

Background: Androgen receptor (AR) inactivation causes telomere dysfunction.
Results: AR-inactivation-induced telomere dysfunction led to the activation of ATM at telomeres, and ATM inhibition blocked repair of damaged telomeric DNA and augmented cell death.
Conclusion: ATM promotes survival of AR-inactivated prostate cancer cells with telomere dysfunction.
Significance: ATM inhibitors may potentiate the efficacy of AR-targeted therapies for the treatment of prostate cancer.
Keywords: androgen receptor, ataxia telangiectasia, DNA damage response, DNA repair, prostate cancer, telomere
Abstract
Androgen receptor (AR) plays a role in maintaining telomere stability in prostate cancer cells, as AR inactivation induces telomere dysfunction within 3 h. Since telomere dysfunction in other systems is known to activate ATM (ataxia telangiectasia mutated)-mediated DNA damage response (DDR) signaling pathways, we investigated the role of ATM-mediated DDR signaling in AR-inactivated prostate cancer cells. Indeed, the induction of telomere dysfunction in cells treated with AR-antagonists (Casodex or MDV3100) or AR-siRNA was associated with a dramatic increase in phosphorylation (activation) of ATM and its downstream effector Chk2 and the presenceof phosphorylated ATM at telomeres, indicating activation of DDR signaling at telomeres. Moreover, Casodex washout led to the reversal of telomere dysfunction, indicating repair of damaged telomeres. ATM inhibitor blocked ATM phosphorylation, induced PARP cleavage, abrogated cell cycle checkpoint activation and attenuated the formation of γH2AX foci at telomeres in AR-inactivated cells, suggesting that ATM inhibitor induces apoptosis in AR-inactivated cells by blocking the repair of damaged DNA at telomeres. Finally, colony formation assay revealed a dramatic decrease in the survival of cells co-treated with Casodex and ATM inhibitor as compared with those treated with either Casodex or ATM inhibitor alone. These observations indicate that inhibitors of DDR signaling pathways may offer a unique opportunity to enhance the potency of AR-targeted therapies for the treatment of androgen-sensitive as well as castration-resistant prostate cancer.
Introduction
Androgen and its derivatives, which act via androgen receptor (AR),2 play an essential role in prostate cancer (1). Hence, androgen-deprivation therapy (ADT) has been the mainstay for the treatment of advanced prostate cancer for over seven decades. However, ADT is not curative; although the disease regresses initially in response to ADT, it eventually progresses to castration-resistant prostate cancer (CRPC) in nearly all patients (2). Therefore, there is an urgent need for novel therapeutic strategies for the treatment of prostate cancer. Our studies point to a novel role of AR in telomere stability (3, 4) that may be harnessed to potentiate the efficacy of currently available AR-targeted therapies for a more effective treatment of prostate cancer.
Telomeres are ribonucleoprotein structures that prevent chromosome ends from being detected as lesions, masking them from constitutive exposure to the DNA damage response (DDR) machinery. Telomeres contain six core proteins, viz., TRF1, TRF2, Rap1, TIN2, POT1, and TPP1, in a complex termed shelterin, whose structural integrity is critical for telomere stability (5). In addition to shelterin, an array of accessory proteins that play a role in DDR signaling (e.g. Mre11 complex, 9-1-1 complex, RAD51, BRCA2), DNA repair (e.g. Ku70/80, XPF/ERCC1, Apollo) or DNA replication (e.g. CTC1-STN1-TEN1 complex, Origin Recognition Complex, RecQ helicase, POLA1/p180, POLA2/p68) are associated with telomeres to ensure timely repair and replication of telomere DNA during the cell cycle (5). In these roles, accessory proteins are only transiently associated with telomeres, whereas shelterin proteins are present at telomeres throughout the cell cycle (5). We recently reported that AR itself is an accessory protein associated with telomeres in prostate cancer cells; AR chromatin immunoprecipitate prepared using AR antibodies (AR-ChIP) contains telomeric DNA, telomeric chromatin isolated using a protocol called “proteomics of isolated chromatin” (PICh) (6) contains AR, and AR immunoprecipitates and colocalizes with telomeric proteins in LNCaP cells (3, 4). A functional role of AR in telomere stability is indicated from the observations that (a) AR inactivation by androgen-depletion, treatment with anti-androgens such as bicalutamide (Casodex) or MDV3100 (Enzalutamide), or treatment with AR-siRNA results in telomere dysfunction, and (b) the synthetic androgen R1881 blocks androgen depletion-mediated telomere dysfunction (3, 4).
Telomere dysfunction represents telomeric DNA damage that triggers DNA damage response (DDR) signaling to activate ATM (ataxia telangiectasia mutated) and ATR (ataxia telangiectasia and Rad3 related) kinases, which in turn activate cell cycle checkpoints that lead to (a) inhibition of cell cycle progression, (b) repair of damaged telomeric DNA, and (c) resumption of cell cycle progression and cell survival (7, 8). In this process, cells are prone to the formation of aberrant telomeres, such as telomere breakages, telomere deletions and sister chromatid telomere fusions, that push cells into breakage-fusion-bridge cycles, resulting in unequal distribution of genetic material to daughter cells and, thereby, genome instability (9). Genomic instability is a salient feature of cancers (10, 11) and it underlies the biological differences between indolent and aggressive prostate cancers (12). In patients with prostate cancer, genomic instability due to telomere shortening is reported to be associated with worse prognosis (13). Interestingly, telomere aberrations of the kind seen in TRF1- or TRF2-deficient cells with telomere dysfunction (14, 15) are also seen in AR-inactivated prostate cancer cells (4). However, it is not known whether AR inactivation-induced telomere dysfunction triggers activation of DDR signaling pathways that promote survival of AR inactivated prostate cancer cells.
ATM is one of the major DDR signaling pathways activated in cells with dysfunctional telomeres (5, 16). ATM is principally activated following DNA double-strand breaks (DSBs) through autophosphorylation of its serine 1981 (17); this leads to the phosphorylation of multiple downstream proteins such as H2AX, p53, Chk2, BRCA1, NBS1, and SMC1 involved in DNA damage recognition, cell cycle checkpoint activation, DNA repair and, under some circumstances, apoptosis (18). ATM stimulated cell cycle checkpoint activation causes G2 arrest, which is believed to provide time needed to repair damaged DNA before mitotic cell division. Thus, ATM activation can provide a survival advantage to cells with DSBs, such as those caused by ionizing radiation and genotoxic agents. Interestingly, the ATM protein level is reported to be higher in prostate cancer cells than in normal tissues (19) and ATM is highly activated in prostatatic intraepithelial neoplasia (PIN), which is regarded as a precursor of prostate cancer (20). These reports raise an intriguing possibility that the presence of high ATM protein levels may have an important role in the maintenance of the shortened telomeres commonly found in prostate cancer cells (21). However, whether ATM plays a role in survival of AR-inactivated prostate cancer cells that have damaged telomeric DNA is not known.
We investigated the status of ATM and its downstream targets in androgen-sensitive LNCaP and castration-resistant 22Rv1 cells subjected to AR-inactivation-induced telomere dysfunction. Our studies show that telomere dysfunction in AR-inactivated or AR-knockdown cells is associated with activation (phosphorylation) of ATM and its downstream target Chk2, and that the inhibition of ATM abrogates G2 arrest, suppresses repair of damaged telomeric DNA, and sensitizes both LNCaP and 22Rv1 cells to AR-inactivation-induced cell death. Thus these studies demonstrate for the first time that inhibitors of DDR signaling pathways potentiate the efficacy AR-targeted therapies for an effective treatment of prostate cancer.
Experimental Procedures
Cell Culture
LNCaP and 22Rv1 cells purchased from ATCC were grown in RPMI (Gibco BRL) containing 10% fetal bovine serum (FBS), 2.5 mm glutamine, 100 μg/ml streptomycin and 100 U/ml penicillin (complete medium). Exponentially growing cells, in complete medium, were treated with or without 10–50 μm bicalutamide (Casodex from LKT Laboratories, MN) or 3–10 μm MDV3100 (Selleck Chemicals, TX) to inhibit AR activity. For AR knockdown, exponentially growing LNCaP cells (1.0–2.0 × 105 cells/well of a 6-well plate) were transfected with 200 pmol of AR-siRNA (Sc-29204, Santa Cruz Biotechnology) or scrambled-siRNA (Santa Cruz Biotechnology) using Lipofectamine 2000 (Invitrogen) following the manufacturer's instructions. Cells were processed 36 h later for immunofluorescence staining or Western blotting.
Indirect Immunofluorescence
The immunofluorescent staining of cells grown on glass slides was performed as described (22). Cells were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100, and incubated at 4 °C overnight with antibodies against TIN2 (22), 53BP1 (Abcam), γH2AX (i.e. phosphorylated-H2AX) (Upstate), p-ATM (Mouse, Cell Signaling), RPA 32 (Ab-1, Calbiochem), or RPA 70 (C21, Santa Cruz Biotechnology). RPA and 53BP1 or RPA and γH2AX co-staining was performed as described by Mirzoeva and Petrini (23). Cells were then washed and stained with goat-anti-rabbit-FITC and/or goat-anti-mouse-Texas Red (Molecular Probes) secondary antibodies for double-immunostaining. For triple-immunostaining, cells were stained with donkey-anti-rabbit Alexa fluor 488, donkey-anti-mouse Alexa fluor 594, and donkey-anti-goat Alexa fluor 350 (Molecular Probes) secondary antibodies. Images of cells were acquired on an LSM-410 confocal microscope (Zeiss).
Telomere dysfunction-induced activation of DDR signaling pathways leads to the recruitment of 53BP1 to, and phosphorylation of H2AX at, telomeres. Therefore, cells containing immunofluorescent foci of 53BP1 and phosphorylated H2AX (γH2AX) colocalized with telomeric protein TIN2, which are referred to as telomere-dysfunction induced foci (TIF), were scored as a measure of DNA damage response as described previously (3, 4). Individual cells with >5 53BP1 or γH2AX foci were considered as being TIF response-positive.
Cell Extracts and Western Blotting
Cells were digested with trypsin, washed with PBS and suspended in Buffer A (50 mm Tris-HCl, pH 7.4, 250 mm NaCl, 0.1% Triton X-100, 5 mm EDTA, 50 mm NaF, and 0.1 mm Na3VO4) supplemented with protease inhibitor mixture (P-8340, Sigma) as described (24). Cells were then subjected twice to 30 pulses of sonication with a Branson Sonifier 250 set at output control 2 and duty cycle 20, with intermittent cooling on ice. The sonicated cell extract was cleared by centrifugation in an Eppendorf centrifuge at 12,500 rpm for 10 min. For Western blotting, membranes were probed with antibodies against AR (AR-N20, Santa Cruz Biotechnology), p-ATM (Cell Signaling Biotechnology), ATM (2C1, Santa Cruz Biotechnology), p-Chk2 (Cell Signaling), Chk2 (Cell Signaling), p-ATR (Cell Signaling), ATR (N19, Santa Cruz), p-Chk1 (Cell Signaling), Chk1(G4, Santa Cruz), β-actin (I-19, Santa Cruz Biotechnology), or GAPDH (AB2302, Millipore). Immunoreactive bands were developed using horseradish peroxidase-conjugated secondary antibodies and SuperSignal WestPico chemiluminescent substrate (Pierce), and visualized using x-ray film.
Real-time qPCR
Total RNA was prepared as described (24). RNA was reverse transcribed using random hexamers and oligo (dT) primer and Transcriptor Reverse Transcriptase (Roche Applied Science) according to the manufacturer's instructions. Real-time qPCR was performed on the Applied Biosystems 7500 Fast Real Time PCR system using the TaqMan Universal Master Mix II with UNG (Applied Biosystems). Cycling was performed using default conditions of the 7900HT Software (Applied Biosystems); 2 min at 50 °C and 10 min at 95 °C, followed by 40 cycles of 15 s at 95 °C and 1 min at 60 °C. The relative quantitative expression of hTERT was calculated using 18S rRNA as the internal control. All real-time qPCR assays were carried out using four technical replicates and two independent cDNA preparations. Primers for hTERT (Assay ID: Hs00972656_m1) and 18S RNA (Assay ID: Hs03928990_g1) were from Applied Biosystems.
Fluorescence in Situ Hybridization (FISH) of Telomeres (Tel-FISH)
Telomeres were visualized by fluorescence in situ hybridization of metaphase chromosome spreads using a telomeric protein nucleic acid (PNA) probe, as described (25). LNCaP cells were treated with 50 μm Casodex for 24 h, then washed in medium without Casodex, and incubated overnight at 37 °C in medium containing colcemid (0.1 μg/ml, Sigma). Cells were trypsinized and collected by centrifugation at 1000 × g (8 min). After hypotonic swelling in 0.075 mm KCl for 25 min at 37 °C, cells were fixed in methanol:acetic acid (3:1) and dropped onto glass slides held at an angle to prepare metaphase spreads. Tel-FISH of metaphase chromosomes was performed as described (25), using a Cy3-OO-(CCCTAA)3-peptide nucleic acid (PNA) probe (Panagene).
Colony Formation Assay
This procedure is essentially as described by Guzman et al. (26). Exponentially growing cells (0.5–1.0 × 104 cells/well of a 6-well plate) were treated with 0, 10, 25, or 50 μm Casodex in the presence or absence of 2 μm KU-60019 for 24 h, then washed to remove drugs and allowed to grow for 10–14 days, then fixed and stained with 0.01% crystal violet (27). The areas of stained surviving cells in each plate were photographed and measured using the ImageJ program (26). The survival fraction was plotted relative to control (vehicle).
FACS Analysis
LNCaP cells (1 × 106) treated with Casodex (50 μm) and/or KU-60019 (2 μm) for 24 h were collected, washed in PBS, and fixed in ice-cold 70% ethanol for at least 24 h. Cells recovered by centrifugation were resuspended in PBS containing 0.1% (v/v) Triton X-100 and 1 μg/ml DAPI (Molecular Probes, Inc.) for 10 min. The samples were then subjected to flow cytometry using BD LSRII (BD Biosciences) at the Cytometry Resources Core, Wayne State University School of Medicine. Flow cytometry data were analyzed with ModFit LT v4.0.5 (Verity, Topsham, ME) software.
Statistics
Data are presented as mean ± S.D. of three or more independent experiments. Statistical significance was calculated using the two-tailed Student's t test, using GraphPad Prism Software. A p value of <0.05 was considered significant.
Results
AR Inactivation-induced Telomere Dysfunction Triggers DDR at Telomeres
DNA damage is marked by the phosphorylation of histone H2AX (γ-H2AX) at the sites of damaged DNA (28), and colocalization of γ-H2AX and TIN2 (a shelterin protein unique to telomeres (22)) indicates telomere DNA damage (4, 29). Fig. 1A shows representative micrographs of cells with TIN2 and γ-H2AX colocalization in control and Casodex treated cultures. As shown in Fig. 1B, there was a 2–3-fold increase in the percentage of cells with TIN2 and γ-H2AX colocalization foci, which is referred to as telomere-dysfunction-induced foci (TIF) response (28), within 10 h of treatment with AR-antagonist Casodex, at a pharmacologically relevant concentration (30), compared with controls (Fig. 1B, top panel). Notably, although AR has been reported to regulate the expression of telomerase (31, 32), under conditions in which Casodex induced telomere dysfunction (TIF response), we found no significant decrease in expression of hTERT, which encodes the catalytic subunit of telomerase as determined by real time qPCR (Fig. 1B, bottom panel). This observation rules out the possibility that telomere dysfunction in AR-inactivated cells is due to down-regulation of telomerase in AR-inactivated LNCaP cells.
FIGURE 1.

AR-inactivation-induced telomere dysfunction triggers the activation of DNA damage response at telomeres. A, AR-inactivation causes DNA damage at telomeres: Exponentially growing LNCaP cells were treated with 50 μm Casodex for 6 h, fixed, and subjected to immunostaining using antibodies against γ-H2AX (green), a marker of DNA double strand breaks, and TIN2 (red), a telomeric protein. Foci with γ-H2AX and TIN2 colocalization (indicated by yellow arrows) are shown in the merge panel. Insets show representative cells in which TIN2 is colocalized with γ-H2AX (Casodex treated) or not (control) at a higher magnification (×1000). B, Casodex induces TIF response, a measure of telomere dysfunction, within 10 h of treatment (top panel) and induction of telomere dysfunction is not due to a decrease in telomerase (bottom panel). Top: LNCaP cells treated with 50 μm Casodex for 0, 3, and 10 h were immunostained using antibodies to γ-H2AX and TIN2 and assessed for TIF response by scoring the percentage of cells with >5 foci of colocalized γ-H2AX and TIN2. ∼80 cells in each treatment group were scored in three separate experiments as described (4). Error bars represent mean ± S.D., n = 3. Bottom: total RNA was isolated from similarly treated parallel cultures described above and relative hTERT mRNA level was measured by real-time qPCR as described under “Experimental Procedures.” Error bars represent mean ± S.D., n = 4. *, p < 0.05; and **, p < 0.01. C, AR-antagonist Casodex induces phosphorylation of ATM and Chk2 in LNCaP cells. LNCaP cells were treated for the indicated period with 50 μm Casodex, cell extracts prepared, and Western blotting performed using p-ATM (S1981), ATM, p-Chk2 (T68), Chk2, and β-actin antibodies. β-Actin was used as a loading control. The data are representative of two independent experiments. D, AR-antagonist MDV3100 induces ATM activation in LNCaP cells. LNCaP cells were treated with 10 μm MDV3100 in the absence or presence of 20 nm R1881, for 24 h. Western blotting of the proteins from 3 independent experiments were performed as described above. *, p < 0.05. E, AR knockdown induces ATM activation in LNCaP cells. Exponentially growing LNCaP cells were treated with AR-siRNA or scrambled (SC)-siRNA for 36 h. Cell extracts were subjected to Western blotting to confirm knockdown of AR protein and phosphorylation of ATM and ChK2 as described above. F, Casodex induces ATM activation in 22Rv1 cells. Top, 22Rv1 cells treated with 50 μm Casodex for 24 h were analyzed for TIF response by scoring the percentage of cells containing >5 foci of colocalized γ-H2AX and TIN2. ∼80 cells in each treatment group were scored in three separate experiments. Error bars are mean ± S.D., n = 3 independent experiments. ***, p < 0.001. Bottom, 22Rv1 cells treated with 50 μm Casodex for 6 h were evaluated for p-ATM, ATM, and β-actin as described in Fig. 1C. G, AR-inactivation induces p-ATM localization at telomeres in LNCaP and 22RV1 cells. LNCaP and 22Rv1 cells treated with 50 μm Casodex for 6 h were immunostained with antibodies to pATM (red) and TIN2 (green). Colocalization (yellow) of pATM and TIN2 is shown in the merge panels.
Since telomere dysfunction activates ATM kinase (16), and activated ATM kinase phosphorylates histone H2AX at telomeres, we tested whether Casodex-induced telomere dysfunction results in activation (phosphorylation) of ATM kinase. Indeed, within 6 h of treatment with Casodex, ATM kinase was activated, as shown by an increase in phosphorylation of ATM at serine 1981 (Fig. 1C). The AR-antagonist MDV3100 (33), which also induces a TIF response (4), also induced phosphorylation of ATM (Fig. 1D). This effect was abrogated by the addition of R1881, a synthetic androgen, indicating that the effect of MDV3100 on ATM phosphorylation was due to AR antagonism and not a nonspecific toxic effect (Fig. 1D).
To provide direct evidence for a role of AR-inactivation in activation of ATM, we used AR-siRNA to suppress AR expression in LNCaP cells. Transfection of cells with AR-siRNA reduced the AR protein level by ∼80%, compared with control transfection with scrambled (SC)-siRNA (Fig. 1E), and the decrease in AR protein was associated with an increase in ATM phosphorylation, compared with SC-siRNA controls; this indicates that AR-inactivation is responsible for activation of ATM.
Activation of ATM kinase by AR inactivation in prostate cancer cells appears to activate ATM-mediated DDR signaling, based on the finding that ATM-downstream-target Chk2 was also activated (as indicated by Chk2 phosphorylation at T68), as shown in Fig. 1C and 1E. Notably, telomere dysfunction and the activation of ATM kinase by AR inactivation also occurs in AR-positive castration-resistant 22Rv1 cells (Fig. 1F) that are resistant to the growth inhibitory effect of AR antagonists such as Casodex (34). This suggests that telomere dysfunction and the activation of DDR signaling caused by AR inactivation are unrelated to the growth inhibitory effect of AR inactivation.
Finally, since telomere dysfunction in other systems is known to recruit and phosphorylate ATM kinase at telomeres (35), we tested whether this also occurs following AR-inactivation in prostate cancer cells. AR inactivation in LNCaP or 22Rv1 cells treated with Casodex for 6 h indeed caused ATM phosphorylation at telomeres, based on colocalization of pATM and TIN2 antibodies (Fig. 1G, merge panels of Casodex-treated cells). Importantly, almost all of the pATM foci colocalized with TIN2 (Fig. 1G), indicating that Casodex-induced DNA damage is confined largely to telomeres and is not genome-wide.
Repair of Casodex-induced Telomere Dysfunction Leads to the Formation of Aberrant Telomeres
Since the activation of DDR signaling gives the cell an opportunity to repair DNA damage before mitosis, we looked for the repair of telomere dysfunction and telomere DNA damage caused by AR inactivation. Since AR inactivation arrests LNCaP cellcycle progression and blocks entry into mitosis (36), we removed Casodex after 24 h treatment to allow the cells to enter S and progress through G2/M. Both the Casodex-treated and “Casodex-removed” cells had similar survival fractions (Fig. 2A), indicating that cells treated with Casodex for 24 h do not die (24). Notably, however, the dose-dependent increase in TIF response during 24 h treatment with Casodex decreased from >55% with 50 μm Casodex to <5% (a basal level) 24 h after the removal of Casodex (Fig. 2B). Thus, when Casodex was removed from the culture, dysfunctional telomeres appear to be repaired. Therefore, we used Tel-FISH to examine the telomeres of Casodex-removed cells. As reported previously with LNCaP cells (4), 22Rv1 cells also exhibited several telomere aberration phenotypes including broken (fragile) telomeres and telomere loss (Fig. 2C), which occur as a result of homologous recombination (HR) of damaged telomeres (37, 38), and sister-chromatid telomere fusion and telomere end-to-end fusion (Fig. 2D), which result from non-homologous end joining (NHEJ) of damaged telomeres (14, 38). While each of these aberrations was present at a low basal level in control cells, Casodex treatment resulted in a significant increase in telomere breakages and loss (Fig. 2C) and sister-telomere fusions (Fig. 2D). Although telomere end-to-end fusions also appeared to be noticeably increased, the increase was not statistically significant. Nevertheless, these results suggest activation of both HR and NHEJ pathways to facilitate repair of damaged telomeric DNA in Casodex-treated cells.
FIGURE 2.

Repair of AR-inactivation-induced telomere dysfunction leads to the formation of aberrant telomeres. A and B, cell survival is unaffected by a 24 h Casodex treatment, but Casodex-induced telomere dysfunction is repaired within 24 h after removal of the drug. LNCaP cells were treated with 0, 10, 25, or 50 μm Casodex for 24 h, and cells treated with 50 μm Casodex were then incubated in medium without Casodex for an additional 6 or 24 h. A, cells stained or not stained with trypan blue were counted using a hemocytometer. The survival fraction was calculated as the percentage of cells not stained by trypan blue; error bars are mean ± S.D., n = 3. ∼100 cells in each treatment were scored in three separate experiments. B, TIF response was assessed as in Fig. 1B; error bars are mean ± S.D., n = 3. ∼80 cells in each treatment group were scored in three separate experiments. C and D, Casodex treatment causes telomere aberrations in AR-positive 22Rv1 prostate cancer cells. 22RV1 cells were treated with or without 50 μm Casodex for 24 h. Cells were washed to remove Casodex, and metaphase spreads were prepared and subjected to Tel-FISH, then scored for the percentage of chromosomes with telomere aberrations. Representative images of chromosomes with (a) more than one Tel-FISH signal indicating telomere DNA breakage (white arrows) or absence of a Tel-FISH signal indicating telomere loss (yellow triangle), or (b) a single Tel-FISH signal between two sister chromatids indicating sister telomere fusion (yellow arrow) or a pair of Tel-FISH signals between two connected chromosomes indicating telomere end-to-end fusion (red arrow). The percentage of chromosomes with telomere breakage, telomere loss, sister telomere fusion, or telomere end-to-end fusion was determined by scoring each type of telomere aberration in more than 1700 chromosomes from control or Casodex-treated cells. Error bars are mean ± S.D.; *, p < 0.05 and ***, p < 0.001.
ATM Inhibitor Blocks the Activation of ATM Kinase and Repair of Damaged Telomeric DNA in AR-inactivated Cells
Since ATM/ATR inhibition or knockdown has been shown to inhibit the repair of telomere DNA damage caused by the disruption of shelterin components (16), we tested the effect of ATM inhibition in AR-inactivated LNCaP cells by using the ATM-kinase-specific inhibitor KU-60019 (27). Casodex alone induced ATM phosphorylation, but co-treatment with KU-60019 blocked this effect of Casodex (Fig. 3A). Interestingly, whereas 24 h treatment with Casodex alone did not kill LNCaP cells (Fig. 2A) or cause PARP cleavage (Fig. 3A), co-treatment with Casodex and KU-60019 for 24 h caused PARP cleavage (Fig. 3A), a marker of apoptotic cell death; this suggests a role of ATM in survival of cells treated with Casodex for 24 h. We also assessed the effect of KU-60019 on Casodex-induced phosphorylation of H2AX at telomeres. Casodex alone induced phosphorylation of H2AX at telomeres (Fig. 3, B and C), but co-treatment with Casodex and KU-60019 attenuated this effect (Fig. 3, B and C). These results indicate that KU-60019 effectively suppresses Casodex-induced activation of ATM kinase at telomeres.
FIGURE 3.
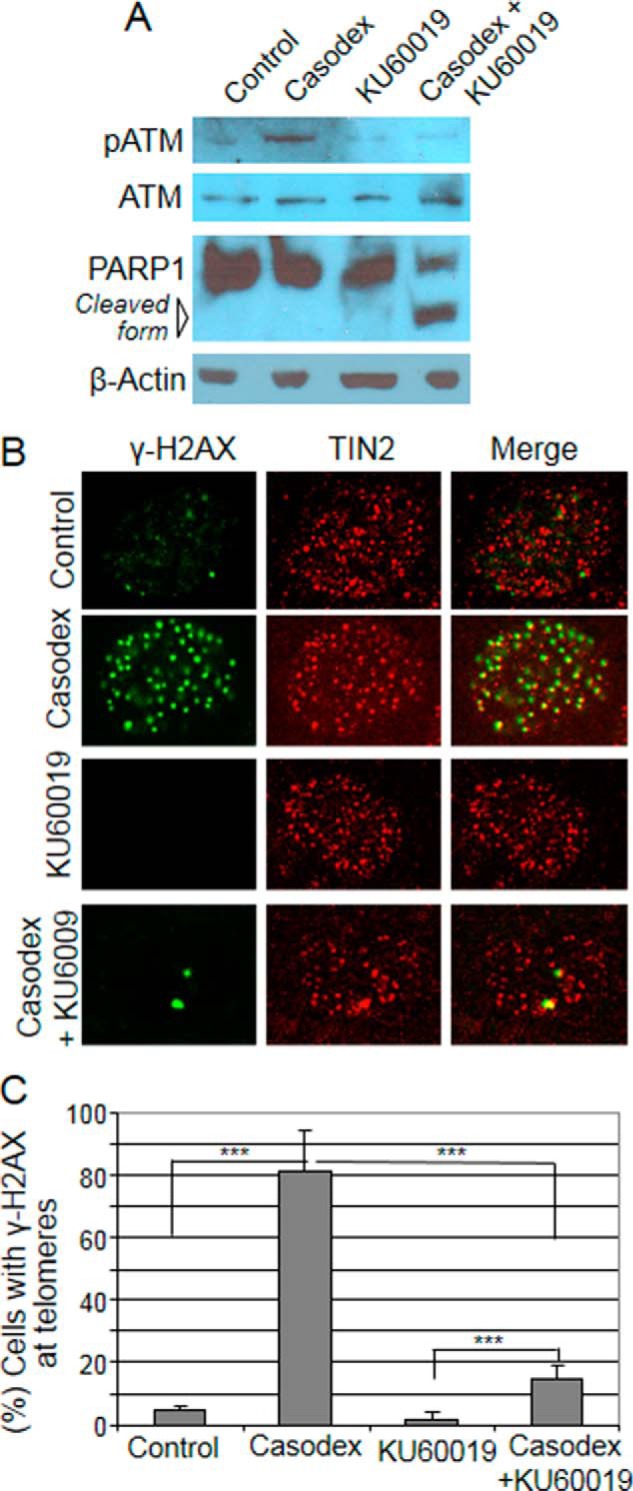
ATM inhibitor blocks activation of ATM kinase in AR-inactivated cells. A, ATM inhibitor suppresses Casodex-induced ATM phosphorylation and induces PARP1 cleavage. LNCaP cells were treated with Casodex (50 μm) and/or KU-60019 (2 μm) for 24 h, and cell extracts were subjected to Western blot analysis using antibodies to p-ATM, ATM, and PARP-1. B and C, ATM inhibitor suppresses Casodex-induced phosphorylation of H2AX at telomeres. B, images of representative LNCaP cells treated with Casodex (50 μm) and/or KU-60019 (2 μm) and immunostained with antibodies to γ-H2AX (green) and TIN2 (red). Colocalization (yellow) of γH2AX and TIN2 is shown in the merge panel. C, data are presented as the percentage of cells with >5 γH2AX foci/cell. ∼80 cells in each treatment group were scored in three separate experiments; mean ± S.D., n = 3. *, p < 0.05; **, p < 0.01; and ***, p < 0.001.
To determine the effect of ATM inhibition on repair of damaged telomeric DNA in AR-inactivated cells, we measured the formation of replication protein A (RPA) foci, which indicate the presence of unrepaired single-stranded DNA (39, 40), at telomeres. RPA foci at telomeres (see merge panels in Fig. 4, A and B) were more common in cells co-treated with Casodex and ATM kinase inhibitor KU-60019 than in control cells (treated with Casodex or KU-60019 alone) (Fig. 4, A and B). In cells co-treated with Casodex and KU-60019, RPA foci colocalized with telomere marker protein TIN2 (Fig. 4A) and DNA damage marker protein 53BP1 (Fig. 4B). Triple-immunostaining using antibodies against RPA, 53BP1 and TIN2 revealed colocalization of RPA with both 53BP1 and TIN2 in cells treated with Casodex and KU-60019 (Fig. 4C, white foci in the “merge” panel of cells treated with Casodex and KU-60019, but not in untreated control cells), indicating the presence of RPA at sites of DNA damage in telomeres. Quantitative analysis showed that cells co-treated with Casodex and ATM inhibitor had a 2-fold increase in RPA and TIN2 colocalization as compared with those treated with Casodex alone and >5-fold increase as compared with those treated with KU60019 alone (Fig. 4D). Similarly, there was a 4-fold increase of RPA and 53BP1 colocalization in cells co-treated with Casodex and KU60019 as compared with those treated with either Casodex alone or ATM inhibitor alone (Fig. 4D). Thus, combined treatment with Casodex plus ATM inhibitor leads to a sustained accumulation of RPA at telomeres, indicating failure to repair damaged telomeric DNA. In addition, cells treated with Casodex plus ATM inhibitor did not show a decreased TIF response relative to Casodex alone, indicating that dysfunctional telomeres caused by Casodex remain unrepaired (Fig. 4D).
FIGURE 4.

ATM inhibitor blocks telomere repair in AR-inactivated cells. A, ATM inhibitor increases RPA foci at telomeres in AR-inactivated LNCaP cells. Images are of representative LNCaP cells treated with Casodex (50 μm) and/or KU-60019 (2 μm) and immunostained with antibodies to RPA32 and TIN2. B, ATM inhibitor increases RPA colocalization with 53BP1 in AR-inactivated LNCaP cells. Images are of representative LNCaP cells treated with Casodex (50 μm) and/or KU-60019 (2 μm) and immunostained with antibodies to RPA32 and 53BP1. Merge panels show colocalization of RPA32 and TIN2 (A) or of RPA32 and 53BP1 (B). C, RPA foci are colocalized with TIN2 and 53BP1 in cells treated with Casodex (50 μm) and KU-60019 (2 μm). Image is a representative LNCaP cell untreated or treated with Casodex (50 μm) and KU-60019 (2 μm) and stained with antibodies to RPA70 (blue), TIN2 (green) and 53BP1 (red). Merge panel of treated, but not untreated (Contol), cell shows triple colocalization (white spots indicated by arrowheads) of TIN2, RPA, and 53BP1. D, ATM inhibitor increases RPA at damaged telomeres in AR-inactivated cells. Data are presented as the percentage of cells with >3 53BP1 and RPA colocalized foci/cell or >3 TIN2 and RPA colocalized foci/cell; mean ± S.D., n = 3. The TIF response was assessed by scoring the percentage of cells with >5 foci that contain colocalized 53BP1 (Abcam) and TIN2 (Santa Cruz Biotechnology). ∼80 cells in each treatment group were scored in three separate experiments. The data are representative of three independent experiments. Error bars show mean ± S.D., n = 3 independent experiments. *, p < 0.05; **, p < 0.01; and ***, p < 0.001.
ATM Inhibitor Augments AR Inactivation-mediated Cell Death
Since ATM activation usually involves activation of the G1 checkpoint to allow repair of damaged telomeric DNA (41), we evaluated the effect of AR inactivation on cell cycle checkpoint activation in LNCaP cells. We examined the cell cycle profile of LNCaP cells treated with Casodex in the presence or absence of KU60019 for 24 h. Surprisingly, Casodex caused a 3-fold increase in the percentage of cells in G2 phase (21%), as compared with controls (7%) (Fig. 5, A and B), indicating activation of the G2/M checkpoint. Since G2/M checkpoint activation is usually mediated by ATR and Chk1 (42), we tested whether Casodex treatment leads to the activation of ATR and Chk1 also. As shown in Fig. 5C, both ATR and Chk1 were phosphorylated (activated) within 6 h of treatment with Casodex, validating G2/M checkpoint activation. As Casodex also activates ATM and Chk2 phosphorylation (Fig. 1C), which are markers of G1 checkpoint activation (41), our data suggests that G2 arrest in Casodex-treated cells is both ATM- and ATR-dependent. Notably, G2/M checkpoint activation by Casodex treatment was abrogated by co-treatment with the ATM inhibitor KU-60019, as Casodex + KU-60019 reduced the percentage of cells in G2 phase from 21% to less than 1% (Fig. 5). This suggests that the activation of ATR and Chk1 is downstream of ATM activation in Casodex-treated cells (42).
FIGURE 5.

ATM inhibitor enhances sub-G1 fraction of AR-inactivated cells in FACS analysis. A, FACS analysis of LNCaP cells treated with Casodex (50 μm) and/or KU-60019 (2 μm) for 24 h. Peaks of cells in sub-G1, G1, and G2 are indicated by arrows. B, percentage of cells in different phases of cell cycle was determined using ModFit LT v4.0.5 (Verity, Topsham, ME). Error bars show mean ± S.D., n = 3 independent experiments. *, p < 0.05; **, p < 0.01; and ***, p < 0.001. C, AR-inactivation induces ATR and Chk1 phosphorylation in LNCaP cells. LNCaP cells were treated for indicated periods with 50 μm Casodex, cell extracts were prepared, and Western blotting was performed. The data are representative of two independent experiments.
ATM inhibitor decreased the G2 fraction of Casodex-treated cells and concomitantly increased the fraction of cells with sub-G1 DNA content, which represent dead cells (see Fig. 5). Thus, ATM inhibitor abrogated the G2 arrest of Casodex-treated cells and led to cell death. As ATM inhibitor had little effect on the sub-G1 fraction of untreated cells, the effect on Casodex-treated cells suggests that ATM inhibitor allowed cells with Casodex-induced telomere DNA damage to bypass the G2/M checkpoint and enter mitosis prematurely without undergoing DNA repair. As failure to repair DNA damage can lead to cell death (7, 8), we infer that Casodex treatment caused telomere dysfunction, telomere DNA damage and activation of the G2/M checkpoint, and that by abrogating this checkpoint, cells accumulate catastrophic telomere loss and undergo cell death (7).
To further assess the effect of ATM inhibition on the death of AR-inactivated LNCaP cells, we measured cell survival using a clonogenic assay design in which cells were treated for 24 h, then washed and allowed to grow for 14 days. Instead of counting colonies as a measure of the percentage of cells that survived the initial treatment, and measuring colony size as a measure of proliferation of the surviving cells, we used the intensity-weighted colony area percentage (26) as a measure of total cell survival. Using this approach, the survival fraction following 24 h treatment with Casodex alone was 75% (Fig. 6, A and B; p = 0.02 compared with no treatment). This likely represents an effect on both colony number and colony size, as we have found in other experiments (not shown) that Casodex treatment decreases both the number of colonies and the size of some surviving colonies. By contrast, the survival fraction following co-treatment with Casodex and KU-60019 was 5% (Fig. 6, A and B). These data are consistent with our observations that co-treatment with Casodex and ATM inhibitor KU-60019 induced PARP cleavage (Fig. 3A) and increased the fraction of cells with sub-G1 DNA content (Fig. 5), and together support the conclusion that ATM inhibitor potentiates the ability of Casodex to kill prostate cancer cells.
FIGURE 6.
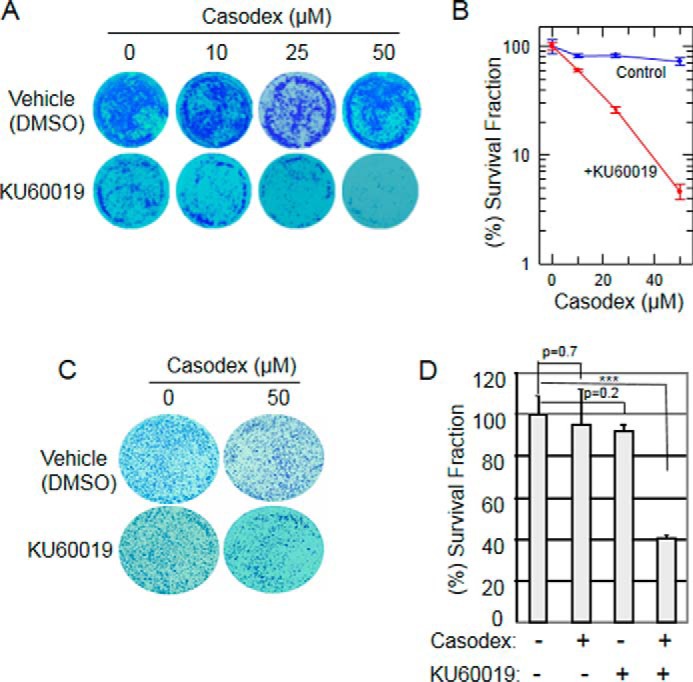
ATM inhibitor promotes death of AR-inactivated prostate cancer cells. A, LNCaP cells treated with 0, 10, 25, or 50 μm Casodex in the presence or absence of 2 μm KU-60019 for 24 h were then washed to remove drugs and allowed to grow for 10–14 days, then fixed and stained with 0.01% crystal violet as described under “Experimental Procedures.” B, area of stained surviving cells in each plate was photographed and measured using the ImageJ program. The survival fraction was plotted relative to control (vehicle); mean ± S.D. of three independent experiments. C and D, 22Rv1 cells were treated for 24 h with 50 μm Casodex and/or 2 μm KU-60019, then washed to remove drugs and subjected to a colony formation assay and analyzed as described in Fig. 6, A and B; mean ± S.D. of three independent experiments. *, p < 0.05;**, p < 0.01; ***, p < 0.001.
It is notable that 22Rv1 cells, which are resistant to the growth inhibitory effect of Casodex but nonetheless exhibit a TIF response to Casodex, became sensitive to cell killing by Casodex in the added presence of ATM inhibitor (Fig. 6, C and D). Thus, the effect of ATM inhibition in killing Casodex-treated cells is not unique to androgen-sensitive LNCaP cells, and therefore may have its greatest application in the treatment of drug-resistant disease. However, even sustained treatment of Casodex-sensitive cells with Casodex alone may not be 100% effective, so co-treatment with Casodex and ATM inhibitor may be better than Casodex alone even for androgen-sensitive disease.
Collectively, these results suggest that ATM inhibitor KU-60019 blocks Casodex-induced DDR signaling, leaving damaged telomeric DNA unrepaired in Casodex-treated cells, preventing checkpoint activation and, thereby, allowing both androgen-sensitive LNCaP and castration-resistant 22Rv1 cells to enter mitosis with too much telomeric DNA damage to sustain survival.
Discussion
DDR signaling pathways can enable tumor cells to survive DNA damage that is induced by chemotherapeutic treatments; therefore, inhibitors of specific DDR pathways prove efficacious when used in combination with DNA-damaging chemotherapeutic drugs (7). In the present study, we demonstrated that 1) the induction of telomere dysfunction in cells treated with AR-antagonists (Casodex or MDV3100) or AR-siRNA is associated with a dramatic increase in phosphorylation of ATM and Chk2 and the presence of phosphorylated ATM at telomeres, indicating that AR inactivation leads to the activation of ATM DDR signaling pathways at telomeres, and that 2) ATM inhibition enhances the death of AR-inactivated prostate cancer cells by blocking the repair of AR-inactivation-induced dysfunctional telomeres in androgen-sensitive (LNCaP) and castration-resistant (22Rv1) prostate cancer cells. Thus, these studies demonstrate for the first time that the role of AR in telomere stability can be exploited to augment the potency of currently available AR-targeted therapies for the treatment of prostate cancer.
We have shown previously that AR plays an important role in maintaining telomere stability through its interaction with shelterin proteins, viz., TRF-1, TRF-2, and TIN-2, and that the disruption of these interactions in AR-inactivated cells leads to telomere dysfunction (3, 4). However, since AR is implicated to regulate the expression of telomerase (31, 32), which is required for maintaining telomere length during each cell division cycle, and since telomere shortening in telomerase-deficient cells can also lead to telomere dysfunction (35), we evaluated whether telomere dysfunction observed in AR-inactivated cells may have resulted from telomerase down-regulation. Our studies show no noticeable change in expression of telomerase catalytic subunit, human telomerase reverse transcriptase (hTERT), under the conditions in which telomere dysfunction was observed (Fig. 1B). Moreover, it was reported that AR-inactivation lasting several days is required in order to notice any significant decrease in telomerase activity (43). Thus telomere dysfunction observed within a few hours of AR inactivation would not have resulted from down-regulation of telomerase activity in Casodex treated LNCaP cells. Furthermore, inhibition of de novo mRNA and protein biosynthesis by actinomycin D and cycloheximide, respectively, does not cause telomere dysfunction nor inhibit the TIF response to Casodex (4). Together, these observations point to a role of AR in telomere stability that is independent of its role as a transcription factor.
Interestingly, telomere aberrations (telomere breakage/fragile telomeres and telomere fusion) caused by AR inactivation in both androgen-sensitive and castration-resistant 22Rv1 cells (Fig. 2) are reminiscent of those that are reported to occur in other cell types with defective shelterin proteins (5). These shelterin proteins are implicated to play a role in telomere DNA replication or repair. For example, in mouse embryo fibroblasts, TRF-1 knock-out interferes with DNA replication, and disruption of telomere replication leads to telomere breakage and the appearance of multiple telomeric signals at the end of a chromatid (phenotype referred to as fragile telomeres) (15, 44). Similarly, telomere fusion, which is commonly seen in cells that are defective in DNA repair signaling pathways, is observed in cells that are TRF-2-negative, implicating a role of TRF-2 in DNA repair (14). These observations raise an intriguing possibility that AR may play a role in telomere DNA replication or repair and that aberrant telomeres in AR-inactivated prostate cancer cells may result from impaired telomere DNA replication or repair. Consistent with this possibility are the observations that AR co-immunoprecipitates with TRF-1 and TRF-2 (3), AR is associated with telomere DNA (4), and AR is implicated to play a non-transcriptional role in DNA replication (36, 45) and possibly in repair of nuclear DNA (46). However, it remains to be determined whether AR inactivation interferes with either replication or repair of telomere DNA.
DDR signaling pathways play an intricate role in preserving genomic DNA integrity by sensing DNA damage as and when it occurs and activating repair mechanisms to repair the lesion (7). It was reported recently that DNA damage caused by ionizing radiation leads to the activation of AR, which in turn induces the expression of DNA repair genes that promote the survival of irradiated prostate cancer cells (47, 48). Based on these reports, we expected that AR-inactivation would down regulate DDR signaling pathways and block the repair of damaged telomeres. Instead, we observed the opposite: we found a dramatic increase in ATM phosphorylation and Chk2 phosphorylation within 6 h of AR-inactivation or AR-knockdown, and during this period there was no noticeable change in either ATM or Chk2 at the protein level in either LNCaP or 22Rv1 cells (Fig. 1). Activation of DNA repair pathways in AR-inactivated cells is also evident from the detection of aberrant telomeres (Fig. 2), which result from NHEJ and/or homologous recombination (HR) DNA repair mechanisms (5). These observations suggest that, at least in the short-term, the repair of damaged telomeric DNA in AR-inactivated cells may not involve AR-dependent transcriptional regulation of DNA repair genes.
We considered the question whether ATM activation in AR-inactivated cells may have also resulted from non-telomeric DNA damage. We evaluated this possibility by monitoring the formation of γ-H2AX foci, which indicate sites of DNA damage, in the context of TIN2 foci, which indicate telomeric regions, in Casodex-treated cells. We found that γ-H2AX foci were outnumbered by TIN2 foci and almost all γ-H2AX foci colocalized with TIN2 foci (Figs. 1A and 3B); this indicates that the majority of the DNA damage was localized to telomeres. Similarly, there were fewer pATM foci than TIN2 foci, and almost all pATM foci colocalized with TIN2 foci in Casodex-treated LNCaP and 22Rv1 cells (Fig. 1G); this indicates that the majority of the DNA damage response to Casodex was localized at telomeres. In addition, in cells treated with Casodex plus ATM inhibitor, triple-immunofluorescence revealed that RPA, which binds to stretches of unrepaired single-stranded DNA (39, 40), colocalized with 53BP1 (purple foci in Fig. 4C, merge panel), and more than 80% of these colocalized foci were confined to telomeres as indicated by their colocalization with TIN2 (white foci in Fig. 4C, “merge” panel); this indicates that the majority of unrepaired DNA was localized at telomeres. Although we cannot completely rule out some DNA damage at non-telomeric sites that may not be readily detectable with the immunofluorescence approaches employed in this study, our data indicate that both DNA damage and the presence of activated ATM (pATM) are confined mostly to telomeres in AR-inactivated cells. It is well known that telomere dysfunction represents telomere DNA damage that leads to the activation of ATM and ATR signaling pathways (5). Likewise, we observed that under conditions in which AR-inactivation caused telomere dysfunction, both ATM and ATR signaling were activated within 6 h of treatment with Casodex (Figs. 1 and 5). It is also well known that activation of ATM/ATR in turn activates cell cycle checkpoints, leading to cell cycle arrest to allow for repair of damaged DNA. Likewise, we observed that treatment with Casodex for no more than 24 h was sufficient to cause marked enrichment of LNCaP cells in G2/M phase (Fig. 5B), indicating G2/M checkpoint activation. G2/M checkpoint activation in Casodex-treated cells was confirmed by finding activation of ATR and Chk1 (Fig. 5C), which promote G2/M checkpoint activation (42). Interestingly, ATM inhibitor KU-60019 abrogated the G2/M checkpoint, suggesting that the activation of ATR and Chk1 is downstream of ATM activation in Casodex-treated cells (42). As failure to repair DNA damage can lead to cell death, activation of ATM/ATR may thereby promote cell survival (7). Conversely, inhibition of ATM/ATR can block this process and promote cell death (27, 49). In the present study we show that co-treatment of LNCaP cells with AR antagonist Casodex plus ATM inhibitor KU-60019 decreased cell survival to a much greater extent than did treatment with Casodex alone (Figs. 5 and 6), and this decrease was associated with inhibition of ATM phosphorylation and induction of PARP cleavage (Fig. 3A).
In summary, as depicted in Fig. 7, our studies suggest that telomere dysfunction caused by AR-inactivation can lead to the activation of cell cycle checkpoints, which may allow repair of damaged telomeric DNA and cell survival, and that inhibitors of ATM DDR signaling can abrogate cell cycle checkpoint activation and promote death of AR-inactivated prostate cancer cells. Interestingly, ATM-inactivation in normal cells has no untoward consequences (50). However, inhibitors of ATM/ATR signaling hypersensitize cancer cells to DNA damaging agents (8). Therefore, several inhibitors of ATM/ATR signaling pathways are currently in clinical trials to enhance the potency of DNA damaging therapeutic agents, such as radiation or genotoxic drugs that cause DSBs, for the treatment of a variety of cancers (8). Accordingly, our results imply that inhibitors of ATM/ATR DDR signaling may offer a unique opportunity to enhance the potency of AR-inactivation therapies for the treatment of androgen-sensitive and castration-resistant prostate cancer.
FIGURE 7.
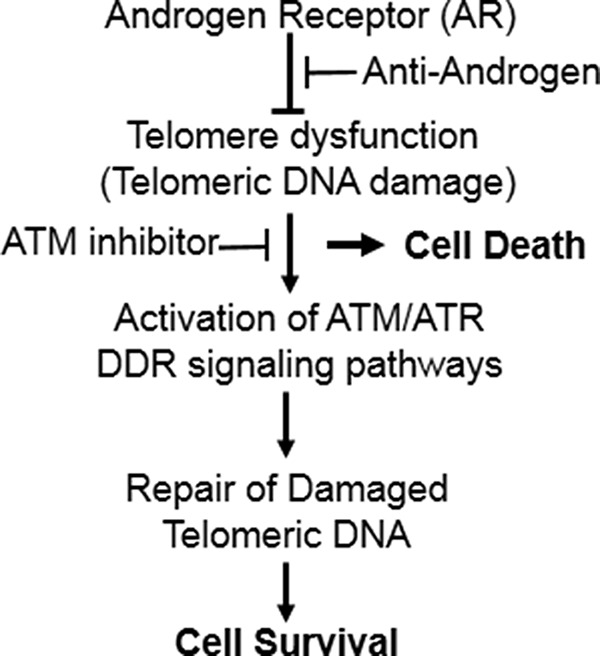
Model showing a role of ATM/ATR signaling in the survival of AR-inactivated prostate cancer cells. Telomere dysfunction caused by AR-inactivation can lead to the activation of ATM/ATR-mediated checkpoints, which may allow repair of damaged telomeric DNA, and promote cell survival. Inhibitors of ATM/ATR signaling pathways can block cell cycle checkpoint activation and thereby promote death of AR-inactivated prostate cancer cells.
Author Contributions
M. M., E. B., P. R., and S. K. conceived and coordinated the study and wrote the paper. V. R. and S. K. designed, performed and analyzed the experiments shown in Figs. 1 to 7. M. W. performed and analyzed the experiments shown in Figs. 1, C, D, E, and F and 3A. N. C. performed and analyzed real time qPCR. N. K. designed, performed, and analyzed the experiments shown in Fig. 5, A and B. All authors reviewed the results and approved the final version of the manuscript.
This work was supported by Department of Defense Prostate Cancer Research Program Grant W81XWH-10-1-0117 (to S. K.) and a grant from Henry Ford Health System (to S. K.). The Cytometry Resources Core is supported in part by NIH Center Grant P30CA022455 to the Karmanos Cancer Institute, Wayne State University, and the Perinatology Research Branch of the National Institutes of Child Health and Development, Wayne State University. The authors declare that they have no conflicts of interest with the contents of this article.
- AR
- androgen receptor
- ATM
- ataxia telangiectasia mutated
- DDR
- DNA damage response
- ADT
- androgen-deprivation therapy
- CRPC
- castration-resistant prostate cancer
- FISH
- fluorescence in situ hybridization
- PNA
- protein nucleic acid
- TIF
- telomere-dysfunction-induced foci
- HR
- homologous recombination
- NHEJ
- non-homologous end joining.
References
- 1. Heinlein C. A., Chang C. (2004) Androgen receptor in prostate cancer. Endocr. Rev. 25, 276–308 [DOI] [PubMed] [Google Scholar]
- 2. Feldman B. J., Feldman D. (2001) The development of androgen-independent prostate cancer. Nature Reviews. Cancer 1, 34–45 [DOI] [PubMed] [Google Scholar]
- 3. Kim S. H., Richardson M., Chinnakannu K., Bai V. U., Menon M., Barrack E. R., Reddy G. P. (2010) Androgen receptor interacts with telomeric proteins in prostate cancer cells. J. Biol. Chem. 285, 10472–10476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhou J., Richardson M., Reddy V., Menon M., Barrack E. R., Reddy G. P., Kim S. H. (2013) Structural and functional association of androgen receptor with telomeres in prostate cancer cells. Aging 5, 3–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Palm W., de Lange T. (2008) How shelterin protects mammalian telomeres. Annu. Rev. Genet. 42, 301–334 [DOI] [PubMed] [Google Scholar]
- 6. Déjardin J., Kingston R. E. (2009) Purification of proteins associated with specific genomic Loci. Cell 136, 175–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Smith J., Tho L. M., Xu N., Gillespie D. A. (2010) The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 108, 73–112 [DOI] [PubMed] [Google Scholar]
- 8. Helleday T., Petermann E., Lundin C., Hodgson B., Sharma R. A. (2008) DNA repair pathways as targets for cancer therapy. Nature Reviews. Cancer 8, 193–204 [DOI] [PubMed] [Google Scholar]
- 9. Maser R. S., DePinho R. A. (2002) Connecting chromosomes, crisis, and cancer. Science 297, 565–569 [DOI] [PubMed] [Google Scholar]
- 10. Murnane J. P. (2010) Telomere loss as a mechanism for chromosome instability in human cancer. Cancer Res. 70, 4255–4259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chin K., de Solorzano C. O., Knowles D., Jones A., Chou W., Rodriguez E. G., Kuo W. L., Ljung B. M., Chew K., Myambo K., Miranda M., Krig S., Garbe J., Stampfer M., Yaswen P., Gray J. W., Lockett S. J. (2004) In situ analyses of genome instability in breast cancer. Nature Genetics 36, 984–988 [DOI] [PubMed] [Google Scholar]
- 12. Taylor B. S., Schultz N., Hieronymus H., Gopalan A., Xiao Y., Carver B. S., Arora V. K., Kaushik P., Cerami E., Reva B., Antipin Y., Mitsiades N., Landers T., Dolgalev I., Major J. E., Wilson M., Socci N. D., Lash A. E., Heguy A., Eastham J. A., Scher H. I., Reuter V. E., Scardino P. T., Sander C., Sawyers C. L., Gerald W. L. (2010) Integrative genomic profiling of human prostate cancer. Cancer Cell 18, 11–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heaphy C. M., Yoon G. S., Peskoe S. B., Joshu C. E., Lee T. K., Giovannucci E., Mucci L. A., Kenfield S. A., Stampfer M. J., Hicks J. L., De Marzo A. M., Platz E. A., Meeker A. K. (2013) Prostate cancer cell telomere length variability and stromal cell telomere length as prognostic markers for metastasis and death. Cancer Discovery 3, 1130–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Celli G. B., de Lange T. (2005) DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nature Cell Biology 7, 712–718 [DOI] [PubMed] [Google Scholar]
- 15. Sfeir A., Kosiyatrakul S. T., Hockemeyer D., MacRae S. L., Karlseder J., Schildkraut C. L., de Lange T. (2009) Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138, 90–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Denchi E. L., de Lange T. (2007) Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 448, 1068–1071 [DOI] [PubMed] [Google Scholar]
- 17. Bakkenist C. J., Kastan M. B. (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421, 499–506 [DOI] [PubMed] [Google Scholar]
- 18. Lavin M. F., Kozlov S. (2007) ATM activation and DNA damage response. Cell Cycle 6, 931–942 [DOI] [PubMed] [Google Scholar]
- 19. Angèle S., Falconer A., Foster C. S., Taniere P., Eeles R. A., Hall J. (2004) ATM protein overexpression in prostate tumors: possible role in telomere maintenance. Am. J. Clin. Pathol. 121, 231–236 [DOI] [PubMed] [Google Scholar]
- 20. Fan C., Quan R., Feng X., Gillis A., He L., Matsumoto E. D., Salama S., Cutz J. C., Kapoor A., Tang D. (2006) ATM activation is accompanied with earlier stages of prostate tumorigenesis. Biochim. Biophys. Acta 1763, 1090–1097 [DOI] [PubMed] [Google Scholar]
- 21. Joshua A. M., Shen E., Yoshimoto M., Marrano P., Zielenska M., Evans A. J., Van der Kwast T., Squire J. A. (2011) Topographical analysis of telomere length and correlation with genomic instability in whole mount prostatectomies. The Prostate 71, 778–790 [DOI] [PubMed] [Google Scholar]
- 22. Kim S. H., Kaminker P., Campisi J. (1999) TIN2, a new regulator of telomere length in human cells [see comments]. Nature Genetics 23, 405–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mirzoeva O. K., Petrini J. H. (2001) DNA damage-dependent nuclear dynamics of the Mre11 complex. Mol. Cell. Biol. 21, 281–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bai V. U., Cifuentes E., Menon M., Barrack E. R., Reddy G. P. (2005) Androgen receptor regulates Cdc6 in synchronized LNCaP cells progressing from G1 to S phase. J. Cell. Physiol. 204, 381–387 [DOI] [PubMed] [Google Scholar]
- 25. Bailey S. M., Meyne J., Chen D. J., Kurimasa A., Li G. C., Lehnert B. E., Goodwin E. H. (1999) DNA double-strand break repair proteins are required to cap the ends of mammalian chromosomes. Proc. Natl. Acad. Sci. U.S.A. 96, 14899–14904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guzmán C., Bagga M., Kaur A., Westermarck J., Abankwa D. (2014) ColonyArea: an ImageJ plugin to automatically quantify colony formation in clonogenic assays. PloS one 9, e92444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Golding S. E., Rosenberg E., Valerie N., Hussaini I., Frigerio M., Cockcroft X. F., Chong W. Y., Hummersone M., Rigoreau L., Menear K. A., O'Connor M. J., Povirk L. F., van Meter T., Valerie K. (2009) Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion Molecular Cancer Therapeutics 8, 2894–2902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Takai H., Smogorzewska A., de Lange T. (2003) DNA damage foci at dysfunctional telomeres. Curr. Biol. 13, 1549–1556 [DOI] [PubMed] [Google Scholar]
- 29. Kim S. H., Beausejour C., Davalos A. R., Kaminker P., Heo S. J., Campisi J. (2004) TIN2 mediates functions of TRF2 at human telomeres. J. Biol. Chem. 279, 43799–43804 [DOI] [PubMed] [Google Scholar]
- 30. Zhan P., Lee E. C., Packman K., Tenniswood M. (2002) Induction of invasive phenotype by Casodex in hormone-sensitive prostate cancer cells. J. Steroid Biochem. Mol. Biol. 83, 101–111 [DOI] [PubMed] [Google Scholar]
- 31. Guo C., Armbruster B. N., Price D. T., Counter C. M. (2003) In vivo regulation of hTERT expression and telomerase activity by androgen. J. Urol. 170, 615–618 [DOI] [PubMed] [Google Scholar]
- 32. Liu S., Qi Y., Ge Y., Duplessis T., Rowan B. G., Ip C., Cheng H., Rennie P. S., Horikawa I., Lustig A. J., Yu Q., Zhang H., Dong Y. (2010) Telomerase as an important target of androgen signaling blockade for prostate cancer treatment. Molecular Cancer Therapeutics 9, 2016–2025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tran C., Ouk S., Clegg N. J., Chen Y., Watson P. A., Arora V., Wongvipat J., Smith-Jones P. M., Yoo D., Kwon A., Wasielewska T., Welsbie D., Chen C. D., Higano C. S., Beer T. M., Hung D. T., Scher H. I., Jung M. E., Sawyers C. L. (2009) Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 324, 787–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yamashita S., Lai K. P., Chuang K. L., Xu D., Miyamoto H., Tochigi T., Pang S. T., Li L., Arai Y., Kung H. J., Yeh S., Chang C. (2012) ASC-J9 suppresses castration-resistant prostate cancer growth through degradation of full-length and splice variant androgen receptors. Neoplasia 14, 74–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. d'Adda di Fagagna F., Reaper P. M., Clay-Farrace L., Fiegler H., Carr P., Von Zglinicki T., Saretzki G., Carter N. P., Jackson S. P. (2003) A DNA damage checkpoint response in telomere-initiated senescence. Nature 426, 194–198 [DOI] [PubMed] [Google Scholar]
- 36. Murthy S., Wu M., Bai V. U., Hou Z., Menon M., Barrack E. R., Kim S. H., Reddy G. P. (2013) Role of androgen receptor in progression of LNCaP prostate cancer cells from G1 to S phase. PloS one 8, e56692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Min J., Choi E. S., Hwang K., Kim J., Sampath S., Venkitaraman A. R., Lee H. (2012) The breast cancer susceptibility gene BRCA2 is required for the maintenance of telomere homeostasis. J. Biol. Chem. 287, 5091–5101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sfeir A., de Lange T. (2012) Removal of shelterin reveals the telomere end-protection problem. Science 336, 593–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gong Y., de Lange T. (2010) A Shld1-controlled POT1a provides support for repression of ATR signaling at telomeres through RPA exclusion. Mol. Cell 40, 377–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Martínez P., Flores J. M., Blasco M. A. (2012) 53BP1 deficiency combined with telomere dysfunction activates ATR-dependent DNA damage response. J. Cell Biol. 197, 283–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Herbig U., Jobling W. A., Chen B. P., Chen D. J., Sedivy J. M. (2004) Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol. Cell 14, 501–513 [DOI] [PubMed] [Google Scholar]
- 42. Jazayeri A., Falck J., Lukas C., Bartek J., Smith G. C., Lukas J., Jackson S. P. (2006) ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nature Cell Biology 8, 37–45 [DOI] [PubMed] [Google Scholar]
- 43. Bouchal J., Baumforth K. R., Sváchová M., Murray P. G., von Angerer E., Kolár Z. (2005) Microarray analysis of bicalutamide action on telomerase activity, p53 pathway and viability of prostate carcinoma cell lines. J. Pharm. Pharmacol. 57, 83–92 [DOI] [PubMed] [Google Scholar]
- 44. Martínez P., Thanasoula M., Muñoz P., Liao C., Tejera A., McNees C., Flores J. M., Fernández-Capetillo O., Tarsounas M., Blasco M. A. (2009) Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev. 23, 2060–2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. D'Antonio J. M., Vander Griend D. J., Isaacs J. T. (2009) DNA licensing as a novel androgen receptor mediated therapeutic target for prostate cancer. Endocrine-related Cancer 16, 325–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mayeur G. L., Kung W. J., Martinez A., Izumiya C., Chen D. J., Kung H. J. (2005) Ku is a novel transcriptional recycling coactivator of the androgen receptor in prostate cancer cells. J. Biol. Chem. 280, 10827–10833 [DOI] [PubMed] [Google Scholar]
- 47. Goodwin J. F., Schiewer M. J., Dean J. L., Schrecengost R. S., de Leeuw R., Han S., Ma T., Den R. B., Dicker A. P., Feng F. Y., Knudsen K. E. (2013) A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discovery 3, 1254–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Polkinghorn W. R., Parker J. S., Lee M. X., Kass E. M., Spratt D. E., Iaquinta P. J., Arora V. K., Yen W. F., Cai L., Zheng D., Carver B. S., Chen Y., Watson P. A., Shah N. P., Fujisawa S., Goglia A. G., Gopalan A., Hieronymus H., Wongvipat J., Scardino P. T., Zelefsky M. J., Jasin M., Chaudhuri J., Powell S. N., Sawyers C. L. (2013) Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discovery 3, 1245–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Prevo R., Fokas E., Reaper P. M., Charlton P. A., Pollard J. R., McKenna W. G., Muschel R. J., Brunner T. B. (2012) The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer Biology Therapy 13, 1072–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jallepalli P. V., Lengauer C., Vogelstein B., Bunz F. (2003) The Chk2 tumor suppressor is not required for p53 responses in human cancer cells. J. Biol. Chem. 278, 20475–20479 [DOI] [PubMed] [Google Scholar]