

Background: PfRh4 binds complement receptor 1 to mediate malaria parasite entry into red blood cells.
Results: Monoclonal antibodies and inhibitors either block or enhance PfRh4 interaction with complement receptor 1.
Conclusion: Identification was made of critical regions and residues within PfRh4 and CR1 that mediate successful P. falciparum entry.
Significance: Understanding functional regions within PfRh4 will aid in design of vaccine subunits.
Keywords: complement, host-pathogen interaction, invasion, malaria, monoclonal antibody, plasmodium, PfRh4, complement receptor 1
Abstract
Plasmodium falciparum parasites must invade red blood cells to survive within humans. Entry into red blood cells is governed by interactions between parasite adhesins and red blood cell receptors. Previously we identified that P. falciparum reticulocyte binding protein-like homologue 4 (PfRh4) binds to complement receptor 1 (CR1) to mediate entry of malaria parasites into human red blood cells. In this report we characterize a collection of anti-PfRh4 monoclonal antibodies and CR1 protein fragments that modulate the interaction between PfRh4 and CR1. We identify an anti-PfRh4 monoclonal that blocks PfRh4-CR1 interaction in vitro, inhibits PfRh4 binding to red blood cells, and as a result abolishes the PfRh4-CR1 invasion pathway in P. falciparum. Epitope mapping of anti-PfRh4 monoclonal antibodies identified distinct functional regions within PfRh4 involved in modulating its interaction with CR1. Furthermore, we designed a set of protein fragments based on extensive mutagenesis analyses of the PfRh4 binding site on CR1 and determined their interaction affinities using surface plasmon resonance. These CR1 protein fragments bind tightly to PfRh4 and also function as soluble inhibitors to block PfRh4 binding to red blood cells and to inhibit the PfRh4-CR1 invasion pathway. Our findings can aid future efforts in designing specific single epitope antibodies to block P. falciparum invasion via complement receptor 1.
Introduction
Malaria parasites are obligate intracellular microbes exquisitely adapted for invasion and survival within the red blood cell of the host. To gain entry into red blood cells, the parasite must recognize and bind to these cells as well as activate a complex series of steps that involve multiple protein-protein interactions between parasite and host cells (for review see Ref. 1). The subsequent cycles of growth, replication of parasites, and egress from infected blood cells are responsible for the symptoms associated with malaria (for review see Ref. 2).
Parasite invasion begins with initial recognition and attachment of merozoites, the invasive form of malaria parasites, to red blood cells (3). This interaction is dynamic and involves considerable deformation of the red blood cell membrane as the parasite rolls across the host cell surface (4). After initial attachment, the merozoite orientates itself to juxtapose its apical prominence with the red blood cell surface. This allows parasite adhesins localized at the apical tip to interact with their cognate receptors to mediate irreversible attachment and commitment to invasion (5, 6). After this, a tight junction is formed between the parasite and the red blood cell membrane (7). Active invasion proceeds through an invagination of the surface and the tight junction moves from the apical to posterior pole of the merozoite, powered by the parasite's acto-myosin motor. Once the merozoite is inside the red blood cell, the red blood cell membrane is sealed behind it, completing invasion. Remarkably, the entire invasion process is accomplished within a few minutes (4).
Of the five human malaria species, Plasmodium falciparum is the most lethal. In P. falciparum, two gene families encode important parasite adhesins utilized for engagement with red blood cell receptors: erythrocyte binding-like antigens (EBAs; EBA175, EBA181, EBA140, EBL-1) (8, 9) and P. falciparum reticulocyte binding-like homolog proteins (PfRhs; Rh1, Rh2a/b, Rh4, Rh5) (10–12). During invasion these adhesins localize to the apical tip of the merozoite and bind specific receptors to initiate parasite entry into human red blood cells. Several red blood cell receptors have been identified as entry points for P. falciparum. The glycophorins were the first identified and exclusively bind members of the PfEBA2 family of proteins (glycophorin A to EBA-175, glycophorin B to EBL-1, and glycophorin C to EBA-140) (13–16). For the PfRh family of proteins, only two cognate adhesion-receptor pairs have been identified: PfRh4 to complement receptor 1 (CR1) and PfRh5 to basigin (17–19).
Molecular studies show that EBA and PfRh proteins are potential vaccine candidates because: (i) they play a crucial role in merozoite invasion; (ii) anti-EBA and anti-PfRhs antibodies inhibit parasite invasion; (iii) epidemiological studies show that most anti-EBA and anti-PfRhs antibodies in human cohort studies correlate with protection from clinical disease (1, 20–25). In addition to clinical applications, monoclonal antibodies (mAbs) have provided unique tools with which to study molecular pathways of parasite invasion, and some of the functional regions of PfRh family members have been recently characterized. Anti-PfRh1 mAbs affect rhoptry secretion and calcium signaling after merozoite attachment to erythrocytes, suggesting that PfRh1 plays a role in downstream signaling events (26). Anti-PfRh5 antibodies that interfere with the PfRh5-basigin interaction completely inhibit the ability of P. falciparum parasites to invade red blood cells (23, 27), and the mechanism by which inhibition occurs has been elucidated by the recent crystal structures of PfRh5 alone and with either its receptor basigin or neutralizing antibodies (28, 29). PfRh5 adopts a novel fold using a α-helical scaffold that provides binding sites at the tips of helices for basigin and some inhibitory monoclonal antibodies (29). The high resolution structures of PfRh5-basigin and PfRh5-mAbs binding interfaces will clearly allow future structure-guided design of inhibitory epitopes for more potent neutralizing mAbs.
Characterization of the PfRh4-CR1 invasion pathway has validated the potential of PfRh4 as a vaccine candidate (for review see Ref. 30). A soluble fragment of the PfRh4 ectodomain (rPfRh4) that encompasses the red blood cell binding region can be successfully expressed in Escherichia coli, and rabbit polyclonal antibodies raised against this fragment are able to inhibit PfRh4 binding to red blood cells (31). Furthermore, affinity-purified anti-rPfRh4 human antibodies from individuals in malaria endemic regions inhibited P. falciparum invasion via the PfRh4-CR1 pathway and correlated with protection (24). Immunization (in rabbits) with a combination of EBA-175, PfRh2a/b, and PfRh4 recombinant proteins induced antibodies that potently blocked merozoite invasion in vitro (22).
Previous work has mapped the PfRh4-interacting region on CR1 and also identified soluble forms of CR1 that are able to act as competitive inhibitors (Refs. 17, 31, and 32 and reviewed in Ref. 30). CR1 is a type one integral membrane glycoprotein composed of an N-terminal ectodomain that has a number of allelic variants, a transmembrane region, and a C-terminal cytoplasmic domain. The most common allelic variant of CR1 is composed of 28–30 structural modules called complement control protein (CCP) modules in the extracellular domain. A truncated form of CR1 (sCR1) lacking the transmembrane and cytoplasmic domain, inhibits PfRh4 binding to CR1 on the red blood cell surface (17). Clinical isolates from Kenya also demonstrated a significant utilization of CR1 for invasion of intact erythrocytes that was inhibited in the presence of sCR1 (34). Initial mapping studies identified the first three modules of CR1 (CCPs 1–3) as the most specific inhibitor of the PfRh4-CR1 invasion pathway (33). Recent work using CCPs 1–3 helped define the role of PfRh4 in the deformation of red blood cell membrane during P. falciparum invasion into red blood cells (35). Further mapping of the PfRh4 binding site on CR1 using truncation and deletion constructs pinpoint CCP 1 as the major binding site for PfRh4, and extensive mutagenesis experiments within this domain clearly delineated the PfRh4 binding site (32). These studies employed ELISA, co-immunoprecipitation and surface plasmon resonance (SPR) to characterize mutations that affected PfRh4-CR1 complex formation and showed that clustered mutations in residues 6–9 or single mutations in residues 18 and 20 resulted in a dramatic loss in affinity for rPfRh4. Park et al. (32) were able to engineer an artificial binding site within CCPs 8–14 by substituting residues within CCP 1 that are critical for PfRh4 interaction to their homologous position in CCP 8. Strikingly, this engineered site within CCPs 8–14 showed a 30-fold higher affinity for rPfRh4. Although the effects of the mutations are well understood in biochemical protein-protein interaction assays, it will be important to determine in a cellular context if any of these mutations lose their ability to block PfRh4-CR1 invasion or, in the case of the engineered site, lead to a potentially better inhibitor of P. falciparum invasion.
The availability of anti-PfRh4 mAbs that interfere with the PfRh4-CR1 interaction would provide an important tool in the identification of inhibitory epitopes in the binding interface. In this paper we generated anti-PfRh4 mAbs and tested their ability to modulate the interaction between PfRh4 and CR1 in vitro and to inhibit P. falciparum invasion. Furthermore, we characterize a collection of CR1-based inhibitors that will be invaluable in determining structure-function relationships between this ligand-receptor pair. Our results will identify distinct functional regions within PfRh4 and CR1 that are important for mediating entry of P. falciparum parasites into human red blood cells.
Experimental Procedures
Anti-PfRh4 Mouse Monoclonal Antibodies Production
Anti-PfRh4 mAbs were produced at the Monoclonal Antibody Facility at the Walter and Eliza Hall Institute. BALB/c and C57Bl6 mice received three immunizations of recombinant PfRh4 purified as described below. At day 0, Complete Freund's adjuvant was mixed with the antigen into an emulsion and injected intraperitoneally. At day 30 and day 60 the antigen was mixed with incomplete Freund's adjuvant, and the emulsion was injected intraperitoneally. Serum ELISA titrations were performed at day 72. The mouse with the best response received a final injection of antigen in saline, and splenocytes were harvested 3 days later. Spleen cells were fused with SP2/0 myeloma cells to form B-cell-myeloma fused cells (hybridomas). Hybridomas were grown in hypoxanthine-aminopterin thymidine growth medium. ELISA was used to select hybridomas producing antibodies specific to PfRh4. Hybridomas were cloned by limiting dilution in multiwell plates aiming for one cell or less per well. The subcloning supernatants were screened by ELISA. Two or more rounds of limiting dilution cloning were generally required before the hybridomas were deemed monoclonal. The antibodies were purified from monoclonal hybridoma supernatants with protein A-Sepharose.
ELISA
96-Well flat-bottomed plates (Maxisorp; Nunc) were coated with rPfRh4 (1 μg/well) resuspended in PBS and incubated for 2 h at room temperature. Plates were incubated with 5% skim milk, 0.01% Tween 20 for 1 h at room temperature to block nonspecific binding. After washing, anti-PfRh4 mAbs were added at 1:1000 dilution for 1 h at room temperature. Plates were washed 3 times before the addition of HRP-conjugated goat anti-mouse secondary antibodies (1:1000 dilution) for 1 h at room temperature. Azino-bis-3-ethylbenthiazoline-6-sulfonic acid (ABTS liquid substrate; Sigma) was used to detect HRP activity. 1% SDS was used to stop the reaction, and absorbance was measured at 405 nm. All washes were done in PBS, 0.01% Tween 20, and dilutions of antibodies were done in 0.5% skim milk, 0.01% Tween 20. All samples were performed in duplicate.
For competitive binding experiments with mAbs, the ELISA protocol was performed as above with the following modifications. Microtiter wells were coated with CCPs 1–3 (1 μg/well). After washing and blocking, rPfRh4 was added at a final concentration of 1 μg/well together with anti-PfRh4 mAbs. Binding of rPfRh4 was detected using an anti-PfRh4 rabbit polyclonal antibody followed by addition of HRP conjugated goat anti-rabbit secondary antibody.
Indirect Immunofluorescence Assay
Indirect immunofluorescence assay samples were prepared from magnet-purified parasites as described previously (36). Briefly, parasites were smeared on glass slides and fixed using 100% ice-cold methanol for 30 s. After fixation, samples were blocked overnight in 3% BSA (Sigma) in PBS at 4 °C. Primary antibodies were diluted in blocking solution and applied to slides. After a 1-h incubation at room temperature, the slides were washed 3 times in PBS. This was followed with a 1-h incubation with secondary antibodies diluted in blocking solution; slides were protected from light during the incubation. The slides were washed three times with PBS and mounted in VectaShield (Vector Laboratories) with 0.1 ng/μl 4′,6-diamidino-2-phenylindole (DAPI, Invitrogen). Primary antibodies used in indirect immunofluorescence assay imaging were diluted in 3% BSA in PBS, and the concentrations were as follows: mouse anti-PfRh4 (2 mg/ml, 1:500), rabbit anti-PfRh2 (2 mg/ml, 1:500). Secondary antibodies were used at the following concentrations: Alexa Fluor 488 goat anti-mouse (1:500) and Alexa Fluor 594 goat anti-rabbit (1:500). Fluorescence images were obtained using DeltaVision Elite widefield fluorescence microscope. Z stacks were taken above and below parasites and processed using Axiovision deconvolution software package.
Immunoblotting and Antibodies
Proteins larger than 100 kDa were run on 3–8% Tris acetate, whereas smaller proteins were run on 4–12% Bis-Tris SDS-PAGE gels (Invitrogen). Western blotting was performed using standard protocols, and the blots were processed with an enhanced chemiluminescence (ECL) system (Amersham Biosciences). Western blots probed with anti-PfRh4 mouse mAbs were conducted 2 mg/ml at 1:1000 dilution followed by HRP-conjugated goat anti-mouse secondary antibody at a 1:1000 dilution.
Immunoprecipitation Assays
In a reaction volume of 100 μl, anti-PfRh4 mAbs were incubated with recombinant PfRh4 at 0.1 and 0.05 mg/ml, respectively, for 1 h at room temperature. 5 μl of packed Protein G-Sepharose beads were added to capture the anti-PfRh4 mAbs; this incubation proceeded for 1 h at room temperature. Beads were washed five times with PBS, and proteins were eluted with equivalent volumes of 2× non-reducing sample buffer and boiled for 5 min before separating on SDS/PAGE gels. Protein eluates were visualized using SimplyBlue SafeStain (Life Technologies), performed according to the manufacturer's protocol.
Fluorescence Resonance Energy Transfer (FRET) Assay
To screen for antibodies that disrupt the PfRh4 interaction with CR1, we labeled rPfRh4 and the CCPs 1 and 2 with NHS ester derivatives of DyLight 594 and DyLight 488 (Life Technologies), respectively, to assess the rPfRh4-CCPs 1 and 2 interaction by FRET. rPfRh4 (2 mg/ml) was labeled at a 1:3 molar ratio with DyLight 594 in 50 mm MES, pH 6.0, 150 mm sodium chloride. Buffer exchange to 50 mm MES pH 6.0 and free dye removal was achieved using a Micro Bio-Spin P-6 column (Bio-Rad). CCPs 1 and 2 were similarly labeled with DyLight 488, and each preparation was assessed by spectrophotometry to determine the average number of dye molecules per protein molecule. After labeling, each rPfRh4 molecule carried an average of 2.6 DyLight-594 molecules, whereas CCPs 1 and 2 carried 1.8 DyLight 488 molecules. 10-μl binding reactions of rPfRh4·DyLight-594 and CCPs 1 and 2·DyLight488, each at 125 nm in 50 mm MES, pH 6.0, 150 mm sodium chloride, were placed in Corning 384-well plates (#3820), and fluorescence intensity at the following wavelengths were measured using an EnVision plate reader (PerkinElmer Life Sciences): DyLight-488 (donor) 485/14-nm excitation filter and 535/25-nm emission filter, DyLight-594 (acceptor) 590/20-nm excitation filter and 615/9-nm emission filter, and sensitized emission 485/14-nm excitation filter and 615/9-nm emission filter. In an ideal situation sensitized emission should only be measured when DyLight-488 and DyLight-594 are in close proximity, but in practice we measured a significant contribution to FRET-mediated sensitized emission from DyLight-594 in the absence of DyLight-488 and chose to correct for this by calculating FRET as a ratio of sensitized emission over DyLight-594 fluorescence intensity. This creates a unit-less FRET ratio that can be used to compare binding of rPfRh4·DyLight-594 to CCPs 1 and 2·DyLight-488 in the presence or absence of candidate inhibitors that disrupt the interaction.
Flow Cytometry-based Erythrocyte Binding Assay
The flow cytometry-based erythrocyte binding assay was performed as described with the following modifications (17). 40 μl of packed erythrocytes were washed twice with 400 μl of 1% BSA/PBS and resuspended to a final volume of 1 × 107 cells/ml. Meanwhile, 1.25 μg of rPfRh4 was preincubated with 10 μg of mAb at room temperature for 10 min. 100 μl of the resuspended erythrocytes was added to the recombinant protein/antibody mix and incubated for 30 min. After binding, erythrocytes were washed three times with 1% BSA, PBS. To detect rPfRh4 binding, anti-PfRh4 rabbit IgG R550 (1:100 dilution) was added and incubated for 30 min followed by 3 washes with 1% BSA, PBS. Alexa Fluor 488 goat anti-rabbit secondary antibody (1:100, Life Technologies) was added and incubated at room temperature and protected from light for 30 min. Erythrocytes were washed twice with 1% BSA. PBS followed by 1 wash in PBS and resuspended in 500 μl of PBS before being transferred to FACS tubes. A total of 50,000 erythrocytes were read on the FACSCalibur flow cytometer (BD Biosciences), and the results were analyzed using FlowJo software (Three Star). The percentage of erythrocytes with bound rPfRh4 was determined by normalizing the number of erythrocytes exhibiting a positive Alexa Fluor 488 signal that is above the background (which is Alexa Fluor 488 signal of erythrocytes without rPfRh4 added) on the total number of erythrocytes.
Parasite Culture and Growth Inhibition Assay
P. falciparum asexual stages were maintained in human O+ erythrocytes in RPMI-HEPES medium with 50 μg/ml hypoxanthine, 25 mm NaHCO3, 20 μg/ml gentamicin, and 0.5% Albumax II (Gibco; Invitrogen) in 1% O2, 4% CO, and 95% N2 at 37 °C and synchronized by standard methods. We used the D10-PHG for the majority of our assays, which are GFP-positive (37).
Growth inhibition assays were performed as described with the following modifications (33). Starting parasitemia for the growth assays was 0.2% with a hematocrit of ∼1%. CR1 constructs, anti-PfRh4 mAbs, bovine serum albumin (BSA, Sigma), and heparin were added at the beginning of the assay. Growth inhibition assays were performed in 96-well round-bottom microtiter plates (BD Biosciences) over 2 cycles of parasite growth. After 96 h the parasitemia was determined by flow cytometry of GFP-positive and EtBr-stained trophozoite-stage parasites using a FACSCalibur (BD Biosciences) and a plate reader. For each well 40,000 cells or more were counted. Growth was expressed as mean parasitemia obtained from triplicate readings. At least two independent assays were performed, each in triplicate. % Growth refers to the % parasitemia in the presence of CR1 constructs or anti-PfRh4 mAbs relative to the % parasitemia with the addition of PBS (no mAb control, which was arbitrarily set to be 100%).
Mapping of mAb Epitopes
An overlapping peptide array consisting of 15-mer biotinylated peptides with a SGSG linker was synthesized by Mimotopes Pty Ltd, Clayton, Australia. Peptides were resuspended and applied to streptavidin-coated plates. ELISA was performed as stated above.
Antibody Affinity Measurements
Binding of rPfRh4 to anti-PfRh4 mAbs 5H12 and 10C9 was monitored by SPR using the Biacore 3000 system (GE Healthcare), where experiments were performed at 25 °C in PBS-p +. CMD-500 (XanTec bioanalytics GmbH, Dusseldorf, Germany) chip surfaces were prepared by immobilization of anti-mouse IgG at 30 μg/ml in 10 mm sodium acetate, pH 5.0, using standard NHS-EDC (EDC, 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide) amine coupling methods, where a final immobilization of 7542.2 resonance units was achieved. The antibody affinity assay was performed using an indirect capture method where antibodies 5H12 and 10C9 were diluted to 30 μg/ml in PBS-p + and injected for 120 s at 5 μl/min with a 300-s stabilization time. After stabilization, rPfRh4 in PBS-p + was injected at 20 μl/min for 420 s followed by a 7200-s dissociation over 2-fold increases in concentration between 1.5625 nm and 50 nm. The chip surface was regenerated before each run with a single 20-s injection of 10 mm glycine, pH 1.7 at 30 μl/min where baseline was achieved. The buffer difference was subtracted from blank cell lanes that had been activated and deactivated using standard amine coupling methods. Each concentration series was performed in duplicate. Data generated were processed using the BIAevaluation software (GE Healthcare), where multicycle kinetics were obtained using a 1:1 Langmuir binding model.
Protein Expression and Purification
Recombinant PfRh4 was previously expressed using expression vector pET-45b(+) as described (17, 31, 33). For this paper we digested this clone using BamHI and XhoI and cloned the DNA-encoding PfRh4 in-frame into a pProEX HTa vector, which contains a hexa-His tag and tobacco etch virus cleavage site at the N-terminal end. The fusion protein was expressed in BL21 (DE3) bacteria cells and purified over a nickel-nitrilotriacetic acid column (Qiagen) under native conditions. The purified protein underwent tobacco etch virus protease cleavage overnight at 4 °C to remove 23 amino acids including the hexa-His tag. Cleaved rPfRh4 was further purified on a Superdex 200 gel filtration column (Hiload 16/60; GE Healthcare), and appropriate fractions concentrated and loaded onto a cation exchange column (Mono S 5/50 GL; GE Healthcare). Recombinant rPfRh4 eluted from the column as a monomer and purity was determined using SDS-PAGE.
Protein constructs CCPs 1 and 2 and CCPs 1–3 were expressed and purified as described before (33). In brief, existent Pichia pastoris clones were fermented as described in Schmidt et al. (38), and the recombinant proteins were secreted into the supernatant and captured by ion chromatography with Sepharose beads. The capture step was followed by cation and size exclusion chromatography. The coding DNA for CCPs 8 and 9 and the mutant versions of CCPs 1 and 2, i.e. m6–9, m18, and m20, as well as the revertant mutant CCPs 8 and 9 m7–9 and 18–20r, were codon-optimized, gene-synthesized (IDT Technologies), and subcloned into the P. pastoris expression vector pPICZαB. Within CCPs 1 and 2 and its mutant versions the N-glycosylation sites were removed by substituting relevant Asn to Gln residues. The novel expression cassettes were transformed into the P. pastoris strain KM71H according to the manufacturer's instructions (Life Technologies). Protein expression and purification was carried out as for CCPs 1 and 2 and yielded between 10 and 52 mg of a highly pure protein preparation per liter fermenter supernatant. N-Glycosylation (CCP 8 and 9 constructs) was removed with the endoglycosidase EndoHf (New England BioLabs) when applicable in between purification steps using a published procedure (38). All CCP 1 and 2 and CCP 8 and 9 constructs were submitted to mass spectrometry analysis. Purified proteins were analyzed on an LTQ-Orbitrap Velos Pro (Thermo Scientific) online coupled to an RSLCnano uPLC (Thermo Scientific) equipped with a Vydac MS C4 column (300 Å, 5 μm, 5 mm × 300 μm) (Grace). After desalting the samples at 5% acetonitrile, 0.1% formic acid with a flow of 10 μl/min for 10 min, proteins were eluted from the column by raising the concentration of acetonitrile to 43% for 5 min. MS spectra were acquired in the Orbitrap part of the instrument at a resolution of 30,000. The acquired data were processed using the QualBrowser (Thermo Scientific) in combination with in-house written VBA-script for MSExcel (Microsoft Corp.). These analyses confirmed the identity of the protein preparations with a maximal deviation from the theoretical molecular mass of 12.5 ppm (or 0.18 Da). Additionally, one-dimensional NMR spectroscopy shows that line widths and signal dispersion (measured at 30 °C in PBS with addition of 7% D2O) are consistent with mono-dispersed, well folded protein molecules.
Surface Plasmon Resonance Measurement of CCP Mutants and rPfRh4
To analyze the interactions between rPfRh4 with recombinant CR1 constructs, SPR experiments were carried out at 25 °C using a Reichert SR7500DC SPR instrument (Reichert Technologies, Buffalo, New York). Recombinant Rh4 was covalently immobilized onto one flow cell of a carboxymethyldextran hydrogel biosensor chip (CMD500m, purchased from XanTec bioanalytics GmbH, Dusseldorf, Germany) by standard amine coupling according to the manufacturer's instructions. Amine coupling in the absence of any protein (dummy coupling) was performed on a second flow cell yielding a reference flow cell. Signals obtained for the rPfRh4 surface were subtracted by signals obtained for the reference flow cell according to standard procedure. Only reference-subtracted sensorgrams are shown throughout. As running buffer 10 mm HEPES, pH 7.4, 150 mm NaCl, 1 mm MgCl2, and 0.005% Tween 20 was used throughout, apart from one protein series of CCPs 1 and 2 (indicated), which was assayed in the same buffer but with MgCl2 substituted with 3 mm EDTA. Proteins were diluted into running buffer before assaying and were injected on the chip for 3 min at a flow rate of 25 μl/min. The dissociation phase consisted of buffer flow at 25 μl/min for 5 min and was followed by a regeneration step with 1 m NaCl. CCPs 1 and 2, CCPs 1 and 2 m6–9, CCPs 1 and 2 m18, CCPs 8 and 9 m7–9,18–20r (CCPs 8 and 9 Rev) and CCPs 8 and 9 were assayed at 1:1 dilution series from 10 μm to 0.61 nm. CCP 1 and 2 m20 and CCP 8 and 9 were assayed at 1:1 dilution series from 10 μm to 9.8 nm. Affinity constants were extracted by plotting the response at steady state against the molar concentration and subsequent fitting the affinity with the TRACEDRAWER software using a 1:1 steady state affinity model. To probe reproducibility the binding of double injections (at same concentration) were probed for all analytes at the highest concentration assayed (apart from CCP 1 and 2 m18 and CCPs 1–3) and at the lowest concentration assayed (for all).
Results
Characterization of Anti-PfRh4 Monoclonal Antibodies
PfRh4 binds to CR1 to mediate entry of P. falciparum parasites into red blood cells (17). We previously generated a recombinant fragment of PfRh4 (rPfRh4) that binds to red blood cells and forms a complex with sCR1 and CCPs 1–3 (17, 31). We immunized mice with rPfRh4, generated 10 murine mAbs that could detect rPfRh4 by ELISA (Fig. 1A), and proceeded to examine whether they bound native PfRH4 or interfered with receptor engagement. 2E8 mAb is specific to the C-terminal end of native PfRh4, which does not include the region encompassed by rPfRh4 and as expected did not show significant reactivity to rPfRh4 (Fig. 1A) (17).
FIGURE 1.

Characterization of monoclonal antibodies specific to PfRh4. A, detection of rPfRh4 by anti-PfRh4 mAbs by ELISA. Anti-PfRh4 mAbs were added to microtiter wells coated with rPfRh4. Bound antigen-antibody complexes were detected with anti-mouse HRP. Error bars represent the mean ± S.E. of three independent assays repeated in triplicate. B, localization of native PfRh4 was assessed by wide-field immunofluorescence assay using the anti-PfRh4 mAbs. PfRh4 (green) was co-stained with rhoptry protein, PfRh2 (red), and nuclei-stain DAPI (blue). Differential interference contrast (DIC) shows the differential interference contrast view of the same field. Scale bar = 5 μm. C, native PfRh4 is detected by all anti-PfRh4 mAbs except for 5H12. Western blot of saponin-lysed schizont pellet lysates from wild type and ΔPfRh4 strains were separated by SDS-PAGE and probed with anti-PfRh4 mAbs under reducing conditions. D, immunoprecipitation of rPfRh4 using anti-PfRh4 mAbs. Recombinant PfRh4 was incubated with anti-PfRh4 mAbs and protein G-Sepharose. Protein eluates were fractionated on SDS-PAGE and visualized using SimplyBlue SafeStain. I, input. U, unbound. E, eluate. E, immunoprecipitation of PfRh4 from culture supernatants using anti-PfRh4 mAbs. Culture supernatants were incubated with anti-PfRh4 mAbs and protein G-Sepharose. Protein eluates were fractionated on SDS-PAGE and probed with anti-PfRh4 polyclonal antibody. The asterisk marks the processed fragment of PfRh4 present in culture supernatants. For all panels, molecular weight is indicated on the left in kDa.
PfRh4 together with the other PfRh family of proteins localize to the rhoptries of P. falciparum parasites (6). Using an indirect immunofluorescence assay we showed that all mAbs except 6F12 detected PfRh4 in an apical localization that predominantly co-localized with PfRh2, another rhoptry protein (Fig. 1B) (39). We tested the ability of the mAbs to detect unfolded PfRh4 from solubilized P. falciparum schizont lysate by Western blotting. Native PfRh4 migrates as a doublet at 180 and 190 kDa (31), which was detected by all the mAbs except for 5H12 in a P. falciparum strain expressing PfRh4 (Fig. 1C). This PfRh4-specific doublet is not present in parasite lysates harvested from a PfRh4 knock-out strain. We hypothesize that 5H12 recognizes a conformational epitope that is not present under SDS-PAGE conditions and which denatures PfRh4.3 To examine if these mAbs are able to immunoprecipitate folded rPfRh4, we incubated rPfRh4 with mAbs and used protein G-Sepharose beads to capture the antigen-antibody complexes. Only 5H12, 6A5, and 10C9 were able to immunoprecipitate rPfRh4 as seen by its presence in the eluate lane (Fig. 1, D and E lanes). We also performed similar assays using parasite culture supernatants, which contain a processed 160-kDa fragment of PfRh4 (31), and were able to show that 2E8, 5H12, 6A5, and 10C9 were able to immunoprecipitate PfRh4 (Fig. 1E). Collectively these results show that all anti-PfRh4 mAbs have specific reactivity to both rPfRh4 and native PfRh4 from P. falciparum parasites. In addition, 5H12 may recognize a conformational epitope on PfRh4.
Modulation of Recombinant PfRh4 and CR1 Interaction with Monoclonal Antibodies
To determine whether anti-PfRh4 mAbs inhibited the interaction between PfRh4 and CR1, we developed a FRET assay where 125 nm rPfRh4 labeled with DyLight-594 could be shown to bind to the same concentration of recombinant CCPs 1 and 2 labeled with DyLight-488 (Fig. 2, A–D, No inh columns). We validated this assay by introducing 50-fold molar excess of unlabeled rPfRh4 and CCPs 1 and 2 to compete with the FRET pair and found a marked decrease in FRET ratio (Fig. 2D, unRh4 and CCPs 1 and 2, respectively). Denaturants such as 3 m guanidinium chloride and 1% sodium dodecyl sulfate also reduced FRET to similar levels. In contrast, proteins that were unable to bind PfRh4 such as CCPs 8 and 9 and bovine serum albumin had no effect on FRET (Fig. 2D) (33). For the following FRET assays, we arbitrarily defined 100% binding as the level of binding measured when no inhibitor was added and 0% binding as the level of binding measured in denaturing concentrations of sodium dodecyl sulfate (Fig. 2D, dotted lines). This assay allowed rapid screening of the mAbs and identified 5H12 mAb as an inhibitor of the rPfRh4-CR1 interaction (Fig. 2E).
FIGURE 2.

Inhibition of the PfRh4-CR1 interaction with anti-PfRh4 mAbs. Novel FRET-based assay to monitor the interaction between PfRh4 and CR1 where CCPs 1 and 2 (A) and rPfRh4 (B) were labeled with Dylight 488 and 594, respectively, and incubated at 1:1 molar concentration (no inh) either in the presence of unlabeled proteins (CCPs 1 and 2, BSA, CCPs 8 and 9, unRh4) or denaturants (guanidine hydrochloride (GndCL) and SDS). The fluorescence intensity (FI) of DyLight-488 (donor) was measured with a 485/14-nm excitation filter and a 535/25-nm emission filter and DyLight-594 was measured with a 590/20-nm excitation filter and 615/9-nm emission filter. C, measurement of FRET fluorescence intensity using the DyLight-488 (donor) 485/14-nm excitation filter and the DyLight 594 (acceptor) 615/9-nm emission filter. D, FRET ratio on the y axis represents “excitation 488/emission 615” value over Dylight 594 value. Unlabeled CCPs 1 and 2 and rPfRh4 (unRh4) were able to inhibit labeled PfRh4-CR1 interaction on a similar level to denaturants, SDS and guanidine hydrochloride (GndCL). Error bars represent the S.E. of three independent repeats. E, 5H12 inhibits the PfRh4-CR1 interaction in the FRET-based assay. Dylight-labeled rPfRh4 and CCPs 1 and 2 were incubated with various anti-PfRh4 mAbs. Percentage binding refers to the FRET ratio relative to “no mAb” and SDS controls. Error bars represent S.E. of three independent repeats. F, 5H12 inhibits PfRh4-CR1 interaction in ELISA-based assay. Microtiter wells were coated with CCPs 1–3. Recombinant PfRh4 was added in the presence of anti-Rh4 mAbs. Bound rPfRh4 was detected with an anti-PfRh4 rabbit polyclonal primary antibody followed by an anti-rabbit-HRP secondary antibody. Error bars represent S.E. of two independent repeats. G, 5H12 disrupts PfRh4-CR1 complex formation. The immunoprecipitation assay was performed by incubating rPfRh4 with 5H12 and 10C9 (left panel) and subsequently in conjunction with CCPs 1–3 (right panel). I, input. U, unbound. E, eluate.
To confirm the results obtained from the FRET-based assay, we examined the effect of mAbs on PfRh4-CR1 interaction by ELISA (Fig. 2F). Recombinant PfRh4 was incubated briefly with each mAb before binding to immobilized CCPs 1–3, and bound PfRh4 was detected using a polyclonal anti-PfRh4 antibody. The addition of CCPs 1–3 as a known inhibitor showed the expected marked reduction in PfRh4-CR1 binding (Fig. 2F) (33). 5H12 showed similar levels of inhibition of the PfRh4-CR1 interaction as in the FRET assay. We observed that two mAbs, 6A5 and 10C9, appeared to enhance the interaction between rPfRh4 and CCPs 1–3 (Fig. 2F).
We examined whether the inhibitory mAb 5H12 and the enhancing mAb 10C9 were able to co-immunoprecipitate PfRh4 and CCPs 1–3. Both 5H12 and 10C9 immunoprecipitate rPfRh4 with similar efficiencies as indicated by the depletion of rPfRh4 in the unbound fraction (lane U) and an enrichment of rPfRh4 in the eluate fraction (Fig. 2G, left panel, lane E). When CCPs 1–3 was added into the assay, 10C9 was able to immunoprecipitate the rPfRh4-CCPs 1–3 complex (Fig. 2G, right panel). In contrast, although 5H12 successfully immunoprecipitated rPfRh4, we did not detect a corresponding CCPs 1–3 protein (Fig. 2G, right panel). 5H12 was less efficient at immunoprecipitating rPfRh4 when CCPs 1–3 were present, suggesting that 5H12 binds to a region of rPfRh4 that is also bound by CCPs 1–3 (Fig. 2G, right panel versus left panel).
Blocking the Red Blood Cell Binding Capabilities of PfRh4
We utilized a flow cytometry-based red blood cell binding assay to determine the effects of the mAbs on PfRh4 interaction with native CR1 on the surface of red blood cells. This method entailed incubating rPfRh4 with red blood cells in the presence of mAbs; rPfRh4 binding was detected using an anti-PfRh4 rabbit polyclonal antibody followed by an anti-rabbit secondary antibody conjugated to a fluorophore, which was detected by flow cytometry. This assay was used previously to show that the levels of rPfRh4 binding to erythrocytes directly correlates with the levels of CR1 on the red blood cell surface (17). For this assay we used an intermediate amount of rPfRh4 that allows both increases and decreases in rPfRh4 binding to be measured. As expected, the addition of soluble CCPs 1 and 2 was able to inhibit rPfRh4 binding to red blood cells (Fig. 3, A and B). 1D11, 5H12, 6A5, and 10C9 were all able to affect PfRh4 binding to red blood cells. Consistent with previous results, 5H12 was able to inhibit rPfRh4 binding to a similar extent as CCPs 1 and 2 (Fig. 3, A and B). On the other hand, 1D11 and 10C9 showed strong enhancement of rPfRh4 erythrocyte binding, whereas 6A5 increased rPfRh4 binding more modestly (3.6, 19.5, and 1.5-fold increase, respectively).
FIGURE 3.

Inhibition of the PfRh4 invasion pathway. A, dot plots show the binding of rPfRh4 to red blood cells in the presence of anti-PfRh4 mAbs. Binding was detected using an anti-PfRh4 rabbit IgG antibody followed by a secondary anti-rabbit Alexa 488 antibody. Numbers outside of the purple box refer to the percentage of red blood cells with bound rPfRh4 relative to the full red blood cell population. B, recombinant PfRh4 binding to erythroid CR1 was inhibited by 5H12. Binding assays were performed by incubating rPfRh4 and red blood cells in the presence of various anti-PfRh4 mAbs, and the level of red blood cell binding was determined by flow cytometry. Percentage RBC binding on the y axis refers to the percentage of rPfRh4-erythroid CR1 binding relative to the no mAb control (arbitrarily set to be 100%). Error bars represent S.E. of three independent repeats. C, anti-PfRh4 mAbs were tested with untreated (black bars) or neuraminidase-treated erythrocytes (gray bars) at a final concentration of 0.2 mg/ml. Growth inhibition assays were performed using the D10-PHG strain of P. falciparum. D, anti-PfRh4 mAb, 5H12, inhibits the PfRh4-CR1 pathway to similar levels as the recombinant CR1 constructs. D10-PHG strain was tested in untreated (black bar) or neuraminidase-treated erythrocytes (gray bar) in the presence of anti-PfRh4 mAbs (at a final concentration of 0.2 mg/ml) and recombinant CR1 constructs (at a final concentration of 0.1 mg/ml). Error bars represent S.E. from five independent repeats. E, titration of 5H12 in growth inhibition assays in the presence of untreated (black bar) or neuraminidase-treated red blood cells (gray bar). Error bars represent S.E. from four independent repeats. For panels C, D, and E, percent growth on the y axis refers to the percentage parasitemia relative to the no mAb control, which is arbitrarily set at 100%.
We tested if the mAbs could inhibit the PfRh4-CR1 invasion pathway in P. falciparum growth assays. As a negative control in these experiments we used the 2E8 mAb, which is specific to the C-terminal end of native PfRh4 (not present in rPfRh4) and has previously been shown to be unable to inhibit parasite growth (17). P. falciparum parasites invade red blood cells via several redundant invasion pathways mediated by EBA and PfRh proteins. To abolish the pathways mediated by the EBA family of proteins, red blood cells were treated with neuraminidase to remove sialic acid residues from the glycophorins, which are cognate receptors for EBA proteins. Upon neuraminidase treatment, 90% of the invasion events are dependent on PfRh4 for entry into red blood cells via CR1 (17, 33). When all 10 mAbs and 2E8 were tested in neuraminidase-treated red blood cells, we observed inhibition of the PfRh4-CR1 pathway only when 5H12 was added to the growth medium (Fig. 3C). In subsequent growth assays we additionally tested CR1 fragments, 5H12, and the enhancing mAb 10C9. The addition of CCPs 1 and 2, CCPs 1–3, sCR1 inhibited >90% of the invasion events in neuraminidase-treated red blood cells as expected (Fig. 3, C and D), whereas the addition of 2E8 and 10C9 mAb did not perturb growth in a statistically significant manner. 5H12 again exhibited similar levels of growth inhibition as soluble forms of CR1 (Fig. 3D). Titration of 5H12 showed dose-dependent inhibition of P. falciparum invasion events and growth in neuraminidase-treated red blood cells (Fig. 3E).
Mapping the Epitopes for Monoclonal Antibody Binding
Having characterized the functional outcomes of mAb binding to PfRh4, we next utilized a panel of overlapping 15-mer peptides completely encompassing rPfRh4 to identify epitopes recognized by the anti-PfRh4 mAbs (Fig. 4). Using ELISA we detected specific binding to either a single peptide or a set of overlapping peptides for all of the mAbs except 1D11, 1H3, and 5H12. Both 1A6 and 3A6 bound two overlapping peptides: N238 (NEKLEKYTNKFEHNI) and E242 (EKYTNKFEHNIKPHI). 5D2 bound a single peptide (Y266, YINNSDCHLTCSKYK). 2A1 bound three overlapping peptides Y522 (YDNIYIILKQKFLNK), Y526 (YIILKQKFLNKLNEI), and K530 (KQKFLNKLNEIIQNH). 10C9 bound two overlapping peptides, N662 (NFTHKTIQQILQQQY) and K666 (KTIQQILQQQYKNNT), whereas 6A5 also recognized peptide K666. It is interesting to note that both 10C9 and 6A5 enhance rPfRh4 red blood cell binding. 6F12 bound to novel epitopes encompassed by two overlapping peptides, N718 (NIEHIKQILINYIQE) and I722 (IKQILINYIQECKPI).
FIGURE 4.
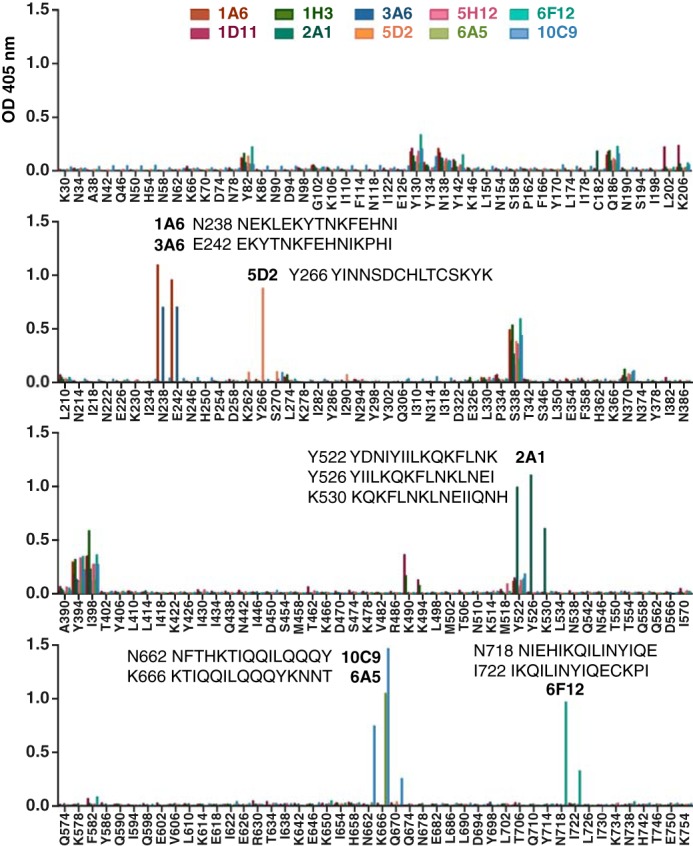
Epitope mapping of PfRh4 monoclonal antibodies. Detection of anti-PfRh4 mAbs binding to an overlapping peptide array encompassing rPfRh4 by ELISA. Peptides are named by the single-letter code and numerical position of their first amino acid within PfRh4; e.g. K30 indicates the peptide KEKKNDPEADSKNSQ starting at amino acid position 30. Significant binding of anti-PfRh4 mAbs to peptides is highlighted with the peptide sequence.
To determine if any of the mAbs share a similar epitope to 5H12, we also performed competition ELISA between 5H12 (which was directly conjugated to HRP, 5H12-HRP) and all the other mAbs. Using rPfRh4-coated microtiter plates, we examined the level of 5H12-HRP binding in the presence of each mAb by ELISA. Although the addition of 5H12 itself successfully competed with 5H12-HRP, none of the other mAbs showed a similar phenotype (data not shown).4 These data show that the 5H12 epitope is unique and does not overlap with other epitopes bound by the collection of anti-PfRh4 mAbs.
Antibody Affinities for PfRh4
The affinity of two mAbs, 5H12 and 10C9, to rPfRh4 was evaluated using SPR. 5H12 and 10C9 showed clear binding (Fig. 5, A and B, respectively). rPfRh4 bound to 5H12 with low nanomolar affinity (KD = 1.34 nm) and with association (ka = 1.29 × 105 m−1s−1) and dissociation (kd = 1.73 × 10−4 s−1) (Table 1). In comparison, 10C9 showed an even higher affinity for rPfRh4 (KD = 0.48 nm), with association (ka = 5.64 × 104 m−1s−1) and dissociation (kd = 3.69 × 10−5 s−1) (Table 1).
FIGURE 5.

Kinetics of mAb-PfRh4 interactions. Sensorgrams of multi-cycle kinetic measurements of 5H12 (A) and 10C9 (B) with PfRh4. The sensorgrams represent duplicate runs obtained on separate days with increasing concentrations of rPfRh4 (1.5625, 3.125, 6.25, 12.5, 25, 50 nm) flowed over 5H12 and 10C9 captured on anti-mouse IgG. Solid lines represent the observed binding, and dots represent 1:1 Langmuir model-fitted binding with kinetic values reported in Table 1. R.U., resonance units.
TABLE 1.
Multicycle kinetic measurements of mAb-PfRh4 interactions
Kinetic parameters (ka and kd) and KD values extracted from 5H12-rPfRh4 and 10C9-rPfRh4 sensogram fitted to a global 1:1 interaction model are reported. Maximum resonance values (Rmax) and χ2 values of goodness of fit (<10% Rmax) for the models are also presented. RU, resonance units.
| mAb | ka | kd | Rmax | KD | χ2 |
|---|---|---|---|---|---|
| m−1 s−1 | 1/s | RU | m | ||
| 5H12 | 1.29E+05 | 1.73E−04 | 236 | 1.34E−9 | 2.7 |
| 10C9 | 5.64E+04 | 3.69E−05 | 111 | 4.82E−10 | 2.3 |
CR1-based Inhibitors That Block PfRh4-CR1 Interaction
Mutagenesis analyses within CCPs 1–7 identified several residues essential for PfRh4 binding within CCP 1 (Fig. 6A) (32). Although these mutations clearly abolished PfRh4-CR1 interaction in vitro as shown by ELISA, immunoprecipitation, and SPR measurements, the mutant proteins were not assessed for their functional effects in red blood cell binding or growth assays. To this end we generated recombinant CCPs 1 and 2 proteins in P. pastoris that contained mutations of these critical residues (Fig. 6B). These mutations (m) are named in relation to their positions in CCP 1 that were substituted with the respective amino acids in CCP 8 (Fig. 6C) (32). We produced correctly folded CCPs 1 and 2 m6–9, CCPs 1 and 2 m18, and CCPs 1 and 2 m20. In addition, we also expressed an engineered PfRh4 binding site in CCPs 8 and 9 by introducing CCP 1 residues at 7–9 and 18–20 to their homologous site within CCP 8 (32). This mutant is called CCPs 8 and 9 m7–9,18–20r but will be referred to as CCPs 8 and 9 Rev for simplicity. The proteins were purified to a high degree of purity, as judged by prominent single protein bands under reducing and non-reducing conditions after SDS-PAGE analysis (Fig. 6B). The faster mobility under non-reducing conditions is indicative of disulfide bridges being formed within these constructs. Protein identity and quality have additionally been confirmed by mass spectrometry with a maximal deviation of 12.5 ppm (equating to 0.18 Da) from the theoretical molecular mass.
FIGURE 6.

CR1-based inhibitors of PfRh4-CR1 interaction. A, schematic of CR1 polypeptide. Each box labeled 1–30 represents a CCP module of 60–70 amino acid residues. The first 28 CCPs are organized based on homology into four long homologous repeats (LHR) A–D, each consist of seven CCPs. The functional sites of CR1 are labeled Site 1 and Site 2 with their respective activities. The transmembrane domain and cytoplasmic tail are represented as a curvy line at the C-terminal end. B, SDS-PAGE gel of purified CCP constructs. 2 μg of each construct were loaded onto a NuPAGE® gradient gel (4–12%) under reducing and non-reducing conditions and stained with Coomassie Brilliant Blue. Molecular weight markers are indicated in kDa in the middle of the gel. C, the first 25 amino acids within CCPs 1 and 2 and CCPs 8 and 9 are shown. Wild type CCP 1 and CCP 8 sequence is in black and orange, respectively. Numbers after the m, for mutants, denote the position of the amino acids. D, immunoprecipitation assay to monitor PfRh4-CR1 complex formation. Anti-PfRh4 antibody 10C9 was incubated with rPfRh4 in the presence of either CCPs 1 and 2, CCPs 1 and 2 m6–9, CCPs 1 and 2 m18, CCPs 1 and 2 m20, CCPs 8 and 9, or CCPs 8 and 9 Rev. Proteins were stained with SimplyBlue SafeStain. I, input. U, unbound. E, eluate. E, inhibitory antibody 5H12 is able to block complex formation between CCPs 8 and 9 Rev and rPfRh4. F, effects of mutagenesis within CCPs 1 and 2 on rPfRh4 binding to red blood cells. Percentage RBC binding of rPfRh4 on the y axis refers to the percentage of rPfRh4-erythroid CR1 binding relative to the rPfRh4 only control (arbitrarily set to be 100%). Error bars represent ± S.E. from three independent repeats. G, growth assays in the presence of CR1 recombinant constructs. Various CCP mutants and soluble CR1 were tested with untreated (black bars) or neuraminidase-treated erythrocytes (gray bars) at a final concentration of 0.1 mg/ml. Growth (percentage of control) on the y axis refers to the percentage parasitemia relative to the PBS control. Error bars represent S.E. from three independent repeats.
Using immunoprecipitation assays, we examined whether these mutants could still form a complex with rPfRh4. We observed that CCPs 1 and 2 was able to form a stable complex with rPfRh4 (Fig. 6, D and E). Unexpectedly CCPs 1 and 2 m6–9 and CCPs 1 and 2 m18 were still able to interact with rPfRh4 (32). Mutation of residue 20 (CCPs 1 and 2 m20) completely abolished complex formation. As expected, CCPs 8 and 9 did not interact with rPfRh4, whereas CCPs 8 and 9 Rev was able to form a stable complex with rPfRh4 (Fig. 6D, right panel), indicating that the substituted residues from CCP 1 to CCP 8 are sufficient to mediate PfRh4 binding.
To determine if CCPs 8 and 9 Rev bound to PfRh4 using the same binding site as recognized by the inhibitory antibody, we examined if the addition of mAb 5H12 would interfere with the rPfRh4-CCPs 8 and 9 Rev complex. 10C9 was able to immunoprecipitate rPfRh4-CCPs 8 and 9 Rev complex (Fig. 6E). However, 5H12 successfully immunoprecipitated rPfRh4, but we did not detect a corresponding CCPs 8 and 9 Rev fragment (Fig. 6E), suggesting that 5H12 inhibits the interaction between rPfRh4 and CCPs 8 and 9 Rev.
We utilized the flow cytometry-based red blood cell binding assay described earlier to determine the effects of the mutant proteins. As expected, the addition of soluble CCPs 1 and 2, CCPs 1 and 2 m6–9, CCPs 1 and 2 m18, and CCPs 8 and 9 Rev was able to inhibit rPfRh4 binding to red blood cells (Fig. 6F). Proteins that did not bind rPfRh4 such as CCPs 8 and 9 and CCPs 1 and 2 m20 were not able to inhibit rPfRh4 binding to red blood cells (Fig. 6F). Consistent with these results, we observed that the addition of CCPs 1 and 2, CCPs 1 and 2 m6–9, CCPs 1 and 2 m18, and CCPs 8 and 9 Rev was able to inhibit the majority of P. falciparum invasion events into neuraminidase-treated red blood cells (Fig. 6G). This was similar to the level of inhibition seen upon the addition of CCPs 1–3 and sCR1. As expected, we did not observe any inhibition of the PfRh4-CR1 invasion pathway with the addition of CCPs 8 and 9 or CCP 1 m20 (Fig. 6G).
To ensure that the protein fragments that were used in all the assays above were properly folded, we also performed one-dimensional NMR spectroscopy. One-dimensional NMR spectroscopy shows that line widths and signal dispersion are consistent with monodispersed, well folded protein molecules (Fig. 7, A–F).
FIGURE 7.
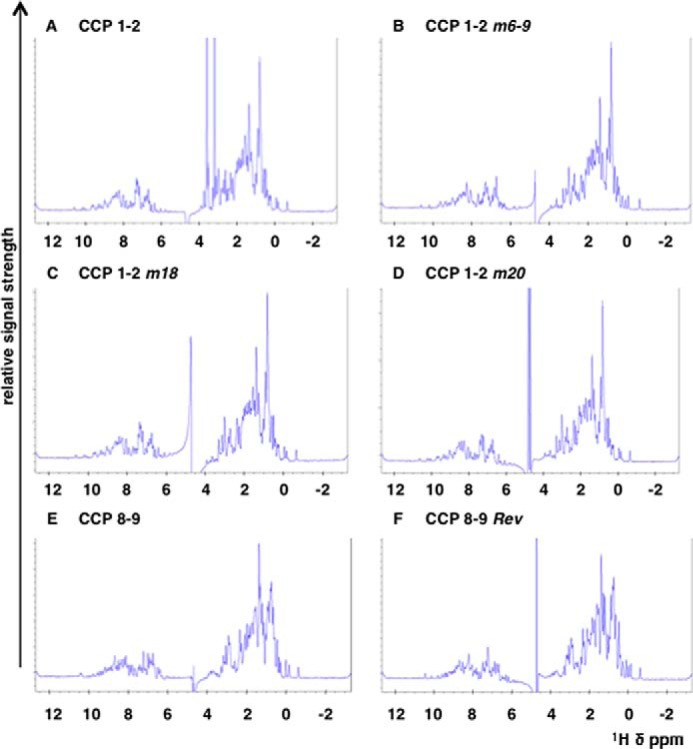
1H NMR spectra of CR1 protein fragments. Spectra for the CR1 constructs CCPs 1 and 2 (A), CCPs 1 and 2 m6–9 (B), CCPs 1 and 2 m18 (C), CCPs 1 and 2 m20 (D), CCPs 8 and 9 (E), and CCPs 8 and 9 Rev (F). The spectra were recorded on a 800-MHz spectrometer from Bruker at 303 K in PBS with the addition of D2O to 7%. All samples were measured at a protein concentration of 50 μm. The dispersion and line shape as well as peak intensities are comparable among the different samples and indicate comparable high quality of the sample preparations. The overall good signal dispersion and the sharp line widths are consistent with mono-dispersed, well folded protein molecules. The spectrum of CCPs 1 and 2 (A) exhibits two strong signals between 3 and 4 ppm. These signals relate to a small contamination with glycerol that derives from the protein concentration process using a commercial spin concentrator (which typically use glycerol to preserve the filtration membrane of the spin concentrator). The signal at 4.8 ppm relates to residual water signal.
Affinity of CR1-based Inhibitors for Binding to PfRh4
We expected that mutations in CCPs 1 and 2 and CCPs 8 and 9 would directly influence each mutant protein's affinity for PfRh4. To determine the binding constants of the wild type and mutant proteins, we employed SPR. rPfRh4 was immobilized onto a carboxymethyldextran hydrogel sensorchip surface, and the CR1 constructs were assayed in HEPES-buffered saline containing 1 mm MgCl2 (Fig. 8). CCPs 1 and 2, CCPs 1 and 2 m6–9, and the revertant mutant CCPs 8 and 9 Rev exhibited the highest affinities for rPfRh4 binding (20.0, 33.5, and 64.9 nm; Fig. 8, A, B, and F, respectively), which is in accordance with their inhibitory potential. CCPs 1 and 2 m18 interacts with rPfRh4 with an affinity of 336 nm (Fig. 8C), which is markedly reduced from the CCPs 1 and 2 binding constant of 20 nm. As expected CCPs 1 and 2 m20 and CCPs 8 and 9 failed to produce any substantial binding response (Fig. 8, D and E). Although the determined affinity constants for the CR1-based inhibitors are in excellent correlation with their biological activities in this study, we noted that the binding constants differ from previous studies, in which CCPs 1–7 or CCPs 1–3 were found to interact with PfRh4 with affinities in the range from 0.5 to 11 μm (32, 33). To rule out that the presence of CCP 3 (within the construct CCPs 1–3) alters the binding affinities to rPfRh4, we also performed affinity measurements using CCPs 1–3 (Fig. 8G). With a KD of 39.2 nm CCPs 1–3 exhibit similar binding properties when compared with CCPs 1 and 2, ruling out a prominent influence of CCP 3 on the underlying interaction. Because the previous studies have assayed the binding behavior in HEPES buffer in the absence of MgCl2, we also interrogated if the presence of magnesium ions could have modified the interaction. However, the presence or absence of magnesium ions did not influence the binding of CCPs 1 and 2 to rPfRh4 with the KD in 1 mm MgCl2 or 3 mm EDTA buffer being fitted to 20.0 or 16.9 nm (Fig. 8, A and H, respectively). Thus, the markedly tighter association between the CR1 constructs and rPfRh4 observed in this study likely originates from a modified purification protocol of the rPfRh4 construct, which is discussed below. Using SPR, we determined that the inhibitory biological activity for the CCPs 1 and 2 and CCPs 8 and 9 mutant constructs correlates remarkably well with the affinities for their binding partner rPfRh4, which are in the lower nanomolar range.
FIGURE 8.

Binding activity of CR1 constructs for rPfRh4 as measured by SPR. A, 2900 resonance units (RU) rPfRh4 were coupled covalently to a carboxymethyldextran hydrogel biosensor chip. Reference subtracted sensorgrams of a 2-fold analyte dilution series for CCPs 1 and 2 binding to rPfRh4 are shown. The right plot is the respective response at steady state versus molar concentration fitted to a 1:1 steady state affinity model. CCPs 1 and 2 m6–9 (B), CCPs 1 and 2 m18 (C), CCPs 1 and 2 m20 (D), CCPs 8 and 9 (E), and CCPs 8 and 9 Rev (F), are as described above. G, 1700 resonance units resonance units of rPfRh4 were coupled covalently to a carboxymethyldextran hydrogel biosensor chip, and a 2-fold concentration series of CCPs 1–3 in running buffer containing 1 mm MgCl2 was probed for binding. Reference-subtracted sensorgrams are shown. The corresponding right plots are of responses achieved in panel G at steady state versus molar concentration fitted to a 1:1 steady state affinity model. H, binding was assayed for CCPs 1 and 2 in running buffer containing 3 mm EDTA MgCl2 over a chip immobilized with 1950 resonance units of rPfRh4. A 2-fold concentration series was probed for binding, and reference subtracted sensorgrams are shown. The right-hand graph shows a plot of responses achieved in panel H at steady state versus molar concentration, which was fitted to a 1:1 steady state affinity model.
Discussion
Parasite entry into red blood cells is an essential component of the P. falciparum life cycle in humans. P. falciparum has evolved to use redundant invasion pathways, and through the ability to switch pathways via differential expression of parasite adhesins, the parasite has more opportunities for successful invasion in the face of the human immune response and red blood cell polymorphisms prevalent in malaria endemic regions. By activating expression of PfRh4, the parasite is able to switch receptor usage from sialic acid-dependent to sialic acid-independent pathways, thus providing a mechanism for the parasite to invade via non-glycophorin mediated entry. In the work reported here we characterize a repertoire of anti-PfRh4 mAbs including the first anti-PfRh4 neutralizing antibody (summarized in Table 2) and describe a collection of CR1-based inhibitors, which are able to block PfRh4 interaction with CR1 and inhibit P. falciparum sialic-acid independent invasion pathways. Collectively, these results will guide future work on the development of single epitope inhibitors of the CR1-PfRh4 invasion pathway.
TABLE 2.
Characterization of anti-PfRh4 monoclonal antibodies
+ and − refer to positive and negative reactivity to PfRh4, respectively, using different detection techniques. For the interaction and functional assays, N, E, and I refer to neutral, enhancing, and inhibitory effects, respectively. WB, Western blot; IP, immunoprecipitation; IFA, indirect immunofluorescence assay; GIA, growth inhibition assay.

5H12 mAb represents the first anti-PfRh4 neutralizing antibody. Using three robust PfRh4-CR1 interaction assays based on ELISA, FRET, and immunoprecipitation, we show that 5H12 is able to disrupt PfRh4 interaction with CR1. The addition of 5H12 also blocks PfRh4 binding to red blood cells and inhibits the PfRh4-CR1 pathway in P. falciparum growth assays. A recent study reporting the atomic resolution structure of PfRh5 in complex with its inhibitory antibodies QA1 and QA5 show that the epitopes for these antibodies overlap with the basigin N- and C-terminal domain binding site, respectively. We hypothesize that 5H12 would bind to a conformation-dependent epitope that overlaps with the CR1 binding site in PfRh4 and in effect blocks the ability for the receptor to bind. Epitope mapping of 5H12 with PfRh4 using x-ray crystallography will allow us to identify the binding interface and guide the design of inhibitory antibody epitopes.
6A5 and 10C9 mAb are able to enhance PfRh4 binding to CR1 on red blood cells. Epitope mapping shows that 10C9 bound two overlapping peptides, N662 (NFTHKTIQQILQQQY) and K666 (KTIQQILQQQYKNNT), whereas 6A5 recognized only peptide K666. The striking feature of these peptides is the presence of polyglutamine repeats. As these epitopes are present outside of the minimal red blood cell binding domain of PfRh4 (31), the binding of the mAbs may elicit an allosteric conformational change that enhances the binding capabilities of PfRh4 to red blood cells. Previously we observed that the addition of rabbit polyclonal antibodies raised to the C-terminal end of native PfRh4 were able to enhance the binding of PfRh4 to red blood cells (data not shown).5 Examination of the protein sequence highlighted the presence of QQXXXXQQQ and a triple QQQ site within the recombinant protein used as immunogen. Antibodies that bind to these sites may elicit an allosteric change in full-length PfRh4, resulting in enhanced binding to CR1, although the molecular mechanism remains to be experimentally verified. It will be important to exclude the epitopes above in future immunizations as not to generate antibodies that enhance the interaction between PfRh4 and CR1.
We also describe four CR1-based inhibitors; CCPs 1 and 2, CCPs 1 and 2 m6–9, CCPs 1 and 2 m18, and CCPs 8 and 9 Rev. These inhibitors competed for PfRh4 binding to erythroid CR1 and block the use of the PfRh4-CR1 invasion pathways in P. falciparum. Their inhibitory effects directly correlate with their affinity for PfRh4. The least potent inhibitor CCPs 1 and 2 m18 has a KD of 336 nm compared with the 33, 65, and 20 nm for CCPs 1 and 2 m6–9, CCPs 8 and 9 Rev, and CCPs 1 and 2, respectively. In contrast to the affinity constants for the rPfRh4-CR1 interaction being in the lower nanomolar range as determined in this study, two previous studies have found the binding constants for the interaction of CR1 CCPs 1–7 or CCPs 1–3 to be in the range between 0.5 and 11 μm (32, 33). This former work has also employed SPR to measure the affinity constants for the binding of sensor chip-immobilized rPfRh4 to CR1 CCPs 1–3 or CCP s1–7. Although the CCPs 1–3 construct was overexpressed by P. pastoris and purified to homogeneity, the CCPs 1–7 wild type and mutant constructs were expressed by HEK293T with the proteins being directly harvested from serum-free supernatants or captured by metal affinity chromatograph facilitated by a hexa-His tag. Despite differences in glycosylation status and purity, the CCPs 1–3 and CCPs 1–7 preparations exhibited an overall similar binding behavior for rPfRh4 (0.5–11 μm).
In our present study we measured the affinity of Pichia-produced CCPs 1–3 for rPfRh4 alongside the novel wild type and mutant constructs within CCPs 1 and 2 and observed a markedly tighter interaction than described before (32, 33). Because the CCP 1 and 3 proteins in all studies have been prepared by the same procedures, a possible reason for this remarkable shift toward higher affinity likely originates in the modification of the purification protocol for rPfRh4, which now includes tobacco etch virus cleavage of the N-terminal hexa-His tag followed by ion exchange chromatography. The fact that the removal of 23 amino acids including the purification tag leads to a substantial increase in affinity argues that accessibility of the N terminus in PfRh4 is critical for its interaction with CR1. In terms of specific amino acids within CCP 1 that are critical for binding PfRh4, previous work identified that m6–9 exhibited a >60% loss of affinity for rPfRh4, whereas m18 and m20 mutants completely abolished the interaction in vitro. Although we are able to establish that m20 is a critical residue involved in mediating PfRh4 interaction, we observed that m6–9 and m18, although lowering the affinity to various degrees, were not essential. We do note, however, that m18 did result in a strong reduction in PfRh4 binding, whereas the effect of the m6–9 substitution is negligible. Possible reasons for the different outcomes may originate from the different preparations of rPfRh4, resulting in the presence or absence of the purification tag.
Structurally, our study refines the previously proposed binding site and suggests that the surface-exposed, hydrophobic residue Phe-20 plays the primary role in binding PfRh4 with some contribution from the negatively charged side chain of Asp-18, which on the surface of CCP 1 lies directly next to the aromatic side chain of Phe-20.
Park et al. (32) were able to engineer an artificial binding site within CCPs 8–14 by substituting residues within CCP 1 that are critical for PfRh4 interaction to their homologous position in CCP 8. This artificial binding site carrying six CCP 1 amino acid substitutions, m7–9,18–20r, bound ∼30-fold better than the wild type binding site as measured by SPR. Consistent with this, our results show that CCPs 8 and 9 Rev was able to form a stable complex with rPfRh4, whereas CCPs 8 and 9 did not. With 20 and 65 nm we found the affinities between rPfRh4 and CCPs 1 and 2 or CCPs 8 and 9 Rev, respectively, to be in a similar range. This observation is in agreement with the comparable biological activity observed for these two constructs. Although we also observed conversion of CCPs 8 and 9 into an active PfRh4 binding partner upon homologous substitution of six amino acids, we were unable to confirm that CCPs 8 and 9 Rev bound substantially tighter to PfRh4 than CCPs 1 and 2. A possible reason for this discrepancy may be that the novel purification method used for rPfRh4 produces a protein that more closely reflects the physiological conformation of the parasite adhesin on merozoites. Another reason may lie in a potential contribution of CCP domains 10–14 in binding to PfRh4. However, this is unlikely as the addition of 5H12 mAb is sufficient to prevent the formation of the complex, suggesting that CCPs 8 and 9 Rev and CCPs 1 and 2 essentially bind in the same mode to the exact same epitope on PfRh4. Furthermore, CCPs 8 and 9 Rev is able to block PfRh4 binding to red blood cells and functions as an inhibitor of P. falciparum PfRh4-CR1 invasion pathway to a similar extent as CCPs 1 and 2, CCPs 1–3, and sCR1.
Our results highlight distinct regions within PfRh4 and CR1 that are important for mediating entry of P. falciparum parasites. We have demonstrated that it is possible to generate anti-PfRh4 neutralizing antibodies and a repertoire of CR1-based inhibitors that both block PfRh4-CR1 interaction and show functional inhibition in P. falciparum growth. Furthermore, we identified distinct epitopes within PfRh4 that should not be included in future vaccinations because of their ability to generate antibodies that enhance the interaction with CR1. This collection of anti-PfRh4 mAbs and CR1 constructs will be invaluable tools in the efforts to obtain an atomic resolution structure of the binding interface between PfRh4 and CR1 that will further inform the rational design of potent inhibitors.
Author Contributions
N. T. Y. L. and W.-H. T. contributed to project concept, acquisition, analyses, and interpretation of the data for the whole manuscript. C. Q. S. and M. J. H. expressed FH fragments in P. pastoris. C. Q. S., M. J. H., and C. S. L. performed surface plasmon resonance experiments. M. J. C. assisted with the design and experiments with FRET assay. A. T. K. performed P. falciparum growth assays. C. W. assisted in protein purification of rPfRh4 for antibody immunizations. A. F. C. also assisted in the design of experiments and interpretation of results, and all other authors were involved in writing and reviewing the manuscript.
Acknowledgments
We acknowledge the Victorian State Government Operational Infrastructure Support and Australian Government, National Health and Medical Research Council (NHMRC) Independent Research Institute Infrastructure Support Scheme (IRIISS). We acknowledge the excellent service at the Core Unit Mass Spectrometry and Proteomics (CUMP) of the University Clinic Ulm and thank Remco Sprangers for kind help in acquiring the 1H NMR spectra.
This work was supported by National Health and Medical Research Council Grant APP1026581 and an Australian Research Council Future Fellowship (to W. H.-T.). The authors declare that they have no conflicts of interest with the contents of this article.
5H12 mAb did not detect native PfRh4 by Western blotting in both reducing and non-reducing conditions.
Using competition ELISA, only unlabelled 5H12 mAb could outcompete 5H12-HRP for binding to immobilized rPfRh4.
The addition of rabbit polyclonal antibodies raised to the C-terminal end of PfRh4 was able to enhance binding of native PfRh4 to red blood cells with no effect on EBA-175 binding.
- EBA
- erythrocyte binding-like antigen
- PfRh
- P. falciparum reticulocyte binding-like homolog protein
- rPfRh
- recombinant PfRh
- CR1
- complement receptor 1
- mAb
- monoclonal antibody
- CCP
- complement control protein
- SPR
- surface plasmon resonance
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1. Tham W.-H., Healer J., Cowman A. F. (2012) Erythrocyte and reticulocyte binding-like proteins of Plasmodium falciparum. Trends Parasitol. 28, 23–30 [DOI] [PubMed] [Google Scholar]
- 2. Miller L. H., Baruch D. I., Marsh K., Doumbo O. K. (2002) The pathogenic basis of malaria. Nature 415, 673–679 [DOI] [PubMed] [Google Scholar]
- 3. Dvorak J. A., Miller L. H., Whitehouse W. C., Shiroishi T. (1975) Invasion of erythrocytes by malaria merozoites. Science 187, 748–750 [DOI] [PubMed] [Google Scholar]
- 4. Gilson P. R., Crabb B. S. (2009) Morphology and kinetics of the three distinct phases of red blood cell invasion by Plasmodium falciparum merozoites. Int. J. Parasitol. 39, 91–96 [DOI] [PubMed] [Google Scholar]
- 5. Miller L. H., Aikawa M., Johnson J. G., Shiroishi T. (1979) Interaction between cytochalasin B-treated malarial parasites and erythrocytes: attachment and junction formation. J. Exp. Med. 149, 172–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Riglar D. T., Richard D., Wilson D. W., Boyle M. J., Dekiwadia C., Turnbull L., Angrisano F., Marapana D. S., Rogers K. L., Whitchurch C. B., Beeson J. G., Cowman A. F., Ralph S. A., Baum J. (2011) Super-resolution dissection of coordinated events during malaria parasite invasion of the human erythrocyte. Cell Host Microbe 9, 9–20 [DOI] [PubMed] [Google Scholar]
- 7. Aikawa M., Miller L. H., Johnson J., Rabbege J. (1978) Erythrocyte entry by malarial parasites: a moving junction between erythrocyte and parasite. J. Cell Biol. 77, 72–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Adams J. H., Sim B. K., Dolan S. A., Fang X., Kaslow D. C., Miller L. H. (1992) A family of erythrocyte binding proteins of malaria parasites. Proc. Natl. Acad. Sci. U.S.A. 89, 7085–7089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Haynes J. D., Dalton J. P., Klotz F. W., McGinniss M. H., Hadley T. J., Hudson D. E., Miller L. H. (1988) Receptor-like specificity of a Plasmodium knowlesi malarial protein that binds to Duffy antigen ligands on erythrocytes. J. Exp. Med. 167, 1873–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Triglia T., Thompson J., Caruana S. R., Delorenzi M., Speed T., Cowman A. F. (2001) Identification of proteins from Plasmodium falciparum that are homologous to reticulocyte binding proteins in Plasmodium vivax. Infect. Immun. 69, 1084–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rayner J. C., Vargas-Serrato E., Huber C. S., Galinski M. R., Barnwell J. W. (2001) A Plasmodium falciparum homologue of Plasmodium vivax reticulocyte binding protein (PvRBP1) defines a trypsin-resistant erythrocyte invasion pathway. J. Exp. Med. 194, 1571–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hayton K., Gaur D., Liu A., Takahashi J., Henschen B., Singh S., Lambert L., Furuya T., Bouttenot R., Doll M., Nawaz F., Mu J., Jiang L., Miller L. H., Wellems T. E. (2008) Erythrocyte binding protein PfRH5 polymorphisms determine species-specific pathways of Plasmodium falciparum invasion. Cell Host Microbe 4, 40–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lobo C.-A., Rodriguez M., Reid M., Lustigman S. (2003) Glycophorin C is the receptor for the Plasmodium falciparum erythrocyte binding ligand PfEBP-2 (baebl). Blood 101, 4628–4631 [DOI] [PubMed] [Google Scholar]
- 14. Maier A. G., Duraisingh M. T., Reeder J. C., Patel S. S., Kazura J. W., Zimmerman P. A., Cowman A. F. (2003) Plasmodium falciparum erythrocyte invasion through glycophorin C and selection for Gerbich negativity in human populations. Nat. Med. 9, 87–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mayer D. C., Mu J.-B., Feng X., Su X. Z., Miller L. H. (2002) Polymorphism in a Plasmodium falciparum erythrocyte-binding ligand changes its receptor specificity. J. Exp. Med. 196, 1523–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sim B. K., Chitnis C. E., Wasniowska K., Hadley T. J., Miller L. H. (1994) Receptor and ligand domains for invasion of erythrocytes by Plasmodium falciparum. Science 264, 1941–1944 [DOI] [PubMed] [Google Scholar]
- 17. Tham W.-H., Wilson D. W., Lopaticki S., Schmidt C. Q., Tetteh-Quarcoo P. B., Barlow P. N., Richard D., Corbin J. E., Beeson J. G., Cowman A. F. (2010) Complement receptor 1 is the host erythrocyte receptor for Plasmodium falciparum PfRh4 invasion ligand. Proc. Natl. Acad. Sci. U.S.A. 107, 17327–17332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Crosnier C., Bustamante L. Y., Bartholdson S. J., Bei A. K., Theron M., Uchikawa M., Mboup S., Ndir O., Kwiatkowski D. P., Duraisingh M. T., Rayner J. C., Wright G. J. (2011) Basigin is a receptor essential for erythrocyte invasion by Plasmodium falciparum. Nature 480, 534–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Spadafora C., Awandare G. A., Kopydlowski K. M., Czege J., Moch J. K., Finberg R. W., Tsokos G. C., Stoute J. A. (2010) Complement receptor 1 is a sialic acid-independent erythrocyte receptor of Plasmodium falciparum. PLoS Pathog. 6, e1000968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Richards J. S., Arumugam T. U., Reiling L., Healer J., Hodder A. N., Fowkes F. J., Cross N., Langer C., Takeo S., Uboldi A. D., Thompson J. K., Gilson P. R., Coppel R. L., Siba P. M., King C. L., Torii M., Chitnis C. E., Narum D. L., Mueller I., Crabb B. S., Cowman A. F., Tsuboi T., Beeson J. G. (2013) Identification and prioritization of merozoite antigens as targets of protective human immunity to Plasmodium falciparum malaria for vaccine and biomarker development. J. Immunol. 191, 795–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Persson K. E., Fowkes F. J., McCallum F. J., Gicheru N., Reiling L., Richards J. S., Wilson D. W., Lopaticki S., Cowman A. F., Marsh K., Beeson J. G. (2013) Erythrocyte-binding antigens of Plasmodium falciparum are targets of human inhibitory antibodies and function to evade naturally acquired immunity. J. Immunol. 191, 785–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lopaticki S., Maier A. G., Thompson J., Wilson D. W., Tham W.-H., Triglia T., Gout A., Speed T. P., Beeson J. G., Healer J., Cowman A. F. (2011) Reticulocyte and erythrocyte binding-like proteins function cooperatively in invasion of human erythrocytes by malaria parasites. Infect. Immun. 79, 1107–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Douglas A. D., Williams A. R., Knuepfer E., Illingworth J. J., Furze J. M., Crosnier C., Choudhary P., Bustamante L. Y., Zakutansky S. E., Awuah D. K., Alanine D. G., Theron M., Worth A., Shimkets R., Rayner J. C., Holder A. A., Wright G. J., Draper S. J. (2014) Neutralization of Plasmodium falciparum merozoites by antibodies against PfRH5. J. Immunol. 192, 245–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reiling L., Richards J. S., Fowkes F. J., Wilson D. W., Chokejindachai W., Barry A. E., Tham W.-H., Stubbs J., Langer C., Donelson J., Michon P., Tavul L., Crabb B. S., Siba P. M., Cowman A. F., Mueller I., Beeson J. G. (2012) The Plasmodium falciparum erythrocyte invasion ligand Pfrh4 as a target of functional and protective human antibodies against malaria. PLoS ONE 7, e45253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reiling L., Richards J. S., Fowkes F. J., Barry A. E., Triglia T., Chokejindachai W., Michon P., Tavul L., Siba P. M., Cowman A. F., Mueller I., Beeson J. G. (2010) Evidence that the erythrocyte invasion ligand PfRh2 is a target of protective immunity against Plasmodium falciparum malaria. J. Immunol. 185, 6157–6167 [DOI] [PubMed] [Google Scholar]
- 26. Gao X., Gunalan K., Yap S. S., Preiser P. R. (2013) Triggers of key calcium signals during erythrocyte invasion by Plasmodium falciparum. Nat. Commun. 4, 2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Douglas A. D., Baldeviano G. C., Lucas C. M., Lugo-Roman L. A., Crosnier C., Bartholdson S. J., Diouf A., Miura K., Lambert L. E., Ventocilla J. A., Leiva K. P., Milne K. H., Illingworth J. J., Spencer A. J., Hjerrild K. A., Alanine D. G., Turner A. V., Moorhead J. T., Edgel K. A., Wu Y., Long C. A., Wright G. J., Lescano A. G., Draper S. J. (2015) A PfRH5-based vaccine is efficacious against heterologous strain blood-stage Plasmodium falciparum infection in Aotus monkeys. Cell Host Microbe. 17, 130–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen L., Xu Y., Healer J., Thompson J. K., Smith B. J., Lawrence M. C., Cowman A. F. (2014) Crystal structure of PfRh5, an essential P. falciparum ligand for invasion of human erythrocytes. Elife 10.7554/eLife.04187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wright K. E., Hjerrild K. A., Bartlett J., Douglas A. D., Jin J., Brown R. E., Illingworth J. J., Ashfield R., Clemmensen S. B., de Jongh W. A., Draper S. J., Higgins M. K. (2014) Structure of malaria invasion protein RH5 with erythrocyte basigin and blocking antibodies. Nature 515, 427–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schmidt C. Q., Kennedy A. T., Tham W.-H. (2015) More than just immune evasion: hijacking complement by Plasmodium falciparum. Mol. Immunol. 67, 71–84 [DOI] [PubMed] [Google Scholar]
- 31. Tham W.-H., Wilson D. W., Reiling L., Chen L., Beeson J. G., Cowman A. F. (2009) Antibodies to reticulocyte binding protein-like homologue 4 inhibit invasion of Plasmodium falciparum into human erythrocytes. Infect. Immun. 77, 2427–2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Park H. J., Guariento M., Maciejewski M., Hauhart R., Tham W.-H., Cowman A. F., Schmidt C. Q., Mertens H. D., Liszewski M. K., Hourcade D. E., Barlow P. N., Atkinson J. P. (2014) Using mutagenesis and structural biology to map the binding site for the Plasmodium falciparum merozoite protein PfRh4 on the human immune adherence receptor. J. Biol. Chem. 289, 450–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tham W.-H., Schmidt C. Q., Hauhart R. E., Guariento M., Tetteh-Quarcoo P. B., Lopaticki S., Atkinson J. P., Barlow P. N., Cowman A. F. (2011) Plasmodium falciparum uses a key functional site in complement receptor type-1 for invasion of human erythrocytes. Blood 118, 1923–1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Awandare G. A., Spadafora C., Moch J. K., Dutta S., Haynes J. D., Stoute J. A. (2011) Plasmodium falciparum field isolates use complement receptor 1 (CR1) as a receptor for invasion of erythrocytes. Mol. Biochem. Parasitol. 177, 57–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Weiss G. E., Gilson P. R., Taechalertpaisarn T., Tham W.-H., de Jong N. W., Harvey K. L., Fowkes F. J., Barlow P. N., Rayner J. C., Wright G. J., Cowman A. F., Crabb B. S. (2015) Revealing the sequence and resulting cellular morphology of receptor-ligand interactions during Plasmodium falciparum invasion of erythrocytes. PLoS Pathog. 11, e1004670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Boyle M. J., Wilson D. W., Richards J. S., Riglar D. T., Tetteh K. K., Conway D. J., Ralph S. A., Baum J., Beeson J. G. (2010) Isolation of viable Plasmodium falciparum merozoites to define erythrocyte invasion events and advance vaccine and drug development. Proc. Natl. Acad. Sci. U.S.A. 107, 14378–14383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wilson D. W., Crabb B. S., Beeson J. G. (2010) Development of fluorescent Plasmodium falciparum for in vitro growth inhibition assays. Malar. J. 9, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schmidt C. Q., Slingsby F. C., Richards A., Barlow P. N. (2011) Production of biologically active complement factor H in therapeutically useful quantities. Protein Expr. Purif. 76, 254–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Triglia T., Tham W.-H., Hodder A., Cowman A. F. (2009) Reticulocyte binding protein homologues are key adhesins during erythrocyte invasion by Plasmodium falciparum. Cell. Microbiol. 11, 1671–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]