

Background: The endo-α-d-N-acetylgalactosaminidase SpGH101 from Streptococcus pneumoniae hydrolyzes the O-linked T-antigen from proteins.
Results: SpGH101 displays an unusual conformational change on substrate binding and a distinctive arrangement of its catalytic machinery.
Conclusion: Substrate hydrolysis proceeds through a retaining mechanism with a proton shuttle.
Significance: This is the first evidence of proton shuttle in a retaining glycoside hydrolase.
Keywords: crystal structure, enzyme catalysis, glycobiology, glycoside hydrolase, mucin, Streptococcus pneumoniae
Abstract
O-Linked glycosylation is one of the most abundant post-translational modifications of proteins. Within the secretory pathway of higher eukaryotes, the core of these glycans is frequently an N-acetylgalactosamine residue that is α-linked to serine or threonine residues. Glycoside hydrolases in family 101 are presently the only known enzymes to be able to hydrolyze this glycosidic linkage. Here we determine the high-resolution structures of the catalytic domain comprising a fragment of GH101 from Streptococcus pneumoniae TIGR4, SpGH101, in the absence of carbohydrate, and in complex with reaction products, inhibitor, and substrate analogues. Upon substrate binding, a tryptophan lid (residues 724-WNW-726) closes on the substrate. The closing of this lid fully engages the substrate in the active site with Asp-764 positioned directly beneath C1 of the sugar residue bound within the −1 subsite, consistent with its proposed role as the catalytic nucleophile. In all of the bound forms of the enzyme, however, the proposed catalytic acid/base residue was found to be too distant from the glycosidic oxygen (>4.3 Å) to serve directly as a general catalytic acid/base residue and thereby facilitate cleavage of the glycosidic bond. These same complexes, however, revealed a structurally conserved water molecule positioned between the catalytic acid/base and the glycosidic oxygen. On the basis of these structural observations we propose a new variation of the retaining glycoside hydrolase mechanism wherein the intervening water molecule enables a Grotthuss proton shuttle between Glu-796 and the glycosidic oxygen, permitting this residue to serve as the general acid/base catalytic residue.
Introduction
Mucins are highly modified glycoproteins that serve as the first line of defense in preserving the integrity of mucosal epithelial cell layers against chemical, physical, and microbial challenge. The high carbohydrate content of mucins contributes to their biophysical and biochemical properties, conferring protection to mucosal membranes by lubricating the cell surface and inhibiting bacterial colonization. The sugars comprising this glycoconjugate are complex highly branched O-glycans that are linked via an α-glycosidic bond to the hydroxyl group of serine and threonine residues.
In animals, the core of these O-linked mucin glycans is an N-acetyl-d-galactosamine (GalNAc) residue. This base structure, referred to as the Tn-antigen, creates the foundation for what has been defined as eight O-glycan cores (1), which are often elaborated to form more complex structures. Only one family of enzyme is known to cleave the glycosidic bond joining GalNAc to serine or threonine: the family 101 glycoside hydrolases (2–6). To date, most GH1016 enzymes that have been characterized are active on the core GalNAc bearing a terminal β-1,3-linked galactose residue, which makes up the core 1 O-glycan (Galβ1–3GalNAcα1-Ser/Thr); a structure that is frequently referred to as the Thomsen-Friedenreich (T or TF) antigen. Thus, these enzymes are classified as endo-α-N-acetylgalactosaminidases. The T-antigen is the preferred substrate for GH101 enzymes from Bifidobacterium longum (EngBF) and Streptococcus pneumoniae (Eng or SpGH101 as we will refer to it here). EngCP from Clostridium perfingens also displays activity on the core 2 O-glycan (Galβ1–3(GlcNAcβ1-6)GalNAcα1-Ser/Thr), albeit at a lower level than on the T-antigen (5). Other examples from Propionibacterium acnes and Enterococcus faecalis are active on the core 3 O-glycan (GlcNAcβ1–3GalNAcα1-Ser/Thr) (6, 7).
EngBF was the founding member of glycoside hydrolase family 101 (3) and uses a retaining catalytic mechanism, involving the transient formation of a glycosyl enzyme intermediate. Structural studies of EngBF and SpGH101 from S. pneumoniae strain R6, here referred to as SpGH101R6, revealed the similarity of the (β/α)8-barrel catalytic module to α-amylases in GH family 13 (3, 8). The identity of the catalytic residues were proposed for the GH101 enzymes based on similar, but not exact, spatial positioning of the GH13 catalytic residues with an aspartate and a glutamate in GH101. Mutagenesis and chemical rescue studies provide strong evidence in favor of residues Asp-764 and Glu-796 in SpGH101R6 acting as the catalytic nucleophile and catalytic general acid/base residues, respectively (9).
The almost strict occurrence of GH101 enzymes in host-adapted bacteria suggests they play a role in either commensalism or pathogenesis. Indeed, for example, deletion of the gene encoding SpGH101 reduced the ability of S. pneumoniae strain 1121 to colonize the upper airways in a mouse model of nasopharyngeal colonization (10). These observations make the molecular bases by which SpGH101 binds and processes its substrates of interest. Such information could prove useful in exploiting these enzymes as tools for biotechnology as well as to design inhibitors that could have value as antimicrobials. Presently, however, experimental studies defining the molecular interactions that enable substrate recognition and turnover are lacking because of the absence of GH101 structures in complex with ligands.
To gain a better understanding of these molecular details, we focused on structural studies of SpGH101 from the S. pneumoniae TIGR4 strain, which has 99% amino acid sequence identity with SpGH101R6, in complex with a series of carbohydrate ligands including substrates, products, and inhibitor. The results collectively suggest an unusual reaction mechanism involving conformational change during substrate binding coupled with catalysis involving the general acid/base catalytic residue functioning from a distance by way of a short Grotthuss proton shuttle mediated by a single intervening water molecule.
Experimental Procedures
Cloning, Protein Production, Purification, Crystallization, and Structure Determination
A truncated form of SpGH101 comprising residues 317–1425 of the full-length protein (S. pneumoniae TIGR4 strain) and with an N-terminal His6 tag, referred to as SpGH101(N) was cloned, expressed, purified, and crystallized as previously described (2). Selenomethionine-labeled SpGH101(N) was produced using Escherichia coli B834 (DE3) as the expression strain (Novagen). The defined media containing selenomethionine was prepared according to the instructions of the manufacturer (Athena Enzyme Systems). Cells were harvested by centrifugation at 6000 × g for 10 min, chemically lysed, the supernatant was cleared by centrifugation at 27,000 × g for 45 min, and the polypeptides were purified from the cell-free extract using immobilized metal affinity chromatography following the methods for the unlabeled protein (2). The purity of fractions was assessed using SDS-PAGE and those deemed to be greater than 95% pure were pooled, concentrated, and buffer exchanged into 20 mm Tris-HCl, pH 8.0, in a stirred ultrafiltration unit (Amicon) using a 10-kDa molecular mass cut-off membrane (Filtron). Selenomethionine-substituted SpGH101(N) protein was further purified by size-exclusion chromatography using Sephacryl S-200 (GE Biosciences) in 20 mm Tris-HCl, pH 8.0. Prior to crystallization, the selenomethionine-substituted SpGH101(N) was concentrated to 15 mg/ml in 20 mm Tris-HCl, pH 8.0.
Selenomethionine-substituted SpGH101(N) crystals were grown by the hanging drop vapor diffusion method in conditions identical to the unlabeled protein: 2-μl drops with 1:1 ratio of protein to 25% (w/v) polyethylene glycol (PEG) 1500 (Hampton Research) at 292 K. Crystals of native and selenomethionine-substituted SpGH101(N) were cryoprotected in 1 μl of 33% (w/v) PEG 1500 supplemented with 6% (v/v) MPD (Hampton Research), and flash-cooled directly in a nitrogen-gas stream at 113 K. Diffraction data for native crystals were collected on beamline 9-2 of the Stanford Synchrotron Light Source and data processed with XDS and scaled with SCALA (11, 12). A single-wavelength anomalous dispersion dataset optimized for selenium was collected for crystals of the selenomethionine-substituted SpGH101(N) at the Canadian Light Source on beamline 08ID-1 and the data processed with MOSFLM and scaled with SCALA (13). Using this data, the heavy atom substructure determination, phasing, and density modification was performed with AutoSHARP (14). Twenty-two selenium positions present in the single SpGH101(N) monomer in the asymmetric unit were used for phasing with the full 2.45-Å resolution dataset (acentric/centric figures of merit 0.35/0.14; phasing power, 0.93). The phases resulting from density improvement were of sufficient quality for BUCCANEER (15) to build a virtually complete model. This initial model was used as a search template for molecular replacement using PHASER and the higher resolution native data set (16). The native structure was iteratively improved with cycles of manual building with COOT and refinement with REFMAC (17, 18).
Crystals of recombinant SpGH101(N) were suitable to determine an initial structure of the protein; however, crystals were difficult to reproduce consistently and were not sufficiently robust to obtain structures in complex with ligands. Therefore, an alternate construct of SpGH101, called SpGH101(C), was cloned into pET22. This gene fusion encoded a methionine immediately preceding the gene fragment encoding amino acids 317–1425, which was followed by a sequence that added a C-terminal His6 tag and stop codon. This pET22 construct was used as a template to generate D764N and E796Q mutants by standard PCR site-directed mutagenesis procedures (see Table 1 for primer sequences). The DNA sequence fidelity of all constructs was verified using bidirectional sequencing.
TABLE 1.
Oligonucleotide primers
| Name | Nucleotide sequence | Used to clone |
|---|---|---|
| GH101pET22For | TATACATATGGAAAAAGAAACAGGTCCTG | SpGH101(C) |
| GH101pET22Rev | CGGCGTCTCGAGCAACATCTTACCTGTTAGGG | SpGH101(C) |
| GH101D764NFor | CTTTATCTATGTGAACGTTTGGGGTAATGG | SpGH101(C)D764N |
| GH101D764NRev | CCATTACCCCAAACGTTCACATAGATAAAG | SpGH101(C)D764N |
| GH101E796QFor | CGCTTTGCGATCCAGTGGGGCCATGGTGG | SpGH101(C)E796Q |
| GH101E796QRev | CCACCATGGCCCCACTGGATCGCAAAGCG | SpGH101(C)E796Q |
Recombinant SpGH101(C) and mutants were produced in E. coli as for SpGH101(N) and purified to homogeneity by nickel-affinity chromatography and anion exchangechromatography. SpGH101(C), SpGH101(C)D764N, and SpGH101(C)E796Q in 20 mm Tris-HCl, pH 8.0, were crystallized by mixing equal volumes of 15 mg/ml of protein with a solution consisting of 12% (w/v) PEG 3350 and 0.15 m ammonium citrate, pH 7.0, using the sitting-drop vapor-diffusion method at 292 K. Plate shaped crystals developed after ∼3 days and were utilized for subsequent microseeding in future hanging-drop setups.
Complexes of SpGH101(C) with T-antigen methyl glycoside (methyl 2-acetamido-2-deoxy-3-O-(β-d-galactopyranosyl)-α-d-galactopyranoside; Galβ(1–3)GalNAcα(1-)OMe; Toronto Research Chemicals) and PugT (3-O-(1-β-d-galactopyrano)-2-N-acetyl-2-deoxy-d-galactopyranosylidene)amino-N-phenylcarbamate; see below for synthesis) were generated by soaking crystals in crystallization solution containing a high excess of compound for ∼5 min. This approach was also used to soak crystals of SpGH101(C)E796Q in excess PNP-T-antigen (4-nitrophenyl 2-acetamido-2-deoxy-3-O-(β-d-galactopyranosyl)-α-d-galactopyranoside; Galβ(1–3)GalNAcα(1-)PNP; Toronto Research Chemicals) and a T-antigen containing glycopeptide (a kind gift from Professor Lai-Xi Wang); SpGH101(C)D764N was soaked in serinyl T-antigen (serinyl 2-acetamido-2-deoxy-3-O-(β-d-galactopyranosyl)-α-d-galactopyranoside; Galβ(1–3)GalNAcα(1-)serine; Toronto Research Chemicals). In all cases, crystals were cryoprotected by rapidly drawing through crystallization solution supplemented with 30% (v/v) ethylene glycol and cooled in a liquid nitrogen stream at 113 K. Data were collected on SSRL beamlines 11-2 and 9-2 or on a home beam comprising a MM002 x-ray generator coupled to Osmic “Blue” Optics, an RAXIS 4++ x-ray detector, and an Oxford Cryostream 700 cryocooler. Data collected at the SSRL was processed with XDS and SCALA, whereas data collected on the home beam was processed with MOSFLM and SCALA. These structures were solved by molecular replacement using the structure of SpGH101(N) as a starting point. The structures were iteratively improved with cycles of manual building with COOT and refinement with REFMAC.
In all cases, waters were added using the FINDWATERS option in COOT and manually inspected prior to the final refinement. Refinement procedures were monitored by flagging 5% of all observation as “free” (19). Model validation was performed with MOLPROBITY (20, 21). All data collection and model statistics are shown in Table 2.
TABLE 2.
Data collection and structure refinement statistics
| SpGH101(N) SeMet | SpGH101(N) | SpGH101(C) + T-antigen methyl glycoside | SpGH101(C) + PUGT | SpGH101(C)D764N + serinyl T-antigen | SpGH101(C)E796Q + glycopeptide T-antigen product | SpGH101(C)E796Q + pNP-T-antigen product | |
|---|---|---|---|---|---|---|---|
| Data collection | |||||||
| Beamline | CLS 08ID-1 | SSRL 9–2 | SSRL 11–1 | SSRL 9–2 | SSRL 9–2 | MM002 | SSRL 9–1 |
| Wavelength | 0.97905 | 0.97946 | 0.97945 | 0.97901 | 0.86700 | 1.5419 | 0.86700 |
| Space group | P21 | P21 | P22121 | P22121 | P22121 | P22121 | P22121 |
| a, b, c (Å) | 78.0, 89.6, 87.2 (β = 111.3) | 76.3, 89.1, 88.6 (β = 110.9) | 86.7, 121.8, 139.4 | 87.1, 122.1, 139.8 | 87.1, 122.0, 139.6 | 87.2, 121.9, 139.3 | 86.9, 121.6, 139.4 |
| Resolution (Å) | 20-2.45 (2.58-2.45) | 30-1.85 (1.95-1.85) | 50-1.80 (1.86-1.80) | 50-1.46 (1.53-1.46) | 50-1.80 (1.86-1.80) | 30-2.50 (2.58-2.50) | 50-1.75 (1.84-1.75) |
| Rmerge | 0.048 (0.442) | 0.043 (0.376) | 0.090 (0.516) | 0.077 (0.375) | 0.059 (0.445) | 0.159 (0.496) | 0.067 (0.398) |
| I/σI | 47.4 (4.4) | 47.1 (4.6) | 22.7 (4.3) | 16.6 (4.9) | 24.8 (4.3) | 6.2 (2.1) | 24.8 (4.4) |
| Completeness (%) | 97.4 (83.4) | 99.1 (98.2) | 99.8 (99.7) | 99.6 (98.1) | 99.8 (99.5) | 99.9 (99.8) | 99.5 (96.6) |
| Redundancy | 7.1 (7.3) | 6.9 (6.5) | 7.8 (7.8) | 6.8 (6.0) | 8.1 (7.6) | 3.6 (3.4) | 8.5 (5.9) |
| Refinement | |||||||
| Resolution (Å) | 1.85 | 1.80 | 1.46 | 1.80 | 2.5 | 1.75 | |
| No. of reflections | 88792 | 129705 | 242256 | 129714 | 49388 | 140912 | |
| Rwork/Rfree | 0.14/0.18 | 0.15/0.18 | 0.14/0.16 | 0.15/0.18 | 0.17/0.23 | 0.15/0.17 | |
| No. of atoms | |||||||
| Protein | 8762 | 8937 | 8983 | 8892 | 8891 | 8901 | |
| Ion | 5 | 4 | 4 | 4 | 4 | 4 | |
| Liganda | NAb | 27 | 36 | 32 | 26 | 26 | |
| Solvent | 1517 | 1536 | 1803 | 1673 | 1036 | 1537 | |
| B-factors | |||||||
| Protein | 11.4 | 16.2 | 11.2 | 15.0 | 19.3 | 17.2 | |
| Ion | 8.8 | 15.7 | 13.5 | 12.3 | 17.6 | 13.4 | |
| Liganda | NA | 21.5 | 14.7 | 16.4 | 19.6 | 15.1 | |
| Solvent | 24.2 | 30.1 | 25.5 | 30.7 | 21.8 | 31.7 | |
| R.m.s. deviation | |||||||
| Bond lengths (Å) | 0.012 | 0.011 | 0.017 | 0.012 | 0.012 | 0.012 | |
| Bond angles (degrees) | 1.467 | 1.467 | 1.793 | 1.422 | 1.507 | 1.449 | |
| Ramachandran | |||||||
| Preferred (%) | 97.4 | 97.0 | 97.4 | 97.4 | 97.0 | 97.3 | |
| Allowed (%) | 2.5 | 2.9 | 2.4 | 2.3 | 2.9 | 2.5 | |
| Disallowed (%) | 0.1 | 0.1 | 0.2 | 0.3 | 0.1 | 0.2 | |
a Carbohydrates and carbohydrate derivatives only.
b NA, not applicable.
General Synthetic Procedures
PUGT was synthesized according to the steps shown in Scheme 1. All solvents were dried prior to use. Synthetic reactions were monitored by TLC using Merck Kieselgel 60 F254 aluminum-backed sheets. Compounds were detected by charring with a 10% concentrated sulfuric acid in ethanol solution and heating. Flash chromatography under a positive pressure was performed with Merck Kieselgel 60 (230–400 mesh) using specified eluants. 1H and 13C NMR spectra were recorded on 600 MHz (150 MHz for 13C) (chemical shifts quoted relative to CDCl3 or CD3OD where appropriate).
SCHEME 1.

Synthesis of PUGT. a, BnOH, AcCl, 60 °C, 80%; b, benzaldehyde, ZnCl2, room temperature, 75%; c, Ac2O, NaOAc, reflux, 53%; d, Hg(CN)2, benzene/MeNO2, 40–45 °C; e, 60% HOAc, 60 °C; f, Ac2O, pyridine, 4-dimethylaminopyridine, room temperature, 61% (3 steps); g, H2, Pd(OH)2, HOAc, room temperature, 90%; h, NH2OH·HCl, pyridine, MeOH, reflux, 86%; i, DBU, NCS, dichloromethane, −40 °C to room temperature, 35%; j, phenyl isocyanate, THF, room temperature, 57%; k, NH3/MeOH, 0 °C, 52%.
Benzyl(2-acetamido-4,6-benzylidene-2-deoxy-α-d-galactopyranoside (1)
2-Acetamido-2-deoxy-d-galactose (22) (8.0 g, 36.2 mmol) was dissolved in BnOH (100 ml) at 60 °C and acetyl chloride (2.0 ml) was added. The resulting reaction mixture was stirred overnight at 60 °C and then was cooled to room temperature. To the mixture was added excess Et2O, and the resulting white solid precipitate was filtered and washed with Et2O, then dried under vacuum to yield benzyl 2-acetamido-2-deoxy-α-d-galactopyranoside as a white solid (9.0 g, 80%). This intermediate glycoside was used directly without further purification (9.0 g, 28.9 mmol) and suspended in benzaldehyde (30 ml) with ZnCl2 (4.5 g). The reaction mixture was stirred at room temperature overnight to yield a clear solution to which excess cold water and hexanes was added. The resulting yellowish precipitate was collected by suction filtration, washed thoroughly with water and hexanes and then dissolved in dichloromethane (300 ml). This organic solution was washed with brine and then dried (MgSO4). After filtration and removal of the solvent the residue was dried under vacuum to yield the desired product 1 as a solid foam (22) (8.7 g, 75%). 1H NMR (600 MHz, CDCl3): δ 7.53–7.50 (m, 2H), 7.40–7.35 (m, 6H), 7.34–7.31 (m, 2H), 5.73 (d, 1H, J = 9.0 Hz), 5.57 (s, 1H), 5.05 (d, 1H, J = 3.6 Hz), 4.73 (d, 1H, J = 12 Hz), 4.55 (d, 1H, J = 12 Hz), 4.47 (ddd, 1H, J = 10.8, 9.6, 3.6 Hz), 4.24 (dd, 1H, J = 12.6, 1.8 Hz), 4.23 (d, 1H, J = 1.2 Hz), 4.04 (dd, 1H, J = 12.6, 1.8 Hz), 3.87 (dd, 1H, J = 10.8, 3.0 Hz), 3.67 (s, 1H), 3.03 (brs, 1H), 1.98 (s, 3H). 13C NMR (150 MHz, CDCl3): δ 171.2, 137.4, 137.1, 129.2, 128.6, 128.2 (2C), 128.0, 126.3, 101.3, 97.9, 75.5, 70.1, 69.3, 69.1, 63.2, 50.3, 23.4.
Benzyl(2,3,4,6-tetra-O-acetyl-β-d-galactopyranosyl)-(1→3)-2-acetamido-4,6-benzylidene-2-deoxy-α-d-galactopyranoside (3) (23)
Acceptor 1 (4.85 g, 12.1 mmol) and Hg(CN)2 (6.50 g, 25.6 mmol) were suspended in a solution of benzene/MeNO2 (1:1, 240 ml). The mixture was distilled until 120 ml of solvent was removed. The temperature was then adjusted to 40–45 °C after which donor 2,3,4,6-tetra-O-acetyl-α-d-galactopyranosyl bromide 2 (24) (10.0 g, 24.3 mmol) in a solution of benzene/MeNO2 (1:1, 48 ml) was added. The resulting mixture was stirred at 40–45 °C overnight, then cooled to room temperature, diluted with benzene (300 ml), and filtered. The organic layer was washed with saturated NaHCO3, 10% KI, and brine, after which it was dried (MgSO4). After filtration and removal of the solvent, the residue was dried under vacuum to yield product 3 as clear syrup (23) that was used in the next step without further purification.
Benzyl(2,3,4,6-Tetra-O-acetyl-β-d-galactopyranosyl)-(1→3)-2-acetamido-2-deoxy-α-d-galactopyranoside (4) (23)
Compound 3 was dissolved in aqueous 60% HOAc (140 ml), stirred at 60 °C overnight, cooled to room temperature, after which the solvent was removed under high vacuum and the residue co-evaporated with toluene (4 × 30 ml). The resulting residue was then dried under vacuum to yield product 4 as a solid foam (23) that was used in the next step reaction without further purification.
Benzyl(2,3,4,6-tetra-O-acetyl-β-d-galactopyranosyl)-(1→3)-2-acetamido-4,6-di-O-acetyl-2-deoxy-α-d-galactopyranoside (5) (22)
Compound 4 and 4-dimethylaminopyridine (610 mg) were dissolved in pyridine (50 ml) after which acetic anhydride (4.60 ml, 48.4 mmol) was added at room temperature. The resulting mixture was stirred at room temperature overnight and MeOH (10 ml) was then added. The solvent was removed under reduced pressure and the resulting residue was co-evaporated with toluene (4 × 30 ml) to obtain a syrupy residue. Gradient silica gel column chromatography using chloroform:acetone (4:1 to 2:1) as the mobile phase afforded product 5 as a solid foam (22) (5.36 g, 61% for 3 steps). 1H NMR (600 MHz, CDCl3): δ 7.36–7.26 (m, 5H), 5.72 (d, 1H, J = 9.0 Hz), 5.34 (d, 1H, J = 2.4 Hz), 5.29 (d, 1H, J = 3.0 Hz), 5.07 (dd, 1H, J = 10.2, 8.4 Hz), 4.98 (d, 1H, J = 3.0 Hz), 4.90 (dd, 1H, J = 10.2, 3.0 Hz), 4.65 (d, 1H, J = 11.4 Hz), 4.55 (d, 1H, J = 8.4 Hz), 4.49 (td, 1H, J = 9.0, 3.6 Hz), 4.42 (d, 1H, J = 11.4 Hz), 4.16–4.11 (m, 2H), 4.08 (t, 1H, J = 6.0 Hz), 3.97 (dd, 1H, J = 12.6, 8.4 Hz), 3.90 (dd, 1H, J = 11.4, 3.6 Hz), 3.83 (t, 1H, J = 6.6 Hz), 2.09 (s, 3H), 2.07 (s, 3H), 2.04 (s, 3H), 2.00 (s, 3H), 1.98 (s, 3H), 1.91 (s, 6H). 13C NMR (150 MHz, CDCl3): δ 170.4, 170.3, 170.2, 170.0 (2C), 169.6, 169.5, 136.6, 128.5, 128.3, 128.2, 100.3, 97.0, 72.6, 70.6 (2C), 70.0, 68.7, 68.4, 67.4, 66.6, 62.5, 61.0, 23.1, 20.6 (3C), 20.5 (2C), 20.4.
2,3,4,6-Tetra-O-acetyl-β-d-galactopyranosyl-(1→3)-2-acetamido-4,6-di-O-acetyl-2-deoxy-d-galactopyranose (6) (25)
Compound 5 (2.57 g, 3.54 mmol) was dissolved in acetic acid (50 ml) and Pd(OH)2 (300 mg) was added to form a suspension. The atmosphere of the reaction was replaced by H2 and the reaction stirred at room temperature overnight. The mixture was then filtered by Celite, washed with MeOH, and the solvent removed under reduced pressure. The residue was co-evaporated with toluene (3 × 15 ml) after which the desired material was purified by gradient silica gel flash column chromatography using as the mobile phase chloroform:acetone (1:1.5 to 1:1) to obtain product 6 as a solid foam (2.02 g, 90%) (25). 1H NMR (600 MHz, CDCl3): δ 6.02 (d, 1H, J = 8.4 Hz), 5.37 (d, 1H, J = 2.4 Hz), 5.34 (dd, 1H, J = 3.6, 0.6 Hz), 5.33 (t, 1H, J = 3.6Hz), 5.11 (dd, 1H, J = 10.2, 7.8 Hz), 4.94 (dd, 1H, J = 10.2, 3.6 Hz), 4.63 (d, 1H, J = 7.8 Hz), 4.40 (ddd, 1H, J = 11.4, 9.0, 3.6 Hz), 4.33 (t, 1H, J = 6.6 Hz), 4.15–4.11 (m, 3H), 4.05 (dd, 1H, J = 10.8, 3.6 Hz), 3.96 (dd, 1H, J = 12.0, 7.2 Hz), 3.89 (td, 1H, J = 7.8, 1.2 Hz), 2.15 (s, 3H), 2.11 (s, 3H), 2.07 (s, 3H), 2.05 (s, 3H), 2.04 (s, 3H), 2.00 (s, 3H), 1.96 (s, 3H). 13C NMR (150 MHz, CDCl3): δ 170.7, 170.6, 170.5, 170.3, 170.1, 170.0, 169.9, 100.3, 92.0, 72.2, 70.8 (2C), 70.0, 68.8, 68.6, 67.0, 66.7, 62.6, 61.0, 49.6, 23.2, 20.8 (2C), 20.7, 20.6 (2C), 20.5.
2,3,4,6-Tetra-O-acetyl-β-d-galactopyranosyl-(1→3)-2-acetamido-4,5-di-O-acetyl-2-deoxy-d-galactose Oxime (7)
An adaptation of the method used by Stubbs et al. (26) for the preparation of 3,4,6-tri-O-acetyl-2-acetamido-2-deoxy-d-galactose oxime was used. To a solution of 6 (2.12 g, 3.34 mmol) in MeOH (5.0 ml) was added pyridine (0.20 ml, 2.3 mmol) and hydroxylamine hydrochloride (348 mg, 5.00 mmol). The resulting solution was stirred at reflux overnight. The solution was cooled to room temperature and then concentrated to provide a residue. Co-evaporation of the residue with toluene (2 × 20 ml) provided a solid residue that was dissolved in EtOAc, washed with water (2 × 10 ml), brine (10 ml), and dried (MgSO4). After filtration and removal of the solvent under reduced pressure silica gel flash column chromatography using chloroform:acetone as the mobile phase (1:1.5 to 1:1) afforded oxime 7 as a solid foam (1.86 g, 85.7%).
2,3,4,6-Tetra-O-acetyl-β-d-galactopyranosyl-(1→3)-2-acetamido-4,5-di-O-acetyl-2-deoxy-d-galactonohydroximo-1,5-lactone (8)
An adaptation of the method used by Lammerts van Bueren et al. (27) for the preparation of 4-O-[2,3,4,6-tetra-O-acetyl-α-d-glucopyranosyl-1,4-tris-(2,3,6-tri-O-acetyl-α-d-glucopyranosyl)-1,4]-2,3,6-tri-O-acetyl-d-gluconohydroximo-1,5-lactone was used. To a solution of oxime 7 (1.00 g, 1.54 mmol) and N-chlorosuccinimide (226 mg, 1.69 mmol) in dichloromethane (15 ml) at −40 °C was added 1,8-Diazabicyclo[5.4.0]undec-7-ene (0.253 ml, 1.69 mmol) such that the temperature did not exceed −40 °C. The resulting mixture was allowed to stir for 30 min at a temperature carefully maintained between −40 and −45 °C. The mixture was then was allowed to warm to room temperature over 3 h in a dry ice bath. Water was then added to the mixture followed by EtOAc (100 ml). The organic layer was separated and washed with water (2 × 10 ml), brine (1 × 10 ml), and dried (MgSO4). After filtration and removal of the solvent under reduced pressure gradient silica gel flash column chromatography of the resultant residue using acetone:chloroform (1:3 to 1:2 to 1:1) as the mobile phase gave product 8 as a solid foam (350 mg, 35%). 1H NMR (600 MHz, CDCl3): δ 7.91 (s, 1H), 6.74 (d, 1H, J = 6.0 Hz), 5.40–5.36 (m, 2H), 5.15 (dd, 1H, J = 10.2, 7.8 Hz), 5.04 (dd, 1H, J = 10.2, 3.0 Hz), 4.83 (d, 1H, J = 7.8 Hz), 4.67 (dd, 1H, J = 6.0, 4.8 Hz), 4.60 (t, 1H, J = 4.2 Hz), 4.32 (dd, 1H, J = 12, 4.8 Hz), 4.29 (t, 1H, J = 4.2 Hz), 4.22 (dd, 1H, J = 12, 6.6 Hz), 4.15 (dd, 1H, J = 11.4, 7.2 Hz), 4.11 (dd, 1H, J = 10.8, 6.0 Hz), 3.98 (t, 1H, J = 7.2, 1.2 Hz), 2.16 (s, 3H), 2.14 (s, 3H), 2.07 (s, 3H), 2.06 (s, 3H), 2.03 (s, 3H), 2.02 (s, 3H), 1.97 (s, 3H). 13C NMR (150 MHz, CDCl3): δ 170.8, 170.6 (2C), 170.5, 170.2, 170.1, 169.9, 100.8, 84.4, 81.7, 71.0, 70.7, 69.4, 68.6, 67.0, 62.2, 61.1, 55.8, 22.8, 20.9, 20.8, 20.7 (2C), 20.6 (2C).
O-{2,3,4,6-Tetra-O-acetyl-β-d-galactopyranosyl-(1→3)-2-acetamido-4,5-di-O-acetyl-2-deoxy-d-galactopyranosylidene}amino N-phenylcarbamate (9)
An adaptation of the method used by Stubbs et al. (26) for the preparation of O-(3,4,6-tri-O-acetyl-2-acetamido-2-deoxy-d-galactopyranosylidene)amino N-phenylcarbamate was used. To a solution of 8 (172 mg, 0.265 mmol) and Et3N (112 μl, 0.795 mmol) in THF (10 ml), phenyl isocyanate was added (43.4 μl, 0.398 mmol). The resulting solution was stirred at room temperature overnight. Removal of the solvent under reduced pressure followed by gradient silica gel flash column chromatography of the resultant residue using acetone:chloroform (1:6 to 1:4) as the mobile phase yielded the product 9 as a solid foam (117 mg, 57%).
O-{β-d-Galactopyranosyl-(1→3)-2-acetamido-2-deoxy-d-galactopyranosylidene}amino N-phenylcarbamate (10)
An adaptation of the method used by Stubbs et al. (26) for the preparation of O-(2-acetamido-2-deoxy-d-galactopyranosylidene)amino-N-phenylcarbamate was used. To a cold (0 °C) solution of carbamate 9 (96.7 mg, 0.126 mmol) in MeOH (10 ml) a saturated solution of ammonia in MeOH was added (2.0 ml). The resulting solution was left to stand (0 °C, 3 h). Removal of the solvent under reduced pressue, followed by gradient silica gel flash column chromatography of the residue using EtOAc/MeOH/H2O as the mobile phase (30:2:1 to 15:2:1) afforded the desired product 10 as a colorless solid (33.5 mg, 52%). 1H NMR (600 MHz, CD3OD): δ 7.46 (d, 2H, J = 7.2 Hz), 7.30 (t-like, 2H, J = 7.2 Hz), 7.06 (t, 1H, J = 9.0 Hz), 5.27 (d, 1H, J = 6.0 Hz), 4.71 (dd, 1H, J = 6.0, 1.8 Hz), 4.65 (t, 1H, J = 6.0 Hz), 4.47 (d, 1H, J = 7.2 Hz), 4.05 (td, 1H, J = 7.2, 1.8 Hz), 3.83–3.76 (m, 2H), 3.83–3.76 (m, 2H), 3.74–3.67 (m, 3H), 3.56–3.52 (m, 2H), 3.48 (dd, 1H, J = 9.6, 3.6 Hz), 2.04 (s, 3H). 13C NMR (150 MHz, CD3OD): δ 173.4, 162.8, 154.7, 139.5, 129.9, 124.7, 120.4, 104.7, 87.7, 82.2, 77.2, 74.8, 72.3, 70.7, 70.4, 63.4, 62.9, 56.3, 22.8. HR-MS: calculated for C21H30N3O12 (M + H)+: 516.1824; found: 516.1832.
Results
SpGH101 Structure
The apo form of a truncated version of GH101 from S. pneumoniae R6 GH101 (SpGH101R6) was previously determined to 2.9-Å resolution; the resulting structure included seven of its 8 protein domains (PDB code 3ECQ; Fig. 1, A and B) (8). In an effort to generate a construct of SpGH101 that would enable determination of structures at higher resolutions, and in complex with ligands, we created a truncation of SpGH101 from S. pneumoniae TIGR4 (2). Overall, the GH101 enzymes from each S. pneumoniae strain have 99% amino acid sequence identity. Our construct of the protein from the TIGR4 strain included five of the central domains (Fig. 1A). The construct with an N-terminal His6 tag, SpGH101(N), crystallized in space group P21 with a single molecule in the asymmetric unit and scattered to 1.85-Å resolution (2). Selenomethionine-labeled SpGH101(N) allowed a preliminary structure to be determined to 2.35-Å resolution by the single-wavelength anomalous dispersion method (these experiments were performed prior to the release of the SpGH101R6 coordinates). An initial model comprising the single monomer in the asymmetric unit was generated by autobuilding with minimal manual intervention. This preliminary model was used to solve the structure of the unlabeled protein at the higher 1.85-Å resolution. The final model comprised 1104 residues, including residues 317–1418 of SpGH101. With the exception of the 3 amino acids immediately preceding the SpGH101 sequence, the N-terminal His6 tag and thrombin cleavage sequence could not be modeled. The structure of SpGH101(N) reveals the distorted (β/α)8-barrel core catalytic domain (domain 3; residues ∼600–890) flanked by four additional domains that are mainly of β-sheet character (domains 2, 4, 5, and 6; Fig. 1, A and B). The structure of SpGH101(N) is, as expected, highly similar to SpGH101R6, aligning with a root mean square deviation of 0.53 Å over 1045 matched Cα positions. A detailed description of the domain architecture of pneumococcal GH101 was previously given by Caines et al. (8) for SpGH101R6.
FIGURE 1.

Structural properties of the SpGH101 endo-α-N-acetylgalactosaminidase. A, a schematic of the domain architecture of SpGH101. The seven domains defined by the structure of SpGH101R6 (PDB ID 3ECQ) are indicated. The approximate number of the domain boundaries are provided above the schematic. The domains present in the SpGH101(N) and SpGH101(C) constructs used in this study is indicated below the schematic. B, schematic representation of the structure of SpGH101(N) comprising domains 2–6 (colored sequentially according to the colors in panel A and labeled). The three bound calcium ion (pink) and one manganese ion (purple) are shown as spheres and labeled. The active site pocket is shown as a gray surface. The structure is overlaid with the structure of SpGH101R6 (3ECQ), which is shown in transparent gray. C, an overlap of the active sites of SpGH101(N) (purple), SpGH101R6 (gray), and EngBF from Bifidobacterium longum (blue; PDB ID 2ZXQ). Relevant residues are shown as sticks and labeled black for the pneumococcal enzymes and blue for EngBF.
There are four cation coordination centers distributed throughout the structure of SpGH101(N) (Fig. 1B). Based on refined atom occupancies, temperature factors, and coordination geometry, three of these positions were modeled as Ca2+ ions and one as a Mn2+ ion. One Ca2+ (Ca1) is coordinated in the turn that precedes the C-terminal α-helix of domain 2. Additional Ca2+ ions, Ca2 and Ca3, are coordinated between two strands of domain 5 and two coil regions of domain six, respectively (Fig. 1B). The Mn2+ ion (Mn1) is coordinated between the catalytic domain (domain 3) and domain 6 (Fig. 1B). In the structure of SpGH101R6 an equivalent ion to Ca1 was not modeled, whereas the position occupied by Mn1 was modeled as a Na+ ion. SpGH101(N) aligns with EngBF from Bifidobacterium longum (determined to 2.0-Å resolution) (3) with a root mean square deviation of 1.00 Å over 929 matched Cα positions. The structure of EngBF contains 4 modeled Mn2+ ions that overlap with the 4 ions modeled in SpGH101(N). Given the presence of these ions at domain interfaces it is likely they play a structural role. Indeed, treatment of EngBF with EDTA has a significant destabilizing effect on the enzyme (3).
At the center of the (β/α)8-barrel core, the catalytic domain is a distinctive pocket that houses the catalytic machinery: Asp-764 as the catalytic nucleophile and Glu-796 as the catalytic general acid/base (Fig. 1C). The proposed catalytic residues are also conserved in EngBF, which possesses the same substrate specificity as the GH101 enzyme from S. pneumoniae. Notable are the presence of two tryptophan residues, Trp-724 and Trp-726, which form an open lid over the active site. These residues are also conserved in EngBF (Fig. 1C) and in this enzyme, on the basis of their position relative to the active site and low activity of site-directed mutants, have been proposed to play a role in substrate recognition (3).
Although the structural analyses of GH101 enzymes performed to date have been important in establishing the general identity of the active site, the molecular details regarding how these enzymes recognize substrates remain unknown because no structures have been determined in complex with any carbohydrate ligands. Thus, we pursued structures of GH101 from S. pneumoniae TIGR4 in complex with an array of carbohydrate ligands. However, the crystal form used to obtain the structure of SpGH101(N) proved to be difficult to reproduce consistently and was not sufficiently robust to withstand physical manipulations (e.g. soaking in solutions with substrates or ligands). By generating a construct encoding the same fragment of GH101 but with a C-terminal His6 tag, referred to as SpGH101(C), we were able to reproducibly obtain robust crystals in space group P22121 with superior diffraction qualities and having a single molecule in the asymmetric unit. Using this construct and versions wherein the proposed residues acting as catalytic nucleophile (Asp-764) and acid/base (Glu-796) were conservatively mutated to generate SpGH101(C)D764N and SpGH101(C)E796Q, we were able to obtain structures of GH101 from S. pneumoniae TIGR4 in complex with various carbohydrate ligands including substrates, products, and inhibitor.
The Structures of SpGH101(C) Mutants in Complex with T-antigen
In SpGH101R6, E796Q and D764N substitutions reduce the activity of the enzyme by 30- and 700-fold, respectively (9). We therefore incorporated these mutations into SpGH101(C) in an effort to obtain non-hydrolyzed substrate complexes. Crystals of SpGH101(C)E796Q were soaked with PNP-T-antigen or with a glycopeptide bearing a T-antigen and data sets to 1.75- and 2.5-Å resolution, respectively, were collected. In both cases, clear electron density was found for the T-antigen disaccharide but no electron density was observed for either the nitrophenol group or the peptide (Fig. 2, A and B). These observations suggested that, despite the introduction of the mutation, over the time during which the crystals were soaked in solutions of the substrates, they had been hydrolyzed in the active site to generate the observed product complexes. Unlike the disaccharide in the crystal soaked with glycopeptide, which could only be modeled as the α-anomer, the disaccharide in the PNP-T-antigen soaked crystal was modeled as a 1:1 mixture of α- and β-anomers (Fig. 2A). Otherwise, within the positional error of these structures (maximum likelihood estimated standard uncertainties of <0.2 Å) the placement of the monsaccharide units was indistinguishable as was the arrangement of the protein side chains within the active site. Binding of this disaccharide product, however, resulted in a change in the conformation of the active site. Relative to the position of the loop containing Trp-724 and Trp-726 in the 1.85-Å ligand-free structure we obtained, this loop in the product complexes was pulled toward the active site while the tryptophan side chains themselves closed over the top of the disaccharide through a movement of ∼3–5 Å and a rotation of ∼50° (Fig. 2B). The resulting active site is a partially closed pocket that very closely complements the shape of the disaccharide yet also accommodates several water molecules (Fig. 2C). This pocket has two subsites, −1 and −2, which accommodate the GalNAc and Gal residues of the T-antigen, respectively.
FIGURE 2.

Complexes of SpGH101(C) mutants with T-antigen. A, the structure of SpGH101(C)E796Q in complex with the T-antigen product after incubation of the crystals with pNP-T-antigen. Relevant side chains are shown as sticks. The sugar, modeled as both anomers, is shown as green sticks and the proposed catalytic water as a red sphere. The gray mesh shows the maximum likelihood/σa-weighted Fo − Fc electron density map (contoured at 3σ) for the carbohydrate and water. Dashed lines show putative hydrogen bonds; arrows show relevant distances. Distances in Å are given. B, as in panel A but for the T-antigen product after soaking the crystals with a T-antigen bearing glycopeptide. The structure is overlaid with the non-complex SpGH101(N) structure, shown in gray with arrows to highlight the conformational change involved in closing the active site lid. C, the active site of the T-antigen complex from the glycopeptide soak shown as a solvent accessible surface. The purple surface indicates the surface from the closed tryptophan lid. Relevant waters are shown as blue spheres. D, divergent stereo view of the hydrogen bonding interactions in the active site. Putative hydrogen bonds are shown as dashed lines. E, the structure of SpGH101(C)D764N in complex with the serinyl T-antigen. Relevant side chains are shown as sticks. The glyco-amino acid is shown as green sticks. The gray mesh shows the maximum likelihood/σa-weighted Fo − Fc electron density map (contoured at 3σ). Dashed lines show putative hydrogen bonds; arrows show relevant distances. Distances in Å are given.
Engagement of the T-antigen by the active site results in an array of hydrogen bonds between the sugar and amino acid side chains in the enzyme, providing a distinct pattern of recognition for the T-antigen (Fig. 2D). In particular are direct and water-mediated hydrogen bonds with the axial C4 hydroxyl groups that likely provide specificity for the two galacto-configured monosaccharides. Although the acetamido group of the GalNAc residue makes only a single water-mediated hydrogen bond, it fits into a well tailored pocket in the active where van der Waals interactions likely provide selectivity for this chemical group.
In both of the complexes, Asp-764 is located ∼3.5 Å immediately below the anomeric carbon and is therefore well positioned to serve as a catalytic nucleophile. Notably, however, O1 in the α-anomer, which would approximate the position of the glycosidic oxygen in the substrate, is over 4.3 Å away from the proposed general acid/base catalytic residue Glu-796 (Fig. 2, A and B), too far to enable this residue to serve directly as a proton donor. In the two product complexes, however, a well ordered water molecule, which is also present in the ligand-free structure of the non-mutated enzyme, occupies this gap, forming a hydrogen bonding chain between the proposed general acid/base residue and O1.
The Structure of SpGH101(C) in Complex with Substrate Analogues
Although SpGH101(C)E796Q appeared to retain enough activity to hydrolyze substrates during the crystal soaking procedure, by using the SpGH101(C)D764N nucleophile mutant we were able to obtain a structure of the intact serinyl T-antigen bound within the active site to 1.8-Å resolution. The electron density of the sugar was clear and continuous with the density for the O-linked serine residue (Fig. 2E). The protein displayed the same structural change as seen for the product complexes, with an identical pattern of hydrogen bonding to the sugar portion (not shown). Again, the glycosidic oxygen linking the serine and GalNAc is 4.9 Å away from the proposed catalytic general acid/base residue, Glu-796. Furthermore, the serine residue is positioned such that its carboxylate oxygen interacts with Oϵ1 of Glu-796, effectively taking the position of the conserved water residue observed in the product complexes. As with the product complexes, Asn-764 is positioned 3.9 Å below C1 to perform its proposed role as nucleophile when present as an aspartate in the wild-type enzyme.
To obtain an alternative substrate complex but with wild-type SpGH101(C) we soaked crystals of SpGH101(C) with T-antigen methyl glycoside, reasoning that the methoxy group would be a poor leaving group because it would lack any aglycon binding interactions and may therefore be a poor substrate for the enzyme. The electron density in the area of the active site indicated two bound forms of the enzyme, with each form binding uncleaved T-antigen methyl glycoside (Fig. 3A). The minor species, modeled with 30% occupancy, matched the closed conformation of the enzyme (Fig. 3B). The T-antigen methyl glycoside in this bound form was not well occupied and could not be modeled with complete confidence; however, the sugar refined in this conformation appeared to be intact and was observed to reside in the active site in a position very close to that observed for the product complexes and the serinyl T-antigen complex. Like the T-antigen product complexes, electron density for a well ordered water molecule was modeled as bridging Glu-796 and O1 of the ligand (Fig. 3, A and B).
FIGURE 3.

Complexes of unmutated SpGH101(C) with T-antigen analogues. A, the structure of SpGH101(C) in complex with the T-antigen methyl glycoside. The gray mesh shows the maximum likelihood/σa-weighted Fo − Fc electron density map (contoured at 3σ). The map was generated by refinement of the structure with Trp-724, Trp-726, and Arg-1256 modeled as alanines and the carbohydrate ligand and putative catalytic water absent. The sticks show the final modeled structures, which represents two major conformations for the carbohydrate and Trp-724, Trp-726, and Arg-1256 residues (see text for details). B, the minor “B” conformation of the T-antigen methyl glycoside complex (purple and green with a water as a red sphere) overlaid with the T-antigen glycopeptide product complex (transparent gray and yellow with a water as a blue sphere). Dashed lines show putative hydrogen bonds; arrows show relevant distances. Distances in Å are given. C, the major “A” conformation of the T-antigen methyl glycoside complex (purple and green) overlaid with the T-antigen glycopeptide product complex (transparent gray and yellow with a water as a blue sphere). Potential hydrogen bonds for the T-antigen methyl glycoside complex are shown as black dashed lines, whereas those for the T-antigen glycopeptide product complex are shown as gray dashed lines. E, the structure of SpGH101(C) in complex with PUGT (purple and green) overlaid with the T-antigen glycopeptide product complex (transparent gray and yellow). The gray mesh shows the maximum likelihood/σa-weighted Fo − Fc electron density map (contoured at 3σ) for PUGT. Dashed lines show putative hydrogen bonds; arrows show relevant distances. Distances in Å are given. The curved arrow highlights the altered positions of the acetamido group in the two ligands. The inset shows the tryptophan residues that interact with the phenylcarbamate aglycon.
The major bound species, modeled with 70% occupancy, matched the open form of the enzyme active site with Trp-724 and Trp-726 rotated away from the active site (Fig. 3C). Remarkably, clear electron density was observed for the intact T-antigen methyl glycoside bound to the underside of the aromatic lid formed by the side chains of the two tryptophans (Fig. 3A). Furthermore, the non-reducing end C3 and C4 hydroxyl groups of the modeled T-antigen methyl glycoside make three direct hydrogen bonds with active site residues (Fig. 3C). Notably, all of these residues are involved in coordinating the deeply bound disaccharide, although in the deeply bound saccharide, two of the direct hydrogen bonds are replaced by a single water-mediated hydrogen bond (Fig. 3C). In addition, the C6 hydroxyl group of the Gal hydrogen bonds with Nϵ of Trp-724, an interaction that is maintained when the aromatic lid closes on the active site (Fig. 2D).
In an effort to better visualize the nature of potential aglycon interactions, we also synthesized a novel endo-α-N-acetylglucosaminidase inhibitor (PUGT) comprising galactose β-1,3-linked to an O-(2-N-acetyl-2-deoxy-d-galactopyranosylidene)amino N-phenylcarbamate residue at the “reducing” terminus. The transition state structure stabilized by glycoside hydrolases is thought to be mimicked by the trigonal C1 of such compounds and, as such, these inhibitors typically bind tightly with the trigonal C1 in the −1 subsite and the oxime group spanning the position normally occupied by the scissile glycosidic bond (28). Although PUGT proved to be a surprisingly poor inhibitor (Ki >10 mm; data not shown), we were able to obtain a complex of SpGH101(C) with this compound at 1.46-Å resolution. The electron density was clear for all but the most distal atoms of the phenyl ring of the compound (Fig. 3D). Again, the active site is found in its closed form. In contrast to all of the other complexes we obtained where the Gal and GalNAc rings were in the relaxed 4C1 conformation, the pyranose ring of PUGT in the −1 subsite refined in the 4H3 conformation, whereas the Gal residue in the −2 subsite was in the 4C1 conformation. Despite the planar conformation of the −1 residue at C2-C1-O5-C5, which pulls this portion of PUGT deeper into the −1 subsite relative to the product complexes, the carbohydrate portion of the inhibitor makes the same set of direct hydrogen bonds observed in all of the other T-antigen complexes (Fig. 3D). In this complex, however, the acetamido group is flipped up relative to its position in all of the other complexes (Fig. 3D). The significance of this position of the acetamido group is unclear, as it does not appear to be induced by a lack of space for the acetamido group. The planarity of C1 in PUGT positions the oxime O and N atoms at distances of 3.0 and 2.8 Å, respectively, from Glu-796 and with appropriate geometry to hydrogen bond. Asp-764 is positioned 3.1 Å immediately below C1. The plane of the PUGT phenyl ring is positioned at roughly right angles to the plane of the carbohydrate portion of the compound and thus extends out of the active site and into solution. Limited interactions are made between the phenylcarbamate group of PUGT and the protein. The indole ring of Trp-810 makes an unusual aromatic ring edge-edge interaction with the aryl group of PUGT (∼4 Å) distance and the edge of the Trp-797 ring interacts at right angles with the carbamate portion of the compound (Fig. 3D, inset). Despite these fortuitous interactions PUGT displayed poor inhibition of the enzyme, which cannot be readily reconciled with the observed structure.
Discussion
Substrate Recognition
Conformational changes in glycoside hydrolases upon substrate recognition are relatively uncommon and typically involve engagement of the active site machinery, such as movement of loops containing the catalytic residues in GH3, GH29, and GH115 enzymes (29–31). Thus, the conformational change in SpGH101 involving the movement of a two-residue tryptophan “lid” over the active site, which is solely involved in substrate recognition rather than positioning of the catalytic machinery, is rare among glycoside hydrolases. In the absence of substrate, the enzyme is present in an “open” form, with a fully accessible active site (Fig. 4A). In this form, the planar guanidine group of Arg-1256 lies above the tryptophan lid, packing against the indole ring of Trp-726 likely forming a cation-π interaction. In the fully engaged substrate “bound” form of the enzyme, the tryptophan lid is closed by an ∼50° rotation through an ∼5 Å arc (Fig. 4B) from the position observed in the open form of the enzyme. In the bound form, Arg-1256 follows the closing of the lid by flipping down to maintain its interaction with Trp-726. In the bound form the active site is partially occluded by the tryptophan lid (Fig. 2C), suggesting the lid must be open to accept substrate and then only after substrate binding does the lid close on the bound substrate.
FIGURE 4.

The trajectory of substrate recognition by SpGH101. A, the open form of the enzyme in its resting, unliganded state. Conserved water molecules are shown as red spheres. B, the bound, or engaged, form of the enzyme with substrate bound deep in the active site. The motion of the tryptophan lid and arginine residue that close the active site are indicated by curved arrows. C, the crystallographically observed open form of SpGH101 bound to substrate. In all panels, relevant residues are shown as sticks with the substrate (aglycon indicated by R) in green. Potential hydrogen bonds are shown as dashed lines. Relevant water molecules are shown as red spheres. The bracketed equilibrium arrows represent a possible step for which there is currently no evidence of its occurrence.
In addition to observing open unbound and closed bound forms of the enzyme, we also fortuitously obtained an unusual, partially occupied form using the T-antigen methyl glycoside as a substrate analogue. In this form, the planes made by carbons C4-C5-C6 of the GalNAc β-face and the same carbons of the β-face of the Gal residue pack against the platform made by tryptophans 724 and 726 in the open form of the enzyme. This binding appears to displace a water molecule coordinated by Glu-1253 and Asp-1254, whereas a set of hydrogen bonds involving the C3, C4, and C6 hydroxyl groups of the terminal Gal residue provide specific interactions with the protein (Fig. 4C). We acknowledge that this bound form of the enzyme may be a fortuitously obtained artifact of the procedure used to obtain the crystal structure of this complex. This interaction, however, has the hallmarks of a specific protein-carbohydrate interaction and thus we cannot completely discount its potential relevance. Indeed, from this potential intermediate state, closing of the tryptophan lid with a ∼1.2 Å slide of the carbohydrate toward the catalytic machinery, would both maintain how the sugar packs against the tryptophan lid and result in full engagement of the active site. This rearrangement would also require reinsertion of the displaced water that interacts with terminal Gal residue to fill the gap formed by movement of the substrate toward the catalytic residues (Fig. 4C). Thus, if this open and bound species is one that is able to form in solution, it may represent an intermediate formed on the way to full engagement of the active site. Alternatively, it is also possible that this intermediate form is a non-productively bound form that does not lead to catalysis.
The conformational changes involved in substrate recognition concludes with a tight fit of the disaccharide into the closed active site. The highly complementary contouring of the active site to the disaccharide sterically prevents accommodation of any glycan structures larger than the T-antigen (Fig. 2C), such as a β-1,6-GlcNAc modification of the core GalNAc (to create a core 2 O-glycan), α-2,6-sialyation of the core GalNAc, or α-2,3-sialylation of the terminal Gal. As noted by Suzuki et al. (3), structural variation of GH101 enzymes in the region where O6 of the core GalNAc binds likely enables the activity of other GH101 family members toward core 3 O-glycans. In SpGH101, whereas the −1 and −2 subsites impart specificity for the T-antigen, the enzyme lacks any definable features that accommodate the protein/peptide aglycon. A surface representation of the protein in the region of the active site shows a wide, shallow trough that is consistent with accommodation of a protein or peptide aglycon (Fig. 5).
FIGURE 5.
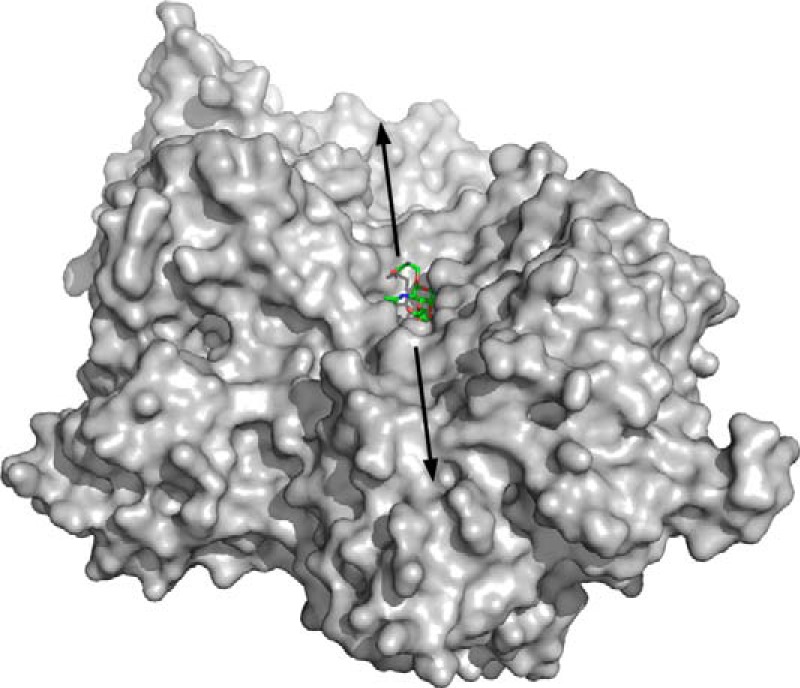
Solvent accessible surface representation of the SpGH101 active site. The solvent accessible surface is shown in gray and the serinyl T-antigen is shown in green sticks. The arrows indicate the approximate extend of the peptide/protein aglycon binding site.
Catalytic Mechanism
The evidence to date supports a catalytic mechanism for GH101 enzymes that retains the stereochemistry at the anomeric carbon and, in SpGH101, uses Asp-764 as the catalytic nucleophile and Glu-796 as the general acid/base residue (Fig. 6A). An overlap of SpGH101(C) with the Thermoactinomyces vulgaris α-amylase (TVAI) in complex with its substrate, as previously noted, shows the structural conservation of the catalytic residues in TVAI (Asp-356 and Glu-396) with Asp-764 and Glu-796 in SpGH101(C) (32). Asp-764 and Glu-796 are also located in proximity to C1 and O1, respectively, of the carbohydrate, and thus the arrangement is largely consistent with the functional assignment of these residues (Fig. 6B). Indeed, in all of our five complexes of SpGH101(C) with the T-antigen and T-antigen analogues, the side chain of Asp-764 is positioned <3.9 Å below C1, poised for an in-line attack and, therefore, consistent with a role as the catalytic nucleophile (Figs. 2, A, B, and E, and 3, B and D). In contrast, however, a more detailed examination of our structures suggests that the role played by Glu-796 is slightly ambiguous. A catalytic general acid/base should be positioned less than ∼3.3 Å away from the glycosidic oxygen, as it is in TVAI (Fig. 6B), to act as an effective proton donor to aid in departure of the leaving group. In all of our complexes the O1 of the GalNAc residue is positioned more than 4.3 Å away from the side chain of Glu-796 (Figs. 2, A, B, and E, and 3B), with no other residues suitably placed to act as a general acid/base. Although mutagenesis and chemical rescue data provide strong support for the identity of Glu-796 as the general acid/base residue, our structural data suggests that Glu-796 is unable to directly donate a proton to the glycosidic oxygen of the substrate, therefore leading to the question of how this residue functions in catalysis.
FIGURE 6.

Proposed catalytic mechanism for SpGH101. A, the mechanism proposed by Willis et al. (9) based on the canonical double displacement mechanism with retention of the stereochemistry at the anomeric carbon. The side chains of Asp-764 and Glu-796 acts as the proposed nucleophile and acid/base, respectively. B, an overlap of the SpGH101(C) T-antigen glycopeptide product complex (purple and green) with the T. vulgaris α-amylase (TVAI) in complex with its substrate (gray, PDB code 2D0H). SpGH101 residues are labeled in purple and the TVAI residues in black. The arrow indicates the distance from the TVAI acid/base residue to the glycosidic oxygen with the distance given in Å. C, an overlap of the proposed catalytic acid/base residue (position 796) from the uncomplexed SpGH101(N) structure (pink side chain and water as red sphere), the product complex of SpGH101(C)E796Q from the pNP-T-antigen soak (purple and water as purple sphere), the product complex of SpGH101(C)E796Q from the glycopeptide soak (yellow and water as yellow sphere), and the SpGH101(C) T-antigen methyl glycoside complex (blue and water as blue sphere). Only the T-antigen from the glycopeptide T-antigen product complex is shown as green sticks. Ranges of distances are give in Å. D, a refined catalytic mechanism for SpGH101 proposed on the basis of structural studies (see text for details).
In both T-antigen product complexes and the T-antigen methyl glycoside complex a well ordered water molecule bridges O1 of the GalNAc and the Glu-796 side chain (Figs. 2, A and B, and 3B). Moreover, the placement of this water molecule is well conserved in these complexes as well as in the absence of substrate (Fig. 6C). The only contradiction to this observation is our structure of the serinyl T-antigen bound to SpGH101(C), which lacks the conserved water. In this structure, a carboxylate oxygen of the serine occupies the position where the conserved water would otherwise be (Fig. 2E). This conformation, however, we argue is an artifact promoted by the presence of only a single amino acid attached to the sugar moiety. Consistent with this interpretation, the conformation we observe is such that the carboxylate of the serine is buried within the active site, which would permit recognition of T-antigen motifs positioned only on the C-terminal residue of a protein or peptide. This scenario is incompatible with the ability of SpGH101 to remove the T-antigen from mucins (33, 34) and our observation of its activity on a glycopeptide with a T-antigen modification positioned in the middle of the peptide. On this basis, we suggest that the conformation of the serinyl T-antigen in the crystal structure is a non-productively bound conformation in which the carboxylate oxygen takes the position of the catalytic bridging water molecule.
From the position of the conserved water molecule that bridges Glu-796 and the O1 of the GalNAc in the bound disaccharide substrates we propose that Glu-796 acts indirectly as the general acid/base through a short Grotthuss chain (Fig. 6D). Catalytic mechanisms involving Grotthuss chains are seen in glycoside hydrolases such as the GH6 and GH124 β-1,4-endoglucanases (35, 36). All of the enzymes previously proposed to utilize such a mechanism, however, are inverting enzymes where the catalytic base residue acts indirectly through a short water chain with the final deprotonated water in the chain acting as a nucleophile that attacks C1 of the sugar. In the proposed mechanism for SpGH101, an enzyme that retains the stereochemistry at the anomeric carbon (retaining mechanism), proton transfer from the acid/base Glu-796 to the glycosidic oxygen is coordinated through the intervening water molecule, thereby aiding departure of the protein leaving group. Two scenarios are plausible for the deglycosylation step. In one, the side chain of Glu-796 acts as a general base, which, through the intervening catalytic water, facilitates attack of an incoming water molecule on the anomeric center of the glycosyl-enzyme intermediate. Alternatively, the intervening catalytic water may shift to directly attack the anomeric center of the glycosyl-enzyme intermediate, ultimately to be replaced by a new water molecule following departure of the product. Given the distance the catalytic water would have to migrate (>∼2 Å), however, we find this alternative less likely.
Overall, substrate recognition by SpGH101 is accompanied by a structural change that comprises the closing of a tryptophan lid on the active site, pinning the substrate in position and causing partial occlusion of the entrance to the active site. Once SpGH101 engages the substrate, the glycosidic oxygen is too far from the acid/base residue to directly accept a proton. As a water molecule bridges this distance in SpGH101 we propose a mechanism whereby the water molecule acts as a proton shuttle to deliver a proton to the glycosidic oxygen during catalysis from the general acid/base residue. Why SpGH101 may have adopted this mechanistic variation of the classical retaining glycoside hydrolase mechanism is unclear. However, such a Grotthuss-like mechanism may enable SpGH101, and other GH101 enzymes, to better accept large protein and peptide substrates having highly variable structures including some that adopt non-optimal conformations. This reasoning implies that SpGH101 may represent a unique example of how a GH catalytic mechanism has adapted to accommodate a specific class of substrate.
Author Contributions
K. G. and M. D. L. S. performed and analyzed experiments. L. D. performed the compound synthesis and analysis. A. B. B. and D. J. V. conceived and coordinated the study and performed analyses. M. D. L. S., D. J. V., and A. B. B. wrote the paper.
Acknowledgments
We thank the beamline staff at the Stanford Synchrotron Research Laboratory BL9-2 and the Canadian Light Source 08ID-1 where diffraction data were collected. The Canadian Light Source is supported by the Natural Sciences and Engineering Research Council of Canada (NSERC), the National Research Council Canada, CIHR, the Province of Saskatchewan, Western Economic Diversification Canada, and the University of Saskatchewan. We thank Professor Lai-Xi Wang for providing us with the glycopeptide bearing the T-antigen.
This work was supported in part by a Canadian Institutes of Health Research Operating Grant MOP 130305. The authors declare that they have no conflicts of interest with the contents of this article.
- GH101
- glycoside hydrolase family 101
- PUGT
- galactose β-1,3-linked to an O-(2-N-acetyl-2-deoxy-d-galactopyranosylidene)amino-N-phenylcarbamate residue at the reducing terminus
- T-antigen
- Galβ1–3GalNAcα1-linked to a Ser or Thr.
References
- 1. Brockhausen I., Schachter H., Stanley P. (2009) O-GalNAc Glycans. in Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., Hart G. W., Etzler M. E., eds) 2nd Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 2. Gregg K. J., Boraston A. B. (2009) Cloning, recombinant production, crystallization and preliminary x-ray diffraction analysis of a family 101 glycoside hydrolase from Streptococcus pneumoniae. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 65, 133–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Suzuki R., Katayama T., Kitaoka M., Kumagai H., Wakagi T., Shoun H., Ashida H., Yamamoto K., Fushinobu S. (2009) Crystallographic and mutational analyses of substrate recognition of endo-α-N-acetylgalactosaminidase from Bifidobacterium longum. J. Biochem. 146, 389–398 [DOI] [PubMed] [Google Scholar]
- 4. Fujita K., Oura F., Nagamine N., Katayama T., Hiratake J., Sakata K., Kumagai H., Yamamoto K. (2005) Identification and molecular cloning of a novel glycoside hydrolase family of core 1 type O-glycan-specific endo-α-N-acetylgalactosaminidase from Bifidobacterium longum. J. Biol. Chem. 280, 37415–37422 [DOI] [PubMed] [Google Scholar]
- 5. Ashida H., Maki R., Ozawa H., Tani Y., Kiyohara M., Fujita M., Imamura A., Ishida H., Kiso M., Yamamoto K. (2008) Characterization of two different endo-α-N-acetylgalactosaminidases from probiotic and pathogenic enterobacteria, Bifidobacterium longum and Clostridium perfringens. Glycobiology 18, 727–734 [DOI] [PubMed] [Google Scholar]
- 6. Koutsioulis D., Landry D., Guthrie E. P. (2008) Novel endo-α-N-acetylgalactosaminidases with broader substrate specificity. Glycobiology 18, 799–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goda H. M., Ushigusa K., Ito H., Okino N., Narimatsu H., Ito M. (2008) Molecular cloning, expression, and characterization of a novel endo-α-N-acetylgalactosaminidase from Enterococcus faecalis. Biochem. Biophys. Res. Commun. 375, 441–446 [DOI] [PubMed] [Google Scholar]
- 8. Caines M. E., Zhu H., Vuckovic M., Willis L. M., Withers S. G., Wakarchuk W. W., Strynadka N. C. (2008) The structural basis for T-antigen hydrolysis by Streptococcus pneumoniae: a target for structure-based vaccine design. J. Biol. Chem. 283, 31279–31283 [DOI] [PubMed] [Google Scholar]
- 9. Willis L. M., Zhang R., Reid A., Withers S. G., Wakarchuk W. W. (2009) Mechanistic investigation of the endo-α-N-acetylgalactosaminidase from Streptococcus pneumoniae R6. Biochemistry 48, 10334–10341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marion C., Limoli D. H., Bobulsky G. S., Abraham J. L., Burnaugh A. M., King S. J. (2009) Identification of a pneumococcal glycosidase that modifies O-linked glycans. Infect. Immun. 77, 1389–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 13. Leslie A. G. (2006) The integration of macromolecular diffraction data. Acta Crystallogr. D Biol. Crystallogr. 62, 48–57 [DOI] [PubMed] [Google Scholar]
- 14. Vonrhein C., Blanc E., Roversi P., Bricogne G. (2007) Automated structure solution with autoSHARP. Methods Mol. Biol. 364, 215–230 [DOI] [PubMed] [Google Scholar]
- 15. Cowtan K. (2006) The Buccaneer software for automated model building: 1. tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 62, 1002–1011 [DOI] [PubMed] [Google Scholar]
- 16. McCoy A. J. (2007) Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 63, 32–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 18. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 19. Brünger A. T. (1992) Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–475 [DOI] [PubMed] [Google Scholar]
- 20. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B. 3rd, Snoeyink J., Richardson J. S., Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Flowers H. M., Hapiro D. (1965) Synthesis of 2-acetamido-2-deoxy-3-O-(β-d-galactopyranosyl)-α-d-galactose. J. Org. Chem. 30, 2041–2043 [Google Scholar]
- 23. Xia J., Alderfer J. L., Piskorz C. F., Locke R. D., Matta K. L. (2000) A convergent synthesis of trisaccharides with α-Neu5Ac-(2→3)-β-d-gal-(1→4)-β-d-GlcNAc and α-Neu5Ac-(2→3)-β-d-gal-(1→3)-α-d-GalNAc sequences. Carbohydr. Res. 328, 147–163 [DOI] [PubMed] [Google Scholar]
- 24. Schroeder L. R., Counts K. M., Haigh F. C. (1974) Stereoselectivity of Koenigs-Knorr syntheses of alkyl β-d-galactopyranoside and β-d-xylopyranoside peracetates promoted by mercuric bromide and mercuric oxide. Carbohydr. Res. 37, 368–372 [Google Scholar]
- 25. Lubineau A., Bienayme H., Le Gallic J. (1989) Synthesis of α-linked derivatives of N-acetyl glucosamine and Gal-β(1–3)GalNac (T-antigen) directly with the natural N-acetyl protecting group. J. Chem. Soc. Chem. Commun. 1918–1919 [Google Scholar]
- 26. Stubbs K. A., Macauley M. S., Vocadlo D. J. (2009) A selective inhibitor Gal-PUGNAc of human lysosomal β-hexosaminidases modulates levels of the ganglioside GM2 in neuroblastoma cells. Angew. Chem. Int. Ed. Engl. 48, 1300–1303 [DOI] [PubMed] [Google Scholar]
- 27. Lammerts van Bueren A., Ficko-Blean E., Pluvinage B., Hehemann J. H., Higgins M. A., Deng L., Ogunniyi A. D., Stroeher U. H., El Warry N., Burke R. D., Czjzek M., Paton J. C., Vocadlo D. J., Boraston A. B. (2011) The conformation and function of a multimodular glycogen-degrading pneumococcal virulence factor. Structure 19, 640–651 [DOI] [PubMed] [Google Scholar]
- 28. Gloster T. M., Vocadlo D. J. (2012) Developing inhibitors of glycan processing enzymes as tools for enabling glycobiology. Nat. Chem. Biol. 8, 683–694 [DOI] [PubMed] [Google Scholar]
- 29. Bacik J. P., Whitworth G. E., Stubbs K. A., Vocadlo D. J., Mark B. L. (2012) Active site plasticity within the glycoside hydrolase NagZ underlies a dynamic mechanism of substrate distortion. Chem. Biol. 19, 1471–1482 [DOI] [PubMed] [Google Scholar]
- 30. Sulzenbacher G., Bignon C., Nishimura T., Tarling C. A., Withers S. G., Henrissat B., Bourne Y. (2004) Crystal structure of Thermotoga maritima α-l-fucosidase: insights into the catalytic mechanism and the molecular basis for fucosidosis. J. Biol. Chem. 279, 13119–13128 [DOI] [PubMed] [Google Scholar]
- 31. Rogowski A., Baslé A., Farinas C. S., Solovyova A., Mortimer J. C., Dupree P., Gilbert H. J., Bolam D. N. (2014) Evidence that GH115 α-glucuronidase activity, which is required to degrade plant biomass, is dependent on conformational flexibility. J. Biol. Chem. 289, 53–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Abe A., Yoshida H., Tonozuka T., Sakano Y., Kamitori S. (2005) Complexes of Thermoactinomyces vulgaris R-47 α-amylase 1 and pullulan model oligossacharides provide new insight into the mechanism for recognizing substrates with α-(1,6)glycosidic linkages. FEBS J. 272, 6145–6153 [DOI] [PubMed] [Google Scholar]
- 33. Bhavanandan V. P., Codington J. F. (1983) Selective release of the disaccharide 2-acetamido-2-deoxy-3-O-(β-d-galactopyranosyl)-d-galactose from epiglycanin by endo-N-acetyl-α-d-galactosaminidase. Carbohydr. Res. 118, 81–89 [DOI] [PubMed] [Google Scholar]
- 34. Umemoto J., Bhavanandan V. P., Davidson E. A. (1977) Purification and properties of an endo-α-N-acetyl-d-galactosaminidase from Diplococcus pneumoniae. J. Biol. Chem. 252, 8609–8614 [PubMed] [Google Scholar]
- 35. Koivula A., Ruohonen L., Wohlfahrt G., Reinikainen T., Teeri T. T., Piens K., Claeyssens M., Weber M., Vasella A., Becker D., Sinnott M. L., Zou J. Y., Kleywegt G. J., Szardenings M., Ståhlberg J., Jones T. A. (2002) The active site of cellobiohydrolase Cel6A from Trichoderma reesei: the roles of aspartic acids D221 and D175. J. Am. Chem. Soc. 124, 10015–10024 [DOI] [PubMed] [Google Scholar]
- 36. Brás J. L., Cartmell A., Carvalho A. L., Verzé G., Bayer E. A., Vazana Y., Correia M. A., Prates J. A., Ratnaparkhe S., Boraston A. B., Romão M. J., Fontes C. M., Gilbert H. J. (2011) Structural insights into a unique cellulase fold and mechanism of cellulose hydrolysis. Proc. Natl. Acad. Sci. U.S.A. 108, 5237–5242 [DOI] [PMC free article] [PubMed] [Google Scholar]