMs and R2 proteins, as it displays some structural and functional features typical of either group of di-metal carboxylate proteins.
In class Ia and Ib R2s, two glutamate side chains, corresponding to Glu-167 and Glu-202 of R2lox, rotate upon oxidation (10, 66–71), whereas in BMMs and R2lox only the C-terminal glutamate ligand (corresponding to Glu-202) moves to make room for the bridging μO(H) ligand (72–79). The same single carboxylate shift occurs in the founding member of the other group of Mn/Fe proteins, the Chlamydia trachomatis class Ic R2 protein (R2c) (20, 80). The residue corresponding to Glu-167 is a monodentate ligand to the site 2 metal ion in BMMs and Mn/Fe proteins, whereas it shifts from bidentate to monodentate ligation upon oxidation in class Ia and Ib R2 proteins. Interestingly, the crystallographic data of reduced state R2lox can also be interpreted such that Glu-167 coordinates the iron ion in bidentate mode (as in class Ia and Ib R2 proteins), and the fatty acid ligand is not bound to the metal ions (Fig. 3F). Although this conformation does not explain the data satisfactorily, it may be present in a minor proportion of molecules, as also suggested by the XAS data. The reduced active site of R2lox displays a significant degree of mobility. This may explain why the protein can activate oxygen in the presence of the fatty acid ligand, as it would prevent oxygen activation if it was coordinated to both metal ions at all times. Although we presume that the mixture of fatty acids that copurifies with the protein from the heterologous expression host may not represent its physiological substrate(s), the structures discussed here are probably physiologically relevant nevertheless. It appears likely that ligand entry is gated by the flexible loop between the two long C-terminal helices αG and αH (Figs. 8C and 9, A and B). The protein may well bind its physiological substrate(s) in the reduced state, and the substrate may similarly be only weakly coordinated to the metal ions, so that oxygen activation is possible. Such a scenario is encountered in the distantly related cyanobacterial aldehyde-deformylating oxygenase, where oxygen activation is triggered by substrate binding (28).
In RNR R2 proteins, the C terminus interacts with the R1 subunit for radical delivery but is disordered in the absence of R1 (5). A similar functional order-disorder switch has been suggested for the C terminus of R2lox. The C terminus of the Mycobacterium tuberculosis R2lox (MtR2lox) is disordered in the original crystal structure (21), but it forms an additional α-helix in a different crystal form (65). In the G. kaustophilus R2lox (GkR2loxI), the last 15 residues are not seen in the electron density. The additional helix in MtR2lox shields a large concave, positively charged surface patch on the protein. The equivalent surface of GkR2loxI is also concave and positively charged (Fig. 9C), suggesting that its C terminus might similarly form a helix interacting with this patch under certain conditions. Packing interactions may prevent it from forming in the crystals. The positively charged concave surface patch on R2lox might interact with other proteins such as a reductase that reduces the metal cofactor after turnover, or the cell membrane, for example for substrate delivery. The C-terminal helix could be used to shield the surface in the absence of the cognate interaction partner to prevent unwanted interactions.
Notably, this basic patch on R2lox surrounds the hydrophobic channel leading from the protein surface to metal site 2 (Figs. 8A and 9C). Channels proposed to allow access of the oxidant to the metal center are present in the equivalent position in class Ia and Ib R2 proteins (81–84). In the recently solved structure of R2c in the reduced state, there is no such channel, but the protein surface displays a cavity in the same position as R2lox, with only Ile-231 (corresponding to Ile-206 in R2lox) blocking access to site 2 (80). Thus, the structures of R2lox presented here are in line with proposals made for other di-metal carboxylate proteins that oxygen first binds to site 2 (10, 67, 80–83, 85). The proposed oxygen channel in R2lox is accessible in the reduced state, but occluded in the oxidized state, suggesting that oxygen access is controlled by the redox state of the metal center and may additionally be gated by the putative C-terminal helix acting as a redox state-dependent switch. It could, for example, only allow interaction with a reductase in the oxidized post-turnover state. The C terminus of R2lox may thus act similarly to the regulatory subunit that controls access of oxygen, substrates, and electrons to the active site of BMMs in an oxidation state-dependent manner (86–89).
The metal ions likely gain access to the active site through other channels, as both the channel leading to site 2 and the ligand-binding tunnel are hydrophobic and occluded in the apoprotein, whereas site 1 is disordered in the absence of metals, leaving the active site solvent accessible from this direction (22).
The metal-metal distance in the different redox states of R2lox is more similar to BMMs than to class Ia and Ib R2s. In these latter two groups, the metal-metal distance increases quite drastically from ∼3.2–3.5 Å in the MIII2 state to ∼3.9 Å in the MII2 state (10, 67–70, 81–83, 90–93). In R2lox, however, it is only slightly larger in the reduced than in the oxidized state (Table 5), as is also the case in BMMs (72–79, 94–96). Similarly, in R2c the metal-metal distance increases by only 0.1–0.2 Å upon reduction, although the metal ions are much closer to each other in this protein than in R2lox, with a distance of 3.2 Å in the MnII/FeII state (20, 21, 55–57, 80). It would appear that the active site geometry is more restrained in the Mn/Fe proteins and BMMs than in class Ia and Ib R2 proteins. This is probably caused by the π-helical geometry of the segment of helix E, which spans residues 159–172 and contains the metal ligand Glu-167 and the cross-link residue Tyr-162 in R2lox (Fig. 9B). Helix E is π-helical in both the oxidized and the reduced state in R2lox and R2c (20, 22, 80), and consequently also in the absence of the ether cross-link, whereas in BMMs, helix E forms a π-helix upon binding of the regulatory subunit (78, 79). The cross-link in R2lox appears to impose additional strain and probably enforces this rigid geometry during catalysis to prevent unwanted side reactions. It is possibly this geometry that prevents Glu-167 from shifting to a bidentate coordination of the iron ion in site 2 in the reduced state. In class Ia and Ib R2s, helix E adopts a regular α-helical structure, and the metal ligand corresponding to Glu-167 shifts to bidentate coordination mode upon reduction (66–71). As noted above, this shift does not occur in R2c or R2lox (20, 22, 80) nor in BMMs (72–79). This difference between class Ia and Ib R2 proteins on the one hand and R2c, R2lox, and BMMs on the other hand correlates with the different nature of their N-terminal metal ligand, an aspartate in the former group and a glutamate in the latter, thus providing the class Ia and Ib R2 proteins with an overall more flexible active site. Notably, these differences do not correlate either with cofactor composition or one-electron versus two-electron redox reactivity of the cofactors. They do, however, correlate with the presence or absence of a tyrosyl radical in the active enzyme. The active state of class Ia and Ib R2 proteins is MIII2-Y• (10, 11, 13, 18, 19, 97), whereas in R2c it is the MnIV/FeIII state (14), and in BMMs and (most likely) R2lox it is the MIV2 state (22, 96, 98). The tyrosine that is oxidized in class Ia and Ib R2s is replaced by a phenylalanine in R2c and R2lox and a glutamine in BMMs. This residue (Phe-109 in R2lox) is situated in helix C, together with the N-terminal (D/E)XXH metal ligand motif. Therefore, it seems likely that the more rigid active site geometry of R2c, R2lox, and BMMs is required to stabilize the cofactor in the active high redox state and prevent unwanted side reactions.
The data presented here demonstrate that the active site structure of R2lox is essentially the same regardless of cofactor composition. R2lox does not display the metal type-dependent carboxylate shifts that are observed in class Ib R2 (NrdF) proteins (10, 70, 81, 84). NrdF proteins are active with a diiron cofactor (19, 70, 99), but most likely use a dimanganese cofactor in vivo (10–13, 100–103). Because this cofactor cannot activate oxygen, they require an additional flavodoxin subunit, NrdI, that delivers a reduced form of oxygen to the dimanganese center in NrdF (19, 81, 104, 105). The metal-dependent carboxylate shifts in NrdF are likely coupled to the different access pathways for the respective oxidant (81, 82, 84). The situation in R2lox is different, as both the Mn/Fe and the diiron cofactor are capable of activating oxygen and catalyzing formation of the ether cross-link. The structural similarity between the two types of cofactors suggests that these reactions proceed via similar mechanisms at the Mn/Fe and the diiron cluster. Formation of the inactive Mn/Mn cluster seems to be strongly disfavored (22), and due to the lack of activation it would remain weakly bound and could easily be displaced. It is therefore possible that in vivo, R2lox functions with either a diiron or a Mn/Fe cluster depending on metal availability. However, given the differences in redox potential between manganese and iron, the different metal clusters might also be utilized for different chemical reactions (53). R2lox proteins may represent a much simpler scaffold capable of hydrocarbon oxidation than the complex multisubunit BMMs. It is therefore of great interest to uncover their catalytic capabilities.
Author Contributions
J. J. G. performed protein production, purification, and crystallization, collected, and analyzed x-ray crystallographic and SAXS data, and prepared samples for spectroscopy. R. K., P. S., and M. Haumann collected and analyzed XAS data. R. M. M. B. and J. L. performed MS analysis of the cross-link. A. N. performed MS analysis of the fatty acid ligand. J. J. G. and M. Högbom designed the study and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank the staff at beamlines PX14.1/BESSY, ID23-2/ESRF, X06SA/SLS, Samba/SOLEIL, and I911-SAXS/Max II for technical assistance. We gratefully acknowledge Gregor Witte and Henryk Korza for help with SAXS data analysis, Pavel Afonine for advice on crystal structure refinements, Daniel Lundin for helpful discussions, and Matthew Bennett for critical reading of the manuscript.
The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Movie S1.
- BMM
- bacterial multicomponent monooxygenase
- BVS
- bond valence sum
- EXAFS
- extended x-ray absorption fine structure
- FA
- formic acid
- FT
- Fourier transform
- GkR2loxI
- R2lox protein I from Geobacillus kaustophilus, referred to as R2lox throughout
- MtR2lox
- R2lox protein from Mycobacterium tuberculosis
- RNR
- ribonucleotide reductase
- R2c
- class Ic RNR R2 protein
- R2lox
- R2-like ligand-binding oxidase
- SAXS
- small angle x-ray scattering
- TXRF
- total reflection X-ray fluorescence
- XANES
- x-ray absorption near edge structure
- XAS
- x-ray absorption spectroscopy
- xpr
- x-ray photoreduction.
References
- 1. Lundin D., Poole A. M., Sjöberg B. M., Högbom M. (2012) Use of structural phylogenetic networks for classification of the ferritin-like superfamily. J. Biol. Chem. 287, 20565–20575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nordlund P., Eklund H. (1995) Di-iron-carboxylate proteins. Curr. Opin. Struct. Biol. 5, 758–766 [DOI] [PubMed] [Google Scholar]
- 3. Theil E. C., Matzapetakis M., Liu X. (2006) Ferritins: iron/oxygen biominerals in protein nanocages. J. Biol. Inorg. Chem. 11, 803–810 [DOI] [PubMed] [Google Scholar]
- 4. Bradley J. M., Moore G. R., Le Brun N. E. (2014) Mechanisms of iron mineralization in ferritins: one size does not fit all. J. Biol. Inorg. Chem. 19, 775–785 [DOI] [PubMed] [Google Scholar]
- 5. Nordlund P., Reichard P. (2006) Ribonucleotide reductases. Annu. Rev. Biochem. 75, 681–706 [DOI] [PubMed] [Google Scholar]
- 6. Sazinsky M. H., Lippard S. J. (2006) Correlating structure with function in bacterial multicomponent monooxygenases and related diiron proteins. Acc. Chem. Res. 39, 558–566 [DOI] [PubMed] [Google Scholar]
- 7. Hofer A., Crona M., Logan D. T., Sjöberg B. M. (2012) DNA building blocks: keeping control of manufacture. Crit. Rev. Biochem. Mol. Biol. 47, 50–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Notomista E., Lahm A., Di Donato A., Tramontano A. (2003) Evolution of bacterial and archaeal multicomponent monooxygenases. J. Mol. Evol. 56, 435–445 [DOI] [PubMed] [Google Scholar]
- 9. Stubbe J. (2003) Di-iron-tyrosyl radical ribonucleotide reductases. Curr. Opin. Chem. Biol. 7, 183–188 [DOI] [PubMed] [Google Scholar]
- 10. Cox N., Ogata H., Stolle P., Reijerse E., Auling G., Lubitz W. (2010) A tyrosyl-dimanganese coupled spin system is the native metalloradical cofactor of the R2F subunit of the ribonucleotide reductase of Corynebacterium ammoniagenes. J. Am. Chem. Soc. 132, 11197–11213 [DOI] [PubMed] [Google Scholar]
- 11. Stolle P., Barckhausen O., Oehlmann W., Knobbe N., Vogt C., Pierik A. J., Cox N., Schmidt P. P., Reijerse E. J., Lubitz W., Auling G. (2010) Homologous expression of the nrdF gene of Corynebacterium ammoniagenes strain ATCC 6872 generates a manganese-metallocofactor (R2F) and a stable tyrosyl radical (Y) involved in ribonucleotide reduction. FEBS J. 277, 4849–4862 [DOI] [PubMed] [Google Scholar]
- 12. Cotruvo J. A., Stubbe J. (2011) Escherichia coli class Ib ribonucleotide reductase contains a dimanganese(III)-tyrosyl radical cofactor in vivo. Biochemistry 50, 1672–1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang Y., Stubbe J. (2011) Bacillus subtilis class Ib ribonucleotide reductase is a dimanganese(III)-tyrosyl radical enzyme. Biochemistry 50, 5615–5623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jiang W., Yun D., Saleh L., Barr E. W., Xing G., Hoffart L. M., Maslak M. A., Krebs C., Bollinger J. M. Jr. (2007) A manganese(IV)/iron(III) cofactor in Chlamydia trachomatis ribonucleotide reductase. Science 316, 1188–1191 [DOI] [PubMed] [Google Scholar]
- 15. Voevodskaya N., Lendzian F., Ehrenberg A., Gräslund A. (2007) High catalytic activity achieved with a mixed manganese-iron site in protein R2 of Chlamydia ribonucleotide reductase. FEBS Lett. 581, 3351–3355 [DOI] [PubMed] [Google Scholar]
- 16. Andersson C. S., Öhrström M., Popović-Bijelić A., Gräslund A., Stenmark P., Högbom M. (2012) The manganese ion of the heterodinuclear Mn/Fe cofactor in Chlamydia trachomatis ribonucleotide reductase R2c is located at metal position 1. J. Am. Chem. Soc. 134, 123–125 [DOI] [PubMed] [Google Scholar]
- 17. Dassama L. M., Boal A. K., Krebs C., Rosenzweig A. C., Bollinger J. M. Jr. (2012) Evidence that the β subunit of Chlamydia trachomatis ribonucleotide reductase is active with the manganese ion of its manganese(IV)/iron(III) cofactor in site 1. J. Am. Chem. Soc. 134, 2520–2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sjöberg B. M., Reichard P. (1977) Nature of the free radical in ribonucleotide reductase from Escherichia coli. J. Biol. Chem. 252, 536–541 [PubMed] [Google Scholar]
- 19. Cotruvo J. A. Jr., Stubbe J. (2010) An active dimanganese(III)-tyrosyl radical cofactor in Escherichia coli class Ib ribonucleotide reductase. Biochemistry 49, 1297–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Högbom M., Stenmark P., Voevodskaya N., McClarty G., Gräslund A., Nordlund P. (2004) The radical site in chlamydial ribonucleotide reductase defines a new R2 subclass. Science 305, 245–248 [DOI] [PubMed] [Google Scholar]
- 21. Andersson C. S., Högbom M. (2009) A Mycobacterium tuberculosis ligand-binding Mn/Fe protein reveals a new cofactor in a remodeled R2-protein scaffold. Proc. Natl. Acad. Sci. U.S.A. 106, 5633–5638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Griese J. J., Roos K., Cox N., Shafaat H. S., Branca R. M., Lehtiö J., Gräslund A., Lubitz W., Siegbahn P. E., Högbom M. (2013) Direct observation of structurally encoded metal discrimination and ether bond formation in a heterodinuclear metalloprotein. Proc. Natl. Acad. Sci. U.S.A. 110, 17189–17194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Högbom M. (2010) The manganese/iron-carboxylate proteins: what is what, where are they, and what can the sequences tell us? J. Biol. Inorg. Chem. 15, 339–349 [DOI] [PubMed] [Google Scholar]
- 24. Schirmer A., Rude M. A., Li X., Popova E., del Cardayre S. B. (2010) Microbial biosynthesis of alkanes. Science 329, 559–562 [DOI] [PubMed] [Google Scholar]
- 25. Warui D. M., Li N., Nørgaard H., Krebs C., Bollinger J. M. Jr., Booker S. J. (2011) Detection of formate, rather than carbon monoxide, as the stoichiometric coproduct in conversion of fatty aldehydes to alkanes by a cyanobacterial aldehyde decarbonylase. J. Am. Chem. Soc. 133, 3316–3319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li N., Nørgaard H., Warui D. M., Booker S. J., Krebs C., Bollinger J. M. Jr. (2011) Conversion of fatty aldehydes to alka(e)nes and formate by a cyanobacterial aldehyde decarbonylase: cryptic redox by an unusual dimetal oxygenase. J. Am. Chem. Soc. 133, 6158–6161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li N., Chang W. C., Warui D. M., Booker S. J., Krebs C., Bollinger J. M. Jr. (2012) Evidence for only oxygenative cleavage of aldehydes to alk(a/e)nes and formate by cyanobacterial aldehyde decarbonylases. Biochemistry 51, 7908–7916 [DOI] [PubMed] [Google Scholar]
- 28. Pandelia M. E., Li N., Nørgaard H., Warui D. M., Rajakovich L. J., Chang W. C., Booker S. J., Krebs C., Bollinger J. M. Jr. (2013) Substrate-triggered addition of dioxygen to the diferrous cofactor of aldehyde-deformylating oxygenase to form a diferric-peroxide intermediate. J. Am. Chem. Soc. 135, 15801–15812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shafaat H. S., Griese J. J., Pantazis D. A., Roos K., Andersson C. S., Popović-Bijelić A., Gräslund A., Siegbahn P. E., Neese F., Lubitz W., Högbom M., Cox N. (2014) Electronic structural flexibility of heterobimetallic Mn/Fe cofactors: R2lox and R2c proteins. J. Am. Chem. Soc. 136, 13399–13409 [DOI] [PubMed] [Google Scholar]
- 30. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., Adams P. D. (2012) Towardautomated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dolinsky T. J., Nielsen J. E., McCammon J. A., Baker N. A. (2004) PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 39. Klockenkämper R. (1997) in Chemical Analysis (Winefordner J. D., ed) Vol. 140, John Wiley & Sons, Inc., New York [Google Scholar]
- 40. Reschke S., Niks D., Wilson H., Sigfridsson K. G., Haumann M., Rajagopalan K. V., Hille R., Leimkühler S. (2013) Effect of exchange of the cysteine molybdenum ligand with selenocysteine on the structure and function of the active site in human sulfite oxidase. Biochemistry 52, 8295–8303 [DOI] [PubMed] [Google Scholar]
- 41. Reschke S., Sigfridsson K. G., Kaufmann P., Leidel N., Horn S., Gast K., Schulzke C., Haumann M., Leimkühler S. (2013) Identification of a bis-molybdopterin intermediate in molybdenum cofactor biosynthesis in Escherichia coli. J. Biol. Chem. 288, 29736–29745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dau H., Liebisch P., Haumann M. (2003) X-ray absorption spectroscopy to analyze nuclear geometry and electronic structure of biological metal centers–potential and questions examined with special focus on the tetra-nuclear manganese complex of oxygenic photosynthesis. Anal. Bioanal. Chem. 376, 562–583 [DOI] [PubMed] [Google Scholar]
- 43. Zabinsky S. I., Rehr J. J., Ankudinov A., Albers R. C., Eller M. J. (1995) Multiple-scattering calculations of x-ray-absorption spectra. Phys. Rev. B Condens. Matter 52, 2995–3009 [DOI] [PubMed] [Google Scholar]
- 44. Liu W. T., Thorp H. H. (1993) Bond valence sum analysis of metal-ligand bond lengths in metalloenzymes and model complexes. 2. Refined distances and other enzymes. Inorg. Chem. 32, 4102–4105 [Google Scholar]
- 45. Wiśniewski J. R., Zougman A., Nagaraj N., Mann M. (2009) Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 [DOI] [PubMed] [Google Scholar]
- 46. Konarev P. V., Petoukhov M. V., Volkov V. V., Svergun D. I. (2006) ATSAS 2.1, a program package for small-angle scattering data analysis. J. Appl. Crystallogr. 39, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Svergun D., Barberato C., Koch M. H. (1995) CRYSOL–A program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 48. Franke D., Svergun D. I. (2009) DAMMIF, a program for rapid ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Svergun D. I., Petoukhov M. V., Koch M. H. (2001) Determination of domain structure of proteins from x-ray solution scattering. Biophys. J. 80, 2946–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kozin M. B., Svergun D. I. (2001) Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 34, 33–41 [Google Scholar]
- 51. Volkov V. V., Svergun D. I. (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wriggers W., Chacon P. (2001) Using Situs for the registration of protein structures with low-resolution bead models from x-ray solution scattering. J. Appl. Crystallogr. 34, 773–776 [Google Scholar]
- 53. Roos K., Siegbahn P. E. (2011) Oxygen cleavage with manganese and iron in ribonucleotide reductase from Chlamydia trachomatis. J. Biol. Inorg. Chem. 16, 553–565 [DOI] [PubMed] [Google Scholar]
- 54. Bollinger J. M., Chen S. X., Parkin S. E., Mangravite L. M., Ley B. A., Edmondson D. E., Huynh B. H. (1997) Differential iron(II) affinity of the sites of the diiron cluster in protein R2 of Escherichia coli ribonucleotide reductase: tracking the individual sites through the O-2 activation sequence. J. Am. Chem. Soc. 119, 5976–5977 [Google Scholar]
- 55. Leidel N., Popović-Bijelić A., Havelius K. G., Chernev P., Voevodskaya N., Gräslund A., Haumann M. (2012) High-valent [MnFe] and [FeFe] cofactors in ribonucleotide reductases. Biochim. Biophys. Acta 1817, 430–444 [DOI] [PubMed] [Google Scholar]
- 56. Sigfridsson K. G., Chernev P., Leidel N., Popovic-Bijelic A., Gräslund A., Haumann M. (2013) Rapid x-ray photoreduction of dimetal-oxygen cofactors in ribonucleotide reductase. J. Biol. Chem. 288, 9648–9661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Voevodskaya N., Lendzian F., Sanganas O., Grundmeier A., Gräslund A., Haumann M. (2009) Redox intermediates of the Mn-Fe Site in subunit R2 of Chlamydia trachomatis ribonucleotide reductase: an x-ray absorption and EPR study. J. Biol. Chem. 284, 4555–4566 [DOI] [PubMed] [Google Scholar]
- 58. Dau H., Haumann M. (2008) The manganese complex of photosystem II in its reaction cycle–basic framework and possible realization at the atomic level. Coord. Chem. Rev. 252, 273–295 [Google Scholar]
- 59. Grabolle M., Haumann M., Müller C., Liebisch P., Dau H. (2006) Rapid loss of structural motifs in the manganese complex of oxygenic photosynthesis by x-ray irradiation at 10–300 K. J. Biol. Chem. 281, 4580–4588 [DOI] [PubMed] [Google Scholar]
- 60. Younker J. M., Krest C. M., Jiang W., Krebs C., Bollinger J. M. Jr., Green M. T. (2008) Structural analysis of the Mn(IV)/Fe(III) cofactor of Chlamydia trachomatis ribonucleotide reductase by extended x-ray absorption fine structure spectroscopy and density functional theory calculations. J. Am. Chem. Soc. 130, 15022–15027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Thelander L. (1973) Physicochemical characterization of ribonucleoside diphosphate reductase from Escherichia coli. J. Biol. Chem. 248, 4591–4601 [PubMed] [Google Scholar]
- 62. Cotruvo J. A., Stubbe J. (2011) Class I ribonucleotide reductases: metallocofactor assembly and repair in vitro and in vivo. Annu. Rev. Biochem. 80, 733–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ando N., Brignole E. J., Zimanyi C. M., Funk M. A., Yokoyama K., Asturias F. J., Stubbe J., Drennan C. L. (2011) Structural interconversions modulate activity of Escherichia coli ribonucleotide reductase. Proc. Natl. Acad. Sci. U.S.A. 108, 21046–21051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 65. Andersson C. S., Berthold C. L., Högbom M. (2012) A dynamic C-terminal segment in the Mycobacterium tuberculosis Mn/Fe R2lox protein can adopt a helical structure with possible functional consequences. Chem. Biodivers. 9, 1981–1988 [DOI] [PubMed] [Google Scholar]
- 66. Nordlund P., Sjöberg B. M., Eklund H. (1990) Three-dimensional structure of the free radical protein of ribonucleotide reductase. Nature 345, 593–598 [DOI] [PubMed] [Google Scholar]
- 67. Nordlund P., Eklund H. (1993) Structure and function of the Escherichia coli ribonucleotide reductase protein R2. J. Mol. Biol. 232, 123–164 [DOI] [PubMed] [Google Scholar]
- 68. Logan D. T., Su X. D., Aberg A., Regnström K., Hajdu J., Eklund H., Nordlund P. (1996) Crystal structure of reduced protein R2 of ribonucleotide reductase: the structural basis for oxygen activation at a dinuclear iron site. Structure 4, 1053–1064 [DOI] [PubMed] [Google Scholar]
- 69. Eriksson M., Jordan A., Eklund H. (1998) Structure of Salmonella typhimurium nrdF ribonucleotide reductase in its oxidized and reduced forms. Biochemistry 37, 13359–13369 [DOI] [PubMed] [Google Scholar]
- 70. Högbom M., Huque Y., Sjöberg B. M., Nordlund P. (2002) Crystal structure of the di-iron/radical protein of ribonucleotide reductase from Corynebacterium ammoniagenes. Biochemistry 41, 1381–1389 [DOI] [PubMed] [Google Scholar]
- 71. Högbom M., Galander M., Andersson M., Kolberg M., Hofbauer W., Lassmann G., Nordlund P., Lendzian F. (2003) Displacement of the tyrosyl radical cofactor in ribonucleotide reductase obtained by single-crystal high-field EPR and 1.4-A x-ray data. Proc. Natl. Acad. Sci. U.S.A. 100, 3209–3214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rosenzweig A. C., Frederick C. A., Lippard S. J., Nordlund P. (1993) Crystal structure of a bacterial non-haem iron hydroxylase that catalyses the biological oxidation of methane. Nature 366, 537–543 [DOI] [PubMed] [Google Scholar]
- 73. Rosenzweig A. C., Nordlund P., Takahara P. M., Frederick C. A., Lippard S. J. (1995) Geometry of the soluble methane monooxygenase catalytic diiron center in two oxidation states. Chem. Biol. 2, 409–418 [PubMed] [Google Scholar]
- 74. Rosenzweig A. C., Brandstetter H., Whittington D. A., Nordlund P., Lippard S. J., Frederick C. A. (1997) Crystal structures of the methane monooxygenase hydroxylase from Methylococcus capsulatus (Bath): implications for substrate gating and component interactions. Proteins 29, 141–152 [PubMed] [Google Scholar]
- 75. Elango N., Radhakrishnan R., Froland W. A., Wallar B. J., Earhart C. A., Lipscomb J. D., Ohlendorf D. H. (1997) Crystal structure of the hydroxylase component of methane monooxygenase from Methylosinus trichosporium OB3b. Protein Sci. 6, 556–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Whittington D. A., Lippard S. J. (2001) Crystal structures of the soluble methane monooxygenase hydroxylase from Methylococcus capsulatus (Bath) demonstrating geometrical variability at the dinuclear iron active site. J. Am. Chem. Soc. 123, 827–838 [DOI] [PubMed] [Google Scholar]
- 77. Sazinsky M. H., Bard J., Di Donato A., Lippard S. J. (2004) Crystal structure of the toluene/o-xylene monooxygenase hydroxylase from Pseudomonas stutzeri OX1. Insight into the substrate specificity, substrate channeling, and active site tuning of multicomponent monooxygenases. J. Biol. Chem. 279, 30600–30610 [DOI] [PubMed] [Google Scholar]
- 78. Sazinsky M. H., Dunten P. W., McCormick M. S., DiDonato A., Lippard S. J. (2006) X-ray structure of a hydroxylase-regulatory protein complex from a hydrocarbon-oxidizing multicomponent monooxygenase, Pseudomonas sp. OX1 phenol hydroxylase. Biochemistry 45, 15392–15404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bailey L. J., McCoy J. G., Phillips G. N. Jr., Fox B. G. (2008) Structural consequences of effector protein complex formation in a diiron hydroxylase. Proc. Natl. Acad. Sci. U.S.A. 105, 19194–19198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Dassama L. M., Krebs C., Bollinger J. M. Jr., Rosenzweig A. C., Boal A. K. (2013) Structural basis for assembly of the Mn(IV)/Fe(III) cofactor in the class Ic ribonucleotide reductase from Chlamydia trachomatis. Biochemistry 52, 6424–6436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Boal A. K., Cotruvo J. A. Jr., Stubbe J., Rosenzweig A. C. (2010) Structural basis for activation of class Ib ribonucleotide reductase. Science 329, 1526–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Boal A. K., Cotruvo J. A. Jr., Stubbe J., Rosenzweig A. C. (2012) The dimanganese(II) site of Bacillus subtilis class Ib ribonucleotide reductase. Biochemistry 51, 3861–3871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Voegtli W. C., Sommerhalter M., Saleh L., Baldwin J., Bollinger J. M. Jr., Rosenzweig A. C. (2003) Variable coordination geometries at the diiron(II) active site of ribonucleotide reductase R2. J. Am. Chem. Soc. 125, 15822–15830 [DOI] [PubMed] [Google Scholar]
- 84. Hammerstad M., Hersleth H. P., Tomter A. B., Røhr A. K., Andersson K. K. (2014) Crystal structure of Bacillus cereus class Ib ribonucleotide reductase di-iron NrdF in complex with NrdI. ACS Chem. Biol. 9, 526–537 [DOI] [PubMed] [Google Scholar]
- 85. Andersson M. E., Hogbom M., Rinaldo-Matthis A., Andersson K. K., Sjoberg B. M., Nordlund P. (1999) The crystal structure of an azide complex of the diferrous R2 subunit of ribonucleotide reductase displays a novel carboxylate shift with important mechanistic implications for diiron-catalyzed oxygen activation. J. Am. Chem. Soc. 121, 2346–2352 [Google Scholar]
- 86. Lee S. J., McCormick M. S., Lippard S. J., Cho U. S. (2013) Control of substrate access to the active site in methane monooxygenase. Nature 494, 380–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wang W., Iacob R. E., Luoh R. P., Engen J. R., Lippard S. J. (2014) Electron transfer control in soluble methane monooxygenase. J. Am. Chem. Soc. 136, 9754–9762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wang W., Lippard S. J. (2014) Diiron oxidation state control of substrate access to the active site of soluble methane monooxygenase mediated by the regulatory component. J. Am. Chem. Soc. 136, 2244–2247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Liang A. D., Lippard S. J. (2014) Component interactions and electron transfer in toluene/o-xylene monooxygenase. Biochemistry 53, 7368–7375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Bunker G., Petersson L., Sjöberg B. M., Sahlin M., Chance M., Chance B., Ehrenberg A. (1987) Extended x-ray absorption fine structure studies on the iron-containing subunit of ribonucleotide reductase from Escherichia coli. Biochemistry 26, 4708–4716 [DOI] [PubMed] [Google Scholar]
- 91. Scarrow R. C., Maroney M. J., Palmer S. M., Que L., Roe A. L., Salowe S. P., Stubbe J. (1987) EXAFS studies of binuclear iron proteins–hemerythrin and ribonucleotide reductase. J. Am. Chem. Soc. 109, 7857–7864 [Google Scholar]
- 92. Scarrow R. C., Maroney M. J., Palmer S. M., Que L., Salowe S. P., Stubbe J. (1986) EXAFS studies of the B2 subunit of the ribonucleotide reductase from Escherichia coli. J. Am. Chem. Soc. 108, 6832–6834 [Google Scholar]
- 93. Dassama L. M., Silakov A., Krest C. M., Calixto J. C., Krebs C., Bollinger J. M. Jr., Green M. T. (2013) A 2.8 A Fe-Fe separation in the Fe2(III/IV) intermediate, X, from Escherichia coli ribonucleotide reductase. J. Am. Chem. Soc. 135, 16758–16761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Dewitt J. G., Rosenzweig A. C., Salifoglou A., Hedman B., Lippard S. J., Hodgson K. O. (1995) X-ray absorption spectroscopic studies of the diiron center in methane monooxygenase in the presence of substrate and the coupling protein of the enzyme-system. Inorg. Chem. 34, 2505–2515 [Google Scholar]
- 95. Shu L. J., Liu Y., Lipscomb J. D., Que L. (1996) X-ray absorption spectroscopic studies of the methane monooxygenase hydroxylase component from Methylosinus trichosporium OB3b. J. Biol. Inorg. Chem. 1, 297–304 [Google Scholar]
- 96. Shu L., Nesheim J. C., Kauffmann K., Münck E., Lipscomb J. D., Que L. Jr. (1997) An Fe2IVO2 diamond core structure for the key intermediate Q of methane monooxygenase. Science 275, 515–518 [DOI] [PubMed] [Google Scholar]
- 97. Atkin C. L., Thelander L., Reichard P., Lang G. (1973) Iron and free radical in ribonucleotide reductase. Exchange of iron and Mossbauer spectroscopy of the protein B2 subunit of the Escherichia coli enzyme. J. Biol. Chem. 248, 7464–7472 [PubMed] [Google Scholar]
- 98. Lee S. K., Fox B. G., Froland W. A., Lipscomb J. D., Munck E. (1993) A transient intermediate of the methane monooxygenase catalytic cycle containing an Fe(Iv)Fe(Iv) cluster. J. Am. Chem. Soc. 115, 6450–6451 [Google Scholar]
- 99. Huque Y., Fieschi F., Torrents E., Gibert I., Eliasson R., Reichard P., Sahlin M., Sjoberg B. M. (2000) The active form of the R2F protein of class Ib ribonucleotide reductase from Corynebacterium ammoniagenes is a diferric protein. J. Biol. Chem. 275, 25365–25371 [DOI] [PubMed] [Google Scholar]
- 100. Vassinova N., Kozyrev D. (2000) A method for direct cloning of fur-regulated genes: identification of seven new fur-regulated loci in Escherichia coli. Microbiology 146, 3171–3182 [DOI] [PubMed] [Google Scholar]
- 101. Monje-Casas F., Jurado J., Prieto-Alamo M. J., Holmgren A., Pueyo C. (2001) Expression analysis of the nrdHIEF operon from Escherichia coli. Conditions that trigger the transcript level in vivo. J. Biol. Chem. 276, 18031–18037 [DOI] [PubMed] [Google Scholar]
- 102. McHugh J. P., Rodríguez-Quinoñes F., Abdul-Tehrani H., Svistunenko D. A., Poole R. K., Cooper C. E., Andrews S. C. (2003) Global iron-dependent gene regulation in Escherichia coli. A new mechanism for iron homeostasis. J. Biol. Chem. 278, 29478–29486 [DOI] [PubMed] [Google Scholar]
- 103. Gon S., Faulkner M. J., Beckwith J. (2006) In vivo requirement for glutaredoxins and thioredoxins in the reduction of the ribonucleotide reductases of Escherichia coli. Antioxid. Redox Signal. 8, 735–742 [DOI] [PubMed] [Google Scholar]
- 104. Cotruvo J. A. Jr., Stich T. A., Britt R. D., Stubbe J. (2013) Mechanism of assembly of the dimanganese-tyrosyl radical cofactor of class Ib ribonucleotide reductase: enzymatic generation of superoxide is required for tyrosine oxidation via a Mn(III)Mn(IV) intermediate. J. Am. Chem. Soc. 135, 4027–4039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Crona M., Torrents E., Røhr A. K., Hofer A., Furrer E., Tomter A. B., Andersson K. K., Sahlin M., Sjöberg B. M. (2011) NrdH-redoxin protein mediates high enzyme activity in manganese-reconstituted ribonucleotide reductase from Bacillus anthracis. J. Biol. Chem. 286, 33053–33060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Dundas J., Ouyang Z., Tseng J., Binkowski A., Turpaz Y., Liang J. (2006) CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 34, W116–W118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Karplus P. A., Diederichs K. (2012) Linking crystallographic model and data quality. Science 336, 1030–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.