

Background: FIT2 is an ER protein implicated in regulating cytosolic LD formation in mammals.
Results: FIT2 deficiency in an inducible whole body knock-out mouse model leads to lethal enteropathy.
Conclusion: FIT2 is required for normal intestinal homeostasis and survival.
Significance: This study provides evidence that FIT2 is essential for murine intestinal health.
Keywords: cell death, intestinal epithelium, lipid droplet, lipoprotein, stem cells
Abstract
Lipid droplets (LDs) are phylogenetically conserved cytoplasmic organelles that store neutral lipids within a phospholipid monolayer. LDs compartmentalize lipids and may help to prevent cellular damage caused by their excess or bioactive forms. FIT2 is a ubiquitously expressed transmembrane endoplasmic reticulum (ER) membrane protein that has previously been implicated in LD formation in mammalian cells and tissue. Recent data indicate that FIT2 plays an essential role in fat storage in an in vivo constitutive adipose FIT2 knock-out mouse model, but the physiological effects of postnatal whole body FIT2 depletion have never been studied. Here, we show that tamoxifen-induced FIT2 deletion using a whole body ROSA26CreERT2-driven FIT2 knock-out (iF2KO) mouse model leads to lethal intestinal pathology, including villus blunting and death of intestinal crypts, and loss of lipid absorption. iF2KO mice lose weight and die within 2 weeks after the first tamoxifen dose. At the cellular level, LDs failed to form in iF2KO enterocytes after acute oil challenge and instead accumulated within the ER. Intestinal bile acid transporters were transcriptionally dysregulated in iF2KO mice, leading to the buildup of bile acids within enterocytes. These data support the conclusion that FIT2 plays an essential role in regulating intestinal health and survival postnatally.
Introduction
Cytosolic lipid droplets (LD)2 are organelles made up of a hydrophobic core of neutral lipids such as triglycerides (TG) and cholesteryl esters surrounded by a phospholipid monolayer and a unique proteome (1). The common view on LD formation is that neutral lipids accumulate between the leaflets of the ER through the action of de novo synthesizing enzymes for TG, phospholipid, and cholesteryl esters leading to the expansion of the nascent LD into the cytoplasm. Indeed, two pools of LDs have been identified, one that has its phospholipid leaflet in continuity with the ER membrane and another that is truly cytosolic and disconnected with the ER (2). The mechanisms that give rise to these two pools of ER-derived LDs are not entirely understood.
Fat storage-inducing transmembrane protein 2 (FIT2/FITM2) is an evolutionarily conserved and ubiquitously expressed ER membrane protein that has been implicated in regulating cytosolic LD formation in mammals and phospholipid metabolism in the yeast Saccharomyces cerevisiae (3, 4). FIT2 is part of a two-gene family with 35% identity with FIT1. FIT1 is conserved from fish to humans and is not expressed in adipose tissue or adipocytes but is expressed primarily in skeletal and cardiac muscle. FIT2 is most highly expressed in adipose tissue and directly regulated by peroxisome proliferator-activated receptor γ, the master transcription factor for adipocyte differentiation (5–7). FIT2 is a 262-amino acid protein in mammals having six transmembrane domains with both N and C termini facing the cytoplasm. Overexpression of FIT2 in cells consistently resulted in the accumulation of TG-rich LDs (3, 8) with a gain-of-function mutation in transmembrane domain 4 found to significantly increase LD size and number (9). Purified FIT2 in detergent micelles binds directly TG and diacylglyceride, and this binding is important for FIT2-mediated LD formation (8, 10). More recently, adipose tissue deficiency of FIT2 in mice was shown to lead to a progressive lipodystrophy associated with adipose tissue necrosis. At a cellular level, FIT2 deficiency in mouse primary adipocytes resulted in a significant reduction in LD number but an increase in LD size (11). Therefore, FIT2 plays a subtle but poorly understood role in LD biology, but it is essential for adipose tissue TG storage and survival.
Given that FIT2 is ubiquitously expressed, we set out to study the function of FIT2 in whole body metabolism by generating a tamoxifen-inducible FIT2 whole body knock-out (iF2KO) mouse model. To our surprise, the induction of FIT2 deletion in 3-week-old iF2KO mice led to a decrease in body weight and severe intestinal injury and malabsorption, leading to death. In view of these unexpected results, we sought to determine the cause of this intestinal phenotype in iF2KO mice.
Here, we show unexpectedly that postnatal FIT2 deletion leads to destruction of the villus and crypt architecture. These changes were accompanied by a disruption of de novo cytosolic LD formation in the small intestine upon acute fat challenge, leading to a shift in the distribution of neutral lipid into the ER, but without changes in chylomicron synthesis upon acute fat challenge. These data indicate that cytosolic LDs are not required for chylomicron assembly and secretion but that FIT2 is essential for maintaining neutral lipid and BA homeostasis within enterocytes, which is in turn important for small intestinal health and function.
Experimental Procedures
Mice
Mice carrying floxed FIT2 (L/L) alleles that were previously generated (11) were crossed to ROSA26CreERT2 mice (12) to derive ROSA26CreERT2 L/L progeny. To generate iF2KO mice, we injected 3-week-old post-weaned ROSA26CreERT2 L/L mice with 100 μg/g tamoxifen (Sigma, T5648) per day for 3 consecutive days. L/L littermates were also injected with tamoxifen and used as controls unless otherwise stated. Villin-cre and VillinCreERT2 strains (13) were crossed to L/L mice to derive Villin-cre L/L and VillinCreERT2 L/L mice. VillinCreERT2 L/L mice were subjected to the same injection protocol as ROSA26CreERT2 L/L mice. Mice were maintained on a standard chow diet composed of 4.8% fat (Specialty Feeds, catalog no. 8310) for all studies. For studies requiring acute fat challenge, mice were gavaged with 200 μl of commercially available olive oil. For lipid absorption studies, mice were injected intraperitoneally with 1 g/kg of Pluronic F-127 (Sigma) lipolysis inhibitor before gavaging with 0.2 μCi of [14C]oleic acid, 0.2 μCi of [3H]triolein, or 0.2 μCi of [3H]palmitate dissolved in 200 μl of 12.7 mm BSA-conjugated oleate per mouse. Serum was collected via the retro-orbital vein. For EdU staining, mice were injected intraperitoneally with 160 μg/g body weight of EdU (Molecular probes, Invitrogen) dissolved in PBS. The mice were sacrificed, and the small intestine was harvested 2 and 24 h later. Study protocols were approved by the Institutional Animal Care and Use Committee of Singhealth.
Tissue Processing and Histology
For paraffin sections, tissues were fixed overnight in 10% neutral buffered formalin (Sigma, HT501128) and stored in 70% ethanol for at least 24 h. Tissues were processed using a Leica TP-1020 tissue processor and a Leica tissue embedding station EG-1160 following standard protocols. Paraffin-embedded sections were stained with H&E (hematoxylin from Vector Laboratories, H-3401; eosin from Sigma, HT110380) using standard protocols and visualized using a Leica DM 2000 microscope with the Leica application suite version 3.5.0. For Plin2 and Plin3 staining, slideswere subject to deparaffinization and heat-mediated antigen retrieval with sodium citrate buffer, pH 6.0, and then incubated with either Plin2 primary antibody, 1:100 dilution (Novus Biologicals, NB110-40878), or Plin3 primary antibody, 1:300 dilution (Abcam, AB118605). Secondary antibody incubation was carried out at 1:250 dilution with Alexa Fluor 594 (Life Technologies, Inc., A-21207) or Alexa Fluor 488 (Life Technologies, Inc., A-11055). Proteins were visualized using a Carl Zeiss LSM 710 confocal microscope. Anti-lysozyme (Abcam, catalog no. ab108508) staining was performed on paraffin sections using primary dilution of 1:200 in 3% BSA/TBST overnight. Endogenous peroxidase activity was blocked in 3% H2O2 in TBST for 30 min at room temperature. Secondary antibody incubation was performed with goat anti-rabbit IgG (H + L)-HRP conjugate (Bio-Rad catalog no. 170-6515) at 1:200 dilution for 1 h. 3,3′-Diaminobenzidine chromogen and substrate buffer (Dako catalog no. 34680) was used to visualize the signal, and the slides were counterstained with hematoxylin. For EdU staining, the small intestine was harvested and embedded in paraffin. The Click-iT® Plus EdU imaging kit (Molecular Probes, Invitrogen) was used according to the manufacturer's protocol. For TUNEL staining, the ApopTag Plus fluorescein in situ apoptosis detection kit (Chemicon, S7111) was used on paraffin sections according to the manufacturer's instructions. Alcian blue-PAS (Merck, catalog no. 1.01646.0001) staining was performed on paraffin sections according to the manufacturer's instructions. Anti-calnexin and BODIPY staining were performed on frozen sections. Tissues were fixed overnight in 10% neutral buffered formalin, stored in 15% sucrose solution for 8 h, and then transferred to 30% sucrose solution overnight. The samples were embedded in OCT (Sakura, MSK-4583-1) and sectioned to 10 μm thickness. Slides were subject to antigen retrieval before BODIPY (Life Technologies, Inc., D3922) and primary antibody staining. Anti-calnexin (Abcam, AB22595) primary antibody was used at a 1:100 dilution followed by Alexa Fluor 555 (Life Technologies, Inc., A-21428) secondary antibody at 1:250 dilution.
Electron Microscopy
Duodenum samples were fixed in 2.5% glutaraldehyde in phosphate-buffered saline at 4 °C for 1 h, before osmication with 1% osmium tetroxide, pH 7.4, for 1 h. The samples were dehydrated through an ascending series of ethanol at room temperature, followed by infiltration with acetone and resin. They were embedded in resin and allowed to polymerize at 60 °C for 24 h. Samples were cut using an ultramicrotome, mounted on Formvar-coated copper grids, and stained with uranyl acetate and lead citrate. Grids were viewed using a JEOL 1010 transmission electron microscope.
Enterocyte Isolation and Culture
Enterocytes were extracted from murine small intestine as described previously (14). Briefly, the intestines were flushed with cold DPBS and immersed in DPBS with 30 μm EDTA and 1.5 mm DTT (Sigma) on ice for 20 min. The intestines were then transferred to DPBS with 30 μm EDTA at 37 °C for 10 min, after which the epithelial cells were released from the basement membrane via vigorous shaking. Epithelial cells were then resuspended in 10% FBS/DMEM with 10 μm Y27632 ROCK inhibitor (Sigma) to prevent anoikis.
Crypt Isolation and Culture
Crypt isolation and culture were performed as described previously (15). Briefly, murine small intestine was opened longitudinally, flushed thoroughly with cold PBS, and macerated. Fragments were incubated in PBS containing 2 mm EDTA with gentle shaking followed by repetitive pipetting and gravity sedimentation. Supernatant fractions were collected and washed with PBS. After repeating three times, the third fraction was enriched in crypts as judged by microscopy. Crypt culture was performed under growth conditions as described previously (16).
Lipid Extraction and Analysis
Mice were fasted for 4 h prior to gavage and sacrifice. Enterocytes were isolated as described above. Isolated enterocytes were homogenized in PBS for determination of protein concentration via BCA protein assay (Thermo-Scientific). Cell homogenates were extracted for lipid using chloroform/methanol (2:1, v/v). Extracted lipids were dried down and resuspended in 1% Triton X-100 for analysis. The triglyceride LiquiColor® test (Stanbio Laboratories, 2200-225) was used to determine total triglyceride content. For measurement of total bile acid content, 15 cm of distal small intestine was homogenized, and the lipids were extracted as above. Extracted lipids were dried down and resuspended as above. Total bile acid content was measured using the mouse total bile acids kit (Crystal Chem Inc., catalog no. 80470).
Measurement of Plasma Analytes
Serum TG levels were determined using the triglyceride LiquiColor® test (Stanbio Laboratories, 2200-225). Serum glucose was measured with a standard glucometer and test strips from OneTouch® Ultra® (LifeScan, 53885). For radioactive transport studies, 10 μl of serum was dissolved in 4 ml of scintillation fluid. 3H and 14C activity were measured by scintillation counting.
Western Blot and Gene Expression Analysis
Tissues were homogenized in RIPA buffer plus EDTA-free protease inhibitor mixture (Roche Applied Science) and phosphatase inhibitor mixtures 2 and 3 (Sigma, P5726 and P0044). Protein concentration was determined by a BCA protein assay (Thermo-Scientific), and 80 μg of protein was used per sample. Samples were separated by SDS-PAGE on 8 or 15% polyacrylamide gels, transferred to nitrocellulose membranes (Bio-Rad), and incubated with the indicated antibodies: Fit2 (3), Plin2 (Novus Biologicals, NB110-40878), Mtp (Santa Cruz Biotechnology SC33116), apoB (Chemicon), and Gapdh (Santa Cruz Biotechnology SC32233). IRDye® 680 and 800 secondary antibodies were obtained from LI-COR. Quantification of signals was performed using the Odyssey infrared scanner (LI-COR).
RNA was purified and extracted from whole intestine and liver samples using the Qiagen RNeasy mini kit (catalog no. 74106). cDNA was synthesized from RNA using the iScript Select cDNA synthesis kit (Bio-Rad, 170-8897). Gene expression was assessed by quantitative real time PCR using SYBR Green PCR master mix (Applied Biosystems catalog no. 4364346) and an Applied Biosystems HT7900 real time PCR system. Primer sequences are available upon request. Target transcripts were normalized to GAPDH.
Microarray Analysis
Affymetrix GeneChip® Mouse Genome 430 2.0 Array was used. Data are accessible at NCBI GEO database, accession GSE70465. Small intestine was harvested from L/L and ROSA26CreERT2 L/L mice at days 2, 3, and 5 post-tamoxifen treatment and subjected to microarray analysis. Pooled intestinal RNA from n biological replicates were used for each genotype and time point as follows: day 2 L/L n = 6 and day 2 KO n = 7; day 3 L/L n = 6 and day 3 KO n = 6; and day 5 L/L n = 8 and day 5 KO n = 7.
Statistical Analysis
All quantitative data are represented as means ± S.E. p values were generated by Student's t test.
Results
Postnatal Deletion of FIT2 Leads to Progressive Weight Loss and Death
A purported function of FIT2 is to regulate the formation of LDs in various cell and tissue types (3, 17).We previously deleted FIT2 in an adipose tissue knock-out mouse model, leading to defects in LD storage of fat in white adipose tissue (WAT), infiltration of WAT with inflammatory macrophages, death of adipocytes, and metabolic dysfunction typical of lipodystrophies, which was significantly exacerbated by high fat feeding (11). As FIT2 is a ubiquitously expressed protein, we sought to understand its function in whole body metabolism by inducing whole body postnatal FIT2 deletion (iF2KO). To carry this out, mice carrying floxed FIT2 (L/L) alleles that were previously generated (11) were crossed to ROSA26CreERT2 mice to derive ROSA26CreERT2 L/L progeny. To generate iF2KO mice, we injected 3-week-old post-weaned ROSA26CreERT2 L/L mice with 100 μg/g tamoxifen per day for 3 consecutive days. L/L littermates were also injected with tamoxifen and used as controls. Quantification of FIT2 protein at day 7 post-tamoxifen injection confirmed that iF2KO mice had a reduction in FIT2 levels in heart, quadriceps, subcutaneous WAT, liver, and enterocytes (Fig. 1A). There was a significant loss of weight in iF2KO mice at day 8 post-tamoxifen treatment, compared with the tamoxifen-injected controls, including tamoxifen-injected ROSA26CreERT2 L/+ and ROSA26CreERT2 +/+ mice (Fig. 1B). Unexpectedly, iF2KO mice died between days 9 and 13 post-injection (Fig. 1C). Upon post-mortem examination, iF2KO mice were found to have bloated gastrointestinal tracts (Fig. 1D), liquid cecal contents (Fig. 1E), and decreased intestinal length (Fig. 1F). We then performed H&E staining to investigate the effects of FIT2 deletion on intestinal morphology. At day 6 post-tamoxifen treatment, obliteration of the intestinal crypts was observed in iF2KO mice, with sparing of the Paneth cells (Fig. 1G). By day 8 post-tamoxifen treatment, widespread villus shortening and blunting were observed in iF2KO intestine (Fig. 1G). At day 9 post-tamoxifen treatment, intestinal absorption of [3H]palmitate was significantly decreased in iF2KO mice (Fig. 1H). In addition, at day 10 post-tamoxifen treatment, fasted serum glucose was significantly lower in iF2KO mice (Fig. 1I). In summary, these data support the conclusion that the death of iF2KO mice was caused by lethal enteropathy and severe malabsorption.
FIGURE 1.

Postnatal whole body deletion of FIT2 leads to decrease in body weight and lethal enteropathy. A, Western blot analysis of FIT2 on the indicated tissues showing FIT2 depletion in the heart, quadriceps (Quad), subcutaneous WAT (SWAT), liver, and enterocytes (Ent) of iF2KO mice. B, progressive body weights of tamoxifen-injected iF2KO (labeled as Cre L/L) and control mice from day 1 to day 14 post-tamoxifen treatment. C, iF2KO (labeled as Cre L/L) mice die between days 9 and 13 post-tamoxifen treatment. D, iF2KO gastrointestinal tract was bloated and filled with liquid contents at day 8 post-tamoxifen treatment. E, iF2KO cecal contents were liquid compared with L/L controls at day 8 post-tamoxifen treatment. F, length of iF2KO intestine at day 8 post-tamoxifen treatment. L/L, n = 6; iF2KO, n = 6. G, H&E staining of iF2KO small intestine at day 6 and day 8 post-tamoxifen treatment. iF2KO intestinal crypts were obliterated with Paneth cell sparing (black arrowheads) at day 6 post-tamoxifen treatment, and there was severe villus shortening and blunting in iF2KO mice by day 8 post-tamoxifen treatment. Magnification, ×40. Scale bar, 100 μm. H, iF2KO and L/L mice at day 9 post-tamoxifen injection were gavaged with [3H]palmitate to measure intestinal lipid absorption. A significant decrease in [3H]palmitate absorption was observed in iF2KO mice at 1 and 2 h post-gavage. I, fasted serum glucose was significantly reduced at day 10 post-tamoxifen injection in iF2KO mice. L/L, n = 4; iF2KO, n = 4. Data are represented as mean with error bars indicating S.E. Each point represents an independent biological replicate. **, p < 0.01; ***, p < 0.001; ****, p < 0.0001, two-tailed t test.
FIT2 Deletion Inhibits Intestinal Stem Cell Renewal Without Suppressing Wnt and Notch Signaling
The intestinal crypt is the stem cell niche in the gut that produces the entire epithelial lineage and is essential for the continued renewal of the absorptive epithelium lining the villi (18). To uncover the cause of the severe enteropathy observed in iF2KO mice, we directly tested whether stem cell proliferation in intestinal crypts was affected in iF2KO mice. To test for stem cell proliferation in vivo, iF2KO and L/L control mice were intraperitoneally injected at day 5 post-tamoxifen treatment with EdU to label proliferating cells, and intestines were collected 2 and 24 h after EdU administration. As expected, L/L control crypts exhibited strong EdU incorporation in crypt cell nuclei at 2 h post-labeling, followed by the movement of EdU-positive cells out of the crypts and up the villi surface at 24 h post-labeling (Fig. 2A). In contrast, iF2KO crypts had decreased EdU labeling relative to L/L controls, and EdU-positive cells failed to leave the crypt (Fig. 2A). These findings indicate that FIT2 deletion leads to defects in intestinal stem cell renewal. Consistent with decreased stem cell proliferation and villus epithelial cell renewal, iF2KO crypts were strongly TUNEL-positive at day 3 post-tamoxifen treatment (Fig. 2B), indicating that FIT2 deletion leads to intestinal crypt cell death.
FIGURE 2.
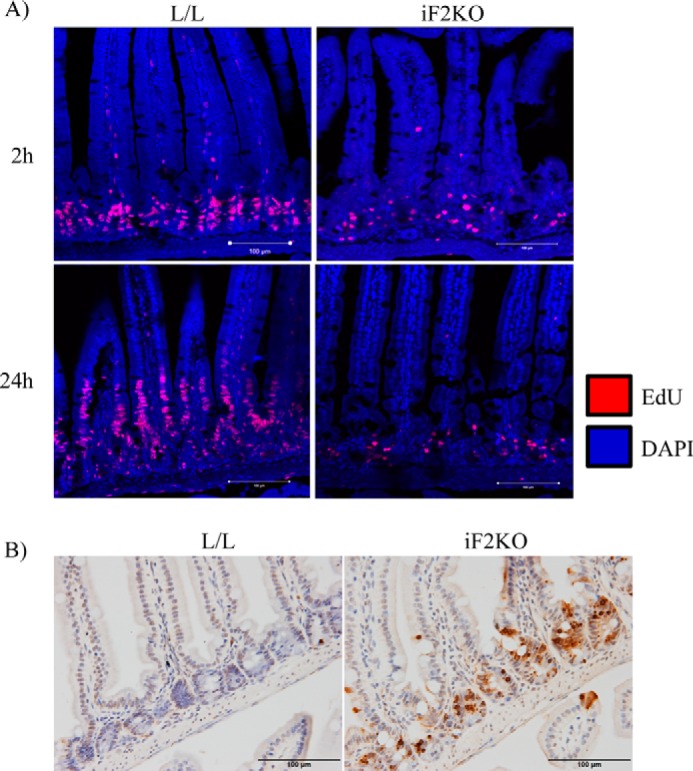
Deletion of FIT2 leads to inhibition of intestinal stem cell renewal and crypt cell death. A, EdU staining of proliferating cells in L/L and iF2KO intestine 2 and 24 h after intraperitoneal injection of 160 μg/g EdU. Mice were injected with EdU at day 5 post-tamoxifen treatment. Magnification, ×40. Scale bar, 100 μm. B, TUNEL staining of L/L and iF2KO small intestine at day 3 post-tamoxifen treatment. Magnification, ×40. Scale bar, 100 μm.
Because murine intestinal stem cell renewal involves the pro-survival Wnt and Notch signaling pathways, we performed RT-PCR on various Wnt and Notch ligands, receptors, and target genes on iF2KO whole intestine at day 5 post-tamoxifen treatment. Contrary to our expectation that Wnt and Notch signaling would be decreased in iF2KO intestines, we saw a significant increase in Wnts 2b, 3, and 4 and a small but insignificant increase in Wnts 2, 5a, 6, and 9a in iF2KO intestine (Fig. 3A). Furthermore, Wnt target genes such as Axin2 and Lgr5 were not significantly changed, whereas c-myc was significantly up-regulated (Fig. 3B). Notch signaling is also essential for maintenance of the intestinal stem cell compartment, and a decrease in Notch signaling inhibits crypt cell renewal and promotes differentiation into Paneth, goblet, and enteroendocrine secretory cell lineages (19–24). Unexpectedly, Notch signaling ligands Dll1 and Dll4 (Fig. 3C) and Notch receptors 1 and 2 (Fig. 3D) were significantly increased in iF2KO intestine. Positively regulated Notch target genes such as Ascl2 and Hes1 were significantly up-regulated (Fig. 3E). The only Notch target gene found to be significantly down-regulated was Olfm4 (Fig. 3E). Down-regulation of Olfm4 could reflect the destruction of a specific population of crypt stem cells that exclusively express this gene (25). There were no significant changes in transcript levels of Atoh1 or p27 and p57 cyclin-dependent kinase inhibitors, all of which are normally repressed by Notch activation. The lack of Atoh1 up-regulation was surprising as there was an increase in anti-lysozyme staining at the iF2KO crypt base (Fig. 4A), suggesting increased Paneth cell production of this enzyme. Microarray analysis indicated an up-regulation of Paneth-specific gene signals in iF2KO small intestine (Fig. 4B). We surmised that these observations were due to the longevity of Paneth cells, which can survive for up to 8 weeks following ablation of crypt stem cells (26). Goblet cells appeared larger (Fig. 4C), but numbers were not significantly different (Fig. 4D), which may indicate a defective secretory pathway instead of increased secretory cell differentiation due to Notch inhibition (27). From these results, we concluded that death of crypt stem cells was not due to defects in Wnt or Notch signaling, but instead it acts via a separate and as yet unknown pathway.
FIGURE 3.

FIT2 deletion inhibits intestinal stem cell renewal without suppressing Wnt and Notch signaling. Quantitative RT-PCR of mRNAs derived from L/L and iF2KO small intestine at day 5 post-tamoxifen treatment. Mice were fasted for 4 h prior to sacrifice. Data are represented as mean with error bars indicating S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001, two-tailed t test.
FIGURE 4.

FIT2 deletion results in increased anti-lysozyme staining, Paneth cell transcripts, and goblet cell size but not number. A, anti-lysozyme staining of L/L and iF2KO small intestine at day 6 post-tamoxifen treatment. Magnification, ×20. Scale bar, 200 μm. B, heat map of Paneth cell transcripts in a microarray performed on small intestine harvested from days 2, 3, and 5 post-tamoxifen-treated mice. Data are calculated from log2 signal intensity values and represented as log2(KO/WT). Pooled intestinal RNA from n biological replicates was used for each genotype and time point. Day 2 L/L, n = 6, and day 2 KO, n = 7; day 3 L/L, n = 6, and day 3 KO, n = 6; day 5 L/L, n = 8, and day 5 KO, n = 7. C, Alcian blue/PAS staining of L/L and iF2KO small intestine to highlight goblet cell phenotypes at day 6 post-tamoxifen treatment. Magnification, ×20. Scale bar, 200 μm. D, quantification of goblet cell number per 100 villi at day 6 post-tamoxifen treatment. Data are represented as mean with error bars indicating S.E. n.s., not significant.
Consistent with increased stem cell death in the intestines of iF2KO mice, microarray profiling on whole intestine from days 2, 3, and 5 post-tamoxifen-injected L/L and ROSA26CreERT2 L/L mice showed that many p53 pathway genes were up-regulated in iF2KO intestine and in particular several pro-apoptotic p53-induced genes, namely Puma (Bbc3), p21 (Cdkn1a), Trp53inp1, and Bax (Fig. 5A).
FIGURE 5.

FIT2 deletion results in increased transcripts in the pro-apoptotic pathway. A heat map of up-regulated transcripts in the p53 pathway from a microarray performed on small intestine harvested from days 2, 3, and 5 post-tamoxifen-treated mice. Data are calculated from log2 signal intensity values and represented as log2(KO/WT). Pooled intestinal RNA from n biological replicates were used for each genotype and time point. Day 2 L/L, n = 6, and day 2 KO, n = 7; day 3 L/L, n = 6, and day 3 KO, n = 6; day 5 L/L, n = 8, and day 5 KO, n = 7.
FIT2 Deletion Results in the Absence of Cytosolic LD Formation in Enterocytes
Because FIT2 has been implicated in LD formation (3, 17), we hypothesized that iF2KO mice would have reduced de novo LD formation in enterocytes during an acute fat challenge, a condition that normally involves massive de novo LD formation (28, 29). To test this idea, we gavaged fasted mice with a bolus of olive oil before histological analysis. Mice at day 5 post-tamoxifen treatment were used as neither villus blunting nor weight loss was observed at this time point (Figs. 1B and 2A). As expected, H&E staining of L/L small intestine showed well defined spherical cytosolic LDs near the apical membrane of enterocytes at 1 h after oil gavage (Fig. 6A). In contrast, there were no visible LDs in the enterocytes of iF2KO mice post-oil gavage. Instead, large spaces were visible near the basolateral membrane of iF2KO enterocytes, and the enterocytes exhibited centrally localized nuclei (Fig. 6A). To further characterize the LD defect seen in gavaged iF2KO enterocytes, we performed immunofluorescence staining for the LD-associated proteins Plin2 and Plin3. Plin2 is primarily found on the surface of LDs, whereas Plin3 is stable in the cytoplasm and targets to the LD surface upon fatty acid loading (30). It has been shown by coherent anti-Stokes Raman scattering microscopy that Plin3 coats LDs in enterocytes after an acute fat challenge, whereas Plin2 coats LDs in enterocytes of mice fed a chronic high fat diet (29). Consistent with the lack of cytosolic LDs in the H&E-stained iF2KO villi (Fig. 6A), iF2KO intestinal epithelium did not form either Plin2- or Plin3-coated LDs 2 h after oil gavage (Fig. 6B). In line with the visual absence of LDs in iF2KO mice, Western blot analysis of enterocytes from oil-gavaged mice showed a decrease in Plin2, ApoB48, and Mtp protein levels in iF2KO compared with L/L mice, which exhibited the expected increase in Plin2 protein levels following oil gavage (Fig. 6C). Despite the absence of LDs in the iF2KO small intestine, we found that there was no significant difference in TG levels between control and iF2KO enterocytes post-oil gavage (Fig. 6D). Given that LDs were absent in iF2KO enterocytes but total TG remained unchanged, we sought to determine the subcellular localization of the TG pool in iF2KO mice post-oil gavage. To this end, we performed immunofluorescence co-staining of murine small intestine from oil-gavaged mice using anti-calnexin antibody, an ER marker, and BODIPY 493/503, a neutral lipid dye for staining LDs. We hypothesized that the neutral lipid must be accumulating in the ER lumen, the site of chylomicron assembly, because cytosolic LDs were absent in iF2KO enterocytes (Fig. 6, A and B). We observed that anti-calnexin antibody and BODIPY stained separate and distinct regions in control L/L enterocytes, whereas both signals clearly co-localized in iF2KO enterocytes (Fig. 6E). These findings indicate a luminal ER localization of TG in iF2KO intestines. Ultrastructural analysis of the enterocytes using electron microscopy confirmed that LDs accumulated within double membrane tubular ER membrane structures in the iF2KO intestine, but they appeared to be largely sequestered normally within spherical cytosolic LDs in the control (Fig. 6F). The ER luminal LDs observed in iF2KO intestinal epithelium is consistent with their identity as chylomicrons (31, 32). Taken together, these data indicate that whole body FIT2 deletion inhibits the formation of LDs in enterocytes and leads to the accumulation of TG within the ER lumen during acute fat challenge.
FIGURE 6.

Changes in enterocyte lipid subcellular localization after acute oil challenge in iF2KO mice. A, H&E staining of L/L and iF2KO small intestine at day 5 post-tamoxifen treatment, 2 h post-oil gavage. Magnification, ×63. Scale bar, 100 μm. B, immunofluorescent staining of Plin2 and Plin3 in L/L and iF2KO small intestine at day 5 post-tamoxifen treatment. Magnification, ×63. Scale bar, 10 μm. C, Western blot analysis of Plin2, ApoB48, and Mtp in enterocytes of gavaged versus non-oil gavaged L/L and iF2KO mice at day 5 post-tamoxifen treatment. D, measurement of total TG content in enterocytes harvested from day 5 post-tamoxifen-treated mice, at 2 h post-oil gavage. n.s., not significant. E, BODIPY and anti-calnexin co-staining of small intestine harvested from day 5 post-tamoxifen-injected mice at 2 h post-oil gavage. Magnification, ×63. Scale bar, 20 μm. F, electron micrographs of L/L and iF2KO small intestine at day 5 post-tamoxifen injection, 2 h post-oil gavage. Magnification, ×110,000. Scale bar, 0.2 μm.
Enterocyte LD Formation Is Not Required for Intestinal TG Absorption
Cytosolic LDs have previously been shown to form in enterocytes after an acute fat challenge (28, 29). However, whether cytosolic LD formation is a necessary step in dietary TG absorption has yet to be established. We have shown that iF2KO mice fail to form cytosolic LDs in their enterocytes after an acute fat challenge (Fig. 6, A and B). Here, we investigated the effects of FIT2 deficiency on dietary TG absorption by gavaging L/L control and iF2KO mice with olive oil, [3H]triolein (trioleoylglycerol), or [14C]oleic acid. Mice at day 5 post-tamoxifen treatment were used because neither villus blunting nor weight loss was observed at this time point (Figs. 1B and 2A). Consistent with the absence of villus blunting in iF2KO intestine at day 5 post-tamoxifen treatment, there was no significant difference in the amount of serum TG (Fig. 7A) or serum 3H and 14C signals (Fig. 7B) post-gavage between iF2KO and L/L mice. Furthermore, we performed Western blot analysis of serum ApoB48 and ApoB100 before and 3 h after acute fat challenge. Both L/L and iF2KO mice had a similar increase in serum ApoB48 post-oil gavage (Fig. 7, C and D). Taken together, these data indicate that in the iF2KO model, cytosolic LD formation is not an essential step for chylomicron production or secretion and that dietary TG absorption can occur normally in the absence of enterocyte cytosolic LD formation.
FIGURE 7.

FIT2 deletion in iF2KO mice does not affect lipid absorption and lipoprotein levels after acute oil challenge. A, quantification of serum TG concentrations 1 and 3 h post-oil gavage in day 5 post-tamoxifen-treated mice. B, quantification of serum 3H and 14C signals at 3 h post-gavage with [3H]triolein and [14C]oleic acid, respectively, in day 5 post-tamoxifen-treated mice. C, Western blot analysis of ApoB48 and ApoB100 levels in serum from post-oil gavaged L/L and iF2KO mice, harvested at day 5 post-tamoxifen treatment. D, densitometry of ApoB levels shown in C. Data are represented as mean with error bars indicating S.E. *, p < 0.05, two-tailed t test. n.s., not significant.
FIT2 Deletion Leads to Dysregulation of Multiple Genes Involved in Bile Acid Transport
To explain the changes in villus and crypt morphology and crypt death in iF2KO mice, we hypothesized that there could be disruption of normal BA transport. BAs are anionic detergents that are postprandially released into the small intestinal lumen where they solubilize fatty acids, cholesterol, and lipophilic vitamins before uptake into the enterocyte (33). More than 90% of BAs are reabsorbed from the small intestine to be recycled at the liver, where less than 10% of BAs are synthesized de novo via the Cyp7a1 pathway. Enterohepatic reabsorption of BAs occurs actively in the ileum and passively down the rest of the intestine (34, 35). BA malabsorption may occur when BAs are unable to enter the lumenal membrane of the enterocyte for reabsorption or when they are unable to leave from the basolateral membrane to re-enter the circulation (36–40). We isolated enterocytes at day 8 post-tamoxifen treatment and quantified the amount of total BAs present. There was a significant increase in total BA content in iF2KO enterocytes compared with controls (Fig. 8A), which could indicate a defect in BA exit from the enterocyte basolateral membrane. BA accumulation in the gut leads to up-regulation of the key intestinal hormone mFgf15 that negatively regulates hepatic BA synthesis by inhibiting the expression of liver Cyp7A1, a rate-limiting enzyme for BA synthesis (41). Consistent with BA accumulation within enterocytes (Fig. 8A), mFgf15 mRNA was significantly induced in intestines of iF2KO mice relative to L/L controls (Fig. 8B), and Cyp7A1 mRNA was significantly reduced in livers of iF2KO mice relative to L/L controls. (Fig. 8C).
FIGURE 8.

Expression of BA transporters is dysregulated in iF2KO small intestine, leading to accumulation of BAs within enterocytes. A, quantification of enterocyte total BA content in day 8 post-tamoxifen-treated mice. L/L, n = 8; iF2KO, n = 8. B and D–G, quantitative RT-PCR of mRNAs derived from L/L and iF2KO small intestine at days 7–8 post-tamoxifen treatment; L/L, n = 5, and iF2KO, n = 6, except for B, iF2KO, n = 5. C, quantitative RT-PCR of mRNAs derived from L/L and iF2KO livers at days 7–8 post-tamoxifen treatment. L/L, n = 5, and iF2KO, n = 5. Mice were fasted for 4 h prior to sacrifice. Data are represented as mean with error bars indicating S.E. Each point represents an independent biological replicate. *, p < 0.05; **, p < 0.01; ***, p < 0.001, two-tailed t test. n.s., not significant.
The complex process of intestinal BA absorption is tightly regulated by FXR-mediated transcriptional control of genes encoding BA transporters, such as the apical ileal sodium-dependent BA transporter (Asbt/Slc10a2), the cytosolic ileal BA-binding protein (I-babp/Fabp6), and the heteromeric organic solute transporter (OSTα-OSTβ) (41–44). Asbt is localized at the brush-border membrane where it mediates the transport of BAs from the intestinal lumen into the enterocyte (45). BAs are then bound to the cytosolic Fabp6, which is thought to facilitate the transcellular transport of BAs to the basolateral membrane and may protect the enterocyte from BA cytotoxicity (37, 46–49). At the enterocyte basolateral membrane, BAs are exported via the heteromeric OSTα-OSTβ transporter (50, 51) into the portal circulation where they bind to albumin. Analysis of distal intestine mRNA transcripts using day 7 and 8 post-tamoxifen-treated intestine showed that FIT2 deletion resulted in a significant decrease in Fabp6 and Ostβ (Fig. 8, D and E) but not Asbt and Ostα transcripts (Fig. 8, F and G). Taken together, our data suggest that the accumulation of BAs in iF2KO enterocytes could be a result of the down-regulation of the basolateral BA exporter, OSTβ, and the cytosolic BA-binding protein, Fabp6, which may in turn lead to the disruption of cellular membranes and the destruction of villus and crypt architecture.
FIT2 Deletion in Intestinal Epithelium Is Insufficient to Cause Lethal Enteropathy
To directly test whether the cause of lethal enteropathy in iF2KO mice was caused by the deletion of FIT2 in intestinal crypts, we generated both a constitutive villin-cre-mediated intestine-specific FIT2 knock-out (VF2KO) mouse model and a tamoxifen-inducible intestine-specific villin-CreERT2 mediated FIT2 knock-out (iVF2KO) mouse model. Villin-cre and villin-CreERT2 are expressed in intestinal stem cells and all lineages derived from crypt stem cells (13, 52). VF2KO mice were found to be viable. FIT2 deletion in VF2KO small intestine was confirmed by Western blot (Fig. 9A). H&E staining of VF2KO small intestine revealed normal villus and crypt architecture (Fig. 9B). To investigate whether FIT2 deletion inhibits intestinal stem cell proliferation in a cell autonomous manner, we used an established intestinal crypt organoid culture technique that recapitulates the anatomy and physiology of the intestinal crypt in tissue culture, without the presence of supportive stromal cells (16). Crypts derived from VF2KO mice were able to form mature organoids (Fig. 9C), indicating that FIT2 deletion in crypts alone does not lead to cell death. Furthermore, Plin2- and Plin3-coated cytosolic LDs were able to form normally upon olive oil challenge of VF2KO mice (Fig. 9D).
FIGURE 9.

FIT2 deletion in intestinal epithelium is insufficient to cause lethal enteropathy. A, Western blot analysis of FIT2 on the small intestine indicates FIT2 deletion in this tissue in VF2KO mice. B, H&E staining indicates normal villus and crypt architecture in VF2KO small intestine. Magnification, ×20. Scale bar, 200 μm. C, light microscopy image of a mature organoid from in vitro culture of VF2KO-derived crypts. Magnification, ×40. Scale bar, 20 μm. D, immunofluorescence staining of Plin2- and Plin3-coated LDs in VF2KO small intestine. Magnification, ×63. Scale bar, 10 μm. E, Western blot analysis of FIT2 at day 8 post-tamoxifen treatment indicates FIT2 deletion in the small intestine of villin-CreERT2 positive L/L (iVF2KO) but not L/L mice. F, body weight of L/L and iVF2KO mice at day 9 post-tamoxifen treatment. G, quantification of serum 3H signal at 3 h post-gavage with [3H]triolein, in day 9 post-tamoxifen-treated mice. Data are represented as mean with error bars indicating S.E. n.s., not significant.
Using the same tamoxifen treatment protocol as for the iF2KO model, Western blot analysis indicated that FIT2 is depleted in iVF2KO intestine at day 8 post-tamoxifen treatment (Fig. 9E). Similar to our observations in the VF2KO model, iVF2KO mice were viable and had normal body weight at day 9 post-tamoxifen (Fig. 9F). To assess the functional lipid absorption capacity of the iVF2KO intestine, we gavaged the mice with [3H]triolein and measured the appearance of 3H in the serum. iVF2KO mice had normal [3H]triolein absorption at day 9 post-tamoxifen (Fig. 9G). Taken together, these results indicate that FIT2 deletion in cell types other than the intestinal epithelium alone was essential for the development of lethal malabsorptive enteropathy.
Discussion
In this study, we show that FIT2 plays an important role in intestinal homeostasis and survival. FIT2 deficiency in the iF2KO mouse model resulted in progressive weight loss, intestinal crypt death, and villus blunting, and eventually it led to lethal malabsorption. Although we did not directly assess the turnover of mature epithelium in iF2KO intestine, it is possible that in addition to reduced crypt stem cell proliferation and survival, FIT2 deletion could have affected survival of mature epithelium. Murine intestinal stem cell renewal involves the pro-survival Wnt and Notch signaling pathways. Here, we found that not only were Wnt and Notch ligands, receptors, and target genes not down-regulated in iF2KO intestine, several of these transcripts were significantly up-regulated by RT-PCR analysis. Although this could be an adaptive attempt by crypt stem cells to regenerate the dying small intestine (53), the simplest explanation for an increase in Wnt- and Notch-related transcripts is enrichment of mesenchymal or stromal cells and long-lived Paneth cells in the intestine because of the death of crypt stem cells. Paneth cells are known to secrete Wnt and Notch ligands (54, 55), and intestinal stromal cells have also been found to secrete many Wnts (15, 55).
The discovery of malabsorptive enteropathy caused by FIT2 deletion is reminiscent of the congenital diarrheal disorder found in humans with an acyl-CoA:diacylglycerol acyltransferase 1 (DGAT1) mutation (56). DGAT1 is a transmembrane protein with its putative C-terminal catalytic site facing the ER lumen (57, 58). DGAT1 catalyzes the final step in TG synthesis (57) and acts upstream of FIT2 during LD biogenesis. Disruption of DGAT1 in humans was thought to result in the buildup of excess diacylglycerol and free fatty acids in enterocytes, leading to toxicity and enteropathy (56). In the iF2KO mouse model, we found that BAs accumulated within enterocytes, in conjunction with a transcriptional down-regulation of basolateral BA transporter OSTβ and ileal BA-binding protein Fabp6. Cytosolic Fabp6 is thought to facilitate the transcellular transport of BAs to the basolateral membrane and may protect the enterocyte from BA cytotoxicity (37, 46–49). At the enterocyte basolateral membrane, BAs are exported via the heteromeric OSTα-OSTβ transporter (50, 51) into the portal circulation. Immunolocalization studies have previously shown that OSTα and OSTβ co-expression was required for plasma membrane expression of the heteromeric transporter (51). To our knowledge, there have been no reports of animal models showing down-regulation of both Fabp6 and OST without a compensatory decrease in Asbt transcription in enterocytes, which is thought to keep BA levels within normal limits. It is likely that the gene expression changes we observed in iF2KO intestine could lead to the toxic accumulation of BAs in enterocytes, with BAs entering the enterocyte via Asbt but not being able to bind Fabp6 or leave the cell via OST, thus contributing to the lethal enteropathy seen in iF2KO mice. Although dysregulation of BA transporters at the transcriptional level is significant and could explain the accumulation of BAs within iF2KO enterocytes, we remain cautious in this conclusion because transporter protein levels or localization might not reflect the changes in their mRNAs that we observed.
Direct evidence of a dynamic cytoplasmic TG pool in enterocytes during the process of dietary fat absorption was shown by Zhu et al. (28), by combining coherent anti-Stokes Raman scattering microscopy and fluorescent imaging of exposed small intestine. Although there is evidence for a dynamic pool of cytosolic LDs in enterocytes during dietary fat absorption (28, 29), the question of whether this LD pool is essential for lipid absorption and for the function and health of the small intestinal epithelium remains unanswered. In this study, we showed that iF2KO mice could not form LDs in their enterocytes after an acute oil challenge, and instead they partitioned the resynthesized neutral lipid into lipoprotein particles in the ER. However, the lack of LDs in iF2KO enterocytes did not suppress intestinal lipid absorption into the bloodstream. iF2KO mice could absorb lipids normally (Fig. 7, A and B) post-oil gavage even though they did not form cytosolic LDs within enterocytes at the same early experimental time point before anatomical changes occurred in the villi of iF2KO mice. Our data suggest that cytosolic LD formation is not an essential step for chylomicron production or secretion and that dietary TG absorption can occur normally even in the absence of enterocyte cytosolic LD formation.
A striking observation from our study was that the lethal intestinal phenotype caused by FIT2 deletion in iF2KO mice could not be recapitulated using the intestine-specific VF2KO and iVF2KO mouse models. This indicates that FIT2 in enterocytes is not important for intestinal stem cell survival and that FIT2 expressed in a cell or tissue type other than the intestinal epithelium could be important for intestinal stem cell survival. The small intestine is made up of not only cells of epithelial origin but also mesenchymal or stromal cells, gut immune cells, and vascular as well as lymphatic endothelial cells. FIT2 deletion in one or more of these cell types in iF2KO mice could contribute to the lethal intestinal phenotype. Indeed, FIT2 protein expression is low in enterocytes (Fig. 1A). Further FIT2 tissue- and cell-specific deficiency models must be generated in the future to address this question.
A similar intestinal phenotype as we described here for iF2KO mice has been recently reported for inducible deficiency of fatty-acid synthase (Fas). Fas catalyzes the first committed step in de novo lipogenesis by using malonyl-CoA and NADPH to synthesize palmitate after acetyl-CoA priming. Lodhi et al. (66) recently discovered that inducible global knock-out of fatty-acid synthase (iFASKO) in mice using the ROSA26CreERT2 model, as we used in this study, was lethal due to damage to the intestinal epithelium and leukopenia. Moreover, and similar to our study here, intestinal epithelium-specific deletion of Fas using villin-cre did not result in intestinal damage. Like FIT2, Fas is ubiquitously expressed and has diverse tissue-specific effects. The similarity between iFASKO and iF2KO mice raises the possibility that FAS and FIT2 function in a linear pathway. The tissue-specific functions of Fas in liver (59, 60), brain (61), macrophages (62), heart (63), vascular endothelium (64), intestinal epithelium (65), and adipose tissue (66) have been thought to arise from the channeling of newly synthesized palmitate to different subcellular compartments, which then transmit relevant signals that modulate lipid metabolism. One possibility is that FIT2 functions at the ER to regulate the downstream metabolism of palmitate for elongation, desaturation, and glycerolipid synthesis or release for other signaling or metabolic processes. FIT2 may mediate these effects by functioning as a scaffold protein for lipid metabolic enzymes, but this idea has yet to be explored. Tissue-specific effects of FIT2 would then be due to the differences in FIT2-binding proteins and the predominance of different lipid metabolic pathways within each cell type.
Author Contributions
V. J. G. and D. L. S. conceived the study and wrote the paper. V. J. G., J. S. Y. T., B. C. T., and C. S. performed experiments. W. Y. O. performed EM experiments. Y. C. L., L. S., and S. G. performed bioinformatics analysis of gene array data.
Acknowledgments
We thank Dr. Ya-Jun Wu for help with electron microscopy and the Duke-NUS Genome Biology Facility for their help in microarray profiling.
This work was supported in part by Singapore Ministry of Health National Medical Research Council Grant CBRG/0012/2012 (to D. L. S.) and a National Science Scholarship award (to V. J. G.). The authors declare that they have no conflicts of interest with the contents of this article.
- LD
- lipid droplet
- BA
- bile acid
- ER
- endoplasmic reticulum
- TG
- triglyceride
- WAT
- white adipose tissue
- OST
- organic solute transporter
- FAS
- fatty-acid synthase
- EdU
- 5-ethynyl-2′-deoxyuridine
- DPBS
- Dulbecco's phosphate-buffered saline
- MTP
- microsomal triglyceride transfer protein
- ASBT
- apical sodium-dependent bile acid transporter.
References
- 1. Wilfling F., Haas J. T., Walther T. C., Farese R. V. Jr. (2014) Lipid droplet biogenesis. Curr. Opin. Cell Biol. 29, 39–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wilfling F., Wang H., Haas J. T., Krahmer N., Gould T. J., Uchida A., Cheng J. X., Graham M., Christiano R., Fröhlich F., Liu X., Buhman K. K., Coleman R. A., Bewersdorf J., Farese R. V. Jr., Walther T. C. (2013) Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Dev. Cell 24, 384–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kadereit B., Kumar P., Wang W. J., Miranda D., Snapp E. L., Severina N., Torregroza I., Evans T., Silver D. L. (2008) Evolutionarily conserved gene family important for fat storage. Proc. Natl. Acad. Sci. U.S.A. 105, 94–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Moir R. D., Gross D. A., Silver D. L., Willis I. M. (2012) SCS3 and YFT2 link transcription of phospholipid biosynthetic genes to ER stress and the UPR. PLoS Genet. 8, e1002890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tontonoz P., Hu E., Graves R. A., Budavari A. I., Spiegelman B. M. (1994) mPPARγ 2: tissue-specific regulator of an adipocyte enhancer. Genes Dev. 8, 1224–1234 [DOI] [PubMed] [Google Scholar]
- 6. Rangwala S. M., Rhoades B., Shapiro J. S., Rich A. S., Kim J. K., Shulman G. I., Kaestner K. H., Lazar M. A. (2003) Genetic modulation of PPARγ phosphorylation regulates insulin sensitivity. Dev. Cell 5, 657–663 [DOI] [PubMed] [Google Scholar]
- 7. Lefterova M. I., Zhang Y., Steger D. J., Schupp M., Schug J., Cristancho A., Feng D., Zhuo D., Stoeckert C. J. Jr., Liu X. S., Lazar M. A. (2008) PPARγ and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev. 22, 2941–2952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gross D. A., Zhan C., Silver D. L. (2011) Direct binding of triglyceride to fat storage-inducing transmembrane proteins 1 and 2 is important for lipid droplet formation. Proc. Natl. Acad. Sci. U.S.A. 108, 19581–19586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gross D. A., Snapp E. L., Silver D. L. (2010) Structural insights into triglyceride storage mediated by fat storage-inducing transmembrane (FIT) protein 2. PLoS One 5, e10796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jackson K. G., Robertson M. D., Fielding B. A., Frayn K. N., Williams C. M. (2002) Olive oil increases the number of triacylglycerol-rich chylomicron particles compared with other oils: an effect retained when a second standard meal is fed. Am. J. Clin. Nutr. 76, 942–949 [DOI] [PubMed] [Google Scholar]
- 11. Miranda D. A., Kim J. H., Nguyen L. N., Cheng W., Tan B. C., Goh V. J., Tan J. S., Yaligar J., Kn B. P., Velan S. S., Wang H., Silver D. L. (2014) Fat storage-inducing transmembrane protein 2 is required for normal fat storage in adipose tissue. J. Biol. Chem. 289, 9560–9572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hameyer D., Loonstra A., Eshkind L., Schmitt S., Antunes C., Groen A., Bindels E., Jonkers J., Krimpenfort P., Meuwissen R., Rijswijk L., Bex A., Berns A., Bockamp E. (2007) Toxicity of ligand-dependent Cre recombinases and generation of a conditional Cre deleter mouse allowing mosaic recombination in peripheral tissues. Physiol. Genomics 31, 32–41 [DOI] [PubMed] [Google Scholar]
- 13. el Marjou F., Janssen K. P., Chang B. H., Li M., Hindie V., Chan L., Louvard D., Chambon P., Metzger D., Robine S. (2004) Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis 39, 186–193 [DOI] [PubMed] [Google Scholar]
- 14. Gracz A. D., Puthoff B. J., Magness S. T. (2012) Identification, isolation, and culture of intestinal epithelial stem cells from murine intestine. Methods Mol. Biol. 879, 89–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kabiri Z., Greicius G., Madan B., Biechele S., Zhong Z., Zaribafzadeh H., Edison, Aliyev J., Wu Y., Bunte R., Williams B. O., Rossant J., Virshup D. M. (2014) Stroma provides an intestinal stem cell niche in the absence of epithelial Wnts. Development 141, 2206–2215 [DOI] [PubMed] [Google Scholar]
- 16. Sato T., Clevers H. (2013) Primary mouse small intestinal epithelial cell cultures. Methods Mol. Biol. 945, 319–328 [DOI] [PubMed] [Google Scholar]
- 17. Miranda D. A., Koves T. R., Gross D. A., Chadt A., Al-Hasani H., Cline G. W., Schwartz G. J., Muoio D. M., Silver D. L. (2011) Re-patterning of skeletal muscle energy metabolism by fat storage-inducing transmembrane protein 2. J. Biol. Chem. 286, 42188–42199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barker N., van de Wetering M., Clevers H. (2008) The intestinal stem cell. Genes Dev. 22, 1856–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sancho R., Cremona C. A., Behrens A. (2015) Stem cell and progenitor fate in the mammalian intestine: Notch and lateral inhibition in homeostasis and disease. EMBO Rep. 16, 571–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Perdigoto C. N., Schweisguth F., Bardin A. J. (2011) Distinct levels of Notch activity for commitment and terminal differentiation of stem cells in the adult fly intestine. Development 138, 4585–4595 [DOI] [PubMed] [Google Scholar]
- 21. Crosnier C., Vargesson N., Gschmeissner S., Ariza-McNaughton L., Morrison A., Lewis J. (2005) Delta-Notch signalling controls commitment to a secretory fate in the zebrafish intestine. Development 132, 1093–1104 [DOI] [PubMed] [Google Scholar]
- 22. Fre S., Huyghe M., Mourikis P., Robine S., Louvard D., Artavanis-Tsakonas S. (2005) Notch signals control the fate of immature progenitor cells in the intestine. Nature 435, 964–968 [DOI] [PubMed] [Google Scholar]
- 23. Ishifune C., Maruyama S., Sasaki Y., Yagita H., Hozumi K., Tomita T., Kishihara K., Yasutomo K. (2014) Differentiation of CD11c+ CX3CR1+ cells in the small intestine requires Notch signaling. Proc. Natl. Acad. Sci. U.S.A. 111, 5986–5991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li H. J., Kapoor A., Giel-Moloney M., Rindi G., Leiter A. B. (2012) Notch signaling differentially regulates the cell fate of early endocrine precursor cells and their maturing descendants in the mouse pancreas and intestine. Dev. Biol. 371, 156–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. VanDussen K. L., Carulli A. J., Keeley T. M., Patel S. R., Puthoff B. J., Magness S. T., Tran I. T., Maillard I., Siebel C., Kolterud Å., Grosse A. S., Gumucio D. L., Ernst S. A., Tsai Y. H., Dempsey P. J., Samuelson L. C. (2012) Notch signaling modulates proliferation and differentiation of intestinal crypt base columnar stem cells. Development 139, 488–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ireland H., Houghton C., Howard L., Winton D. J. (2005) Cellular inheritance of a Cre-activated reporter gene to determine Paneth cell longevity in the murine small intestine. Dev. Dyn. 233, 1332–1336 [DOI] [PubMed] [Google Scholar]
- 27. van Es J. H., van Gijn M. E., Riccio O., van den Born M., Vooijs M., Begthel H., Cozijnsen M., Robine S., Winton D. J., Radtke F., Clevers H. (2005) Notch/γ-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 435, 959–963 [DOI] [PubMed] [Google Scholar]
- 28. Zhu J., Lee B., Buhman K. K., Cheng J. X. (2009) A dynamic, cytoplasmic triacylglycerol pool in enterocytes revealed by ex vivo and in vivo coherent anti-Stokes Raman scattering imaging. J. Lipid Res. 50, 1080–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee B., Zhu J., Wolins N. E., Cheng J. X., Buhman K. K. (2009) Differential association of adipophilin and TIP47 proteins with cytoplasmic lipid droplets in mouse enterocytes during dietary fat absorption. Biochim. Biophys. Acta 1791, 1173–1180 [DOI] [PubMed] [Google Scholar]
- 30. Ohsaki Y., Maeda T., Maeda M., Tauchi-Sato K., Fujimoto T. (2006) Recruitment of TIP47 to lipid droplets is controlled by the putative hydrophobic cleft. Biochem. Biophys. Res. Commun. 347, 279–287 [DOI] [PubMed] [Google Scholar]
- 31. Raabe M., Véniant M. M., Sullivan M. A., Zlot C. H., Björkegren J., Nielsen L. B., Wong J. S., Hamilton R. L., Young S. G. (1999) Analysis of the role of microsomal triglyceride transfer protein in the liver of tissue-specific knockout mice. J. Clin. Invest. 103, 1287–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xie Y., Newberry E. P., Young S. G., Robine S., Hamilton R. L., Wong J. S., Luo J., Kennedy S., Davidson N. O. (2006) Compensatory increase in hepatic lipogenesis in mice with conditional intestine-specific Mttp deficiency. J. Biol. Chem. 281, 4075–4086 [DOI] [PubMed] [Google Scholar]
- 33. Wolkoff A. W., Cohen D. E. (2003) Bile acid regulation of hepatic physiology: I. Hepatocyte transport of bile acids. Am. J. Physiol. Gastrointest. Liver Physiol. 284, G175–G179 [DOI] [PubMed] [Google Scholar]
- 34. Krag E., Phillips S. F. (1974) Active and passive bile acid absorption in man. Perfusion studies of the ileum and jejunum. J. Clin. Invest. 53, 1686–1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schiff E. R., Small N. C., Dietschy J. M. (1972) Characterization of the kinetics of the passive and active transport mechanisms for bile acid absorption in the small intestine and colon of the rat. J. Clin. Invest. 51, 1351–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jung D., Inagaki T., Gerard R. D., Dawson P. A., Kliewer S. A., Mangelsdorf D. J., Moschetta A. (2007) FXR agonists and FGF15 reduce fecal bile acid excretion in a mouse model of bile acid malabsorption. J. Lipid Res. 48, 2693–2700 [DOI] [PubMed] [Google Scholar]
- 37. Kramer W., Girbig F., Gutjahr U., Kowalewski S., Jouvenal K., Müller G., Tripier D., Wess G. (1993) Intestinal bile acid absorption. Na+-dependent bile acid transport activity in rabbit small intestine correlates with the coexpression of an integral 93-kDa and a peripheral 14-kDa bile acid-binding membrane protein along the duodenum-ileum axis. J. Biol. Chem. 268, 18035–18046 [PubMed] [Google Scholar]
- 38. Rao A., Haywood J., Craddock A. L., Belinsky M. G., Kruh G. D., Dawson P. A. (2008) The organic solute transporter α-β, Ostα-Ostβ, is essential for intestinal bile acid transport and homeostasis. Proc. Natl. Acad. Sci. U.S.A. 105, 3891–3896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ballatori N., Fang F., Christian W. V., Li N., Hammond C. L. (2008) Ostα-Ostβ is required for bile acid and conjugated steroid disposition in the intestine, kidney, and liver. Am. J. Physiol. Gastrointest. Liver Physiol. 295, G179–G186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li H., Xu G., Shang Q., Pan L., Shefer S., Batta A. K., Bollineni J., Tint G. S., Keller B. T., Salen G. (2004) Inhibition of ileal bile acid transport lowers plasma cholesterol levels by inactivating hepatic farnesoid X receptor and stimulating cholesterol 7 α-hydroxylase. Metab. Clin. Exp. 53, 927–932 [DOI] [PubMed] [Google Scholar]
- 41. Kliewer S. A., Mangelsdorf D. J. (2015) Bile acids as hormones: the FXR-FGF15/19 pathway. Digest. Dis. 33, 327–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Klaassen C. D., Aleksunes L. M. (2010) Xenobiotic, bile acid, and cholesterol transporters: function and regulation. Pharmacol. Rev. 62, 1–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gadaleta R. M., van Mil S. W., Oldenburg B., Siersema P. D., Klomp L. W., van Erpecum K. J. (2010) Bile acids and their nuclear receptor FXR: relevance for hepatobiliary and gastrointestinal disease. Biochim. Biophys. Acta 1801, 683–692 [DOI] [PubMed] [Google Scholar]
- 44. Zollner G., Wagner M., Moustafa T., Fickert P., Silbert D., Gumhold J., Fuchsbichler A., Halilbasic E., Denk H., Marschall H. U., Trauner M. (2006) Coordinated induction of bile acid detoxification and alternative elimination in mice: role of FXR-regulated organic solute transporter-α/β in the adaptive response to bile acids. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G923–G932 [DOI] [PubMed] [Google Scholar]
- 45. Dawson P. A., Haywood J., Craddock A. L., Wilson M., Tietjen M., Kluckman K., Maeda N., Parks J. S. (2003) Targeted deletion of the ileal bile acid transporter eliminates enterohepatic cycling of bile acids in mice. J. Biol. Chem. 278, 33920–33927 [DOI] [PubMed] [Google Scholar]
- 46. Oelkers P., Dawson P. A. (1995) Cloning and chromosomal localization of the human ileal lipid-binding protein. Biochim. Biophys. Acta 1257, 199–202 [DOI] [PubMed] [Google Scholar]
- 47. Lin M. C., Kramer W., Wilson F. A. (1990) Identification of cytosolic and microsomal bile acid-binding proteins in rat ileal enterocytes. J. Biol. Chem. 265, 14986–14995 [PubMed] [Google Scholar]
- 48. Kramer W., Wess G., Bewersdorf U., Corsiero D., Girbig F., Weyland C., Stengelin S., Enhsen A., Bock K., Kleine H., Le Dreau M. A., Schäfer H. L. (1997) Topological photoaffinity labeling of the rabbit ileal Na+/bile-salt-cotransport system. Eur. J. Biochem. 249, 456–464 [DOI] [PubMed] [Google Scholar]
- 49. Praslickova D., Torchia E. C., Sugiyama M. G., Magrane E. J., Zwicker B. L., Kolodzieyski L., Agellon L. B. (2012) The ileal lipid binding protein is required for efficient absorption and transport of bile acids in the distal portion of the murine small intestine. PLoS One 7, e50810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ballatori N., Christian W. V., Lee J. Y., Dawson P. A., Soroka C. J., Boyer J. L., Madejczyk M. S., Li N. (2005) OSTα-OSTβ: a major basolateral bile acid and steroid transporter in human intestinal, renal, and biliary epithelia. Hepatology 42, 1270–1279 [DOI] [PubMed] [Google Scholar]
- 51. Dawson P. A., Hubbert M., Haywood J., Craddock A. L., Zerangue N., Christian W. V., Ballatori N. (2005) The heteromeric organic solute transporter α-β, Ostα-Ostβ, is an ileal basolateral bile acid transporter. J. Biol. Chem. 280, 6960–6968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Madison B. B., Dunbar L., Qiao X. T., Braunstein K., Braunstein E., Gumucio D. L. (2002) Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J. Biol. Chem. 277, 33275–33283 [DOI] [PubMed] [Google Scholar]
- 53. Roth S., Franken P., Sacchetti A., Kremer A., Anderson K., Sansom O., Fodde R. (2012) Paneth cells in intestinal homeostasis and tissue injury. PLoS One 7, e38965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sato T., van Es J. H., Snippert H. J., Stange D. E., Vries R. G., van den Born M., Barker N., Shroyer N. F., van de Wetering M., Clevers H. (2011) Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 469, 415–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Farin H. F., Van Es J. H., Clevers H. (2012) Redundant sources of Wnt regulate intestinal stem cells and promote formation of Paneth cells. Gastroenterology 143, 1518–1529 [DOI] [PubMed] [Google Scholar]
- 56. Haas J. T., Winter H. S., Lim E., Kirby A., Blumenstiel B., DeFelice M., Gabriel S., Jalas C., Branski D., Grueter C. A., Toporovski M. S., Walther T. C., Daly M. J., Farese R. V. Jr. (2012) DGAT1 mutation is linked to a congenital diarrheal disorder. J. Clin. Invest. 122, 4680–4684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cases S., Smith S. J., Zheng Y. W., Myers H. M., Lear S. R., Sande E., Novak S., Collins C., Welch C. B., Lusis A. J., Erickson S. K., Farese R. V. Jr. (1998) Identification of a gene encoding an acyl CoA:diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc. Natl. Acad. Sci. U.S.A. 95, 13018–13023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. McFie P. J., Stone S. L., Banman S. L., Stone S. J. (2010) Topological orientation of acyl-CoA:diacylglycerol acyltransferase-1 (DGAT1) and identification of a putative active site histidine and the role of the N terminus in dimer/tetramer formation. J. Biol. Chem. 285, 37377–37387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chakravarthy M. V., Lodhi I. J., Yin L., Malapaka R. R., Xu H. E., Turk J., Semenkovich C. F. (2009) Identification of a physiologically relevant endogenous ligand for PPARα in liver. Cell 138, 476–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chakravarthy M. V., Pan Z., Zhu Y., Tordjman K., Schneider J. G., Coleman T., Turk J., Semenkovich C. F. (2005) “New” hepatic fat activates PPARα to maintain glucose, lipid, and cholesterol homeostasis. Cell Metab. 1, 309–322 [DOI] [PubMed] [Google Scholar]
- 61. Chakravarthy M. V., Zhu Y., López M., Yin L., Wozniak D. F., Coleman T., Hu Z., Wolfgang M., Vidal-Puig A., Lane M. D., Semenkovich C. F. (2007) Brain fatty acid synthase activates PPARα to maintain energy homeostasis. J. Clin. Invest. 117, 2539–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schneider J. G., Yang Z., Chakravarthy M. V., Lodhi I. J., Wei X., Turk J., Semenkovich C. F. (2010) Macrophage fatty-acid synthase deficiency decreases diet-induced atherosclerosis. J. Biol. Chem. 285, 23398–23409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Razani B., Zhang H., Schulze P. C., Schilling J. D., Verbsky J., Lodhi I. J., Topkara V. K., Feng C., Coleman T., Kovacs A., Kelly D. P., Saffitz J. E., Dorn G. W. 2nd, Nichols C. G., Semenkovich C. F. (2011) Fatty acid synthase modulates homeostatic responses to myocardial stress. J. Biol. Chem. 286, 30949–30961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wei X., Schneider J. G., Shenouda S. M., Lee A., Towler D. A., Chakravarthy M. V., Vita J. A., Semenkovich C. F. (2011) De novo lipogenesis maintains vascular homeostasis through endothelial nitric-oxide synthase (eNOS) palmitoylation. J. Biol. Chem. 286, 2933–2945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wei X., Yang Z., Rey F. E., Ridaura V. K., Davidson N. O., Gordon J. I., Semenkovich C. F. (2012) Fatty acid synthase modulates intestinal barrier function through palmitoylation of mucin 2. Cell Host Microbe 11, 140–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lodhi I. J., Yin L., Jensen-Urstad A. P., Funai K., Coleman T., Baird J. H., El Ramahi M. K., Razani B., Song H., Fu-Hsu F., Turk J., Semenkovich C. F. (2012) Inhibiting adipose tissue lipogenesis reprograms thermogenesis and peroxisome proliferator-activated receptor γ activation to decrease diet-induced obesity. Cell Metab. 16, 189–201 [DOI] [PMC free article] [PubMed] [Google Scholar]