

Background: G protein-coupled receptors (GPCRs) are thought to exist as homo- or heterodimers, but the molecular determinants of their dimerization remain undercharacterized.
Results: TM peptides disrupted the dimerization of rhodopsin (Rho), decreasing its thermal stability and the binding of Gt without affecting its rate of G protein activation.
Conclusion: Both the TM1,2 and TM4,5 domains reflect the Rho dimer/oligomer interface.
Significance: Multiple Rho association interfaces affect this GPCR function.
Keywords: G protein, G protein-coupled receptor (GPCR), oligomerization, receptor, receptor structure-function
Abstract
Although homo- and heterodimerizations of G protein-coupled receptors (GPCRs) are well documented, GPCR monomers are able to assemble in different ways, thus causing variations in the interactive interface between receptor monomers among different GPCRs. Moreover, the functional consequences of this phenomenon, which remain to be clarified, could be specific for different GPCRs. Synthetic peptides derived from transmembrane (TM) domains can interact with a full-length GPCR, blocking dimer formation and affecting its function. Here we used peptides corresponding to TM helices of bovine rhodopsin (Rho) to investigate the Rho dimer interface and functional consequences of its disruption. Incubation of Rho with TM1, TM2, TM4, and TM5 peptides in rod outer segment (ROS) membranes shifted the resulting detergent-solubilized protein migration through a gel filtration column toward smaller molecular masses with a reduced propensity for dimer formation in a cross-linking reaction. Binding of these TM peptides to Rho was characterized by both mass spectrometry and a label-free assay from which dissociation constants were calculated. A BRET (bioluminescence resonance energy transfer) assay revealed that the physical interaction between Rho molecules expressed in membranes of living cells was blocked by the same four TM peptides identified in our in vitro experiments. Although disruption of the Rho dimer/oligomer had no effect on the rates of G protein activation, binding of Gt to the activated receptor stabilized the dimer. However, TM peptide-induced disruption of dimer/oligomer decreased receptor stability, suggesting that Rho supramolecular organization could be essential for ROS stabilization and receptor trafficking.
Introduction
During the past three decades, the concept of GPCR4 dimerization/oligomerization has captivated researchers engaged in this field (reviewed in Refs. 1–14). This topic is interesting from several perspectives including: (i) possible expansion of the pool of unique GPCR signaling clusters resulting from homo- and heterodimerization; (ii) more complex pharmacology when only one of two monomers is activated, with plausible intermolecular modulation within the dimer; (iii) novel pharmacology that permits allosteric regulators to occupy the interface engaged in complex formation; (iv) an increase in local receptor concentrations to allow a longer dwelling time for ligands; (v) the fact that some GPCRs, typically from family C, are covalently linked and their dimers, essential for activity, interact with the same set of G proteins and arrestins as other GPCRs including family A and B members sharing a common mechanism of activation; (vi) their structural complementarity with partner heterotrimeric G proteins with GPCR-binding surfaces larger than that of a monomeric receptor; (vii) their structural complementarity to partner arrestins with bipartite structures that fit GPCR dimers; and (viii) the high level of phosphorylation, particularly of Rho, wherein activation of one receptor produces phosphorylation of many others in a process called “high-gain phosphorylation.”
This di/oligomerization concept is supported by several imaging techniques (1–4), biochemical and biophysical approaches including fluorescence resonance energy transfer (FRET) and bioluminescence resonance energy transfer (BRET) assays, pharmacological investigations, structural studies, genetics, mutagenesis, and structural modeling (5–14). But a different perspective also has been advanced, namely that GPCRs are monomers because: (i) the monomeric receptor is often, but not always, functional (15–18); (ii) interpretation of BRET and FRET experiments in transformed cells is not always reliable (19, 20); and (iii) some structural studies are interpreted as proof of a GPCR monomeric structure (see Ref. 21 for a recent debate). Finally, it should be noted that oligomerization of GPCRs has been implicated not only in signaling but also in the trafficking and stability of many GPCRs (22–24). In other cases, the functional consequences of GPCR dimerization remain obscure (25).
Rho in the retina presents a unique opportunity to study the di/oligomerization problem. Rho has a high (mm) concentration in specific compartments called rod outer segments (ROS) allowing advanced high resolution imaging techniques to be used. Initial biophysical techniques were complicated by insufficient resolution of Rho oligomers in native membranes (26, 27). However, the development of cutting edge, high resolution imaging techniques such as atomic force microscopy (AFM) has revealed the existence of densely packed rows of Rho dimers in native rod photoreceptor disk membranes (1, 28–30), which these could be extracted as separate dimers or entire rows of dimers with mild detergents (31). The initial study involving cryoelectron microscopy lacked the resolution required to image Rho (32), but advances in this technique have changed the situation. The AFM findings were confirmed by cryoelectron tomographic studies of Rho organization in intact rod photoreceptors (33). These seminal studies identified tracks, comprised of rows of Rho dimers aligned parallel to disk membrane incisures, which together with precomplexed Gt could account for the biphasic kinetics of phototransduction needed for rapid signaling. This especially important observation identified Rho oligomerization by the mildest method currently available without the isolation of cellular membranes.
Rho self-associates with an estimated 1/Keq = 1010 molecules/μm2, implying that at its density in ROS, 87% of the total Rho population would be found in a dimeric complex (34). However, only limited evidence is available regarding the involved Rho self-interacting interfaces. As reported for several GPCRs including Rho, specific TM helices are likely involved in the formation of contacting surfaces within the dimer. An initial model of the Rho oligomer organization was derived from the AFM image of a Rho oligomer found within disk membranes isolated from mouse eyes. This model suggests that helices TM4 and TM5 form a primary dimer interface, whereas TM1 and TM2 are involved in weaker interactions between rows of dimers (1, 30). Direct structural evidence of Rho dimerization emanating from two- and three-dimensional crystallography of Rho in its photoactivated states together with opsin indicates an interface involving TM1, TM2, and H8 (17, 35, 36). Moreover, x-ray structures of several other GPCRs also have revealed the existence of two dimer interfaces, a primary interface including TM1, TM2, and helix H8 with a secondary interface involving either TM4 and TM5 found in the structure of the β1-adrenergic receptor (37) or TM5 and TM6 in chemokine type 4 (CXCR4) and μ-opioid receptors (38, 39). The presence of two distinct interfaces in crystal structures of several GPCRs other than Rho raises the possibility that the formation of oligomer arrays could result from the common feature of interfaces embedded into GPCR receptor sequences. However, different dimerization interfaces also could exist due to the dynamic equilibria of these receptors (40) and/or possible activation-dependent rearrangements of TM domains (41).
Different strategies have been used to map the GPCR dimer interface and its functional significance. These strategies include: (i) structure-based molecular modeling to predict residues potentially involved in dimer formation (42–44); (ii) cysteine mutagenesis of amino acids predicted to be involved in formation of the dimer followed by cross-linking, which successfully mapped several amino acids at the dimer interface of opsin, dopamine D2, and serotonin 5HT2c receptors (45–47); and (iii) disruption of the dimer interface by mutation of certain dimer-contacting residues (48, 49) or with synthetic peptides derived from selected TM domains, resulting in specific alterations of receptor function, which for example was demonstrated for CXCR4 and formyl peptide receptors targeting TM4 (50, 51) and for β2AR and leukotriene B4 (BLT1) receptors targeting TM6 (52, 53). However, TM peptides corresponding to TM6 and TM7 of the cholecystokinin receptor inhibited receptor dimerization without altering its function, as assessed by ligand binding and agonist-stimulated intracellular calcium concentration assays (25).
To define the physiological role of the Rho dimer, we experimentally disrupted dimer contacts with synthetic peptides corresponding to specific transmembrane domains. The disruptive effects of these TM peptides were evaluated first in vitro by cross-linking, gel filtration, mass spectrometry and a label-free analysis and then in live cells by employing the BRET assay. The effect of selected TM peptides on the function of Rho was tested in vitro by a tryptophan (Trp) fluorescence-based Gt activation assay and in cultured cells with a cAMP accumulation assay. Our results indicated a reduced Rho dimerization in the presence of peptides corresponding to TM1, TM2, TM4, and TM5, confirming the existence of two dimerization interfaces. The binding of Gt to photoactivated Rho promoted and/or stabilized Rho self-association, an observation implying that Rho dimerization enhances its in vivo function. However, disruption of the dimer contact interface decreased the thermal stability of Rho without affecting the rates of its G protein activation.
Materials and Methods
Chemicals
Asp-N endoproteinase was purchased from Sigma. Bradford ULTRA was obtained from Novexin (Cambridge, United Kingdom). Coelenterazine h, purchased from NanoLight Technology (Pinetop, AZ), was dissolved in ethanol to make a 5 mg/ml stock solution stored at −80 °C. The cAMP Direct Biotrak EIA kit was purchased from GE Healthcare Life Sciences. Forskolin, bought from Tocris Bioscience (Bristol, United Kingdom), was dissolved in DMSO to obtain a 24 mm stock solution, stored at −20 °C. GTPγS and 9-cis-retinal were purchased from Sigma. n-Dodecyl-β-d-maltoside (DDM) was obtained from Affymetrix Inc. (Maumee, OH). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), ProLong Gold mounting solution and 4′,6-diamidino-2-phenylindole (DAPI) were purchased from Life Technologies, Inc. Ro-20-1724 was obtained from Calbiochem.
Peptide Synthesis
Peptides derived from bovine Rho transmembrane domains were custom synthesized (EZBbiolab, Carmel, IN), and their primary sequences are shown in Table 1. A highly polar tag sequence was added to the N or C termini of TM peptides to improve their solubility and monitor their insertion into a lipid bilayer (54). TM peptides were acetylated at the N terminus or amidated at the C terminus to increase their propensity to form helices. In TM4, TM5, and TM6, Cys residues were substituted by Ser residues to prevent the formation of disulfide bonds and peptide aggregation. A Trp residue was added to the N terminus of TM2 and the C termini of TM5 and TM7 to evaluate the insertion of TM peptides into detergent micelles with fluorescence spectroscopy. The sequences and homogeneity of synthetic peptides were verified by mass spectrometry.
TABLE 1.
Amino acid sequences of synthetic peptides derived from the transmembrane domains of bovine Rho
| TM peptide | Bovine sequencesa | Mol. massb | Similarity (identity) to mouse sequencec |
|---|---|---|---|
| Da | % | ||
| TM1 | SKSKSKWQFSMLAAYMFLLIMLGFPINFLTLY-NH2 | 3789.70 | 100.0 (96.2) |
| TM2 | Ac-WYILLNLAVADLFMVFGGFTTTLYTSKSKSK | 3558.23 | 100.0 (100.0) |
| TM3 | SKSKSKNLEGFFATLGGEIALWSLVVLAI-NH2 | 3080.08 | 100.0 (100.0) |
| TM4 | Ac-ENHAIMGVAFTWVMALASAAPPLVGWSKSKSK | 3428.07 | 92.3 (88.5) |
| TM5 | SKSKSKNESFVIYMFVVHFIIPLIVIFFSYGQLVFW-NH2 | 4314.25 | 93.1 (89.7) |
| TM6 | Ac-AEKEVTRMVIIMVIAFLISWLPYAGVAFYIFSKSKSK | 4281.24 | 90.3 (90.3) |
| TM7 | SKSKSKFMTIPAFFAKTSAVYNPVIYW-NH2 | 3111.73 | 100.0 (80.0) |
| TM4 scrambled | Ac-VNFEAAPMLAMWIAAVPLSGHAGTWVSKSKSK | 3428.07 | NAd |
a Residues that differ from those in native bovine rhodopsin are underlined. In TM2, TM5, and TM7, Trp residues were added at the N or C terminus. In TM4 and TM6, Cys residues were replaced by Ser residues. A highly polar tag, SKSKSK (bold face), was added at the N terminus of odd-numbered TM helices and at the C terminus of even-numbered TM helices.
b Molecular mass.
c Local similarities and identities (in parentheses) of TM peptide sequences were calculated with EMBOSS Matcher in the default mode, which employs a rigorous algorithm based on Pearson's align application, v. 2.0u4 (90).
d NA, not applicable.
Fluorescence Spectroscopy
To determine the insertion of TM peptides into DDM micelles, fluorescence measurements of fractions obtained from gel filtration of TM peptides were carried out at room temperature with a PerkinElmer L55 luminescence spectrophotometer at 20 °C. Emission spectra were recorded between 300 and 480 nm after excitation at 260 nm, with excitation and emission slit bands set at 5 and 10 nm, respectively.
Purification of Rho
Bovine ROS membranes were prepared from fresh retinas under dim red light (55). Samples were either treated with TM peptides or solubilized in DDM and used for Rho purification by a ZnCl2-opsin precipitation method (56). ZnCl2 then was removed by means of a 48-h dialysis in 10 mm bis-Tris propane (BTP), 100 mm NaCl, and 0.2 mm DDM, pH 7.5. Rho concentrations were measured with a UV-visible spectrophotometer (Cary 50, Varian, Palo Alto, CA) by using the absorption coefficient ϵ500 nm = 40,600 m−1cm−1 (57).
Treatment of Rho in ROS Membranes with TM Peptides
To assess the effect of different TM peptides on Rho dimerization, synthetic TM peptides were first dissolved in a 2.5 mm stock solution of DMSO and then diluted with 20 mm BTP, 100 mm NaCl, and 1 mm DDM, pH 7.5, to a final concentration of 500 μm. After a 30-min incubation at room temperature, single TM peptides or their mixtures (prepared in a 1:1 ratio) were added to ROS membranes resuspended in 20 mm BTP, 100 mm NaCl, pH 7.5, to achieve final concentrations of Rho and TM peptides of 25 and 250 μm, respectively, in 0.2 mm DDM. Alternatively, ROS were treated with increasing concentrations of a four-peptide mixture (TM1, TM2, TM4, and TM5) ranging from 31.25 to 250 μm. Following a 1-h incubation at room temperature, the effect of TM peptides on Rho dimers was assessed by either gel filtration or cross-linking.
Cross-linking of ROS Membranes
After 1 mm disuccinimidyl glutarate (DSG) cross-linker (Thermo Scientific, Waltham, MA) was added to ROS samples treated with TM peptides, the cross-linking reaction was allowed to proceed for 2 h on ice. The reaction was terminated with 1 m Tris-HCl, pH 8.0, added to a final concentration of 50 mm and incubated for 15 min. Cross-linked Rho (10 μg) was separated on a 10% SDS-PAGE, and the abundance of the dimer was evaluated by the densitometry of the protein bands.
Gel Filtration
ROS membranes incubated with TM peptides were solubilized with 10 mm DDM and, after centrifugation at 100,000 × g for 30 min, samples were loaded onto a Superdex 200 10/300 GL (GE Healthcare Life Sciences) gel filtration column equilibrated with 20 mm BTP, 100 mm NaCl, and 1 mm DDM, pH 7.5. Separation profiles of Rho treated with TM peptides were compared with those of untreated Rho.
Mass Spectrometry
Mass spectrometry analyses were performed to determine the identities of TM peptides in Rho samples treated with TM peptides and purified by gel filtration. TM peptide standards and Rho samples treated with TM peptides were analyzed with a LTQ Velos mass spectrometer (Thermo Scientific) equipped with an electrospray ionization source operated in a positive mode. The capillary temperature was set to 350 °C. The mass spectrometer was coupled to an Accela 600 chromatography system equipped with an Accela Autosampler (Thermo Scientific). The chromatography setup employed a two-pump-controlled reverse-phase elution system with an aqueous phase composed of 0.1% formic acid in H2O (A) and an organic phase composed of 0.1% formic acid in acetonitrile (B). Peptide standards and Rho samples (∼0.2 μg of standard and 15 μl of Rho 40 μg/ml, respectively) were loaded onto an Onyx Monolytic C18 100 × 3.0-mm column (Phenomenex, Torrance, CA) equilibrated previously with 98% A and 2% B for 20 min. Peptides were eluted with the following gradients: 0–4 min, 98% A and 2% B; 4–48 min, 2% A and 98% B; 48–50 min, 98% A. MS2 spectra were collected by use of collision-induced dissociation with the normalized collision energy set to 35 kV. Selected ion monitoring was set up for the following ions: TM1, m/z = 1263.50; TM2, m/z = 890.30; TM3, m/z = 1026.90; TM4, m/z = 1143.40; TM5, m/z = 1079.30; TM6, m/z = 1070.80; TM7, m/z = 778.70; scrambled TM4, m/z = 1143.72. Spectra were analyzed and interpreted with Xcalibur software (version 2.1.0.1139).
Binding of TM Peptides to Immobilized Rho
To evaluate the binding of TM peptides to Rho, we also used a label-free assay that measures changes in the refractive index upon the addition of a ligand (58). Rho (75 μg/ml) dissolved in 20 mm HEPES, pH 8.0, containing 1 mm DDM was immobilized on the surface of an EnSpire-LFB 384-well plate (a label-free biochemical sensor plate with amine coupling preactivated (PerkinElmer Life Sciences)) for 1 h at room temperature before overnight incubation at 4 °C. Next day, excess Rho was removed, and the plate was washed four times with PBS (10 mm Na2HPO4, 1.76 mm KH2PO4, pH 7.4, 137 mm NaCl, and 2.7 mm KCl) containing 0.4 mm DDM and 0.08% DMSO. After a 2-h equilibration at room temperature, the baseline was measured, and then various dilutions of TM peptides dissolved in the same buffer were dispensed into the plate containing immobilized Rho and the binding signal recorded with 30 replicates by an EnSpire multi-mode plate reader (PerkinElmer Life Sciences).
Gt Activation
Gt was purified after extraction from ROS membranes isolated from 200 dark-adapted bovine retinas, as described previously (59, 60). The activation properties of Rho treated or untreated with TM peptides and separated by gel filtration were tested in the Trp fluorescence Gt activation assay. Gt was mixed with Rho at a 10:1 ratio, with Gt at 250 nm and Rho at 25 nm concentrations, and the sample was bleached for 30 s with a Fiber-Light illuminator (Dolan Jenner Industries Inc., Boxborough, MA) through a band-pass wavelength filter (480–520 nm) followed by a 5-min incubation with continuous low-speed stirring. The intrinsic fluorescence increase from Gtα upon the addition of 5 μm GTPγS was measured with a PerkinElmer LS 55 luminescence spectrophotometer employing excitation and emission wavelengths of 300 nm and 345 nm, respectively (61–63). No signals from Rho without Gt were detected in the control experiments.
ROS Cleavage with Asp-N Endoproteinase
ROS membranes (2 mg/ml) suspended in 10 mm Tris-HCl, pH 7.5, were treated with Asp-N endoproteinase (Sigma) at 0.06 μg/ml concentration for 16 h at room temperature to obtain about 50% digestion of Rho. Adding 1 mm DTT and 1 mm EDTA terminated this reaction. Membranes were solubilized by adding DDM to a final concentration of 10 mm followed by a 1-h incubation at room temperature on a rotating platform and then centrifugation at 100,000 × g for 30 min. The supernatant then was incubated with 1D4 immunoaffinity resin equilibrated with 20 mm BTP, 120 mm NaCl, and 2 mm DDM, pH 7.5, for 1 h at room temperature. ΔRho329 was collected in the flow-through fraction, and full-length Rho, after an extensive resin wash, was eluted from the resin with the same buffer containing 1D4 peptide (0.6 mg/ml) derived from the Rho C terminus. Equal amounts of ΔRho329 and full-length Rho were mixed and then combined with Gt in about a 1:1 molar ratio. Additionally, a mixture of equal amounts of TM1, TM2, TM4, and TM5 peptides was added. These samples were illuminated through a 480–520 nm band-pass wavelength filter for 5 min and then incubated with 1D4 affinity resin for 1 h at room temperature. After a wash with 20 mm BTP, 120 mm NaCl, and 2 mm DDM, pH 7.5, and elution with 20 mm BTP, 120 mm NaCl, and 2 mm DDM, pH 7.5, containing 1D4 peptide (0.6 mg/ml), the eluents were analyzed by immunoblotting with the N-terminal B6-30 anti-Rho antibody, and the intensity of protein bands was quantified by densitometric analysis.
Stability
Five μm DDM-solubilized Rho dissolved in 1 ml of 20 mm BTP, 120 mm NaCl, and 1 mm DDM, pH 7.5, was mixed with TM peptides added from 2.5 mm DMSO stock solutions. Mixtures of four peptides, namely TM1, TM2, TM4, and TM5, or peptide pairs of either TM1/TM2 or TM4/TM5 were added to Rho samples at 2.5 μm each. In the control experiment, an equivalent concentration of either DMSO or TM7 was used. Samples were incubated at 55 °C in the dark, and their spectra were recorded every 15 min for 2 h. To determine the effect of TM peptides on Rho stability, the absorbance at 504 nm was plotted against time. Each sample incubated with TM peptides was compared with a control sample without TM peptide, and all samples were measured in triplicate.
Generation of a Stable Opsin-expressing HEK-293 Cell Line and Cell Culture
Constructs of mouse opsin fused to Venus (mOpsin·Venus) and Renilla luciferase (mOpsin·Rluc) in the pCDNA3.1Zeo vector were a generous gift from Dr. N. A. Lambert (Georgia Regents University, Augusta, GA). A stable HEK-293 cell line expressing both mOpsin·Rluc and mOpsin·Venus was generated by sequential incorporation of the mOpsin·Rluc pCDNA3.1Zeo and mOpsin·Venus pCDNA3.1Zeo by Zeocin selection (500 μg/ml) for Rluc fluorescence and flow cytometry selection for Venus fluorescence, respectively. Expression of the mOpsin·Rluc fusion protein was confirmed by immunoblots with rabbit anti-Rluc antibody (Medical and Biological Laboratories Co., Ltd., Nagoya, Japan) and mouse monoclonal B630 anti-Rho antibody (molecular mass of mOpsin·RLuc, 75 kDa). Expression of mOpsin·Venus fusion protein was confirmed by immunoblots with the B630 anti-Rho antibody (molecular mass mOpsin·Venus, 66 kDa), and by Venus fluorescence under a fluorescent microscope.
HEK-293 (mOpsin·Rluc and mOpsin·Venus) stable cells were cultured in DMEM with 10% FBS (Hyclone, Logan, UT), 5 μg/ml plasmocin (InvivoGen, San Diego, CA), and 1 unit/ml penicillin with 1 μg/ml streptomycin (Life Technologies) at 37 °C under 5% CO2 per instructions from the ATCC® Animal Cell Culture Guide.
Immunoblotting
HEK-293 (mOpsin·Rluc and mOpsin·Venus) stable cells were collected, pelleted at 1,000 × g, and washed twice with PBS. Cell pellets were resuspended in PBS supplemented with Complete protease inhibitor mixture (Roche Diagnostics) containing 10 mm DDM to solubilize membranes followed by sonication at room temperature in a water bath for 5 min. This solution then was centrifuged at 16,000 × g for 15 min at 4 °C. Fifty μg of total cell extract was separated on a 10% SDS-PAGE followed by transfer onto a PVDF membrane. The PVDF membrane, after blocking with 5% unsaturated milk, was incubated with B6-30 anti-Rho monoclonal antibody and anti-Rluc polyclonal antibody, each at a dilution of 1:1000. Immunoblots were developed with a chemiluminescent substrate for goat anti-mouse- or anti-rabbit-coupled horseradish peroxidase antisera. Total expression and the ratio of both mOpsin·Rluc and mOpsin·Venus proteins were evaluated by densitometric analyses of the protein bands. A 2.5–10-ng calibration curve was prepared by using serial dilutions of a known amount of Rho standard.
Immunostaining
HEK-293 cells expressing mOps·Rluc and mOps·Venus were cultured overnight on a coverslip coated with poly-l-lysine (Sigma). Cells were washed once with PBS and then fixed with 4% paraformaldehyde for 20 min followed by three washes with PBS with 0.1% Triton X-100. Next, cells were incubated with 10% goat serum in PBS with 0.1% Tween 20 (PBST) for 30 min and washed again three times with PBST. Then cells were incubated with rabbit anti-Rluc polyclonal antibody at a 1:1000 dilution overnight at 4 °C. After three washes with PBST, the cells were incubated for 1 h at room temperature with a 1:200 dilution of the secondary antibody labeled with Cy3-conjugated goat-anti-rabbit IgG antibody (Invitrogen). After cells were washed three times with PBST, a 20-μl drop of Prolong Gold mounting solution with DAPI (Life Technologies) was placed on a glass slide before the coverslip was positioned on top. Images were taken with a Leica upright fluorescence microscope. Three channels were used to obtain fluorescent images from Cy3 (200 ms), Venus (200 ms), and DAPI (15 ms).
The BRET Assay
On day 1, HEK-293 (mOpsin·Rluc and mOpsin·Venus) stable cells were detached from the tissue culture flask by 0.05% trypsin and resuspended in culture medium. Cells were counted with a hemocytometer (Thermo Scientific) and then diluted to 80 × 104 cells/ml. The cell diluent was dispensed into a Falcon 96-well tissue culture plate (Corning Life Sciences) at 200 μl/well with a multichannel pipette. The plate was centrifuged at 300 × g for 30 s and cultured at 37 °C with 5% CO2 and 90% humidity. On day 2, each well of the 96-well plate was treated with 10 μl of 200 μm 9-cis-retinal dissolved in culture medium to achieve a 10 μm final concentration. The plate was covered with aluminum foil and cultured overnight at 37 °C in 5% CO2 with 90% humidity. On day 3, TM peptides were freshly diluted from 2.5 mm DMSO stock solutions to a 50 μm concentration with PBS containing 10 mm DDM and incubated for 30 min at room temperature to allow helix formation. Four μl of these solutions was added to achieve a final concentration of 1 μm TM peptide and 0.2 mm DDM in the cell cultures. Cells were treated either with single peptides or with a combination of TM1/TM2, TM4/TM5, or all four of these peptides added equally, each at 0.25 μm to achieve a final concentration totaling 1 μm or 1 μm of each peptide. Alternatively, the cells were treated with decreasing concentrations of TM peptides (1.5–0.01 μm) to determine the half-maximal concentration (EC50) of TM peptides disrupting the Rho dimer. Each condition was replicated four times. The plate with peptide-treated cells was incubated at 37 °C for 1 h in the dark. Then under dim red light, the culture medium was aspirated and replaced with 200 μl PBS/well. Cells were resuspended and transferred from the 96-well tissue culture plate to a white-walled opaque 96-well plate (Corning Life Sciences). Coelenterazine h was diluted to 25 μm in PBS. Each well of the 96-well plate was injected with 25 μl of diluted coelenterazine h followed by dual luminescence readings at 480 and 530 nm, 5 s after each injection, by the SpectraMax plate reader with the BRET1 filter set (Molecular Devices, Sunnyvale, CA). The BRET1 signal was calculated as the emission ratio at 530 compared with 480 nm. To compare and average three independent experiments, the BRET signal was scored with the following equation,
 |
where
BRETx is the BRET signal at a specific peptide concentration, BRET0 is the BRET signal at the lowest, non-effective peptide concentration, and BRET−100 is the BRET signal at the most effective peptide concentration.
cAMP Assay
HEK-293 (mOpsin·Rluc and mOpsin·Venus) stable cells were plated in 96-well plates at a density of 100,000 cells/well in 90 μl of medium. After 8 h, 9-cis-retinal was added to each well to achieve a final concentration of 10 μm. The plates then were wrapped with aluminum foil and kept in a cell culture incubator overnight. The next morning, TM peptides prepared identically to those for the BRET assay were added to these cells to achieve a 1 μm final concentration, and the plates were incubated at 37 °C for 1 h. Next, a phosphodiesterase inhibitor (Ro-20-1724) was added to each well at a final concentration of 100 μm, and the plate was incubated for 20 min at room temperature. Then, forskolin (5 μm final concentration) was added to each well, after which one plate was immediately illuminated with bright light (150-watt bulb) for 15 min at room temperature while the second plate was kept in the dark. The levels of accumulated cAMP were detected with anti-cAMP antiserum from the cAMP Direct Biotrak EIA kit (GE Healthcare Life Sciences) and the absorbance readout at 630 nm by the Flexstation plate reader (Molecular Devices) at the final step of the assay, according to the protocol provided.
Cell Survival Evaluation
The cytotoxicity of each TM peptide was examined with the MTT cell proliferation assay. HEK-293 (mOpsin·Rluc and mOpsin·Venus) stable cells were seeded in a 96-well plate at 50,000 cells/well in 100 μl of medium. After 24 h, TM peptides at 1 μm concentration, or the 0.2 mm DDM control, were added to these cells and incubated at 37 °C for 1 h. Then, the medium containing TM peptides was replaced with fresh medium, and cells were cultured until the next day when 100 μl of phenol-free medium and 10 μl of the 12 mm MTT stock solution were added to each well, after which the cells were incubated at 37 °C for 4 h. Then, 100 μl of 0.1% SDS in 0.01 m HCl solution was added to each well and mixed thoroughly with the pipette, and the plate was incubated at 37 °C for another 4 h. Finally, each sample was mixed again, and the absorbance at 570 nm was read with a Flexstation plate reader (Molecular Devices).
Results
Design of TM Peptides
TM peptide sequences were derived from the transmembrane regions of the bovine Rho sequence (GenBankTM accession number AAA30674) (see Fig. 1; primary sequences are shown in Table 1). A highly polar tag with the SKSKSK amino acid sequence was added to the N termini of the odd-numbered TM helices and to the C termini of even-numbered TM helices to improve peptide solubility and control the direction of their insertion into the membrane bilayer (54). TM peptides were acetylated at the N terminus or amidated at the C terminus to increase their propensity to form helical structures. In TM4 and TM6, the Cys residues were replaced with Ser residues to avoid the formation of internal disulfide bonds. When absent in the peptide sequence, a Trp residue was added to the N terminus or C terminus of the peptide to allow evaluation of the TM peptide insertion into micelles by fluorescence spectroscopy. This strategy included TM2, TM5, and TM7. Additionally, the aromatic amino acids, including the Trp residues, have a propensity to localize at the membrane-water interfaces, where they provide a driving force for membrane protein folding and stability (64).
FIGURE 1.
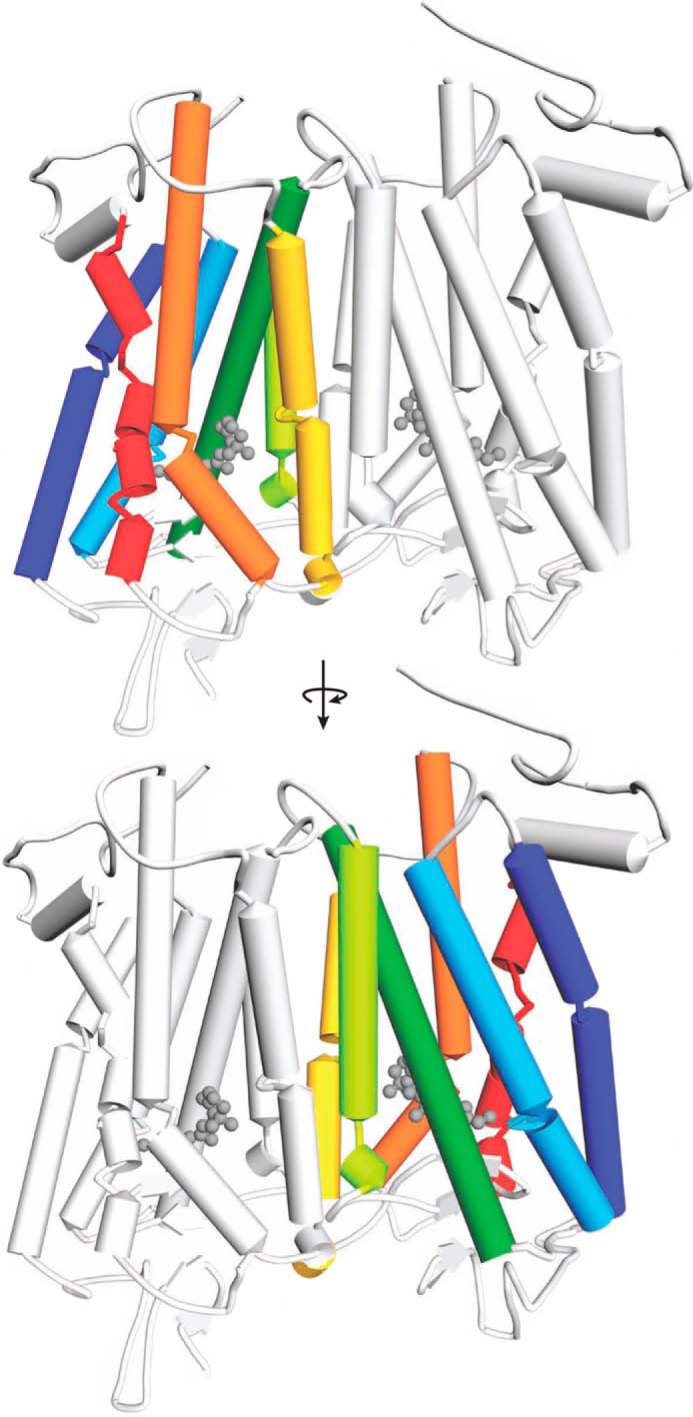
Model of the Rho dimer. Transmembrane helices in each Rho monomer are colored as follows: TM1, dark blue; TM2, light blue; TM3, dark green; TM4, light green; TM5, yellow; TM6, orange; and TM7, red. The same color code is also used in Figs. 2–5 and 7–9. The dimer-contacting surface is based on modeling studies involving helices TM4 and TM5 (28). The second monomer within the Rho dimer is colored light gray.
We used TM-derived peptides to determine the interaction surface within the Rho dimer. Because of the high complexity of protein-protein and protein-peptide interactions, we used multiple complementary methods to evaluate the effects of TM peptides on Rho dimerization.
Binding of TM Peptides to Rho
The binding of the reconstituted TM peptides to Rho was examined by two independent techniques, initially by evaluating TM peptide co-migration with Rho by size exclusion chromatography and then by the label-free assay, to assess the binding of TM peptides to immobilized Rho. Each TM peptide was incubated with Rho in ROS and then solubilized with DDM. TM peptide-Rho mixtures or TM peptides alone were loaded on a S200 gel filtration column equilibrated with buffer containing 1 mm DDM. Because of their high hydrophobicity, TM peptides associated with detergent micelles, and with their Trp fluorescence used as a marker, these were detected in almost all fractions eluted from the gel filtration column when loaded alone. Thus, to assure the specificity of the TM peptide-Rho interaction, equivalent fractions of TM peptides alone separated by gel filtration and incubated with Rho were analyzed by mass spectrometry. Although detectable by their Trp fluorescence, none of the TM peptides were detected upon gel filtration alone, indicating that their concentrations were below the MS detection limit. However, in the Rho samples eluted from the gel filtration column, ions identified as TM1, TM2, TM3, TM4, and TM5 were present, indicating that these peptides associated with full-length Rho and that these interactions increased the TM peptide concentrations in fractions corresponding to the Rho peak (Fig. 2). In contrast, peptides corresponding to TM6 and TM7 were not detected in the Rho samples, suggesting that these peptides were less likely to be involved in forming the interaction surface between Rho molecules (Fig. 2). Moreover, the scrambled TM4 peptide was not detected as well. To confirm these results, we also performed a label-free binding assay of the TM peptides to plate-immobilized Rho. By using various dilutions of TM peptides, we not only identified those TM peptides that bound to Rho but also determined their dissociation constants (Kd) (Table 2 and Fig. 3). Five of the TM peptides (TM1–5) demonstrated effective binding to immobilized Rho; TM4 had the greatest affinity, with a calculated apparent Kd of 0.47 ± 0.27 μm. However, binding of TM6, TM7, and the scrambled TM4 to immobilized Rho was not detected (Table 2 and Fig. 3).
FIGURE 2.

Analysis of Rho-TM peptide complexes by mass spectrometry. Rho samples incubated with TM peptides and purified by gel filtration were analyzed by mass spectrometry. TM1, TM2, TM3, TM4, and TM5 were co-purified with Rho. Their LC-MS elution profiles are shown as colored chromatograms, and their masses were identical to those of authentic peptide standards (black chromatograms). However, TM6, TM7, and scrambled TM4 were not detected in the Rho samples. Sequences of Rho-derived identified TM peptides are indicated with the appropriate color (as shown in Fig. 1). The scrambled TM4 sequence is colored gray. Substituted Cys → Ser and added Trp residues are underlined. The SKSKSK polar tag is shown in black.
TABLE 2.
Binding of TM peptides to immobilized Rho
Freshly prepared TM peptides in concentrations ranging from 0.006 to 2 μm were added to an EnSpire-LFB 384-well plate with Rho immobilized at the bottom of each well. Binding responses were measured over a time course by an EnSpire plate reader integrated with a Corning label-free module. Binding responses were stabilized for about 30 min after peptides were added, and 10 data points were averaged to obtain the binding response for each well. Averages and standard deviations of the binding responses calculated from three repeats of each peptide concentration were plotted as y values and error bars in the binding chart, with the concentrations of peptide tested as the x values. Binding curves were fitted by the Hill function from Origin 8.1 software. NA, not associated.
| TM peptides | Kd |
|---|---|
| μm | |
| TM1 | 0.78 ± 0.44 |
| TM2 | 1.18 ± 0.53 |
| TM3 | 0.64 ± 0.13 |
| TM4 | 0.47 ± 0.27 |
| TM5 | 0.62 ± 0.03 |
| TM6 | NA |
| TM7 | NA |
| TM4 scrambled | NA |
FIGURE 3.

Binding of TM peptides to immobilized Rho. Examples of the binding response curves are shown for TM4, TM5, and scrambled TM4. Rho was immobilized onto an EnSpire-LFB 384-well plate, and binding responses of TM peptides delivered to the plate wells in concentrations ranging from 0.006 to 2 μm were measured with an EnSpire plate reader integrated with a Corning label-free module. After reactions reached equilibria, the responses from 10 measurements were averaged for each data point. Averaged data from three independent experiments were plotted as y values with corresponding concentrations of tested peptide as x values. Binding curves were fitted by the Hill function with Origin 8.1 software.
Effect of Rho-derived TM Peptides on the Rho Size Exclusion Chromatography Profile
To assess the effects of TM peptides on Rho oligomeric organization, Rho samples incubated with TM peptides were loaded on a S200 gel filtration column equilibrated with buffer containing 1 mm DDM, which retains the dimeric organization of Rho (65). Elution profiles of Rho samples treated with and without TM peptides and monitored at 280 nm indicated changes in Rho migration in the presence of most of the TM peptides (Fig. 4). TM4 caused the most pronounced shift in the protein migration toward smaller molecular masses, with less prominent shifts noted for TM1, TM2, TM3, and TM5. A small change in Rho migration toward a higher molecular mass was exhibited by TM6, whereas no shift was observed for the TM7 peptide and the scrambled TM4 peptide. However, the largest difference in the Rho elution profile was observed in the presence of a higher (3 mm) DDM concentration, which fully disrupted Rho dimerization and shifted the equilibrium toward Rho monomers. These changes in the Rho gel filtration elution profile indicate that TM peptides not only associated with Rho but also affected its oligomeric organization, shifting the equilibrium toward the monomeric state. However, increasing the concentration of DDM was far more disruptive than TM peptides in this respect.
FIGURE 4.

Changes in Rho gel filtration profile upon binding of TM peptides. Recorded at 280 nm, gel filtration elution profiles of bovine Rho were carried out in buffer containing 1 mm DDM to maintain the Rho dimeric state both in the absence (black lines) and presence (colored lines, code as shown in Fig. 1) of TM peptides. An increased concentration of DDM (3 mm) shifted Rho from the dimeric to the monomeric state in the control (bottom row, middle panel) experiment.
Effect of Rho-derived TM Peptides on Rho Cross-linking
Rho molecules have a propensity to self-associate, forming both dimers and oligomers (66). Chemical cross-linking of Rho in ROS membranes caused an increased level of Rho dimers and a decreased level of monomers (Fig. 5A). Because the TM regions of GPCRs can interact at the dimer interface, we tested whether the synthetic peptides that share identity with amino acid sequences derived from specific TM domains could affect dimer formation. Peptides corresponding to TM1 or the TM1/TM2 pair had a modest effect in preventing Rho cross-linking (Fig. 5, A and B). A greater dimer disruptive effect was observed for TM4 and TM5 and a mixture of both. An even more pronounced effect in preventing dimer formation during cross-linking was found when all four peptides, namely TM1, TM2, TM4, and TM5, were incubated with Rho. These results support the idea that multiple GPCR dimer interfaces exist. Moreover, the blocking of dimer formation was dose-dependent. Increasing concentrations of the TM1, TM2, TM4, and TM5 mixture resulted in a gradual reduction of the dimer from ∼65 to 40%, accompanied by an increase in monomer levels (Fig. 5, C and D). No changes in Rho cross-linking were observed in the presence of TM3, TM6, and TM7 (Fig. 5, A and B) or scrambled TM4 (Fig. 5E), indicating that the disruptive effect of TM peptide on the Rho dimer is sequence-specific.
FIGURE 5.

Effect of TM peptides on Rho cross-linking. A, effects of TM peptides on formation of the DSG-cross-linked Rho dimer. ROS membranes (25 μm Rho)were incubated with TM peptides (final concentration, 250 μm) and then cross-linked in the dark with DSG. Ten μg of Rho was loaded on each SDS-PAGE. B, quantification of DSG-cross-linked dimers. The abundance of DSG-cross-linked Rho dimers was calculated from densitometric analyses of protein bands corresponding to the Rho monomer and dimer from three independent experiments. Results are presented as means ± SD. Asterisks indicate a statistically significant decrease (p < 0.05, Student's t test) in Rho dimer formation. C, dose-dependent effects of TM peptides (TM1, TM2, TM4, and TM5 mixed in equal amounts) on Rho DSG cross-linking. ROS membranes (25 μm Rho) were incubated with increasing concentrations of a TM1, TM2, TM4, and TM5 peptide mixture (31.25, 62.5, 125, and 250 μm) and then cross-linked in the dark with DSG. Ten μg of Rho was loaded on each SDS-PAGE. D, densitometric analyses and quantification of the Rho dimer from gels shown in C are plotted here. E, comparison of the effects of TM4 and the scrambled TM4 peptides on formation of the DSG-cross-linked Rho dimer. The abundance of DSG-cross-linked Rho dimers was calculated from densitometric analyses of protein bands corresponding to the Rho monomer and dimer from three independent experiments. Results are presented as means ± SD.
Effect of Rho-derived TM Peptides on Rho BRET
GPCR dimerization in living cells can be demonstrated using bioluminescence resonance energy transfer between two populations of a given receptor tagged with either a fluorescent protein or Rluc expressed in the same cells. To generate a reliable basal BRET signal due to Rho dimerization, we generated a stable cell line, namely HEK-293 (mOpsin·Rluc and mOpsin·Venus) cells expressing both mouse Opsin·Rluc and mouse Opsin·Venus. Expression of these markers was detected by immunoblot analyses with a specific monoclonal antibody against the Rho N terminus and a polyclonal antibody against Rluc (Fig. 6A). All results were confirmed by fluorescence microscopy. Both mOpsin·Rluc and mOpsin·Venus co-localized at the plasma membrane (Fig. 6B). Densitometric analysis of the protein bands revealed a donor to acceptor (mOpsin·Rluc to mOpsin·Venus) expression ratio of 1 to 2.4 ± 0.4 (Fig. 6A). This ratio indicated an excess of the acceptor commonly used for the BRET assay with transiently expressed receptors (67). To determine whether the increase in the BRET signal resulted from Rho dimerization, we first did a control experiment in HEK-293 cells transiently transfected with vectors only expressing mOpsin·Rluc (donor) or with vectors expressing both mOpsin·Rluc (donor) and mOpsin·Venus (acceptor) as a positive control and mOpsin·Rluc (donor) and Kras·Venus (acceptor) as a negative control (Fig. 6C). The BRET increase in cells expressing mOpsin·Rluc and Kras·Venus was due to co-localization of the BRET donor and acceptor on the cell membrane, but this increase was significantly lower than the increased BRET signal in cells co-expressing mOpsin·Rluc and mOpsin·Venus due to Rho dimerization (Fig. 6C, light gray bars). In the stable HEK-293 (mOpsin·Rluc and mOpsin·Venus) cell line, a much higher BRET signal was detected due to Rho dimerization (Fig. 6C, left, dark gray bar), and this was disrupted with increasing concentrations of DDM (Fig. 6C, right, dark gray bar, and D).
FIGURE 6.

Stable expression of mOpsin·Rluc and mOpsin·Venus in HEK-293 cells. A stable cell line, namely HEK-293 (mOpsin·Rluc and mOpsin·Venus), was generated by sequential genome integration of mOpsin·Rluc and mOpsin·Venus. A, expression of mOpsin·Rluc and mOpsin·Venus was assessed in 50 μg of total protein cell lysate by immunoblotting with a monoclonal antibody recognizing the N terminus of Rho (B6-30) and a polyclonal antibody against Rluc, respectively. The ratio of mOpsin·Rluc to mOpsin·Venus was calculated to be 1:2.4. Note that the sample was run on the same gel and transferred onto PVDF membranes developed individually with different antibodies. B, membrane localization of mOpsin·Venus was determined by detecting Venus fluorescence, and mOpsin·Rluc was detected by immunostaining with anti-Rluc antibody. Merging of the two images indicates co-localization of both receptors. C, specificity of the BRET signal. Left, light gray vertical bars: the BRET signal was recorded in HEK-293 cells transiently transfected with vectors expressing mOpsin·Rluc (donor) only or mOpsin·Rluc (donor) with Kras·Venus (acceptor) used as a negative control (here the BRET increase was due to co-localization of the BRET donor and acceptor on cell membranes), and mOpsin·Rluc (donor) and mOpsin·Venus (acceptor) (in this case, the BRET increase was due to Rho dimerization, an effect greater than that achieved by co-localization). Right, dark gray vertical bars: the BRET signal was recorded in the stable HEK-293 (mOpsin·Rluc and mOpsin·Venus) cell line before and after treatment with 1 mm DDM. D, the decrease of the BRET signal with increasing concentrations of DDM shown is due to the disruption of opsin dimers.
To further probe the role of TM regions in Rho dimerization, we used a competitive BRET assay. Because of the high sequence homology (93%) between mouse and bovine Rho (Table 1), the same bovine Rho-derived TM peptides were employed in these experiments. Single peptides corresponding to TM1, TM2, TM4, and TM5 significantly inhibited the BRET signal of receptors present in both opsin and isorhodopsin (isoRho) conformations, the latter formed after regeneration with 9-cis-retinal chromophore (Fig. 7, A and B). The incubation of cells with mixtures of TM1 with TM2 and TM4 with TM5 peptides or all four TM peptides failed to enhance BRET inhibition. Moreover, the BRET signal was not affected in cells incubated with TM3, TM6, or TM7. Thus, only TM peptides derived from TM1, TM2, TM4, or TM5, predicted to be involved in the formation of the dimer interfaces, modulated the BRET signal of opsin and/or isoRho. Because of insolubility of these hydrophobic TM peptides, they were all reconstituted in a low concentration of DDM (achieving a 0.2 mm final concentration in cell culture) for the BRET assay. These low DDM concentrations did not inhibit the BRET signal (Fig. 6D) but actually caused a small increase (Fig. 7, A and B), possibly because of increased receptor internalization in the presence of an unfavorable environment. In fact, we observed increased amounts of vesicular structures near the cell membrane in cells treated with 0.2 mm DDM or TM peptides (Fig. 7C), whereas the overall membrane localization of opsin was not affected. Replacing the medium containing TM peptide with fresh medium after a 1-h treatment allowed cells to maintain their proper growth. Thus, their viability measured 24 h later was unchanged as compared with that of untreated cells (Fig. 7D). Disruption of the opsin and isoRho BRET signal with TM1, TM2, TM4, and TM5 was dose-dependent (Fig. 8). EC50 values calculated from dose-dependent curves fitted to experimental data points indicated that TM4 was the most potent inhibitor of Rho self-association (EC50 of 0.28 μm for opsin and 0.35 μm for isoRho) (see Fig. 8, C–F). TM5 inhibited opsin and isoRho BRET with an EC50 of 0.36 μm for opsin and 0.65 μm for isoRho. Similar EC50 values of ∼0.5 μm (see Fig. 8, A and B) were obtained for TM1 and TM2. The BRET signal was not affected in cells incubated with different doses of TM3, TM6, and TM7 and the scrambled TM4 peptides (Fig. 8, E and F). These results strongly indicate that the TM4 and most likely TM5 transmembrane regions provide the most pronounced receptor-receptor interacting surfaces.
FIGURE 7.

Effects of TM peptides on both opsin and isoRho dimerization in a cell membrane. HEK-293 (mOpsin·Rluc and mOpsin·Venus) cells were either incubated with 9-cis-retinal, resulting in a light-sensitive isoRho at the cell membrane (A), or the receptor remained in an opsin, light-insensitive state (B). The BRET signal was recorded after cells were treated with 1 μm TM peptides for 1 h at 37 °C in the dark. BRET signals were decreased by TM1, TM2, TM4, and TM5 but not by TM3, TM6, or TM7. Changes in cell morphology and Rho localization in cells treated with TM peptides or 0.2 mm DDM were monitored by fluorescence microscopy (C), and a cell proliferation assay was used to determine cytotoxicity (D). Statistically significant (p < 0.05, Student's t test) decreases in the BRET signal are indicated by an asterisk.
FIGURE 8.

Dose-dependent effects of TM peptides on both opsin and isoRho dimerization in live cells. Effects of increasing concentrations of TM peptides on theBRET signal. HEK-293 (mOpsin·Rluc and mOpsin·Venus) cells were either incubated with 9-cis-retinal resulting in a light-sensitive isoRho at the cell membrane (A, C, and E) or without 9-cis-retinal so that the receptor remained in its opsin, light-insensitive state (B, D, and F). The BRET signal was measured after cells were treated with TM peptides at concentrations ranging from 0.02 to 2 μm for 1 h at 37 °C. BRET signals derived from three independent experiments obtained for TM1, TM2, TM4, and TM5 were scored as described under “Materials and Methods,” and a dose-response curve was fitted by the Hill function in SigmaPlot software. Calculated half-maximal BRET signal inhibition values are presented as EC50 values (A–D). E and F, overlays of ΔBRET determined in TM peptide-treated cells indicate the inhibitory efficacies of these peptides. TM3, TM6, and TM7 peptides and the scrambled TM4 peptide had no inhibitory effect on the BRET signal.
Functional Consequences of Rho Dimerization
Because TM-derived peptides can modulate the activity of some GPCRs (50, 52, 53, 68), we investigated whether disruption of the Rho dimer interface with TM peptides affects Rho activation. First, we performed an in vitro Gt activation assay (Fig. 9A) with bovine Rho that was incubated with TM peptides and purified by gel filtration chromatography. Here we found no major differences in Gt activation rates for all TM domains tested. We also checked whether disruption of the dimer by TM peptides affects in situ cAMP signaling. Interestingly, Rho can bind not only to Gt but also to Gi in the absence of Gt to induce signaling responses (69). Thus, we evaluated cAMP accumulation levels in response to light activation in HEK-293 (mOpsin·Rluc and mOpsin·Venus) cells regenerated with 9-cis-retinal and treated with TM peptides. Cells were incubated with TM1, TM2, TM4, and TM5, which most potently inhibited the BRET signal, with TM7 used as a control. The accumulation of cAMP was inhibited after exposure to light only in cells regenerated with 9-cis-retinal, which contained the isoRho pigment. Similar levels of cAMP reduction were found for all cells treated or untreated with TM peptides (Fig. 9B). Therefore, treatment with dimer-disruptive TM peptides did not affect light-induced Gi signaling by isoRho, suggesting that Rho dimerization possibly is not critical for G protein-dependent responses. However, another explanation could be that structural changes that occur in Rho upon light activation cause a specific rearrangement of the dimer interface. Consistent with that hypothesis, the binding of the Rho dimer to its Gt heterotrimer and the consequent formation of a pentameric complex has been reported previously (65, 70, 71).
FIGURE 9.
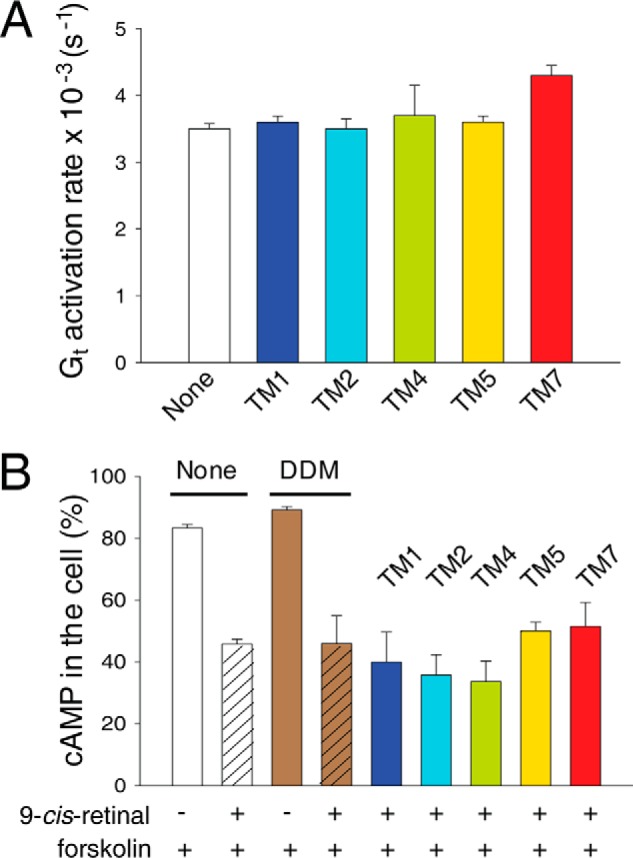
Effects of TM peptides on Rho function. A, rates of in vitro Gt activation with Rho purified by gel filtration after treatment with TM peptides. B, effects of selected TM peptides on light-stimulated cAMP accumulation in cells expressing opsin. HEK-293 (mOpsin·Rluc and mOpsin·Venus) cells were regenerated with 9-cis-retinal overnight (control cells were not incubated with 9-cis-retinal) and treated with 1 μm TM peptides for 1 h at 37 °C following stimulation with forskolin and light illumination. Control cells, not exposed to 9-cis-retinal, underwent the same protocol. cAMP levels were detected as described under “Materials and Methods.” Each experiment was performed in triplicate.
Thus, we tested whether the Gt protein stabilizes the Rho dimer upon light stimulation and if such stabilization could be inhibited by TM peptides. To obtain two distinct populations of Rho, we performed a digestion of Rho with Asp-N endoproteinase to cleave Rho specifically at the C terminus between Gly329 and Asp330, producing a shorter but fully functional Δ329Rho. Rho was digested with about 50% efficiency, and both populations were separated by 1D4 immunoaffinity chromatography (Fig. 10A). The formation of the Δ329RhoRho dimer was analyzed in a Rho and Δ329Rho 1:1 mixture into which either Gt or Gt plus TM peptides (TM1, TM2, TM4, and TM5) were added and then, after light activation, loaded on a 1D4 immunoaffinity column. Retention of Δ329Rho on such a column was possible only upon association with full-length Rho. Interestingly, the binding of Gt resulted in an increased amount of Δ329Rho in the eluate. This amount was slightly but significantly decreased in the presence of TM peptides that interfered with the dimer interface. Thus, Gt stabilized the dimeric conformation of Rho (Fig. 10, B and C).
FIGURE 10.

Stabilization of the Rho dimer by Gt binding. A, SDS-PAGE of Asp-N endoproteinase-digested ROS membranes (left panel). Digestion conditions were chosen to obtain 50% full-length Rho and 50% Δ329Rho. SDS-PAGE of Rho and Δ329Rho separated by 1D4 immunoaffinity chromatography (right panel). Δ329Rho lacking the 1D4 antibody epitope was in the unbound fraction (Ub), and full-length Rho was located in the elution fraction (El). B, effects of Gt binding on RhoΔ329Rho dimer stability. Gt was added to a Rho and Δ329Rho mixture in a 1:1 molar ratio (+Gt). Additionally, a mixture containing TM1, TM2, TM4, and TM5 peptides was added (+TM+Gt). Samples then were either light-exposed or kept in the dark. The resulting samples were subjected to 1D4 immunoaffinity chromatography. Elution fractions were analyzed by immunoblotting with B6-30 N-terminal anti-Rho antibodies (left panel) and anti-Gtα and anti-Gtβ antibodies (right panel). C, quantification of Δ329Rho. Protein bands were analyzed by densitometry, and the retention of Δ329Rho was quantified with respect to both Rho and Δ329Rho. The presence of TM peptides decreased the retention of Δ329Rho and thus the formation of the RhoΔ329Rho dimer. Data derived from three independent experiments are presented as means ± SD. Statistically significant differences (p < 0.05, Student's t test) are indicated with an asterisk.
Effect of Rho-derived TM Peptides on Rho Thermal Stability
The oligomeric assembly of Rho is critical for its stability (31, 72). Rho is a kinetically stable protein, although tightly packed in the membrane bilayer of rod outer segment disks. However, disruption of this protein-protein and protein-lipid supramolecular organization by detergent solubilization significantly decreases its stability and increases the rates of thermal denaturation in a detergent type- and concentration-dependent manner (73, 74). Monomeric Rho is prompted to release chromophore in the dark at 50 °C much faster (∼40% more protein decayed after 2 h of incubation) than Rho oligomers or synthetic dimers produced by Rho cross-linking (31, 72). Thus, we used a thermal stability assay to assess the effects of TM peptides that interfered with the Rho dimer interface. Loss of chromophore at 55 °C in the dark was monitored spectrophotometrically in the absence or presence of TM peptides. A significant reduction in Rho stability was observed in the presence of both TM1 with TM2 and TM4 with TM5 (∼15% more Rho decayed over 2 h of incubation at 55 °C), and this effect was enhanced in the presence of all four peptides (∼20% more Rho decayed over 2 h of incubation at 55 °C). In contrast, TM7 only slightly affected Rho stability (Fig. 11).
FIGURE 11.

Effects of TM peptides on Rho thermal stability. The thermal stability of Rho solubilized in 1 mm DDM in the absence or presence of TM peptides was determined. Samples were incubated at 55 °C, their absorbance spectra were recorded every 15 min, and the absorbance at 504 nm was then plotted as a function of time. A, thermal stability of Rho alone versus Rho in the presence of TM1, TM2, TM4, and TM5. B, thermal stability of Rho alone versus Rho in the presence of TM7. C, thermal stability of Rho alone versus Rho in the presence of TM1 and TM2. D, thermal stability of Rho alone versus Rho in the presence of TM4 and TM5. Changes in Rho thermal stability at 120 min observed in the presence of TM1, TM2, TM4, and TM5 and each peptide pair, namely TM1 with TM2 and TM4 with TM5, were statistically significant (p < 0.05, Student's t test, calculated using individual points obtained at 120 min). Each experiment was performed in triplicate.
Discussion
Evidence accumulated for more than a decade indicates that GPCRs form dimers and/or higher oligomers; the functional importance of these structures in affecting ligand recognition and signaling has been suggested for several of them (12, 14, 75, 76). Indeed, asymmetry with positive or negative cooperativity between monomers within the dimer where binding of ligand to one GPCR monomer increases or decreases the ligand binding affinity of the other monomer has been demonstrated (12, 41). Because GPCRs comprise one of the most important signaling mediators and pharmacologic targets, understanding the physiological importance of GPCR self-association is critical. The structural basis of GPCR dimerization has yet to be clarified, and in many cases the monomer-monomer interaction interface is predicted based on experimental observations and computational modeling (28, 43, 77, 78). Interestingly, many predictions have agreed with the structural details derived from the recently solved crystal structures of several GPCRs, which suggest the existence of two GPCR dimerization modes, indicating TM1, TM2, and cytoplasmic helix H8 as one and TM4/TM5 or TM5/TM6 as a second dimer interface (37–39, 79). Available x-ray structures of Rho also provide evidence for its dimerization. Parallel dimers were found in both two-dimensional crystals of Rho in the dark (80, 81) and a Rho Meta I state (82) as well as in three-dimensional crystals of photoactivated Rho (35), opsin (17), and Rho Meta II (36). Although the original crystallographic dimer of Rho was in an antiparallel orientation, it was also detected in the first crystal structure of bovine Rho (83). Structural data revealed contacts between TM1, helix H8, and TM2 as comprising a more stable dimer interface. But the most compelling evidence for Rho dimerization in native membranes comes from AFM images of Rho dimers organized in rows on the surface of isolated disk membranes from mouse eyes (1). More importantly, this groundbreaking discovery was confirmed 12 years later by independent tomographic studies of Rho supramolecular organization in cryosections of mouse rod photoreceptors (33).
TM domains, as a tool for disrupting GPCR dimerization of homo- as well as heterodimers to test dimer functions, were successfully applied to several GPCRs (25, 48, 52, 53, 68). These synthetic TM peptides not only associated with full-length receptors but also had a propensity to self-associate and form a functional receptor (84, 85). In the present study we focused on determining the interfaces of Rho dimerization and their physiological roles by using synthetic peptides derived from bovine Rho TM regions to inhibit Rho dimer formation. We examined the effects of all seven TM segments and a scrambled sequence of TM4 on Rho dimerization. Using size exclusion chromatography in combination with mass spectrometry analyses, we found first that five of seven TM peptides, TM1–TM5, but not TM6, TM7, or scrambled TM4, associated with Rho. These TM peptides caused a shift in the migration of Rho at the gel filtration column toward smaller molecular masses, suggesting their influence on the Rho oligomeric state. The small effect of some TM peptides on the Rho gel filtration profile could potentially be caused by peptide dissociation during gel filtration, because no peptides were present in the gel filtration buffer. The most significant change was observed with TM4, but its migration shift was still smaller than that in the presence of 3 mm DDM. Although the added mass produced by the attached TM peptide could be responsible for this effect, nevertheless the association of TM peptides with Rho was confirmed by their binding to immobilized Rho. By applying both cross-linking and BRET approaches, we found that TM1 and TM2 as well as TM4 and TM5 peptides interfered with the formation of the Rho dimer and reduced its abundance after incubation of ROS membranes with the DSG cross-linker. Interestingly, a symmetrical Rho dimer interface involving TM1 and helix H8 was identified previously in rod outer segment disk membranes by using a specific Cys316–Cys316 cross-linking approach (86). Opsin expressed heterologously exists in the membrane of live cells in an equilibrium between dimers and monomers (34, 45). Thus, by treating cells stably expressing either opsin or isoRho regenerated from 9-cis-retinal with TM peptides and using the BRET assay, we found that the same four peptides, i.e. TM1, TM2, TM4, and TM5, reduced the BRET signal from the receptor by blocking the association of both isoRho and opsin molecules to form dimers. These results support the existence of two dimerization interfaces, in agreement with earlier predictions based on Rho oligomer organization within the membrane (1, 30) and the crystallographic organization of several other GPCRs (37–39, 79).
The GPCR TM-derived peptides in many cases not only disrupted this receptor dimerization but also inhibited its ligand-induced signaling. As observed for the BLT1 receptor, its interaction with G protein was possible only when both protomers were loaded with an agonist, which allowed the formation of a pentameric complex composed of a receptor dimer and G protein heterotrimer. However, this binding was completely prevented by the presence of excess TM6 peptide (53). Signaling of the β2AR was similarly affected by the TM6 peptide derived from the β2AR primary sequence, resulting in reduced basal and isoproterenol-stimulated adenylate cyclase activity (52). Treatment of cells expressing both angiotensin II and secretin with peptide TM4 derived from either GPCR caused reduced cAMP signaling, indicating the involvement of TM4 in G protein-dependent responses. However, the TM1 of angiotensin II and TM2 of secretin, which reduced BRET signaling from the heterocomplex had no significant effect on cAMP signaling (68). Thus, experimental approaches using TM domains that disrupt specific dimer interaction interfaces have proved a powerful tool for examining the importance of GPCR dimerization on cellular physiology.
Our previous studies revealed that an asymmetric Rho dimer binds to a Gt heterotrimer forming a heteropentameric complex (65, 70, 71, 87). In these current studies, we tested the effect of TM peptides on the disruption of the Rho dimer and its consequences for G protein activation in vitro and in cultured cells expressing opsin. We found that Gt activation rates by Rho samples incubated with single TM peptides (TM1, TM2, TM4, and TM5) and purified by gel filtration did not significantly differ from those of the untreated control samples. Because heterologously expressed Rho can couple to the Gi cascade in mammalian cells we tested the effect of TM peptides on cAMP signaling in cells expressing either opsin or regenerated isoRho. Cells treated with the same TM peptides that blocked dimerization found in the cross-linking and BRET assays failed to affect cAMP accumulation. A similar lack of signaling inhibition, with the most relevance for the dimerization of the TM6-derived peptide, was found for the cholecystokinin receptor (25). Nevertheless, we observed that Gt stabilized the Rho dimer upon light activation and that this stabilization was reduced in the presence of TM peptides, which disrupt Rho self-association. Moreover, disruption of the Rho dimer with TM peptides strongly decreased the thermal stability of Rho, indicating the importance of Rho oligomeric organization for its stability.
In conclusion, the results of this study clearly show that the Rho molecule has two self-association interfaces, namely one at TM1 and TM2 and the second, more important interface at the TM4 and TM5 region. Disruption of Rho dimerization by synthesized and self-assembled TM peptides did not affect Rho-G protein coupling either in vitro or in situ, suggesting that Rho self-association is not critical for Rho-G protein activation. The illumination of single Rho molecules leads to neuronal responses in the brain and activates Gt in vitro (17, 88). However, the oligomeric organization of Rho in membranes is compatible with its physiological kinetics, and together with the formation of inactive precoupled complexes, this can provide a specific regulatory mechanism for reliable signal transduction (89). Dimerization of Rho may serve two important objectives: first, the formation of a stable Rho-Gt complex, and second, the stabilization of Rho pigment structure in the dark.
Author Contributions
B. J. and K. P. conceived the study and wrote the paper. B. J., Y. C., H. J., T. O., and L. H. designed, performed, and analyzed the experiments shown.
Acknowledgments
We thank Dr. Leslie T. Webster, Jr. and members of the Palczewski laboratory for helpful comments on this manuscript. Xiaoyu Li contributed to the stable cell line generation and BRET assays.
This work was supported, in whole or in part, by National Institutes of Health Grants EY008061 and EY019478. The authors declare that they have no conflicts of interest with the contents of this article.
- GPCR
- G protein-coupled receptor
- BRET
- bioluminescence resonance energy transfer
- Rho
- rhodopsin
- ROS
- rod outer segment
- TM
- transmembrane
- AFM
- atomic force microscopy
- DMSO
- dimethyl sulfoxide
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- DDM
- n-dodecyl-β-d-maltoside
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- BTP
- bis-Tris propane (1,3-bis[tris(hydroxymethyl)methylamino]propane)
- DSG
- disuccinimidyl glutarate
- PBST
- PBS with 0.1% Tween 20
- β2AR
- β2 adrenergic receptor.
References
- 1. Fotiadis D., Liang Y., Filipek S., Saperstein D. A., Engel A., Palczewski K. (2003) Atomic-force microscopy: rhodopsin dimers in native disc membranes. Nature 421, 127–128 [DOI] [PubMed] [Google Scholar]
- 2. Vobornik D., Rouleau Y., Haley J., Bani-Yaghoub M., Taylor R., Johnston L. J., Pezacki J. P. (2009) Nanoscale organization of β2-adrenergic receptor-Venus fusion protein domains on the surface of mammalian cells. Biochem. Biophys. Res. Commun. 382, 85–90 [DOI] [PubMed] [Google Scholar]
- 3. Ianoul A., Grant D. D., Rouleau Y., Bani-Yaghoub M., Johnston L. J., Pezacki J. P. (2005) Imaging nanometer domains of β-adrenergic receptor complexes on the surface of cardiac myocytes. Nat. Chem. Biol. 1, 196–202 [DOI] [PubMed] [Google Scholar]
- 4. Albizu L., Cottet M., Kralikova M., Stoev S., Seyer R., Brabet I., Roux T., Bazin H., Bourrier E., Lamarque L., Breton C., Rives M. L., Newman A., Javitch J., Trinquet E., Manning M., Pin J. P., Mouillac B., Durroux T. (2010) Time-resolved FRET between GPCR ligands reveals oligomers in native tissues. Nat. Chem. Biol. 6, 587–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Breitwieser G. E. (2004) G protein-coupled receptor oligomerization: implications for G protein activation and cell signaling. Circ. Res. 94, 17–27 [DOI] [PubMed] [Google Scholar]
- 6. Ng H. K., Chow B. K. (2015) Oligomerization of family B GPCRs: exploration in inter-family oligomer formation. Front. Endocrinol. (Lausanne) 6, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ferré S., Casadó V., Devi L. A., Filizola M., Jockers R., Lohse M. J., Milligan G., Pin J. P., Guitart X. (2014) G protein-coupled receptor oligomerization revisited: functional and pharmacological perspectives. Pharmacol. Rev. 66, 413–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bouvier M. (2001) Oligomerization of G-protein-coupled transmitter receptors. Nat. Rev. Neurosci. 2, 274–286 [DOI] [PubMed] [Google Scholar]
- 9. Milligan G. (2008) A day in the life of a G protein-coupled receptor: the contribution to function of G protein-coupled receptor dimerization. Br. J. Pharmacol. 153, Suppl. 1, S216–S229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ferré S. (2015) The GPCR heterotetramer: challenging classical pharmacology. Trends Pharmacol. Sci. 36, 145–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tena-Campos M., Ramon E., Rivera D., Borroto-Escuela D. O., Romero-Fernandez W., Fuxe K., Garriga P. (2014) G-protein-coupled receptors oligomerization: emerging signaling units and new opportunities for drug design. Curr. Protein Pept. Sci. 15, 648–658 [DOI] [PubMed] [Google Scholar]
- 12. Rivero-Müller A., Jonas K. C., Hanyaloglu A. C., Huhtaniemi I. (2013) Di/oligomerization of GPCRs: mechanisms and functional significance. Prog. Mol. Biol. Transl. Sci. 117, 163–185 [DOI] [PubMed] [Google Scholar]
- 13. Selent J., Kaczor A. A. (2011) Oligomerization of G protein-coupled receptors: computational methods. Curr. Med. Chem. 18, 4588–4605 [DOI] [PubMed] [Google Scholar]
- 14. Maurice P., Kamal M., Jockers R. (2011) Asymmetry of GPCR oligomers supports their functional relevance. Trends Pharmacol. Sci. 32, 514–520 [DOI] [PubMed] [Google Scholar]
- 15. White J. F., Grodnitzky J., Louis J. M., Trinh L. B., Shiloach J., Gutierrez J., Northup J. K., Grisshammer R. (2007) Dimerization of the class A G protein-coupled neurotensin receptor NTS1 alters G protein interaction. Proc. Natl. Acad. Sci. U.S.A. 104, 12199–12204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chabre M., le Maire M. (2005) Monomeric G-protein-coupled receptor as a functional unit. Biochemistry 44, 9395–9403 [DOI] [PubMed] [Google Scholar]
- 17. Whorton M. R., Jastrzebska B., Park P. S., Fotiadis D., Engel A., Palczewski K., Sunahara R. K. (2008) Efficient coupling of transducin to monomeric rhodopsin in a phospholipid bilayer. J. Biol. Chem. 283, 4387–4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Whorton M. R., Bokoch M. P., Rasmussen S. G., Huang B., Zare R. N., Kobilka B., Sunahara R. K. (2007) A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc. Natl. Acad. Sci. U.S.A. 104, 7682–7687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Milligan G., Ramsay D., Pascal G., Carrillo J. J. (2003) GPCR dimerisation. Life Sci. 74, 181–188 [DOI] [PubMed] [Google Scholar]
- 20. Cottet M., Faklaris O., Maurel D., Scholler P., Doumazane E., Trinquet E., Pin J. P., Durroux T. (2012) BRET and time-resolved FRET strategy to study GPCR oligomerization: from cell lines toward native tissues. Front. Endocrinol. (Lausanne) 3, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lambert N. A., Javitch J. A. (2014) CrossTalk opposing view: weighing the evidence for class A GPCR dimers, the jury is still out. J. Physiol. 592, 2443–2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Salahpour A., Espinoza S., Masri B., Lam V., Barak L. S., Gainetdinov R. R. (2012) BRET biosensors to study GPCR biology, pharmacology, and signal transduction. Front. Endocrinol. (Lausanne) 3, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Salon J. A., Lodowski D. T., Palczewski K. (2011) The significance of G protein-coupled receptor crystallography for drug discovery. Pharmacol. Rev. 63, 901–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Milligan G. (2009) G protein-coupled receptor hetero-dimerization: contribution to pharmacology and function. Br. J. Pharmacol. 158, 5–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Harikumar K. G., Dong M., Cheng Z., Pinon D. I., Lybrand T. P., Miller L. J. (2006) Transmembrane segment peptides can disrupt cholecystokinin receptor oligomerization without affecting receptor function. Biochemistry 45, 14706–14716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chabre M., Deterre P., Antonny B. (2009) The apparent cooperativity of some GPCRs does not necessarily imply dimerization. Trends Pharmacol. Sci. 30, 182–187 [DOI] [PubMed] [Google Scholar]
- 27. Chabre M., Cone R., Saibil H. (2003) Biophysics: is rhodopsin dimeric in native retinal rods? Nature 426, 30–31 [DOI] [PubMed] [Google Scholar]
- 28. Fotiadis D., Liang Y., Filipek S., Saperstein D. A., Engel A., Palczewski K. (2004) The G protein-coupled receptor rhodopsin in the native membrane. FEBS Lett. 564, 281–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Filipek S., Krzysko K. A., Fotiadis D., Liang Y., Saperstein D. A., Engel A., Palczewski K. (2004) A concept for G protein activation by G protein-coupled receptor dimers: the transducin/rhodopsin interface. Photochem. Photobiol. Sci. 3, 628–638 [DOI] [PubMed] [Google Scholar]
- 30. Liang Y., Fotiadis D., Filipek S., Saperstein D. A., Palczewski K., Engel A. (2003) Organization of the G protein-coupled receptors rhodopsin and opsin in native membranes. J. Biol. Chem. 278, 21655–21662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jastrzebska B., Fotiadis D., Jang G. F., Stenkamp R. E., Engel A., Palczewski K. (2006) Functional and structural characterization of rhodopsin oligomers. J. Biol. Chem. 281, 11917–11922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nickell S., Park P. S., Baumeister W., Palczewski K. (2007) Three-dimensional architecture of murine rod outer segments determined by cryoelectron tomography. J. Cell Biol. 177, 917–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gunkel M., Schöneberg J., Alkhaldi W., Irsen S., Noé F., Kaupp U. B., Al-Amoudi A. (2015) Higher-order architecture of rhodopsin in intact photoreceptors and its implication for phototransduction kinetics. Structure 23, 628–638 [DOI] [PubMed] [Google Scholar]
- 34. Comar W. D., Schubert S. M., Jastrzebska B., Palczewski K., Smith A. W. (2014) Time-resolved fluorescence spectroscopy measures clustering and mobility of a G protein-coupled receptor opsin in live cell membranes. J. Am. Chem. Soc. 136, 8342–8349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Salom D., Lodowski D. T., Stenkamp R. E., Le Trong I., Golczak M., Jastrzebska B., Harris T., Ballesteros J. A., Palczewski K. (2006) Crystal structure of a photoactivated deprotonated intermediate of rhodopsin. Proc. Natl. Acad. Sci. U.S.A. 103, 16123–16128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Choe H. W., Kim Y. J., Park J. H., Morizumi T., Pai E. F., Krauss N., Hofmann K. P., Scheerer P., Ernst O. P. (2011) Crystal structure of metarhodopsin II. Nature 471, 651–655 [DOI] [PubMed] [Google Scholar]
- 37. Huang J., Chen S., Zhang J. J., Huang X. Y. (2013) Crystal structure of oligomeric β1-adrenergic G protein-coupled receptors in ligand-free basal state. Nat. Struct. Mol. Biol. 20, 419–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wu B., Chien E. Y., Mol C. D., Fenalti G., Liu W., Katritch V., Abagyan R., Brooun A., Wells P., Bi F. C., Hamel D. J., Kuhn P., Handel T. M., Cherezov V., Stevens R. C. (2010) Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 330, 1066–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Manglik A., Kruse A. C., Kobilka T. S., Thian F. S., Mathiesen J. M., Sunahara R. K., Pardo L., Weis W. I., Kobilka B. K., Granier S. (2012) Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature 485, 321–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McMillin S. M., Heusel M., Liu T., Costanzi S., Wess J. (2011) Structural basis of M3 muscarinic receptor dimer/oligomer formation. J. Biol. Chem. 286, 28584–28598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xue L., Rovira X., Scholler P., Zhao H., Liu J., Pin J. P., Rondard P. (2015) Major ligand-induced rearrangement of the heptahelical domain interface in a GPCR dimer. Nat. Chem. Biol. 11, 134–140 [DOI] [PubMed] [Google Scholar]
- 42. Nemoto W., Toh H. (2005) Prediction of interfaces for oligomerizations of G-protein coupled receptors. Proteins 58, 644–660 [DOI] [PubMed] [Google Scholar]
- 43. Periole X., Knepp A. M., Sakmar T. P., Marrink S. J., Huber T. (2012) Structural determinants of the supramolecular organization of G protein-coupled receptors in bilayers. J. Am. Chem. Soc. 134, 10959–10965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Simpson L. M., Taddese B., Wall I. D., Reynolds C. A. (2010) Bioinformatics and molecular modelling approaches to GPCR oligomerization. Curr. Opin. Pharmacol. 10, 30–37 [DOI] [PubMed] [Google Scholar]
- 45. Kota P., Reeves P. J., Rajbhandary U. L., Khorana H. G. (2006) Opsin is present as dimers in COS1 cells: identification of amino acids at the dimeric interface. Proc. Natl. Acad. Sci. U.S.A. 103, 3054–3059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Guo W., Shi L., Javitch J. A. (2003) The fourth transmembrane segment forms the interface of the dopamine D2 receptor homodimer. J. Biol. Chem. 278, 4385–4388 [DOI] [PubMed] [Google Scholar]
- 47. Mancia F., Assur Z., Herman A. G., Siegel R., Hendrickson W. A. (2008) Ligand sensitivity in dimeric associations of the serotonin 5HT2c receptor. EMBO Rep. 9, 363–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Harikumar K. G., Pinon D. I., Miller L. J. (2007) Transmembrane segment IV contributes a functionally important interface for oligomerization of the Class II G protein-coupled secretin receptor. J. Biol. Chem. 282, 30363–30372 [DOI] [PubMed] [Google Scholar]
- 49. Marsango S., Caltabiano G., Pou C., Varela Liste M. J., Milligan G. (2015) Analysis of human dopamine D3 receptor quaternary structure. J. Biol. Chem. 290, 15146–15162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hayashi R., Osada S., Yoshiki M., Sugiyama D., Fujita I., Hamasaki Y., Kodama H. (2006) Superoxide production in human neutrophils is enhanced by treatment with transmembrane peptides derived from human formyl peptide receptor. J. Biochem. 139, 981–988 [DOI] [PubMed] [Google Scholar]
- 51. Wang J., He L., Combs C. A., Roderiquez G., Norcross M. A. (2006) Dimerization of CXCR4 in living malignant cells: control of cell migration by a synthetic peptide that reduces homologous CXCR4 interactions. Mol. Cancer Ther. 5, 2474–2483 [DOI] [PubMed] [Google Scholar]
- 52. Hebert T. E., Moffett S., Morello J. P., Loisel T. P., Bichet D. G., Barret C., Bouvier M. (1996) A peptide derived from a β2-adrenergic receptor transmembrane domain inhibits both receptor dimerization and activation. J. Biol. Chem. 271, 16384–16392 [DOI] [PubMed] [Google Scholar]
- 53. Banères J. L., Parello J. (2003) Structure-based analysis of GPCR function: evidence for a novel pentameric assembly between the dimeric leukotriene B4 receptor BLT1 and the G-protein. J. Mol. Biol. 329, 815–829 [DOI] [PubMed] [Google Scholar]
- 54. Wimley W. C., White S. H. (2000) Designing transmembrane α-helices that insert spontaneously. Biochemistry 39, 4432–4442 [DOI] [PubMed] [Google Scholar]
- 55. Papermaster D. S. (1982) Preparation of retinal rod outer segments. Methods Enzymol. 81, 48–52 [DOI] [PubMed] [Google Scholar]
- 56. Okada T., Tsujimoto R., Muraoka M., Funamoto C. (2005) Methods and results in X-ray crystallography of bovine rhodopsin, in G Protein-Coupled Receptors: Structure, Function, and Ligand Screening (Haga T., ed) pp. 245–261, CRC Press LLC, Boca Raton, FL [Google Scholar]
- 57. Wald G., Brown P. K. (1953) The molecular excitation of rhodopsin. J. Gen. Physiol. 37, 189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Vela L., Lowe P. N., Gerstenmaier J., Laing L. G., Stimmel J. B., Orband-Miller L. A., Martin J. J. (2011) Validation of an optical microplate label-free platform in the screening of chemical libraries for direct binding to a nuclear receptor. Assay Drug Dev. Technol. 9, 532–548 [DOI] [PubMed] [Google Scholar]
- 59. Goc A., Angel T. E., Jastrzebska B., Wang B., Wintrode P. L., Palczewski K. (2008) Different properties of the native and reconstituted heterotrimeric G protein transducin. Biochemistry 47, 12409–12419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jastrzebska B. (2015) Oligomeric state of rhodopsin within rhodopsin-transducin complex probed with succinylated concanavalin A. Methods Mol. Biol. 1271, 221–233 [DOI] [PubMed] [Google Scholar]
- 61. Fahmy K., Sakmar T. P. (1993) Regulation of the rhodopsin-transducin interaction by a highly conserved carboxylic acid group. Biochemistry 32, 7229–7236 [DOI] [PubMed] [Google Scholar]
- 62. Farrens D. L., Altenbach C., Yang K., Hubbell W. L., Khorana H. G. (1996) Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science 274, 768–770 [DOI] [PubMed] [Google Scholar]
- 63. Heck M., Hofmann K. P. (2001) Maximal rate and nucleotide dependence of rhodopsin-catalyzed transducin activation: initial rate analysis based on a double displacement mechanism. J. Biol. Chem. 276, 10000–10009 [DOI] [PubMed] [Google Scholar]
- 64. Müller D. J., Wu N., Palczewski K. (2008) Vertebrate membrane proteins: structure, function, and insights from biophysical approaches. Pharmacol. Rev. 60, 43–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Jastrzebska B., Ringler P., Palczewski K., Engel A. (2013) The rhodopsin-transducin complex houses two distinct rhodopsin molecules. J. Struct. Biol. 182, 164–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mansoor S. E., Palczewski K., Farrens D. L. (2006) Rhodopsin self-associates in asolectin liposomes. Proc. Natl. Acad. Sci. U.S.A. 103, 3060–3065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. James J. R., Oliveira M. I., Carmo A. M., Iaboni A., Davis S. J. (2006) A rigorous experimental framework for detecting protein oligomerization using bioluminescence resonance energy transfer. Nat. Methods 3, 1001–1006 [DOI] [PubMed] [Google Scholar]
- 68. Lee L. T., Ng S. Y., Chu J. Y., Sekar R., Harikumar K. G., Miller L. J., Chow B. K. (2014) Transmembrane peptides as unique tools to demonstrate the in vivo action of a cross-class GPCR heterocomplex. FASEB J. 28, 2632–2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chen Y., Jastrzebska B., Cao P., Zhang J., Wang B., Sun W., Yuan Y., Feng Z., Palczewski K. (2014) Inherent instability of the retinitis pigmentosa P23H mutant opsin. J. Biol. Chem. 289, 9288–9303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Jastrzebska B., Ringler P., Lodowski D. T., Moiseenkova-Bell V., Golczak M., Müller S. A., Palczewski K., Engel A. (2011) Rhodopsin-transducin heteropentamer: three-dimensional structure and biochemical characterization. J. Struct. Biol. 176, 387–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jastrzebska B., Orban T., Golczak M., Engel A., Palczewski K. (2013) Asymmetry of the rhodopsin dimer in complex with transducin. FASEB J. 27, 1572–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jastrzebska B., Maeda T., Zhu L., Fotiadis D., Filipek S., Engel A., Stenkamp R. E., Palczewski K. (2004) Functional characterization of rhodopsin monomers and dimers in detergents. J. Biol. Chem. 279, 54663–54675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ramon E., Marron J., del Valle L., Bosch L., Andrés A., Manyosa J., Garriga P. (2003) Effect of dodecyl maltoside detergent on rhodopsin stability and function. Vision Res. 43, 3055–3061 [DOI] [PubMed] [Google Scholar]
- 74. Corley S. C., Sprangers P., Albert A. D. (2011) The bilayer enhances rhodopsin kinetic stability in bovine rod outer segment disk membranes. Biophys. J. 100, 2946–2954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Fanelli F., Seeber M., Felline A., Casciari D., Raimondi F. (2013) Quaternary structure predictions and structural communication features of GPCR dimers. Prog Mol. Biol. Transl. Sci. 117, 105–142 [DOI] [PubMed] [Google Scholar]
- 76. Jastrzebska B., Tsybovsky Y., Palczewski K. (2010) Complexes between photoactivated rhodopsin and transducin: progress and questions. Biochem. J. 428, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gorinski N., Kowalsman N., Renner U., Wirth A., Reinartz M. T., Seifert R., Zeug A., Ponimaskin E., Niv M. Y. (2012) Computational and experimental analysis of the transmembrane domain 4/5 dimerization interface of the serotonin 5-HT(1A) receptor. Mol. Pharmacol. 82, 448–463 [DOI] [PubMed] [Google Scholar]
- 78. Hernanz-Falcón P., Rodríguez-Frade J. M., Serrano A., Juan D., del Sol A., Soriano S. F., Roncal F., Gómez L., Valencia A., Martínez-A C., Mellado M. (2004) Identification of amino acid residues crucial for chemokine receptor dimerization. Nat. Immunol. 5, 216–223 [DOI] [PubMed] [Google Scholar]
- 79. Wu H., Wacker D., Mileni M., Katritch V., Han G. W., Vardy E., Liu W., Thompson A. A., Huang X. P., Carroll F. I., Mascarella S. W., Westkaemper R. B., Mosier P. D., Roth B. L., Cherezov V., Stevens R. C. (2012) Structure of the human κ-opioid receptor in complex with JDTic. Nature 485, 327–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Schertler G. F., Hargrave P. A. (1995) Projection structure of frog rhodopsin in two crystal forms. Proc. Natl. Acad. Sci. U.S.A. 92, 11578–11582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Schertler G. F., Villa C., Henderson R. (1993) Projection structure of rhodopsin. Nature 362, 770–772 [DOI] [PubMed] [Google Scholar]
- 82. Ruprecht J. J., Mielke T., Vogel R., Villa C., Schertler G. F. (2004) Electron crystallography reveals the structure of metarhodopsin I. EMBO J. 23, 3609–3620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Palczewski K., Kumasaka T., Hori T., Behnke C. A., Motoshima H., Fox B. A., Le Trong I., Teller D. C., Okada T., Stenkamp R. E., Yamamoto M., Miyano M. (2000) Crystal structure of rhodopsin: a G protein-coupled receptor. Science 289, 739–745 [DOI] [PubMed] [Google Scholar]
- 84. MacKenzie K. R., Prestegard J. H., Engelman D. M. (1997) A transmembrane helix dimer: structure and implications. Science 276, 131–133 [DOI] [PubMed] [Google Scholar]
- 85. Lee W. K., Han J. J., Jin B. S., Boo D. W., Yu Y. G. (2009) Functional reconstitution of the human serotonin receptor 5-HT(6) using synthetic transmembrane peptides. Biochem. Biophys. Res. Commun. 390, 815–820 [DOI] [PubMed] [Google Scholar]
- 86. Knepp A. M., Periole X., Marrink S. J., Sakmar T. P., Huber T. (2012) Rhodopsin forms a dimer with cytoplasmic helix 8 contacts in native membranes. Biochemistry 51, 1819–1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Jastrzebska B. (2013) GPCR: G protein complexes: the fundamental signaling assembly. Amino Acids 45, 1303–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Baylor D. A., Lamb T. D., Yau K. W. (1979) Responses of retinal rods to single photons. J. Physiol. 288, 613–634 [PMC free article] [PubMed] [Google Scholar]
- 89. Dell'Orco D., Koch K. W. (2011) A dynamic scaffolding mechanism for rhodopsin and transducin interaction in vertebrate vision. Biochem. J. 440, 263–271 [DOI] [PubMed] [Google Scholar]
- 90. Rice P., Longden I., Bleasby A. (2000) EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16, 276–277 [DOI] [PubMed] [Google Scholar]