

Background: Autoantibodies against complement C3 are found in patients with the autoimmune disease systemic lupus erythematosus.
Results: C3 autoantibodies are found in 30% of lupus nephritis patients and inhibit C3 interaction with the regulatory proteins Factor H and CR1.
Conclusion: C3 autoantibodies have overt capability to overactivate complement.
Significance: C3 autoantibodies can contribute to the pathological process in lupus nephritis.
Keywords: antibody, autoimmune disease, complement system, protein-protein interaction, surface plasmon resonance (SPR)
Abstract
Lupus nephritis (LN) is a complication of the autoimmune disease systemic lupus erythematosus. Because the complement system plays a critical role in orchestrating inflammatory and immune responses as well as in the clearance of immune complexes, autoreactivity to complement components may have considerable pathological consequences. Autoantibodies against the central complement component C3 have been reported in systemic lupus erythematosus, but their molecular mechanism and functional relevance are not well understood. The objective of this study was to evaluate the frequency and the functional properties of the anti-C3 autoantibodies. Anti-C3 autoantibodies were measured in plasma of 39 LN patients, and identification of their epitopes on the C3 molecule was performed. By using surface plasmon resonance, we analyzed the influence of patient-derived IgG antibodies on the interaction of C3b with Factor B, Factor H, and complement receptor 1. The capacity of these antibodies to dysregulate the C3 convertase on the surface of endothelial cell was measured by flow cytometry. Here we report that the frequency of anti-C3 autoantibodies in LN is ∼30%. They inhibited interactions of the negative complement regulators Factor H and complement receptor 1 with C3b. An enhanced C3 deposition was also observed on human endothelial cells in the presence of C3 autoantibodies. In addition, anti-C3 autoantibody levels correlated with disease activity. In conclusion, the anti-C3 autoantibodies in LN may contribute to the autoimmune pathology by their capacity to overactivate the complement system.
Introduction
Lupus nephritis (LN)4 is a complication of systemic lupus erythematosus (SLE) that significantly increases patients' morbidity and mortality. The disease progression is associated with the presence of autoantibodies that recognize various self-molecules, including dsDNA, nuclear proteins, ribosomal proteins, and complement component C1q (1–3). These autoantibodies may appear as a result of an immune response induced by an inefficient clearance of apoptotic cells and cellular debris, which serve as a source of autoantigens. Because the complement system plays a critical role in orchestrating inflammatory and immune responses and clearance of immune complexes and apoptotic cells (4–6), autoreactivity to complement may have considerable pathological consequences (7–9). Despite the predominant role of the classical pathway in the initiation of complement activation in SLE, complement-mediated damage is often caused by the alternative pathway amplification loop (10). C3 is a convergent point of the complement system, common between the classical, lectin, and alternative pathways. Antibodies against C3 have been described in patients with autoimmune diseases, such as SLE (11–14) or Crohn disease (15), in some nephrotic kidney diseases (16, 17), and in dense deposit disease (DDD) (7) as well as in autoimmune-prone mice (12). Based on the limited functional studies of these antibodies, it is difficult to conclude whether they are a disease-relevant factor or a simple epiphenomenon, one among many, arising from the dysregulated immune response in the autoimmune diseases (18, 19). To date, anti-C3 antibodies have been shown to inhibit the inactivation of C3b to iC3b by Factor I (FI) in the presence of complement receptor 1 (CR1) as a cofactor (14) and to prevent the clearance of apoptotic cells by mouse but not human macrophages (12). No study of the anti-C3 antibodies has been carried out for SLE patients with LN. Therefore, we sought to investigate the prevalence of anti-C3 antibodies in patients with LN and to characterize their functional properties in order to evaluate their pathological relevance.
We have demonstrated that anti-C3 antibodies are present in ∼30% of the studied cohort of SLE patients with LN. They interfere with the regulation of the alternative pathway C3 convertase and therefore could have pathological consequences in LN.
Experimental Procedures
Cohort Description
Thirty-nine Bulgarian patients with SLE and biopsy-proven LN, who were attended during a period from August 2011 to June 2014 at the Nephrology Clinic of University Hospital “Tzaritza Ioanna-ISUL,” Medical University (Sofia, Bulgaria) were enrolled in the study. All patients provided written informed consent. The cohort consisted of 32 females (82.1%) and 7 males (17.9%) with a median age of 39 years (range 23–65 years) and a median LN duration of 9 years (range 0.1–29 years). The patients were distributed according to the World Health Organization classification of LN as follows: 1 patient (2.6%) had LN class I, 12 (30.7%) had LN class II, 3 (7.7%) had LN class III, 17 (43.6%) had LN class IV, 5 (12.8%) had LN class V, and 1 (2.6%) had LN class VI. At the time of plasma sampling, the LN patients were categorized according to the British Islet Lupus Assessment Group (BILAG) renal score as follows: 12 patients (30.7%) with category A LN, 9 (23.1%) with category B, 5 (12.8%) with category C, and 13 (33.3%) with category D. There were no patients with category E LN in our cohort. The BILAG renal score assesses the activity of LN according to the presence of specific clinical, laboratory, and histology features (20, 21). BILAG renal score (22) category A means that LN is active and requires steroid or immunosuppressive treatment. Category B means that the activity of LN requires only symptomatic therapy. Category C indicates mild and stable LN. Category D indicates previous renal disease, and category E indicates no previous renal disease. During the study period of 3 years, 21 of the patients visited the Nephrology Clinic more than once and were monitored by multiple collections of plasma and urine samples. A total of 84 plasma samples were analyzed.
ELISA for Identification of Anti-C3 Antibodies
Coating of ELISA plates was performed at 20 μg/ml C3 (Complement Technology (Tyler, TX) or Calbiochem) in PBS for 1 h, followed by a blocking of the plates by PBS plus 0.25% Tween 20. Plasma was diluted 1:100 in PBS plus 0.05% Tween 20 and was applied for 1 h, and bound IgG was revealed by an anti-human IgG antibody conjugated with HRP (Southern Biotech) diluted 1:1000 in PBS plus 0.05% Tween 20, followed by a 3,3′,5,5′-tetramethylbenzidine substrate system. Alternatively, plasma samples were serially diluted starting from 1:50 and applied on coated and blocked plates to evaluate the dose response of the binding of the autoantibodies.
For epitope mapping of the anti-C3 autoantibodies, different C3 fragments (C3b, C3c, and C3d) or proteins homologous to C3, such as C4 and C5 (all from Complement Technology), were coated as described for C3, and a 1:100 dilution of plasma was applied. Screening of anti-FB antibodies was done by the same approach.
IgG Purification
IgG was purified from plasma of patients positive or negative for anti-C3 autoantibodies or from plasma of healthy donors by using Protein G beads (GE Healthcare), as recommended by the manufacturer. The concentration of the IgG was determined by a Nanodrop spectrophotometer, and the purity of IgG was determined by SDS-PAGE on 10% precast gels (Invitrogen, Life Technologies, Inc., and Novex), followed by silver staining of the gel (Bio-Rad, Marne-la-coquette, France).
Characterization of the Interaction of the Anti-C3 Autoantibodies with C3b by Surface Plasmon Resonance (SPR)
The interaction of the anti-C3 autoantibodies with C3b was analyzed in real time using ProteOn XPR36 SPR equipment (Bio-Rad). C3b (Complement Technology) was covalently immobilized to a GLC sensor chip (Bio-Rad) following the manufacturer's procedure. Protein G-purified IgG from LN patients or healthy donors was injected for 300 s at six different concentrations (300, 150, 75, 35, 17.5, and 0 μg/ml) diluted in PBS plus 0.005% Tween running buffer. The dissociation was followed for 300 s. The signal from the interspots, reflecting the background binding, was subtracted, as recommended by the manufacturer. A channel coated with C5 was used as a negative control because no patient from the cohort showed binding to this protein. Because the exact proportion of the anti-C3 antibodies among the patients' IgG was unknown, kinetic analyses that determine the precise binding constants and affinity were unfeasible. In order to evaluate the stability of the formed complexes, the dissociation rates were calculated by using ProteOn Manager software.
Effect of the Anti-C3 Autoantibodies on the Interaction of FH and Soluble CR1 with C3b
First, C3b was immobilized on a sensor chip as described above, and patients' or normal donors' IgG was injected at 300 μg/ml. PBS with 0.005% Tween 20 was used as a running and sample dilution buffer. Following 300 s of dissociation, FH (Complement Technology) at 50 μg/ml or soluble CR1 (R&D Systems) at 10 μg/ml was injected. The interaction of FH with C3b was monitored for 200 s of association and 300 s of dissociation. The interaction of CR1 with C3b was followed for 300 s of association and 600 s of dissociation. Background signal from a mock-immobilized line of the chip or the interspots was subtracted by ProteOn Manager software. The FH or sCR1 injection was aligned at 0 RU at the moment of injection in order to evaluate the effect of IgG on FH and sCR1 binding.
Effect of the Anti-C3 Autoantibodies on the Interaction of FB with C3b and the Formation of a C3 Convertase
To study the effect of the anti-C3 IgGs on the interaction of FB with C3b and the C3 convertase formation, Protein G-purified IgG from LN patients or healthy donors was injected for 300 s at concentration of 300 μg/ml in 10 mm Hepes, 75 mm NaCl, 1 mm MgCl, 0.005% Tween 20, pH 7.4, buffer, on a C3b-coupled biosensor chip. The same volume of running buffer was separately injected to serve as a control. Following 300 s of dissociation, FB at 80 μg/ml in the presence or in the absence of FD at 8 μg/ml (Complement Technology) was injected for 300 s of association, followed by 300 s of dissociation. The background signal from the interspots was subtracted. Data were analyzed as for FH and CR1.
C3 Convertase Activity Assay
To test the capacity of the anti-C3 antibodies to modify the efficacy of the C3 convertase, 10 μl of C3 (25 μg/ml) was preincubated with 10 μl of healthy donor or autoantibody-positive patient IgG (1 mg/ml) for 10 min on ice. Subsequently, the mix was incubated with 4 μl of FB (25 μg/ml) and FD (0.25 μg/ml) for 0, 2, or 10 min at 37 °C. The reaction buffer was 10 mm Tris, 40 mm NaCl, 10 mm MgCl2, pH 7.4. The reaction was stopped by the addition of 12 μl of a reducing sample buffer (3×). The cleavage of C3 to C3b was probed by Western blot. The cleavage efficiency was evaluated by the appearance of the α′ band (with lower molecular weight due to the loss of the C3a part) and the disappearance of the α band. To test the capacity of FH to prevent the formation and to dissociate the C3 convertase, FH at 2 μg/ml was added to the mix and incubated for 10 min.
FI Cofactor Assay
To test the capacity of the anti-C3 autoantibodies to modify the cofactor activity of FH for FI, resulting in C3b inactivation, a cofactor assay was performed. C3b (2 μl, 20 μg/ml) was incubated on ice for 10 min with purified IgG (20 μl, 1 mg/ml) from healthy controls and anti-C3 antibody-positive patients on ice. FH (2 μl, 20 μg/ml) was then added to the mix, followed by FI (2 μl, 20 μg/ml). The mix was incubated at 37 °C for 0 s, 30 s, 2 min, and 10 min in a TBS buffer (10 mm Tris, 150 mm NaCl, pH 7.4), and the reaction was stopped by the addition of 13 μl of reducing sample buffer (3×). The cleavage of C3b to iC3b was probed by Western blot. The cleavage efficiency was evaluated by the appearance of the α43 band and the disappearance of the α band.
Alternative Pathway Activation in Serum
Purified IgG from normal donors and autoantibody-positive patients was added to the normal human serum diluted 1:3 in 10 mm MgCl2 and 10 mm EGTA TBS buffer and incubated for 30 min at 37 °C. The released Ba, a measure of the amount of formed C3 convertases, was quantified by the MicroVue Ba kit (Quidel).
C3 Deposition on Endothelial Cells
Resting primary human endothelial cells were isolated from an umbilical vein and cultured as described (23–25). At passage 3, the cells were seeded to 24-well plates and incubated with a normal human serum alone or supplemented with 500 μg/ml IgG purified from healthy donors or from LN patients with or without anti-C3 autoantibodies. The serum was diluted 1:3 in M199 medium, in the presence or in the absence of 10 mm EGTA and 10 mm MgCl2. EGTA-Mg served to inhibit the classical and the lectin pathways and to allow activation of the alternative complement pathway only. After 30 min of incubation with the serum at 37 °C in 5% CO2, the cells were detached with PBS containing 1% BSA, 0.1% sodium azide, and 10 mm EDTA and labeled. Labeling with anti-C3c monoclonal mouse antibody (Quidel) at 10 μg/ml, followed by a phycoerythrin-labeled secondary antibody (BD Biosciences), was used to detect C3 fragment deposition, and labeling with anti-human IgG-phycoerythrin antibody (BD Biosciences) was used to measure the presence of antibodies that bind to endothelial cells. The cells were analyzed by FACS on an LSRII machine (BD Biosciences) and with FCS Express software. Gating was performed on the entire cell population.
Results
Prevalence of Anti-C3 Autoantibodies in Patients with LN
The screening revealed that 31% of the patients (12 of 39; Fig. 1A) and 33 of 84 samples were positive for anti-C3 antibodies. The cut-off for evaluating of the positivity was set as the average + 3 S.D. values of the binding, obtained with the plasma from the 30 tested healthy donors. The titers of the autoantibody-positive patients varied over time (Fig. 1B). Because in patients with DDD, positive for anti-C3 antibodies, anti-Factor B antibodies were also detected (7), all LN patients in this study were screened for anti-FB antibodies as well. No anti-FB-positive plasma was detected in any patient (data not shown).
FIGURE 1.
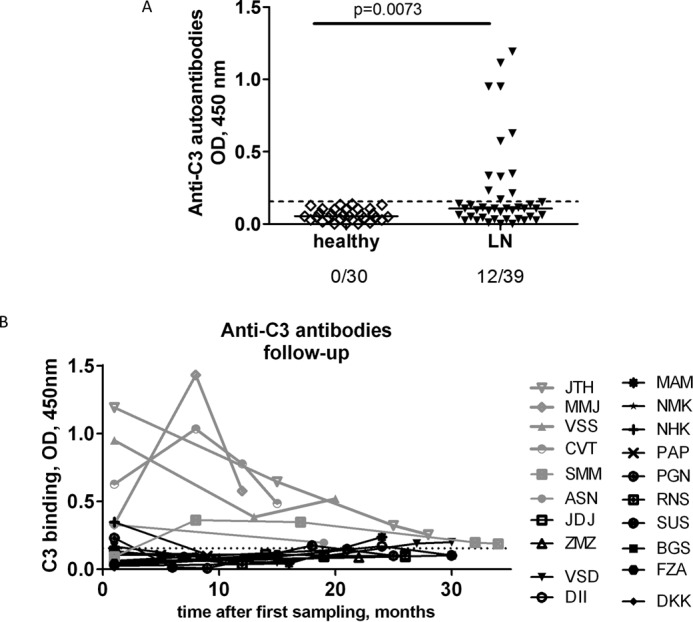
Presence of anti-C3 autoantibodies in LN. A, reactivity against C3 from plasma of patients with LN at the moment of the first sampling, compared with healthy donors, measured by ELISA. The dotted line represents the positivity cut-off, determined as average +3SD of the signal, obtained from the plasma of 20 healthy donors. B, dynamic of the levels of anti-C3 autoantibodies over time. Only the patients for whom two or more samples were available are included in the figure.
Correlation of the Anti-C3 Autoantibody Titers with Disease Activity
Anti-C3 autoantibodies were more frequent in the patients with severe disease, classified as category A according to the BILAG renal score, compared with patients with milder disease, classified as category B, C, or D (p = 0.019; Fig. 2A). The levels of these antibodies were significantly higher in anti-dsDNA-positive patients compared with the negative ones (p = 0.0003) (Fig. 2B). Patients positive for anti-C3 antibodies were significantly younger as compared with the negative ones (p = 0.0086; Fig. 2C). In the samples positive for anti-C3 autoantibodies, the levels of circulating C3 were significantly lower, as compared with the negative samples (p = 0.01; Fig. 2D). Patients positive for anti-C3 also have a tendency to have lower C4 levels compared with the negative cases (p = 0.057; Fig. 2E).
FIGURE 2.

Association of the presence of anti-C3 autoantibodies with the disease parameters. A, levels of anti-C3 autoantibodies according to disease activity. Patients are classified according to BILAG criteria. B, levels of the anti-C3 autoantibodies in patients positive or negative for anti-dsDNA autoantibodies. C, levels of the anti-C3 autoantibodies according to the age of the patients. Shown are C3 (D) and C4 (E) levels in LN patients positive or negative for anti-C3 autoantibodies. Correlation of the levels of anti-C3 autoantibodies with the renal function (eGFR) for two patients for whom samples were available at diagnosis and in follow up: JTH (F) and CVT (G). The statistical analysis was performed by Mann-Whitney t test. Wherever applicable, the dotted line represents the cut-off for the normal range.
The glomerular filtration rate (eGFR) and the creatinine levels of the anti-C3 antibody-positive patients were not significantly different compared with the negative patients (data not shown). Nevertheless, for two of the patients, strongly positive for anti-C3 antibodies at the moment of diagnosis of the LN (JTH and CVT), an alteration of the kidney function was detected during the study period, measured by the eGFR. In both cases, there was a close inverse correlation between the titers of anti-C3 antibodies and the eGFR levels (Fig. 2, F and G, for JTH and CVT, respectively). The other two patients with high anti-C3 titers (MMJ and VSS) retained a normal eGFR throughout the study period despite the dynamic of the antibody titers (Fig. 1B).
Binding Characteristics and Epitope Mapping of Anti-C3 Autoantibodies in LN Patients
The binding of anti-C3 antibodies from patients' plasma to immobilized C3 was dose-dependent, as measured by ELISA (Fig. 3A). Pronounced binding was also detected to immobilized C3b, as measured in real time by SPR-based technology, using purified IgG from plasma of autoantibody-positive patients (Fig. 3B). The level of binding detected by SPR correlated with the ELISA results. For antibodies from patients JTH and CVT, the SPR binding correlated with the eGFR in the same way as the binding detected by ELISA.
FIGURE 3.

Binding of anti-C3 antibodies to C3 and C3b. A, dose dependences of the binding of IgG from different follow up plasma samples to immobilized C3, measured by ELISA. B, binding of purified IgG from the highest positive samples to C3b in real time, measured by SPR. N, normal control IgG, purified from healthy donors. N1, N2, N3, and N4 indicate the number of available consecutive samples from the given patient. The percentage of plasma reflects the sample dilution.
To determine the location of the binding epitopes of the anti-C3 antibodies within the C3 molecule, the reactivities of the patients' plasma were probed toward immobilized C3, C3b, iC3b, C3c, and C3d as well as to C3-homologous proteins C4 and C5. A strong binding was detected to immobilized C3 and C3b in all cases and in all samples (Fig. 4). In a group of patients (MMJ, VSS, and CVT shown), binding was also observed toward iC3b and C3c, whereas JTH showed no recognition of iC3b and C3c. The antibodies from no patient recognized the C3d fragment. The binding profiles varied among different patients, but generally, the profiles of the recognized epitopes were conserved over time. Among the anti-C3-positive patients, in 5 of 12, a recognition of C4 was also detected at least in one sample, whereas no patient plasma was positive for anti-C5 autoantibodies (data not shown).
FIGURE 4.

Epitope mapping of the anti-C3 antibodies. The binding of IgG from patients' plasma to C3 and its different fragments (C3b, iC3b, C3c, and C3d) and to C4 was measured by ELISA. The inset gives schematic representation of C3 and the cleavage process to obtain the different functional fragments used for the epitope mapping. Results are presented for the patients JTH (A), MMJ (B), CVT (C), and VSS (D).
To evaluate the repertoire of recognized autoantigens by autoantibodies in the patients, an immunoblot to endothelial antigens was performed (data not shown). The repertoire was restricted to a limited number of self-proteins present in these cells. Therefore, the presence of anti-C3 antibodies is not associated with indiscriminant and nonspecific binding to multiple autoantigens.
Functional Properties of Anti-C3 Antibodies on C3b Protein Interactions
Anti-C3 antibody-positive samples from the patients who showed the strongest reactivity were used for functional studies. The presence of the antibodies resulted in a decreased capacity of C3b to bind its negative regulators FH and CR1, as demonstrated by SPR-based technology (Fig. 5, A and B). Moreover, this reduced FH binding was associated with a reduced cofactor activity for FI, measured by Western blot (Fig. 5C). When samples from the same patient were tested over time, the level of the inhibition of the FH binding, as well as the reduction of the cofactor activity, correlated with the C3b binding level (Fig. 5) (data not shown). The immobilized C3b in the different experiments performed was always ∼5000 RU. Due to the use of the amine coupling technology, not all immobilized C3b molecules were in a functionally active conformation. Because the experimentally determined Rmax (maximal binding response at saturation) for FH and CR1 (∼500 and 700 RU, respectively), was ∼10% of the theoretically expected value, it can be roughly estimated that about 10% of the immobilized C3b molecules were functionally active. Taking into account the response after the introduction of patients' IgG, it can be estimated that only half of functionally active C3 molecules will be covered by the anti-C3 antibodies present in the sample. In addition, the profiles of the FH and CR1 binding curves in the presence of patients' IgG (with exception of the C3b-CR1 interaction in the presence of IgG from CVT1) were very similar to profiles of IgG from healthy donors, despite the decreased response.
FIGURE 5.

Functional consequences of the anti-C3 antibodies. Shown is binding of Factor H (A) and binding of soluble CR1 (B) to a C3b-coated SPR chip, exposed to IgG from healthy donors, IgG from LN patients, or buffer only, measured by SPR. Black line, buffer control; green line, healthy donor IgG. Results from two independent experiments with different samples are presented for CR1. A, left, phase of the interaction of IgG with the immobilized C3b is depicted, followed by the introduction of FH. Right, phase of introduction of FH, where the curves are aligned at the moment of FH injection. C, evaluation of the cofactor activity of FH in the presence of IgG from healthy donor and from an anti-C3 autoantibody-positive patient. C3b, FH, FI, and IgG were incubated for the indicated times. The reaction was stopped by the addition of reducing sample buffer, and the cleavage pattern of C3b was evaluated by a Western blot. The appearance of the α43 band (an indicator for the cleavage of C3b to iC3b by FI) was measured over time. The results for MMJ are shown. Purified IgG from the plasma of the patients JTH, MMJ, CVT, and VSS was used for this study.
In terms of FB binding, the antibodies had a heterogeneous behavior. In two cases, when bound to C3b, the autoantibodies induced formation of a more potent and rapidly cycling C3 convertase (having faster apparent association but also a faster dissociation rate), resulting in overall more efficient cleavage of C3 to C3a and C3b, as measured by Western blot (Fig. 6). For one tested patient, the IgGs did not modify the formation of the C3 convertase and the rate of C3 cleavage. Finally, for autoantibodies from two patients, a reduction of the FB binding, C3 convertase formation, and C3 cleavage rate was detected (Fig. 6, B, C, and D). No major differences were observed in the dissociation of the C3 convertase by FH (Fig. 6, C and D).
FIGURE 6.

Formation and dissociation of the C3 convertase. A, C3b-coated SPR chip was exposed to IgG from healthy donors or LN patients. Subsequently, C3 convertase was formed by injection of FB and FD in Mg2+-containing buffer. Left, raw data with the step of the interaction of IgG with the immobilized C3b, followed by the introduction of FB and FD. Right, phase of interaction of FB + FD, where the curves are aligned at the moment of injection. B, formation of the C3 convertase and dissociation by FH in the presence of IgG from a healthy donor, measured by SPR. The spontaneous dissociation is shown in black, and the FH-mediated dissociation is shown in green. C, formation and FH-mediated dissociation of the C3 convertase in the presence of IgG from healthy donor (green) and IgG from two patients (JTH (red) and MMJ (blue)), measured by SPR. D, formation and FH-mediated dissociation of the C3 convertase in the presence of IgG from healthy donor and a IgG from patient. C3, FB, FD, and IgG were incubated in the absence or in the presence of FH for the indicated time. The reaction was stopped by the addition of a reducing sample buffer, and the cleavage pattern of C3 (the appearance of the α′ band, indicating the activity of the convertase and the generation of C3b) was evaluated by Western blot. The results for patient MMJ are shown.
Functional Properties of Anti-C3 Antibodies When Added to Normal Human Serum
To explore further the overall functional effect of the anti-C3 autoantibodies, the activity of the alternative pathway was measured by the release of Ba, when the purified IgG from healthy donors or the autoantibody-positive patients was added to normal serum in the presence of EGTA-Mg to block the classical and lectin pathway and allow only the activity of the alternative pathway. The level of released Ba was higher in the patients' IgG group, as compared with the normal IgGs (Fig. 7A). Moreover, in the presence of anti-C3 autoantibodies, stronger C3 deposition on resting endothelial cells, incubated with normal human serum, supplemented or not with EGTA-Mg, was detected (Fig. 7, B and C). In alternative pathway-favoring conditions (EGTA-Mg), the C3 deposition was significantly stronger in the presence of IgG from anti-C3 autoantibody-positive samples, compared with IgG from normal donors and IgG from LN patients, negative for anti-C3 autoantibodies (Fig. 7D). Anti-endothelial cell antibodies were present with no difference in the anti-C3 autoantibody-positive and -negative samples (Fig. 7E).
FIGURE 7.

Effect of the anti-C3 autoantibodies on complement activity in normal human serum. A, generation of Ba after incubation of IgG from heathy donors and anti-C3 antibody-positive patients with a normal human serum in the presence of EGTA-Mg. B–D, C3 deposition on the endothelial cells from normal human serum, supplemented with IgG from LN patients or healthy donors (Normal), measured by flow cytometry. B, representative histogram from FACS analysis of the deposition of C3 in the presence of anti-C3 autoantibody-positive IgG. C, C3 deposition from normal serum, supplemented with IgG from all positive patients for whom a sufficient amount of IgG was available (n = 9), compared with IgG from healthy donors (n = 6). D, C3 deposition on endothelial cells from normal human serum, supplemented with EGTA-Mg2+ (to block the classical and lectin pathways) and IgG from healthy donors (n = 6) or LN patients, positive (LN+; n = 9) or negative (LN−; n = 5) for anti-C3 autoantibodies. Factor H-depleted serum served as a positive control, and the absence of IgG (medium only) was the negative control for C3 deposition in C and D. E, the presence of anti-endothelial cell IgG antibodies in patients with LN, positive (LN+) or negative (LN−) for anti-C3 autoantibodies, compared with healthy donors. The statistical analyses were performed by Mann-Whitney t test. HUVEC, human umbilical vein endothelial cells.
Discussion
This study demonstrated that anti-C3 autoantibodies are present in ∼30% of SLE patients with lupus nephritis and particularly in those with active disease. These antibodies have an overt capacity to dysregulate the alternative complement pathway in vitro, which may contribute to the C3 consumption found frequently in positive patients.
The observed frequency of 31% of the anti-C3 antibodies in the studied LN cohort is similar to the 32% (11 of 34), 30% (6 of 20), and 25% (13 of 53) found previously in patients with SLE (11, 12, 14). Using an ELISA-based approach, anti-C3 autoantibodies were found only rarely in other diseases, including in 2 of 20 and 1 of 18 patients with rheumatoid arthritis in two different cohorts (12, 14), in two isolated patients with DDD (7), and one isolated patient with atypical hemolytic uremic syndrome (aHUS) (26), and no patient was positive in a cohort of primary biliary cirrhosis (14). Early studies performed by an agglutination technique (27, 28) detected the presence of immunoconglutinins, which turned out to be anti-C3 and/or anti-C4 autoantibodies, in patients with acute nephritis (29), with nephrotic syndrome (16, 17), in cases with ulcerative colitis, and in Crohn disease patients (15). The titers of immunoconglutinins decreased with the achievement of remission in patients with steroid-sensitive nephrotic syndrome (16). The anti-C3 autoantibody titers, measured by ELISA, in a SLE cohort (11, 13) and in the DDD patients (7) also decreased with improvement of the clinical condition and normalization of the C3 levels. In agreement with these results, our data indicate an association of the level of C3-binding antibodies with the disease activity. The levels of these antibodies were higher in patients with an active disease, classified BILAG category A, compared with the rest of the patients. Moreover, improvement of the renal function, as evaluated by decrease in the creatinine levels and increase of the eGFR in two patients for whom samples at the moment of diagnosis of the LN as well as during follow up were available, correlated with a reduction in the titers of C3 autoantibodies. Also, the anti-C3 autoantibodies were found in relatively younger patients (less than 43 years in age). These antibodies were detected only in patients positive for anti-dsDNA autoantibodies, which correlates with the results from autoimmune-prone mice that have been also reported to develop anti-C3 autoantibodies (12). Patients with anti-C3 autoantibodies had significantly lower C3 levels, compared with the negative patients, suggesting an impact on the complement consumption. The analysis of the limited number of patients in our cohort and the data from the literature suggest that the anti-C3 autoantibodies are present in about one-third of the SLE cases, including the ones with renal involvement, and that their presence correlates with the disease activity and with the complement consumption. Therefore, anti-C3 autoantibodies may be active players in the pathological process and not just an epiphenomenon, as may be the case for many other SLE-associated autoantibodies (18). To clarify the potential contribution of the anti-C3 autoantibodies to the complement overactivation in these patients, we characterized their binding epitopes and dissected their functional activity.
By using epitope mapping, we localized the domains of C3 recognized by the autoantibodies. The antibodies of all but one of the studied patients recognized C3, C3b, iC3b, and C3c with variable intensity, in agreement with the results for SLE patients (13) and for a mixed cohort of inflammatory rheumatic diseases (including SLE patients), where C3c was used as an antigen (30). Therefore, for these patients, the recognized epitopes should be localized within the C3c portion of the molecule, common to the four well recognized molecules, namely C3, C3b, iC3b, and C3c. Notably, antibodies from these patients reacted also with C4, suggesting that a common epitope may exist. This epitope should be exposed at the surface of the immobilized C3 and C3b protein surfaces in order to be recognized by the antibodies, present in C3c fragment and preserved in C4 but absent in C5, because no C5 reactivity was detected. The antibodies of the patient JTH recognized primarily C3 and C3b, suggesting the presence of a conformational epitope, located in the C3b portion, lost after the cleavage of C3b. No patient was positive for anti-C3d antibodies, similar to the patients with SLE (13), DDD (7), and aHUS (26). Interestingly, the plasma of only one of the two DDD patients recognized C3c, and neither were positive for iC3b (7), contrary to the aHUS patient, whose antibodies recognized iC3b. The fact that the anti-C3 autoantibodies are associated with distinct pathologies, the observed differences in the recognized epitopes, and the association with anti-FB autoantibodies in DDD and with anti-C4 autoantibodies in SLE and LN suggest that these antibodies may have different properties and warrant further comparative studies.
To understand whether anti-C3 autoantibodies contribute to complement overactivation in LN and hence have pathological significance, their functional consequences were characterized. Anti-C3 autoantibodies from LN patients inhibited the interaction of the negative complement regulators FH and CR1 with C3b. In the presence of IgG from anti-C3 autoantibody-positive patients, the FH binding and cofactor activity was reduced compared with the case of normal IgG, implying a significant impairment of the C3b regulation by FH. Indeed, the addition of IgG from autoantibody-positive patients to normal serum resulted in increased Ba generation, compared with IgG from healthy donors, reflecting the overactivation of the alternative pathway. Also, the addition of normal human serum supplemented with IgG from autoantibody-positive patients to resting endothelial cells resulted in an increase in C3 deposition compared with IgG from healthy donors and IgG from LN patients negative for anti-C3 autoantibodies. An increase in the C3 deposition in the same setting was obtained for FH-depleted serum and sera of patients with FH mutations or gain of function mutations in C3 or FB, found in aHUS patients (23–25, 31). The anti-C3 autoantibodies perturbed specifically the alternative complement pathway, because the increased C3 deposition and Ba release were detected in the presence of EGTA-Mg. The presence of anti-endothelial cell antibodies was detected in both anti-C3-positive and -negative patients. Their occurrence could also contribute to the initiation of the complement cascade via the classical pathway, thus feeding the alternative pathway amplification loop. Therefore, the reduced FH binding to C3b could induce inefficient complement regulation, similarly to cases of reduced FH levels in the plasma, found in LN (32). A mouse model also showed that FH deficiency accelerated the development of lupus nephritis (33). Our results contribute to the understanding that the alternative complement pathway activation may play an important role in the kidney damage in the context of LN, similar to DDD, C3 glomerulonephritis, certain cases of membranoproliferative glomerulonephritis type I, or the thrombotic microangiopathy renal disease aHUS (34, 35).
Anti-C3 autoantibodies also inhibited the interaction of C3b with another complement regulator, CR1. CR1 dissociates the C3 convertases and serves as a cofactor for FI in the cleavage of both C3b and C4b (36). It is the only cofactor able to assist FI in the cleavage of iC3b to C3dg. The observed decrease in the CR1 binding reported here may explain the perturbation of the CR1 cofactor activity for the inactivation of C3b by Factor I in the presence of anti-C3 autoantibodies described by Nilsson et al. (14). It has been shown that in SLE patients, there is a marked decline in the levels of CR1 expressed by erythrocytes (37–39), leukocytes (40, 41), and glomerular podocytes (42, 43). The reduced expression of CR1 results in C3 activation and inefficient clearance of the immune complexes (36, 44). Therefore, the inhibition of the binding of CR1 to C3b by anti-C3 autoantibodies described in this study suggests an impairment of regulatory functions of CR1. The reactivity toward both C3b and C4 implies that the C4b inactivation by CR1 may be affected as well. Similar to the decreased expression of CR1 in SLE patients, the inefficient binding of CR1 to complement-opsonized immune complexes might impair their clearance. This inevitably would contribute to the disease physiopathology. Of note, the defective clearance of immune complexes and their accumulation in the kidneys is a hallmark of LN.
Taking into account that we obtain between 10 and 50% inhibition of the FH or CR1 binding (depending on the titer of the antibodies), we can hypothesize that if an antibody binds to the C3b, it will prevent completely the interaction with FH or CR1. The residual binding that we observe will be due to interaction of FH with the free C3b molecules (not bound to IgG). Based on these considerations, we favor a scenario in which some C3b molecules are not bound by autoantibody, and they are active in binding of complement regulators. The scenario in which the autoantibody-bound C3b still binds FH or sCR1 but with a somewhat lower affinity is less likely because in all but one case the binding pattern and complex stability of the C3b-FH or C3b-CR1 complexes were similar (albeit with a lower response) in the presence of patient IgG compared with control IgG. If we assume the first scenario, we can hypothesize that the anti-C3 IgG binds to an area of the C3b molecule carrying the FH and CR1 binding sites and will compete for the binding with these two regulators. The location of the FH binding sites has been well described (45–47), and data are available for the approximate location of the CR1 binding site because it overlaps with the binding site of the first two domains of FH (46, 48, 49). This part of the C3b molecule is located in the C3c fragment and is available in the immobilized C3, C3b, C3c, and iC3b but not in C3d. The hypothesis that the anti-C3 autoantibodies will bind to this same area is supported by epitope mapping, showing strong binding to immobilized C3 and C3b, weaker binding to C3c and iC3b, and no binding to C3d.
Anti-C3 autoantibodies may also perturb the clearance of apoptotic bodies in lupus patients. Kenyon et al. (12) reported that anti-C3 autoantibodies from autoimmune-prone mice bind to C3 fragment-opsonized apoptotic cells and prevent their phagocytosis by macrophages. Therefore, these antibodies may contribute to accumulation of the apoptotic cells and debris and generation of autoimmune response, similar to the case of quantitative and functional C1q deficiency (50, 51).
Anti-C3 autoantibodies from part of the positive LN patients induced formation of a stronger but rapidly cycling C3 convertase, measured by SPR. This convertase was more efficient, cleaving a higher amount of C3 to C3a and C3b. This behavior is similar to some gain of function Factor B mutations, found in aHUS (52), and different from the stabilization of the C3 convertase in the presence of a C3 nephritic factor (C3Nef). C3Nef is an autoantibody, found in C3 glomerulopathies (34), that binds to the C3 convertase C3bBb but not to its isolated fragments C3b and Bb (53, 54). C3Nef is very rare in SLE and is detected in some patients with a partial lipodystrophy (55–58). The positive patients here showed no sign of partial lipodystrophy. The C3 antibodies described in this study were not C3Nef, because they recognized C3 and C3b, contrary to C3Nef, and the presence of C3Nef was also excluded in the sample of patient JTH by a reference C3 convertase stabilization hemolytic assay (34). This direct effect on the C3 convertase is not shared by the anti-C3 antibodies from all patients. In some cases, no effect on the C3 convertase was found, and in others, the effect was opposite. This result is in line with the previously reported decrease in the C3/C5 convertase activity detected in the presence of anti-C3 autoantibodies from two of three tested positive patients (14). Despite the lower efficacy of the C3 convertase, the anti-C3 autoantibodies from these patients perturbed the binding of FH and CR1 to the C3b, thus acting on the regulation, rather than on the activation step of the convertase. Therefore, despite the lower level of C3 convertase formation, the decreased activity of FH tips the balance to overall overactivation of the system.
Conclusion
The role of the overactivation of the classical complement pathway in systemic lupus erythematosus and lupus nephritis is well characterized, but little is known about the contribution of the alternative pathway. Although autoantibodies against the central complement component C3 have been described in systemic lupus erythematosus patients, their mechanism of action and influence over the alternative pathway were unclear. In this study, we describe the molecular mechanism by which the anti-C3 autoantibodies from lupus nephritis patients dysregulate the alternative complement pathway and contribute to the pathological process. These results suggest that anti-C3 autoantibodies may be a biomarker for lupus nephritis severity and provide evidence for the contribution of the alternative complement pathway in this disease.
Author Contributions
L. T. R., J. D. D., and V. V. V. conceived and coordinated the study and wrote the paper. R. N., S. C., L. T. R., J. D. D., M.-A. D.-D., and V. F.-B. designed, performed, and analyzed the experiments. V. V. V., V. J. L., and B. P. D. took care of the patients and collected clinical data. V. V. V. analyzed the clinical data. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
The cytometric analysis was performed at the “Centre d'Imagerie Cellulaire et de Cytomètrie,” (CICC), Centre de Recherche des Cordeliers UMR S 1138 (Paris, France). CICC is a member of the UPMC Flow Cytometry Network (RECYF).
This work was supported by INSERM and by Agence Nationale de la Recherche Grant ANR-13-JCV1-006-01. The authors declare that they have no conflicts of interest with the contents of this article.
- LN
- lupus nephritis
- SLE
- systemic lupus erythematosus
- DDD
- dense deposit disease
- CR1
- complement receptor 1
- BILAG
- British Islet Lupus Assessment Group
- FB
- FD, FH, and FI, Factor B, D, H, and I, respectively
- eGFR
- glomerular filtration rate
- C3Nef
- C3 nephritic factor
- RU
- response units.
References
- 1. Tsokos G. C. (2011) Systemic lupus erythematosus. N. Engl. J. Med. 365, 2110–2121 [DOI] [PubMed] [Google Scholar]
- 2. Radanova M., Vasilev V., Deliyska B., Kishore U., Ikonomov V., Ivanova D. (2012) Anti-C1q autoantibodies specific against the globular domain of the C1qB-chain from patient with lupus nephritis inhibit C1q binding to IgG and CRP. Immunobiology 217, 684–691 [DOI] [PubMed] [Google Scholar]
- 3. Vanhecke D., Roumenina L. T., Wan H., Osthoff M., Schaller M., Trendelenburg M. (2012) Identification of a major linear C1q epitope allows detection of systemic lupus erythematosus anti-C1q antibodies by a specific peptide-based enzyme-linked immunosorbent assay. Arthritis Rheum. 64, 3706–3714 [DOI] [PubMed] [Google Scholar]
- 4. Cole D. S., Morgan B. P. (2003) Beyond lysis: how complement influences cell fate. Clin. Sci. 104, 455–466 [DOI] [PubMed] [Google Scholar]
- 5. Merle N. S., Church S. E., Fremeaux-Bacchi V., Roumenina L. T. (2015) Complement system part I: molecular mechanisms of activation and regulation. Front. Immunol. 6, 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Merle N. S., Noe R., Halbwachs-Mecarelli L., Fremeaux-Bacchi V., Roumenina L. T. (2015) Complement system part II: role in immunity. Front. Immunol. 6, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen Q., Muller D., Rudolph B., Hartmann A., Kuwertz-Broking E., Wu K., Kirschfink M., Skerka C., Zipfel P. F. (2011) Combined C3b and factor B autoantibodies and MPGN type II. N. Engl. J. Med. 365, 2340–2342 [DOI] [PubMed] [Google Scholar]
- 8. Dragon-Durey M. A., Loirat C., Cloarec S., Macher M. A., Blouin J., Nivet H., Weiss L., Fridman W. H., Frémeaux-Bacchi V. (2005) Anti-Factor H autoantibodies associated with atypical hemolytic uremic syndrome. J. Am. Soc. Nephrol. 16, 555–563 [DOI] [PubMed] [Google Scholar]
- 9. Pickering M. C., Botto M. (2010) Are anti-C1q antibodies different from other SLE autoantibodies? Nat. Rev. Rheumatol. 6, 490–493 [DOI] [PubMed] [Google Scholar]
- 10. Lachmann P. J. (2009) The amplification loop of the complement pathways. Adv. Immunol. 104, 115–149 [DOI] [PubMed] [Google Scholar]
- 11. Durand C. G., Burge J. J. (1984) A new enzyme-linked immunosorbent assay (ELISA) for measuring immunoconglutinins directed against the third component of human complement: findings in systemic lupus erythematosus. J. Immunol. Methods 73, 57–66 [DOI] [PubMed] [Google Scholar]
- 12. Kenyon K. D., Cole C., Crawford F., Kappler J. W., Thurman J. M., Bratton D. L., Boackle S. A., Henson P. M. (2011) IgG autoantibodies against deposited C3 inhibit macrophage-mediated apoptotic cell engulfment in systemic autoimmunity. J. Immunol. 187, 2101–2111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nilsson B., Ekdahl K. N., Sjöholm A., Nilsson U. R., Sturfelt G. (1992) Detection and characterization of immunoconglutinins in patients with systemic lupus erythematosus (SLE): serial analysis in relation to disease course. Clin. Exp. Immunol. 90, 251–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nilsson B., Ekdahl K. N., Svarvare M., Bjelle A., Nilsson U. R. (1990) Purification and characterization of IgG immunoconglutinins from patients with systemic lupus erythematosus: implications for a regulatory function. Clin. Exp. Immunol. 82, 262–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Potter B. J., Brown D. J., Watson A., Jewell D. P. (1980) Complement inhibitors and immunoconglutinins in ulcerative colitis and Crohn's disease. Gut 21, 1030–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ngu J. L., Barratt T. M., Soothill J. F. (1970) Immunoconglutinin and complement changes in steroid sensitive relapsing nephrotic syndrome of children. Clin. Exp. Immunol. 6, 109–116 [PMC free article] [PubMed] [Google Scholar]
- 17. Ngu J. L., Blackett K. (1970) Complement and immunoconglutinin changes in the nephrotic syndrome of adult Africans. J. Trop. Med. Hyg. 73, 250–254 [PubMed] [Google Scholar]
- 18. Rekvig O. P., Putterman C., Casu C., Gao H. X., Ghirardello A., Mortensen E. S., Tincani A., Doria A. (2012) Autoantibodies in lupus: culprits or passive bystanders? Autoimmun. Rev. 11, 596–603 [DOI] [PubMed] [Google Scholar]
- 19. Yaniv G., Twig G., Shor D. B., Furer A., Sherer Y., Mozes O., Komisar O., Slonimsky E., Klang E., Lotan E., Welt M., Marai I., Shina A., Amital H., Shoenfeld Y. (2015) A volcanic explosion of autoantibodies in systemic lupus erythematosus: a diversity of 180 different antibodies found in SLE patients. Autoimmun. Rev. 14, 75–79 [DOI] [PubMed] [Google Scholar]
- 20. Churg J., Bernstien J., Glassock R. (1995) Renal Disease: Classification and Atlas of Glomerular Diseases, 2nd Ed., p. 152, Igaku-Shoin, Ltd., New York [Google Scholar]
- 21. Marto N., Bertolaccini M. L., Calabuig E., Hughes G. R., Khamashta M. A. (2005) Anti-C1q antibodies in nephritis: correlation between titres and renal disease activity and positive predictive value in systemic lupus erythematosus. Ann. Rheum. Dis. 64, 444–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hay E. M., Bacon P. A., Gordon C., Isenberg D. A., Maddison P., Snaith M. L., Symmons D. P., Viner N., Zoma A. (1993) The BILAG index: a reliable and valid instrument for measuring clinical disease activity in systemic lupus erythematosus. Q. J. Med. 86, 447–458 [PubMed] [Google Scholar]
- 23. Frimat M., Tabarin F., Dimitrov J. D., Poitou C., Halbwachs-Mecarelli L., Fremeaux-Bacchi V., Roumenina L. T. (2013) Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood 122, 282–292 [DOI] [PubMed] [Google Scholar]
- 24. Roumenina L. T., Frimat M., Miller E. C., Provot F., Dragon-Durey M. A., Bordereau P., Bigot S., Hue C., Satchell S. C., Mathieson P. W., Mousson C., Noel C., Sautes-Fridman C., Halbwachs-Mecarelli L., Atkinson J. P., Lionet A., Fremeaux-Bacchi V. (2012) A prevalent C3 mutation in aHUS patients causes a direct C3 convertase gain of function. Blood 119, 4182–4191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Roumenina L. T., Jablonski M., Hue C., Blouin J., Dimitrov J. D., Dragon-Durey M. A., Cayla M., Fridman W. H., Macher M. A., Ribes D., Moulonguet L., Rostaing L., Satchell S. C., Mathieson P. W., Sautes-Fridman C., Loirat C., Regnier C. H., Halbwachs-Mecarelli L., Fremeaux-Bacchi V. (2009) Hyperfunctional C3 convertase leads to complement deposition on endothelial cells and contributes to atypical hemolytic uremic syndrome. Blood 114, 2837–2845 [DOI] [PubMed] [Google Scholar]
- 26. Józsi M., Reuter S., Nozal P., López-Trascasa M., Sánchez-Corral P., Prohászka Z., Uzonyi B. (2014) Autoantibodies to complement components in C3 glomerulopathy and atypical hemolytic uremic syndrome. Immunol. Lett. 160, 163–171 [DOI] [PubMed] [Google Scholar]
- 27. Lachmann P. J. (1967) Conglutinin and immunoconglutinins. Adv. Immunol. 6, 479–527 [DOI] [PubMed] [Google Scholar]
- 28. Lachmann P. J., Liske R. (1966) The preparation and properties of alexinated intermediates that react with conglutinin. II. Equine, rabbit and human complement. Immunology 11, 255–262 [PMC free article] [PubMed] [Google Scholar]
- 29. Ngu J. L., Soothill J. F. (1969) Immunoconglutinin and complement changes in children with acute nephritis. Clin. Exp. Immunol. 5, 557–566 [PMC free article] [PubMed] [Google Scholar]
- 30. Hautanen A., Gripenberg M., Salonen E. M., Julkunen I., Laitinen O. (1984) C3c-binding in inflammatory rheumatic diseases. Clin. Exp. Rheumatol. 2, 125–130 [PubMed] [Google Scholar]
- 31. Marinozzi M. C., Vergoz L., Rybkine T., Ngo S., Bettoni S., Pashov A., Cayla M., Tabarin F., Jablonski M., Hue C., Smith R. J., Noris M., Halbwachs-Mecarelli L., Donadelli R., Fremeaux-Bacchi V., Roumenina L. T. (2014) Complement factor B mutations in atypical hemolytic uremic syndrome-disease: relevant or benign? J. Am. Soc. Nephrol. 25, 2053–2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang F. M., Yu F., Tan Y., Song D., Zhao M. H. (2012) Serum complement factor H is associated with clinical and pathological activities of patients with lupus nephritis. Rheumatology 51, 2269–2277 [DOI] [PubMed] [Google Scholar]
- 33. Bao L., Haas M., Quigg R. J. (2011) Complement factor H deficiency accelerates development of lupus nephritis. J. Am. Soc. Nephrol. 22, 285–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Servais A., Noël L. H., Roumenina L. T., Le Quintrec M., Ngo S., Dragon-Durey M. A., Macher M. A., Zuber J., Karras A., Provot F., Moulin B., Grünfeld J. P., Niaudet P., Lesavre P., Frémeaux-Bacchi V. (2012) Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int. 82, 454–464 [DOI] [PubMed] [Google Scholar]
- 35. Sethi S., Fervenza F. C. (2012) Membranoproliferative glomerulonephritis: a new look at an old entity. N. Engl. J. Med. 366, 1119–1131 [DOI] [PubMed] [Google Scholar]
- 36. Khera R., Das N. (2009) Complement receptor 1: disease associations and therapeutic implications. Mol. Immunol. 46, 761–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Birmingham D. J., Gavit K. F., McCarty S. M., Yu C. Y., Rovin B. H., Nagaraja H. N., Hebert L. A. (2006) Consumption of erythrocyte CR1 (CD35) is associated with protection against systemic lupus erythematosus renal flare. Clin. Exp. Immunol. 143, 274–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Corvetta A., Pomponio G., Bencivenga R., Luchetti M. M., Spycher M., Spaeth P. J., Danieli G. (1991) Low number of complement C3b/C4b receptors (CR1) on erythrocytes from patients with essential mixed cryoglobulinemia, systemic lupus erythematosus and rheumatoid arthritis: relationship with disease activity, anticardiolipin antibodies, complement activation and therapy. J. Rheumatol. 18, 1021–1025 [PubMed] [Google Scholar]
- 39. Ross G. D., Yount W. J., Walport M. J., Winfield J. B., Parker C. J., Fuller C. R., Taylor R. P., Myones B. L., Lachmann P. J. (1985) Disease-associated loss of erythrocyte complement receptors (CR1, C3b receptors) in patients with systemic lupus erythematosus and other diseases involving autoantibodies and/or complement activation. J. Immunol. 135, 2005–2014 [PubMed] [Google Scholar]
- 40. Fyfe A., Holme E. R., Zoma A., Whaley K. (1987) C3b receptor (CR1) expression on the polymorphonuclear leukocytes from patients with systemic lupus erythematosus. Clin. Exp. Immunol. 67, 300–308 [PMC free article] [PubMed] [Google Scholar]
- 41. Wilson J. G., Ratnoff W. D., Schur P. H., Fearon D. T. (1986) Decreased expression of the C3b/C4b receptor (CR1) and the C3d receptor (CR2) on B lymphocytes and of CR1 on neutrophils of patients with systemic lupus erythematosus. Arthritis Rheum. 29, 739–747 [DOI] [PubMed] [Google Scholar]
- 42. Arora M., Arora R., Tiwari S. C., Das N., Srivastava L. M. (2000) Expression of complement regulatory proteins in diffuse proliferative glomerulonephritis. Lupus 9, 127–131 [DOI] [PubMed] [Google Scholar]
- 43. Raju K. R., Sivasankar B., Anand V., Luthra K., Tiwari S. C., Dinda A. K., Das N., Srivastava L. M. (2001) Use of complement receptor 1 (CD35) assay in the diagnosis and prognosis of immune complex mediated glomerulopathies. Asian Pac. J. Allergy Immunol. 19, 23–27 [PubMed] [Google Scholar]
- 44. Kavai M. (2008) Immune complex clearance by complement receptor type 1 in SLE. Autoimmun. Rev. 8, 160–164 [DOI] [PubMed] [Google Scholar]
- 45. Kajander T., Lehtinen M. J., Hyvärinen S., Bhattacharjee A., Leung E., Isenman D. E., Meri S., Goldman A., Jokiranta T. S. (2011) Dual interaction of factor H with C3d and glycosaminoglycans in host-nonhost discrimination by complement. Proc. Natl. Acad. Sci. U.S.A. 108, 2897–2902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schramm E. C., Roumenina L. T., Rybkine T., Chauvet S., Vieira-Martins P., Hue C., Maga T., Valoti E., Wilson V., Jokiranta S., Smith R. J., Noris M., Goodship T., Atkinson J. P., Fremeaux-Bacchi V. (2015) Mapping interactions between complement C3 and regulators using mutations in atypical hemolytic uremic syndrome. Blood 125, 2359–2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wu J., Wu Y. Q., Ricklin D., Janssen B. J., Lambris J. D., Gros P. (2009) Structure of complement fragment C3b-factor H and implications for host protection by complement regulators. Nat. Immunol. 10, 728–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lambris J. D., Lao Z., Oglesby T. J., Atkinson J. P., Hack C. E., Becherer J. D. (1996) Dissection of CR1, factor H, membrane cofactor protein, and factor B binding and functional sites in the third complement component. J. Immunol. 156, 4821–4832 [PubMed] [Google Scholar]
- 49. Oran A. E., Isenman D. E. (1999) Identification of residues within the 727–767 segment of human complement component C3 important for its interaction with factor H and with complement receptor 1 (CR1, CD35). J. Biol. Chem. 274, 5120–5130 [DOI] [PubMed] [Google Scholar]
- 50. Leffler J., Bengtsson A. A., Blom A. M. (2014) The complement system in systemic lupus erythematosus: an update. Ann. Rheum. Dis. 73, 1601–1606 [DOI] [PubMed] [Google Scholar]
- 51. Roumenina L. T., Sène D., Radanova M., Blouin J., Halbwachs-Mecarelli L., Dragon-Durey M. A., Fridman W. H., Fremeaux-Bacchi V. (2011) Functional complement C1q abnormality leads to impaired immune complexes and apoptotic cell clearance. J. Immunol. 187, 4369–4373 [DOI] [PubMed] [Google Scholar]
- 52. Goicoechea de Jorge E., Harris C. L., Esparza-Gordillo J., Carreras L., Arranz E. A., Garrido C. A., López-Trascasa M., Sánchez-Corral P., Morgan B. P., Rodríguez de Córdoba S. (2007) Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc. Natl. Acad. Sci. U.S.A. 104, 240–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Paixão-Cavalcante D., López-Trascasa M., Skattum L., Giclas P. C., Goodship T. H., de Córdoba S. R., Truedsson L., Morgan B. P., Harris C. L. (2012) Sensitive and specific assays for C3 nephritic factors clarify mechanisms underlying complement dysregulation. Kidney Int. 82, 1084–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dragon-Durey M. A., Blanc C., Marinozzi M. C., van Schaarenburg R. A., Trouw L. A. (2013) Autoantibodies against complement components and functional consequences. Mol. Immunol. 56, 213–221 [DOI] [PubMed] [Google Scholar]
- 55. Walport M. J., Davies K. A., Botto M., Naughton M. A., Isenberg D. A., Biasi D., Powell R. J., Cheung N. T., Struthers G. R. (1994) C3 nephritic factor and SLE: report of four cases and review of the literature. QJM 87, 609–615 [PubMed] [Google Scholar]
- 56. Jasin H. E. (1979) Systemic lupus erythematosus, partial lipodystrophy and hypocomplementemia. J. Rheumatol. 6, 43–50 [PubMed] [Google Scholar]
- 57. Kitahara M., Koike K., Kurokawa Y., Sawai N., Mori T., Nakazawa K., Shigematsu H., Komiyama A. (1999) Lupus nephritis in juvenile myelomonocytic leukemia. Clin. Nephrol. 51, 314–318 [PubMed] [Google Scholar]
- 58. Ben Ghorbel I., Ben Salem T., Lamloum M., Braham A., Khanfir M., Miled M., Houman M. H. (2010) [Barraquer-Simons syndrome in a patient with systemic lupus erythematosus]. Rev. Med. Interne. 31, 372–374 [DOI] [PubMed] [Google Scholar]