

Background: A newly synthesized 2-hydroxyethyl 5-chloro-4,5-didehydrojasmonate (J11-Cl) has anti-inflammatory effects in carrageenan-induced paw edema.
Results: J11-Cl ameliorates colitis through activation of PPARγ and modulation of NF-κB and MAPK signaling.
Conclusion: J11-Cl acts as an effective anti-inflammatory agent.
Significance: A PPARγ agonist is a novel candidate to treat inflammatory bowel disease.
Keywords: colitis, cytokine, inflammatory bowel disease (IBD), mitogen-activated protein kinase (MAPK), NF-kappa B (NF-κB), peroxisome proliferator-activated receptor (PPAR)
Abstract
Inflammatory bowel disease (IBD) is a chronic inflammatory disease with increasing incidence and prevalence worldwide. Here we investigated the newly synthesized jasmonate analogue 2-hydroxyethyl 5-chloro-4,5-didehydrojasmonate (J11-Cl) for its anti-inflammatory effects on intestinal inflammation. First, to test whether J11-Cl can activate peroxisome proliferator-activated receptors (PPARs), we performed docking simulations because J11-Cl has a structural similarity with anti-inflammatory 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), one of the endogenous ligands of PPARγ. J11-Cl bound to the ligand binding domain of PPARγ in the same manner as 15d-PGJ2 and rosiglitazone, and significantly increased transcriptional activity of PPARγ. In animal experiments, colitis was significantly reduced in mice with J11-Cl treatment, determined by analyses of survival rate, body weight changes, clinical symptoms, and histological evaluation. Moreover, J11-Cl decreased production of pro-inflammatory cytokines including IL-6, IL-8, and G-CSF as well as chemokines including chemokine (C-C motif) ligand (CCL)20, chemokine (C-X-C motif) ligand (CXCL)2, CXCL3, and chemokine (C-X3-C motif) ligand 1 (CX3CL1) in colon tissues, and LPS or TNF-α-stimulated macrophages and epithelial cells. In contrast, production of anti-inflammatory cytokines including IL-2 and IL-4 as well as the proliferative factor, GM-CSF, was increased by J11-Cl. Furthermore, inhibition of MAPKs and NF-κB activation by J11-Cl was also observed. J11-Cl reduced intestinal inflammation by increasing the transcriptional activity of PPARγ and modulating inflammatory signaling pathways. Therefore, our study suggests that J11-Cl may serve as a novel therapeutic agent against IBD.
Introduction
Inflammatory bowel disease (IBD),3 including Crohn disease and ulcerative colitis, is a chronic inflammatory disorder affecting the gastrointestinal tract and is characterized by relapsing and remitting inflammation with epithelial damage (1, 2). The incidence and prevalence of IBD have been increasing worldwide (3). Although the exact cause of IBD remains to be explored, it appears that several factors may contribute to the development of IBD, including genetic predisposition, an altered and dysregulated immune response, and environmental factors such as food intake and smoking (4, 5). Some limitations in drug efficacy may exist because of the combination of factors leading to IBD and varied responses to drugs in individual patients. Therefore, further studies in IBD pathogenesis and development of new drugs for IBD treatment continue to represent important areas of investigation.
Methyl jasmonate is a member of the plant stress hormone family of jasmonates, the most potent regulator of defense-related mechanisms in plants (6). Because of similar partial structure to anti-inflammatory cyclopentenone prostaglandins, methyl jasmonate was selected as the starting material for the synthesis of a series of enone fatty acid-based anti-inflammatory compounds (7). Although one of the α-haloenone analogues, J7, showed enhanced in vitro anti-inflammatory potency, its activity was not as effective as diclofenac or indomethacin, well-known anti-inflammatory agents in the in vivo study because it quickly degraded to an inactive form. Thereafter, stability-improved analogues including α-chlorinated hydroxy alkyl ester, 2-hydroxyethyl 5-chloro-4,5-didehydrojasmonate (J11-Cl), and α-chlorinated branched alkyl ester, tert-butyl 5-chloro-4,5-didehydrojasmonate were evaluated for their potency. One of these analogues, J11-Cl, displayed higher in vivo efficacy than other methyl jasmonate derivatives in the carrageenan-induced paw edema assay (8).
The peroxisome proliferator-activated receptors (PPARs) are ligand-activated nuclear receptors, including three isoforms of PPARα, PPARβ/δ, and PPARγ. They can activate transcription of target genes by binding to PPAR response element (PPRE) after heterodimerization with retinoid X receptor. It has various roles in regulation of genes related to lipid metabolism, insulin sensitization, cell proliferation, and inflammation (9, 10). One of many functions of PPAR is inhibiting inflammatory actions such as cytokine and chemokine secretion when they are activated by endogenous or synthetic ligands (11). Of importance, PPARγ is highly expressed in the colon; the major tissue expression in epithelial cells and lower expression in macrophages and lymphocytes (12).
Important signaling molecules, which mediate various inflammatory processes include mitogen-activated protein kinases (MAPKs) and nuclear factor kappa B (NF-κB). Several studies have reported enhanced expression and phosphorylation of MAPKs in the intestine of IBD patients, suggesting that MAPK inhibitors can be used as anti-inflammatory drugs in this disease (13–15). Activation of the NF-κB pathway plays a crucial role in inflammation by inducing transcription of pro-inflammatory genes (16, 17). Increased activation of NF-κB is observed in macrophages and epithelial cells isolated from IBD patients, which correlates with the severity of intestinal inflammation (18).
Based on these considerations, we aimed to investigate the anti-inflammatory effects of the newly synthesized methyl jasmonate analogue J11-Cl in intestinal inflammation. We studied anti-inflammatory effect of J11-Cl in a murine experimental model of colitis in vivo and assessed its molecular mechanism in murine macrophages and intestinal epithelial cells exposed to inflammatory insults, including lipopolysaccharides (LPS) and tumor necrosis factor alpha (TNF-α).
Experimental Procedures
Docking Simulation
Docking experiments were performed using AutoDock Vina 1.1.2 software (The Scripps Research Institute, La Jolla, CA) against crystal structures of PPARγ and PPARδ. Protein coordinates were downloaded from the Protein Data Bank. The protein crystal structure was prepared for docking by removing native ligand from the ligand binding domain. Then polar hydrogens were added using MGLTools 1.5.4 (The Scripps Research Institute). All ligands were prepared by Chemdraw 12.0 (CambridgeSoft Corp., Cambridge, MA) and MGLTools 1.5.4. The Center of Grid box was calculated by native ligand using Chimera 1.10.1. The size of the grid box (docking space) was confined by the native ligand. Electrostatic charges of the assigned atoms were calculated as Gasteiger charges. After docking simulation, the analysis and visual investigation of ligand-protein interactions of docking schemes were performed using PyMol v1.7 (Schrodinger LLC, New York, NY). Also, LigandScout 3.12 (Inte:Ligand, Austria, Europe) was used to study pharmacophores. Before exploring the amino acid that forms hydrogen bonds with ligand, MMFF94 minimization was performed. We focused on key amino acid residues of the hydrophilic region of the ligand binding domain (PPARγ: Tyr-473, Ser-289 etc, PPARβ/δ: Tyr-437, His-413, His-287) for successful docking. Docking success was evaluated by the lowest affinity value.
Luciferase Assay
The NCM460 cells (3 × 104 cells per well seeded in 48-well plates) were transfected with 0.03 μg of PPRE-X3-TK luc plasmid and 0.3 μg of PPARα, PPARβ/δ, PPARγ expression vector (all constructs were kindly provided by Dr. Hae Young Chung, Pusan National University, Busan, Korea) using Lipofectamine 3000 reagent (Invitrogen) according to the manufacturer's instructions. After incubation for 24 h, cells were treated with vehicle (DMSO), 50 μm J11-Cl, 10 μm WY14643, GW0742, or rosiglitazone (Sigma-Aldrich) for 5 h. Luciferase activities were measured using One-Glo Luciferase Assay System (Promega) with a luminometer (Berthold Technologies GmbH & Co. KG, Bad Wildbad, Germany).
Experimental Colitis
Male C57BL/6 mice (6–7-week-old) were purchased from Samtako Co. Ltd. (Osan, Korea) and acclimatized to the animal care facility for at least 7 days before the experiments. Animals were housed in an air-conditioned atmosphere under a 12-h light/dark cycle and given free access to standard rodent chow (Samtako) and water. The animal study protocols used in this study were approved by the Institutional Animal Care and Use Committee of Pusan National University. For induction of dextran sodium sulfate (DSS)-induced colitis, 7–8-week-old mice were given 3% (w/v) DSS (MP Biomedicals, Irvine, CA) in drinking water for 11 days. Mice were treated daily with J11-Cl (50 mg·kg−1) or vehicle (1% DMSO (Duchefa, Haarlem, Netherlands) and 5% cremophor (Sigma-Aldrich) in saline) by intraperitoneal injection. The mice were monitored daily for mortality and clinical symptoms such as rectal bleeding and diarrhea. The degree of symptoms was graded on a scale of 0–4. The mice were euthanized when clinical symptoms of colitis were manifested during the 11-day period.
Hematoxylin and Eosin (H&E) Staining
After euthanasia, the mouse colon was excised, and segments of the transverse colon were fixed immediately in 10% buffered formalin solution. Tissues were embedded in paraffin and stained with H&E to study histological changes and observed using microscope (Olympus Corp., Tokyo, Japan) and photographed with Moticam 3.0 MP Color Digital Camera (Motic, Causeway Bay, Hong Kong) and motic images plus 2.0 software.
Immunohistochemistry
Formalin-fixed colon tissues were embedded in paraffin and sectioned. After deparaffinized with xylene (Duksan Pure Chemicals, KyungKi-do, Korea), tissues on the slides were rehydrated with ethanol (Merk Millipore Corp., Billerica, MA). For antigen retrieval, slides were placed in heated Tris/EDTA buffer (pH 9.0) (Bioworld Technology, St. Souis Park, MN). After blocking for 1 h with normal rabbit serum (Vector Laboratories), tissues were incubated for 2 h with 1/50 diluted anti-neutrophil antibody, NIMP-R14 (Abcam, Cambridge, MA). Antibody binding were detected using a biotinylated secondary antibody and ABC reagent from Vectastain Elite ABC kit. Next, sections were treated with peroxidase substrate solution and counterstained with hematoxylin. Finally, sections were mounted using VectaMount mounting medium (all purchased from Vector Laboratories). Stained tissues were observed using microscope (Olympus) and photographed with Moticam 3.0 MP Color Digital Camera (Motic) and motic images plus 2.0 software.
Immunofluorescence Staining
Formalin-fixed colon tissues were embedded in paraffin and sectioned. After deparaffinized with xylene (Duksan Pure Chemicals), sections were rehydrated with ethanol (Merck Millipore Corp.) and then placed in heated Tris/EDTA buffer (pH 9.0) (Bioworld Technology) for antigen retrieval. After blocking for 1 h with normal goat serum (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA), sections were incubated overnight at 4 °C with 1/200 diluted anti-CD3 antibody (Abcam) and then incubated for 1 h at room temperature with 1/2500 diluted FITC-conjugated goat anti-rabbit IgG (Bethyl Laboratories, Inc., Montgomery, TX). Finally, sections were mounted using Vectashield mounting medium with DAPI (Vector Laboratories). Stained tissues were observed using Axioskop (Carl Zeiss, Oberkochen, Germany) and photographed with MetaMorph® software.
Cell Culture
Murine macrophage cell line RAW264.7 was obtained from American Type Tissue Culture (Rockville, MD), human non-transformed human colonic epithelial cells NCM460 were obtained from INCELL Corp. (San Antonio, TX), and human leukemia cells HL-60 were obtained from Korean Cell Line Bank (KCLB®, Seoul, Korea). RAW264.7 and NCM460 were cultured in DMEM with high glucose and HL-60 was cultured in RPMI, supplemented with 10% heat-inactivated FBS, 100 units·ml−1 penicillin, and 100 μg·ml−1 streptomycin (all from Hyclone Inc.), at 37 °C in a 5% CO2 atmosphere.
Immunoblot Analysis
Total protein extracts from homogenized mouse colon tissues and whole cell lysates of RAW264.7 and NCM460 cells were prepared in lysis buffer with protease and phosphatase inhibitor cocktails. Protein concentration was measured by the Pierce® BCA Protein Assay Kit (Thermo Scientific, Rockford) and equal amounts of protein extracts were subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membrane. The membranes were blocked with 5% bovine serum albumin in 1× Tris-buffered saline with Tween-20 buffer (TBS-T) for 1 h at room temperature and then incubated overnight at 4 °C with one of following primary antibodies: COX-2 rabbit polyclonal (sc-7951, Santa Cruz Biotechnology), pThr202/Tyr204-ERK1/2 rabbit polyclonal (no. 9101, Cell Signaling), ERK1/2 (no. 9102, Cell Signaling), pThr183/Tyr185-SAPK/JNK rabbit polyclonal (no. 9251, Cell Signaling), SAPK/JNK rabbit polyclonal (no. 9252, Cell Signaling), pThr180/Tyr182-p38 rabbit polyclonal (no. 9211, Cell Signaling), p38 rabbit polyclonal (no. 9212, Cell Signaling), pSer536-NF-κB p65 rabbit monoclonal (no. 3033, Cell Signaling), β-actin (A5441, Sigma-Aldrich). After the membranes were washed for 40 min with 1× TBS-T, membranes were incubated with an appropriate horseradish peroxidase-conjugated secondary antibody (Enzo Life Sciences, NY) for 2 h at room temperature. Bound antibodies were detected by the enhanced chemiluminescence detection system (SuperSignal West Pico Chemiluminescent Substrate Kit, Thermo Fisher Scientific).
Multi-analyte ELISArray
Murine inflammatory cytokines were quantified using a multi-analyte ELISArray (SABiosciences, Qiagen, Inc.). In brief, 6–7 colon tissues from each group of mice were lysed with protein lysis buffer, and the lysates were incubated with capture antibodies for 2 h at room temperature. After three washes, samples were re-incubated with biotinylated detection antibodies for 1 h at room temperature. After another three washes, samples were incubated with avidin-horseradish peroxidase conjugate for 30 min in the dark. After addition of stop solution, optical density was evaluated on a spectrophotometer (iMarkTM Microplate Reader, Bio-Rad Labs.) at 450 nm.
Quantitative Real-time PCR Analysis
Total RNA was prepared from cultured cells using the RNeasy Plus Mini Kit (Qiagen) and from colon tissues of mice using TRIzol® Reagent (Ambion). An equal amount of RNA (1 μg) was transcribed into cDNA, using RT-&GOTM Mastermix (MP Biomedicals, Solon, OH) with an oligo (dT) primer (ELPIS Biotech Inc., Daejeon, Korea). Real-time PCR was performed on a Thermal Cycler Dice Real Time System using SYBR® Premix Ex TaqTM (Takara Bio Inc., Otsu, Shiga, Japan). Each reaction was performed in triplicate or quadruplicate. The PCR primer sequences were designed according to the gene sequences reported in GenBankTM and were chemically synthesized. Expression of mouse GAPDH, IL-6, IL-8, and human GAPDH, CCL20, CXCL2, CXCL3, and CX3CL1 was estimated by quantitative real-time PCR using the following primers. Mouse GAPDH forward primer: 5′-CTC ACT GGC ATG GCC TTC CG-3′ and reverse primer: 5′-ACC ACC CTG TTG CTG TAG CC-3′; mouse IL-6 forward primer: 5′-GGA CTG ATG CTG GTG ACA AC-3′ and reverse primer: 5′-TCC ACG ATT TCC CAG AGA AC-3′; mouse IL-8 forward primer: 5′-ACG TGT CCC CAA GTA ACG GA-3′ and reverse primer: 5′-TCA GAA GCC AGC GTT CAC C-3′; human GAPDH forward primer: 5′-CGC TCT CTG CTC CTC CTG TT-3′ and reverse primer: 5′-CCA TGG TGT CTG AGC GAT GT-3′; human CCL20 forward primer: 5′-GAC TGC TGT CTT GGA TAC AC-3′ and reverse primer: 5′-GAT TTG CGC ACA CAG ACA AC-3′; human CXCL2 forward primer: 5′-CCA ACT GAC CAG AAG GAA GG-3′ and reverse primer: 5′-GAT GCT CAA ACA CAT TAG GCG-3′; human CXCL3 forward primer: 5′-TCA CTG AAC TGC GCT GCC AG-3′ and reverse primer: 5′-TCT TCC CAT TCT TGA GTG TGG C-3′; human CX3CL1 forward primer: 5′-TAT CAC TCC TGT CCC TGA CG-3′ and reverse primer: 5′-GCT CTG GTA GGT GAA CAT GG-3′. Relative gene expression was determined by the difference in the CT values of the target genes after normalization to the RNA input level, using a CT value of GAPDH. Relative quantification was represented by the 2−▵▵CT calculation.
Migration Assay
Migration assay was performed using Cultrex® Cell Migration Assay kits (Trevigen Inc., Gaithersburg, MD) according to the manufacturer's instruction. In brief, NCM460 cells (1 × 105 cells per well seeded in 24-well plate) were stimulated with 10 ng·ml−1 TNF-α after pretreatment of 50 μm J11-Cl for 30 min. HL-60 cells were loaded in uncoated Boyden chambers (5 × 104 cells per chamber). After 24-h incubation, migrated cells were dissociated by cell dissociation buffer with calcein-AM and relative fluorescence unit was measured using GLOMAX multi detection system (Promega).
Statistical Analysis
Statistical analysis was performed using GraphPad Prism version 5.03 for Windows (GraphPad Software). Data are presented as the mean ± S.E. Difference in survival was shown by Kaplan-Meier plot. The log-rank test was used to compare significant survival difference. Group data were compared by t test or 1-way or 2-way analysis of variance followed by multiple comparisons test. The nonparametric Mann Whitney U test was used to compare clinical symptom of colitis.
Results
Interaction between J11-Cl and PPARγ
Considering structural similarity between J11-Cl and the endogenous PPARγ ligand 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) (19), we investigated whether J11-Cl can bind to PPARγ in the same manner as 15d-PGJ2. Docking simulation of J11-Cl to PPARγ showed that the polar side chain including a hydroxyl group is located in the hydrophilic domain and the other non-polar alkyl chain is located in the hydrophobic region of the ligand binding domain of PPARγ (Fig. 1A), which displays very similar topology with that of 15d-PGJ2 (Fig. 1B). It was reported that the cocrystal structure of 15d-PGJ2 exhibits a hydrogen bond between the carboxyl group of 15d-PGJ2 and the amino acid residue Tyr-473. A covalent bond between the β-carbon of the enone moiety of 15d-PGJ2 and the amino acid residue Cys-285 is also indicated. Docking simulation of J11-Cl also suggested formation of hydrogen bonds with Tyr-473 and His-449. The amino acid residues Tyr-473 and His-449 are also key residues for hydrogen bonding of rosiglitazone in the ligand binding domain of PPARγ (Fig. 1C). Therefore, docking simulation indicated that J11-Cl binds to the ligand binding domain of PPARγ in a same manner as 15d-PGJ2 and rosiglitazone, and key amino acid residues Tyr-473 and His-449 are involved in hydrogen bonding between J11-Cl and PPARγ.
FIGURE 1.

Interactions of rosiglitazone, 15d-PGJ2, GW0742, and J11-Cl with the ligand binding domain of either PPARγ or PPARβ/δ. A, docking simulation of J11-Cl to the ligand binding domain of PPARγ. B, cocrystal structure of 15d-PGJ2 with PPARγ (PDB code number 2ZK1). C, cocrystal structure of rosiglitazone with PPARγ (PDB code number 2PRG). D, docking simulation of J11-Cl to the ligand binding domain of PPARβ/δ. E, cocrystal structure of GW0742 with PPARβ/δ (PDB code number 3TKM).
Interaction between J11-Cl and PPARβ/δ
The cocrystal structure of GW0742 and PPARβ/δ (PDB 3TKM) was selected as a control model to investigate the interaction between J11-Cl and PPARβ/δ. Docking simulation of J11-Cl suggested formation of hydrogen bonds of the hydroxyl group of J11-Cl with Tyr-437, His-413, and His-287 of PPARβ/δ (Fig. 1D). Similar to the case of docking simulation to PPARγ, the polar side chain of J11-Cl is located in the hydrophilic domain and the other non-polar alkyl chain is located in the hydrophobic region of the ligand binding domain of PPARβ/δ. It was reported that the carboxyl group of GW0742 forms hydrogen bonds with Tyr-437, His-413, and His-287 of PPARβ/δ (Fig. 1E). Tyr-437 is the common key amino acid residue shared both in PPARγ and PPARβ/δ.
When the docking topologies of rosiglitazone and GW0742 were simulated to be identical to each cocrystal structures, the affinity scores were −8.3 kcal/mol (Fig. 1C) and −11.0 kcal/mol (Fig. 1E), respectively. Whereas those of J11-Cl to PPARγ and PPARβ/δ were −6.3 kcal/mol (Fig. 1A) and −6.6 kcal/mol (Fig. 1D), respectively. Therefore, J11-Cl may be expected to show higher relative docking affinity to PPARγ (−6.3 kcal/mol) than PPARβ/δ (−6.6 kcal/mol) compared with their standard control rosiglitazone (−8.3 kcal/mol) and GW0742 (−11.0 kcal/mol), respectively.
Increased Transcriptional Activity of PPARγ by J11-Cl
Next we tested whether J11-Cl regulates transcriptional activity of PPARs, luciferase assays for three PPARs were performed. Human colonic epithelial cells (NCM460) transfected with PPRE-X3-TK luc plasmid (PPRE-luc) and PPARα, PPARβ/δ or PPARγ expression vector were treated with vehicle, J11-Cl, or positive controls for each PPAR (WY14643 for PPARα, GW0742 for PPARβ/δ, rosiglitazone for PPARγ). When the cells were transfected with PPRE-luc and PPARα or PPARβ/δ, luciferase activities were significantly increased by only positive controls. Although docking simulation of J11-Cl suggested formation of hydrogen bonds of J11-Cl with PPARβ/δ (Fig. 1D), luciferase activities of PPARβ/δ were not significantly increased by J11-Cl. However, when the cells were transfected with PPRE-luc and PPARγ, luciferase activities were significantly increased by J11-Cl as well as rosiglitazone (∼2.34-fold increase in J11-Cl and 2.54-fold in rosiglitazone) (Fig. 2). These results indicate that J11-Cl increases transcriptional activity of PPARγ.
FIGURE 2.
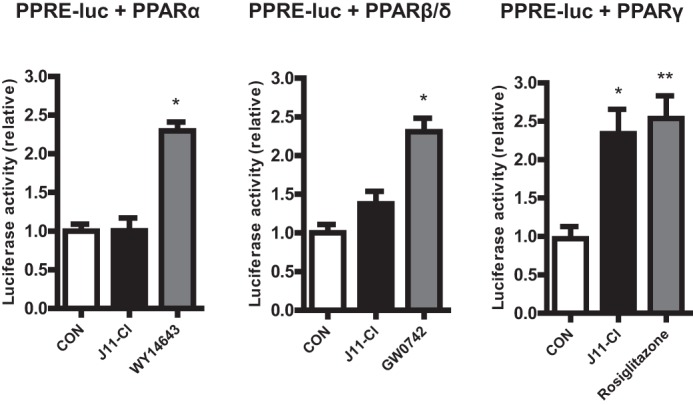
J11-Cl increased transcriptional activity of PPARγ. Transcriptional activity of PPARγ, not PPARα and PPRAβ/δ, was significantly increased by J11-Cl. Data are shown as the mean ± S.E. A single asterisk (*) indicates p < 0.05; double asterisks (**) indicate p < 0.01.
Ameliorated Symptoms of DSS-induced Colitis by J11-Cl
To evaluate anti-inflammatory effects of J11-Cl in vivo, we used a mouse model of colitis. One group of mice was treated with vehicle, and the other group of mice was treated with J11-Cl with the supply of 3% DSS in drinking water. Fig. 3A indicated that J11-Cl significantly increased survival of mice compared with the vehicle control. Moreover, body weight loss of J11-Cl-treated mice was less severe than that of vehicle-treated mice, and rectal bleeding and diarrhea were relieved by J11-Cl treatment (Fig. 3, B–D). Histological analysis indicated that tissue damage was less severe in the colon from J11-Cl-treated mice than that from the vehicle control (Fig. 4A). Moreover, infiltration of neutrophils and T cells into the inflamed site was inhibited in J11-Cl-treated mice compared to the vehicle control (Fig. 4, B and C). Taken together, these results suggest that J11-Cl has a protective role against colonic inflammation.
FIGURE 3.

J11-Cl significantly increased survival of mice and alleviated symptoms of colitis. Mice were supplied with 3% DSS in drinking water for 11 days. One group of mice was treated with J11-Cl (50 mg·kg−1) and the other group of mice was treated with vehicle (1% DMSO and 5% cremophor in saline) daily by intraperitoneal injection. The mice were monitored for rectal bleeding and diarrhea (scaled 0–4) for the duration of the experiment. A, analysis of survival rate using the log-rank test indicated that J11-Cl significantly increased survival of mice (p = 0.0013). B, body weight loss in mice treated with J11-Cl was less severe than that in the control group (vehicle treated mice). C, rectal bleeding was relieved by J11-Cl compared with the control group. D, diarrhea was relieved by J11-Cl compared with the control group. Data are shown as the mean ± S.E.; n = 12 (control group), n = 14 (J11-Cl group). A single asterisk (*) indicates p < 0.05; double asterisks (**) indicate p < 0.01; triple asterisks (***) indicate p < 0.001.
FIGURE 4.

Tissues from J11-Cl treated mice showed less severe colonic inflammation. A, colon tissues were stained for H&E. Control mice showed more severe tissue damage with loss of crypt architecture and greater infiltration of inflammatory cells than J11-Cl-treated mice. Bar, 100 μm. B, colon tissues were stained for neutrophil surface marker, Ly-6G. Infiltration of neutrophils (brown) was significantly decreased by J11-Cl treatment. Bar, 30 μm. The number of infiltrated neutrophils was counted. C, colon tissues were stained for T cell marker, CD3. Infiltration of T cells (green) was significantly decreased by J11-Cl treatment. Arrowheads indicate CD3-positive cells. Bar, 10 μm. The number of CD3 positive cells was counted. For counting neutrophil and CD3-positive cell number, cells were counted in four randomly picked fields from two independent colon tissues in each group. Data are shown as the mean ± S.E. A single asterisk (*) indicates p < 0.05; triple asterisks (***) indicate p < 0.001.
Modulation of Pro-inflammatory Signaling Pathways by J11-Cl
Increased activities of MAPK and NF-κB contribute to intestinal inflammation. Moreover, MAPK including p38, JNK, and ERK1/2 are significantly activated in IBD (13–15, 18). Thus we investigated whether J11-Cl can modulate the activity of MAPKs and NF-κB. Phosphorylation of ERK1/2 and the expression of representative pro-inflammatory mediator COX-2 were decreased in J11-Cl-treated colon tissues (Fig. 5A). In addition, J11-Cl pretreatment inhibited LPS-mediated NF-κB p65 activation and phosphorylation of ERK1/2, JNK, and p38 in macrophage cell line RAW264.7, although disruption of p38 activity was relatively less significant compared with the other two MAPK (Fig. 5B). Next we checked whether J11-Cl inhibits inflammatory signaling in human colonic epithelial cells. J11-Cl pretreatment also inhibited TNF-α-mediated NF-κB p65 activation and phosphorylation of ERK1/2 and JNK in NCM460 (Fig. 5C). Taken together, J11-Cl shows anti-inflammatory effects through down-regulation of COX-2 expression, inhibition of NF-κB activation, and inhibition of the activity of MAPKs, especially ERK1/2 and JNK.
FIGURE 5.

J11-Cl modulated pro-inflammatory signaling pathways. A, expression of COX-2 and p-ERK1/2 in the colon tissues was determined by immunoblotting, using total ERK1/2 as an internal control. B, RAW264.7 cells were pretreated with 50 μm J11-Cl for 30 min and stimulated with 20 ng·ml−1 LPS for 15 min. Expression of phosphorylated NF-κB p65 and MAPKs (p-ERK1/2, p-JNK, and p-p38) was determined by immunoblotting, using β-actin and total MAPKs as an internal control. C, NCM460 cells were pretreated with 50 μm J11-Cl for 30 min and stimulated with 1 ng·ml−1 TNF-α for 15 min. Expression of p-NF-κB p65, p-ERK1/2, and p-JNK was determined by immunoblotting, using β-actin, ERK1/2, and JNK as an internal control.
Increase in Production of Anti-inflammatory Cytokines and Proliferative Factor but Decrease in Production of Pro-inflammatory Cytokines by J11-Cl
To investigate the mechanism of which J11-Cl exerts anti-inflammatory effect during colitis, we determined whether J11-Cl regulates production of several cytokines. The mRNA and protein levels of pro-inflammatory cytokines including IL-6, IL-8, and G-CSF were decreased in J11-Cl-treated colon tissues compared with the control (Fig. 6A). However, the protein levels of anti-inflammatory cytokines, IL-2 and IL-4, were increased by J11-Cl (Fig. 6B). GM-CSF produced by epithelial cells is associated with epithelial cell proliferation and repair in damaged colonic mucosa (20, 21). In Fig. 6C, the protein level of GM-CSF was increased in J11-Cl-treated colon tissues suggesting a proliferative role of J11-Cl in inflamed mucosa. To further elucidate an immune-modulatory effect of J11-Cl, the mRNA expression levels of pro-inflammatory cytokines were measured in LPS-treated RAW264.7 cells. Pretreatment of J11-Cl significantly reduced LPS-induced IL-6 and IL-8 expression (Fig. 6D). Therefore, these results indicate that J11-Cl may confer anti-inflammatory effects through modulation of cytokine expression.
FIGURE 6.
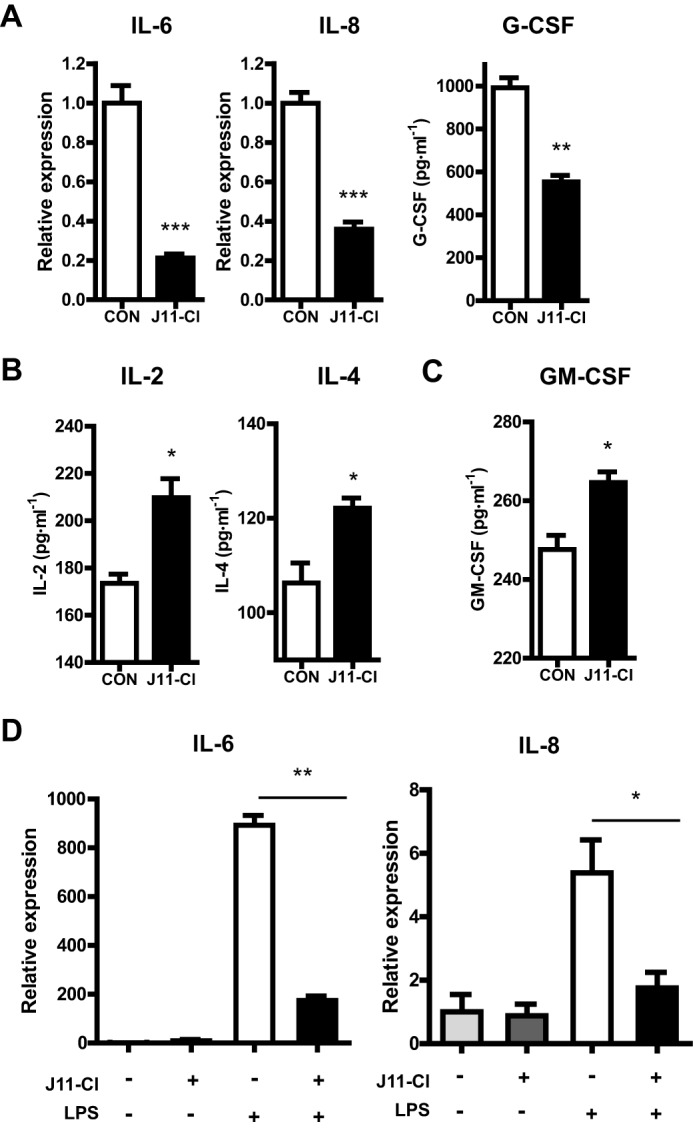
J11-Cl modulated expression of pro-inflammatory, anti-inflammatory and proliferative cytokines. A, expression of pro-inflammatory cytokines in mouse colon tissues. The protein level of G-CSF and mRNA levels of IL-6 and IL-8 were decreased by J11-Cl. B, expression of anti-inflammatory cytokines in the mouse colon tissues. The protein levels of IL-2 and IL-4 were increased by J11-Cl. C, expression of proliferative cytokine in the colon tissues from mice. The protein level of GM-CSF was increased by J11-Cl. D, expression of pro-inflammatory cytokines in LPS-treated RAW264.7 cells. The cells were pretreated with 50 μm J11-Cl for 30 min and stimulated with 20 ng·ml−1 LPS for 3 h. The mRNA levels of IL-6 and IL-8 were decreased by J11-Cl. Data are shown as the mean ± S.E. A single asterisk (*) indicates p < 0.05; double asterisks (**) indicate p < 0.01; triple asterisks (***) indicate p < 0.001.
Decreased Inflammatory Chemokine Expression by J11-Cl
Previous studies revealed that several chemokines, such as CXCL1, CXCL2, CXCL3, and CCL20 are up-regulated specifically in the inflamed colon of IBD patients (22). In NCM460 cells, TNF-α-mediated increase of the inflammatory chemokines CCL20, CXCL2, CXCL3, and CX3CL1 was significantly reduced by pretreatment with J11-Cl (Fig. 7A). Furthermore, when NCM460 cells were co-cultured with leukocytes, increased number of migrated leukocytes by TNF-α stimulation was also reduced by J11-Cl treatment (Fig. 7B). This result further suggests potent anti-inflammatory effects of J11-Cl.
FIGURE 7.
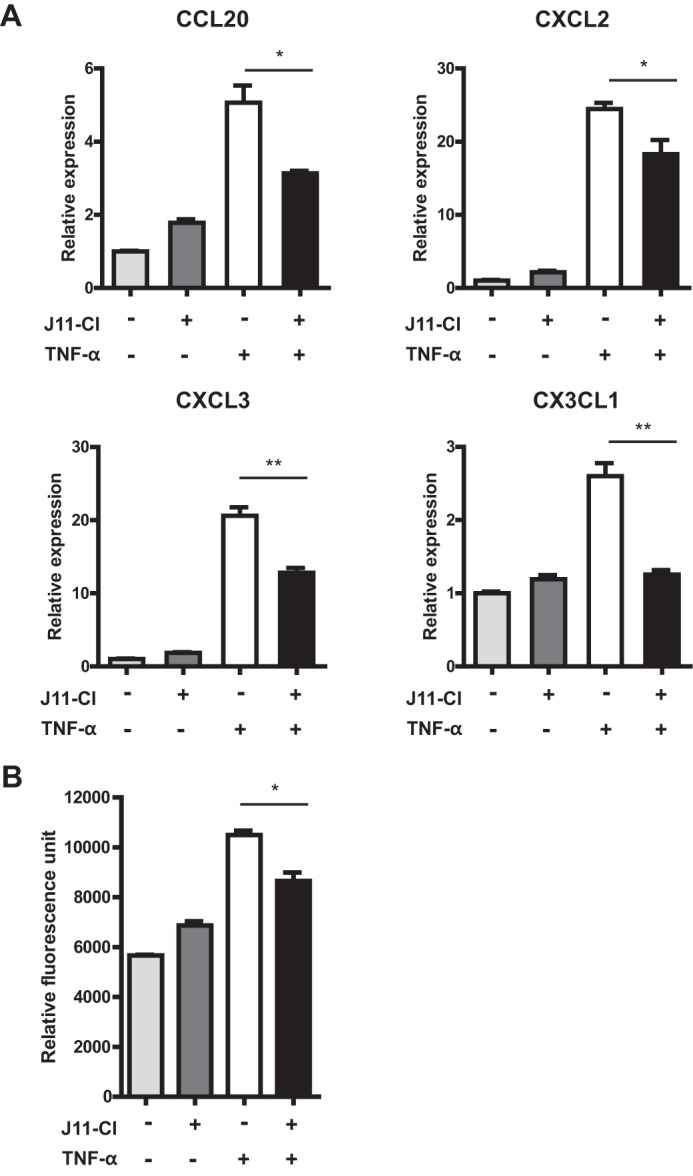
The mRNA expression of inflammatory chemokines in colonic epithelial cells and the number of migrated leukocytes was decreased by J11-Cl. A, NCM460 cells were pretreated with 50 μm J11-Cl for 30 min and stimulated with 1 ng·ml−1 TNF-α for 3 h. Relative gene expression of CCL20, CXCL2, CXCL3, and CX3CL1 was significantly decreased by J11-Cl treatment in TNF-α-treated NCM460 cells. B, NCM460 cells were pretreated with 50 μm J11-Cl for 30 min and stimulated with 10 ng·ml−1 TNF-α for 24 h. Relative fluorescence unit indicates migrated HL-60 cells. Data are shown as the mean ± S.E. A single asterisk (*) indicates p < 0.05; double asterisks (**) indicate p < 0.01.
Discussion
In this study, we investigated the anti-inflammatory effect of the newly synthesized jasmonate analogue J11-Cl and elucidated its molecular mechanisms. Based on structural similarity of J11-Cl with anti-inflammatory cyclopentenone prostaglandin 15d-PGJ2, we investigated whether J11-Cl can bind to and activate PPARγ. J11-Cl and PPARγ agonist rosiglitazone significantly increased PPARγ transcriptional activity, albeit binding affinity of J11-Cl to PPARγ was slightly lower than that of rosiglitazone. It is the evidence that J11-Cl may exert anti-inflammatory effects by binding to PPARγ, which may inhibit NF-κB-mediated signaling pathways (11, 12).
Next, we observed strong anti-inflammatory effects of J11-Cl in a murine and well-accepted model of colitis in vivo. Survival of DSS-exposed mice was significantly increased, and symptoms of colitis including body weight loss, rectal bleeding, and diarrhea were significantly alleviated by J11-Cl treatment, suggesting a protective role of this compound in colonic inflammation. Histological analysis using colon tissues showed less damage in the crypt architecture in the J11-Cl-treated mice than in control, also indicating anti-inflammatory effects of J11-Cl.
As discussed above inhibition of MAPKs represents a promising therapeutic potential in IBD. In an in vitro study using Caco-2 intestinal epithelial cells, involvement of MAPKs in pro-inflammatory cytokine production is demonstrated by the decrease of IL-6 and IL-8 through specific inhibitor treatment (23). In an in vivo study using the murine DSS-induced colitis model, the p38 inhibitor SB203580 not only ameliorates DSS-induced colitis but also inhibits pro-inflammatory cytokine expression and NF-κB activation (24). Another study demonstrated that the JNK inhibitor SP600125 has protective effects against DSS-induced colitis and reduces production of pro-inflammatory cytokines (25) and apoptosis. In another study using the DSS-induced colitis model, apostain, a marker of apoptosis, is reduced with concomitant TNF-α decrease in JNK inhibitor-treated mice, and the JNK inhibitor shows protective effects against DSS-induced colitis (26). But, relatively few studies have been conducted related to the role of ERK1/2 in IBD. ERK1/2 is overexpressed and phosphorylated in IBD tissues (14). Additionally, macrophage inflammatory protein 3 alpha synthesis by IL-21 in epithelial cells and IL-1β release after DSS administration in murine peritoneal macrophages are mediated by ERK1/2 activation (27, 28). The ability of J11-C1 to inhibit MAPK activation, especially ERK1/2 and JNK may explain the anti-inflammatory action of J11-Cl in vivo and in vitro.
In addition to its regulatory effects on MAPK activity, J11-C1 also modulated NF-κB activity. As previously mentioned, the NF-κB pathway plays a central role in inflammation (16, 17). Persistent COX-2 inhibition through long-term treatment of celecoxib down-regulates NF-κB activity in enterocytes (29). Therefore, COX-2 inhibition by J11-C1 in inflamed colon tissues may contribute to NF-κB inhibition.
Next we measured the expression level of several cytokines as the first step to elucidate the anti-inflammatory mechanism of J11-Cl. IL-2, IL-4, and GM-CSF were significantly increased whereas IL-6, IL-8, and G-CSF were significantly decreased by J11-Cl treatment.
IL-2 is necessary for maintenance of regulatory T cells, and neutralization of IL-2 is associated with autoimmune disease. IL-2 also regulates inflammation by disrupting IL-6-dependent signaling, resulting in inhibition of T helper 17 differentiation (30, 31). IL-4 is a representative anti-inflammatory cytokine possessing immunosuppressive activities against colitis (32). In addition, macrophages exposed to IL-4 switches their phenotype to the M2 subtype, which possesses immunoregulatory functions (33). Therefore, increased production of IL-2 and IL-4 by J11-Cl suggests its anti-inflammatory property. Furthermore, Suttles et al. reported that ERK1/2 phosphorylation in monocyte activation was reduced by IL-4 (34), suggesting that inhibition of ERK1/2 activation and increased IL-4 production may be closely linked to the anti-inflammatory effects of J11-Cl.
Our histologic evidence suggests that J11-Cl treatment might protect the intestinal epithelium from injury. Protecting the epithelial barrier of the inflamed intestine is important in preventing hyperactivation of the mucosal immune system since the integrity of the intestinal epithelium is crucial for maintaining intestinal homeostasis (35, 36). Recombinant GM-CSF treatment reduces the severity of colitis in mice and GM-CSF produced by epithelial cells plays a key role in epithelial cell proliferation and repair in damaged colonic mucosa (20, 21). Additionally, phosphorylation of ERK1/2 was also increased when the cells were stimulated with J11-Cl alone. Since the ERK pathway has an important role in GM-CSF-dependent cell proliferation (37), it is possible that increased phosphorylation of ERK1/2 by J11-Cl alone may contribute to enhanced epithelial cell proliferation by producing GM-CSF when there is no inflammation. However, this requires further study to clarify the proliferative effect of J11-Cl in colonocytes. In summary, J11-Cl may preserve the epithelial barrier by increasing GM-CSF.
The expression level of the pro-inflammatory cytokine IL-6 is higher in IBD patients and IL-6 blockade is suggested as effective treatment for IBD (38). A previous report showed that IL-6 production was mediated by phosphorylation of ERK1/2 (39). Therefore, reduced ERK1/2 activity by J11-Cl treatment may result in down-regulation of IL-6. Moreover, increased IL-8 expression is observed in the colonic mucosa of patients with IBD (40). It has also known that G-CSF recruits neutrophils rapidly into inflamed sites (41). In our result, neutrophil infiltration significantly decreased in J11-Cl-treated colon tissues compared with the vehicle control. This result suggests that decrease of G-CSF by J11-Cl may inhibit neutrophil infiltration into inflamed sites.
Dysregulated cytokine production has been implicated in the pathogenesis of IBD (42), and chemokines such as CXCL1, CXCL2, CXCL3, and CCL20 are expressed specifically in inflamed areas of IBD colon (22). NF-κB is also activated in macrophages and epithelial cells of inflamed intestinal mucosa and its activation induces transcription of pro-inflammatory cytokines and chemokines, suggesting a crucial role in inflammatory diseases (16, 18). Moreover, chemokine expression can be induced by cytokines, which involves NF-κB activation (43, 44). Based on these studies and our evidence it is possible that inhibition of NF-κB activation and suppressed mRNA expression of pro-inflammatory cytokines and chemokines may be involved in the protective effects of J11-C1.
In conclusion, J11-Cl may exert anti-inflammatory effects by enhancing the production of anti-inflammatory cytokines, inhibiting the expression of pro-inflammatory cytokines and chemokines, and increasing epithelial cell proliferation by regulating MAPKs and NF-κB activation. Our study reveals the potential of methyl jasmonate derivative J11-Cl as a new anti-inflammatory drug for IBD treatment.
Author Contributions
J. C. performed the experiments and wrote the manuscript. Y. L. performed the experiments. X. Y., S. J. K., and S. S. synthesized the compound. T. H. N. performed docking simulation. CP helped data interpretation. H. R. M. supervised the synthesis of the compound and helped data interpretation. J. H. J. supervised the synthesis of the compound and wrote the manuscript. E. I. designed and supervised the study and wrote the manuscript. All authors analyzed the results and approved the final version of the manuscript.
This work was supported by National Research Foundation of Korea (NRF) Grant 2009-0083538 and 2014R1A1A2057432. The authors declare that they have no conflicts of interest with the contents of this article.
- IBD
- inflammatory bowel disease
- 15d-PGJ2
- 15-deoxy-Δ12,14-prostaglandin J2
- CCL
- chemokine (C-C motif) ligand
- CXCL
- chemokine (C-X-C motif) ligand
- CX3CL1
- chemokine (C-X3-C motif) ligand
- G-CSF
- granulocyte colony-stimulating factor
- GM-CSF
- granulocyte macrophage colony-stimulating factor
- J11-Cl
- 2-hydroxyethyl 5-chloro-4,5-didehydrojasmonate
- LPS
- lipopolysaccharide
- MAPK
- mitogen-activated protein kinase
- NF-κB
- nuclear factor kappa B
- PPAR
- peroxisome proliferator-activated receptor
- PPRE
- PPAR response element
- TNF-α
- tumor necrosis factor α.
References
- 1. Danese S., Fiocchi C. (2011) Ulcerative colitis. NE J. Med. 365, 1713–1725 [DOI] [PubMed] [Google Scholar]
- 2. Baumgart D. C., Sandborn W. J. (2012) Crohn's disease. Lancet 380, 1590–1605 [DOI] [PubMed] [Google Scholar]
- 3. Molodecky N. A., Soon I. S., Rabi D. M., Ghali W. A., Ferris M., Chernoff G., Benchimol E. I., Panaccione R., Ghosh S., Barkema H. W., Kaplan G. G. (2012) Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 142, 46–54 [DOI] [PubMed] [Google Scholar]
- 4. Shih D. Q., Targan S. R. (2009) Insights into IBD Pathogenesis. Curr. Gastroenterol. Rep. 11, 473–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ananthakrishnan A. N. (2013) Environmental risk factors for inflammatory bowel disease. Gastroenterol. Hepatol. 9, 367–374 [PMC free article] [PubMed] [Google Scholar]
- 6. Cesari I. M., Carvalho E., Figueiredo Rodrigues M., Mendonça Bdos S., Amôedo N. D., Rumjanek F. D. (2014) Methyl jasmonate: putative mechanisms of action on cancer cells cycle, metabolism, and apoptosis. Int. J. Cell Biol. 2014, 572097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dang H. T., Lee H. J., Yoo E. S., Hong J., Bao B., Choi J. S., Jung J. H. (2008) New jasmonate analogues as potential anti-inflammatory agents. Bioorganic Medicinal Chemistry 16, 10228–10235 [DOI] [PubMed] [Google Scholar]
- 8. Dang H. T., Lee Y. M., Kang G. J., Yoo E. S., Hong J., Lee S. M., Lee S. K., Pyee Y., Chung H. J., Moon H. R., Kim H. S., Jung J. H. (2012) In vitro stability and in vivo anti-inflammatory efficacy of synthetic jasmonates. Bioorganic Medicinal Chemistry 20, 4109–4116 [DOI] [PubMed] [Google Scholar]
- 9. Kota B. P., Huang T. H., Roufogalis B. D. (2005) An overview on biological mechanisms of PPARs. Pharmacol. Res. 51, 85–94 [DOI] [PubMed] [Google Scholar]
- 10. Michalik L., Auwerx J., Berger J. P., Chatterjee V. K., Glass C. K., Gonzalez F. J., Grimaldi P. A., Kadowaki T., Lazar M. A., O'Rahilly S., Palmer C. N., Plutzky J., Reddy J. K., Spiegelman B. M., Staels B., Wahli W. (2006) International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol. Rev. 58, 726–741 [DOI] [PubMed] [Google Scholar]
- 11. Straus D. S., Glass C. K. (2007) Anti-inflammatory actions of PPAR ligands: new insights on cellular and molecular mechanisms. Trends Immunol. 28, 551–558 [DOI] [PubMed] [Google Scholar]
- 12. Dubuquoy L., Rousseaux C., Thuru X., Peyrin-Biroulet L., Romano O., Chavatte P., Chamaillard M., Desreumaux P. (2006) PPARγ as a new therapeutic target in inflammatory bowel diseases. Gut 55, 1341–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kaminska B. (2005) MAPK signalling pathways as molecular targets for anti-inflammatory therapy–from molecular mechanisms to therapeutic benefits. Biochim. Biophys. Acta 1754, 253–262 [DOI] [PubMed] [Google Scholar]
- 14. Waetzig G. H., Seegert D., Rosenstiel P., Nikolaus S., Schreiber S. (2002) p38 mitogen-activated protein kinase is activated and linked to TNF-α signaling in inflammatory bowel disease. J. Immunol. 168, 5342–5351 [DOI] [PubMed] [Google Scholar]
- 15. Broom O. J., Widjaya B., Troelsen J., Olsen J., Nielsen O. H. (2009) Mitogen activated protein kinases: a role in inflammatory bowel disease? Clin. Exp. Immunol. 158, 272–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tak P. P., Firestein G. S. (2001) NF-κB: a key role in inflammatory diseases. J. Clin. Investig. 107, 7–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Atreya I., Atreya R., Neurath M. F. (2008) NF-κB in inflammatory bowel disease. J. Int. Med. 263, 591–596 [DOI] [PubMed] [Google Scholar]
- 18. Rogler G., Brand K., Vogl D., Page S., Hofmeister R., Andus T., Knuechel R., Baeuerle P. A., Schölmerich J., Gross V. (1998) Nuclear factor κB is activated in macrophages and epithelial cells of inflamed intestinal mucosa. Gastroenterology 115, 357–369 [DOI] [PubMed] [Google Scholar]
- 19. Rotondo D., Davidson J. (2002) Prostaglandin and PPAR control of immune cell function. Immunology 105, 20–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sainathan S. K., Hanna E. M., Gong Q., Bishnupuri K. S., Luo Q., Colonna M., White F. V., Croze E., Houchen C., Anant S., Dieckgraefe B. K. (2008) Granulocyte macrophage colony-stimulating factor ameliorates DSS-induced experimental colitis. Inflammatory Bowel Diseases 14, 88–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Egea L., McAllister C. S., Lakhdari O., Minev I., Shenouda S., Kagnoff M. F. (2013) GM-CSF produced by nonhematopoietic cells is required for early epithelial cell proliferation and repair of injured colonic mucosa. J. Immunol. 190, 1702–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Puleston J., Cooper M., Murch S., Bid K., Makh S., Ashwood P., Bingham A. H., Green H., Moss P., Dhillon A., Morris R., Strobel S., Gelinas R., Pounder R. E., Platt A. (2005) A distinct subset of chemokines dominates the mucosal chemokine response in inflammatory bowel disease. Alimentary Pharmacology Therapeutics 21, 109–120 [DOI] [PubMed] [Google Scholar]
- 23. Garat C., Arend W. P. (2003) Intracellular IL-1Ra type 1 inhibits IL-1-induced IL-6 and IL-8 production in Caco-2 intestinal epithelial cells through inhibition of p38 mitogen-activated protein kinase and NF-κB pathways. Cytokine 23, 31–40 [DOI] [PubMed] [Google Scholar]
- 24. Hollenbach E., Neumann M., Vieth M., Roessner A., Malfertheiner P., Naumann M. (2004) Inhibition of p38 MAP kinase- and RICK/NF-κB-signaling suppresses inflammatory bowel disease. FASEB J. 18, 1550–1552 [DOI] [PubMed] [Google Scholar]
- 25. Mitsuyama K., Suzuki A., Tomiyasu N., Tsuruta O., Kitazaki S., Takeda T., Satoh Y., Bennett B. L., Toyonaga A., Sata M. (2006) Pro-inflammatory signaling by Jun-N-terminal kinase in inflammatory bowel disease. Int. J. Mol. Med. 17, 449–455 [PubMed] [Google Scholar]
- 26. Assi K., Pillai R., Gómez-Munoz A., Owen D., Salh B. (2006) The specific JNK inhibitor SP600125 targets tumour necrosis factor-α production and epithelial cell apoptosis in acute murine colitis. Immunology 118, 112–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Caruso R., Fina D., Peluso I., Stolfi C., Fantini M. C., Gioia V., Caprioli F., Del Vecchio Blanco G., Paoluzi O. A., Macdonald T. T., Pallone F., Monteleone G. (2007) A functional role for interleukin-21 in promoting the synthesis of the T-cell chemoattractant, MIP-3α, by gut epithelial cells. Gastroenterology 132, 166–175 [DOI] [PubMed] [Google Scholar]
- 28. Kwon K. H., Ohigashi H., Murakami A. (2007) Dextran sulfate sodium enhances interleukin-1 beta release via activation of p38 MAPK and ERK1/2 pathways in murine peritoneal macrophages. Life Sciences 81, 362–371 [DOI] [PubMed] [Google Scholar]
- 29. Carothers A. M., Davids J. S., Damas B. C., Bertagnolli M. M. (2010) Persistent cyclooxygenase-2 inhibition downregulates NF-κB, resulting in chronic intestinal inflammation in the min/+ mouse model of colon tumorigenesis. Cancer Research 70, 4433–4442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Banchereau J., Pascual V., O'Garra A. (2012) From IL-2 to IL-37: the expanding spectrum of anti-inflammatory cytokines. Nature Immunology 13, 925–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Setoguchi R., Hori S., Takahashi T., Sakaguchi S. (2005) Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J. Exp. Med. 201, 723–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Müzes G., Molnár B., Tulassay Z., Sipos F. (2012) Changes of the cytokine profile in inflammatory bowel diseases. World Journal of Gastroenterology 18, 5848–5861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stein M., Keshav S., Harris N., Gordon S. (1992) Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J. Exp. Med. 176, 287–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Suttles J., Milhorn D. M., Miller R. W., Poe J. C., Wahl L. M., Stout R. D. (1999) CD40 signaling of monocyte inflammatory cytokine synthesis through an ERK1/2-dependent pathway. A target of interleukin (IL)-4 and IL-10 anti-inflammatory action. J. Biol. Chem. 274, 5835–5842 [DOI] [PubMed] [Google Scholar]
- 35. Xavier R. J., Podolsky D. K. (2007) Unravelling the pathogenesis of inflammatory bowel disease. Nature 448, 427–434 [DOI] [PubMed] [Google Scholar]
- 36. Kim J. Y., Kajino-Sakamoto R., Omori E., Jobin C., Ninomiya-Tsuji J. (2009) Intestinal epithelial-derived TAK1 signaling is essential for cytoprotection against chemical-induced colitis. PloS one 4, e4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kolonics A., Apáti A., Jánossy J., Brózik A., Gáti R., Schaefer A., Magócsi M. (2001) Activation of Raf/ERK1/2 MAP kinase pathway is involved in GM-CSF-induced proliferation and survival but not in erythropoietin-induced differentiation of TF-1 cells. Cellular Signalling 13, 743–754 [DOI] [PubMed] [Google Scholar]
- 38. Mudter J., Neurath M. F. (2007) Il-6 signaling in inflammatory bowel disease: pathophysiological role and clinical relevance. Inflammatory Bowel Diseases 13, 1016–1023 [DOI] [PubMed] [Google Scholar]
- 39. Leonard M., Ryan M. P., Watson A. J., Schramek H., Healy E. (1999) Role of MAP kinase pathways in mediating IL-6 production in human primary mesangial and proximal tubular cells. Kidney Int. 56, 1366–1377 [DOI] [PubMed] [Google Scholar]
- 40. Daig R., Andus T., Aschenbrenner E., Falk W., Schölmerich J., Gross V. (1996) Increased interleukin 8 expression in the colon mucosa of patients with inflammatory bowel disease. Gut 38, 216–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kolaczkowska E., Kubes P. (2013) Neutrophil recruitment and function in health and inflammation. Nature Reviews. Immunology 13, 159–175 [DOI] [PubMed] [Google Scholar]
- 42. Neurath M. F. (2014) Cytokines in inflammatory bowel disease. Nature Reviews. Immunology 14, 329–342 [DOI] [PubMed] [Google Scholar]
- 43. Fujiie S., Hieshima K., Izawa D., Nakayama T., Fujisawa R., Ohyanagi H., Yoshie O. (2001) Proinflammatory cytokines induce liver and activation-regulated chemokine/macrophage inflammatory protein-3α/CCL20 in mucosal epithelial cells through NF-κB [correction of NK-κB]. Int. Immunol. 13, 1255–1263 [DOI] [PubMed] [Google Scholar]
- 44. Baker M. S., Chen X., Rotramel A., Nelson J., Kaufman D. B. (2003) Proinflammatory cytokines induce NF-κB-dependent/NO-independent chemokine gene expression in MIN6 beta cells. J. Surgical Res. 110, 295–303 [DOI] [PubMed] [Google Scholar]