

Background: M. tuberculosis LdtMt5 is an LdtMt2 paralog that cross-links peptidoglycan stem peptides.
Results: LdtMt5 is structurally divergent, strains lacking LdtMt5 are more susceptible to chemical and environmental stresses, and LdtMt2 cannot compensate for its loss.
Conclusion: LdtMt2 and LdtMt5 serve non-redundant roles in peptidoglycan maintenance.
Significance: LdtMt5 is necessary for properly maintaining cell wall integrity and should be pursued as a drug target.
Keywords: antibiotics, biosynthesis, cell wall, enzyme kinetics, enzyme structure, Mycobacterium tuberculosis, peptidoglycan
Abstract
The final step of peptidoglycan (PG) biosynthesis in bacteria involves cross-linking of peptide side chains. This step in Mycobacterium tuberculosis is catalyzed by ld- and dd-transpeptidases that generate 3→3 and 4→3 transpeptide linkages, respectively. M. tuberculosis PG is predominantly 3→3 cross-linked, and LdtMt2 is the dominant ld-transpeptidase. There are four additional sequence paralogs of LdtMt2 encoded by the genome of this pathogen, and the reason for this apparent redundancy is unknown. Here, we studied one of the paralogs, LdtMt5, and found it to be structurally and functionally distinct. The structures of apo-LdtMt5 and its meropenem adduct presented here demonstrate that, despite overall architectural similarity to LdtMt2, the LdtMt5 active site has marked differences. The presence of a structurally divergent catalytic site and a proline-rich C-terminal subdomain suggest that this protein may have a distinct role in PG metabolism, perhaps involving other cell wall-anchored proteins. Furthermore, M. tuberculosis lacking a functional copy of LdtMt5 displayed aberrant growth and was more susceptible to killing by crystal violet, osmotic shock, and select carbapenem antibiotics. Therefore, we conclude that LdtMt5 is not a functionally redundant ld-transpeptidase, but rather it serves a unique and important role in maintaining the integrity of the M. tuberculosis cell wall.
Introduction
Virtually all bacteria possess a peptidoglycan (PG)4 layer that encapsulates the cytoplasmic membrane and provides shape and rigidity to the cell. Considered as a vital bacterial “organelle,” the PG is essential for cellular growth and viability. β-Lactams, the class of drugs with the highest impact in treating bacterial infections in humans (1), act by inhibiting PG biosynthesis (2).
Mature PG comprises aminosugar strands with alternating N-acetylmuramic acid and N-acetylglucosamine units and peptide moieties that are cross-linked to one another (3). Lipid II, a precursor to PG, is synthesized intracellularly and consists of a single disaccharide-peptide and lipid moiety. In Mycobacterium tuberculosis, the infectious pathogen that causes tuberculosis, the pathway for lipid II biosynthesis comprises more than 20 genes, and all are essential for the viability of this pathogen (4). Lipid II is transported across the membrane, and units are linked together by transglycosylases that extend the aminosugar strand. The final step in PG maturation involves cross-linking the peptide moieties to one another by both dd- and ld-transpeptidases.
Until recently, the PG was considered to be largely cross-linked by dd-transpeptidases that generate 4→3 transpeptide linkages between the fourth amino acid (d-alanine) of one chain and the third amino acid (meso-diaminopimelic acid in M. tuberculosis) of an adjacent chain. Interestingly, the majority of transpeptide linkages in Mycobacterium spp. are between meso-diaminopimelic acid residues of two peptide chains (5). These 3→3 linkages are formed by ld-transpeptidases (6, 7), and the dominant ld-transpeptidase in M. tuberculosis is LdtMt2 (8). Unlike dd-transpeptidases that utilize a catalytic serine and pentapeptide substrates (9), ld-transpeptidases require a catalytic cysteine residue (10, 11) and utilize tetrapeptide substrates. ld-Transpeptidation has now been identified in a range of pathogenic bacteria (11–13), highlighting the importance of this fundamental mechanism across bacterial species.
In M. tuberculosis, loss of LdtMt2 results in altered cell size, growth, and virulence as well as loss of the ability of the organism to secrete low molecular weight proteins and increased susceptibility to amoxicillin (8, 14). The genome of M. tuberculosis encodes four additional paralogs of LdtMt2. On the basis of in vitro cross-linking activity or sequence similarity, they have been annotated as LdtMt1 (Rv0116c), LdtMt3 (Rv1433), LdtMt4 (Rv0192), and LdtMt5 (Rv0483) and share amino acid sequence identity of 36, 34, 35, and 28% with LdtMt2, respectively. It is unclear whether the five sequence paralogs are functionally redundant.
We used a combination of biophysical, biochemical, and genetic approaches to study LdtMt5. Here, we report the first crystal structures of apo- and meropenem-bound LdtMt5 and describe the phenotypic effects on M. tuberculosis lacking this enzyme. Our data indicate that LdtMt5 is structurally divergent compared with other M. tuberculosis ld-transpeptidases and that this protein serves a critical and distinct role in proper maintenance of M. tuberculosis cell wall integrity, highlighting its potential as a novel drug target.
Experimental Procedures
General Methods
All reagents were obtained from commercial sources. Spectrophotometric analyses were performed on a Shimadzu UV-1800 UV-visible spectrophotometer. Primers were purchased from Integrated DNA Technologies. Isothermal titration calorimetry (ITC) experiments were performed using a high precision VP-ITC titration calorimeter system (Microcal Inc.). Ultraperformance liquid chromatography (LC)-high resolution MS samples were analyzed on a Waters Acquity H-Class ultraperformance LC system equipped with a multiwavelength ultraviolet-visible diode array detector in conjunction with a Waters Acquity BEH-300 ultraperformance LC column packed with a C4 stationary phase (2.1 × 50 mm; 1.7 μm) in tandem with high resolution MS analysis by a Waters Xevo-G2 quadrupole-TOF electrospray ionization mass spectrometer. Molecular graphics and analyses were performed with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by National Institutes of Health NIGMS Grant P41-GM103311).
Cloning, Overexpression, and Purification of LdtMt5
A truncated version of ldtMt5 (encoding amino acids 55–451) was amplified by PCR (1× New England Biolabs GC reaction buffer, 200 μm dNTPs, 2 ng/μl CDC1551 M. tuberculosis genomic DNA, 500 nm primers (Table 1), 1 unit of Phusion polymerase, and 3% DMSO), digested with NdeI and XhoI, and cloned into a modified pET28a vector that encodes for a TEV-cleavable N-terminal His6 tag (10). Escherichia coli BL21(DE3) cells harboring the ldtMt5-pET28a plasmid were grown to an A600 of ∼0.5 at 37 °C. Flasks were then cooled on ice with periodic shaking. Protein overexpression was induced with 100 μm isopropyl 1-thio-β-d-galactopyranoside, and flasks were returned to an incubator shaker at 16 °C for an additional 24 h. Cells were harvested at 4 °C and stored overnight at −20 °C. Thawed cells were resuspended in protein purification buffer (25 mm Tris, pH 8.0, 400 mm NaCl, 10% glycerol, and 1 mm tris(2-carboxyethyl)phosphine (TCEP)) and protease inhibitor mixture (Roche Applied Science) and lysed by ultrasonication, and cell debris was removed by centrifugation at 4 °C. The supernatant was incubated with nickel-nitrilotriacetic acid resin at 4 °C for 90 min, and His6-tagged LdtMt5 was eluted from the resin over a stepwise gradient of 5–500 mm imidazole. Fractions containing LdtMt5 (as determined by SDS-PAGE) were combined, and protein concentration was determined using the Bio-Rad Protein Assay with bovine serum albumin (BSA) as a standard. The sample was then subjected to dialysis overnight at 4 °C against 1 liter of 25 mm Tris, pH 8.0, 100 mm NaCl, 10% glycerol, and 1 mm TCEP in the presence of TEV protease (1:100 TEV:LdtMt5). Following dialysis, the TEV-treated sample was incubated with fresh nickel-nitrilotriacetic acid resin at 4 °C for 90 min. Cleaved LdtMt5 was collected as flow-through, whereas the His-tagged TEV protease and cleaved His6 tag remained bound to the resin. LdtMt5 was subjected to a second dialysis against 1 liter of 25 mm Tris, pH 8.0, 100 mm NaCl, and 1 mm TCEP for 4 h at 4 °C. The concentration of LdtMt5 was determined using the Bio-Rad Protein Assay with BSA as a standard, and LdtMt5 was concentrated to 12.8 mg/ml prior to being flash frozen in liquid N2. Protein was stored at −80 °C.
TABLE 1.
Summary of primers used
Restriction sites are underlined. F, forward primer; R, reverse primer.
| Gene/variant | Primers |
|---|---|
| ldtMt5 | F, 5′-TATTGCCATATGAAGCTGGCCGAGAAGAGG-3′ |
| R, 5′-TATTGCCTCGAGCTACCCACCCGGTCCGTT-3′ | |
| H342A LdtMt5 | F, 5′-CAACAACGGCGAGTTCATCGCTG CCAACCCTATGAGCGCC-3′ |
| R, 5′-GGCGCTCATAGGGTTGGCAGCGATGAACTCGCCGTTGTTG-3′ | |
| H342Q LdtMt5 | F, 5′-CAACAACGGCGAGTTCATCCAGGCCAACCCTATGAGCGCC-3′ |
| R, 5′-GGCGCTCATAGGGTTGGCCTGGATGAAC TCGCCGTTGTTG-3′ | |
| M346W LdtMt5 | F, 5′-GAGTTCATCCATGCCAACCCTTGGAGCGCCGGTGCCCAGGGCAAC-3′ |
| R, 5′-GTTGCCCTGGGCACCGGCGCTCCAAGGGTTGGCATGGATGAACTC-3′ | |
| T357V LdtMt5 | F, 5′-CAGGGCAACAGCAATGTCGTCAACGGCTGTATCAACCTGTCG-3′ |
| R, 5′-CGACAGGTTGATACAGCCGTTGACGACATTGCTGTTGCCCTG-3′ | |
| N358A LdtMt5 | F, 5′-GGCAACAGCAATGTCACCGCCGGCTGTATCAACCTGTCGACG-3′ |
| R, 5′-CGTCGACAGGTTGATACAGCCGGCGGTGACATTGCTGTTGCC-3′ | |
| N358H LdtMt5 | F, 5′-CAGGGCAACAGCAATGTCACCCACGGCTGTATCAACCTGTCGACG-3′ |
| R, 5′-CGTCGACAGGTTGATACAGCCGTGGGTGACATTGCTGTTGCCCTG-3′ | |
| C360A LdtMt5 | F, 5′-CAGCAATGTCACCAACGGCGCTATCAACCTGTCGACGGAG-3′ |
| R, 5′-CTCCGTCGACAGGTTGATAGCGCCGTTGGTGACATTGCTG-3′ | |
| N362A LdtMt5 | F, 5′-GTCACCAACGGCTGTATCGCCCTGTCGACGGAGAACGCC-3′ |
| R, 5′-GGCGTTCTCCGTCGACAGGGCGATACAGCCGTTGGTGAC-3′ |
Site-directed Mutagenesis Studies
Site-directed mutagenesis reactions were performed as described previously with minor modifications (15). Briefly, two PCRs (25 μl) containing either the forward or reverse primer were set up in parallel. Each PCR contained 1× New England Biolabs GC reaction buffer, 200 μm dNTPs, 1 ng/μl template, a 500 nm concentration of either the forward or reverse primer, 1 unit of Phusion polymerase, and 3% DMSO. The ldtMt5-pET28a plasmid was used as the template to generate each LdtMt5 variant (see Table 1 for primers). Sufficient elongation of primer at 68 °C occurred over 15 min. Forward and reverse PCRs were then combined (new volume of 50 μl), and complementary strands were reannealed following gradual cooling (95 °C, 5 min; 90 °C, 1 min; 80 °C, 1 min; 70 °C, 30 s; 60 °C, 30 s; 50 °C, 30 s; and 40 °C, 30 s). Samples were then incubated at 37 °C, and template DNA was digested with 1 unit of DpnI for 3 h. All constructs were fully sequenced, and competent bacteria were transformed with mutagenic plasmid. All variants were purified as described above.
Kinetic Analyses
The nitrocefin hydrolytic activities of LdtMt2, LdtMt5, and LdtMt5 variants were measured spectrophotometrically as reported previously for LdtMt2 (10) but with modifications. Briefly, reaction mixtures containing 1× tribuffer (100 mm MES, 50 mm N-ethylmorpholine, and 50 mm diethanolamine, pH 10), 0.1 mg/ml BSA, 100 mm NaCl, 1 mm TCEP, 5% DMSO, and 10 μm LdtMt5 or LdtMt5 variant were preincubated at 37 °C for 5 min. Nitrocefin (Calbiochem) was added to initiate each reaction, and the rate of nitrocefin hydrolysis was measured at 486 nm at 37 °C. For each set of reactions, the rate of nitrocefin hydrolysis in the absence of enzyme was observed and was subtracted from the initial rate of nitrocefin hydrolysis in the presence of enzyme at each substrate concentration. Initial rates of nitrocefin hydrolysis were measured over 3 min. An extinction coefficient (ϵM) of 20,500 m−1 cm−1 was used in determining the concentration of hydrolyzed nitrocefin as it was experimentally determined that ϵM does not change with changing pH under these buffering conditions (data not shown). Non-linear regression analyses of initial velocities were performed using GraphPad Prism (version 5). Reaction mixtures containing nitrocefin (100 μm) and LdtMt5 or LdtMt2 (5 μm) at varying pH values in 1× tribuffer and the conditions described above were monitored spectrophotometrically for determining the optimal pH for each enzyme. To rule out the possibility that residual TEV incompletely removed during protein purification could be contributing to nitrocefin hydrolysis at pH 10, we evaluated nitrocefin as a substrate for TEV at pH 10. Only baseline levels of hydrolysis were observed, indicating that TEV was not significantly contributing to the observed rates of hydrolysis by LdtMt5 and LdtMt5 variants (data not shown). Conversely, when we evaluated nitrocefin as a substrate for His6-LdtMt5, we observed rates of hydrolysis that were comparable with TEV-cleaved LdtMt5 (data not shown).
Mass Spectrometry Analyses
LdtMt5 or N358H/M346W LdtMt5 (2 μm) in 12.5 mm Tris buffer, pH 8 was incubated in the presence or absence of 50 μm carbapenem for 5 h at room temperature. Reactions were quenched by the addition of trifluoroacetic acid (TFA; final concentration, 0.1%). Samples were filtered through a 0.2-μm filter and analyzed by ultraperformance LC/MS at 60 °C. The mobile phase was as follows: 0–1 min, isocratic 90% water + 10% ACN + 0.1% formic acid; 1–7.5 min, gradient to 20% water + 80% ACN + 0.1% formic acid; 7.5–8.4 min, isocratic 20% water + 80% ACN + 0.1% formic acid; 8.4–8.5 min, gradient to 90% water + 10% ACN + 0.1% formic acid; 8.5–10 min, isocratic 90% water + 10% ACN + 0.1% formic acid. The flow rate was 0.3 ml min−1.
Crystallization Conditions
Crystals of LdtMt5 were obtained by the hanging drop vapor diffusion method at 20 °C. Drops of 2 μl of protein (12.8 mg/ml) and 1 μl of reservoir solution were equilibrated against a reservoir containing 85 mm sodium citrate, pH 5.6, 25.5% polyethylene glycol (PEG) 4,000, 170 mm ammonium acetate, and 15% glycerol. Crystals suitable for data collection grew within 1 week. Crystals of LdtMt5 in complex with meropenem were obtained with protein incubated with the ligand (645 μm) using crystallization conditions identical to that of the apo crystal.
Data Collection, Structure Determination, and Refinement
All diffraction data were collected at beamline X6A of the National Synchrotron Light Source of the Brookhaven National Laboratory (Table 2). X-ray diffraction experiments were carried out with crystals frozen in their respective mother liquor without addition of cryoprotectant. The crystal structure of apo-LdtMt5 was determined by molecular replacement with the program MOLREP (16) using previously determined LdtMt2 structures of individual domains as searching models (Protein Data Bank code 3TUR, catalytic domain (CD) and bacterial Ig-like (BIg) B domains; Protein Data Bank code 4HU2, BIgA domain). After 20 cycles of refinement of the three separate domains as rigid bodies with REFMAC (CCP4), the structure was rebuilt with the molecular modeling program Coot (17) and further refined with the program PHENIX using restrained and translation, libration, and screw anisotropic refinement protocols with four translation, libration, and screw groups for each BIg domain and two for the CD (18). The apo-LdtMt5 structure was refined to a final Rwork of 0.21 and an Rfree of 0.255 with 96.1% of the residues in favored stereochemistry regions (Table 2) and solved to 1.98 Å.
TABLE 2.
Statistics of x-ray data collection and structure refinement
| Apo-LdtMt5 | Meropenem-bound LdtMt5 | |
|---|---|---|
| Wavelength (Å) | 1.0 | 1.0 |
| Resolution range (last shell) (Å) | 28.92–1.98 (2.051–1.98) | 29.04–2.8 (2.9–2.8) |
| Space group | P622 | P622 |
| Unit cell (Å; °) | 101.0, 101.0, 192.7; 90, 90, 120 | 99.4, 99.4, 193.4; 90, 90, 120 |
| Total reflections | 3,529,565 | 1,838,131 |
| Unique reflections | 41,056 (3,985) | 14,522 (1,400) |
| Multiplicity | 7.2 (7.0) | 22.4 (22.9) |
| Completeness (%) | 99.6 (99.15) | 99.4 (98.73) |
| Mean I/σ(I) | 57.4 (3.9) | 53.3 (6.3) |
| Wilson B-factor | 37.81 | 61.89 |
| Rsym | 0.048 (0.513) | 0.09 (0.9) |
| Reflections used for Rfree | 2,046 | 734 |
| Rwork | 0.23 (0.28) | 0.23 (0.29) |
| Rfree | 0.255 (0.36) | 0.275 (0.36) |
| Number of non-hydrogen atoms | 2,947 | 2,750 |
| Protein | 2,740 | 2,669 |
| Ligands | 39 | 32 |
| Water | 168 | 49 |
| Protein residues | 355 | 345 |
| r.m.s. | 0.006 | 0.011 |
| Bonds | ||
| Angles | 1.07 | 1.43 |
| Ramachandran favored (%) | 96 | 93 |
| Ramachandran outliers (%) | 0.85 | 2.6 |
| Clashscore | 3.5 | 9.45 |
| Average B-factor | 80 | 77.0 |
| Protein | 80.1 | 79.8 |
| Ligands | 114.3 | 76 |
| Solvent | 70.9 | 76 |
The crystal structure of meropenem-bound LdtMt5 was determined by molecular replacement and refined using a protocol similar to that used when solving the apo-LdtMt5 structure (Table 2). The partial meropenem adduct was built inside the positive σA (mFo − DFc) electron density difference map. Weak electron density for the sulfur atom of meropenem was observed, and no electron density for the pyrrolidine ring extension of meropenem was observed. The meropenem adduct structure was refined to an Rwork of 0.23 and an Rfree of 0.275 with 93% of the residues in Ramachandran favored regions (Table 2). The structure was solved to 2.8 Å. r.m.s.d. calculations and alignments were performed using the program Molecular Operating Environment (MOE; v.2014.09, GCC Inc.). Additional structural figures were completed using PyMOL (v.1.7.1.3, ©2009–2014 Schrödinger, LLC) and Chimera, v.1.9 (19). Buried surface area calculations were performed using the Protein Interfaces, Surfaces, and Assemblies' Service (PISA) at the European Bioinformatics Institute (20). Coordinates and structure factors were deposited in the Protein Data Bank under the codes 4Z7A (apo-LdtMt5) and 4ZFQ (meropenem-bound LdtMt5).
Calorimetric Studies
Freshly thawed LdtMt5 protein was dialyzed overnight in 1 liter of buffer containing 25 mm Tris-HCl, pH 7.5, 100 mm NaCl, and 1 mm TCEP at 4 °C. Dialyzed protein solution was then filtered through a 0.22-μm filter and diluted to 10 μm. The protein concentration was determined by UV absorption at 280 nm using a calculated extinction coefficient of 78,840 m−1 cm−1. Freshly thawed aliquots of 20 mm carbapenem stock solutions were diluted to 1 mm in protein dialysis buffer. Protein and ligand solutions were degassed for 20 min in a ThermaVac®. Ligand injections (10 μl) into the cell containing LdtMt5 were performed with 240-s equilibrations between injections. Data were analyzed with Origin 7 software (OriginLab). All calorimetry experiments were carried out at 27 °C.
M. tuberculosis Strains and Culture Conditions
M. tuberculosis CDC1551 (21) (herein referred to as “wild type”) was used as the host strain to generate a transposon insertion mutant in the MT0501 (ldtMt5::Tn) gene as described previously (22). All strains were grown in Middlebrook 7H9 liquid medium supplemented with 0.2% glycerol, 0.05% Tween 80, 10% (v/v) oleic acid/albumin/dextrose/catalase, 50 μg/ml cycloheximide (herein referred to as 7H9 complete medium), and when indicated 2.0 μg/ml crystal violet. The ldtMt5::Tn strain was grown in the presence of 20 μg/ml kanamycin. Middlebrook selective 7H11 solid medium (BD Biosciences) was used for enumerating colony forming units (cfus) in in vitro growth studies. M. tuberculosis H37Rv was used in generating meropenem-resistant strains.
In Vitro Growth and Crystal Violet Studies
All M. tuberculosis strains were grown to midexponential phase with an A600 of ∼0.8 in 7H9 complete medium at 37 °C. Cultures were then diluted to an A600 of 0.1 in 7H9 complete medium in the presence or absence of 2.0 μg/ml crystal violet, and turbidity of the cultures was determined daily. Appropriate dilutions of each strain were cultured on Middlebrook 7H11 medium to determine surviving bacilli by enumerating the cfus.
Osmotic Tolerance Studies
Wild-type or ldtMt5::Tn strains were grown to late exponential phase (A600 ∼ 2–3) in 7H9 complete medium. Cultures were diluted to an A600 of 0.5, and cells were pelleted at low speed. Cells were resuspended in 150 mm NaCl or double distilled H2O (0 mm NaCl) solutions containing 0.05% Tween 80. Cells were incubated in these conditions for 1 h with shaking at 37 °C. Viability was determined by culturing and enumerating the cfus on 7H11 selective agar.
Determination of Minimum Inhibitory Concentration
Carbapenem minimum inhibitory concentrations were determined using the standard broth dilution method (23). Briefly, 105 M. tuberculosis bacilli were inoculated into 2.5 ml of 7H9 broth supplemented with 0.2% glycerol, 10% (v/v) oleic acid/albumin/dextrose/catalase, and 50 μg/ml cycloheximide, and the drug was added at different concentrations in the μm–mm range. The cultures were incubated at 37 °C without shaking and evaluated for growth by visual inspection of the broth at 14 and 21 days. Minimum inhibitory concentration values are representative of three independent experiments.
Electron Microscopy Experiments
Field emission scanning electron microscopy and transmission electron microscopy experiments were performed as described previously (14).
Results
LdtMt5 Structure
The apo and meropenem adduct structures of an N-terminally truncated LdtMt5 protein lacking the hydrophobic domain predicted to be a membrane anchor for this protein (amino acids 55–451) were determined using x-ray crystallography (Fig. 1). This truncated protein displays higher sequence identity to LdtMt2 (31%) than the full-length protein does (28%) and includes the proline-rich extension of the C-terminal subdomain (ex-CTSD) comprising residues 417–451 that is absent in all other M. tuberculosis ld-transpeptidases (Fig. 2A).
FIGURE 1.

Crystal structure of LdtMt5. Left, apo-LdtMt5; right, rotated 90°. LdtMt5 is composed of two BIg domains and a CD. The semitransparent volume is surface-accessible by a 3.5-Å radius probe. Residues of the active site (His342, Thr357, Asn358, and Cys360) are represented as sticks within the CD. Tryptophan residues of the CTSD are also represented as sticks and interact with a hydrophobic patch at the interface of the BIgB domain. The prominent outer cavity that is observed in LdtMt2 is absent in apo-LdtMt5 but is indicated as a reference (10). The LdtMt5 secondary structure schematic is colored as a rainbow from blue (N terminus) to red (C-terminus). Orange dashes represent the disordered portion of loop LC, and red dashes represent the disordered ex-CTSD. This figure was made using Chimera (19).
FIGURE 2.

Comparison of M. tuberculosisld-transpeptidases. A, the sequence alignment based on the structural superposition. The observed secondary structures are noted above the amino acid sequences. Starred residues are those highlighted in the text (blue, coordinating His342; cyan, BIgA and BIgB interface; green, CTSD-BIgB-CD core interaction; red, catalytic residues; yellow, loop LD). Named loops are also marked, and the characteristic ld-transpeptidase motif is boxed in blue rectangle. B, overlay of the apo structures of LdtMt2 (Protein Data Bank code 3VYN; in cyan) and LdtMt5 (this work; in red). Observed differences in the CD are concentrated in the β-hairpin and loops LC–LE. Disordered regions are represented as dashed lines. C, the LdtMt5 adduct structure displays the largest CD conformational changes among characterized ld-transpeptidases. The EYY-folded CD cores of apo-LdtMt5 (red), holo-LdtMt5 (pink), and the apo and holo crystal structures of LdtMt1 (Protein Data Bank code 4JMN, apo, in light green; Protein Data Bank code 4JMX, imipenem adduct, in dark green), and LdtMt2 (Protein Data Bank code 3VYN, apo, in cyan; Protein Data Bank code 3VYP, meropenem adduct, in blue) are shown as smoothed Cα traces. Structural elements with high r.m.s.d. are shown in full opacity; the remaining structural elements are displayed as transparent Cα traces to demonstrate conservation of the EYY-folded CD cores. The largest changes among structurally characterized ld-transpeptidases are observed in the β-hairpin, and in the case of LdtMt5, there is a dramatic displacement of loop LC that occurs after adduct formation (indicated with red curved arrows). The C-terminal portion of the CTSDs was excluded for clarity. D, accessible surface map of apo-LdtMt5 colored by the magnitude of the observed atomic temperature factors from low (green) to high (magenta) motility. The flexibility of the β-hairpin as indicated by the high atomic temperature factor correlates with its large displacement upon adduct formation. These images, the sequence alignment, and structural superpositions were performed using the program MOE. The sequence representation was performed using ESPript3 (48).
Apo-LdtMt5 and meropenem adduct crystals belong to the P6222 space group with similar cell dimensions and one molecule in the asymmetric unit (Table 2). Electron density for residues 56–346 and 353–416 was observed in the apo crystal form. Residues 347–352 within loop LC (residues 344–362; nomenclature from Ref. 10) and residues 417 to the C-terminus (residue 451) are disordered; their electron density was not observed in either apo- or meropenem adduct-LdtMt5 crystal structures. LdtMt5 possesses three globular domains (Fig. 1) as observed in its paralog LdtMt2 (Fig. 2B) (10, 24–26). The structure reveals an N terminus span of long-chain hydrophilic residues (residues 56–60) that precede two in-tandem antiparallel β-barrels resembling BIg domains joined by a short three-residue linker (BIgA, residues 61–153; BIgB, residues 156–252) followed by a C-terminal CD (residues 253–417). The CD can be divided into a core (residues 253–385) that contains an ErfK/YbiS/YhnG fold (EYY-fold) (27) consisting of a β-sandwich with two mixed β-sheets and one helix (α2) and a helically folded C-terminal subdomain (CTSD; residues 386–416). ld-Transpeptidases are characterized by the presence of this EYY-folded domain, which contains the characteristic HXX14–17(S/T)HGChN motif (h represents a hydrophobic residue) where cysteine is the catalytic residue (Fig. 2A). By extension, we presume Cys360 to be the catalytic residue of LdtMt5.
The overall structural overlap between apo-LdtMt5 and LdtMt2 (Protein Data Bank code 3VYN) has an r.m.s. deviation of 2.2 Å for 225 aligned Cα atoms, including 79 identical residues, highlighting their overall structural similarity (Fig. 2, B and C). There are small differences with regard to the orientation of the domains among other structurally characterized ld-transpeptidases. The equivalent BIg domains of LdtMt5 and LdtMt2 are similar; the BIgA domains display a small r.m.s. deviation of 1.0 Å among 65 pairs of Cα atoms aligned, and the BIgB domains display a r.m.s. deviation of 0.94 Å among 103 pairs of aligned Cα atoms. The EYY-folded CD cores of LdtMt1, LdtMt2, and LdtMt5 overlap well (Fig. 2C). However, differences localized to the β-hairpin flap (residues 312–330), loop LC, and the C-terminal region of loop LD increased the pairwise r.m.s.d. An r.m.s.d. of 1.25 Å for the 67 Cα atom pairs within the CD core was observed compared with r.m.s.d. values of 2.3 and 2.6 Å for 115 and 103 Cα atom pairs when the entire CD of LdtMt5 was compared with that of LdtMt2 and LdtMt1 (Protein Data Bank code 4JMN), respectively (the LdtMt1/LdtMt2 overlap is 0.66 Å for 67 Cα atom pairs). Interestingly, the imipenem adduct structure of LdtMt1 (Protein Data Bank code 4JMX) minimally affects placement of the β-hairpin in comparison with its position in apo-LdtMt1, whereas the meropenem adduct-LdtMt5 structure displays the largest changes in the β-hairpin (Fig. 2C).
The Conformation of the LdtMt5 BIg Domains Is Mostly Maintained by Unspecified Hydrophobic Interactions
A small, solvent-accessible area (312 Å2) is buried in the interface between the BIg domains in LdtMt5. A short, three-residue linker (Ala153, Pro154, and Val155) joins BIgA and BIgB. BIgA is rotated ∼30° around the axis passing through the interdomain linker compared with BIgA of LdtMt2 (Fig. 2B). Two hydrophobic patches comprising Pro80, Tyr125, and Pro154 of BIgA and Tyr239 and Tyr248 from BIgB and main-chain atoms of β-barrel loops from both of the domains make an interdomain contact. These proline and tyrosine residues are well conserved among three-domain ld-transpeptidases (Fig. 2A). Tyr125 (from BIgA) and Tyr239 (from BIgB) exchange hydrogen bonds with main-chain atoms of the opposite domain. This greasy and weak contact may provide flexibility in the orientation of the domains. In the LdtMt5 crystal form, a PEG molecule from the crystallization buffer is bound to an exposed hydrophobic patch (Pro122 and Tyr239) at the interface of the BIgA and BIgB domains, apparently stabilizing the observed relative orientations of the domains.
The Conformations of the BIgB Domain and EYY-fold Are Maintained by the CTSD
The LE loop of the LdtMt5 CTSD is slightly longer in comparison with that of LdtMt2 (Fig. 2, B and C). This loop wedges between the EYY-fold and the BIgB domain (Fig. 1). The LdtMt5 CTSD is rich in tryptophan residues (Trp398, Trp400, Trp404, and Trp407). Extensive hydrophobic contacts among the CTSD, BIgB, and CD domains increase the rigidity of the BIgB/CD assembly. In addition to regular contacts between Tyr392 and Trp398 in loop LE with Leu209 and Pro210 in BIgB, the aromatic rings of Trp400, Trp404, and Trp407 in the α3 helix of the CTSD form a “zipper-like” interaction with the aromatic ring of Tyr225 and aliphatic portions of the side chains of Arg223 and Arg221 of the BIgB domain. This structure provides 1336 Å2 of additional area buried in the BIgB/EYY-fold assembly, which itself only contributes 433 Å2.
The LdtMt5 CD Displays Large Structural Differences Relative to LdtMt1 and LdtMt2
The CD of LdtMt5 displays marked differences in comparison with those of LdtMt1 and LdtMt2. The largest differences are seen 1) within the fold and placement of a β-hairpin flap that includes loop LF (the shortest among homologs), 2) in the conformation and partial disorder of loop LC, and 3) in the size of loop LD and loop LE (of the CTSD) (Fig. 2). All of these structural differences are in close proximity to the LdtMt5 active site. The β-hairpin flap covers the active site and is the structural feature that distinguishes some ld-transpeptidases (10, 24, 28) from the first structurally characterized protein containing the EYY-fold (27). This flap displays the largest temperature factors relative to the remainder of LdtMt5 (Fig. 2D), is nine residues shorter in LdtMt5, and displays low homology to other M. tuberculosis ld-transpeptidase β-hairpin flaps (Fig. 2C). In LdtMt5, loop LC displays considerable disorder. Electron density for residues 347–353 in apo-LdtMt5 and residues 348–356 in the meropenem adduct structure was not observed. However, the residues of loop LC that are ordered display fold differences relative to LdtMt1 and LdtMt2 (Fig. 2C).
In previously solved ld-transpeptidase structures, the catalytic site is exposed through two connected cavities, the outer and inner cavities (Fig. 2, B and C). Compared with LdtMt1 and LdtMt2, the small footprint and placement of this β-hairpin in LdtMt5 lead to greater exposure of the catalytic site from the inner cavity (Fig. 2). The β-hairpin flap and loop LC of apo-LdtMt5 are shifted toward the outer cavity, closing it (Figs. 2C and 3A). In our meropenem adduct-LdtMt5 structure, the hairpin and loop are partially disordered; however, the ordered portions appear to shift away from the catalytic site, thereby exposing it (Fig. 3B). Thus, the acylation of LdtMt5 by meropenem appears to “create” an outer cavity reminiscent of that observed in LdtMt1 and LdtMt2 (Fig. 3C).
FIGURE 3.

Comparison of the LdtMt5 and LdtMt2 active sites. A, apo-LdtMt5; B, meropenem-bound LdtMt5 (meropenem adduct shown as purple sticks); C, LdtMt2-PG fragment complex (Protein Data Bank code 3TUR; PG fragment shown as cyan sticks). The left panels show secondary structure schematic representations, and residues within the active site are represented as sticks. The right panels show the probe-accessible surface (3.5-Å radius). Surface zones related to the β-hairpin and loop LC that display the largest structural differences among apo and holo structures are colored purple. Acylation of LdtMt5 by meropenem causes displacements of these structural elements as indicated by the green arrows (right panel) that “restore” the outer cavity.
Loop LD (residues 289–302) within the CD core of LdtMt5 is larger compared with Loop LD of LdtMt1 and LdtMt2 (Fig. 2A-C). The LdtMt5 LD loop has a three-residue insertion that includes a bulky arginine residue (Arg297) and forms a protruding insertion (Fig. 2C). Although most of this loop fold remains unperturbed, the insertion displaces the adjacent LC loop (residues 338–358), thereby closing the active site outer cavity and dramatically modifying the fold and placement of the LC loop.
The LdtMt5 Active Site Is Structurally Divergent Relative to LdtMt1 and LdtMt2
The structural differences within the LdtMt5 CD have dramatic effects on the active site architecture and the readiness of catalytic residues to participate in enzymatic reactions and presumably PG stem recognition. In LdtMt1 and LdtMt2, a conserved methionine residue (Met175 and Met303, respectively) on the internal side of the β-hairpin flap limits the space around the catalytic cysteine. The unique placement of this loop in LdtMt5 results in displacement of this methionine (Met316 in LdtMt5) by the non-conserved Glu328 (Fig. 3). Glu328 is substituted with smaller hydrophobic residues in LdtMt2 (Val322) and LdtMt1 (Ala195). Interestingly, the electron density surrounding this glutamate residue indicates that Glu328 is present in three alternative conformations in the apo-LdtMt5 structure but shows only one conformation in the meropenem adduct structure (Fig. 3, A and B). The most populated conformation of Glu328 and the conserved motif Asn362 (implicated in PG stem recognition) form hydrogen bonds with His342, thereby orienting His342 in such a way that it is rotated 180° from the orientation required to deprotonate Cys360 (Fig. 4, A and B). Furthermore, Cys360 and His342 make a strong contact with one another; electron density connecting the sulfur atom to the imidazole ring carbon is visible in the experimental electron density map (Fig. 4A). However, no covalent bond is present: the coordination geometry of the Cϵ1–Sγ–Cβ bond angle is much smaller than 109°, which is expected for a direct bond. It is likely that this strong contact and coordination of the histidine ring by Asn362 and Glu328 make it difficult for the imidazole ring to rotate into a position that is better poised for catalysis (Fig. 4B). Thus, it is clear that His342 is not optimally poised to act as a catalytic base in LdtMt5 as is the equivalent residue in LdtMt2 (His336) is (Fig. 3C).
FIGURE 4.

An “active” conformation of LdtMt5 is achieved when meropenem binds. A, a nonproductive orientation of His342 is observed in apo-LdtMt5 (σA-weighted electron density map contoured at 1.0 σ level). Residues of the refined structures are shown as stick representations. His342 is in close proximity to Cys360 as suggested by the electron density connecting these two residues. In addition, His342 hydrogen bonds with the most populated alternative conformation of Glu328 and Asn362 (dashed lines). Collectively, these interactions fix the imidazole ring in a nonproductive orientation that is not present in the meropenem adduct structure. B, mechanism of acylation of LdtMt5 by meropenem. C, overlay of active sites of apo and meropenem adduct structures. The red arrows indicate the large displacements of Met346 (Sδ–Sδ, 6.6 Å), Thr357 (Cβ–Cβ, 8.2 Å), and Asn358 (Cβ–Cβ, 4.1 Å) relative to the apo structure. The active site of apo-LdtMt5 is colored gray, and the meropenem adduct structure is colored brown. The ∼180° rotation of His342 is noted with a curved red arrow. Meropenem is represented as magenta sticks. Disordered residues are represented as dashed lines. This figure was made using Chimera (19).
The LC loop of LdtMt5 is fully embedded in the conserved HXX14–17(S/T)HGChN motif that characterizes this family of transpeptidases. LdtMt5 has two variations in the conserved motif: a motif alternative Thr357 of LdtMt5 replaces the LdtMt2 serine (Ser351), and Asn358 replaces the characteristic motif histidine (His352 in LdtMt2). The C-terminus of loop LC forms the “anion hole” at the catalytic site of LdtMt2. Thr357 occludes the outer entrance to the active site (Figs. 3A and 4C) where the PG stem binds to LdtMt2 (Fig. 3C) (10). The loop, which contains the anion hole that comprises a large quantity of positively charged atoms, is folded differently relative to LdtMt2, and Met346 of loop LC replaces a tryptophan residue that is conserved in all other M. tuberculosis ld-transpeptidases (Fig. 2A).
LdtMt5 Is Acylated by Meropenem during Crystallization
The crystal form grown in the presence of meropenem shows electron density for LdtMt5 residues 56–317, 327–348, and 356–416. Electron density for most of the β-hairpin flap is missing in this crystal form, and like apo-LdtMt5, most of loop LC and the ex-CTSD are disordered. The carbapenem core of meropenem was fitted in additional electron density near the catalytic cysteine, which forms an adduct with Cys360 (Fig. 3B); however, no electron density for the 3-[5-(dimethylcarbamoyl)pyrrolidin-2-yl] group of meropenem was observed. Interestingly, the presence of this adduct restores the anion hole and other portions of LC to a similar fold previously observed in other ld-transpeptidases, placing residues with a probable role in catalysis (Met346, Asn358, and Thr357) in positions equivalent to those observed in active ld-transpeptidases (Fig. 4C). Thus, meropenem binding induces a conformational change that enables access to the catalytic site from the outer cavity as observed in other ld-transpeptidases (Fig. 3). In addition, this change promotes release of His342 from its nonproductive contact such that it now hydrogen bonds with Cys360 (distance of Nϵ–Sγ, 3.2 Å; Figs. 3B and 4C).
The most stable tautomer of the carbapenem core is observed where the ring nitrogen is deprotonated (double bond between C3 and N4) and C2 is sp3 hybridized and is in agreement with previously reported LdtMt2-meropenem adduct structures (24, 25). The meropenem core lies with its most apolar side facing a hydrophobic patch formed by Gly338, the aliphatic portion of the side chain of Glu339, and Phe340 at the inner cavity. The C-terminal portion of the main chain of loop LC (Gly359) provides apolar contacts with the other side of the carbapenem core. Four hydrophilic interactions are also observed between the carbapenem core and LdtMt5: 1) Asn358 and 2) the main chain nitrogen atom of Cys360 hydrogen bond to the carbonyl of the opened penem ring, 3) Glu328 hydrogen bonds to the meropenem hydroxyethyl group, and 4) a water molecule (W601) mediates interaction between the meropenem core carboxylate and the carboxylate of Glu339 (Fig. 3B).
We evaluated a series of β-lactams, including the carbapenems listed in Table 3, and measured the thermodynamics of β-lactam binding to LdtMt5 using ITC. Despite the presence of a meropenem adduct on LdtMt5, no significant heat exchange associated with binding was measured by ITC, and no adduct was detected by mass spectrometry after a 5-h incubation of meropenem and LdtMt5 (Table 3).
TABLE 3.
Summary of mass spectrometry data obtained after preincubation of LdtMt5 and different carbapenems
| Enzyme | Carbapenem | Calculated mass of inhibited species | Mass of carbapenem | Observed massa |
|---|---|---|---|---|
| Da | Da | Da | ||
| LdtMt5 | None | 42,603 | 42,605 | |
| Meropenem | 42,986.5 | 383.5 | 42,605 | |
| Imipenem | 42,902.4 | 299.4 | 42,605 | |
| Doripenem | 43,023.1 | 420.1 | 42,604 | |
| Biapenem | 42,953.4 | 350.4 | 42,606 | |
| Tebipenem | 42,986.5 | 383.5 | 42,605 | |
| N358H/M346W LdtMt5 | None | 42,681 | 42,683 | |
| Meropenem | 43,064.5 | 383.5 | 42,684 | |
| Imipenem | 42,980.4 | 299.4 | 42,683 | |
| Doripenem | 43,101.1 | 420.1 | 42,683 | |
| Biapenem | 43,031.4 | 350.4 | 42,683 | |
| Tebipenem | 43,064.5 | 383.5 | 42,684 |
a Reported masses are those that comprise the largest species in the mass spectrum at 100% abundance.
LdtMt5-catalyzed Nitrocefin Hydrolysis Is Optimal at Basic pH
LdtMt5 was probed for transpeptidase/β-lactamase activity using nitrocefin as a substrate. A pH rate profile analysis revealed that LdtMt5 is optimally active at pH >9 (Fig. 5) even after correcting for spontaneous ring opening at basic pH in the absence of enzyme, although its activity was not significantly different from that observed for LdtMt2, which optimally catalyzes nitrocefin hydrolysis at pH 7 (10) (Fig. 5). We also measured nitrocefin binding at pH 8 where little LdtMt5-catalyzed nitrocefin hydrolysis was observed (Fig. 5), but no detectable heat of exchange was observed using ITC (data not shown).
FIGURE 5.
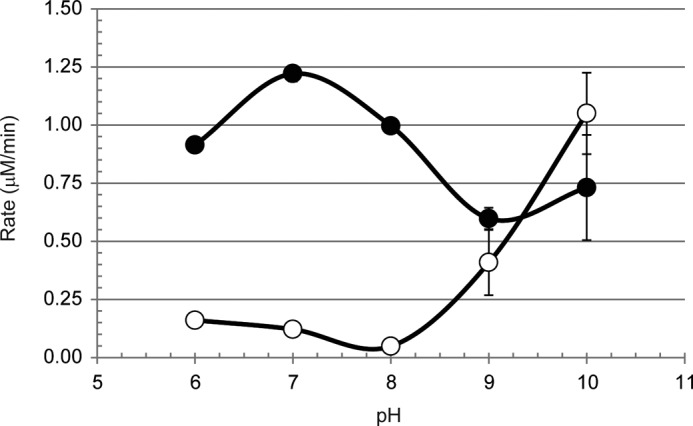
pH rate profile analysis of LdtMt5 (open circles) and LdtMt2 (closed circles). Error bars represent S.E.
Conserved Active Site Residues Are Not Required for Nitrocefin Hydrolysis
On the basis of the apo-LdtMt5 structure, we rationally designed and purified LdtMt5 putative active site variants in an attempt to identify residues responsible for nitrocefin hydrolysis at pH >9. Surprisingly, all of the LdtMt5 variants evaluated, including C360A LdtMt5, hydrolyzed nitrocefin (Table 4 and Fig. 6) with the rates of hydrolysis ordered as follows: N358H > wild type > H342Q > H342A > T357V > N358A > N362A > C360A. Although the rates of hydrolysis were relatively low, conservative mutations (N358H and H342Q) had the lowest impact on kcat/Km specificity constants, whereas C360A LdtMt5 was the least active variant we tested. Interestingly, the Km(nitrocefin) for the N358H variant was 2-fold lower than that of wild type, whereas the kcat values were relatively comparable.
TABLE 4.
Summary of kinetic parameters for nitrocefin hydrolysis by LdtMt5 and LdtMt5 variants at pH 10
The LdtMt5 variants are listed in order of decreasing catalytic efficiency.
| Enzyme | Km | kcat | kcat/Km |
|---|---|---|---|
| μm | min−1 | min−1·m−1 | |
| LdtMt5 N358H | 50 ± 3 | 0.61 ± 0.03 | 12.3 ± 0.4 × 103 |
| LdtMt5 | 97 ± 19 | 0.79 ± 0.09 | 8.7 ± 1.9 × 103 |
| LdtMt5 H342Q | 151 ± 11 | 0.63 ± 0.01 | 4.2 ± 0.2 × 103 |
| LdtMt5 H342A | 153 ± 8 | 0.49 ± 0.01 | 2.9 ± 0.1 × 103 |
| LdtMt5 T357V | 262 ± 72 | 0.55 ± 0.08 | 2.4 ± 0.6 × 103 |
| LdtMt5 N358A | 125 ± 25 | 0.32 ± 0.02 | 2.4 ± 0.3 × 103 |
| LdtMt5 N362A | 175 ± 16 | 0.40 ± 0.01 | 2.2 ± 0.1 × 103 |
| LdtMt5 C360A | 211 ± 40 | 0.33 ± 0.02 | 1.1 ± 0.2 × 103 |
FIGURE 6.

LdtMt5 active site variants catalyze nitrocefin hydrolysis at pH 10. Michaelis-Menten curves for LdtMt5 and LdtMt5 variants are shown. Error bars represent S.E.
Two residues at the ends of loop LC that interact with the PG stem in the outer cavity of LdtMt2 (10), His352 and Trp340, are substituted with Asn358 and Met346, respectively, in LdtMt5. Asn358 replaces this conserved motif histidine (His352) in LdtMt2 that participates in recognition of the donor PG stem (10) and in LdtMt5 that participates in recognition of the meropenem adduct (Fig. 3B). Trp340 in LdtMt2 is proposed to provide tetrapeptide substrate specificity (10). In an attempt to make the LdtMt5 active site more LdtMt2-like, we generated the LdtMt5 N358H/M346W double variant. Contrary to our expectations, mass spectrometry data indicate that the double variant was not acylated by the carbapenems tested (Table 3), and the double variant behaved like wild-type LdtMt5 when probed for its ability to catalyze nitrocefin hydrolysis in a pH rate profile analysis (data not shown).
Loss of ldtMt5 Modestly Enhances Susceptibility to Doripenem and Faropenem
Recent studies have reported that, in addition to inhibiting dd-transpeptidase and carboxypeptidase activities, carbapenems and penems bind to and inhibit ld-transpeptidases (10, 24, 25, 29–31). We hypothesized that loss of LdtMt5 may alter sensitivity to carbapenems as the mutant lacking this protein would have one less target for carbapenems to inhibit. Minimum inhibitory concentration studies were performed to evaluate whether or not loss of ldtMt5 affected the susceptibility of M. tuberculosis to carbapenems (Table 5). The ldtMt5::Tn strain reproducibly had modestly enhanced susceptibility to doripenem and faropenem (a penem) compared with wild type, but neither strain was susceptible to ertapenem or meropenem under the conditions that were tested. Both strains displayed similar susceptibilities to tebipenem pivoxil.
TABLE 5.
Susceptibility of M. tuberculosis strains to isoniazid and carbapenems (and faropenem)
Values are reported as μg/ml.
| Strain | Isoniazid | Ertapenem | Meropenem | Doripenem | Faropenem | Tebipenem pivoxil |
|---|---|---|---|---|---|---|
| Wild type | 0.1 | 20 | 12.5 | 2.5 | 5 | 10 |
| ldtMt5::Tn | 0.1 | 20 | 12.5 | 1.2 | 2.5 | 10 |
Mutations in the ldtMt5 Locus Could Not be Detected in Meropenem-resistant Mutants
We tested the hypothesis that LdtMt5 is a target of meropenem and that meropenem-resistant M. tuberculosis strains would harbor a mutation in the gene encoding this enzyme. Toward this end, we generated genetically resistant strains by isolating mutants grown in the presence of 400 μg/ml meropenem. Nine independent strains were isolated, their genomic DNA was purified, and the ldtMt5 loci (which included ∼100 bp in both 5′- and 3′-UTRs) were PCR-amplified and sequenced. In addition, we sequenced the locus of the parent M. tuberculosis H37Rv that was used to generate the mutants. The DNA sequences of the ldtMt5 loci in all nine meropenem-resistant strains were identical to the parent M. tuberculosis H37Rv and to the sequence of the reference M. tuberculosis H37Rv genome (data not shown) (32).
Loss of ldtMt5 Enhances Sensitivity to Crystal Violet and Osmotic Shock
To determine the effects of loss of functional LdtMt5, we probed the cell wall integrity of wild-type and ldtMt5::Tn strains using crystal violet and osmotic shock. In comparison with wild type, ldtMt5::Tn M. tuberculosis displayed a minor growth defect when grown in complete medium (Fig. 7A). When complete medium was supplemented with crystal violet, ldtMt5::Tn M. tuberculosis behaved similarly to cells lacking the dominant ld-transpeptidase LdtMt2 (ldtMt2::Tn) as both strains were more susceptible to killing by the dye (Fig. 7B). Furthermore, ldtMt5::Tn cells or ldtMt2::Tn cells were 2–3 times less viable than wild-type cells when subjected to osmotic challenge (Fig. 7C). These findings suggest that loss of ldtMt5 alters cell wall permeability and sensitivity to crystal violet and compromises cell wall integrity. We also examined the cell morphology of M. tuberculosis lacking ldtMt5 by electron microscopy. Interestingly, no observable changes in cell size and morphology between wild-type and ldtMt5::Tn strains were observed (Fig. 8). The gene encoding LdtMt5 is in an operon downstream of murB, another PG biosynthetic enzyme. We attempted to complement our ldtMt5::Tn strain with a wild-type copy of ldtMt5 under the control of its native promoter. We designed and tested eight different complemented strains, but none were able to restore growth phenotypes (data not shown).
FIGURE 7.
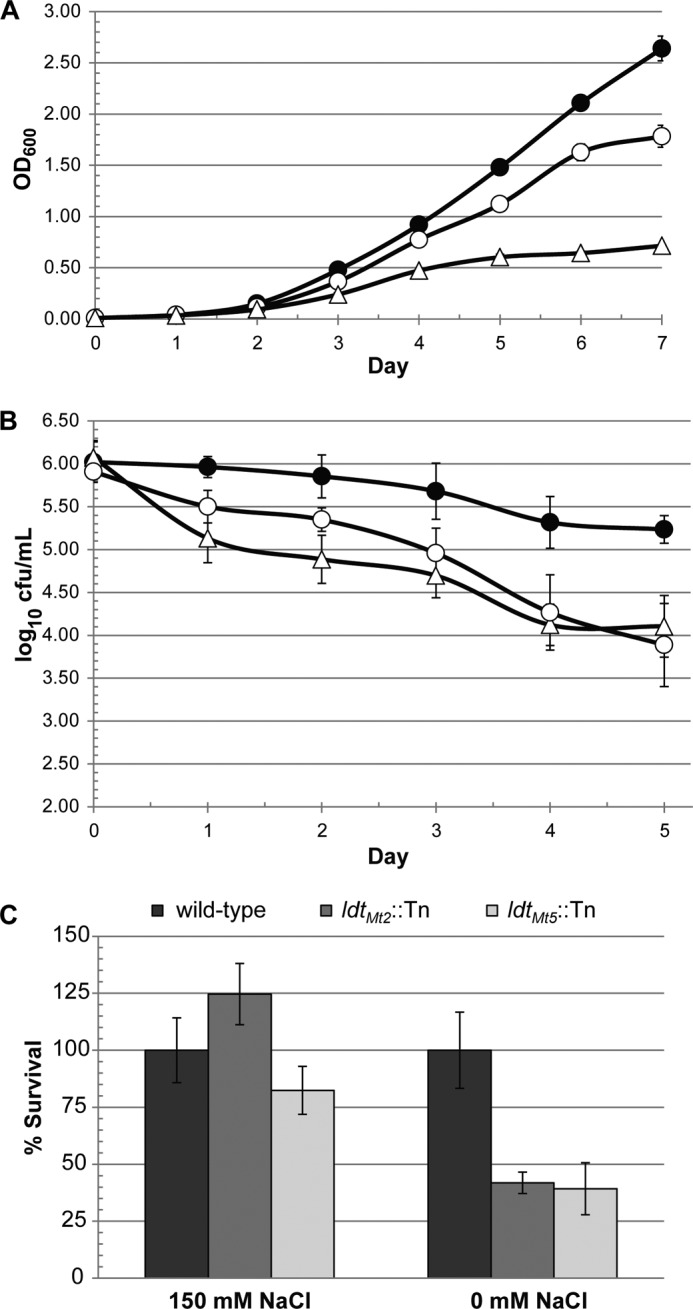
Loss of LdtMt5 sensitizes M. tuberculosis to crystal violet and osmotic stress. Wild-type (closed circles), ldtMt5::Tn (open circles), or ldtMt2::Tn (triangles) M. tuberculosis were grown in 7H9 complete medium (A) or 7H9 medium supplemented with crystal violet (B). C, strains lacking LdtMt2 or LdtMt5 are less tolerant to osmotic shock. Error bars represent S.E.
FIGURE 8.

Transmission (A and C) and scanning (B and D) electron microscopy reveals no significant changes in M. tuberculosis cell morphology upon loss of a functional copy of LdtMt5. A and B, wild-type M. tuberculosis; C and D, ldtMt5::Tn M. tuberculosis. Scale bars for transmission EM images (A and C) and scanning EM images (B and C) represent 100 and 200 nm, respectively.
Discussion
Recently, 3→3 cross-links have been identified in the PG of a variety of bacterial species (5, 33–36), and ld-transpeptidases have been identified as the enzymes that catalyze synthesis of this type of transpeptide bond (6–8, 12, 33, 37, 38). LdtMt5 is a paralog of LdtMt2 from M. tuberculosis and has been reported to catalyze formation of 3→3 transpeptide linkages in PG substrates derived from Mycobacterium abscessus (30).
There are significant structural differences within the CD of LdtMt5 and consequently the active site despite overall architectural similarity to LdtMt2. The β-hairpin flap that covers the active site is the smallest among paralogs and exhibits high mobility (high B factors in the apo structure and partial disorder observed in the meropenem-bound structure; Fig. 2D). This hairpin and loop LC display the largest structural differences among M. tuberculosis ld-transpeptidases (Fig. 2A), and the distinctive structural changes observed upon meropenem adduct formation suggest that their mobility and flexibility could play a role in the catalytic mechanism. The outer cavity that is present in other characterized ld-transpeptidases is absent in apo-LdtMt5; however, such an outer cavity is restored when meropenem acylates LdtMt5. Upon acylation by meropenem, residues from the conserved motif, including His342, Asn358, Cys360, and Asn362, shift to positions resembling those occupied by equivalent residues in LdtMt2 (Figs. 3, B and C, and 4A) and LdtMt1, lining a cavity that could accommodate a PG stem (Fig. 3).
We observed catalytic residues Cys360 and His342 in a nonproductive contact in our apo crystal form, and His342 is not optimally poised for catalysis (Fig. 4A); however, the nonproductive contact appears to be released upon adduct formation (Fig. 4B). We did not observe acylated LdtMt5 by mass spectrometry, likely the result of the presence of this nonproductive contact. However, we clearly observed a meropenem adduct on LdtMt5 in our crystal form. Thus, if given enough time, meropenem will acylate LdtMt5 over the extended incubation period that is required for co-crystallization. Alternatively, a component of the crystallization buffer may promote acylation of LdtMt5 by meropenem.
The pKa of a cysteine side chain is 8.3. Nitrocefin is a poor substrate for LdtMt5, but we observed LdtMt5-catalyzed nitrocefin hydrolysis at pH >9 (Fig. 5). It is feasible that the nonproductive contact of the catalytic residues may be released at pH ≥9 by weakening of the hydrogen bonds holding the residues in this conformation (Fig. 4A). None of the active site variants we designed fully abolished this activity, including C360A LdtMt5; however,C360A LdtMt5 was the least active variant. Furthermore, the N358H substitution affected nitrocefin recognition (KmN358H < KmWT). It has been demonstrated previously that substitutions to any of the catalytic residues of serine proteases significantly reduce the rate of peptide bond cleavage but do not completely abolish it (39), indicating that the remaining catalytic site environment after residue substitutions can still promote turnover albeit slowly. It is feasible that, under basic conditions, Cys360 is deprotonated and can hydrolyze nitrocefin and that excess hydroxide in the LdtMt5 active site will still promote turnover of this unnatural substrate even in the absence of the catalytic cysteine. Alternatively, different LdtMt5 residues may mediate nitrocefin hydrolysis.
In addition to catalyzing 3→3 transpeptidation in PG, ld-transpeptidases incorporate non-canonical d-amino acids into PG during stationary growth phase and catalyze attachment of Braun lipoprotein in some Gram-negative bacteria (12, 40). Unlike LdtMt2, LdtMt5 has a 33-residue ex-CTSD (residues 417–451). The ex-CTSD (Fig. 1) is disordered and contains proline-rich stretches (Fig. 2A). Proline-rich regions have been observed in other mycobacterial PG biosynthetic enzymes, including putative dd-transpeptidases PonA1, PonA2, and PonA3 and LdtMt4, another paralog of LdtMt2 (41, 42). Although these proline-rich regions are seemingly common among these M. tuberculosis cell wall biosynthetic enzymes, their role in M. tuberculosis physiology is still largely unknown. Interestingly, proline-rich sequence stretches frequently mediate protein-protein interactions (43). The proline-rich ex-CTSD of LdtMt5 is in close proximity to the catalytic site. Thus, it is plausible that the LdtMt5 ex-CTSD participates in the recognition of protein substrates and/or binding partners, and these interactions may drive the conformational changes required to release His342 and Cys360 from their nonproductive contact. Likewise, it is reasonable to speculate that the active site of LdtMt5 may have evolved to accommodate large substrates like proteins and play a role in anchoring them to the PG reminiscent of the role ld-transpeptidases serve in Gram-negative species in anchoring Braun lipoprotein (12, 40). Taken together, the major structural differences and divergent catalytic site suggest that LdtMt5 and LdtMt2 evolved to serve different functions in M. tuberculosis (Fig. 9).
FIGURE 9.

Loss of LdtMt2 or LdtMt5 compromises M. tuberculosis cell wall integrity. The M. tuberculosis PG is 3→3 and 4→3 cross-linked by ld- and dd-transpeptidases (not shown), respectively. Both LdtMt2 (green) and LdtMt5 (blue) have two BIg domains, whereas LdtMt1 (red) has one, which likely differentially positions the ld-transpeptidases within the periplasm. Although loss of LdtMt1 alone results in no discernible phenotype, loss of LdtMt2 results in compromised cell wall integrity as a result of loss of 3→3 cross-links in PG. LdtMt5 has a structurally distinct active site relative to both LdtMt1 and LdtMt2, and when LdtMt5 is lost, M. tuberculosis is sensitized to chemical probes and osmotic stress similarly to when LdtMt2 is lost. Although LdtMt5 can catalyze 3→3 cross-link formation in vitro, its physiological function remains unclear.
It has been demonstrated that YbiS, an E. coli ld-transpeptidase, is a substrate of the thioreductase DsbG (44). In E. coli, DsbG reduces the catalytic cysteine of YbiS that is prone to sulfenylation in the periplasm. We have previously reported a crystal structure of LdtMt2 that shows Cys354 oxidized to the sulfenic acid (10), suggesting that M. tuberculosis ld-transpeptidases are also susceptible to oxidation. Although we did not observe any sulfur adducts in our apo-LdtMt5 structure, it is conceivable that LdtMt5 requires binding of a protein partner to maintain the correct oxidation state of its catalytic cysteine in vivo.
Although all β-lactam antibiotics target dd-transpeptidases involved in 4→3 cross-link formation in PG maturation, only the carbapenem class of β-lactams (and faropenem, a penem) inhibit ld-transpeptidases. Furthermore, the genome of M. tuberculosis encodes for BlaC, an extended spectrum class A β-lactamase (45, 46). For these historical reasons, β-lactams are seldom considered for treatment of M. tuberculosis infection. However, carbapenems have been recently identified as poor substrates for BlaC (47). We have previously demonstrated that M. tuberculosis lacking LdtMt2 is more susceptible to killing by β-lactams (8, 14). Sanders et al. (42) have reported that LdtC (homologous to LdtMt5 in M. tuberculosis on the basis of sequence) is the primary ld-transpeptidase in Mycobacterium smegmatis. Strains lacking ldtC are hypersusceptible to imipenem, and ldtMt5 from M tuberculosis fully complements this phenotype in an ldtC mutant, suggesting that these enzymes are equivalent (42). We observed a modest enhancement in susceptibility of the ldtMt5::Tn strain to select carbapenems (Table 5) presumably due to synthetic lethality as these β-lactams may inactivate other targets. Although our meropenem adduct crystal form supported very slow acylation of LdtMt5 over many days, we cannot rule out the possibility that LdtMt5 was more rapidly inactivated by this class of β-lactams in vivo, particularly in the event that LdtMt5 requires a protein-protein interaction for productive catalysis. To date, studies examining acylation of LdtMt5 by carbapenems, including the data presented here, have been in vitro, and interestingly, LdtMt5 is the only LdtMt2 paralog that is not inactivated by carbapenems. The increased susceptibility of ldtMt5::Tn strains to osmotic shock and crystal violet coupled with the observed modest enhancement in susceptibility to carbapenems and our meropenem-LdtMt5 crystal form suggest that LdtMt5 is worth pursuing as a drug target.
Author Contributions
L. A. B. B. and G. L. conceived the study, and L. A. B. B., G. L., and M. A. B. designed the study. L. A. B. B. and G. L. designed and performed the biochemical and genetic experiments and analyzed the results. L. A. B. B. prepared all proteins and together with A. G. and M. A. B. performed the crystallization experiments and preliminary crystal quality analysis; J. J. collected the crystallographic data at synchrotron facilities. A. G. and M. A. B. carried out the crystallographic and structural analyses. Y. P. and M. A. B. performed the ITC experiments. L. A. B. B. and G. L. prepared samples for mass spectrometry, and E. P. L. and C. A. T. performed and analyzed the mass spectrometry data. L. A. B. B., G. L., and M. A. B. wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
We kindly thank Amit Kaushik for assistance with genetic experiments and Maia Schoonmaker Arnold and Mike Delanoy for assistance with electron microscopy. Research was carried out at X6A beamline funded by the National Institute of General Medical Sciences, National Institute of Health under Agreement GM-0080. The National Synchrotron Light Source, Brookhaven National Laboratory is supported by the United States Department of Energy under Contract DE-AC02-98CH10886.
This work was supported, in whole or in part, by National Institutes of Health Grant DP2OD008459 (to G. L.). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (codes 4Z7A and 4ZFQ) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PG
- peptidoglycan
- ITC
- isothermal titration calorimetry
- TCEP
- tris(2-carboxyethyl)phosphine
- BIg
- bacterial Ig-like
- CTSD
- C-terminal subdomain
- CD
- catalytic domain
- ex-CTSD
- extension of the C-terminal subdomain
- TEV
- tobacco etch virus
- r.m.s.
- root mean square
- r.m.s.d.
- root mean square deviation
- Tn
- transposon
- EYY
- ErfK/YbiS/YhnG.
References
- 1. Walsh C. (2003) Antibiotics: Actions, Origins, Resistance, ASM Press, Washington, D. C. [Google Scholar]
- 2. Cho H., Uehara T., Bernhardt T. (2014) β-Lactam antibiotics induce a lethal malfunctioning of the bacterial cell wall synthesis machinery. Cell 159, 1300–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Meroueh S. O., Bencze K. Z., Hesek D., Lee M., Fisher J. F., Stemmler T. L., Mobashery S. (2006) Three-dimensional structure of the bacterial cell wall peptidoglycan. Proc. Natl. Acad. Sci. U.S.A. 103, 4404–4409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lamichhane G., Freundlich J. S., Ekins S., Wickramaratne N., Nolan S. T., Bishai W. R. (2011) Essential metabolites of Mycobacterium tuberculosis and their mimics. mBio 2, e00301–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wietzerbin J., Das B. C., Petit J. F., Lederer E., Leyh-Bouille M., Ghuysen J. M. (1974) Occurrence of d-alanyl-(d)-meso-diaminopimelic acid and meso-diaminopimelyl-meso-diaminopimelic acid interpeptide linkages in the peptidoglycan of mycobacteria. Biochemistry 13, 3471–3476 [DOI] [PubMed] [Google Scholar]
- 6. Lavollay M., Arthur M., Fourgeaud M., Dubost L., Marie A., Veziris N., Blanot D., Gutmann L., Mainardi J. (2008) The peptidoglycan of stationary-phase Mycobacterium tuberculosis predominantly contains cross-links generated by l,d-transpeptidation. J. Bacteriol. 190, 4360–4366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lavollay M., Fourgeaud M., Herrmann J. L., Dubost L., Marie A., Gutmann L., Arthur M., Mainardi J. L. (2011) The peptidoglycan of Mycobacterium abscessus is predominantly cross-linked by l,d-transpeptidases. J. Bacteriol. 193, 778–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gupta R., Lavollay M., Mainardi J. L., Arthur M., Bishai W. R., Lamichhane G. (2010) The Mycobacterium tuberculosis protein LdtMt2 is a nonclassical transpeptidase required for virulence and resistance to amoxicillin. Nat. Med. 16, 466–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ghuysen J. M. (1991) Serine β-lactamases and penicillin-binding proteins. Annu. Rev. Microbiol. 45, 37–67 [DOI] [PubMed] [Google Scholar]
- 10. Erdemli S. B., Gupta R., Bishai W. R., Lamichhane G., Amzel L. M., Bianchet M. A. (2012) Targeting the cell wall of Mycobacterium tuberculosis: structure and mechanism of l,d-transpeptidase 2. Structure 20, 2103–2115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mainardi J. L., Fourgeaud M., Hugonnet J. E., Dubost L., Brouard J. P., Ouazzani J., Rice L. B., Gutmann L., Arthur M. (2005) A novel peptidoglycan cross-linking enzyme for a β-lactam-resistant transpeptidation pathway. J. Biol. Chem. 280, 38146–38152 [DOI] [PubMed] [Google Scholar]
- 12. Magnet S., Dubost L., Marie A., Arthur M., Gutmann L. (2008) Identification of the l,d-transpeptidases for peptidoglycan cross-linking in Escherichia coli. J. Bacteriol. 190, 4782–4785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sanders A. N., Pavelka M. S. (2013) Phenotypic analysis of Escherichia coli mutants lacking l,d-transpeptidases. Microbiology 159, 1842–1852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schoonmaker M. K., Bishai W. R., Lamichhane G. (2014) Nonclassical transpeptidases of Mycobacterium tuberculosis alter cell size, morphology, the cytosolic matrix, protein localization, virulence, and resistance to β-lactams. J. Bacteriol. 196, 1394–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Edelheit O., Hanukoglu A., Hanukoglu I. (2009) Simple and efficient site-directed mutagenesis using two single-primer reactions in parallel to generate mutants for protein structure-function studies. BMC Biotechnol. 9, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 20. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 21. Valway S. E., Sanchez M. P., Shinnick T. F., Orme I., Agerton T., Hoy D., Jones J. S., Westmoreland H., Onorato I. M. (1998) An outbreak involving extensive transmission of a virulent strain of Mycobacterium tuberculosis. N. Engl. J. Med. 338, 633–639 [DOI] [PubMed] [Google Scholar]
- 22. Lamichhane G., Zignol M., Blades N. J., Geiman D. E., Dougherty A., Grosset J., Broman K. W., Bishai W. R. (2003) A postgenomic method for predicting essential genes at subsaturation levels of mutagenesis: application to Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 100, 7213–7218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cynamon M. H., Speirs R. J., Welch J. T. (1998) In vitro antimycobacterial activity of 5-chloropyrazinamide. Antimicrob. Agents. Chemother. 42, 462–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim H. S., Kim J., Im H. N., Yoon J. Y., An D. R., Yoon H. J., Kim J. Y., Min H. K., Kim S. J., Lee J. Y., Han B. W., Suh S. W. (2013) Structural basis for the inhibition of Mycobacterium tuberculosis l,d-transpeptidase by meropenem, a drug effective against extensively drug-resistant strains. Acta Crystallogr. D Biol. Crystallogr. 69, 420–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li W. J., Li D. F., Hu Y. L., Zhang X. E., Bi L. J., Wang D. C. (2013) Crystal structure of l,d-transpeptidase LdtMt2 in complex with meropenem reveals the mechanism of carbapenem against Mycobacterium tuberculosis. Cell Res. 23, 728–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Böth D., Steiner E. M., Stadler D., Lindqvist Y., Schnell R., Schneider G. (2013) Structure of LdtMt2, an l,d-transpeptidase from Mycobacterium tuberculosis. Acta Crystallogr. D Biol. Crystallogr. 69, 432–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bielnicki J., Devedjiev Y., Derewenda U., Dauter Z., Joachimiak A., Derewenda Z. S. (2006) B. subtilis ykuD protein at 2.0 Å resolution: insights into the structure and function of a novel, ubiquitous family of bacterial enzymes. Proteins 62, 144–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Biarrotte-Sorin S., Hugonnet J. E., Delfosse V., Mainardi J. L., Gutmann L., Arthur M., Mayer C. (2006) Crystal structure of a novel β-lactam-insensitive peptidoglycan transpeptidase. J. Mol. Biol. 359, 533–538 [DOI] [PubMed] [Google Scholar]
- 29. Correale S., Ruggiero A., Capparelli R., Pedone E., Berisio R. (2013) Structures of free and inhibited forms of the l,d-transpeptidase LdtMt1 from Mycobacterium tuberculosis. Acta Crystallogr. D Biol. Crystallogr. 69, 1697–1706 [DOI] [PubMed] [Google Scholar]
- 30. Cordillot M., Dubée V., Triboulet S., Dubost L., Marie A., Hugonnet J. E., Arthur M., Mainardi J. L. (2013) In vitro cross-linking of Mycobacterium tuberculosis peptidoglycan by l,d-transpeptidases and inactivation of these enzymes by carbapenems. Antimicrob. Agents Chemother. 57, 5940–5945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kumar P., Arora K., Lloyd J. R., Lee I. Y., Nair V., Fischer E., Boshoff H. I., Barry C. E. 3rd (2012) Meropenem inhibits d,d-carboxypeptidase activity in Mycobacterium tuberculosis. Mol. Microbiol. 86, 367–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cole S. T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S. V., Eiglmeier K., Gas S., Barry C. E. 3rd, Tekaia F., Badcock K., Basham D., Brown D., Chillingworth T., Connor R., Davies R., Devlin K., Feltwell T., Gentles S., Hamlin N., Holroyd S., Hornsby T., Jagels K., Krogh A., McLean J., Moule S., Murphy L., Oliver K., Osborne J., Quail M. A., Rajandream M. A., Rogers J., Rutter S., Seeger K., Skelton J., Squares R., Squares S., Sulston J. E., Taylor K., Whitehead S., Barrell B. G. (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544 [DOI] [PubMed] [Google Scholar]
- 33. Mainardi J. L., Morel V., Fourgeaud M., Cremniter J., Blanot D., Legrand R., Frehel C., Arthur M., Van Heijenoort J., Gutmann L. (2002) Balance between two transpeptidation mechanisms determines the expression of β-lactam resistance in Enterococcus faecium. J. Biol. Chem. 277, 35801–35807 [DOI] [PubMed] [Google Scholar]
- 34. Pisabarro A. G., de Pedro M. A., Vázquez D. (1985) Structural modifications in the peptidoglycan of Escherichia coli associated with changes in the state of growth of the culture. J. Bacteriol. 161, 238–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Peltier J., Courtin P., El Meouche I., Lemée L., Chapot-Chartier M. P., Pons J. L. (2011) Clostridium difficile has an original peptidoglycan structure with a high level of N-acetylglucosamine deacetylation and mainly 3-3 cross-links. J. Biol. Chem. 286, 29053–29062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hugonnet J. E., Haddache N., Veckerlé C., Dubost L., Marie A., Shikura N., Mainardi J. L., Rice L. B., Arthur M. (2014) Peptidoglycan cross-linking in glycopeptide-resistant Actinomycetales. Antimicrob. Agents Chemother. 58, 1749–1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Magnet S., Arbeloa A., Mainardi J. L., Hugonnet J. E., Fourgeaud M., Dubost L., Marie A., Delfosse V., Mayer C., Rice L. B., Arthur M. (2007) Specificity of l,d-transpeptidases from Gram-positive bacteria producing different peptidoglycan chemotypes. J. Biol. Chem. 282, 13151–13159 [DOI] [PubMed] [Google Scholar]
- 38. Cava F., de Pedro M. A., Lam H., Davis B. M., Waldor M. K. (2011) Distinct pathways for modification of the bacterial cell wall by non-canonical d-amino acids. EMBO J. 30, 3442–3453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Corey D. R., Craik C. S. (1992) An investigation into the minimum requirements for peptide hydrolysis by mutation of the catalytic triad of trypsin. J. Am. Chem. Soc. 114, 1784–1790 [Google Scholar]
- 40. Magnet S., Bellais S., Dubost L., Fourgeaud M., Mainardi J. L., Petit-Frère S., Marie A., Mengin-Lecreulx D., Arthur M., Gutmann L. (2007) Identification of the l,d-transpeptidases responsible for attachment of the Braun lipoprotein to Escherichia coli peptidoglycan. J. Bacteriol. 189, 3927–3931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Patru M. M., Pavelka M. S. Jr. (2010) A role for the class A penicillin-binding protein PonA2 in the survival of Mycobacterium smegmatis under conditions of nonreplication. J. Bacteriol. 192, 3043–3054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sanders A. N., Wright L. F., Pavelka M. S. (2014) Genetic characterization of mycobacterial l,d-transpeptidases. Microbiology 160, 1795–1806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schillinger C., Boisguerin P., Krause G. (2009) Domain interaction footprint: a multi-classification approach to predict domain-peptide interactions. Bioinformatics 25, 1632–1639 [DOI] [PubMed] [Google Scholar]
- 44. Depuydt M., Leonard S. E., Vertommen D., Denoncin K., Morsomme P., Wahni K., Messens J., Carroll K. S., Collet J. (2009) A periplasmic reducing system protects single cysteine residues from oxidation. Science 326, 1109–1111 [DOI] [PubMed] [Google Scholar]
- 45. Flores A. R., Parsons L. M., Pavelka M. S. (2005) Genetic analysis of the β-lactamases of Mycobacterium tuberculosis and Mycobacterium smegmatis and susceptibility to β-lactam antibiotics. Microbiology 151, 521–532 [DOI] [PubMed] [Google Scholar]
- 46. Hugonnet J. E., Blanchard J. S. (2007) Irreversible inhibition of the Mycobacterium tuberculosis β-lactamase by clavulanate. Biochemistry 46, 11998–12004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hugonnet J. E., Tremblay L. W., Boshoff H. I., Barry C. E. 3rd, Blanchard J. S. (2009) Meropenem-clavulanate is effective against extensively drug-resistant Mycobacterium tuberculosis. Science 323, 1215–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gouet P., Courcelle E., Stuart D. I., Métoz F. (1999) ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15, 305–308 [DOI] [PubMed] [Google Scholar]