

Background: Pancreatic-islet β-cell tumors (insulinomas) that lack menin express the phospho-isoform of the differentiation factor HLXB9.
Results: Phospho-HLXB9 interacts with the survival factor p54nrb/Nono and also activates the oncogenic c-Met pathway by down-regulating the c-Met inhibitor Cblb.
Conclusion: Targeting the HLXB9-Nono interaction and c-Met in insulinomas can be therapeutic.
Significance: β-Cell proliferation mechanisms propose tumor therapy and strategies to alleviate β-cell loss in diabetes.
Keywords: gene regulation, menin (MEN1), pancreatic islet, pathogenesis, tumor, PNET, c-Met, neuroendocrine, p54nrb/Nono, phospho-HLXB9
Abstract
Pancreatic islet β-cells that lack the MEN1-encoded protein menin develop into tumors. Such tumors express the phosphorylated isoform of the β-cell differentiation transcription factor HLXB9. It is not known how phospho-HLXB9 acts as an oncogenic factor in insulin-secreting β-cell tumors (insulinomas). In this study we investigated the binding partners and target genes of phospho-HLXB9 in mouse insulinoma MIN6 β-cells. Co-immunoprecipitation coupled with mass spectrometry showed a significant association of phospho-HLXB9 with the survival factor p54nrb/Nono (54-kDa nuclear RNA-binding protein, non-POU-domain-containing octamer). Endogenous phospho-HLXB9 co-localized with endogenous Nono in the nucleus. Overexpression of HLXB9 decreased the level of overexpressed Nono but not endogenous Nono. Anti-phospho-HLXB9 chromatin immunoprecipitation followed by deep sequencing (ChIP-Seq) identified the c-Met inhibitor, Cblb, as a direct phospho-HLXB9 target gene. Phospho-HLXB9 occupied the promoter of Cblb and reduced the expression of Cblb mRNA. Cblb overexpression or HLXB9 knockdown decreased c-Met protein and reduced cell migration. Also, increased phospho-HLXB9 coincided with reduced Cblb and increased c-Met in insulinomas of two mouse models of menin loss. These data provide mechanistic insights into the role of phospho-HLXB9 as a pro-oncogenic factor by interacting with a survival factor and by promoting the oncogenic c-Met pathway. These mechanisms have therapeutic implications for reducing β-cell proliferation in insulinomas by inhibiting phospho-HLXB9 or its interaction with Nono and modulating the expression of its direct (Cblb) or indirect (c-Met) targets. Our data also implicate the use of pro-oncogenic activities of phospho-HLXB9 in β-cell expansion strategies to alleviate β-cell loss in diabetes.
Introduction
Human or mouse pancreatic islet β-cells that lack the MEN1-encoded tumor suppressor protein menin develop into pancreatic neuroendocrine tumors (PanNETs or PNETs)2 (1–3). Inactivating MEN1 mutations has been observed in 40% of human sporadic non-functioning PNETs (not associated with hormone hypersecretion) (4). However, sporadic functioning PNETs, the insulin-secreting pancreatic islet β-cell tumors (insulinomas), rarely show MEN1 mutations (5–7). Thus, although menin loss/inactivation specifically in the β-cells can lead to tumor formation, there are many PNETs such as the insulinomas that can form without menin loss/inactivation.
Various roles of menin, a predominantly nuclear protein, in transcriptional regulation and other critical cellular processes have been reported (8). However, it is unclear why menin loss leads to tumors in only specific cell types, for instance, menin loss in the whole pancreas of mice results in only insulinomas (9). To decipher the pathogenesis of insulinomas, we recently investigated the targets of menin in β-cells as potential candidates in the tumorigenesis process. Menin deficiency coincided with up-regulation of a β-cell differentiation factor HLXB9, particularly its phospho isoform, in cell culture experiments and in tumors (10, 11). Also, insulinomas without menin loss expressed phospho-HLXB9, underscoring the importance of menin-independent regulation of this protein in such tumors (10). However, how HLXB9 elicits pro-oncogenic activities in insulinomas is not known.
HLXB9 (also known as HB9, MNR2, and MNX1) is a transcription factor with a dual expression profile during pancreas and β-cell development in embryogenesis and later in adult β-cells (12). The expression of HLXB9 in the adult pancreas is β-cell-specific (13). Hlxb9−/− mice are viable but show dorsal pancreas agenesis and small islets in the rest of the ventral pancreas (13, 14). HLXB9 is phosphorylated by glycogen synthase kinase 3β (GSK-3β) at Ser-78 and Ser-80, and the level of phospho-HLXB9 is increased upon menin knockdown in mouse insulinoma MIN6 β-cells (10). HLXB9 is pro-apoptotic in MIN6 cells; however, phospho-HLXB9 is not pro-apoptotic because HLXB9 expression together with menin knockdown in MIN6 cells does not cause apoptosis (11). This indicates unique activities of phospho-HLXB9 in insulinoma cells.
c-MET (also known as HGFR or MET) is a receptor for hepatocyte growth factor. Aberrant activation of hepatocyte growth factor/MET signaling (through c-MET mutation or increased expression) has been observed in a variety of tumors (15). Although activating c-MET mutations have not been reported in PNETs, they show increased c-MET expression (16, 17). c-Met knock-out mice are embryonic lethal, but conditional loss of c-Met in β-cells results in smaller islets and impaired insulin secretion (18–20). Mouse studies have also shown that c-Met expression or signaling could be utilized for β-cell regeneration strategies in diabetes (21). Thus, proper regulation of c-MET is critical for normal growth and proliferation of β-cells. CBLB (Casitas B-lineage lymphoma b) is an E3 ubiquitin ligase that belongs to the CBL protein family (CBL, CBL-b, and CBL-c) (22). CBL proteins can ubiquitinate activated receptor-tyrosine kinases, such as c-MET, and target them for lysosomal or proteasomal degradation, thus negatively regulating various signaling pathways (22, 23). Loss of CBL protein function predicts increased receptor-tyrosine kinase levels and activity that could lead to increased growth and cancer.
Studying the activities of HLXB9 in insulinomas can have dual benefits: understanding β-cell tumorigenesis and unraveling endogenous β-cell replication mechanisms to replace β-cells in conditions of β-cell loss such as in diabetes. To study the mechanisms by which HLXB9 functions in insulinomas, we investigated the binding partners and direct target genes of phospho-HLXB9 in mouse insulinoma MIN6 β-cells. Phospho-HLXB9 interacted with Nono (Non-POU domain-containing octamer-binding protein) in the nucleus. Nono, also known as p54nrb (54-kDa nuclear RNA binding protein), is associated with RNA processing, DNA repair, and transcriptional regulation, and it has been shown to act as a survival factor in melanoma cells (24–26). Using Phospho-HLXB9 ChIP-Seq, we found that the c-Met inhibitor, Cblb, is a direct target of phospho-HLXB9, another possible reason for pro-oncogenic effects from HLXB9. We also found an inverse correlation between Cblb and c-Met expression in mouse insulinomas. Our findings support therapeutic implications from modulating phospho-HLXB9 or its targets in insulinomas.
Experimental Procedures
Plasmids, shRNA, siRNA, and Antibodies
The following mammalian expression plasmids were used: pcDNA3.1-Myc-His vector (pcDNA3.1-mh) (Invitrogen), mouse-HLXB9 (pcDNA3.1-mh-HB9-WT, and pcDNA3.1-mh-HB9-AA (phospho-dead mutant of HLXB9 with alanine substitution at serine 78 and serine 80)) (11), pcDNA3.1-mh-menin (27), pCMV6-XL4-CBLB (Origene, SC107022), pFLAG-p54 (Nono) (Addgene, plasmid 35379), control and MEN1 shRNA (28), and control and Nono shRNA (29). The following siRNAs were used: negative control (Qiagen, 1027280) and mouse HLXB9 (Dharmacon, L-049859-01). For luciferase reporter assays the promoter-less pEZX-PG02 vector and the promoter constructs pEZX-PG02-Arid1b and pEZX-PG02-Cblb were purchased and confirmed by sequencing (GeneCopoeia). pEZX-PG02-Cblb-SDM2 was constructed by site-directed mutagenesis of the two HLXB9 binding motifs at −735 and −722 in the mouse Cblb promoter (Agilent, QuikChange site-directed mutagenesis kit). The following antibodies were used: mouse anti-HB9 (DSHB, 81.5C10), rabbit anti-HB9 (Bethyl, A303183A), rabbit anti-HB9-PO4 (10), rabbit anti-menin (Bethyl, A300-105A), mouse anti-myc-tag (Millipore, 05-724), rabbit anti-myc-tag (Millipore, 06-549), mouse anti-p54 (Nono) (Millipore, 05-950), mouse anti-Cblb (Santa Cruz, sc-8006), rabbit anti-c-Met (Santa Cruz, sc-10), rabbit anti-H3K4me3 (Active Motif, 39159), rabbit anti-H3K27me3 (Millipore, 07-449), mouse anti-FLAG-tag (Sigma, F3165), mouse anti-HA-tag (Cell Signaling, 2367), mouse anti-β-actin (Sigma, A1978), rabbit anti-histone H3 (Millipore, 06-755), rabbit anti-HSP90 (Cell Signaling, 4877), mouse anti-p84 (Gene Tex, GTX70220), chicken anti-insulin (Abcam, Ab14042), mouse secondary antibody (HRP) (Santa Cruz, sc-2055), rabbit secondary antibody (HRP) (Santa Cruz, sc-2054), Alexa Fluor 594 goat anti-chicken secondary antibody (Life Technologies, A11042), Alexa Fluor 594 goat anti-mouse secondary antibody (Life Technologies, A21132), Alexa Fluor 488 goat anti-rabbit secondary antibody (Life Technologies, A11008).
Cell Culture and Transfection
A mouse insulinoma cell line, MIN6-4N, was cultured in low glucose DMEM supplemented with 15% FBS and 1× antibiotic/antimycotic (Invitrogen, Gemini) (complete DMEM) at 37 °C and 5% CO2. MIN6-4N is a tetraploid (4N) cell line that was generated by isolating single cell clones from the mixed ploidy (2N and 4N) MIN6 cell line (10, 30). Plasmids or siRNA were transfected using Lipofectamine 2000 (Invitrogen) or nucleofection (AMAXA/Lonza). RNA, chromatin, or protein was isolated 48 h or 96 h post-transfection. For menin knockdown combined with HLXB9 overexpression, shRNA plasmids were transfected by nucleofection first into MIN6-4N cells, then 48 h post-transfection the HLXB9 expression plasmids were transfected using Lipofectamine 2000 followed by RNA and protein isolation after 48 h.
Co-immunoprecipitation (Co-IP) and Mass Spectrometry
Large scale co-IP was performed by using the Profound Myc-tag IP/Co-IP kit (Pierce). Whole cell extract (WCE) was prepared in IP buffer (25 mm Tris, pH 7.6, 150 mm NaCl, 0.05% Tween 20, and EDTA-free protease inhibitor mixture (Roche Applied Science)) from MIN6-4N cells transfected with mh-Vector, mh-HB9-WT, or mh-HB9-AA. WCE (2 mg) were mixed with anti-myc-tag agarose beads and rotated end-to-end overnight at 4 °C. The beads were washed in IP buffer three times to remove unbound proteins using spin columns and centrifugation. Bound proteins were eluted in low pH elution buffer, pH 2.8, and the eluates were immediately neutralized with 1 m Tris-Cl, pH 9.5. Proteins in the eluates were separated on 4–20% SDS-PAGE and detected with a silver staining kit (Pierce). Protein bands were carefully excised followed by mass spectrometry and protein identification (Taplin Mass Spectrometry Facility at Harvard). Proteins uniquely found to co-IP with mh-HB9-WT and not with the phospho-dead mutant mh-HB9-AA were used for further analysis. Conventional IP and co-IP assays were performed as described (31) using the WCE of untransfected or transfected MIN6-4N cells with the appropriate antibodies. Proteins after IP or co-IP were detected by Western blot.
Western Blot Analysis
WCE was prepared in IP buffer. A subcellular protein fractionation kit (Pierce) was used to prepare cytoplasmic extract (CE), nuclear soluble extract (NE), and chromatin-bound/pellet extract (CB/PE). Protein extracts were subjected to SDS/PAGE and Western blot with the indicated antibodies. Western blots were performed as per standard protocols for detection by enhanced chemiluminescence (Pierce).
Chromatin Immunoprecipitation (ChIP) and ChIP-Sequencing (ChIP-Seq)
For chromatin preparation, cells were cross-linked with 1% formaldehyde (Sigma) for 15 min at room temperature, quenched with 2.5 mm glycine (Sigma), and washed twice with 1× DPBS. Cross-linked cells were processed for chromatin lysates and sonication (Diagenode Biorupter) to obtain 200–300-bp DNA fragments. ChIP was performed using specific antibodies, with the reagents and protocol from a ChIP assay kit (Millipore).
For ChIP-Seq, chromatin from MIN6-4N cells (40 × 106) transfected with mh-HB9-WT was immunoprecipitated with the phospho-HLXB9 antibody (anti-HB9-PO4) (10). Purified input DNA and anti-HB9-PO4-ChIP DNA were used to prepare ChIP-Seq libraries (Illumina), and the two libraries were sequenced for 51 bp (Illumina, NIDDK Genomics Core). Sequencing produced 156,970,757 tags for the input DNA library and 128,377,001 tags for the anti-HB9-PO4 ChIP library. Further analysis of the library was performed at Genomatix Inc. The “general annotation and statistics for genomic regions” tool (Genomatix) was used to map the tags to the mouse genome (mm9). All the tags mapping to promoter regions were filtered using the following parameters: >5-fold enrichment, <5% false discovery rate, and >100 tags, which resulted in 63 unique regions overlapping with the promoter of total 61 genes. Pathway analysis of these 61 genes using Genomatix software placed 49 genes in some pathways. Ten genes were located in cancer pathways (apoptosis, cell cycle, and genome stability). Chromatin from independent preps of untransfected or mh-HB9-WT-transfected MIN6-4N cells was used to validate the occupancy of phospho-HLXB9 at these regions by ChIP-PCR. Identification of a common DNA sequence motif in the anti-HB9-PO4 ChIP-Seq data set was performed using Genomatix software.
RNA Isolation and Quantitative Real-time PCR
Total RNA was isolated using the RNeasy kit (Qiagen) and treated with DNase I (Ambion). For RNA/cDNA reverse transcriptase PCR (RT-PCR), first strand cDNA was synthesized using an oligo-dT primer and Superscript-III (Invitrogen) and then subjected to quantitative real-time PCR with gene-specific primers. Gapdh was used as the internal control. Quantitative real-time PCR for first strand cDNA and for ChIP DNA were performed with the Brilliant SYBR Green quantitative real-time PCR Master Mix (Agilent) and Mx3000p thermal cycler (Agilent) followed by data analysis as per the manufacturer.
ChIP-PCR primer pairs (mouse) used were as follows: Arid1b (CGGAGTGGTGAATCATAAGCAA and CAAAAGCACTCGGTCTCTCTCA), Cblb (GCTAGCCTAGGTCCATTTCCCAAC and CGCCGGAGGAGGAGACCACTCAC), E2f5 (GAACACTAAAGGCAGGTAAGGAAA and TGATCAGGTGCAAGTATTGTAAGG), Igf2bp2 (TTCAGTAGGTGAAGTCGAGGAGAT and CTAGACCCTTCTAGTACCCCCTTT), Itgam (ACCTGCTAGCATGAACCTGAAT and CAGTGGGCGTCCCAAACAT), Kpna1 (TCATCAGACCCCAGAAAAGG and CCTTCTGCGCTTGTCTAACC), Polr3e (CTTTCGGAAGAGTGGGAAGG and CCACTCTGCTTAAACACAGATCAA), Tbx1 (CACAGAACGCACGTGGACAG and GTCAAGGCTCCGGTGAAGAAG), Top3b (CCGGAAGTGAGTTCGTGAGTG and AGGGCTGGATGGCGAAAAAG), Ub2z (ACTTAAGGCCCCCGTCAGTAG and TACTTCGGATTAAGCGGTGAGC).
RNA/cDNA RT-PCR primer pairs (mouse) used were as follows: rt-Arid1b (AAAGACCATCGAGTCTACCAGACC and AGAGCCAACAGGAGAAGGTGAC), rt-Cblb (GAGCTTTTTGCACGGACTAAGATT and TTACCACTTTGTCCATGAGTTTCC), rt-E2f5 (ACCATGGCTGCTCAAAACCT and CATCTGCTGGGGTAGGAGAAAG), rt-Igf2bp2 (TAATCCCAAAGAAGAAGTGAAGC and CTGTACAATTTCCCTGATCTTGC), rt-Itgam (GTGTGACCTTCCATCCTTCAAC and TTAGACCTCACATACGACTCCTG), rt-Kpna1 (TGCAGTTATTTTCAAAGCAAAACC and TCCATCTGACAGGTATGAGAGAGC), rt-Polr3e (CAAGTCACCCAGTGAATACCTCAT and ACCCAGTTTCCTTGGACTAACATC), rt-Tbx1 (ATGTGGACCCTCGAAAAGACAG and GTCGCAATCCCGGAAGCCTTTG), rt-Top3b (CTCATGGTAGCAGAAAAGCCATC and TTGTACTTCCCCAGAAAATCCAGT), rt-Ub2z (GATGACTGAGAATCCCTACCACAA and CAGGCCACCTCATAGAAGTCATAA), rt-Gapdh (ATCACTGCCACCCAGAAGAC and CAACCTGGTCCTCAGTGTAG).
Promoter Luciferase Reporter Assays
MIN6-4N cells were transfected with the GLuc luciferase reporter plasmids (pEZX-PG02, pEZX-PG02-Arid1b, pEZX-PG02-Cblb, or pEZX-PG02-Cblb-SDM2) alone or together with different amounts of the mh-HB9-WT expressing plasmid. The pcDNA3.1-mh plasmid was used to equalize the total amount of transfected DNA. Cells were harvested 48 h post-transfection to make WCE for Western blot analysis, and the cell culture medium was collected to measure luciferase activity with a luminometer (Berthold) in relative luciferase units (RLU) by using the Secrete-Pair Gaussia Luciferase Assay kit (GeneCopoeia) followed by data analysis as per the manufacturer.
Immunofluorescence (IF) and Immunohistochemistry (IHC)
MIN6-4N cells were cultured in chamber slides followed by detection of endogenous phospho-HLXB9 and Nono proteins by double-IF as described (31). Mouse experiments were conducted under the guidelines of the National Institutes of Health animal care and use committee and under an approved animal study protocol (K070-MDB-12). Formalin-fixed paraffin-embedded pancreas tissue sections from pancreatic islet tumor-bearing18-month-old Men1+/− mice (32), 12-month-old Men1f/f (with germ-line homozygous floxed Men1), and 12-month-old pancreatic islet tumor-bearing RIP-Cre-Men1f/f mice (β-cell-specific Men1 knock-out; germ-line homozygous floxed Men1 with conditional expression of Cre-recombinase in β-cells from the Rat insulin promoter) (33) were stained for insulin using standard IF procedures. For IHC, formalin-fixed paraffin-embedded pancreas sections were processed as previously described (10, 17) for HB9, HB9-PO4, Cblb, and c-Met staining. After IF and IHC, images were captured on the BZ9000 microscope (Keyence).
Cell Proliferation, Migration, and Soft Agar Colony-forming Assays
For assessing proliferation, immediately after nucleofection 10,000 cells were plated in triplicate in three 96-well plates and cultured for 144 h. Viable cells were assessed at 24, 96, and 144 h by the MTT assay (Promega).
Cell migration/invasion assay was performed with the CytoSelect 24-well Cell Invasion Assay kit (Cell Biolabs) using polycarbonate membrane trans-well inserts (8-μm pore size) (Basement Membrane, Colorimetric Format). After culturing for 2 days of post-nucleofection, cells were counted and seeded in complete DMEM at 30,000 cells per trans-well insert in duplicate for 48 h. Cells that invaded the membrane inserts were stained with crystal violet, treated with an extracting solution, and optical density of the solution was measured at 560 nm (Molecular Devices) to measure the relative number of migrating/invasive cells.
Soft agar colony formation assays were performed in duplicate. Immediately after nucleofection, 10,000 cells were resuspended in 3 ml of molten 0.33% agarose (made in complete DMEM) and poured into a 60-mm dish containing a 5-ml solidified layer of 0.66% agarose (made in complete DMEM). Dishes were incubated for 6 weeks and observed for colony formation. Images were captured on the Axiovert 40CFL microscope (Zeiss). All assays were performed as at least three independent experiments at separate times.
Statistics
Data from at least three independent experiments were considered and plotted as the mean ± S.D. Differences between groups were compared by Student's t tests. A p value <0.05 was considered significant (indicated by an * in the graphs).
Results
Identification of Nono as a Phospho-HLXB9 Interacting Protein
To examine the potential pro-oncogenic role of the phosphorylated isoform of HLXB9 (phospho-HLXB9), we used a subtractive proteomic approach to identify HLXB9 interacting proteins specific for phospho-HLXB9. WCE were prepared from mouse insulinoma MIN6 β-cells (MIN6-4N) transfected with empty vector or plasmids expressing normal myc-His-tagged HLXB9 (mh-HB9-WT) or its phospho-dead mutant (mh-HB9-AA, where serine 78 and serine -80 were substituted by alanine). Western blot analysis with an antibody against the myc-tag or a phospho-HLXB9-specific antibody showed that the top band of the doublet detected for mh-HB9-WT was absent in mh-HB9-AA (Fig. 1A). Analysis of proteins obtained after anti-myc-tag co-IP from mh-HB9-WT and mh-HB9-AA transfected WCE by silver staining also showed the same distinct double and single band, respectively (Fig. 1B). These results indicated that cells transfected with mh-HB9-WT expressed both the phospho isoform of HLXB9 and the unphosphorylated isoform of HLXB9, whereas cells transfected with mh-HB9-AA expressed only the unphosphorylated (phospho-dead) isoform of HLXB9. Bands in the silver-stained gel were excised and subjected to mass spectrometry analysis to identify proteins co-precipitating with mh-HB9-WT and mh-HB9-AA. First we prepared two lists of those proteins that were represented by five or more unique peptides present in the bands from each of the mh-HB9-WT and the mh-HB9-AA co-IP. Then we looked in either co-IP for the presence of four or less peptides belonging to any of these proteins and added that information into the two lists. Finally, both lists were compared to identify candidate proteins for further analysis that were uniquely present in the mh-HB9-WT co-IP that would correspond to binding partners of phospho-HLXB9 (Fig. 1C). There was only one such protein, Nono (Non-POU domain-containing octamer-binding protein), also known as p54nrb (54-kDa nuclear RNA-binding protein). In vivo interaction of HLXB9 with Nono was confirmed by co-IP and Western blot analysis of WCE prepared from MIN6-4N cells transfected with mh-HB9-WT (Fig. 1D) and also from untransfected cells (Fig. 1E). Thus, these data demonstrate that endogenous Nono can interact with both transfected and endogenous phospho-HLXB9.
FIGURE 1.

Identification of Nono as a phospho-HLXB9 interacting protein. A, overexpression of HLXB9 shows both its phosphorylated and unphosphorylated isoform. WCE were prepared from MIN6-4N cells transfected with myc-His-tag empty vector (mh-Vector) or plasmids expressing myc-his-tagged HLXB9 (mh-HB9-WT) or the phospho-dead mutant of HLXB9 with alanine substitution at Ser-78 and Ser-80 (mh-HB9-AA). WCE were run on the same gel to generate two Western blots to probe with anti-myc-tag or anti-HB9-PO4 (phospho-HLXB9 antibody). To analyze the bands, the blots were placed side-by-side (indicated by the dotted line). The top band of the doublet in the lane marked mh-HB9-WT corresponds to phospho-HLXB9 because it is not detected with the myc-tag antibody in mh-HB9-AA, and it is detected specifically with anti-HB9-PO4. The bottom band of the doublet corresponds to the unphosphorylated isoform of HLXB9 (HB9-unPO4) because it is not detected with anti-HB9-PO4. β-Actin was used as the loading control. B, large scale co-IP shows the two isoforms of HLXB9 and co-immunoprecipitating proteins. Silver-stained gel of proteins separated on SDS-PAGE after large scale co-IP with a myc-tag antibody using WCE prepared in A. As also seen by Western blot analysis in A, the bands marked SDMS1 and SDMS3 show the doublet in mh-HB9-WT Co-IP (PO4-HB9 and unPO4-HB9) and a single band in mh-HB9-AA corresponding to unPO4-HB9. Bands marked SDMS1, SDMS2, SDMS3, and SDMS4 (that were absent in the mh-Vector lane) were excised from the gel and subjected to mass spectrometry analysis. C, Nono uniquely co-immunoprecipitates with phospho-HLXB9. The number of peptides (total and unique) in the bands excised from the gel shown in B and their corresponding proteins is shown. Several proteins were present in both the mh-HB9-WT and mh-HB9-AA immunoprecipitates. The protein Nono emerged as a phospho-HLXB9-specific partner found only in the co-IP of mh-HB9-WT and not in the co-IP of the phospho-dead mutant of HLXB9 (mh-HB9-AA). D, overexpressed HLXB9 can Co-IP with endogenous Nono. Western blot (WB) probed with anti-myc-tag showing specific co-IP of HLXB9 with Nono using WCE of MIN6-4N cells transfected with mh-HB9-WT. Anti-HA-tag was used as a negative control. Higher exposure (top panel) and lower exposure (bottom panel) of the blot are shown to clearly visualize the input HLXB9 bands. IP, immunoprecipitation. E, endogenous phospho-HLXB9 can co-immunoprecipitate with endogenous Nono. Western blots probed with anti-Nono and anti-HB9 show specific co-IP of endogenous Nono with endogenous phospho-HLXB9 using WCE of MIN6-4N cells. An anti-HA-tag was used as a negative control.
HLXB9 Co-localizes with Nono in the Nucleus, and Overexpression of HLXB9 Decreases the Overexpression of Nono
Endogenous phospho-HLXB9 localizes in the nucleus, and Nono has been shown to localize in the nucleus in the paraspeckles (11, 26). To determine the co-localization of Nono and HLXB9, we analyzed the expression of these proteins in vivo by IF in MIN6-4N cells. IF detection of endogenous phospho-HLXB9 and Nono showed that, as expected, Nono localized to the nuclear paraspeckles (Fig. 2A, red); phospho-HLXB9 showed speckled staining that co-localized with Nono and also some brightly stained spots that did not co-localize with Nono (Fig. 2A, green and Merge). Western blot analysis of WCE prepared from MIN6-4N cells transfected with mh-HB9-WT, FLAG-Nono, or both together revealed that FLAG-Nono overexpression was detected when it was transfected alone; however, when co-transfected with HLXB9, the level of transfected FLAG-Nono was significantly reduced (Fig. 2B). Analysis of Western blots probed with anti-Nono showed that the expression of endogenous Nono was not affected upon HLXB9 overexpression or by knockdown of HLXB9 (Fig. 2, B and C). Therefore, overexpressed HLXB9 could suppress the overexpression of Nono.
FIGURE 2.
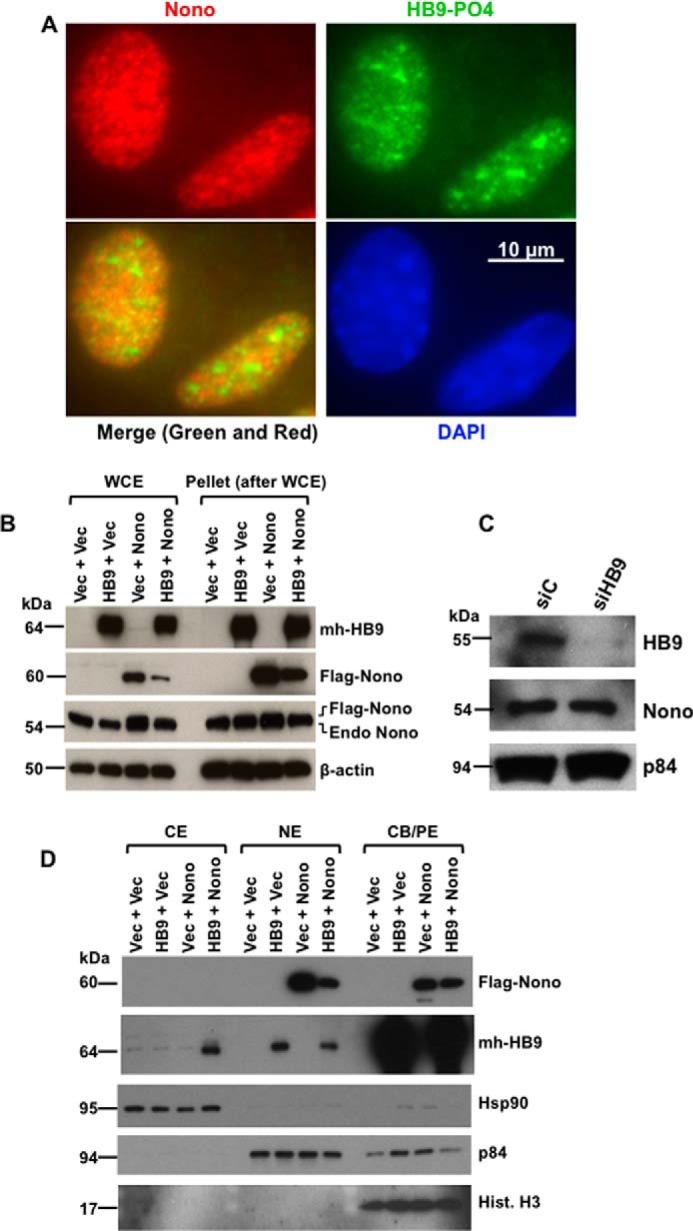
HLXB9 co-localizes with Nono in the nucleus, and co-overexpression of HLXB9 and Nono decreases the overexpression of Nono with translocation of HLXB9 into the cytoplasm. A, endogenous phospho-HLXB9 co-localizes with endogenous Nono in the nucleus. IF images of MIN6-4N cells show endogenous Nono (red) and phospho-HLXB9 (green). DAPI was used to detect the nuclei (blue). A merged image of the red and green IF shows co-localization of Nono and phospho-HLXB9 in subnuclear spots and some regions with phospho-HLXB9 (green) that did not co-localize with Nono. B, overexpression of HLXB9 decreases the level of overexpressed Nono protein. Western blots are shown of WCE and the pellet leftover after WCE preparation from MIN6-4N cells expressing mh-HB9-WT, FLAG-Nono, or both together. Empty vector DNA (Vec) was used to maintain the same amount of DNA in the transfections. The expression of transfected HLXB9 was detected with the anti-myc-tag; Nono was detected with the anti-FLAG-tag and with anti-Nono to detect both endogenous and transfected FLAG-tagged Nono. β-Actin was used as the loading control. Endogenous Nono levels were not affected by HLXB9 overexpression. However, the level of transfected FLAG-Nono was reduced upon HLXB9 overexpression. A similar pattern of bands was seen in the pellet leftover after WCE preparation, indicating that the reduced level of Nono upon HLXB9 overexpression was not due to differential cell lysis in the WCE preparation. C, HLXB9 did not reduce the expression of endogenous Nono protein. Western blot analysis to detect endogenous HLXB9 and Nono using WCE prepared from MIN6-4N cells transfected with control siRNA (siC) or HLXB9 siRNA (siHB9) is shown. p84 was used as the loading control. HLXB9 was significantly knocked down, but that did not affect the level of endogenous Nono. D, co-overexpression of HLXB9 and Nono reduces the level of Nono protein in the nucleus with translocation of HLXB9 to the cytoplasm. Shown is Western blot analysis of subcellular fractionation of CE, NE, and CB/PE from MIN6-4N cells expressing mh-HB9-WT, FLAG-Nono, or both together. Empty vector DNA (Vec) was used to maintain the same amount of DNA in the transfections. The expression of transfected HLXB9 was detected with the anti-myc-tag; Nono was detected with the anti-FLAG-tag; detection of marker proteins (Hsp90 for CE, p84 for NE, and histone H3 for CB/PE) showed minimal cross-contamination of the fractions and also served as loading controls for each fraction. Overexpressed Nono was found in the NE and in the CB/PE, but its level in NE was reduced by HLXB9 overexpression. Overexpressed HLXB9 was mostly located in the CB/PE and with a significant amount in the nucleus, but it was also detected in the CE by Nono overexpression.
To determine whether co-overexpression of these two proteins affected their subcellular localization, Western blots were performed using CE, NE, and CB/PE) (Fig. 2D). Nono was exclusively detected in the NE with a small fraction in the CB/PE. A small fraction of HLXB9 was detected in the NE, and a vast majority of HLXB9 was detected in the CB/PE, underscoring its function as a transcriptional regulator. As observed in Western blots of WCE, the level of transfected Nono was significantly reduced when Nono and HLXB9 were co-transfected, but a small fraction of transfected HLXB9 was also detected in the CE. Thus, overexpressed HLXB9 could suppress the overexpression of Nono in the nucleus. Taken together, these data indicate that co-overexpression of HLXB9 and Nono leads to translocation of HLXB9 into the cytoplasm and reduction of transfected Nono expression. These findings also suggest that the interaction and co-localization of Nono and phospho-HLXB9 occurs exclusively in the nucleus.
Identification of Cblb as a Phospho-HLXB9 Target Gene
Given that HLXB9 is a homeodomain-containing transcription factor, examining the direct target genes of phospho-HLXB9 could help explain its pro-oncogenic potential. Therefore, we performed chromatin immunoprecipitation followed by deep sequencing (ChIP-Seq) using chromatin isolated from mh-HB9-WT transfected MIN6-4N cells. Western blot analysis of anti-HB9-PO4 immunoprecipitates showed the specificity of this antibody in isolating phospho-HLXB9 (Fig. 3A) (top band of the doublet from mh-HB9-WT expression). Sequencing of the input and anti-HB9-PO4 ChIP-Seq library showed that after normalization with the input library, the anti-HB9-PO4 ChIP yielded 83676 tags. Mapping of the tags to the mouse genome showed that 20% of the tags were located near promoter regions and, therefore, were considered for further analysis (Fig. 3B). The promoter region ChIP-Seq tags mapped to 63 unique regions from 61 genes. Of these, 10 genes were located in pathways associated with cancer (apoptosis, cell cycle, and genome stability). Occupancy of phospho-HLXB9 at the promoter of these 10 genes was validated by ChIP-PCR assays showing the highest occupancy (>0.01% of input) at two genes, Arid1b (AT-rich interactive domain-containing protein 1B) and Cblb (Casitas B-lineage lymphoma b) in cells overexpressing HLXB9 (Fig. 3C). However, endogenous anti-HB9-PO4 ChIP showed the highest occupancy (>0.01% of input) only at Cblb (Fig. 3D).
FIGURE 3.

Identification of Cblb as a phospho-HLXB9 target gene. A, the anti-HB9-PO4 antibody specifically recognizes the phosphorylated isoform of HLXB9. WCE and chromatin were prepared from MIN6-4N cells transfected with a plasmid expressing myc-his-tagged HLXB9 (mh-HB9-WT). WCE was used IP with rabbit antibodies anti-myc-tag or anti-HB9-PO4 and detected by Western blot (WB) with mouse anti-myc-tag. Rabbit anti-HA-tag was used as the negative control. The input WCE and anti-myc-tag IP display a doublet corresponding to phospho-HLXB9 and unphosphorylated HLXB9. Anti-HB9-PO4 could specifically immunoprecipitate phospho-HLXB9 corresponding to the top band of the doublet. B, significant enrichment of promoter regions among the anti-HB9-PO4 ChIP-Seq tags. Chromatin prepared from MIN6-4N cells transfected in A was used for ChIP with anti-HB9-PO4. DNA obtained before and after ChIP was used for preparing libraries followed by deep sequencing (ChIP-Seq) and mapping of the anti-HB9-PO4-specific ChIP-Seq tags to the mouse genome. The pie chart shows the percent distribution of tags in the mouse genome (a typical input library, Genomatix) and in the anti-HB9-PO4 ChIP-Seq at the indicated regions; 20% of the anti-HB9-PO4 ChIP-Seq tags were located near promoter regions and selected for further analysis. C, phospho-HLXB9 occupancy is highest at the Arid1b and Cblb gene in cells overexpressing HLXB9. ChIP-quantitative PCR assay for validating the 10 phospho-HLXB9 targets is shown as the percent of input chromatin DNA PCR for each primer pair. Chromatin prepared from MIN6-4N cells expressing mh-HB9-WT was used for anti-HB9-PO4 ChIP. Also shown is a Western blot confirming overexpression of HLXB9 (myc-tag antibody) and β-actin as the loading control. D, endogenous phospho-HLXB9 occupancy is highest at the Cblb gene. ChIP-quantitative PCR assay of the 10 phospho-HLXB9 targets is shown as percent of input chromatin DNA PCR for each primer pair. Chromatin prepared from MIN6-4N cells was used for endogenous anti-HB9-PO4 ChIP. Endogenous phospho-HLXB9 occupancy was only detected at Cblb. E and F, H3K4me3 at Cblb unaffected but reduced H3K27me3 upon HLXB9 knockdown. ChIP-quantitative PCR assay of the 10 phospho-HLXB9 targets is shown as the percent of input chromatin DNA PCR for each primer pair. Chromatin prepared from MIN6-4N cells transfected with control siRNA (siC) or HLXB9 siRNA (siHB9) was used for endogenous anti-H3K4me3 ChIP (E) or H3K27me3 ChIP (F). Also shown is a Western blot confirming knockdown of HLXB9 (HLXB9 antibody) and β-actin as the loading control. In siC versus siHB9, reciprocal H3K4me3 or H3K27me3 at only Cblb was HLXB9 binding-dependent because endogenous phospho-HLXB9 was only found to occupy Cblb (D).
To determine whether phospho-HLXB9 occupancy in chromatin co-related with gene activation or repression, histone marks corresponding with active (H3K4me3) or silenced (H3K27me3) chromatin were examined at the 10 HLXB9 target genes using chromatin from cells expressing a normal level of HLXB9 or upon HLXB9 knockdown (Fig. 3, E and F). Knockdown of HLXB9 showed increased H3K4me3 at two genes (Arid1b and Polr3e), reduced H3K4me3 at one gene (Ub2z), and H3K4me3 unchanged at 2 genes (Cblb and Tbx1) and not detected at 4 genes (E2f5, Igfbp2, Itgam, and Kpna1). A significant level of H3K27me3 was observed at only two genes (Cblb and Top3b), and at both genes HLXB9 knockdown showed significantly less H3K27me3. Because occupancy of endogenous phospho-HLXB9 was observed at only Cblb (Fig. 3D), we deduced that the changes in histone marks at the other genes were HLXB9 binding-independent. Therefore, reduced HLXB9 occupancy at the Cblb promoter showed a reduction in the repressive chromatin mark. Altogether, the ChIP-Seq and ChIP-PCR analyses show that Cblb is a phospho-HLXB9 target gene, and HLXB9 occupancy at the Cblb promoter coincides with regulation of a repressive chromatin mark.
HLXB9 Suppresses the Expression of Cblb mRNA by Suppressing Cblb Promoter Activity
To determine the effect of HLXB9 on the expression of the target genes identified by ChIP-Seq, RT-PCR was performed using RNA isolated from cells with HLXB9 knockdown or overexpression (Fig. 4A). The relative mRNA level of only two genes was affected upon HLXB9 knockdown or overexpression (Arid1b and Cblb) (Fig. 4B). The expression of Arid1b was unaffected by HLXB9 knockdown and reduced upon HLXB9 overexpression. The expression of Cblb was increased upon HLXB9 knockdown and reduced by HLXB9 overexpression.
FIGURE 4.

HLXB9 suppresses the expression of Cblb mRNA by suppressing Cblb promoter activity. A and B, among targets identified by anti-HB9-PO4 ChIP-Seq, the expression of only Arid1b and Cblb was affected by HLXB9. Quantitative RT-PCR is shown of the indicated genes using RNA prepared from MIN6-4N cells transfected with control siRNA (siC) or HLXB9 siRNA (siHB9), empty vector, or mh-HB9-WT. A, Western blot confirming knockdown or overexpression of HLXB9 with anti-HLXB9 or anti-myc-tag, respectively, and β-actin as the loading control. B, HLXB9 knockdown or overexpression regulated the relative mRNA level of only two genes (Arid1b and Cblb). Both were reduced upon HLXB9 overexpression, but only Cblb was increased upon HLXB9 knockdown. Error bar = mean and S.D. from three experiments. * = p < 0.05. C and D, Nono did not regulate the expression of HLXB9 target genes. Shown is quantitative RT-PCR of the indicated genes using RNA prepared from MIN6-4N cells transfected with control shRNA (shC) or Nono shRNA (shNono), FLAG-vector, or FLAG-Nono. A, Western blot confirming knockdown or overexpression of Nono with anti-Nono or anti-FLAG-tag, respectively, and β-actin as the loading control. B, Nono knockdown or overexpression did not regulate the relative mRNA level of any gene. Error bar = mean and S.D. from three experiments. E and F, Cblb promoter activity is suppressed by HLXB9. The promoter region of Cblb (−1078 to +219) located near the sequence identified by anti-HB9-PO4 ChIP-Seq was cloned in the promoter-less luciferase reporter vector PG02 (GeneCopoeia) and analyzed for promoter activity in MIN6-4N cells. RLU for each of the transfections are shown. Compared with the empty vector PG02, the PG02-Cblb plasmid showed significantly high RLU, and co-expression of increasing amounts of HLXB9 suppressed the Cblb promoter activity. Error bar = mean and S.D. from three experiments. * = p < 0.05. A representative Western blot shows the expression of HLXB9 (with anti-myc-tag) in the MIN6-4N cells analyzed for luciferase activity. p84 was used as the loading control. G and H, Cblb expression is down-regulated by the phosphorylated isoform of HLXB9. WCE and RNA were prepared from MIN6-4N cells transfected with control-shRNA (C−) or Men1-shRNA (M−) together with empty vector or mh-HB9-WT. G, Western blot shows the extent of menin knockdown (anti-menin blot) and transfected HLXB9 and increased phospho-HLXB9 in M− (anti-myc-tag blot, top band of the doublet band). H, quantitative RT-PCR shows significantly reduced Cblb mRNA upon menin knockdown in mh-HB9-WT-transfected cells. Error bar = mean and S.D. of a representative experiment performed in triplicate. * = p < 0.05.
The phospho-HLXB9 interacting protein Nono has been shown to participate in transcriptional regulation (34). Therefore, to determine whether the HLXB9 and Nono interaction was relevant at HLXB9 target genes, RT-PCR was performed using RNA isolated from cells with Nono knockdown or overexpression (Fig. 4C). The relative mRNA level of all 10 genes was unaffected upon Nono knockdown or overexpression, indicating that these genes are not regulated by the HLXB9-Nono interaction (Fig. 4D). Also, Nono occupancy was not detected at the Cblb promoter by ChIP-PCR (data not shown).
We next looked at the ability of HLXB9 to directly regulate the promoter activity of Arid1b and Cblb. Promoter analysis of Arid1b showed that the Arid1b region identified by ChIP-Seq was not located in the promoter because no significant activity was detected in promoter-reporter assays (data not shown). It is possible that the annotation of the Arid1b promoter was flawed in the mouse genome database. Because the HLXB9-occupied Arid1b region was not at the promoter, for further study we focused only on Cblb. The Cblb region identified by ChIP-Seq showed significant promoter activity that was significantly suppressed by HLXB9 (Fig. 4, E and F).
To determine whether Cblb was targeted specifically by phospho-HLXB9, we examined the effect of HLXB9 on Cblb mRNA expression in a menin knockdown background where the level of phospho-HLXB9 is up-regulated (Fig. 4G) (top band of the doublet from mh-HB9-WT expression). The level of Cblb mRNA was further reduced by HLXB9 in a menin knockdown background (Fig. 4H). These data indicate that phospho-HLXB9 down-regulates the expression of Cblb by directly suppressing the promoter activity of Cblb.
HLXB9 Binding Motif in the Cblb Promoter
To determine whether the regions identified by anti-HB9-PO4 ChIP-Seq contained a common DNA sequence motif, the promoter region tag sequences were subjected to de novo motif analysis, and a core sequence ATTTTA was identified that resembled homeodomain binding consensus (Fig. 5A), which is probably bound by the C-terminal homeodomain region of HLXB9 (35). A similar motif (ATTTA) was observed in the Cblb promoter region at −735 and at −722 nucleotides upstream of the transcriptional start site (Fig. 5B). Mutation of both sites resulted in loss of suppression of Cblb promoter activity by HLXB9 (Fig. 5, C and D). Therefore, the ATTTTA or ATTTA motif could serve as an HLXB9-responsive element to control promoter activity.
FIGURE 5.
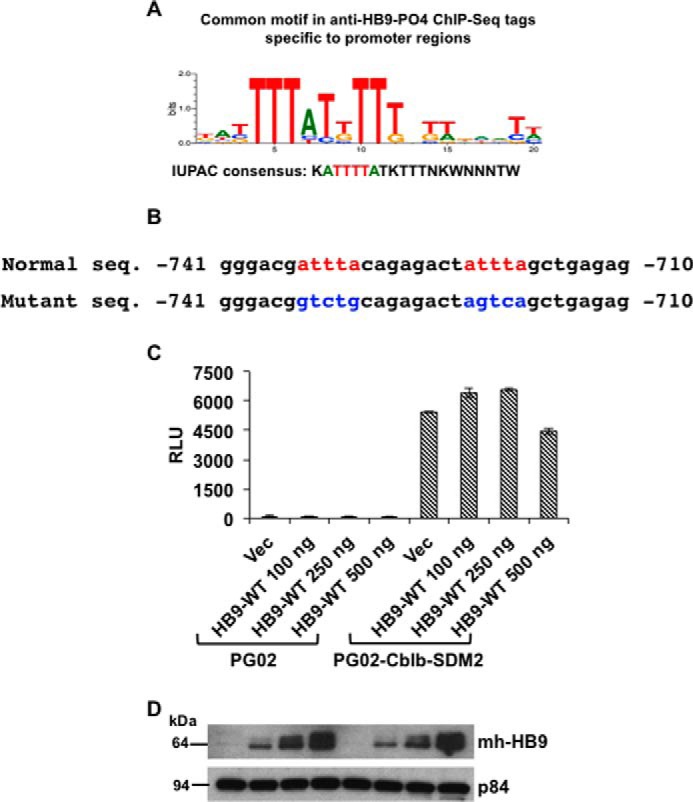
HLXB9 binding motif in the Cblb promoter. A, consensus motif in anti-HB9-PO4 ChIP-Seq tags located at promoter regions. De novo motif analysis from anti-HB9-PO4 ChIP-Seq tag sequences located at promoter regions was performed using Genomatix software. A core sequence ATTTTA was identified that resembles homeodomain-binding consensus (35). B, sequence of the HLXB9 binding motifs in the Cblb promoter. The top line shows the putative HLXB9 binding motifs (red) in the DNA sequence of the Cblb promoter (−741 to −710 region from the transcriptional start site is shown). The bottom line shows nucleotide substitutions (blue) to mutate the motifs by site-directed mutagenesis in the Cblb-promoter construct used in C. C and D, HLXB9 did not suppress the activity of the Cblb promoter containing mutations at the HLXB9 binding motifs. The putative HLXB9 binding motifs shown in B were mutated by site-directed mutagenesis of the PG02-Cblb promoter construct and analyzed for promoter activity in MIN6-4N cells. RLU for each of the transfections are shown. Compared with the empty vector PG02, the PG02-Cblb-SDM2 plasmid showed significantly high RLU and co-expression of increasing amounts of HLXB9 did not suppress the Cblb promoter activity. Error bar = Mean and S.D. from 3 experiments, * = p < 0.05. A representative Western blot shows expression of HLXB9 (with anti-myc-tag) in the MIN6-4N cells analyzed for luciferase activity. p84 was used as the loading control.
HLXB9 Down-regulation Inactivates the Oncogenic c-Met Pathway
Cblb has been shown to target the oncogenic receptor-tyrosine kinase c-Met for ubiquitination and degradation (23). Therefore, we examined whether modulation of endogenous Cblb by HLXB9 would affect the oncogenic c-Met pathway. As expected, overexpression of Cblb in MIN6-4N cells significantly reduced c-Met protein (Fig. 6A). Similarly, knockdown of HLXB9 increased the expression of Cblb protein with a reduction in c-Met protein (Fig. 6A). Cell proliferation was slightly reduced upon Cblb overexpression, but cell proliferation was unaffected upon HLXB9 knockdown (Fig. 6B). Overexpression of Cblb or knockdown of HLXB9, both, resulted in significantly reduced migration of cells but did not affect the ability of cells to form colonies in soft agar (Fig. 6, C and D). Down-regulation of endogenous Nono did not affect cell proliferation, cell migration, or soft agar colony formation (Fig. 6), indicating that the interaction of HLXB9 with Nono to control growth or migration may require additional partners or regulatory events. These data demonstrate that HLXB9, through its control of Cblb expression, is capable of regulating c-Met and its downstream oncogenic effect on cell migration.
FIGURE 6.

Cblb overexpression or HLXB9 knockdown inactivates the oncogenic c-Met pathway. A, overexpression of Cblb or knockdown of HLXB9 decreases c-Met levels. Western blot analysis of the indicated proteins using WCE prepared from MIN6-4N cells transfected (by nucleofection) with empty vector or Cblb expression plasmid, control siRNA (siC), or HLXB9 siRNA (siHB9) and control shRNA (shC) or Nono shRNA (shNono). Cblb overexpression reduced the level of endogenous c-Met. HLXB9 knockdown increased the level of endogenous Cblb and reduced the level of endogenous c-Met. p84 was used as the loading control. The upper band marked pro-c-Met is the glycosylated c-Met precursor form that is cleaved and processed into mature c-Met (lower band) (44). B, reduced cell proliferation from Cblb overexpression but not from HLXB9 or Nono knockdown. An MTT assay assessed cell proliferation of MIN6-4N cells transfected in A. Overexpression of Cblb caused a slight but significant reduction in cell proliferation, but cell proliferation was unaffected upon HLXB9 or Nono knockdown. Note that the Western blots in A were performed using WCE prepared at 96 h post-transfection. Error bar = mean and S.D. from 3 experiments, * = p < 0.05. vec, vector. C, reduced cell migration from Cblb overexpression or HLXB9 knockdown but not from Nono knockdown. Cell migration assay of MIN6-4N cells transfected in A assessed by staining for cells that migrate across a polycarbonate membrane in a Boyden chamber. Stain from the cells was extracted and measured at 560 nm as an index of cell migration. Cblb overexpression or HLXB9 knockdown significantly reduced cell migration. Error bar = mean and S.D. from three experiments. * = p < 0.05. D, soft agar colony formation is unaffected from Cblb overexpression, HLXB9 knockdown, or Nono knockdown. Bright-field microscopy images of colonies (white dots) formed in soft agar by MIN6-4N cells transfected in A. MIN6-4N cells make very small colonies after 4–6 weeks. The number of days to form colonies or the number and size of the colonies was unaffected by Cblb overexpression, HLXB9 knockdown, or Nono knockdown.
Reciprocal Co-relation of Cblb and c-Met Expression in Insulinomas
Next, we further studied the physiological relevance of the pro-oncogenic features of phospho-HLXB9 related to Cblb regulation. Two different mouse models of menin loss develop insulinomas, the conventional Men1+/− mice and the β-cell conditional Men1 knock-out mice (RIP-Cre-Men1f/f), generated and characterized by various groups (32, 33, 36–40). Tumor and corresponding normal islets from the two mouse models (32, 33) were examined for insulin by IF and for HLXB9, phospho-HLXB9, Cblb, and c-Met by IHC. Compared with normal islets, the insulin-positive islet tumor from both mouse models showed increased staining for HLXB9, phospho-HLXB9, and c-Met but almost no or low staining for Cblb (Figs. 7 and 8). This reciprocal expression of Cblb and c-Met demonstrates the importance of elevated c-Met signaling in insulinomas as a potential target for therapy.
FIGURE 7.
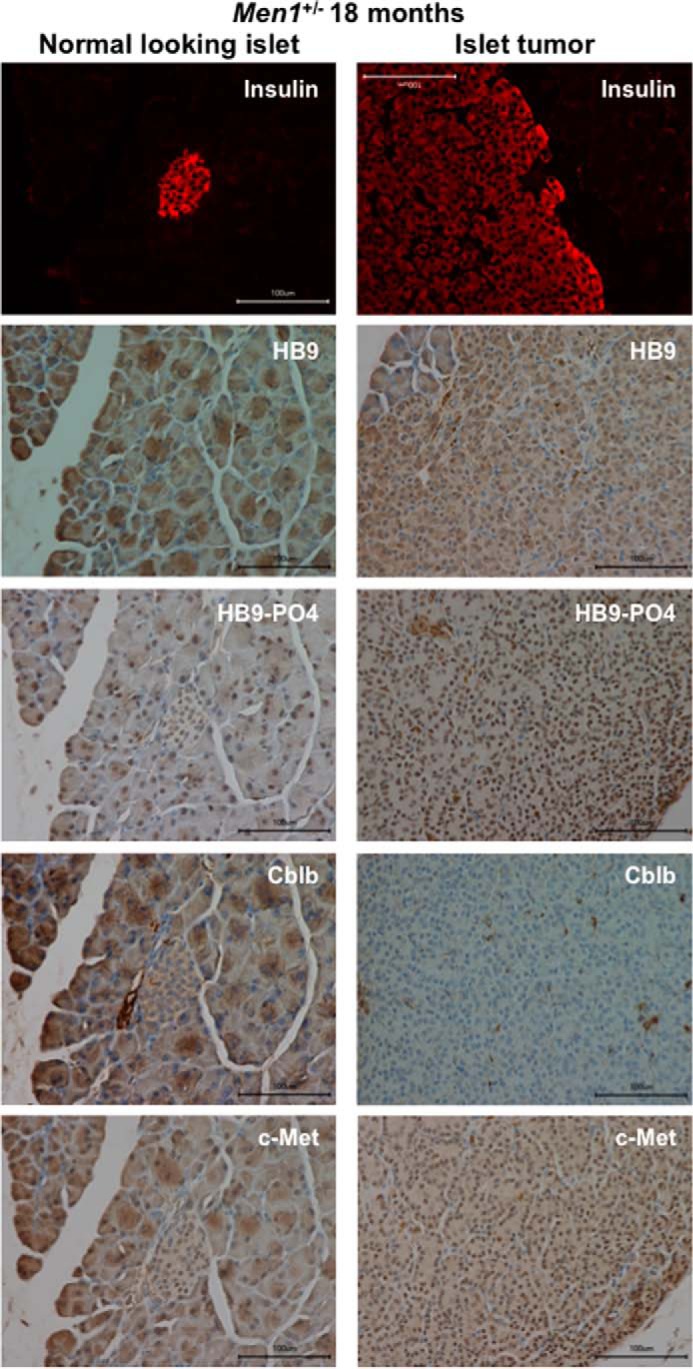
Increased phospho-HLXB9, decreased Cblb, and increased c-Met in insulinoma from the conventional mouse model of menin loss (Men1+/−). Shown are images of immunofluorescence for insulin and IHC for the indicated proteins in the pancreas section of an 18-month-old Men1+/− mouse. Insulin staining shows the location of the normal-looking islet (panels on the left) and the large islet tumor that covers the entire viewing field (panels on the right). Compared with a normal-looking insulin-positive islet in the same section, the insulin-positive islet tumor shows increased nuclear staining for HB9 and HB9-PO4 and increased nuclear and cytoplasmic staining for c-Met but almost no staining for Cblb (cytoplasm).
FIGURE 8.
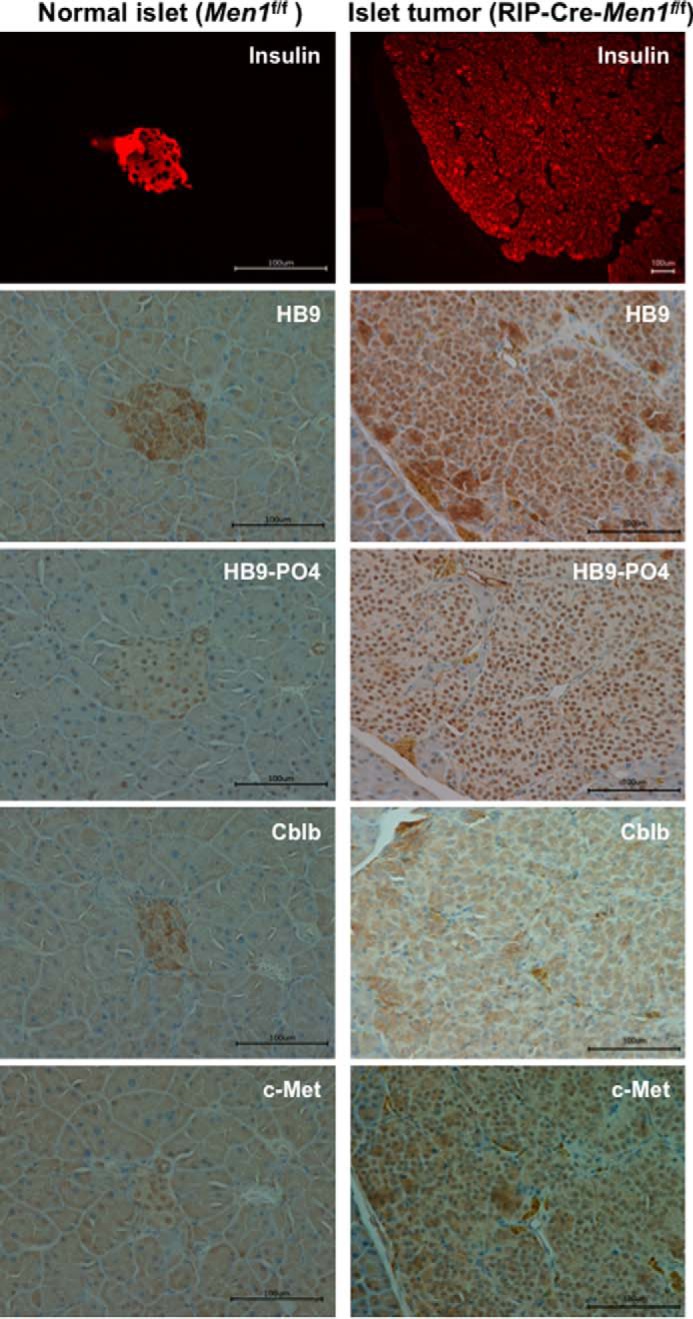
Increased phospho-HLXB9, decreased Cblb, and increased c-Met in insulinoma from the conditional mouse model of menin loss (RIP-Cre-Men1f/f). Images of immunofluorescence for insulin and IHC for the indicated proteins in pancreas sections from a 12-month-old Men1f/f mouse and RIP-Cre-Men1f/f mouse. Compared with the insulin-positive normal islet (panels on the left), the large insulin-positive islet tumor that covers the entire viewing field (panels on the right) shows increased nuclear staining for HB9 and HB9-PO4, increased nuclear and cytoplasmic staining for c-Met, and decreased staining for Cblb (cytoplasm).
Discussion
HLXB9 is a transcription factor that is essential for pancreatic islet β-cell differentiation (12–14). It is known to undergo phosphorylation at amino acid residues serine 78 and serine 80, and an increased level of the phospho isoform of HLXB9 is observed in tumors of the pancreatic islet β-cells (10, 41). Heretofore, the functional relevance of phospho-HLXB9 to tumorigenesis was unknown. In this study we provide evidence for the mechanism by which phospho-HLXB9 functions as a potential pro-oncogenic factor in insulin-secreting pancreatic islet β-cell tumors (insulinomas). Our study systematically identified phospho-HLXB9 binding partners in MIN6 insulinoma β-cells, and we demonstrate that it interacts with and localizes with a survival factor Nono in the nucleus. Co-overexpression of HLXB9 and Nono caused a reduction in the level of overexpressed Nono. However, reduction of endogenous HLXB9 did not affect the level of endogenous Nono. Although the overall level of transfected HLXB9 was unaffected upon Nono overexpression, HLXB9 translocated to the cytoplasm when co-overexpressed with Nono. It is possible that the nucleus maintains a critical level of both proteins such that in times of excess, the protein capable of surviving outside of the nucleus translocates to the cytoplasm, and the expression is reduced of the protein incapable of surviving outside the nucleus. Further studies are warranted to understand the interplay of HLXB9 and Nono in the nucleus and how it relates to tumorigenesis.
HLXB9 can cause apoptosis in mouse insulinoma MIN6 β-cells and neuronal cells (11, 42). However, HLXB9 does not cause apoptosis under conditions of menin deficiency, because in such cells the level of phospho-HLXB9 is up-regulated and in excess of its unphosphorylated isoform, indicating that phospho-HLXB9 is not pro-apoptotic (11). In the current study we found that overexpression of HLXB9 in mouse insulinoma MIN6 β-cells (containing normal endogenous menin), a condition that coincides with apoptosis, showed reduced level of transfected Nono protein. It has been previously noted that Nono is also down-regulated in melanoma cells undergoing apoptosis where Nono acts as a survival factor promoting a response to counteract apoptosis (24, 25, 43). Therefore, it appears that ectopic Nono expression is not tolerated in cells undergoing HLXB9-induced apoptosis. We identified Nono from a subtractive proteomic approach, displaying interaction with phospho-HLXB9 but not with the phospho-dead mutant of HLXB9. This raises the possibility that disrupting the HLXB9-Nono interaction such as by blocking HLXB9 phosphorylation or by a competitive peptide could lead to apoptosis in insulinoma cells. Further studies of the HLXB9-Nono interaction will be essential to determine which of the processes controlled by Nono are relevant targets in β-cells, and it will be of interest to identify the role of this interaction in other endocrine cell types.
Mutations or aberrant expression and activities of receptor-tyrosine kinases such as c-MET occur in various tumor types including breast, liver, lung, kidney, thyroid, and ovary serving as excellent targets for therapeutic inhibitors of c-MET (15). c-MET amplification and mutations have not been observed in the exome sequencing efforts of PNETs and insulinomas (5–7). However, increased c-MET level in human PNETs and insulinomas has been observed (16, 17). We previously showed in mouse insulinoma MIN6 β-cells that c-Met mRNA could be down-regulated by a menin target, the long non-coding RNA Meg3, and a commonly used c-Met inhibitor (PHA-665752) suppressed the growth and migration of mouse insulinoma MIN6 β-cells (17). In the present study we show that c-Met up-regulation in MIN6 cells is mediated indirectly by another menin target, phospho-HLXB9, through down-regulation of the c-Met inhibitor Cblb. In analyzing the occupancy of phospho-HLXB9 at promoter regions of genes associated with cancer pathways, we showed that it down-regulates the expression of the E3 ubiquitin ligase Cblb that is known to target the oncogenic protein c-Met for degradation. The oncogenic c-Met pathway was inactivated upon overexpression of Cblb or increased expression of Cblb upon knockdown of endogenous HLXB9, because both resulted in decreased c-Met protein and significantly reduced migration of mouse insulinoma MIN6 β-cells. Supporting these in vitro and in vivo observations, insulinomas that develop in mouse models of menin loss showed enhanced phospho-HLXB9, with less Cblb and more c-Met, underscoring the importance of the oncogenic c-Met pathway acting downstream of phopho-HLXB9. Further investigations to tie Cblb and c-Met with menin loss, by conducting experiments in menin knock-out β-cells and mouse models, would help to understand whether inhibition of the c-Met oncogenic pathway would suppress insulinoma tumorigenesis. Generation of normal and menin knock-out β-cells will be very useful for such investigations. These findings not only provide valuable insights into the pathobiology of insulinomas but also unravel the possible use of phospho-HLXB9 in β-cell expansion strategies to help alleviate β-cell loss in diabetes.
Although the individual contribution of the menin targets, Meg3 and HLXB9, in insulinoma pathogenesis is not known, their convergence on c-Met expression and signaling highlights the importance of the oncogenic c-Met pathway in insulinoma cells and justifies consideration as a potential therapeutic target. The participation of other menin targets and interacting proteins in c-Met regulation and the expression pattern of c-Met in other tumor types that display menin loss remains to be determined.
Cblb was identified as a direct target of phospho-HLXB9 by ChIP-Seq analysis, and RT-PCR analysis showed that the expression of Cblb mRNA was reduced by HLXB9. Reduced HLXB9 occupancy at the Cblb promoter coincided with a reduction in the repressive chromatin mark H3K27me3. Further studies directed at genome-wide co-occupancy of phospho-HLXB9 with active or repressive chromatin marks and histone modifications associated with enhancer regions such as H3K27Ac and H3K4me1 could shed light on how HLXB9 acts as a transcriptional repressor.
In conclusion, phospho-HLXB9 may promote tumorigenesis by interacting with a survival factor Nono and by activating the oncogenic c-Met pathway. Our studies elucidating the HLXB9-Nono interaction axis and HLXB9-mediated regulation of Cblb and c-Met have therapeutic implications for suppressing β-cell proliferation in insulinomas. Inhibiting the phosphorylation of HLXB9 or its interaction with Nono and/or modulating the expression of the direct (Cblb) and indirect target (c-Met) of HLXB9 offers opportunities for potential therapeutic intervention.
Author Contributions
S. S. D. and S. K. A. designed and performed the experiments, analyzed the data, and wrote the manuscript. V. I. P. carried out all the mouse-related work. S. S. K. and S. I. participated in data interpretation and manuscript writing. All authors discussed the data and the manuscript revisions.
Acknowledgments
We thank Dr. Peter Scacheri (Case Western Reserve University, Cleveland, OH) for Men1f/f and RIP-Cre- Men1f/f mouse tissue formalin-fixed paraffin-embedded blocks and the NIDDK, National Institutes of Health, genomics core facility (Harold Smith and Weiping Chen) for advice and help in generating ChIP-Seq data.
This work was supported, in whole or in part, by National Institutes of Health Grant 1ZIADK075035-03 (NIDDK; Intramural Research Program; to S. K. A.). The authors declare that they have no conflicts of interest with the contents of this article.
ChIP-Seq library sequencing data generated in this study have been deposited in the NCBI GEO repository under the accession number GSE61432.
- PNET
- pancreatic neuroendocrine tumor
- CBLB
- Casitas B-lineage lymphoma b
- WCE
- whole cell extract(s)
- CE
- cytoplasmic extract
- NE
- nuclear extract
- PE
- pellet extract
- CB/PE
- chromatin-bound/pellet extract
- ChIP-Seq
- ChIP sequencing
- RLU
- relative luciferase units
- IF
- immunofluorescence
- IHC
- immunohistochemistry
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- IP
- immunoprecipitation.
References
- 1. Thakker R. V. (2010) Multiple endocrine neoplasia type 1 (MEN1). Best Pract. Res. Clin. Endocrinol. Metab. 24, 355–370 [DOI] [PubMed] [Google Scholar]
- 2. Agarwal S. K. (2014) Exploring the tumors of multiple endocrine neoplasia type 1 in mouse models for basic and preclinical studies. Int. J. Endocr. Oncol. 1, 153–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shin J. J., Gorden P., Libutti S. K. (2010) Insulinoma: pathophysiology, localization and management. Future Oncol. 6, 229–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jiao Y., Shi C., Edil B. H., de Wilde R. F., Klimstra D. S., Maitra A., Schulick R. D., Tang L. H., Wolfgang C. L., Choti M. A., Velculescu V. E., Diaz L. A. Jr., Vogelstein B., Kinzler K. W., Hruban R. H., Papadopoulos N. (2011) DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 331, 1199–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cao Y., Gao Z., Li L., Jiang X., Shan A., Cai J., Peng Y., Li Y., Jiang X., Huang X., Wang J., Wei Q., Qin G., Zhao J., Jin X., Liu L., Li Y., Wang W., Wang J., Ning G. (2013) Whole exome sequencing of insulinoma reveals recurrent T372R mutations in YY1. Nat. Commun. 4, 2810. [DOI] [PubMed] [Google Scholar]
- 6. Cromer M. K., Choi M., Nelson-Williams C., Fonseca A. L., Kunstman J. W., Korah R. M., Overton J. D., Mane S., Kenney B., Malchoff C. D., Stalberg P., Akerström G., Westin G., Hellman P., Carling T., Björklund P., Lifton R. P. (2015) Neomorphic effects of recurrent somatic mutations in Yin Yang 1 in insulin-producing adenomas. Proc. Natl. Acad. Sci. U.S.A. 112, 4062–4067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lichtenauer U. D., Di Dalmazi G., Slater E. P., Wieland T., Kuebart A., Schmittfull A., Schwarzmayr T., Diener S., Wiese D., Thasler W. E., Reincke M., Meitinger T., Schott M., Fassnacht M., Bartsch D. K., Strom T. M., Beuschlein F. (2015) Frequency and clinical correlates of somatic Ying Yang 1 mutations in sporadic insulinomas. J. Clin. Endocrinol. Metab. 100, E776–E782 [DOI] [PubMed] [Google Scholar]
- 8. Hendy G. N., Kaji H., Canaff L. (2009) Cellular functions of menin. Adv Exp. Med. Biol. 668, 37–50 [DOI] [PubMed] [Google Scholar]
- 9. Shen H. C., He M., Powell A., Adem A., Lorang D., Heller C., Grover A. C., Ylaya K., Hewitt S. M., Marx S. J., Spiegel A. M., Libutti S. K. (2009) Recapitulation of pancreatic neuroendocrine tumors in human multiple endocrine neoplasia type I syndrome via Pdx1-directed inactivation of Men1. Cancer Res. 69, 1858–1866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Desai S. S., Modali S. D., Parekh V. I., Kebebew E., Agarwal S. K. (2014) GSK-3β protein phosphorylates and stabilizes HLXB9 protein in insulinoma cells to form a targetable mechanism of controlling insulinoma cell proliferation. J. Biol. Chem. 289, 5386–5398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shi K., Parekh V. I., Roy S., Desai S. S., Agarwal S. K. (2013) The embryonic transcription factor Hlxb9 is a menin interacting partner that controls pancreatic β-cell proliferation and the expression of insulin regulators. Endocr. Relat. Cancer 20, 111–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Conrad E., Stein R., Hunter C. S. (2014) Revealing transcription factors during human pancreatic β cell development. Trends Endocrinol. Metab. 25, 407–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Harrison K. A., Thaler J., Pfaff S. L., Gu H., Kehrl J. H. (1999) Pancreas dorsal lobe agenesis and abnormal islets of Langerhans in Hlxb9-deficient mice. Nat. Genet. 23, 71–75 [DOI] [PubMed] [Google Scholar]
- 14. Li H., Arber S., Jessell T. M., Edlund H. (1999) Selective agenesis of the dorsal pancreas in mice lacking homeobox gene Hlxb9. Nat. Genet. 23, 67–70 [DOI] [PubMed] [Google Scholar]
- 15. Gherardi E., Birchmeier W., Birchmeier C., Vande Woude G. (2012) Targeting MET in cancer: rationale and progress. Nat. Rev. Cancer 12, 89–103 [DOI] [PubMed] [Google Scholar]
- 16. Hansel D. E., Rahman A., House M., Ashfaq R., Berg K., Yeo C. J., Maitra A. (2004) Met proto-oncogene and insulin-like growth factor binding protein 3 overexpression correlates with metastatic ability in well-differentiated pancreatic endocrine neoplasms. Clin. Cancer Res. 10, 6152–6158 [DOI] [PubMed] [Google Scholar]
- 17. Modali S. D., Parekh V. I., Kebebew E., Agarwal S. K. (2015) Epigenetic regulation of the lncRNA MEG3 and its target c-MET in pancreatic neuroendocrine tumors. Mol. Endocrinol. 29, 224–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bladt F., Riethmacher D., Isenmann S., Aguzzi A., Birchmeier C. (1995) Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature 376, 768–771 [DOI] [PubMed] [Google Scholar]
- 19. Roccisana J., Reddy V., Vasavada R. C., Gonzalez-Pertusa J. A., Magnuson M. A., Garcia-Ocaña A. (2005) Targeted inactivation of hepatocyte growth factor receptor c-met in β-cells leads to defective insulin secretion and GLUT-2 down-regulation without alteration of β-cell mass. Diabetes 54, 2090–2102 [DOI] [PubMed] [Google Scholar]
- 20. Mellado-Gil J., Rosa T. C., Demirci C., Gonzalez-Pertusa J. A., Velazquez-Garcia S., Ernst S., Valle S., Vasavada R. C., Stewart A. F., Alonso L. C., Garcia-Ocaña A. (2011) Disruption of hepatocyte growth factor/c-Met signaling enhances pancreatic β-cell death and accelerates the onset of diabetes. Diabetes 60, 525–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alvarez-Perez J. C., Ernst S., Demirci C., Casinelli G. P., Mellado-Gil J. M., Rausell-Palamos F., Vasavada R. C., Garcia-Ocaña A. (2014) Hepatocyte growth factor/c-Met signaling is required for β-cell regeneration. Diabetes 63, 216–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mohapatra B., Ahmad G., Nadeau S., Zutshi N., An W., Scheffe S., Dong L., Feng D., Goetz B., Arya P., Bailey T. A., Palermo N., Borgstahl G. E., Natarajan A., Raja S. M., Naramura M., Band V., Band H. (2013) Protein tyrosine kinase regulation by ubiquitination: critical roles of Cbl-family ubiquitin ligases. Biochim. Biophys. Acta 1833, 122–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Peschard P., Ishiyama N., Lin T., Lipkowitz S., Park M. (2004) A conserved DpYR motif in the juxtamembrane domain of the Met receptor family forms an atypical c-Cbl/Cbl-b tyrosine kinase binding domain binding site required for suppression of oncogenic activation. J. Biol. Chem. 279, 29565–29571 [DOI] [PubMed] [Google Scholar]
- 24. Li S., Kuhne W. W., Kulharya A., Hudson F. Z., Ha K., Cao Z., Dynan W. S. (2009) Involvement of p54(nrb), a PSF partner protein, in DNA double-strand break repair and radioresistance. Nucleic Acids Res. 37, 6746–6753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schiffner S., Zimara N., Schmid R., Bosserhoff A. K. (2011) p54nrb is a new regulator of progression of malignant melanoma. Carcinogenesis 32, 1176–1182 [DOI] [PubMed] [Google Scholar]
- 26. Shav-Tal Y., Zipori D. (2002) PSF and p54(nrb)/NonO–multi-functional nuclear proteins. FEBS Lett. 531, 109–114 [DOI] [PubMed] [Google Scholar]
- 27. Guru S. C., Goldsmith P. K., Burns A. L., Marx S. J., Spiegel A. M., Collins F. S., Chandrasekharappa S. C. (1998) Menin, the product of the MEN1 gene, is a nuclear protein. Proc. Natl. Acad. Sci. U.S.A. 95, 1630–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ang Y. S., Tsai S. Y., Lee D. F., Monk J., Su J., Ratnakumar K., Ding J., Ge Y., Darr H., Chang B., Wang J., Rendl M., Bernstein E., Schaniel C., Lemischka I. R. (2011) Wdr5 mediates self-renewal and reprogramming via the embryonic stem cell core transcriptional network. Cell 145, 183–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yadav S. P., Hao H., Yang H. J., Kautzmann M. A., Brooks M., Nellissery J., Klocke B., Seifert M., Swaroop A. (2014) The transcription-splicing protein NonO/p54nrb and three NonO-interacting proteins bind to distal enhancer region and augment rhodopsin expression. Hum. Mol. Genet. 23, 2132–2144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Miyazaki J., Araki K., Yamato E., Ikegami H., Asano T., Shibasaki Y., Oka Y., Yamamura K. (1990) Establishment of a pancreatic β-cell line that retains glucose-inducible insulin-secretion-special reference to expression of glucose transporter isoforms. Endocrinology 127, 126–132 [DOI] [PubMed] [Google Scholar]
- 31. Sukhodolets K. E., Hickman A. B., Agarwal S. K., Sukhodolets M. V., Obungu V. H., Novotny E. A., Crabtree J. S., Chandrasekharappa S. C., Collins F. S., Spiegel A. M., Burns A. L., Marx S. J. (2003) The 32-kilodalton subunit of replication protein A interacts with menin, the product of the MEN1 tumor suppressor gene. Mol. Cell. Biol. 23, 493–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Crabtree J. S., Scacheri P. C., Ward J. M., Garrett-Beal L., Emmert-Buck M. R., Edgemon K. A., Lorang D., Libutti S. K., Chandrasekharappa S. C., Marx S. J., Spiegel A. M., Collins F. S. (2001) A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proc. Natl. Acad. Sci. U.S.A. 98, 1118–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Crabtree J. S., Scacheri P. C., Ward J. M., McNally S. R., Swain G. P., Montagna C., Hager J. H., Hanahan D., Edlund H., Magnuson M. A., Garrett-Beal L., Burns A. L., Ried T., Chandrasekharappa S. C., Marx S. J., Spiegel A. M., Collins F. S. (2003) Of mice and MEN1: insulinomas in a conditional mouse knockout. Mol. Cell. Biol. 23, 6075–6085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Amelio A. L., Miraglia L. J., Conkright J. J., Mercer B. A., Batalov S., Cavett V., Orth A. P., Busby J., Hogenesch J. B., Conkright M. D. (2007) A coactivator trap identifies NONO (p54nrb) as a component of the cAMP-signaling pathway. Proc. Natl. Acad. Sci. U.S.A. 104, 20314–20319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gehring W. J., Qian Y. Q., Billeter M., Furukubo-Tokunaga K., Schier A. F., Resendez-Perez D., Affolter M., Otting G., Wüthrich K. (1994) Homeodomain-DNA recognition. Cell 78, 211–223 [DOI] [PubMed] [Google Scholar]
- 36. Bertolino P., Tong W. M., Galendo D., Wang Z. Q., Zhang C. X. (2003) Heterozygous Men1 mutant mice develop a range of endocrine tumors mimicking multiple endocrine neoplasia type 1. Mol. Endocrinol. 17, 1880–1892 [DOI] [PubMed] [Google Scholar]
- 37. Bertolino P., Tong W. M., Herrera P. L., Casse H., Zhang C. X., Wang Z. Q. (2003) Pancreatic β-cell-specific ablation of the multiple endocrine neoplasia type 1 (MEN1) gene causes full penetrance of insulinoma development in mice. Cancer Res. 63, 4836–4841 [PubMed] [Google Scholar]
- 38. Biondi C. A., Gartside M. G., Waring P., Loffler K. A., Stark M. S., Magnuson M. A., Kay G. F., Hayward N. K. (2004) Conditional inactivation of the MEN1 gene leads to pancreatic and pituitary tumorigenesis but does not affect normal development of these tissues. Mol. Cell. Biol. 24, 3125–3131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Harding B., Lemos M. C., Reed A. A., Walls G. V., Jeyabalan J., Bowl M. R., Tateossian H., Sullivan N., Hough T., Fraser W. D., Ansorge O., Cheeseman M. T., Thakker R. V. (2009) Multiple endocrine neoplasia type 1 knockout mice develop parathyroid, pancreatic, pituitary, and adrenal tumours with hypercalcaemia, hypophosphataemia, and hypercorticosteronaemia. Endocr. Relat. Cancer 16, 1313–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Loffler K. A., Biondi C. A., Gartside M., Waring P., Stark M., Serewko-Auret M. M., Muller H. K., Hayward N. K., Kay G. F. (2007) Broad tumor spectrum in a mouse model of multiple endocrine neoplasia type 1. Int. J. Cancer 120, 259–267 [DOI] [PubMed] [Google Scholar]
- 41. Dephoure N., Zhou C., Villén J., Beausoleil S. A., Bakalarski C. E., Elledge S. J., Gygi S. P. (2008) A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 105, 10762–10767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Miguel-Aliaga I., Thor S., Gould A. P. (2008) Postmitotic specification of Drosophila insulinergic neurons from pioneer neurons. PLos Biol. 6, e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zanfardino M., Spampanato C., De Cicco R., Buommino E., De Filippis A., Baiano S., Barra A., Morelli F. (2013) Simvastatin reduces melanoma progression in a murine model. Int. J. Oncol. 43, 1763–1770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Giordano S., Di Renzo M. F., Narsimhan R. P., Cooper C. S., Rosa C., Comoglio P. M. (1989) Biosynthesis of the protein encoded by the c-met proto-oncogene. Oncogene 4, 1383–1388 [PubMed] [Google Scholar]