

Background: The synthetic pathway and mechanism by which ceramide synthases (CerSs) mediate cell death are not fully understood.
Results: N-Acylation of recycled sphingosine by CerSs regulates pro-apoptotic events, such as caspase-7 activation, loss of FAK, and plasma membrane rupture.
Conclusion: The salvage pathway of ceramide production is essential for TNFα-induced apoptosis.
Significance: Understanding how CerS/ceramide mediates tumor cell death may reveal new targets for therapy.
Keywords: cell death, ceramide synthase, plasma membrane, PTK2 protein tyrosine kinase 2 (PTK2) (focal adhesion kinase) (FAK), sphingolipid, Ceramide, fumonisin b1
Abstract
Ceramide synthases (CerS1–CerS6), which catalyze the N-acylation of the (dihydro)sphingosine backbone to produce (dihydro)ceramide in both the de novo and the salvage or recycling pathway of ceramide generation, have been implicated in the control of programmed cell death. However, the regulation of the de novo pathway compared with the salvage pathway is not fully understood. In the current study, we have found that late accumulation of multiple ceramide and dihydroceramide species in MCF-7 cells treated with TNFα occurred by up-regulation of both pathways of ceramide synthesis. Nevertheless, fumonisin B1 but not myriocin was able to protect from TNFα-induced cell death, suggesting that ceramide synthase activity is crucial for the progression of cell death and that the pool of ceramide involved derives from the salvage pathway rather than de novo biosynthesis. Furthermore, compared with control cells, TNFα-treated cells exhibited reduced focal adhesion kinase and subsequent plasma membrane permeabilization, which was blocked exclusively by fumonisin B1. In addition, exogenously added C6-ceramide mimicked the effects of TNFα that lead to cell death, which were inhibited by fumonisin B1. Knockdown of individual ceramide synthases identified CerS6 and its product C16-ceramide as the ceramide synthase isoform essential for the regulation of cell death. In summary, our data suggest a novel role for CerS6/C16-ceramide as an upstream effector of the loss of focal adhesion protein and plasma membrane permeabilization, via the activation of caspase-7, and identify the salvage pathway as the critical mechanism of ceramide generation that controls cell death.
Introduction
Programmed cell death, namely apoptosis, is a precisely regulated form of cell death, which is considered to play a critical role in the development and homeostasis of normal tissues (1). In multicellular organisms, apoptosis contributes to elimination of unnecessary or harmful cells to maintain the balance between cell survival and cell death. Programmed cell death is of exceptional clinical importance because its dysregulation promotes aberrant cell proliferation and accumulation of genetic defects, not only leading to tumor formation but also conferring drug resistance to cancer cells (2). Apoptosis can be triggered by extrinsic signals, such as the binding of a death ligand to its cell surface death receptors (e.g. TNFα to TNFR1) or by intrinsic signals, such as genotoxic stress. Nevertheless, both pathways converge on common key control points involving mitochondrial outer membrane permeabilization (MOMP)2 and caspase activation. In vitro, apoptotic cells eventually undergo secondary necrosis (3), characterized by “rounding up” of the cell, calcium overload, and plasma membrane rupture (4–6). Secondary necrosis has also been observed in vivo when clearance of apoptotic cells by macrophages is impaired (4).
Several lines of evidence have demonstrated the regulation of programmed cell death by the bioactive lipid ceramide (Cer). First, intracellular levels of Cer accumulate in response to a myriad of pro-apoptotic insults (7–10). Second, inhibition of Cer-metabolizing enzymes and subsequent Cer generation by pharmacological agents or small interference RNA are observed to delay or abrogate cell death (11). Third, exogenous Cer or Cer analogs are sufficient to induce cell death (12, 13). Cer can be generated through de novo synthesis mediated by the serine palmitoyltransferase and ceramide synthases (CerSs), directly from the hydrolysis of sphingomyelin catalyzed by sphingomyelinase (SMase), or indirectly from breakdown of complex sphingolipids by the N-acylation of sphingosine catalyzed by CerS or the reverse action of some ceramidases in the salvage (or recycling) pathway (14, 15). Some inducers of cell death, such as UV radiation, appear to activate more than one Cer-generating pathway (7, 16). Tumor necrosis factor-α (TNFα) also appears to regulate Cer through multiple pathways, although these have not been dissected carefully, especially the role of the CerSs in the de novo versus the salvage pathways.
CerS are integral membrane proteins of the endoplasmic reticulum and catalyze the N-acylation of the (dihydro)sphingosine backbone to produce (dihydro)ceramide (17). In mammals, six different CerSs have been identified (CerS1–CerS6), each of which synthesizes Cers with different acyl chain lengths. For instance, CerS1 uses C18-acyl-CoA (18), CerS2 uses C22- or C24-acyl-CoAs (19), CerS4 uses C20-acyl-CoA, and CerS5 and CerS6 use C14- and C16-acyl-CoA (20, 21). As mentioned above, CerSs take part in both the de novo synthetic pathway of Cer and the salvage pathway.
For the last 20 years, CerSs have been widely described as modulating cell death triggered by pro-apoptotic inducers, such as chemotherapeutics agents (22, 23), TNFα (24), or ionizing radiation. In the literature, specific roles for CerSs and Cer have been suggested in some processes related to cell death. For instance, CerSs have been described as controlling MOMP (25), caspase-3/7 activation (10), or caspase-3 translocation to the nuclei (26). However, the molecular and biochemical mechanisms of the regulation of programmed cell death by CerSs are not fully understood. Results from the current study now implicate a specific CerSs in the control of cell death by regulating focal adhesion kinase (FAK).
FAK is a 125-kDa cytoplasmic non-receptor tyrosine kinase, localized at focal adhesions. FAK contains three main domains: the catalytic kinase domain, a large N-terminal domain comprising the FERM region, and a C-terminal domain harboring the focal adhesion targeting region (27). Upon growth factor activation or integrin clustering, FAK is autophosphorylated on Tyr-397 (28), docking several other intracellular signaling molecules (29). Activation of FAK has been associated with regulation of a variety of cellular functions, such as cell spreading, migration, cell proliferation, apoptosis, and cell survival (30). Furthermore, FAK has been reported to be overexpressed in various tumor cells, including breast and colon cancer (31, 32).
During apoptosis, FAK plays an important role in cell rounding, loss of focal contacts, and apoptotic membrane formations, such as blebbing (33, 34). Overexpression of FAK blocks cell death, whereas its inhibition induces cell detachment and death (anoikis) in various cell types (30, 35, 36). Moreover, it has been demonstrated that FAK is cleaved by executioner caspases, such as caspase-3, -6, and -7, during apoptosis (34, 37–39).
In this study, we investigate the synthetic pathway and the mechanism by which CerSs mediate TNFα-induced accumulation of Cer and subsequent cell death in MCF-7 breast cancer cells. The results show that the CerS inhibitor fumonisin B1 (FB1) as well as knockdown of CerS6 blocked TNFα-induced late accumulation of Cer, caspase-7 activation, FAK down-regulation, and subsequent secondary necrosis measured by plasma membrane permeabilization. Thus, our data suggest a novel role for CerS6/Cer as an upstream effector of the loss of FAK and apoptosis.
Experimental Procedures
Reagents
Human TNFα was purchased from PeproTech (Rocky Hill, NJ). FB1 was from Enzo Life Sciences (Farmingdale, NY). Myriocin (Myr) was from Sigma-Aldrich. z-VAD-fmk and z-DEVD-fmk were from R&D Systems (Minneapolis, MN). 17-Carbon sphingosine and C6-ceramide were purchased from Avanti Polar Lipids (Alabaster, AL). Anti-caspase-8, anti-caspase-7, anti-phospho-FAK, anti-total FAK, Bid, and anti-GAPDH antibodies were from Cell Signaling Technology (Danvers, MA). Anti-PARP-1 and focal adhesion kinase inhibitor 14 (Y15) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-CerS6 was from Novus Biologicals (Littleton, CO).
Cell Culture and Treatment Conditions
MCF-7 human breast adenocarcinoma cells were originally purchased from American Type Culture Collection (Manassas, VA). RPMI 1640 medium, from Life Technologies, Inc., was supplemented with l-glutamine and 10% (v/v) fetal bovine serum (FBS). Cells were kept in a humidified incubator at 37 °C with 5% CO2. Cells were seeded and treated as needed 24 h after plating. Cells were grown to a final confluence of 80%. In all cases, the media were changed on cells to fresh new medium before treatment. Pretreatments were carried out 1 h prior to the addition of TNFα unless otherwise indicated.
Cell Viability Assay
Cell viability was measured using the oxidation of the aqueous soluble 3-(4,5- dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to the insoluble formazan (Sigma), which was quantified by measuring absorbance at 570 nm (SpectraMax M5, Molecular Devices). Cells were seeded and treated as needed. At the end of the treatment period, medium was removed and replaced with fresh medium. MTT (5 mg/ml) was added to each well, and incubation was carried out at 37 °C for 2 h. Formazan product was solubilized in DMSO and expressed as a percentage of the values obtained from control cells in each matched time point.
Lactate Dehydrogenase Release
Lactate dehydrogenase (LDH) release was determined in the medium using a colorimetric assay kit commercially available (Biovision, Milpitas, CA) following the manufacturer's instructions.
Intracellular Calcium Assay
Calcium overload was determined in the cells using a fluorometric assay kit commercially available (Abcam, Cambridge, MA), following the manufacturer's instructions. Cellular calcium levels were normalized by protein content.
Immunoblotting
Cell proteins were collected by direct lysis in 1× Laemmli buffer (Bio-Rad) supplemented with β-mercaptoethanol (Sigma). Cell lysates were transferred to 1.5-ml microcentrifuge tube, vortexed, and boiled for 5–10 min. Proteins were separated by SDS-PAGE using the Criterion system (Bio-Rad) and transferred to nitrocellulose membranes. Primary antibodies (1:1,000 dilution) were incubated overnight at 4 °C, and horseradish peroxidase-conjugated secondary antibodies (1:5,000 dilution) were incubated for 1 h at room temperature. Immunoreactive proteins were visualized by chemiluminescence via ECL reagent (Thermo Scientific, Rockford, IL). All Western blots were quantified by densitometric analysis using the National Institutes of Health ImageJ version 1.44p software.
Flow Cytometric Analysis
Cell apoptosis was assayed by annexin V/propidium iodide staining assay using Alexa Fluor® 488 annexin V and propidium iodide detection kit (Life Technologies, Grand Island, NY) according to the manufacturer's protocol. Briefly, cells were trypsinized at the end of treatment, collected, washed with ice-cold PBS, and resuspended in buffer containing Alexa Fluor® 488 annexin V and propidium iodide. Cells were stained for 15 min (room temperature) in darkness and immediately analyzed using a BD Biosciences FACSCalibur. Ten thousand events were acquired on the FACSCalibur (BD Biosciences) and analyzed with CellQuest (BD Biosciences) software.
Caspase Activity
Caspase-3/7 activity was determined in cell lysates using a commercially available kit (R&D Systems) according to the manufacturer's instructions. Briefly, after treatments, cells were washed three times with ice-cold PBS. The cell pellets were incubated with lysis buffer and then centrifuged. Supernatant was used for the activity assay and protein determination. The caspase activity was carried out in a 96-well plate using a caspase-3/7 fluorogenic substrate, DEVD-7-amino-4-trifluoromethylcoumarin. After the incubation period (2 h), fluorescence was measured (SpectraMax M5, Molecular Devices). Caspase activity was expressed as -fold change versus control and normalized by protein content.
Real-time Quantitative PCR Analysis
Total RNA was extracted using a Qiagen RNeasy Kit (Qiagen, Valencia, CA) according to the manufacturer's protocol. The concentration and quality of total RNA preparations were evaluated using a NanoDropTM2000c spectrophotometer (Thermo Scientific). Complementary DNA was synthesized from 1 μg of the total RNA using the Superscript III kit for first-strand synthesis (Invitrogen/Life Technologies). Real-time reverse transcription-PCR was performed on an Applied Biosystems® 7500 system. TaqMan® primer/probe for CERS1, CERS2, CERS4, CERS5, CERS6, and β-actin PCR primers were purchased from Invitrogen and were used according to the manufacturer's protocol. Using Q-Gene software, threshold cycle (Ct) values were normalized to β-actin and displayed as percentage expression versus control.
siRNA Transfection
24 h after plating, cells were transfected with double-stranded RNA oligomers using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's protocol. 48 h post-transfection, media were changed, and cells were treated as indicated. siRNA sequences used in this study were as follows: siCerS1 (sense, r(GGU CCU GUA UGC CAC CAG U)dTdT; antisense, r(ACU GGU GGC AUA CAG GAC C)dTdT), siCerS5 (sense, r(GGU UCU UUC AGU AAU GUU A)dTdT; antisense, r(UAA CAU UAC UGA AAG AAC C)dTdG), siCerS6 (sense, r(GGU CUU ACU GUA UUA UGA A)dTdT; antisense, r(UUC ACA AUC AAG UAA GAC C)dAdG). siCerS2 and siCerS4 were purchased from Life Technologies. siBak and siBax were from GE Dharmacon (Lafayette, CO).
Mass Spectrometry
Following treatments, cells were washed three times with ice-cold phosphate-buffered saline (PBS). Cellular pellets were frozen at −80 °C and then submitted for sphingolipidomic analysis by the Lipidomics Core Facility at the Stony Brook University of New York using HPLC/MS determination of sphingolipid mass levels as described previously (40).
Statistical Analysis
Data are represented as means ± S.E. Non-Gaussian distribution was assumed, and data were analyzed by one-way ANOVA and two-way ANOVA with Bonferroni post-test using GraphPad Prism software.
Results
Ceramide Synthases Regulate TNFα-induced Programmed Cell Death in MCF-7 Cells
We first sought to characterize TNFα-induced cell death in our system. MCF-7 cells undergo cell death upon TNFα treatment, as measured by the MTT activity assay and LDH release into the medium (data not shown). Cell viability was significantly decreased in a time-dependent manner, with a 50% decrease at 24 h after treatment. Significant plasma membrane permeabilization, measured by LDH release, was observed at 24 h (and not at 15 h), consistent with this effect being a late event of programmed cell death or secondary necrosis. In addition, reduced inactive and increased active forms of caspase-8 were first detectable at 6 h post-treatment and increased in a time-dependent manner. Caspase-8 activation preceded the cleavage of caspase-7 and of its downstream target poly(ADP-ribose) polymerase (PARP) (data not shown), detectable by Western blotting as early as 15 h after TNFα stimulation. Therefore, these results demonstrate that TNFα treatment induces caspase activation followed by plasma membrane permeabilization and pinpoint the optimal time points when maximal effects can be observed.
Next, we evaluated the potential roles of CerS in these responses. Cer generation mediated by CerSs is implicated in two different pathways: the de novo pathway and the salvage or recycling pathway. To investigate the specific contributions of each in TNFα-induced Cer generation and cell death, MCF-7 cells were preincubated either with Myr, an inhibitor of the enzyme l-serine palmitoyltransferase that catalyzes the first step of the de novo pathway, or with the CerSs inhibitor FB1, which inhibits the N-acylation of dhSph in the de novo pathway and the N-acylation of both dhSph and Sph in the recycling or salvage pathway. Interestingly, the loss of cell viability upon TNFα treatment was partially inhibited by pretreatment of cells with FB1, whereas Myr showed no effect (Fig. 1A). Moreover, FB1 completely inhibited LDH release, whereas Myr failed to do so (Fig. 1B). In addition, TNFα-treated cells exhibited increased intracellular levels of calcium, a hallmark of secondary necrosis, at the same time points when LDH release was observed (15 and 24 h) (Fig. 1C). Most importantly, calcium overload was completely abolished in cells pretreated with FB1 (Fig. 1C). Similarly to cell viability, cells showed significant positivity for annexin V-FITC staining by 24 h of treatment with TNFα (and not at 15 h), which was greatly inhibited only by FB1. Annexin V-FITC positive cells exceeded 25% of the whole population at the 24 h time point but were <10% with FB1 pretreatment. In contrast, Myr pretreatment did not show an inhibitory effect, with >35% of cells being positive (Fig. 1, D and E). Taken together, these data suggest that CerSs control cell death induced by TNFα, probably through a Cer recycling pathway.
FIGURE 1.

The ceramide synthase inhibitor FB1 but not the serine palmitoyltransferase inhibitor Myr protects from TNFα-induced programmed cell death in MCF-7 cells. A, MCF-7 cells were pretreated with 50 μm FB1 or 100 nm Myr for 1 h and then treated with either vehicle (PBS) or 1 nm TNFα for the times indicated. Cell viability was measured by the MTT assay. B, plasma membrane permeabilization was measured by LDH release into the medium. C, intracellular levels of calcium were measured as described under “Experimental Procedures.” D, to quantify TNFα-induced apoptosis and the effects of FB1 and Myr (15 and 24 h post-treatment), flow cytometry was used. Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide staining were used to analyze the percentage of apoptotic cells. E, representation of the average of four independent experiments. A–D, data are the average of at least three independent experiments, and error bars represent S.E. Statistical significance was determined by two-way (A–C) or one-way (D) ANOVA with Bonferroni post-test. **, p < 0.01; ***, p 0.001; ****, p < 0.0001 versus vehicle.
Effects of TNFα Treatment on Sphingolipid Levels
Next, the effects of Myr and FB1 on generation of dhCer and Cer were investigated on untreated and TNFα-treated MCF-7 cells. In TNFα-treated cells, dhCerSs levels were significantly elevated as early as 6 h and remained elevated as compared with control cells (Fig. 2, A and B). FB1 completely blocked the elevation of dhCers in TNFα-treated cells. Similarly, generation of dhCers was also Myr-sensitive, suggesting altogether an induction of de novo synthesis. On the other hand, accumulation of Cer occurred as early as 15 h post-treatment in TNFα-treated cells and continued to increase at 24 h when compared with untreated cells (Fig. 2, C and D).
FIGURE 2.

TNFα-induced accumulation of a variety of sphingolipids. MCF-7 cells were pretreated with 50 μm FB1 or 100 nm Myr for 1 h and then treated with either vehicle (PBS) or 1 nm TNFα for the times indicated. Sphingolipids were quantified by mass spectrometry and normalized to total inorganic phosphates. Effects of FB1 and Myr on total dihydroceramide (A), individual dihydroceramide species (B), total ceramide (C), and individual ceramide species (D) are shown. Data are the average of three independent experiments. Error bars, S.E. For statistical analysis, a two-way ANOVA was performed with a Bonferroni post-test. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
In the absence of TNFα treatment, as expected, both Myr and FB1 decreased basal levels of Cer below untreated (vehicle) cells. Myr inhibition was greater than that of FB1 at the earliest time points (6 h); however, at the later time points (15 and 24 h), both inhibitors decreased Cer to the same extent, suggesting a delayed kinetics for FB1. Pretreatment with either FB1 or Myr reduced the elevation of Cer observed in TNFα-treated cells, although the inhibition was not complete as compared with Myr- or FB1-treated cells, respectively. Most importantly, FB1 inhibited Cer production in TNFα-treated cells significantly more than Myr at both 15 and 24 h post-treatment (Fig. 2C), suggesting additional involvement of the salvage pathway in the response to TNFα.
Next, the levels of sphingoid bases and their phosphates were analyzed. TNFα did not affect the levels of these sphingoid bases or their phosphates at the time points examined. FB1 pretreatment caused, as expected, an accumulation of Sph (Fig. 3A) and sphingosine 1-phosphate (Fig. 3B), and their levels remained unchanged also in the presence of TNFα (24 h time point). Pretreatment with Myr, on the other hand, did not have any appreciable effect on Sph and sphingosine 1-phosphate, either in the absence or in the presence of TNFα. Interestingly, dhSph (Fig. 3C) and dihydrosphingosine 1-phosphate (Fig. 3D) did not change in the presence of Myr or Myr plus TNFα (as compared with vehicle). In contrast, the levels of dhSph and dihydrosphingosine 1-phosphate were increased significantly more in the FB1 plus TNFα compared with the FB1 alone treatments. The latter results are consistent with the activation of the de novo pathway (which results in the formation of dhSph but not Sph) by TNFα such that dhSph is channeled into dhCer in the absence of FB1 but accumulates much more in the presence of FB1.
FIGURE 3.

Effects of TNFα on sphingoid bases. MCF-7 cells were pretreated with 50 μm FB1 or 100 nm Myr for 1 h and then treated with either vehicle (PBS) or 1 nm TNFα for 24 h. Sphingoid bases were quantified by mass spectrometry and normalized to total inorganic phosphates. Effects of FB1 and Myr on Sph (A), sphingosine 1-phosphate (B), dhSph (C), and dhSph 1-phosphate (D) are shown. Data represent the mean and S.E. (error bars) of at least three independent experiments. Statistical significance was determined by one-way ANOVA with Bonferroni post-test. **, p < 0.01; ***, p 0.001; ****, p < 0.0001 versus vehicle.
Differential Effects of Myriocin and Fumonisin B1 on the Accumulation of Long-chain Ceramides
In the last several years, Cers with specific chain lengths have been described as mediating cell death in response to cell stressors in a variety of cancer cells (41–43). Thus, we determined the specific Cers and dhCers elevated upon TNFα treatment. In treated cells, all of the individual Cer species were significantly elevated at the 24 h time point (Fig. 2D). Thus, TNFα induced elevation of 2–3-fold on C14-, C16-, C24:1-, C18:1-, and C22:1-Cers, whereas C20-Cer was increased as high as 6-fold. TNFα also induced elevation of the very long C24- and C24:1-Cer species, the latter being the most abundant species in MCF7 cells.
Notably, FB1 was significantly more effective than Myr in differentially preventing the late accumulation of long-chain species (C14-, C16-, C18-, and C18:1-Cer). The different effects of FB1 and Myr on the elevated C20- and C22:1-Cer were very modest. On the other hand, elevated very long-chain Cers (C24 and C24:1) were equally inhibited by FB1 and Myr (Fig. 2D).
Similarly, dhCer species also accumulated in TNFα-treated cells (Fig. 2B). Pretreatment with FB1 and Myr showed more effective inhibition of long- and medium long-chain dhCers than very long-chain species. Furthermore, Myr inhibited the elevation of long-chain species slightly more efficiently than FB1. Thus, the different effects of FB1 and Myr in preventing the TNFα-induced accumulation of Cer and dhCer species were chain length-specific. Thus, FB1 (which would inhibit both the salvage and de novo pathways) showed more robust effects, especially on the long-chain Cers, than Myr, which selectively inhibits the de novo pathway.
Taken together, these data demonstrate differential effects of FB1 on Cer (but not dhCer accumulation) and on inhibition of cell death. These results suggest that at least two distinct pools (produced by the de novo and salvage pathways) contribute to the accumulation of Cer in TNFα-treated cells. The results also strongly suggest that the long-chain Cer species (the FB1-sensitive pools) may play a more specific role in TNFα-induced cell death.
Caspase Activation Is Necessary for Ceramide Generation and Cell Death Progression
Programmed cell death is a very well coordinated process that involves key steps, such as MOMP, which is controlled by the pro-apoptotic Bcl-2 proteins Bak and Bax, and caspase activation. In order to examine whether MOMP is required for TNFα-induced cell death, we knocked down Bak and Bax and evaluated its effects. In our system, caspase-7 activation and LDH release were not affected by siBak or siBax (data not shown), suggesting that TNFα-induced plasma membrane permeabilization might occur independently of MOMP. Thus, we next tested whether activation of late caspases, a downstream event of MOMP, is necessary for TNFα-induced programmed cell death. To this end, MCF-7 cells were pretreated with either the pan-caspase inhibitor z-VAD-fmk or the caspase-3/7 inhibitor z-DEVD-fmk. Both z-VAD-fmk and z-DEVD-fmk greatly inhibited TNFα-induced loss of cell viability (Fig. 4A), plasma membrane permeabilization (Fig. 4B), calcium overload (data not shown), and phosphatidylserine externalization (Fig. 4, C and D), confirming that cell death induced by TNFα requires caspase activation.
FIGURE 4.
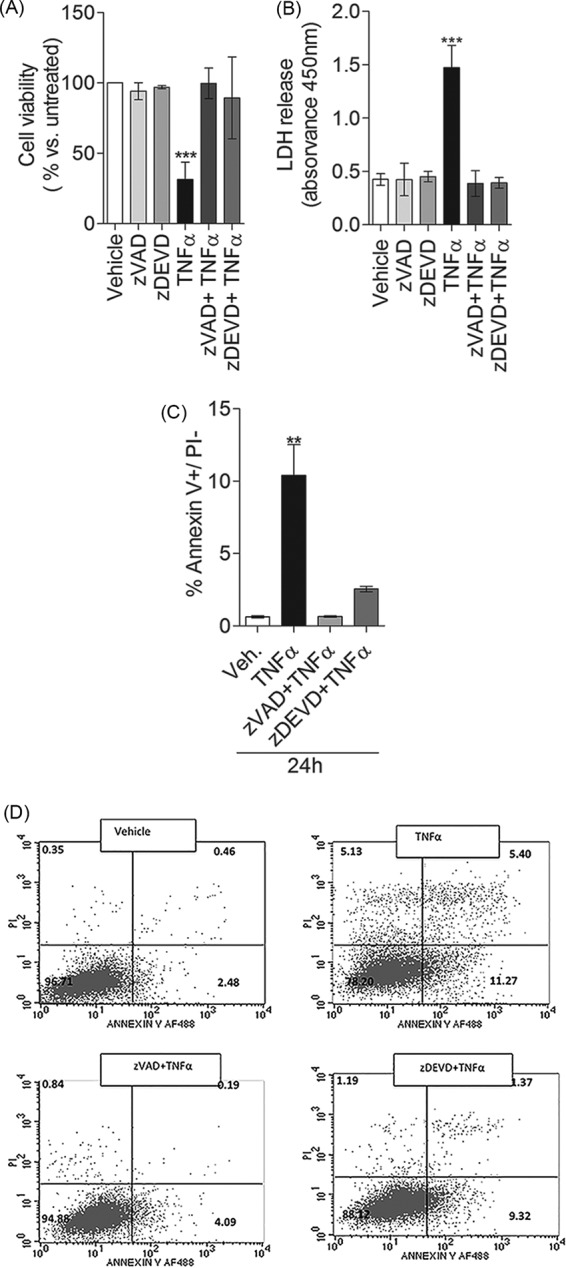
Both the pan-caspase inhibitor z-VAD and the caspase-3/7 inhibitor z-DEVD protect from TNFα-induced programmed cell death. MCF-7 cells were pretreated with either 10 μm z-VAD or 10 μm z-DEVD for 1 h and then treated with vehicle or 1 nm TNFα for 24 h. Effects of z-VAD and z-DEVD on cell viability were measured by the MTT assay (A), and effects on plasma membrane permeabilization were measured by LDH release into the medium (B). C, annexin V-fluorescein isothiocyanate (FITC) and propidium iodide staining were used to analyze the percentage of apoptotic cells. D, representation of the average of three independent experiments. A–C, data are the average of at least three independent experiments, and error bars represent S.E. For statistical analysis, a one-way ANOVA with Bonferroni post-test was used. **, p < 0.01; ***, p 0.001 versus vehicle.
The results thus far show that, in this model, Cer generated through the salvage pathway (Fig. 1) and caspase activities (Fig. 4) are necessary for cell death. Thus, we next tested whether caspase activation and Cer generation are dependent or independent events. To tackle this question, MCF-7 cells were pretreated with the pan-caspase inhibitor z-VAD-fmk and the caspase-3/7 inhibitor z-DEVD-fmk, and Cer generation was quantified by HPLC/MS. The inhibitor z-VAD-fmk or z-DEVD-fmk alone did not decrease Cer levels below those of untreated cells (Fig. 5, A and B). Pretreatment of MCF-7 cells with the pan-caspase inhibitor z-VAD-fmk totally abrogated TNFα-induced accumulation of Cer (Fig. 5A). In contrast, the inhibition of elevated Cers by caspase-3/7 inhibitor z-DEVD-fmk was not as complete because it inhibited ∼70% of TNFα-induced Cer (Fig. 5B). Interestingly, z-VAD-fmk and z-DEVD-fmk effects were not chain length-specific, because accumulation of long-chain and very long-chain Cers was inhibited to the same extent (Table 1). For instance, z-DEVD-fmk pretreatment depleted the accumulation of C16-Cer, C18-Cer, or C24:1-Cer by 70–75% (Fig. 5C and Table 1). Taken together, these data suggest the involvement of caspases in the generation of Cer following TNFα treatment. In addition, the partial inhibition by z-DEVD-fmk of the TNFα-induced Cer indicates the existence of two pools of Cer: one, which accounts for ∼70% of TNFα-induced Cer, regulated by late caspases (caspase-7) and one, which accounts for the remaining 30% of Cer, regulated by early caspases (but not late caspases). Thus, one pool of Cer must be upstream of caspase-7 or at least independent of caspase-7 and regulated by early caspases, whereas a second pool of Cer must function downstream of caspase-7 activity.
FIGURE 5.
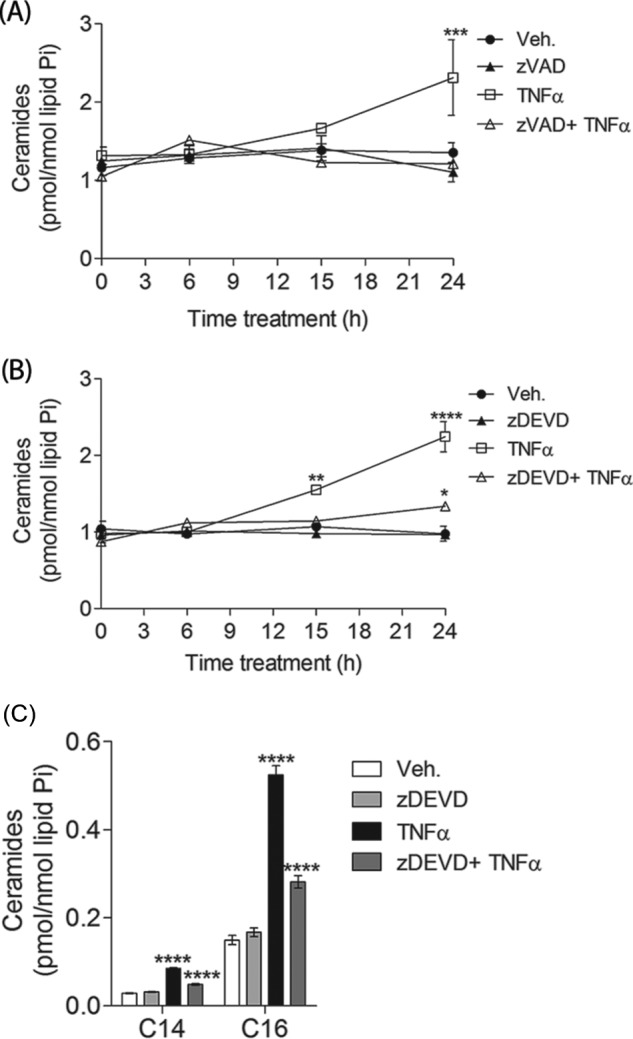
TNFα-induced accumulation of total ceramides is inhibited by the pan-caspase inhibitor z-VAD-fmk and the caspase-3/7 inhibitor z-DEVD-fmk. MCF-7 cells were pretreated with either 10 μm z-VAD or 10 μm z-DEVD for 1 h and then treated with either vehicle (PBS) or 1 nm TNFα for the times indicated. Sphingolipids were quantified by mass spectrometry and normalized to total inorganic phosphates. A, effects of z-VAD-fmk on total ceramides. Effects of z-DEVD-fmk on total ceramides (B) and C14- and C16-ceramide (C) are shown. A–C, data are the average of three independent experiments. Error bars, S.E. For statistical analysis, a two-way ANOVA was performed with a Bonferroni post-test. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
TABLE 1.
Individual species of ceramides are inhibited by the pan-caspase inhibitor z-VAD-fmk and the caspase-3/7 inhibitor z-DEVD-fmk
MCF-7 cells were pretreated with either 10 μm z-VAD or 10 μm z-DEVD for 1 h and then treated with either vehicle (PBS) or 1 nm TNFα for the times indicated. Sphingolipids were quantified by mass spectrometry and normalized to total inorganic phosphates. Shown are the effects of z-VAD-fmk and z-DEVD-fmk on C16-, C18-, C20-, C22-, C24-, and C24:1-ceramides. Data are the average of three independent experiments ± S.E. For statistical analysis, a two-way ANOVA was performed with a Bonferroni post-test. *, p < 0.05; ***, p < 0.001; ****, p < 0.0001.

Ceramide Synthase Activation/Ceramide Generation Function Upstream of Caspase-7
The inhibition of cell death by FB1 (and not Myr) points to a key role of CerS activation in cell death (Fig. 1). On the other hand, the effects of the caspase-3/7 inhibitor z-DEVD-fmk on Cer generation have identified a specific pool of Cer independent of caspase-7 activation, and this pool may act upstream of and regulate caspase-7 activity. To test whether the inhibitory effect of FB1 on cell death occurred upstream of caspase-7, we pretreated cells with FB1 and Myr and then tested their effects on early and late caspases. Neither FB1 nor Myr affected TNFα-induced cleavage of procaspase-8, at neither 15 nor 24 h after treatment (Fig. 6A); nor did they prevent cleavage of Bid (Fig. 6A), a caspase-8 target, in response to TNFα treatment (44, 45). In contrast, FB1 but not Myr reduced the cleavage of procaspase-7 (Fig. 5A). Next, caspase-3/7 activation was examined. Because MCF-7 cells lack caspase-3, the activity measured is most likely due to caspase-7. Interestingly, both FB1 and Myr inhibited caspase-7 activation in TNFα-treated cells at 15 h post-treatment, but only FB1 reduced it at 24 h post-treatment (Fig. 6B). These findings demonstrate that CerS/Cer might function proximal to the effector caspase-7.
FIGURE 6.

The ceramide-synthase inhibitor FB1 protects from TNFα-induced caspase-7 cleavage and activation in MCF-7 cells. A, MCF-7 cells were treated as indicated for 15 or 24 h. The cleavage of caspase-8, caspase-7, and Bid was analyzed by Western blot. A representative experiment is shown. Similar results were obtained in at least two additional experiments. GAPDH was used as gel loading control. B, MCF-7 cells were pretreated with FB1 or Myr for 1 h and then treated with either vehicle (PBS) or 1 nm TNFα for the times indicated. The caspase-3/7-like DEVDase activity assay was performed in treated and untreated cells. Data are the average of three independent experiments. Error bars, S.E. For statistical analysis, a two-way ANOVA was performed with a Bonferroni post-test. *, p < 0.05; **, p < 0.01.
Fumonisin B1 Inhibits TNFα-induced Loss of FAK
During the execution phase of apoptosis, effector caspases (e.g. caspase-7) cleave essential cellular nuclear and cytoskeleton proteins responsible for the morphological changes that characterize apoptosis, such as cell shrinkage, cell detachment, and loss of cell-cell interaction (46). Lamins, gelsolin, α-fodrin, and FAK, among others, are well known targets of caspases (34, 37, 39, 47). In this system, TNFα-treated cells exhibited a time-dependent altered morphology, with many cells rounding up (data not shown). Inhibition of FAK with dominant negative FAK (FAK C-terminal domain) or antisense oligonucleotides has been reported to cause cell rounding, loss of adhesion, and apoptosis in several cancer cell lines, including BT474 human breast cancer cells, C8161 human melanoma cells, and MCF-7 breast carcinoma cells (31, 48–50). Thus, we investigated the possible involvement of FAK in our system. Because FAK activity is regulated by autophosphorylation at Tyr-397 and phosphorylation at this site regulates FAK-mediated downstream signaling (51), we measured total FAK levels and Tyr-397 phosphorylation in untreated and treated cells. A dose- and time-dependent decrease of phosphorylated and total FAK was observed following TNFα (Fig. 7, A and C, respectively), coinciding with the morphologic changes (data not shown). Indeed, phosphorylated FAK was robustly decreased as early as 15 h post-treatment and continued decreasing up to 24 h, as observed by Western blot (Fig. 7C). On the other hand, total FAK levels were slightly decreased at the same time points (Fig. 7C). Furthermore, pretreatment with either z-VAD-fmk or z-DEVD-fmk completely blocked the TNFα-induced effect on total and autophosphorylated FAK at the 24 h time point (Fig. 7B), suggesting that the FAK levels are down-regulated by TNFα treatment and that the process depends on caspase activity.
FIGURE 7.

TNFα-induced dephosphorylation of FAK (Tyr-397) and cleavage of FAK are caspase-dependent events. Fumonisin B1 inhibited the loss of FAK (Tyr(P)-397). MCF-7 cells were treated with the indicated concentration of TNFα for 24 h (A), with 10 μm z-VAD or z-DEVD for 1 h prior to 24-h treatment with TNFα (1 nm) (B), or with 50 μm FB1 or 100 nm Myr for 1 h followed by treatment with either vehicle or TNFα for the times indicated (C). The levels of phosphorylated and total FAK were analyzed by Western blot. A representative experiment is shown. Similar results were obtained in at least two additional experiments. GAPDH was used as a gel loading control. D, quantitative analysis of effects of fumonisin B1 and myriosin on the loss of phosphorylated (Tyr(P)-397) and total FAK upon 24 h of TNFα treatment. Data represent the mean and S.E. (error bars) of at least three independent experiments. Statistical significance was determined by one-way ANOVA with Bonferroni post-test. *, p < 0.05; **, p < 0.01 versus vehicle.
Because we have identified that Cer generated through CerSs regulates cell death upstream of caspase-7, we next asked whether CerSs also control the down-regulation of FAK associated with caspase-7. To this end, FB1 or Myr were added to the medium 1 h prior to TNFα treatment, and FAK protein levels were measured by Western blot up to 24 h post-treatment. As shown in Fig. 7C, FB1 pretreatment significantly protected from TNFα-induced loss of autophosphorylated FAK (Tyr(P)-397) and total FAK, whereas Myr showed no discernable protective effects. In addition, FB1 but not Myr partially overcame the TNFα effects on total FAK levels. Quantification of the phosphorylated and total FAK levels statistically confirmed these differences (Fig. 7D). Collectively, these data demonstrate that CerS activity might control TNFα-induced loss of total and phosphorylated FAK, probably through caspase-7 activation.
Focal Adhesion Kinase Inhibitor Y15 Induces Plasma Membrane Permeabilization Independently of Caspase-7
Apoptosis initiated by the intrinsic or the extrinsic pathway comprises two phases. The first phase, prenecrotic, is characterized by morphological changes in cells, caspase activation, and intact plasma membrane integrity. On the other hand, the second phase, or secondary necrosis, is characterized by cytoplasmic swelling, plasma membrane rupture, and release of intracellular content (4, 46, 52). The events leading to plasma membrane permeabilization are not well understood and are thought to involve ATP depletion, loss of osmotic homeostasis, and cleavage of cortex and plasma membrane proteins, such as paxillin or focal adhesion kinase (4, 52–54). Our data demonstrate that both caspase-7 activation and CerS activity are necessary for the loss of FAK as well as plasma membrane rupture. Furthermore, decreased autophosphorylated and total FAK occurred earlier than LDH release in our system. Thus, we hypothesized that the loss of FAK, downstream of caspases and CerSs, regulates plasma membrane permeabilization in TNFα-treated cells. To tackle this question, the effects of the FAK inhibitor Y15 or inhibitor 14 on cell death were tested. The inhibitor Y15 is known to specifically inhibit the autophosphorylation of FAK (at Tyr-397) and decrease cancer cell viability and block tumor growth (55–57). As expected, Y15-treated cells showed a decrease in phosphorylated FAK (Tyr(P)-397) that occurred as early as 15 min post-treatment (Fig. 8A). Tyr(P)-397-FAK was more effectively and rapidly decreased by Y15 than total FAK, which started to occur at a later time (6 h post-treatment) (Fig. 8A). To test the effect of Y15 on cell viability, MCF-7 cells were treated for different lengths of time, and the results from the MTT assay indicated that viability was reduced by 60% as early as 3 h after treatment and by 80% at 6 h post-Y15 treatment (Fig. 8B). Most importantly, plasma membrane permeabilization was examined in the absence or presence of the FAK inhibitor Y15. Interestingly, LDH release was significantly increased upon Y15 at the latest time point (6 h) (Fig. 8C), concomitantly with reduced total FAK levels and preceded by the loss of phosphorylated FAK. Furthermore, Y15 treatment (24 h) has been shown to decrease pro-caspase-8 and increase cleaved caspase-3 and PARP in DBTRG and U87 cells (56). In our model, MCF-7 cells treated with the FAK inhibitor Y15 showed a decrease of pro-caspase-8 and pro-caspase-7 forms (inactive form) as early as 6 h post-treatment, as analyzed by Western blot analysis (Fig. 8A). Neither cleaved caspase-8 nor cleaved caspase-7 fragments were detected in treated cells, suggesting that caspases are not activated by Y15 treatment. Indeed, this hypothesis was confirmed when caspase-7 activity was measured and no increase was detected in Y15-treated cells at the time points studied (Fig. 8D). In addition, the caspase-7 inhibitor z-DEVD-fmk had no effect on LDH release induced by Y15, as shown in Fig. 7E. Thus, our findings suggest that down-regulation of FAK in the absence of caspase activation is sufficient to induce plasma membrane permeabilization.
FIGURE 8.

The inhibition of the autophosphorylation of FAK on Tyr-397 by FAK inhibitor 14 (Y15) is sufficient to trigger cell death. MCF-7 cells were treated with a 10 μm concentration of the focal adhesion kinase inhibitor Y15 for the time indicated. A, the levels of phosphorylated and total FAK and pro-caspase-7 were analyzed by Western blot upon Y15 treatment for the indicated time. A representative experiment is shown. Similar results were obtained in at least two additional experiments. GAPDH was used as a gel loading control. B, cell viability was measured by the MTT assay. C, plasma membrane permeabilization was measured by the LDH release assay. D, a caspase-3/7-like DEVDase activity assay was performed in the time indicated. E, MCF-7 cells were pretreated with 10 μm z-DEVD for 1 h and then treated with Y15 for 6 h. Plasma membrane permeabilization was measured by LDH release into the medium. Data represent mean and S.E. (error bars) of at least three independent experiments. B–E, statistical significance was determined by one-way ANOVA with Bonferroni post-test. ***, p < 0.001; ****, p < 0.0001 versus vehicle.
Knockdown of Ceramide Synthases 6 Protects from Cell Death Induced by TNFα
Because we have shown that treatment of MCF-7 cells with TNFα triggered accumulation of the sphingolipid Cer followed by cell death and that the CerS inhibitor FB1 greatly inhibited the accumulation of long-chain Cers (C14-, C16-, C18-, and C18:1-Cer) and protected from TNFα-induced cell death, we investigated the involvement of individual CerSs in mediating cell death induced by TNFα using siRNA-mediated knockdown. First, the efficacy of the siRNA- induced silencing of the individual CerS isoforms (CerS1, -2, -4, -5, and -6) was established. As shown in Table 2, expression of CerS2, CerS4, and CerS6 was greatly decreased. siCerS1 and siCerS5 were less effective, albeit the remaining CerS1 and CerS5 expression were 23 and 25% of control, respectively. As reported elsewhere (58), down-regulation of certain CerSs resulted in the up-regulation of untargeted CerSs. For instance, siCerS6 up-regulated the gene expression of CerS1 (2-fold), CerS2 (1.4-fold), and CerS5 (2-fold), whereas siCerS5 up-regulated the expression of CerS1 and CerS6 (data not shown).
TABLE 2.
PCR analysis of individual CerS knockdowns
The gene expression of CerSs in siRNA-treated cells was measured as described under “Experimental Procedures.”
| siRNA | siRNA concentration | Gene expression versus mock |
|---|---|---|
| nm | % | |
| siCerS1 | 20 | 23 ± 13 |
| siCerS2 | 20 | 12 ± 4 |
| siCerS4 | 20 | 3 ± 0 |
| siCerS5 | 5 | 26 ± 9 |
| siCerS6 | 5 | 6 ± 1 |
Next, the effects of the gene silencing on plasma membrane permeabilization were investigated. As depicted in Fig. 9A, siCerS2 and siCerS4 significantly increased basal levels of LDH in the medium as compared with mock, whereas siCerS1, siCerS5, and siCerS6 did not alter LDH levels in untreated (vehicle) cells. Importantly, upon TNFα treatment, down-regulation of CerS6 significantly protected from LDH release. Knockdown of CerS1 also showed a trend to decrease LDH release in treated cells, but it failed to meet our criteria for statistical significance. Furthermore, siCerS1 showed a defect in cell growth that might contribute to lower LDH release in treated cells (data not shown) but not in untreated cells.
FIGURE 9.
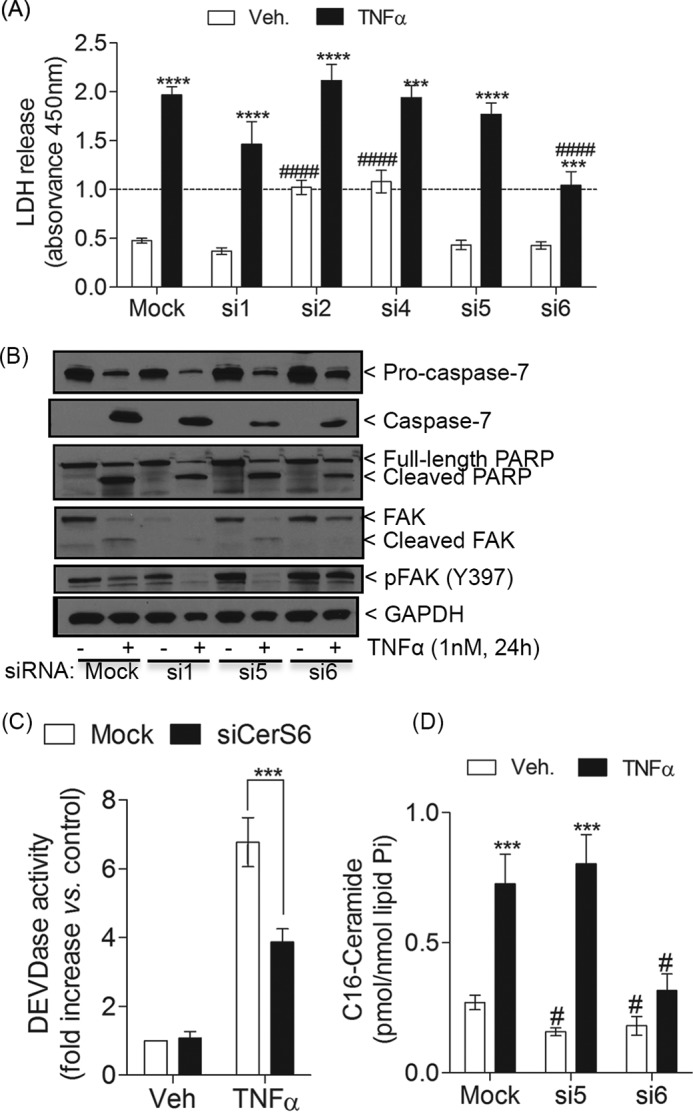
Silencing of CerS6 but not other isoforms protects from cell death. MCF-7 cells were transfected with siCerS1, -2, -4, -5, or -6 or mock (siControl) for 48 h. After changing the medium, cells were treated with TNFα or vehicle for 24 h. A, LDH release was measured as described earlier. B, effects of mock, siCerS1, siCerS5, and siCerS6 on the levels of procaspase-7 and total and phosphorylated FAK were analyzed by Western blot. A representative experiment is shown. Similar results were obtained in at least three additional experiments. GAPDH was used as gel loading control. C, effects of mock and siCerS6 on the caspase-3/7-like DEVDase activity assay. D, effects of mock, siCerS5, and siCerS6 on C16-ceramide. A, C, and D, error bars, S.E. For statistical analysis, a one-way (C) or two-way ANOVA (A and D) was performed with multiple Bonferroni post-tests. ***, p < 0.001; ****, p < 0.0001 versus siRNA-matched untreated cells. #, p < 0.05; ####, p < 0.0001 versus treatment-matched siControl.
Next, we asked whether knockdown of CerS6 could overcome the effects of TNFα on caspase-7 and FAK. Indeed, siCerS6 significantly reduced caspase-7 activation in response to TNFα (Fig. 9, B and C) and partially prevented PARP cleavage (Fig. 9B). The results were most clear in showing that knockdown of CerS6 protected from TNFα-induced cleavage of FAK and loss of phosphorylated FAK. As a control, down-regulation of CerS5 was compared with the effects of siCerS6, because siCerS5 was not able to prevent LDH release in response to TNFα (Fig. 9A), although these CerSs share the same fatty acyl-CoA preference. As for LDH release, siCerS5 was not effective in blocking caspase-7 activation, PARP cleavage, and loss of FAK (phosphorylated and total) (Fig. 9B). In addition, to rule out a possible role for CerS1 in the regulation of TNFα-induced cell death, we also examined the effects of siCerS1. Similar to siCerS5, siCerS1 did not prevent activation of caspase-7; nor did it prevent PARP cleavage or loss of phosphorylated FAK (Fig. 9B). Interestingly, knockdown of CerS1 reduced the levels of inactive FAK in untreated cells, which were further decreased by TNFα. These results demonstrate a key role for CerS6 in regulating caspase-7 and even more so the loss and dephosphorylation of FAK.
Next, the effects of down-regulation of CerS6 on Cer levels were determined. Overall, knockdown of CerS6 caused a slight but not significant increase in total Cers as compared with mock. This basal accumulation of total Cers was mostly due to the increase of very long-chain Cers (C20-, C22-, and C24-Cers) (Table 3), probably because of up-regulation of CerS2 in response to siCerS6. Likewise, knockdown of CerS5 altered basal levels of long (e.g. C18-Cer) and very long-chain CerSs most likely due to its effects on CerS1 and CerS2 expression, respectively. Similar to Fig. 2D, TNFα-treated cells showed a significant elevation of all individual Cers in control cells. On the other hand, siCerS6 treatment blocked the increase of all Cer species induced by TNFα, except for C24-Cer (Table 3). Most importantly, CerS6 knockdown exhibited a robust effect on C16-Cer levels (Fig. 9D and Table 3) by decreasing C16-Cer in vehicle-treated cells as compared with mock and by completely preventing its accumulation upon TNFα treatment, despite up-regulation of CerS5. Of note, siCerS5 basally decreased C14 and C16-Cers, but it showed no inhibitory effects on the elevation of these species upon TNFα treatment (Table 3 and Fig. 9D, respectively). Taken together, our data implicate a very specific pool of C16-Cer regulated by CerS6 in controlling key events in the execution phase of apoptosis, such as the loss of FAK and plasma membrane permeabilization via regulation of caspase-7 activity.
TABLE 3.
Knockdown of CerS6 inhibits the elevation of C16-ceramide upon TNFα treatment
MCF-7 cells were treated as described in the legend to Fig. 8. Sphingolipids were quantified by mass spectrometry and normalized to total inorganic phosphates. Shown are the effects of mock and siCerS6 on ceramides. Data are the average of three independent ± S.E. For statistical analysis, a two-way ANOVA was performed. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus siRNA-matched untreated cells. #, p < 0.05; ###, p < 0.001 versus treatment-matched mock.
| Sphingolipid | Inorganic phosphate ± S.E. |
|||||
|---|---|---|---|---|---|---|
| Mock |
siCerS5 |
siCerS6 |
||||
| Vehicle | TNFα | Vehicle | TNFα | Vehicle | TNFα | |
| pmol/nmol | ||||||
| Total Cer | 1.229 ± 0.495 | 3.496 ± 1.127** | 1.893 ± 0.968 | 4.981 ± 1.514** | 1.970 ± 0.184 | 3.388 ± 1.278 |
| C16:0-Cer | 0.271 ± 0.062 | 0.725 ± 0.256*** | 0.158 ± 0.027# | 0.803 ± 0.194*** | 0.181 ± 0.072# | 0.317 ± 0.126# |
| C14:0-Cer | 0.033 ± 0.004 | 0.090 ± 0.036** | 0.018 ± 0.019# | 0.087 ± 0.032** | 0.040 ± 0.008 | 0.083 ± 0.042 |
| C18:0-Cer | 0.023 ± 0.014 | 0.133 ± 0.100* | 0.023 ± 0.020 | 0.188 ± 0.091** | 0.043 ± 0.016 | 0.117 ± 0.048 |
| C18:1-Cer | 0.010 ± 0.014 | 0.024 ± 0.012* | 0.007 ± 0.006 | 0.042 ± 0.024** | 0.006 ± 0.002 | 0.016 ± 0.004 |
| C20:0-Cer | 0.013 ± 0.010 | 0.080 ± 0.068 | 0.009 ± 0.008 | 0.132 ± 0.116 | 0.019 ± 0.022 | 0.048 ± 0.043 |
| C22:0-Cer | 0.102 ± 0.051 | 0.380 ± 184* | 0.177 ± 0.127 | 0.486 ± 0.176* | 0.202 ± 0.056# | 0.460 ± 0.263 |
| C22:1-Cer | 0.024 ± 0.010 | 0.092 ± 052 | 0.026 ± 0.014 | 0.164 ± 0.104** | 0.040 ± 0.019 | 0.087 ± 0.066 |
| C24:0-Cer | 0.287 ± 0.131 | 0.635 ± 0.123** | 0.358 ± 0.125 | 0.796 ± 0.045* | 0.722 ± 0.045### | 1.167 ± 0.301#* |
| C24:1-Cer | 0.322 ± 0.144 | 0.837 ± 0.275* | 0.533 ± 0.318 | 1.283 ± 0.463** | 0.550 ± 0.286 | 0.845 ± 0.340 |
C6-ceramide-induced Accumulation of Long-chain Ceramides and Cell Death Occurred via the Salvage Pathway
To further demonstrate the role of Cer in the regulation of FAK and plasma membrane permeabilization during cell death, MCF-7 cells were treated with exogenous short-chain C6-Cer (25 μm). First, MCF-7 cells were treated with or without C6-Cer for various times. C6-Cer-treated cells underwent drastic morphological changes occurring as early as 18 h after treatment (Fig. 10A). Indeed, rounding of the cells preceded plasma membrane permeabilization, which was significantly increased as early as 24 h (compared with vehicle control) and robustly continued increasing up to 48 h post-treatment (Fig. 10B). Tyr(P)-397 FAK was significantly reduced as early as 24 h post-treatment (data not shown), whereas both total and phospho-FAK levels were robustly lost at the 48 h time point (Fig. 10D), as examined by Western blot. In addition, C6-Cer treatment was able to induce LDH release to the same extent as TNFα (included as a positive control), as shown in Fig. 10C. Because in this model cell death was prevented by the caspase-7 inhibitor z-DEVD-fmk, the involvement of caspase-7 activation downstream of C6-Cer-induced cell death was next addressed. Similar to the effects on TNFα, inhibition of caspase-7 blocked LDH release following C6-Cer treatment (Fig. 10C), suggesting that caspase-7 activation is required for cell death induced by exogenous C6-Cer treatment. In line with this observation, C6-Cer treatment caused partial loss of pro-caspase-7 (Fig. 10E) and a significant increase of caspase-7 activity (Fig. 10F). Therefore, these results show that C6-Cer is sufficient to induce caspase-7 activation, cleavage of FAK, and plasma membrane permeabilization.
FIGURE 10.

C6-ceramide-induced cell death is inhibited by the ceramide synthase inhibitor FB1. A, MCF-7 cells were treated with or without C6-Cer (25 μm, 18 h), and cell morphology images were taken by phase-contrast microscopy. B, plasma membrane permeabilization was measured by the LDH release assay in cells treated with C6-ceramide for the time indicated. C, cells were treated with EtOH (48 h), C6-Cer (48 h), C6-Cer and z-DEVD (10 μm; added 1 h prior to C6-Cer), or TNFα (1 nm; 24 h). The LDH released into the medium was measured. D, the levels of pro-caspase-7, phosphorylated FAK (Tyr(P)-397), and total FAK were analyzed by Western blot. Two representative experiments are shown. GAPDH was used as gel loading control. E, the caspase-3/7-like DEVDase activity was assayed as described under “Experimental Procedures.” Cells were treated with FB1 1 h prior to the addition of either vehicle (EtOH, 48 h) or C6-Cer (48 h). F, the LDH released into the medium was measured. Ceramides were quantified by mass spectrometry and normalized to total inorganic phosphates. Effects of C6-Cer and of FB1 plus C6-Cer on total ceramide levels (G) and C16-ceramide (H) are shown. B, C, E, F, G, and H, data represent mean and S.E. (error bars) of at least three independent experiments. Statistical significance was determined by one-way ANOVA with Bonferroni post-test (G and H), two-way ANOVA with Bonferroni post-test (B, D, and F), or unpaired t test (E). *, p < 0.05; **, p < 0.01; ***, p 0.001; ****, p < 0.0001 versus EtOH.
Next, the biochemical mechanism by which exogenous C6-Cer induced cell death was investigated. To answer the question of whether exogenous C6-Cer induces cell death due to intracellular accumulation of exogenously added C6-Cer per se or due to the generation of endogenous Cer, cells were pretreated with the CerS inhibitor FB1, and its effects on the Cer levels were examined. As depicted in Fig. 10G, endogenous levels of Cer levels were robustly increased in C6-Cer-treated cells. Treatment of MCF-7 cells with C6-Cer (48 h) resulted in accumulation of total level of Cers by 15-fold as compared with vehicle (EtOH)-treated cells. Likewise, individual long-chain Cer species were robustly increased in treated cells (Fig. 10F and Table 4). For instance, C6-Cer treatment induced an increase of C16-Cer by 22-fold (Fig. 10G). Most importantly, inhibition of CerS by FB1 completely abrogated the accumulation of Cer species (Fig. 10F and Table 4). Interestingly, C6-Cer treatment caused an accumulation of Sph (Table 4), which was further elevated by FB1 pretreatment. Collectively, our data demonstrate that exogenous C6-Cer resulted in increased endogenous long-chain Cers due to deacylation of the short-chain C6-Cer to produce Sph, which is further reacylated by CerS to generate Cers rather than elongation of the acyl chain. More importantly, LDH released was significantly diminished in FB1-pretreated cells as compared with the C6-Cer-treated group (Fig. 10F), suggesting that plasma membrane permeabilization induced by exogenous C6-Cer treatment involves biochemical recycling of short-chain Cer. Collectively, our results define the salvage or recycling pathway of synthesis of a very specific pool of Cer as the source of endogenous Cer that controls cell death and its downstream target, FAK, whose down-regulation via caspase-7 activation leads to plasma membrane rupture.
TABLE 4.
Effects of the inhibition of ceramide synthases by FB1 on sphingolipids upon C6-ceramide treatment
MCF-7 cells were treated as described in the legend to Fig. 9. Sphingolipids were quantified by mass spectrometry and normalized to total inorganic phosphates. Data are the average of three independent experiments ± S.E. For statistical analysis, a one-way ANOVA was performed. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 versus EtOH-treated cells. #, p < 0.05; ##, p < 0.01; ####, p < 0.0001 versus C6-Cer.
| Sphingolipid | Inorganic phosphate ± S.E. |
||
|---|---|---|---|
| Vehicle | C6-Cer | FB1 + C6-Cer | |
| pmol/nmol | |||
| C16:0-Cer | 0.288 ± 0.066 | 5.681 ± 2.159** | 0.310 ± 0.113## |
| C14:0-Cer | 0.024 ± 0.006 | 0.699 ± 0.357* | 0.025 ± 0.010# |
| C18:0-Cer | 0.019 ± 0.004 | 0.688 ± 0.314* | 0.064 ± 0.023# |
| C18:1-Cer | 0.010 ± 0.002 | 0.055 ± 0.007**** | 0.009 ± 0.002#### |
| Sphingosine | 0.034 ± 0.007 | 0.397 ± 0.090** | 0.656 ± 0.089*** # |
Discussion
In the present study, we have defined a novel role for CerS in TNFα-induced cell death, specifically CerS6 and its product C16-Cer, in regulating postmitochondrial events, such as the loss of FAK protein and plasma membrane permeabilization, via the activation of caspase-7. Our results have demonstrated that CerS activity is essential for programmed cell death progression and that the pool of Cer involved derives from the salvage pathway rather than de novo synthesis (summarized in Fig. 11).
FIGURE 11.
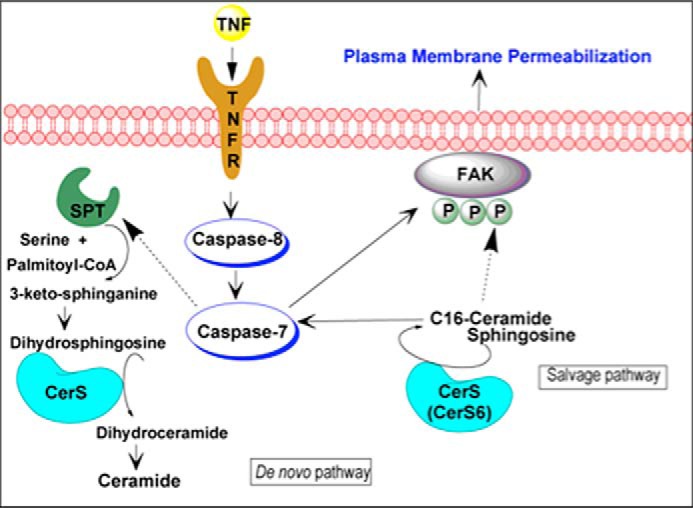
Proposed model of regulation of TNFα-induced cell death by ceramide synthase. TNFα stimulation induces ceramide generation by at least two pathways: a de novo pathway and the salvage pathway, which occurred downstream of the apical caspase-8. CerSs, which control both pathways, catalyze the N-acylation of (dihydro)sphingosine to produce (dihydro)ceramide. In turn, ceramide synthase-derived ceramide from the salvage pathway, especially CerS6 and its product C16-ceramide, activates caspase-7, which cleaves FAK. The loss of Tyr(P)-397-FAK and total FAK preceded cell death and subsequent secondary necrosis. SPT, serine palmitoyltransferase.
TNFα is known to increase intracellular levels of Cer by an earlier activation of neutral and acid SMase and a later activation of the de novo pathway via CerS (7, 59). Significant evidence has accumulated describing the role of acid and neutral SMase in TNFα-induced cell death (13, 59–61). However, much less is known about the role of CerS. In mammals, six different CerSs have been identified (CerS1–CerS6), each of which synthesizes Cers with distinct acyl chain lengths. For instance, CerS1 mostly uses C18-acyl-CoA (18), CerS2 uses C22-C24-acyl-CoAs (19), CerS4 uses C20-acyl-CoA, and CerS5 and CerS6 use C14 and C16-acyl-CoA (20, 21). The role of specific CerSs isoforms/Cer species has been studied in the context of apoptosis and cancer. Over the years, C16-Cer via CerS5/CerS6 activity has been proposed to be pro-apoptotic (10, 62). However, the role of individual CerS isoforms and, therefore, of specific Cers with different acyl chain length might be cell type-specific. In the current study, we have found that TNFα induced accumulation of multiple Cer and dhCer species of different chain lengths and that at the late time points examined, the de novo biosynthesis and the recycling pathway of Cer were both activated. In fact, whereas the difference in the effect of FB1 versus Myr on the late accumulation of total Cers and dhCers is rather small, the enhanced inhibitory effect of FB1 was clearly noticeable when looking at specific species. Indeed, FB1 was specifically more effective in inhibiting the elevation of long-chain species (such as C14 and C16-Cer) and medium long-chain species (C18 and C18:1-Cer) of Cer in response to TNFα as compared with Myr, suggesting that both pathways contribute to the elevation of C14-C18:1-Cers. Moreover, the up-regulation of de novo synthesis is supported by the fact that TNFα induced a further increase in dhSph and dihydrosphingosine 1-phosphate in cells pretreated with FB1. Nevertheless, the de novo pathway is unlikely to be involved in secondary necrosis because Myr failed to prevent any of the events involved, such as caspase-7 activation, loss of FAK, and plasma membrane permeabilization. In addition, the fact that C16-Cer levels in Myr-pretreated cells were at the level of vehicle treatment upon TNFα, whereas LDH release was at the level of the TNFα-treated group, further suggests that only a subset of Cer generated by CerS or the biosynthetic pathway (salvage pathway versus de novo pathway) may regulate downstream events, such as plasma membrane permeabilization.
In addition to the differential effects on late Cer inhibition, we also observed that FB1 per se elevated Sph and dhSph. However, a protective role of elevated sphingoid bases is unlikely because simultaneous treatment with FB1 and Myr, which is known to prevent the accumulation of dhSph (63), did not reverse the effects of FB1 on plasma membrane permeabilization (data not shown). Overall, our data strongly suggest that the synthesis of salvaged Cer from the N-acylation of Sph is the primary source for the specific pool of Cer involved in secondary necrosis induced by TNFα.
The specific mechanism of Cer accumulation is still not completely understood. We found slightly decreased CerS6 mRNA levels that, ultimately, did not affect CerS6 protein levels (data not shown). Thus, we cannot rule out an increase in CerS activity due to post-transcriptional modification. Indeed, several publications from recent years have shown that CerS1 can be regulated by several modes, such as translocation from the endoplasmic reticulum to the Golgi apparatus, ubiquitination and proteasomal degradation, and phosphorylation by protein kinase C (PKC) (64, 65). Another possibility is that CerS activity in response to TNFα is regulated by the formation of heterodimers. For instance, CerS2 activity is enhanced by co-expression of CerS2 with another CerS (19).
Another interesting finding was that exogenously added C6-Cer was able to mimic the effects of TNFα and induced accumulation of long-chain Cers and subsequent cell death in a time- and dose-dependent fashion. Many studies have used short-chain Cers (e.g. C6-Cers) in order to identify biological targets of endogenous Cer as well as Cer signaling pathways (13, 66, 67). In the attempt to identify the specific enzymes involved in the generation of endogenous Cer upon C6-Cer treatment, we found that CerS inhibitor FB1 was able to robustly prevent the accumulation of long-chain Cers as well as plasma membrane permeabilization, thus suggesting that synthesis of Cers in response to C6-Cer treatment involves the Cer recycling pathway, in which short-chain C6-Cer would be deacylated, producing Sph, and then reacylated to Cers via CerSs and not due to the elongation of the fatty acid chain.
The salvage or recycling pathway involves a complex multistep and multilocalization mechanism. First, Cer is generated from the hydrolysis of SM by aSMase in the lysosomes (68). Second, also in the lysosomes, Cer is further hydrolyzed by acid ceramidase to produce Sph (69), which is now able to exit the lysosomes and subsequently be N-acylated by the action of CerSs in the endoplasmic reticulum (20, 70). In the process of evaluating the contribution of some other key enzymes involved in the recycling pathway, we knocked down aSMase, and this had no effect on LDH release upon TNFα treatment (data not shown), suggesting that the pool of Cer generated by aSMase does not control plasma membrane permeabilization. Most importantly, our preliminary data showed that TNFα treatment increased the activity of alkaline ceramide 2 (data not shown), which is localized in the Golgi apparatus, and it is known to catalyze the hydrolysis of Cers and therefore to elevate both cellular Sph and dhSph (71). On the other hand, knockdown of acid ceramidase showed no protective effect in TNFα-induced plasma membrane permeabilization, which correlates with the fact that activity of acid ceramidase was not affected by TNFα (data not shown). Thus, a role for alkaline ceramide 2 in the context of cell death will be a subject for future experiments. Furthermore, Cer in the recycling or salvage pathway can be also generated from the breakdown of glucosylceramide by the lysosomal enzyme glucosylceramidase (68, 72). Indeed, our group has previously published the involvement of PKCδ and glucosylceramidase in the formation of salvaged Cer induced by phorbol 12-myristate 13-acetate (73). Thus, a role for glucosylceramidase in Cer generation and cell death induced by TNFα should not be fully excluded, and it is our intention to test this possibility in future experiments.
It is well established that TNFα induces rapid activation of aSMase, as early as 15–30 min after treatment (74, 75), and subsequent decrease in SM levels. However, SM levels (total and/or individual species) were not altered significantly at time points ranging from 15 min to 3 h (data not shown), suggesting that generation of ceramide via SMase activation may not occur at these early time points in our system. Furthermore, we have also observed accumulation of SM at late time points (data not shown), which was specifically FB1-sensitive; thus, a role for SM in cell death cannot be fully ruled out. On the other hand, the generation of Cer due to hydrolysis of SM by action of the enzyme nSMase has been observed upon a variety of cell death inducers, including TNFα (7, 59, 74). Furthermore, previously, our group has shown that TNFα treatment results in the activation of two distinct pathways of Cer generation: one pathway mediated by nSMase activation and inhibitable by glutathione (GSH) and a second pathway mediated by CerS and inhibitable by FB1 (7). Thus, the contribution of the nSMase-dependent pathway was also examined. Interestingly, preincubation with the nSMase inhibitor GW4869 showed protective effects on LDH release in a dose-dependent fashion. Whereas pretreatment with 10 μm GW4869 caused a decrease in LDH release of ∼50%, 20 μm GW4869 completely abrogated the effects of TNFα (data not shown), thus suggesting that a pool of ceramide generated by nSMase activation at the plasma membrane may also contribute to the late phase of apoptosis and subsequently plasma membrane permeabilization, most likely after recycling within the salvage pathway.
Several components of the death machinery have been implicated in the regulation of pro-apoptotic Cer. In our model, we observed that caspase activity is essential for Cer generation because inhibition of caspases with the pan-caspase inhibitor z-VAD-fmk completely abrogated Cer accumulation and cell death, suggesting that CerS activity and Cer generation occurred downstream of caspase-8. Most importantly, the caspase-3/7 inhibitor z-DEVD-fmk did not completely abolish Cer generation upon TNFα, but it completely prevented cell death. Conversely, we found that CerS activity in the salvage pathway regulates caspase-7 activation because FB1-preteated cells showed reduced cleavage of pro-caspase-7 and reduced active caspase-7 upon TNFα treatment (whereas Myr pretreatment showed no effect). Thus, our results suggest that CerS/Cer generated specifically by the salvage pathway controls caspase-7 activity and therefore the execution phase of apoptosis. Indeed, our results are supported by previous publications that suggested that pro-apoptotic Cer generation might occur downstream of caspase-8 but upstream of caspase-7 activity. For instance, Dbaibo et al. (76) demonstrated that caspase-8 was necessary for Cer formation because CrmA (cytokine response modifier A), an inhibitor of caspase-8, prevented Cer generation and subsequent TNFα-induced death. That same publication also showed cleavage of PARP upon the addition of exogenous C6-Cer in MCF-7 cells, which was also observed in our system. Moreover, the regulation of caspase-7 by CerS activity has been described as occurring upon a variety of cell death stimuli. For instance, Lee et al. (25) found that FB1 inhibited caspase-7 activation by controlling Bax insertion into mitochondrial membranes and cytochrome c release. On the other hand, Cer generated by the salvage pathway was shown to regulate caspase-7 activity without affecting Bax translocation or cytochrome c release in a model of UV-C-induced cell death (10). We have found that MOMP contributes to the amplification of TNFα-induced cell death because knockdown of Bax partially protected from caspase-7 activation and plasma membrane permeabilization (data not shown). Indeed, MCF-7 cells have been described as type I (caspase-8 directly activates caspase-3) and type II (caspase-3 activation requires mitochondrial signal amplification initiated by caspase-8 cleavage), most likely due to the lack of caspase-3. Nevertheless, whether CerS activity/Cer elevation is upstream or downstream of MOMP in our system remains elusive. Collectively, our data demonstrated that CerS/Cer generation regulates apoptosis proximal to the executioner caspase-7.
More importantly, we have found that TNFα induced the accumulation of two different pools of Cer: 1) a pool of Cer upstream of caspase-7 that is z-DEVD-fmk-insensitive and 2) a pool of Cer downstream of caspase-7 that is z-DEVD-fmk-sensitive. Our data strongly indicate that CerS might be responsible for the generation of the z-DEVD-fmk-insensitive pool, which controls caspase-7 activation and cell death. However, the source of the second pool described remains unknown. Considering the quantitative difference of Cer changes induced by FB1 versus Myr upon TNFα treatment and the similar quantitative difference between the z-DEVD-fmk-sensitive and -insensitive Cer pools, we propose that this second pool of Cer that is downstream of caspase-7 and unlikely to regulate plasma membrane permeabilization is contributed by the de novo synthesis of Cer. Another possible source of this second pool could be sphingosine kinase 1, which our group reported to be proteolytically degraded via the caspase-8/cathepsin D signaling pathway within the lysosomes (77). However, these hypotheses are merely speculative, and they require further investigation.
In the attempt to better understand the mechanism of action of CerS/Cer in the regulation of programmed cell death, we found that CerS activity is necessary for postmitochondrial events downstream of caspase-7. Thus, we observed that TNFα-induced loss of autophosphorylated and total FAK protein could be partially prevented by FB1, whereas Myr showed no effect. Furthermore, the levels of FAK were significantly down-regulated by exogenous C6-Cer, which was also able to induce caspase-7 activation and plasma membrane permeabilization. Interestingly, a role for the sphingolipid Cer in the regulation of FAK protein has not been studied in detail. In fact, Marushige and Marushige (78) for the first time showed that exogenously Cer-treated trigeminal neurinoma 476-16 cells showed decreased tyrosine phosphorylation of FAK, which preceded cell rounding and subsequent cell death. More recently, Di Bartolomeo and Spinedi (79) reported that Cer treatment induced decreased FAK and cell detachment, which was not inhibited by the pan-caspase inhibitor z-VAD-fmk, suggesting that these events occurred independently of or upstream of caspase activation. On the other hand, our data indicate that the fate of FAK during cell death is regulated, in part, by CerS activity through regulation of caspase-7 activation, because z-DEVD-fmk completely protected from decreased FAK protein in treated cells. Because we observed down-regulated levels of both total and phosphorylated FAK (Tyr(P)-397) in TNFα-treated cells, which was inhibited by z-DEVD-fmk, we believe that the effect of TNFα on phosphorylated FAK is due to caspase-7 activity rather than a tyrosine phosphatase activity per se. However, the action of a tyrosine phosphatase on Tyr(P)-397-FAK in response to Cer cannot be ruled out. In the literature, only a few proteins have been described as being directly activated by Cer. Protein phosphatases 1, 2A (PPP family), and 2C (PPM family) have been found to be activated by Cer in vitro as well as in vivo (also known as Cer-activated protein phosphatases). For instance, PP2A has been established to regulate the cell cycle and apoptosis and to be activated by Cer (80). Nevertheless, PPPs and PP2C are serine-threonine phosphatases, and although they could be involved in upstream signaling, it is very unlikely that they directly dephosphorylate the tyrosine site of FAK in response to TNFα, although an investigation of the role of Cer-activated protein phosphatases in dephosphorylation of FAK could be an interesting future study. Thus, our results implicate, for the first time, the enzyme CerS as a regulator of FAK protein during cell death, and they identify CerS/Cer generation as a point of cross-talk between FAK and the sphingolipid pathway. Furthermore, our data not only uncover FAK as a downstream target for CerS/Cer in the context of tumor cell death, but they also provide the regulation of caspase-7 activity as a likely mechanism of action.
FAK plays an important role in vital cellular processes, such as cell proliferation, cell survival, apoptosis, motility, and invasion. FAK is overexpressed in various tumor cells, including colon, breast, ovarian, and thyroid cancers (81–83), and it is associated with resistance to anoikis (84). The potential role of FAK in tumor growth and cancer progression has led to the development of several therapeutic agents that target FAK (for a review, see Refs. 30 and 85). Recently, a small molecule, 1,2,4,5-benzenetetraamine tetrahydrochloride (focal adhesion kinase inhibitor 14 or Y15) has been shown to specifically bind the Tyr-397 site, inhibiting the autophosphorylation of FAK in response to integrin signal and therefore inhibiting its activation and downstream signaling (30). FAK inhibitor Y15 has been reported to decrease cell survival and cell adhesion and induce apoptosis in pancreatic (86, 87), colon (88), and thyroid cancer (57); human neuroblastoma (89); and human glioblastoma (56) alone or in combination with chemotherapeutics. We observed that FAK inhibitor Y15 effectively decreased cell viability and plasma membrane permeabilization in a caspase-independent manner in MCF-7 human breast adenocarcinoma cells. Thus, our data demonstrate that inhibition of the autophosphorylation site (Tyr-397) of FAK induces cell death in MCF-7 cells, and it is consistent with the involvement of FAK in plasma membrane permeabilization.
CerS6 was identified as the CerS critical for the regulation of the TNFα-induced execution phase of apoptosis. Indeed, down-regulation of CerS6 greatly protected from plasma membrane permeabilization. Furthermore, siCerS6 highly protected from the TNFα-induced loss of FAK and decreased caspase-7 activation. Interestingly, cells lacking CerS6 showed decreased pro-caspase-8 cleavage (data not shown), suggesting that CerS6 effects on caspase-7 might be key for the amplification of the apoptotic signaling cascade. Indeed, caspase-3, another executioner caspase with similar activity to caspase-7 toward natural and synthetic peptides (90, 91) but absent in MCF-7, has been reported to cleave and activate caspase-8 (92). Although knockdown of CerS1 did show a trend toward inhibition of LDH release in treated cells, we ruled out the possibility that this decrease is via FAK or caspase-7, because siCerS1 had no protective effect on any of them. Moreover, knockdown of CerS5, which also generates C16-Cers, had no protective effect on cell death. Importantly, knockdown of CerS6 was accompanied by multiple changes in Cers levels. The most dramatic effect was the complete inhibition of TNFα-induced accumulation of C16-Cer, even if siCerS6 up-regulated CerS5. Indeed, siCerS6 cells showed decreased basal levels of C16-Cer, which were not affected by TNFα treatment.
In summary, our results have demonstrated that CerS6 and its product C16-Cer regulate events that lead to secondary necrosis, such as caspase-7 activation, loss of FAK, and plasma membrane rupture (Fig. 11). The understanding of how secondary necrosis affects tumor cells has increasingly become a target of interest due to its beneficial implications for anticancer treatment. Secondary necrosis induced by chemotherapeutic agents or ionizing irradiation has been associated with the activation of the immune system and subsequently the clearance of tumor cells (4, 93, 94). Consequently, improved understanding of the mechanisms of CerS/Cer-mediated tumor cell apoptosis at the molecular level is likely to reveal new targets for the development of therapeutic intervention strategies.
Author Contributions
M. J. H.-C. and D. C. conceived and designed the experiments. M. J. H.-C., D. C., C. E. S., M. L., M. M. A., and J. K. Y. performed the experiments. M. J. H.-C. and D. C. analyzed the data. C. M. contributed reagents and HPLC technique. M. J. H.-C., C. L., Y. A. H., and L. M. O. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank all members of the Obeid and Hannun laboratories. We thank Dr. Marc Golightly and Corinne Leombruno at the Flow Cytometry Laboratory as well as Dr. Antonius Koller and Izolda Mileva at the Lipidomics Core Facility of Stony Brook University for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants GM097741 and PO1CA097132 (to L. M. O.). This work was also supported by a Veterans Affairs Merit Award. The authors declare that they have no conflicts of interest with the contents of this article.
- MOMP
- mitochondrial outer membrane permeabilization
- CerS
- ceramide synthase
- dhCerS
- dihydro-CerS
- Cer
- ceramide
- dhCer
- dihydro-Cer
- SMase
- sphingomyelinase
- aSMase
- acid SMase
- nSMase
- neutral SMase
- SM
- sphingomyelin
- Sph
- sphingosine
- dhSph
- dihydro-Sph
- FAK
- focal adhesion kinase
- FB1
- fumonisin B1
- Myr
- myriocin
- LDH
- lactate dehydrogenase
- PARP
- poly(ADP-ribose) polymerase
- z
- benzyloxycarbonyl
- fmk
- fluoromethyl ketone
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- ANOVA
- analysis of variance.
References
- 1. Kerr J. F., Wyllie A. H., Currie A. R. (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26, 239–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fulda S. (2009) Tumor resistance to apoptosis. Int. J. Cancer 124, 511–515 [DOI] [PubMed] [Google Scholar]
- 3. Wyllie A. H., Kerr J. F., Currie A. R. (1980) Cell death: the significance of apoptosis. Int. Rev. Cytol. 68, 251–306 [DOI] [PubMed] [Google Scholar]
- 4. Silva M. T. (2010) Secondary necrosis: the natural outcome of the complete apoptotic program. FEBS Lett. 584, 4491–4499 [DOI] [PubMed] [Google Scholar]
- 5. Robertson A. M., Bird C. C., Waddell A. W., Currie A. R. (1978) Morphological aspects of glucocorticoid-induced cell death in human lymphoblastoid cells. J. Pathol. 126, 181–187 [DOI] [PubMed] [Google Scholar]
- 6. Oropesa-Ávila M., Fernández-Vega A., de la Mata M., Maraver J. G., Cordero M. D., Cotán D., de Miguel M., Calero C. P., Paz M. V., Pavón A. D., Sánchez M. A., Zaderenko A. P., Ybot-González P., Sánchez-Alcázar J. A. (2013) Apoptotic microtubules delimit an active caspase free area in the cellular cortex during the execution phase of apoptosis. Cell Death Dis. 4, e527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dbaibo G. S., El-Assaad W., Krikorian A., Liu B., Diab K., Idriss N. Z., El-Sabban M., Driscoll T. A., Perry D. K., Hannun Y. A. (2001) Ceramide generation by two distinct pathways in tumor necrosis factor α-induced cell death. FEBS Lett. 503, 7–12 [DOI] [PubMed] [Google Scholar]
- 8. Taha T. A., Mullen T. D., Obeid L. M. (2006) A house divided: ceramide, sphingosine, and sphingosine-1-phosphate in programmed cell death. Biochim. Biophys. Acta 1758, 2027–2036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pettus B. J., Chalfant C. E., Hannun Y. A. (2002) Ceramide in apoptosis: an overview and current perspectives. Biochim. Biophys. Acta 1585, 114–125 [DOI] [PubMed] [Google Scholar]
- 10. Mullen T. D., Jenkins R. W., Clarke C. J., Bielawski J., Hannun Y. A., Obeid L. M. (2011) Ceramide synthase-dependent ceramide generation and programmed cell death: involvement of salvage pathway in regulating postmitochondrial events. J. Biol. Chem. 286, 15929–15942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mullen T. D., Obeid L. M. (2012) Ceramide and apoptosis: exploring the enigmatic connections between sphingolipid metabolism and programmed cell death. Anti-cancer Agents Med. Chem. 12, 340–363 [DOI] [PubMed] [Google Scholar]
- 12. Novgorodov S. A., Szulc Z. M., Luberto C., Jones J. A., Bielawski J., Bielawska A., Hannun Y. A., Obeid L. M. (2005) Positively charged ceramide is a potent inducer of mitochondrial permeabilization. J. Biol. Chem. 280, 16096–16105 [DOI] [PubMed] [Google Scholar]
- 13. Obeid L. M., Linardic C. M., Karolak L. A., Hannun Y. A. (1993) Programmed cell death induced by ceramide. Science 259, 1769–1771 [DOI] [PubMed] [Google Scholar]
- 14. Kitatani K., Idkowiak-Baldys J., Hannun Y. A. (2008) The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell. Signal. 20, 1010–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hannun Y. A., Obeid L. M. (2011) Many ceramides. J. Biol. Chem. 286, 27855–27862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Radin N. S. (2003) Killing tumours by ceramide-induced apoptosis: a critique of available drugs. Biochem. J. 371, 243–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stiban J., Tidhar R., Futerman A. H. (2010) Ceramide synthases: roles in cell physiology and signaling. Adv. Exp. Med. Biol. 688, 60–71 [DOI] [PubMed] [Google Scholar]
- 18. Venkataraman K., Riebeling C., Bodennec J., Riezman H., Allegood J. C., Sullards M. C., Merrill A. H. Jr., Futerman A. H. (2002) Upstream of growth and differentiation factor 1 (uog1), a mammalian homolog of the yeast longevity assurance gene 1 (LAG1), regulates N-stearoyl-sphinganine (C18-(dihydro)ceramide) synthesis in a fumonisin B1-independent manner in mammalian cells. J. Biol. Chem. 277, 35642–35649 [DOI] [PubMed] [Google Scholar]
- 19. Laviad E. L., Albee L., Pankova-Kholmyansky I., Epstein S., Park H., Merrill A. H. Jr., Futerman A. H. (2008) Characterization of ceramide synthase 2: tissue distribution, substrate specificity, and inhibition by sphingosine 1-phosphate. J. Biol. Chem. 283, 5677–5684 [DOI] [PubMed] [Google Scholar]
- 20. Riebeling C., Allegood J. C., Wang E., Merrill A. H. Jr., Futerman A. H. (2003) Two mammalian longevity assurance gene (LAG1) family members, trh1 and trh4, regulate dihydroceramide synthesis using different fatty acyl-CoA donors. J. Biol. Chem. 278, 43452–43459 [DOI] [PubMed] [Google Scholar]
- 21. Mizutani Y., Kihara A., Igarashi Y. (2005) Mammalian Lass6 and its related family members regulate synthesis of specific ceramides. Biochem. J. 390, 263–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bose R., Verheij M., Haimovitz-Friedman A., Scotto K., Fuks Z., Kolesnick R. (1995) Ceramide synthase mediates daunorubicin-induced apoptosis: an alternative mechanism for generating death signals. Cell 82, 405–414 [DOI] [PubMed] [Google Scholar]
- 23. Rath G., Schneider C., Langlois B., Sartelet H., Morjani H., Btaouri H. E., Dedieu S., Martiny L. (2009) De novo ceramide synthesis is responsible for the anti-tumor properties of camptothecin and doxorubicin in follicular thyroid carcinoma. Int. J. Biochem. Cell Biol. 41, 1165–1172 [DOI] [PubMed] [Google Scholar]
- 24. Laouar A., Glesne D., Huberman E. (1999) Involvement of protein kinase C-β and ceramide in tumor necrosis factor-α-induced but not Fas-induced apoptosis of human myeloid leukemia cells. J. Biol. Chem. 274, 23526–23534 [DOI] [PubMed] [Google Scholar]
- 25. Lee H., Rotolo J. A., Mesicek J., Penate-Medina T., Rimner A., Liao W. C., Yin X., Ragupathi G., Ehleiter D., Gulbins E., Zhai D., Reed J. C., Haimovitz-Friedman A., Fuks Z., Kolesnick R. (2011) Mitochondrial ceramide-rich macrodomains functionalize Bax upon irradiation. PLoS One 6, e19783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. White-Gilbertson S., Mullen T., Senkal C., Lu P., Ogretmen B., Obeid L., Voelkel-Johnson C. (2009) Ceramide synthase 6 modulates TRAIL sensitivity and nuclear translocation of active caspase-3 in colon cancer cells. Oncogene 28, 1132–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zachary I. (1997) Focal adhesion kinase. Int. J. Biochem. Cell Biol. 29, 929–934 [DOI] [PubMed] [Google Scholar]
- 28. Burridge K., Turner C. E., Romer L. H. (1992) Tyrosine phosphorylation of paxillin and pp125FAK accompanies cell adhesion to extracellular matrix: a role in cytoskeletal assembly. J. Cell Biol. 119, 893–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guan J. L. (1997) Focal adhesion kinase in integrin signaling. Matrix Biol. 16, 195–200 [DOI] [PubMed] [Google Scholar]
- 30. Golubovskaya V. M. (2014) Targeting FAK in human cancer: from finding to first clinical trials. Front. Biosci. 19, 687–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Golubovskaya V. M., Zheng M., Zhang L., Li J. L., Cance W. G. (2009) The direct effect of focal adhesion kinase (FAK), dominant-negative FAK, FAK-CD and FAK siRNA on gene expression and human MCF-7 breast cancer cell tumorigenesis. BMC Cancer 9, 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hao H., Naomoto Y., Bao X., Watanabe N., Sakurama K., Noma K., Motoki T., Tomono Y., Fukazawa T., Shirakawa Y., Yamatsuji T., Matsuoka J., Wang Z. G., Takaoka M. (2009) Focal adhesion kinase as potential target for cancer therapy (Review). Oncol. Rep. 22, 973–979 [DOI] [PubMed] [Google Scholar]
- 33. Mehlen P., Puisieux A. (2006) Metastasis: a question of life or death. Nat. Rev. Cancer 6, 449–458 [DOI] [PubMed] [Google Scholar]
- 34. Levkau B., Herren B., Koyama H., Ross R., Raines E. W. (1998) Caspase-mediated cleavage of focal adhesion kinase pp125FAK and disassembly of focal adhesions in human endothelial cell apoptosis. J. Exp. Med. 187, 579–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang J., Zu J., Xu G., Zhao W., Jinglong Y. (2014) Inhibition of focal adhesion kinase induces apoptosis in human osteosarcoma SAOS-2 cells. Tumour Biol. 35, 1551–1556 [DOI] [PubMed] [Google Scholar]
- 36. Beviglia L., Golubovskaya V., Xu L., Yang X., Craven R. J., Cance W. G. (2003) Focal adhesion kinase N-terminus in breast carcinoma cells induces rounding, detachment and apoptosis. Biochem. J. 373, 201–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wen L. P., Fahrni J. A., Troie S., Guan J. L., Orth K., Rosen G. D. (1997) Cleavage of focal adhesion kinase by caspases during apoptosis. J. Biol. Chem. 272, 26056–26061 [DOI] [PubMed] [Google Scholar]
- 38. Schlaepfer D. D., Hauck C. R., Sieg D. J. (1999) Signaling through focal adhesion kinase. Prog. Biophys. Mol. Biol. 71, 435–478 [DOI] [PubMed] [Google Scholar]
- 39. Gervais F. G., Thornberry N. A., Ruffolo S. C., Nicholson D. W., Roy S. (1998) Caspases cleave focal adhesion kinase during apoptosis to generate a FRNK-like polypeptide. J. Biol. Chem. 273, 17102–17108 [DOI] [PubMed] [Google Scholar]
- 40. Bielawski J., Szulc Z. M., Hannun Y. A., Bielawska A. (2006) Simultaneous quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods 39, 82–91 [DOI] [PubMed] [Google Scholar]
- 41. Panjarian S., Kozhaya L., Arayssi S., Yehia M., Bielawski J., Bielawska A., Usta J., Hannun Y. A., Obeid L. M., Dbaibo G. S. (2008) De novo N-palmitoylsphingosine synthesis is the major biochemical mechanism of ceramide accumulation following p53 up-regulation. Prostaglandins Other Lipid Mediat. 86, 41–48 [DOI] [PubMed] [Google Scholar]
- 42. Senkal C. E., Ponnusamy S., Rossi M. J., Bialewski J., Sinha D., Jiang J. C., Jazwinski S. M., Hannun Y. A., Ogretmen B. (2007) Role of human longevity assurance gene 1 and C18-ceramide in chemotherapy-induced cell death in human head and neck squamous cell carcinomas. Mol. Cancer Ther. 6, 712–722 [DOI] [PubMed] [Google Scholar]
- 43. Hoeferlin L. A., Fekry B., Ogretmen B., Krupenko S. A., Krupenko N. I. (2013) Folate stress induces apoptosis via p53-dependent de novo ceramide synthesis and up-regulation of ceramide synthase 6. J. Biol. Chem. 288, 12880–12890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhao Y., Li S., Childs E. E., Kuharsky D. K., Yin X. M. (2001) Activation of pro-death Bcl-2 family proteins and mitochondria apoptosis pathway in tumor necrosis factor-α-induced liver injury. J. Biol. Chem. 276, 27432–27440 [DOI] [PubMed] [Google Scholar]
- 45. Gross A., Yin X. M., Wang K., Wei M. C., Jockel J., Milliman C., Erdjument-Bromage H., Tempst P., Korsmeyer S. J. (1999) Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death. J. Biol. Chem. 274, 1156–1163 [DOI] [PubMed] [Google Scholar]
- 46. Elmore S. (2007) Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35, 495–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chang H. Y., Yang X. (2000) Proteases for cell suicide: functions and regulation of caspases. Microbiol. Mol. Biol. Rev. 64, 821–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xu L. H., Yang X., Bradham C. A., Brenner D. A., Baldwin A. S. Jr., Craven R. J., Cance W. G. (2000) The focal adhesion kinase suppresses transformation-associated, anchorage-independent apoptosis in human breast cancer cells: involvement of death receptor-related signaling pathways. J. Biol. Chem. 275, 30597–30604 [DOI] [PubMed] [Google Scholar]
- 49. Xu L. H., Yang X., Craven R. J., Cance W. G. (1998) The COOH-terminal domain of the focal adhesion kinase induces loss of adhesion and cell death in human tumor cells. Cell Growth Differ. 9, 999–1005 [PubMed] [Google Scholar]
- 50. Golubovskaya V. M., Gross S., Kaur A. S., Wilson R. I., Xu L. H., Yang X. H., Cance W. G. (2003) Simultaneous inhibition of focal adhesion kinase and SRC enhances detachment and apoptosis in colon cancer cell lines. Mol. Cancer Res. 1, 755–764 [PubMed] [Google Scholar]
- 51. Ciccimaro E., Hevko J., Blair I. A. (2006) Analysis of phosphorylation sites on focal adhesion kinase using nanospray liquid chromatography/multiple reaction monitoring mass spectrometry. Rapid Commun. Mass Spectrom. 20, 3681–3692 [DOI] [PubMed] [Google Scholar]
- 52. Vanden Berghe T., Vanlangenakker N., Parthoens E., Deckers W., Devos M., Festjens N., Guerin C. J., Brunk U. T., Declercq W., Vandenabeele P. (2010) Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 17, 922–930 [DOI] [PubMed] [Google Scholar]
- 53. Ricci J. E., Muñoz-Pinedo C., Fitzgerald P., Bailly-Maitre B., Perkins G. A., Yadava N., Scheffler I. E., Ellisman M. H., Green D. R. (2004) Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell 117, 773–786 [DOI] [PubMed] [Google Scholar]
- 54. Oropesa-Ávila M., Fernández-Vega A., de la Mata M., Garrido-Maraver J., Cotán D., Paz M. V., Pavón A. D., Cordero M. D., Alcocer-Gómez E., de Lavera I., Lema R., Zaderenko A. P., Sánchez-Alcázar J. A. (2014) Apoptotic cells subjected to cold/warming exposure disorganize apoptotic microtubule network and undergo secondary necrosis. Apoptosis 19, 1364–1377 [DOI] [PubMed] [Google Scholar]
- 55. Golubovskaya V., Curtin L., Groman A., Sexton S., Cance W. G. (2015) In vivo toxicity, metabolism and pharmacokinetic properties of FAK inhibitor 14 or Y15 (1,2,4,5-benzenetetramine tetrahydrochloride). Arch. Toxicol. 89, 1095–1101 [DOI] [PubMed] [Google Scholar]
- 56. Golubovskaya V. M., Huang G., Ho B., Yemma M., Morrison C. D., Lee J., Eliceiri B. P., Cance W. G. (2013) Pharmacologic blockade of FAK autophosphorylation decreases human glioblastoma tumor growth and synergizes with temozolomide. Mol. Cancer Ther. 12, 162–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. O'Brien S., Golubovskaya V. M., Conroy J., Liu S., Wang D., Liu B., Cance W. G. (2014) FAK inhibition with small molecule inhibitor Y15 decreases viability, clonogenicity, and cell attachment in thyroid cancer cell lines and synergizes with targeted therapeutics. Oncotarget 5, 7945–7959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mullen T. D., Spassieva S., Jenkins R. W., Kitatani K., Bielawski J., Hannun Y. A., Obeid L. M. (2011) Selective knockdown of ceramide synthases reveals complex interregulation of sphingolipid metabolism. J. Lipid Res. 52, 68–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Luberto C., Hassler D. F., Signorelli P., Okamoto Y., Sawai H., Boros E., Hazen-Martin D. J., Obeid L. M., Hannun Y. A., Smith G. K. (2002) Inhibition of tumor necrosis factor-induced cell death in MCF7 by a novel inhibitor of neutral sphingomyelinase. J. Biol. Chem. 277, 41128–41139 [DOI] [PubMed] [Google Scholar]
- 60. Neumeyer J., Hallas C., Merkel O., Winoto-Morbach S., Jakob M., Thon L., Adam D., Schneider-Brachert W., Schütze S. (2006) TNF-receptor I defective in internalization allows for cell death through activation of neutral sphingomyelinase. Exp. Cell Res. 312, 2142–2153 [DOI] [PubMed] [Google Scholar]
- 61. Kim W. J., Okimoto R. A., Purton L. E., Goodwin M., Haserlat S. M., Dayyani F., Sweetser D. A., McClatchey A. I., Bernard O. A., Look A. T., Bell D. W., Scadden D. T., Haber D. A. (2008) Mutations in the neutral sphingomyelinase gene SMPD3 implicate the ceramide pathway in human leukemias. Blood 111, 4716–4722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jin J., Mullen T. D., Hou Q., Bielawski J., Bielawska A., Zhang X., Obeid L. M., Hannun Y. A., Hsu Y. T. (2009) AMPK inhibitor Compound C stimulates ceramide production and promotes Bax redistribution and apoptosis in MCF7 breast carcinoma cells. J. Lipid Res. 50, 2389–2397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Enongene E. N., Sharma R. P., Bhandari N., Miller J. D., Meredith F. I., Voss K. A., Riley R. T. (2002) Persistence and reversibility of the elevation in free sphingoid bases induced by fumonisin inhibition of ceramide synthase. Toxicol. Sci. 67, 173–181 [DOI] [PubMed] [Google Scholar]
- 64. Sridevi P., Alexander H., Laviad E. L., Pewzner-Jung Y., Hannink M., Futerman A. H., Alexander S. (2009) Ceramide synthase 1 is regulated by proteasomal mediated turnover. Biochim. Biophys. Acta 1793, 1218–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sridevi P., Alexander H., Laviad E. L., Min J., Mesika A., Hannink M., Futerman A. H., Alexander S. (2010) Stress-induced ER to Golgi translocation of ceramide synthase 1 is dependent on proteasomal processing. Exp. Cell Res. 316, 78–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ogretmen B., Pettus B. J., Rossi M. J., Wood R., Usta J., Szulc Z., Bielawska A., Obeid L. M., Hannun Y. A. (2002) Biochemical mechanisms of the generation of endogenous long chain ceramide in response to exogenous short chain ceramide in the A549 human lung adenocarcinoma cell line: role for endogenous ceramide in mediating the action of exogenous ceramide. J. Biol. Chem. 277, 12960–12969 [DOI] [PubMed] [Google Scholar]
- 67. Takeda S., Mitsutake S., Tsuji K., Igarashi Y. (2006) Apoptosis occurs via the ceramide recycling pathway in human HaCaT keratinocytes. J. Biochem. 139, 255–262 [DOI] [PubMed] [Google Scholar]
- 68. Kolter T., Sandhoff K. (2005) Principles of lysosomal membrane digestion: stimulation of sphingolipid degradation by sphingolipid activator proteins and anionic lysosomal lipids. Annu. Rev. Cell Dev. Biol. 21, 81–103 [DOI] [PubMed] [Google Scholar]
- 69. Riboni L., Bassi R., Caminiti A., Prinetti A., Viani P., Tettamanti G. (1998) Metabolic fate of exogenous sphingosine in neuroblastoma neuro2A cells: dose-dependence and biological effects. Ann. N.Y. Acad. Sci. 845, 46–56 [DOI] [PubMed] [Google Scholar]
- 70. Tidhar R., Futerman A. H. (2013) The complexity of sphingolipid biosynthesis in the endoplasmic reticulum. Biochim. Biophys. Acta 1833, 2511–2518 [DOI] [PubMed] [Google Scholar]
- 71. Xu R., Jin J., Hu W., Sun W., Bielawski J., Szulc Z., Taha T., Obeid L. M., Mao C. (2006) Golgi alkaline ceramidase regulates cell proliferation and survival by controlling levels of sphingosine and S1P. FASEB J. 20, 1813–1825 [DOI] [PubMed] [Google Scholar]
- 72. Dinur T., Osiecki K. M., Legler G., Gatt S., Desnick R. J., Grabowski G. A. (1986) Human acid β-glucosidase: isolation and amino acid sequence of a peptide containing the catalytic site. Proc. Natl. Acad. Sci. U.S.A. 83, 1660–1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kitatani K., Sheldon K., Anelli V., Jenkins R. W., Sun Y., Grabowski G. A., Obeid L. M., Hannun Y. A. (2009) Acid β-glucosidase 1 counteracts p38delta-dependent induction of interleukin-6: possible role for ceramide as an anti-inflammatory lipid. J. Biol. Chem. 284, 12979–12988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ségui B., Cuvillier O., Adam-Klages S., Garcia V., Malagarie-Cazenave S., Lévêque S., Caspar-Bauguil S., Coudert J., Salvayre R., Krönke M., Levade T. (2001) Involvement of FAN in TNF-induced apoptosis. J. Clin. Invest. 108, 143–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Jayadev S., Linardic C. M., Hannun Y. A. (1994) Identification of arachidonic acid as a mediator of sphingomyelin hydrolysis in response to tumor necrosis factor α. J. Biol. Chem. 269, 5757–5763 [PubMed] [Google Scholar]
- 76. Dbaibo G. S., Perry D. K., Gamard C. J., Platt R., Poirier G. G., Obeid L. M., Hannun Y. A. (1997) Cytokine response modifier A (CrmA) inhibits ceramide formation in response to tumor necrosis factor (TNF)-α: CrmA and Bcl-2 target distinct components in the apoptotic pathway. J. Exp. Med. 185, 481–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Taha T. A., Kitatani K., Bielawski J., Cho W., Hannun Y. A., Obeid L. M. (2005) Tumor necrosis factor induces the loss of sphingosine kinase-1 by a cathepsin B-dependent mechanism. J. Biol. Chem. 280, 17196–17202 [DOI] [PubMed] [Google Scholar]
- 78. Marushige Y., Marushige K. (1998) Alterations in focal adhesion and cytoskeletal proteins during apoptosis. Anticancer Res. 18, 301–307 [PubMed] [Google Scholar]
- 79. Di Bartolomeo S., Spinedi A. (2002) Ordering ceramide-induced cell detachment and apoptosis in human neuroepithelioma. Neurosci. Lett. 334, 149–152 [DOI] [PubMed] [Google Scholar]
- 80. Perrotti D., Neviani P. (2013) Protein phosphatase 2A: a target for anticancer therapy. Lancet Oncol. 14, e229–e238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Cance W. G., Harris J. E., Iacocca M. V., Roche E., Yang X., Chang J., Simkins S., Xu L. (2000) Immunohistochemical analyses of focal adhesion kinase expression in benign and malignant human breast and colon tissues: correlation with preinvasive and invasive phenotypes. Clin. Cancer Res. 6, 2417–2423 [PubMed] [Google Scholar]
- 82. Golubovskaya V. M., Conway-Dorsey K., Edmiston S. N., Tse C. K., Lark A. A., Livasy C. A., Moore D., Millikan R. C., Cance W. G. (2009) FAK overexpression and p53 mutations are highly correlated in human breast cancer. Int. J. Cancer 125, 1735–1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Oktay M. H., Oktay K., Hamele-Bena D., Buyuk A., Koss L. G. (2003) Focal adhesion kinase as a marker of malignant phenotype in breast and cervical carcinomas. Hum. Pathol. 34, 240–245 [DOI] [PubMed] [Google Scholar]
- 84. Frisch S. M., Vuori K., Ruoslahti E., Chan-Hui P. Y. (1996) Control of adhesion-dependent cell survival by focal adhesion kinase. J. Cell Biol. 134, 793–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Dunn K. B., Heffler M., Golubovskaya V. M. (2010) Evolving therapies and FAK inhibitors for the treatment of cancer. Anticancer Agents Med. Chem. 10, 722–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hochwald S. N., Nyberg C., Zheng M., Zheng D., Wood C., Massoll N. A., Magis A., Ostrov D., Cance W. G., Golubovskaya V. M. (2009) A novel small molecule inhibitor of FAK decreases growth of human pancreatic cancer. Cell Cycle 8, 2435–2443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Zheng D., Golubovskaya V., Kurenova E., Wood C., Massoll N. A., Ostrov D., Cance W. G., Hochwald S. N. (2010) A novel strategy to inhibit FAK and IGF-1R decreases growth of pancreatic cancer xenografts. Mol. Carcinog. 49, 200–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Heffler M., Golubovskaya V. M., Conroy J., Liu S., Wang D., Cance W. G., Dunn K. B. (2013) FAK and HAS inhibition synergistically decrease colon cancer cell viability and affect expression of critical genes. Anticancer Agents Med. Chem. 13, 584–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Beierle E. A., Ma X., Stewart J., Nyberg C., Trujillo A., Cance W. G., Golubovskaya V. M. (2010) Inhibition of focal adhesion kinase decreases tumor growth in human neuroblastoma. Cell Cycle 9, 1005–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Thornberry N. A., Rano T. A., Peterson E. P., Rasper D. M., Timkey T., Garcia-Calvo M., Houtzager V. M., Nordstrom P. A., Roy S., Vaillancourt J. P., Chapman K. T., Nicholson D. W. (1997) A combinatorial approach defines specificities of members of the caspase family and granzyme B: functional relationships established for key mediators of apoptosis. J. Biol. Chem. 272, 17907–17911 [DOI] [PubMed] [Google Scholar]
- 91. Stennicke H. R., Renatus M., Meldal M., Salvesen G. S. (2000) Internally quenched fluorescent peptide substrates disclose the subsite preferences of human caspases 1, 3, 6, 7 and 8. Biochem. J. 350, 563–568 [PMC free article] [PubMed] [Google Scholar]
- 92. Ozören N., El-Deiry W. S. (2003) Cell surface death receptor signaling in normal and cancer cells. Semin. Cancer Biol. 13, 135–147 [DOI] [PubMed] [Google Scholar]
- 93. Herr I., Debatin K. M. (2001) Cellular stress response and apoptosis in cancer therapy. Blood 98, 2603–2614 [DOI] [PubMed] [Google Scholar]
- 94. Jonathan E. C., Bernhard E. J., McKenna W. G. (1999) How does radiation kill cells? Curr. Opin. Chem. Biol. 3, 77–83 [DOI] [PubMed] [Google Scholar]