

Background: Recombinant tissue-type plasminogen activator (tPA) is a potent fibrinolytic agent used in clinics and is inactivated by endogenous PAI-1.
Results: The crystal structure of the tPA·PAI-1 Michaelis complex was determined.
Conclusion: Differences of inhibition of tPA and uPA by PAI-1 are revealed.
Significance: This study offers important clues to design a newer generation of tPA thrombolytics with reduced PAI-1 inactivation.
Keywords: crystal structure, fibrinolysis, serine protease, serpin, structural biology, Michaelis complex, PAI-1, tPA, thrombolytic agents
Abstract
Thrombosis is a leading cause of death worldwide. Recombinant tissue-type plasminogen activator (tPA) is the Food and Drug Administration-approved thrombolytic drug. tPA is rapidly inactivated by endogenous plasminogen activator inhibitor-1 (PAI-1). Engineering on tPA to reduce its inhibition by PAI-1 without compromising its thrombolytic effect is a continuous effort. Precise details, with atomic resolution, of the molecular interactions between tPA and PAI-1 remain unknown despite previous extensive studies. Here, we report the crystal structure of the tPA·PAI-1 Michaelis complex, which shows significant differences from the structure of its urokinase-type plasminogen activator analogue, the uPA·PAI-1 Michaelis complex. The PAI-1 reactive center loop adopts a unique kinked conformation. The structure provides detailed interactions between tPA 37- and 60-loops with PAI-1. On the tPA side, the S2 and S1β pockets open up to accommodate PAI-1. This study provides structural basis to understand the specificity of PAI-1 and to design newer generation of thrombolytic agents with reduced PAI-1 inactivation.
Introduction
Thrombosis is a major contributor to disability and mortality and is estimated to be the cause of one in four deaths worldwide (1). Current thrombolytic therapy consists of administration of a high dose of plasminogen activators (PAs),3 e.g. recombinant human tissue-type plasminogen activator (tPA) or streptokinase (2). Human tPA, along with urokinase-type plasminogen activator (uPA), are the only two physiological PAs in human. They catalyze the conversion of plasminogen (∼2 μm human plasma concentration) to the proteolytic active protease, plasmin, which then degrades fibrin. Recombinant tPA (Alteplase) is the Food and Drug Administration-approved treatment for ischemic strokes, administered intravenously to dissolve the clot and improve blood flow. Recombinant tPA is also used for myocardial infarction and pulmonary embolism (3). A high dose of recombinant tPA (up to 100 mg/50 kg) is typically needed in clinical applications, in part because of the rapid inactivation of recombinant tPA by endogenous plasminogen activators inhibitor 1 (PAI-1 or SERPINE1), a member of serine protease inhibitor (SERPIN) family. Such high dosage leads to dangerous side effects, e.g. intracranial hemorrhage and neurotoxicity (4, 5). Newer generation of recombinant tPA (2) or improved formulation of tPA (6), are continuously being developed. Tenecteplase (TNK-tPA) is a variant of tPA (7) showing slower inhibition by PAI-1.
Physiologically, PAI-1 inhibits PA rapidly and irreversibly, and is the primary negative regulator of the fibrinolytic system. PAI-1 itself has been recognized as a potential therapeutic target for treatment of thrombotic disorders (8, 9) and a marker for prognostic evaluation and disease monitoring (10). High levels of PAI-1 prevent formation of plasmin, resulting in fibrin accumulation and thrombosis.
PAI-1, and also PAI-2 (11), inhibit tPA and uPA by a suicide substrate mechanism (12, 13) common to all SERPIN members (see Fig. 1A). In this SERPIN mechanism, a long flexible loop of PAI-1 (reaction center loop, or RCL) inserts into the active site of PAs to form a transient Michaelis complex. The RCL is cleaved by PAs through the classical serine proteolytic mechanism. PAs form covalent acyl-enzyme intermediates with PAI-1 by cleaving the scissible bond of PAI-1 RCL, following the Michaelis complex. Before the hydrolysis of this acyl-enzyme intermediate, the PAI-1 RCL undergoes major conformational changes and inserts itself into the PAI-1 β sheet A. At the same time, the tPA in the intermediate is pulled to the other side of PAI-1, distorted, and deactivated before the hydrolysis of the acyl-enzyme intermediate can take place. Alternatively, PAI-1 can also spontaneously deactivate by inserting its intact, uncleaved RCL into β sheet A, resulting in the more stable but inactive, so-called latent conformation (14).
FIGURE 1.

Proposed PAI-1 inhibitory mechanism and the overall structure of tPA·PAI-1 Michaelis complex. A, proposed PAI-1 inhibitory mechanism (12). In this scheme, a noncovalent tPA·PAI-1 Michaelis complex is formed during the initial encounter (k1/k−1) of tPA with PAI-1, followed by a reversible scissile bond cleavage step (k2/k−2) that generates a loop-bound acyl-enzyme intermediate, where the distal cleaved RCL remains in the active pocket of tPA. A slow displacement of the distal cleaved RCL within tPA (k3/k−3) results in formation of a loop-displaced acyl-enzyme complex, which has two possible outcomes (branched mechanism): 1) the RCL inserts into the β sheet A, bringing tPA to another end of PAI-1, forming a covalent complex with tPA deactivated; and 2) in some cases, tPA can complete the hydrolysis of PAI-1 without being inactivated, which results in cleaved PAI-1 and active tPA. B, the domain structure of tPA (magenta) and PAI-1 (blue). The interdomain disulfide bond is also shown. K, kringle domain; G, growth factor domain; F, fibronectin type II domain. The tPA-SPD variant containing three mutations (C122A, N173Q, and S195A) and a stable PAI-1 variant (18) containing quadruple mutations (N150H, K154T, Q319L, and M354I) are used to form the Michaelis complex and for crystallization. C, crystal structure of Michaelis complex structure of tPA-SPD·PAI-1 in two orthogonal orientations.
As a protease inhibitor, PAI-1 is very specific to tPA and uPA; its inhibition of these two PAs is at least 1000 time faster than of any other proteases (12). The structural origin of PAI-1 specificity to uPA was revealed by the crystal structure of the uPA·PAI-1 Michaelis complex that we determined in 2011 (15). Compared with uPA, tPA is more susceptible to PAI-1 inhibition with a faster second order rate constant (2.6 × 107 m−1 s−1 versus 4.8 × 106 m−1 s−1) (16).
In the present study, we determined the crystal structure of the Michaelis complex between tPA and PAI-1, which identifies key residues of tPA in the interface and provides insight on the specificity of PAI-1 for tPA and uPA. This structure also explains the PAI-1 resisting property of tenecteplase and offers important clues to design newer generation of PAI-1-resistant tPA variants.
Experimental Procedures
Materials
Pichia pastoris strain X-33, Top10F′, E. coli strain BL21, plasmid pT7-PL, and plasmid pPICZαA were purchased from Invitrogen. All enzymes were from Takara except that Pfu DNA polymerase was from Stratagene. Synthetic DNA oligonucleotides and clones sequencing were from Shanghai Sangon. YNB was from Sigma. Sepharose fast flow and Superdex 75 HR 10/30 size exclusion columns were from GE Healthcare. Other chemicals were purchased from Shanghai Chemical, Inc. Mass spectrometry for human tPA-serine protease domain (tPA-SPD) recombinant was carried out at the State Key Laboratory of Structural Chemistry of Fujian Institute of Research on the Structure of Matter (Chinese Academy of Sciences, Fuzhou, Fujian, China).
Construction of Expression Plasmids of tPA-SPD for Expression in P. pastoris
Human tPA-SPD cDNA (276–527, or Ile16–Pro244 in the chymotrypsin numbering) containing S478A, C395A, and N448Q mutations (or S195A, C122A, and N173Q in the chymotrypsin numbering) was generated by PCR. This tPA-SPD cDNA was cloned into the secretory expression vector pPICαA (Invitrogen) between the XhoI and SalI sites, right after α factor signal peptide. In this construct, a dipeptide (Lys-Arg, Kex2 cleavage site) is inserted after the N-terminal restricted enzyme sites of XhoI. The expression host, P. pastoris, contains the Kex2 protease (Kex2p), which recognizes sequences of -Lys-Arg-↓-Xaa- and -Arg-Arg-↓-Xaa- (where ↓ is the site of cleavage). During recombinant protein expression, P. pastoris Kex2p enzyme will cleave the secreted tPA-SPD and generate a new N terminus of tPA-SPD, allowing the N terminus insertion into the serine protease and the formation of active enzyme.
Transformation and Expression of tPA-SPD in P. pastoris
The tPA-SPD DNA construct was transformed into Top10F′, which was plated on LLB plate (1% yeast extract, 2% tryptone, 0.5% NaCl, and 2% agar) containing 25 μg/ml Zeocin to select positive colonies. The resistant strains were sequenced to identify the right clones. The pPICZαA plasmids containing the right insert were linearized with BstX1 restricted enzyme and electroporated (1500V, 25μF, 200Ω) into P. pastoris strain X-33 (EasySelect Pichia expression kit; Invitrogen). The transfected yeast X-33 were cultured on YPDS plate containing 100 μg/ml Zeocin at 28 °C for 3 days to screen for colonies that have the transformed gene integrated into the host chromosomal DNA.
The P. pastoris clones were analyzed for expression of tPA-SPD insert. For protein expression, the expression clones were inoculated into 5 ml of BMGY medium (1% yeast extract, 2% tryptone, 1% glycerol, and 100 mm potassium phosphate, pH 6.0) until the culture reached an A600 of 6. The cells were harvested by centrifuging at 1500 × g for 5 min at room temperature and resuspended in 10 ml of BMMY medium (1% yeast extract, 2% tryptone, 0.5% methanol, and 100 mm potassium phosphate, pH 6.0) for expression. Then the cultures were induced every 24 h with 1% (v/v) methanol. After expression was confirmed by SDS-PAGE and optimized, a 4-liter scale tPA-SPD expression was followed.
Purification of tPA-SPD Expressed from P. pastoris Strain X-33
After 4 days of induction with 1% methanol, the culture medium was collected by centrifuging at 12,000 × g for 5 min at room temperature. The recombinant protein was captured from the expression medium by a cation exchange column Sepharose fast flow and eluted with a NaCl gradient (0–1.5 m, 300 ml) in 20 mm acetate buffer, pH 4.5, followed by concentration to 1 ml and then purified on a gel filtration chromatography (Superdex 75 HR 10/30 column) equilibrated with 20 mm MES, pH 6.5, containing 1 m NaCl. The recombinant tPA-SPD was characterized by SDS-PAGE and mass spectrometry after trypsin digestion as described previously (17).
Expression and Purification of PAI-1 Mutant 14-1B
The recombinant PAI-1 mutant 14-1B (18) containing four point mutations (N150H, K154T, Q319L, and M354I), and a hexahistidine tag was expressed in Esherichia coli, using the expression vector pT7-PL and BL21 cells as previously described (19). In brief, cells were grown at 37 °C until mid-log phase followed by isopropyl-d-thiogalactoside induction at 16 °C overnight. Cells were harvested the next day and resuspended in buffer 20 mm MES, pH 6.1, 1 m NaCl, followed by ultracentrifugation. After sonication, cell pellets were separated by centrifugation, and the supernatant was loaded onto a 5-ml nickel affinity column (GE Healthcare). Subsequent Superdex75 gel filtration chromatography resulted in PAI-1 of greater than 95% purity.
Crystallization and Data Collection
The tPA-SPD·PAI-1 Michaelis complex was formed by mixing tPA-SPD and PAI-1 in a 1:1 molar ratio at low concentration (∼0.5 mg/ml), followed by dialysis into 20 mm Tris-HCl, pH 7.4, and 150 mm NaCl, concentration to 0.5 ml volume for a further gel filtration chromatography purification, which yielded to a complex of greater than 99% purity. The purified complex was then concentrated to 10 mg/ml before setting up crystallization trials. Crystals of the tPA-SPD·PAI-1 Michaelis complex were grown at 20 °C with the sitting drop method by mixing equal volumes of protein solution and precipitant solution (8% PEG-6K and 0.1 m Tris, pH 7.4), and appeared quickly within 1 day. However, the crystals always appeared as very thin plates and decayed rapidly in the x-ray beam, posing great difficulty for x-ray data collection. Most crystals diffracted to only 4–5 Å at Shanghai Synchrotron Radiation Facility BL-17U Beamline, and the diffracting spots often appeared as elongated or splitted shapes. After many crystallization and data collection trials for 1.5 years, one 3.16 Å data set was finally obtained at Shanghai Synchrotron Radiation Facility Beamline BL17U using 25% glycerol as cryoprotectant at a wavelength of 0.979 Å. The data were processed and scaled using the HKL2000 program package (20). The crystal belongs to P212121 space group with one complex in the crystallographic asymmetric unit. The relatively high Rmerge (0.22) was justified by the high redundancy (∼7) and high completeness (99.9%). Furthermore, the precision-indicating merging R factor Rpim (21) showed a decent value (0.10). The Rpim was previously used in the analysis of plasminogen structure and thrombin·PN-1 structure (22, 23).
Phasing and Refinement
The structure of the tPA-SPD·PAI-1 Michaelis complex was solved by molecular replacement method using MolRep program (24), which gave very strong and unambiguous solutions. A tPA was first positioned inside the crystal lattice using the structure of the tPA-SPD catalytic domain (PDB code 1A5H) (25) as a searching model and all the x-ray data up to 3.3 Å. The molecular replacement gave a contrast of 12.33, a signal to sigma ratio for translational function of 16.02, and a correlation coefficient of 0.365. Next, the position of PAI-1 was searched using the model of active stable variant of PAI-1 (Protein Data Bank code 1DVM) (26) while fixing the already positioned tPA-SPD model, giving only one translational function with a signal to sigma ratio of 19.4 and a correlation coefficient of 0.538. The molecular replacement model was subjected to iterative refinement and manual model rebuilding using Refmac (27) and Coot (28), respectively, giving a final R factor and Rfree factor of 0.20 and 0.27, respectively. The structure was validated with PROCHECK (29) and analyzed by PyMOL (30) and PISA (31).
Results and Discussion
Overall Structure of tPA·PAI-1 Michaelis Complex
Human tPA contains a fibronectin type II domain (amino acids 1–50), a growth factor domain (amino acids 51–91), two kringle domains (amino acids 92–261), an interdomain linker (amino acids 262–275), and a SPD (amino acids 276–527; Fig. 1B). The tPA-SPD domain is responsible for its plasminogen activation function and its inhibition by PAI-1. Thus, we used the recombinant tPA-SPD domain to form the Michaelis complex with PAI-1. We generated three mutations in tPA-SPD: S478A (or S195A in the chymotrypsin numbering) to render the tPA-SPD catalytically inactive, so the Michaelis complex does not proceed to the stable, covalent complex; N448Q (or N173Q in the chymotrypsin numbering) to remove the glycosylation on tPA-SPD, increasing the homogeneity of the recombinant protein and facilitating protein crystallization; and C395A (or C122A in the chymotrypsin numbering that will be used throughout the rest of text) mutation to remove the disulfide bond linked to K2 domain (Fig. 1B). The recombinant tPA-SPD was expressed in P. pastoris and characterized by SDS-PAGE and mass spectrometry after trypsin digestion. The crystal structure of the tPA-SPD·PAI-1 Michaelis complex was determined at 3.16 Å resolution and refined to reasonable R factors (R factor of 20.6%, Rfree of 27.0%) and good geometry (Table 1). The final refined model (Fig. 1C) was comprised of residues 8–379 of PAI-1-14-1B (18) and residues 16–243 of tPA-SPD. The flexible RCL of PAI-1 and the surface loops of tPA-SPD, which were often disordered in previously reported structures (25, 32), are well defined in this structure by the electron density maps (Fig. 2). The tPA-SPD adopts a typical protease conformation without major differences when compared with previous tPA-SPD structures, e.g. the tPA-SPD of PDB code 1RTF (32), giving a root mean square deviation of 0.57 Å for all 908 main chain atoms.
TABLE 1.
X-ray data collection and model refinement statistics for tPA-SPD·PAI-1 crystal
The numbers in parentheses are for the highest resolution shell.
| Data collection statistics | |
|---|---|
| Space group | P212121 |
| Cell dimensions | a = 49.74 Å, b = 73.94 Å, c = 201.8 Å, α = β = γ = 90.00° |
| Wavelength (Å) | 0.979 |
| Resolution (Å) | 35.43-3.16 (3.27-3.16) |
| <I/σ(I)> | 6.97 (3.12) |
| Completeness (%) | 99.9 (99.8) |
| Multiplicity | 6.9 (7.2) |
| Rmergea | 0.22 (0.82) |
| Rpimb | 0.10 (0.32) |
| Model refinement statistics | |
| Rwork/Rfreec | 0.206/0.270 |
| Root mean square deviations of bonds (Å)/angles (°) from ideality | 0.013/1.46 |
| Residues in most favored/outlier regions in Ramachandran plot | 0.90/0.03 |
| Average B-factor (Å2) of model | 47.30 |
a Rmerge = Σ|Ii − <I>|/ΣIi, where Ii is the intensity of the ith observation, and <I> is the mean intensity of the reflections.
b Rpim is a redundancy-independent and precision-indicating merging R factor value (21).
c Rwork = Σ|Fo − Fc|/Σ|Fo|, where Fo and Fc are the observed and calculated structure factor amplitudes, respectively. Rfree was calculated using 5% of the data excluded from refinement.
FIGURE 2.

The RCL of PAI-1 and the surface loops of tPA-SPD are well defined in the current structure by the refined 2Fo − Fc electron densities countered at 1 σ. The refined 2Fo − Fc electron density for RCL (A, carbons in yellow, nitrogens in blue, and oxygens in red) of PAI-1 and 37-loop (B), 60-loop (C), and 99-loop (D) of tPA (carbons in magenta, nitrogens in blue, and oxygens in red).
The Structure of tPA·PAI-1 Michaelis Complex Is Distinct from the uPA·PAI-1 Michaelis Complex Homologue
The structure of tPA·PAI-1 Michaelis complex reveals that the RCL, from residue Ser-I331 to Arg-I356 (“I” represents PAI-1 residues, and “E” represents tPA-S195A residues) inserts into the active site of tPA in an orientation quite different from that in the uPA-SPD·PAI-1 complex (15) (more on this below). As a result of the complex formation, an interface of 1202 Å2 (solvent-inaccessible area) between PAI-1 and tPA-SPD is formed, which is higher than that of the uPA-SPD·PAI-1 complex (1058 Å2). Of the total area of PAI-1, 8% is involved in interacting with tPA, whereas for uPA, only 6.9% is at the interface. In addition, the relative orientation between tPA and PAI-1 is quite different from its uPA homologue (Fig. 3) (15).
FIGURE 3.
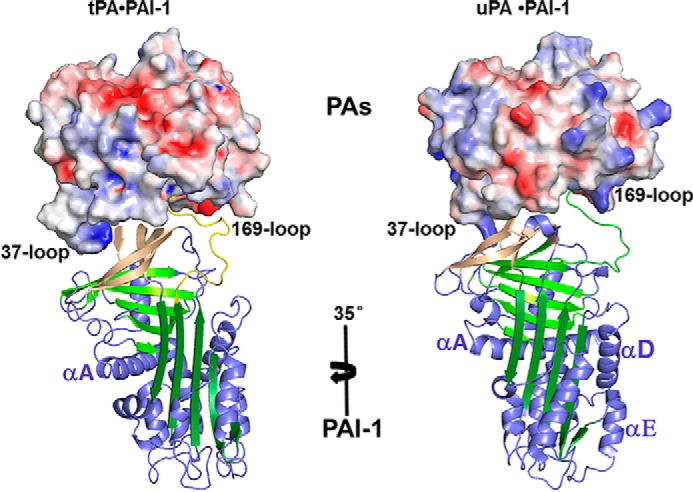
PAI-1 undergoes a 35-degree rotation when the protease of the current structure is overlapped to uPA·PAI-1 Michaelis complex (PDB ID 3PB1) (15). The protease SPD is shown in charged surface, whereas the PAI-1 is in cartoon representation.
An Unusual Conformation of PAI-1 RCL upon Binding to tPA
The RCL was previously (19, 26, 33) observed to be flexible and consistently adopt an extended conformation just above β-sheet C in those uncomplexed active PAI-1 structures, regardless of the crystal packing and crystallization conditions (cyan in Fig. 4A). This orientation is close to the 147 loop of PA (cyan in Fig. 4B). Upon binding to uPA, PAI-1 RCL flips to the other side of the protease (green in Fig. 4B), near the 99-loop of PA, by moving 29.8 Å (distance between Ser-I338 in the two structures). In the current tPA-SPD·PAI-1 structure, the RCL of PAI-1 is at a location between free PAI-1 and uPA-bound PAI-1 (yellow in Fig. 4, A and B).
FIGURE 4.
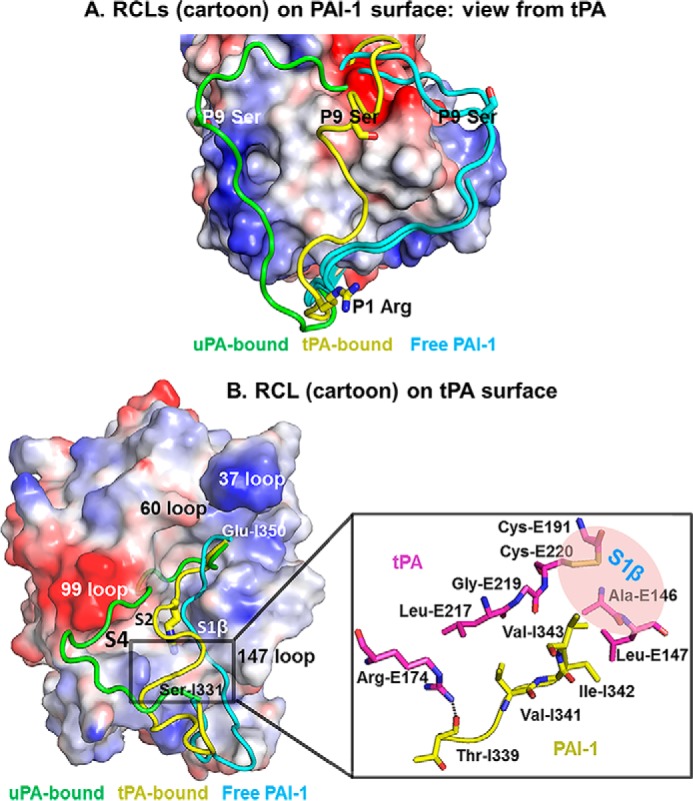
Conformation of the RCL in the tPA·PAI-1 is different from in free PAI-1 and in uPA·PAI-1 Michaelis complexes. A, alignment of active PAI-1 RCL in free form and in PA-bound form. The charged surface representation is the PAI-1 with its RCL removed from the PDB. The RCLs (cyan) of active free PAI-1 (PDB code 1B3K and 1DVM) (26, 33) were observed to adopt an extended conformation just above β-sheet C regardless of the crystal packing and crystallization conditions. The RCL (green) of uPA-bound PAI-1 was observed to flip to another side of PA (PDB code 3PB1) (15). The RCL (yellow) of tPA-bound PAI-1 is at a location between free PAI-1 and uPA-bound. B, left panel, the relative conformations of the RCL in the tPA-SPD·PAI-1 and the uPA-SPD·PAI-1 (PDB-ID 3PB1) Michaelis complexes are displayed on a charged surface representation of tPA-SPD. The RCL in the current structure (yellow) has a localization between the RCL of free PAI-1 (cyan, PDB code 1B3K (33)) and the RCL in the uPA-bound form (green). Right panel, the detailed interactions leading to the kinked conformation of PAI-1. Arg-E174 of tPA (magenta) and Thr-I339 of PAI-1 (yellow) forms a hydrogen bond, which fixes one side of the RCL. The PAI-1 segment Val-341–Ile-342–Val-343 interacts with tPA through hydrophobic interaction, fixing the other end of PAI-1 RCL.
Interestingly, the segment P8–P4 (The number after “P” indicates the position of the residue N-terminal to the scissile bond; the prime indicates residues C-terminal to the scissile bond) of RCL (Thr-Ala-Val-Ile-Val) was observed to adopt a kinked conformation (Fig. 4B), seemingly related to the following two reasons. On one hand, the PAI-1 P8 Thr-I339 residue makes a strong hydrogen bond (2.3 Å) to a residue on the tPA 169-loop (Arg-E174), pulling the RCL toward that loop. On another hand, a hydrophobic segment of PAI-1 (Val-341–Ile-342–Val-343) binds to the hydrophobic S1β pocket of tPA-SPD, which is impossible in the case of uPA because of the change of two residues (Ala-146–Leu-147 in tPA and Ser-146–Thr-147 in uPA). This hydrophobic interaction pulls the RCL away from the 169-loop and toward the 147-loop (Fig. 4B), leading to an overall kinked conformation of the segment P8-P4 of RCL. For PAI-1 in the uPA-bound form, this hydrophobic segment interacts with a hydrophobic residue of uPA 99-loop (Leu97b), which is inaccessible for tPA because of a two-residue shorter 99-loop and the absence of hydrophobic residue in the 99-loop. Consequently, the P4 Val-I343 does not occupy the S4 pocket but fits into the opposite S1β pocket above the S1 pocket, through a complementary interface of 102 Å2 (Figs. 4B and 5C).
FIGURE 5.

The residue Gln-E192 acts as a switch to control the entrance to S1β pocket of tPA. The opening up of tPA S1β binding pocket (B), compared with the free tPA (PDB code 1A5H), which does not have S1β pocket (A) (25), is due to the rotation of tPA Gln-E192 residue. The S1β then accommodates P4 Val-I343 of PAI-1 (C).
tPA Gln-E192 Is a Switch to Control the Entrance to S1β
We observed that the tPA S1β specificity pocket is occupied by PAI-1, which is unique among the published protease-serpin Michaelis complex structures (15, 23, 34–37), except the Michaelis complex between rat trypsin and Manduca sexta antitrypsin (38), in which the S1β pocket of trypsin is partially occupied by P9-Ala of antitrypsin. The tPA S1β pocket is a small hydrophobic pocket, formed by residues Ala-E146, Gln-E192, and Gly-E219 at the edges and the disulfide bond between Cys-E191 and Cys-E220 at the base. The tPA S1β pocket was previously observed to be blocked by the Gln-E192, which forms hydrogen bonds with two residues of the 147-loop (Lys-E143, 3.2 Å, and Ala-E146, 3.4 Å) (Fig. 5A) (25, 32, 39). In the present Michaelis complex structure, Gln-E192 interacts with a residue at the β-s3C (Thr-I205, by a hydrogen bond of 3.1Å) and the P3′ residue (Pro-I349, by a van der Waals contact of 3.5Å) of PAI-1 (Fig. 5B). It seems these two PAI-1 residues displaced the tPA 147-loop residues (Lys-E143 and Ala-E146) for their interactions with Gln-E192. As a result, the tPA S1β pocket is opened up to harbor P4 Val-I343 (Fig. 5C). Thus, Gln-E192 undergoes a conformational change upon PAI-1 binding and appears as a switch to control the entrance to S1β (40). It appears that the S1β pocket of uPA is less hydrophobic than that of tPA, because of two residue differences in the 147-loop (Ala-146–Leu-147 in tPA and Ser-146–Thr-147 in uPA). This interaction site between tPA and PAI-1 may be a target site for the further improvement of recombinant tPA for thrombolysis.
tPA 37-Loop Is a Key Region Making Extensive Interactions with PAI-1
In the Michaelis complexes, the 37-loop (residues 36KHRRSPGER39) of tPA has twice as large a contact area with PAI-1 (247 Å2) than uPA (124 Å2) (15), highlighting the importance of this loop in its inhibition by PAI-1 (41, 42).
On the PAI-1 side, three regions have direct interactions with the tPA 37-loop (Fig. 6A). The first region is a negatively charged patch formed by the side chains of Tyr-I210, Glu-I212, and Asp-I222 on β sheet B and Tyr-I241 on the loop connecting β-s3B and hG. This patch electrostatically attracts the positively charged 37-loop on tPA and makes extensive interactions (two hydrogen bonds between Tyr-I210 and Arg-E37a and Arg-E37b, respectively; two salt bridges between Glu-I212 and Arg-E37a and Arg-E37b, respectively; one hydrogen bond between Glu-I212 and Arg-E37a; and one hydrogen bond and one salt bridge between Asp-I222 and Arg-E37b). The importance of this negatively charged patch in the tPA interaction is a new discovery previously unobserved from extensive biochemical studies. A second region is the P4′ Glu-I350 residue of PAI-1, which is close (3.6 Å) to the tPA di-arginines (Arg-E37a-Arg-E37b) moiety, forming two salt bridges with Arg-37a. Glu-I350 also interacts with another arginine residue (Arg-E39) of tPA by a salt bridge. These three salt bridges are partially shielded away from solvent by the long 37-loop, potentially leading to lower dielectric constant in this region and thus enhancing these charged interactions. The importance of PAI-1 Glu-I350 in tPA binding was also previously identified by site-directed mutagenesis (43, 44). The third region of PAI-1 interacting with tPA 37-loop is the P2′ Ala-I348 of PAI-1, which forms a hydrogen bond with the side chain amino group of tPA Arg-E39. This interaction is at the center of tPA-SPD·PAI-1 interface and is likely more insulated from solvent. Together, the tPA-SPD·PAI-1 interactions on the 37-loop are mainly ionic in nature. As a result, the 37-loop may play a role in orienting tPA toward PAI-1 at a long distance before the formation of the encounter complex.
FIGURE 6.
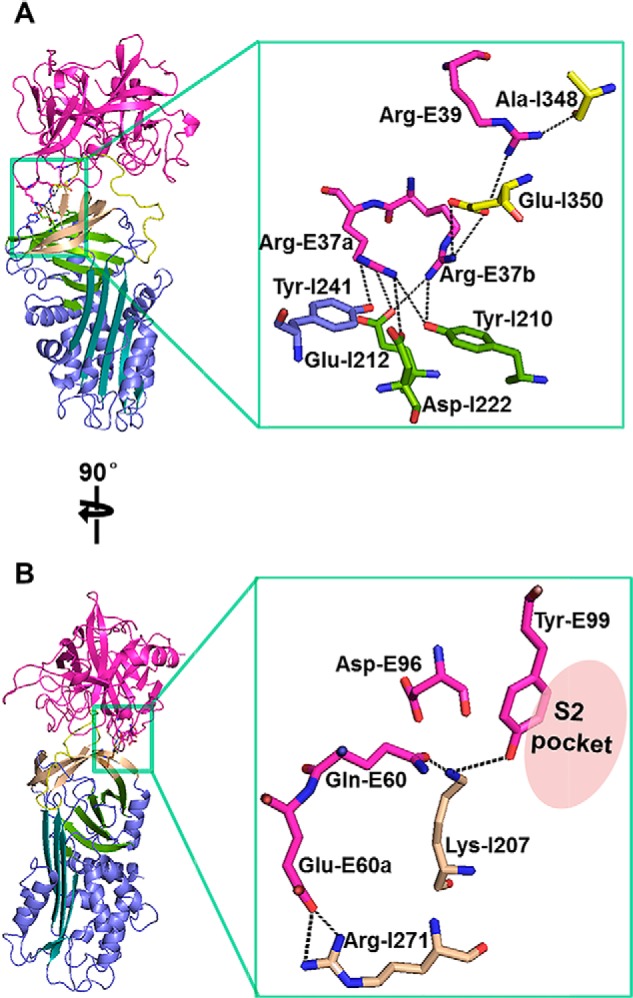
Direct exosite interactions between tPA 37-loop and PAI-1 (A) and between tPA 60-loop and PAI-1 (B). tPA 37-loop makes extensive interactions with PAI-1, especially with a negatively charged patch on PAI-1, comprised of Tyr-I210, Glu-I212, Asp-I222, and Tyr-I241. At tPA 60-loop, the side chains of Gln-E60, Asp-E96, and Tyr-E99 assemble into a negatively charged patch to interact with PAI-1 Lys-I207.
The residue Glu-I351 of PAI-1 was previously shown by mutational studies to implicate in the tPA binding (43), although the opposite conclusion was obtained in a different study (45). Our structure shows that this residue does not interact with the 37-loop and may have a weak van der Waals contact (4.2 Å to Pro-E149) with the 147-loop of tPA-SPD. For comparison, Glu-I351 is critical in the uPA·PAI-1 Michaelis complex formation (15), forming a strong (2.7Å) hydrogen bond with the side chain phenolic group of Tyr-E149.
The tPA 37-loop residues responsible for the direct interactions with PAI-1 comprise Arg-E37a, Arg-E37b, and Arg-E39. On uPA, the corresponding residues are less charged: Arg-E37a, Gly-E37b, and Thr-E39. The current structure demonstrates stronger P′ exosite interaction of PAI-1 to tPA when compared with uPA. These results support the proposed PAI-1 inhibitory mechanism, where release of the P′ side of the cleaved RCL from the substrate pocket of the protease is thought to be rate-limiting (18), and tPA exhibited a much slower rate of formation of the final covalent complex than uPA. This proposed mechanism was also supported by previous charge-reversal mutations on the 37-loop of tPA (R37aE,R37bE,R39E), yielding a tPA mutant that showed much slower association rate (by 25,000 times) to PAI-1 compared with the wild type tPA (47). The grafting of the tPA 37-loop to thrombin rendered the inhibition of the fusion protein by PAI-1, giving a second order rate constants of 106 m−1 s−1, much higher than the wild type thrombin (103 m−1 s−1) (48).
Exosite Interactions between tPA 60-Loop and PAI-1
In the current Michaelis complex structure, tPA 60-loop has a large contact surface (152 Å2) and makes strong interaction with PAI-1 (Fig. 6B) mainly by residues Gln-E60 and Glu-E60a of tPA. The tPA Gln-E60, together with tPA residues at the 99-loop (Asp-E96 and Tyr-E99), constitutes a negatively charged patch. This patch interacts with the positively charged Lys-I207 on β-s3C of PAI-1 (forming two hydrogen bonds: one between the side chain hydroxyl group of Gln-E60 and the side chain amino group of Lys-I207 within 2 Å and another one between the side chain hydroxy of Tyr-E99 and the side chain amino of Lys-I207 within 3.4 Å). It should be pointed out the 2 Å hydrogen bound is unusually short but was well supported by the electron density map, although the resolution of the structure was limited (3.16 Å). The short hydrogen bond was previously reported in other cases (49). This patch on tPA likely contributes to the fast association between tPA and PAI-1. Such conclusion is supported by an earlier study, where a Y99L mutation on tPA reduced inhibition by PAI-1 by more than 10-fold (50).
The tPA Glu-E60a interacts with the Arg-I271 on PAI-1 β-s2C by two salt bridges and one strong hydrogen bond (2.4 Å). The interactions with Glu-E60a are buried in a shallow pocket surrounded by Leu-I269, Pro-I270, and Glu-I350, yielding charge complementary interfaces.
In the free tPA structures without PAI-1 (25, 32), Gln-E60 and Glu-E60a adopt the conformations similar to the current structure of Michaelis complex, regardless of crystal packing. In these conformations, Gln-E60 and Glu-E60a locate at the edge of tPA active pocket and are ready to interact with the Lys-I207 and Arg-I271 of PAI-1 without any conformational changes. Therefore, the tPA 60-loop also contributes to the fast association between tPA and PAI-1. In uPA, a noncharged Ile locates at position 60, making it unable to form strong interactions with the Lys-I207 on PAI-1 β-s3C. The 60a residue of uPA is an aspartate and does not have direct interaction with PAI-1 Arg-I271 (15). It appears that the uPA 60-loop has less interaction with PAI-1 compared with that of tPA.
Enlargement of tPA S2 Pocket upon PAI-1 Binding
Previous structural studies (25, 32, 39) showed that the S2 pocket of tPA has a relatively small size (Fig. 7A) because of the presence of the large residue at position 99 (Tyr-E99), leading to a typical preference for small residues such as Gly at position P2. On the other hand, tPA was found to be able to hydrolyze substrates with larger P2 residues including Pro, Ser, or Ala (51). How does tPA accommodate PAI-1 which has a larger P2 residue (Ala)? We observed in the current structure that the S2 pocket of tPA is enlarged through a 90° rotation of Tyr-E99 phenolic side chain along its Cβ-Cγ axis (Fig. 7B), leading to the accommodation of PAI-1 P2 Ala at the S2 pocket (Fig. 7C).
FIGURE 7.

How does tPA with a small S2 pocket accommodate a large P2 residue? A, the uncomplexed tPA (PDB code 1A5H) has a small S2 subsite because of the presence of a large Tyr-E99. B and C, in the current Michaelis complex structure (B), Tyr-E99 was observed to rotate by ∼90°, leading to a larger S2 subsite, which can accommodate the PAI-1 P2 Ala residue (C).
For comparison, uPA has a His at position 99. The side chain imidazole of the His-E99 is not rotatable because the imidazole group has polar directional interaction with the surrounding residues Tyr-94 (3.3 Å) and Leu-97b (3.3 Å to main chain hydroxyl group) (52). Hence, uPA has strict preference for a Gly residue at the P2 position of its substrates or inhibitors. In a H99Y variant of uPA, the rotation of Tyr-99 was also observed (53). Therefore, the tPA S2 pocket is intrinsically expandable but not induced by PAI-1, which is different from the tPA S1β pocket. Activated coagulation factor IX (FIXa) has a very low activity toward its substrate in the absence of its cofactor factor VIIIa. It was found that loop 99 of FIXa adopts a locked conformation that interferes with canonical substrate binding in S2-S4 pockets and hinders access of substrate to the catalytic site in the absence, but not in the presence, of FVIIIa (54).
Interactions between P1 Arg-I346 and the S1 Pocket
The P1 residue (Arg-I346) of PAI-1 occupies the tPA S1 specificity pocket and has a strong interaction with tPA (Fig. 8A) by making a total of six hydrogen bonds (three main chain hydrogen bonds from the carbonyl to Ala-E195 and oxyanion hole residues Gly-E193 and Asp-E194, one hydrogen bond from the nitrogen atom to Ser-E214, and two bifurcated hydrogen bonds from the side chain guanidine group to the carboxylate of Asp-E189) and three salt bridges (three charged hydrogen bonds from the side chain guanidine group to the carboxylate of Asp-E189) with the surrounding tPA residues. These interactions between PAI-1 P1 residue and tPA S1 pocket are different from that (Fig. 8B) in the uPA·PAI-1 Michaelis complex (15), because of one residue difference at position 190 (Ser-190 in uPA and Ala-190 in tPA).
FIGURE 8.
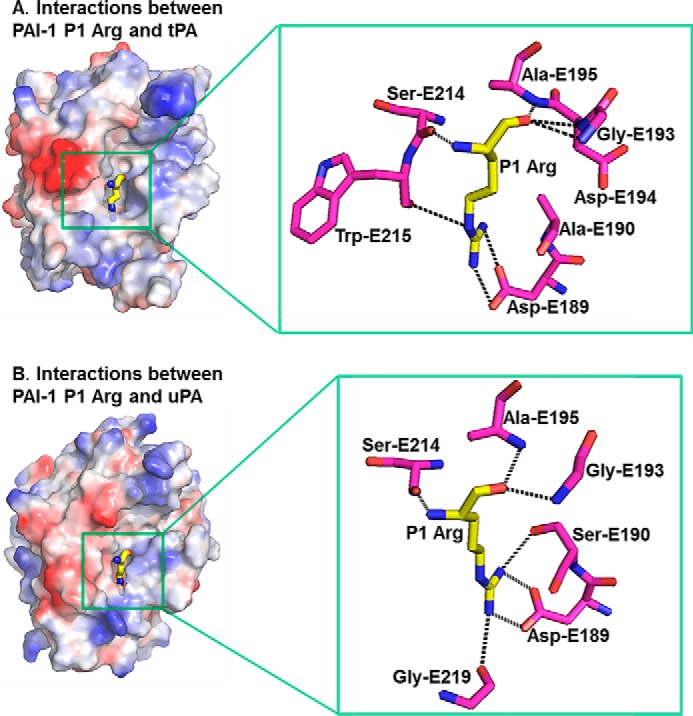
S1-P1 interactions in the tPA·PAI-1 Michaelis complex structure show interactions in the S1 pocket different from those in the uPA·PAI-1 Michaelis complex structure (PDB code 3PB1). A, interactions between PAI-1 P1 Arg and tPA. B, interactions between PAI-1 P1 Arg and uPA. There is one residue difference between tPA and uPA (Ala-190 and Ser-190, respectively).
Mutations on Recombinant tPA to Evade Inactivation by Endogenous PAI-1
Recombinant tPA (or its variants) is the Food and Drug Administration-approved drug for ischemic strokes. It is administered intravenously to improve blood flow by dissolving clots and to maintain patency of blood vessels. However, the tPA treatment for ischemic strokes has a short therapeutic window (<3 h within occlusion) and the high incidence of post-treatment complications, including intracerebral hemorrhage (55, 56), leading to its limited clinical use (only 3–8% of patients eligible for such therapy) (3, 4, 57). Newer generation of recombinant tPA is being continuously developed (2). One idea is to generate tPA variants that is resistant to inactivation by its endogenous inhibitor PAI-1, thus reducing the dose of recombinant tPA used.
Previous mutagenesis studies identified that the residues at positions 36, 37, and 39 of tPA-SPD are pivotal for the inactivation by PAI-1 but play no role in activation of plasminogen (41, 42). A tPA variant in which four alanines replaced the amino acids in positions 36–37b (KHRR36–37bAAAA) has a 90-fold increase in resistance to PAI-1 without compromising fibrin specificity (46, 58). These SPD mutations were combined with the mutations at the kringle 1 domain of tPA, and the resulting tPA variant is named TNK-tPA (tenecteplase) (7) and was shown to be the most effective thrombolytic agent approved by Food and Drug Administration so far, although the bleeding risk is still present. The current structure of tPA·PAI-1 Michaelis complex shows the direct interactions between tPA residues R37a-R37b and PAI-1, providing a structural rationale for the resistance of TNK-tPA for PAI-1 inactivation.
The inhibition of tPA by PAI-1 involves multiple steps (Fig. 1A), and each step can be exploited to design of recombinant tPA resisting PAI-1 inactivation. Even though the formation of Michaelis complex represents only the first step, the current structure of the Michaelis complex seems can explain the results from previous studies. One study showed the tPA variant with R39A mutation has reduced PAI-1 inactivation (47), which is consistent with the observation in this structure that tPA Arg-39 indeed has directly interactions with PAI-1 Ala-348 and Glu-350. The tPA-Y99L mutant had less binding to PAI-1, without compromising plasminogen activation (50). This tPA Tyr-99 residue defines the S2 pocket and interacts with PAI-1 Lys-207.
Along this line, our current structure can provide further clues to design new PAI-1-resisitant tPA variants. As described above, tPA residues Gln-60 and Glu-60a of the 60-loop, Tyr-151 of the 147-loop, and Arg-174 of the 169-loop all participate in extensive exosite interactions with PAI-1. Site-directed mutations on these residues may maximize tPA resistance to inhibition by PAI-1. In addition, the tPA S1β pocket was occupied by PAI-1 P4 Val-343, and mutations around the pocket may enhance tPA resistance to inhibition by PAI-1. The advantage of the mutations at this pocket is not to perturb plasminogen activation activity, because this pocket has some distance away from the catalytic triad. The effects of these mutations on fibrin specificity and fibrin degradation must, of course, be evaluated.
Author Contributions
L. G. is involved throughout the whole project from protein preparation, crystallization, structure determination, and result analysis. M. L., T. Z., and X. S. participated in protein preparation. C. Y. participated in structure determination. P. A. participated in manuscript preparation. M. H. participated in project initiation, result analysis, and manuscript preparation.
Acknowledgments
We thank the Shanghai Synchrotron Radiation Facility for the x-ray beam time and the scientists at Beamline BL17U for assistance with x-ray data collection.
This work was supported by Grants 31170707 and 31370737 from the National Natural Science Foundation of China and the CAS/SAFEA. International Partnership Program for Creative Research Teams. The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 5BRR) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PA
- plasminogen activator
- tPA
- tissue type plasminogen activator
- uPA
- urokinase-type plasminogen activator
- PAI-1
- plasminogen activators inhibitor
- RCL
- reactive center loop
- SPD
- serine protease domain
- serpin
- serine protease inhibitor
- PDB
- Protein Data Bank.
References
- 1.Raskob G. E., Angchaisuksiri P., Blanco A. N., Buller H., Gallus A., Hunt B. J., Hylek E. M., Kakkar A., Konstantinides S. V., McCumber M., Ozaki Y., Wendelboe A., and Weitz J. I. (2014) Thrombosis: A major contributor to global disease burden. Thromb. Res. 134, 931–938 [DOI] [PubMed] [Google Scholar]
- 2.Baruah D. B., Dash R. N., Chaudhari M. R., and Kadam S. S. (2006) Plasminogen activators: a comparison. Vascul. Pharmacol. 44, 1–9 [DOI] [PubMed] [Google Scholar]
- 3.Hatcher M. A., and Starr J. A. (2011) Role of tissue plasminogen activator in acute ischemic stroke. Ann. Pharmacother 45, 364–371 [DOI] [PubMed] [Google Scholar]
- 4.Grotta J. C., Burgin W. S., El-Mitwalli A., Long M., Campbell M., Morgenstern L. B., Malkoff M., and Alexandrov A. V. (2001) Intravenous tissue-type plasminogen activator therapy for ischemic stroke: Houston experience 1996 to 2000. Arch. Neurol. 58, 2009–2013 [DOI] [PubMed] [Google Scholar]
- 5.Hamsten A., Wiman B., de Faire U., and Blombäck M. (1985) Increased plasma levels of a rapid inhibitor of tissue plasminogen activator in young survivors of myocardial infarction. N. Engl. J. Med. 313, 1557–1563 [DOI] [PubMed] [Google Scholar]
- 6.Korin N., Kanapathipillai M., Matthews B. D., Crescente M., Brill A., Mammoto T., Ghosh K., Jurek S., Bencherif S. A., Bhatta D., Coskun A. U., Feldman C. L., Wagner D. D., and Ingber D. E. (2012) Shear-activated nanotherapeutics for drug targeting to obstructed blood vessels. Science 337, 738–742 [DOI] [PubMed] [Google Scholar]
- 7.Keyt B. A., Paoni N. F., Refino C. J., Berleau L., Nguyen H., Chow A., Lai J., Peña L., Pater C., and Ogez J. (1994) A faster-acting and more potent form of tissue plasminogen activator. Proc. Natl. Acad. Sci. U.S.A. 91, 3670–3674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kohler H. P., and Grant P. J. (2000) Plasminogen-activator inhibitor type 1 and coronary artery disease. N. Engl. J. Med. 342, 1792–1801 [DOI] [PubMed] [Google Scholar]
- 9.Vaughan D. E. (2011) PAI-1 antagonists: the promise and the peril. Trans. Am. Clin. Climatol. Assoc. 122, 312–325 [PMC free article] [PubMed] [Google Scholar]
- 10.Cesari M., Pahor M., and Incalzi R. A. (2010) Plasminogen activator inhibitor-1 (PAI-1): a key factor linking fibrinolysis and age-related subclinical and clinical conditions. Cardiovasc. Ther. 28, e72–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Croucher D. R., Saunders D. N., Lobov S., and Ranson M. (2008) Revisiting the biological roles of PAI2 (SERPINB2) in cancer. Nat. Rev. Cancer 8, 535–545 [DOI] [PubMed] [Google Scholar]
- 12.Dupont D. M., Madsen J. B., Kristensen T., Bodker J. S., Blouse G. E., Wind T., and Andreasen P. A. (2009) Biochemical properties of plasminogen activator inhibitor-1. Front. Biosci. (Landmark Ed.) 14, 1337–1361 [DOI] [PubMed] [Google Scholar]
- 13.Van De Craen B., Declerck P. J., and Gils A. (2012) The biochemistry, physiology and pathological roles of PAI-1 and the requirements for PAI-1 inhibition in vivo. Thromb. Res. 130, 576–585 [DOI] [PubMed] [Google Scholar]
- 14.Cazzolli G., Wang F., Beccara S. A., Gershenson A., Faccioli P., and Wintrode P. L. (2014) Serpin latency transition at atomic resolution. Proc. Natl. Acad. Sci. U.S.A. 111, 15414–15419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin Z., Jiang L., Yuan C., Jensen J. K., Zhang X., Luo Z., Furie B. C., Furie B., Andreasen P. A., and Huang M. (2011) Structural basis for recognition of urokinase-type plasminogen activator by plasminogen activator inhibitor-1. J. Biol. Chem. 286, 7027–7032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keijer J., Linders M., van Zonneveld A. J., Ehrlich H. J., de Boer J. P., and Pannekoek H. (1991) The interaction of plasminogen activator inhibitor 1 with plasminogen activators (tissue-type and urokinase-type) and fibrin: localization of interaction sites and physiologic relevance. Blood 78, 401–409 [PubMed] [Google Scholar]
- 17.Zhao G. X., Yuan C., Bian C. B., Hou X. M., Shi X. L., Ye X. M., Huang Z. X., and Huang M. D. (2006) Protein expression and preliminary crystallographic analysis of amino-terminal fragment of urokinase-type plasminogen activator. Protein Express. Purif. 49, 71–77 [DOI] [PubMed] [Google Scholar]
- 18.Olson S. T., Swanson R., Day D., Verhamme I., Kvassman J., and Shore J. D. (2001) Resolution of Michaelis complex, acylation, and conformational change steps in the reactions of the serpin, plasminogen activator inhibitor-1, with tissue plasminogen activator and trypsin. Biochemistry 40, 11742–11756 [DOI] [PubMed] [Google Scholar]
- 19.Lin Z., Jensen J. K., Hong Z., Shi X., Hu L., Andreasen P. A., and Huang M. (2013) Structural insight into inactivation of plasminogen activator inhibitor-1 by a small-molecule antagonist. Chem. Biol. 20, 253–261 [DOI] [PubMed] [Google Scholar]
- 20.Otwinowski Z., and Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 21.Weiss M. S. (2001) Global indicators of x-ray data quality. J. Appl. Crystallogr. 34, 130–135 [Google Scholar]
- 22.Xue Y., Bodin C., and Olsson K. (2012) Crystal structure of the native plasminogen reveals an activation-resistant compact conformation. J. Thromb. Haemost. 10, 1385–1396 [DOI] [PubMed] [Google Scholar]
- 23.Li W., and Huntington J. A. (2012) Crystal structures of protease nexin-1 in complex with heparin and thrombin suggest a 2-step recognition mechanism. Blood 120, 459–467 [DOI] [PubMed] [Google Scholar]
- 24.Vagin A., and Teplyakov A. (1997) MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025 [Google Scholar]
- 25.Renatus M., Bode W., Huber R., Stürzebecher J., Prasa D., Fischer S., Kohnert U., and Stubbs M. T. (1997) Structural mapping of the active site specificity determinants of human tissue-type plasminogen activator: implications for the design of low molecular weight substrates and inhibitors. J. Biol. Chem. 272, 21713–21719 [DOI] [PubMed] [Google Scholar]
- 26.Stout T. J., Graham H., Buckley D. I., and Matthews D. J. (2000) Structures of active and latent PAI-1: a possible stabilizing role for chloride ions. Biochemistry 39, 8460–8469 [DOI] [PubMed] [Google Scholar]
- 27.Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta. Crystallogr. D. Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 28.Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta. Crystallogr. D. Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 29.Laskowski R. A., MacArthur M. W., Mass D. S., and Thornton J. M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 30.DeLano W. L. (2004) The PyMol Molecular Graphics System, DeLano Scientific, San Carlos, CA [Google Scholar]
- 31.Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 32.Lamba D., Bauer M., Huber R., Fischer S., Rudolph R., Kohnert U., and Bode W. (1996) The 2.3 A crystal structure of the catalytic domain of recombinant two-chain human tissue-type plasminogen activator. J. Mol. Biol. 258, 117–135 [DOI] [PubMed] [Google Scholar]
- 33.Sharp A. M., Stein P. E., Pannu N. S., Carrell R. W., Berkenpas M. B., Ginsburg D., Lawrence D. A., and Read R. J. (1999) The active conformation of plasminogen activator inhibitor 1, a target for drugs to control fibrinolysis and cell adhesion. Structure 7, 111–118 [DOI] [PubMed] [Google Scholar]
- 34.Li W., Johnson D. J., Esmon C. T., and Huntington J. A. (2004) Structure of the antithrombin-thrombin-heparin ternary complex reveals the antithrombotic mechanism of heparin. Nat. Struct. Mol. Biol. 11, 857–862 [DOI] [PubMed] [Google Scholar]
- 35.Dementiev A., Simonovic M., Volz K., and Gettins P. G. (2003) Canonical inhibitor-like interactions explain reactivity of alpha1-proteinase inhibitor Pittsburgh and antithrombin with proteinases. J. Biol. Chem. 278, 37881–37887 [DOI] [PubMed] [Google Scholar]
- 36.Johnson D. J., Li W., Adams T. E., and Huntington J. A. (2006) Antithrombin-S195A factor Xa-heparin structure reveals the allosteric mechanism of antithrombin activation. EMBO J. 25, 2029–2037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson D. J., Langdown J., and Huntington J. A. (2010) Molecular basis of factor IXa recognition by heparin-activated antithrombin revealed by a 1.7-A structure of the ternary complex. Proc. Natl. Acad. Sci. U.S.A. 107, 645–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ye S., Cech A. L., Belmares R., Bergstrom R. C., Tong Y., Corey D. R., Kanost M. R., and Goldsmith E. J. (2001) The structure of a Michaelis serpin-protease complex. Nat. Struct. Biol. 8, 979–983 [DOI] [PubMed] [Google Scholar]
- 39.Renatus M., Engh R. A., Stubbs M. T., Huber R., Fischer S., Kohnert U., and Bode W. (1997) Lysine 156 promotes the anomalous proenzyme activity of tPA: X-ray crystal structure of single-chain human tPA. EMBO J. 16, 4797–4805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nienaber V., Wang J., Davidson D., and Henkin J. (2000) Re-engineering of human urokinase provides a system for structure-based drug design at high resolution and reveals a novel structural subsite. J. Biol. Chem. 275, 7239–7248 [DOI] [PubMed] [Google Scholar]
- 41.Madison E. L., Goldsmith E. J., Gerard R. D., Gething M. J., and Sambrook J. F. (1989) Serpin-resistant mutants of human tissue-type plasminogen activator. Nature 339, 721–724 [DOI] [PubMed] [Google Scholar]
- 42.Madison E. L., Goldsmith E. J., Gerard R. D., Gething M. J., Sambrook J. F., and Bassel-Duby R. S. (1990) Amino-acid-residues that affect interaction of tissue-type plasminogen-activator with plasminogen-activator inhibitor-1. Proc. Natl. Acad Sci. U.S.A. 87, 3530–3533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ibarra C. A., Blouse G. E., Christian T. D., and Shore J. D. (2004) The contribution of the exosite residues of plasminogen activator inhibitor-1 to proteinase inhibition. J. Biol. Chem. 279, 3643–3650 [DOI] [PubMed] [Google Scholar]
- 44.Madison E. L., Goldsmith E. J., Gething M. J., Sambrook J. F., and Gerard R. D. (1990) Restoration of serine protease-inhibitor interaction by protein engineering. J. Biol. Chem. 265, 21423–21426 [PubMed] [Google Scholar]
- 45.Wang Q., and Shaltiel S. (2003) Distal hinge of plasminogen activator inhibitor-1 involves its latency transition and specificities toward serine proteases. BMC Biochem. 4, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Refino C. J., Paoni N. F., Keyt B. A., Pater C. S., Badillo J. M., Wurm F. M., Ogez J., and Bennett W. F. (1993) A variant of T-Pa (T103N, KHRR-296–299 AAAA) that, by bolus, has increased potency and decreased systemic activation of plasminogen. Thromb. Haemost. 70, 313–319 [PubMed] [Google Scholar]
- 47.Tachias K., and Madison E. L. (1997) Variants of tissue-type plasminogen activator that display extraordinary resistance to inhibition by the serpin plasminogen activator inhibitor type 1. J. Biol. Chem. 272, 14580–14585 [DOI] [PubMed] [Google Scholar]
- 48.Dekker R. J., Eichinger A., Stoop A. A., Bode W., Pannekoek H., and Horrevoets A. J. (1999) The variable region-1 from tissue-type plasminogen activator confers specificity for plasminogen activator inhibitor-1 to thrombin by facilitating catalysis: release of a kinetic block by a heterologous protein surface loop. J. Mol. Biol. 293, 613–627 [DOI] [PubMed] [Google Scholar]
- 49.Katz B. A., Elrod K., Luong C., Rice M. J., Mackman R. L., Sprengeler P. A., Spencer J., Hataye J., Janc J., Link J., Litvak J., Rai R., Rice K., Sideris S., Verner E., and Young W. (2001) A novel serine protease inhibition motif involving a multi-centered short hydrogen bonding network at the active site. J. Mol. Biol. 307, 1451–1486 [DOI] [PubMed] [Google Scholar]
- 50.Vindigni A., Winfield M., Ayala Y. M., and Di Cera E. (2000) Role of residue Y99 in tissue plasminogen activator. Protein Sci. 9, 619–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Butenas S., Kalafatis M., and Mann K. G. (1997) Analysis of tissue plasminogen activator specificity using peptidyl fluorogenic substrates. Biochemistry 36, 2123–2131 [DOI] [PubMed] [Google Scholar]
- 52.Yu H. Y., Gao D., Zhang X., Jiang L. G., Hong Z. B., Fang X., Yuan C., Wang J. D., and Huang M. D. (2013) Synthesis of a weak basic uPA inhibitor and crystal structure of a complex with uPA. Chinese J. Struct. Chem. 32, 961–968 [Google Scholar]
- 53.Zhao B. Y., Xu P., Jiang L. G., Paaske B., Kromann-Hansen T., Jensen J. K., Sorensen H. P., Liu Z., Nielsen J. T., Christensen A., Hosseini M., Sorensen K. K., Nielsen N. C., Jensen K. J., Huang M. D., and Andreasen P. A. (2014) A cyclic peptidic serine protease inhibitor: increasing affinity by increasing peptide flexibility. PLoS One 9, e115872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sichler K., Kopetzki E., Huber R., Bode W., Hopfner K. P., and Brandstetter H. (2003) Physiological fIXa activation involves a cooperative conformational rearrangement of the 99-loop. J. Biol. Chem. 278, 4121–4126 [DOI] [PubMed] [Google Scholar]
- 55.Festoff B. W., Rao J. S., Rayford A., and Hantaï D. (1990) Plasminogen activators and their inhibitors in the neuromuscular system: 2. Serpins and serpin-protease complex receptors increase during in vitro myogenesis. J. Cell Physiol. 144, 272–279 [DOI] [PubMed] [Google Scholar]
- 56.Busso N., Belin D., Failly-Crépin C., and Vassalli J. D. (1987) Glucocorticoid modulation of plasminogen activators and of one of their inhibitors in the human mammary-carcinoma cell-line Mda-Mb-231. Cancer Res. 47, 364–370 [PubMed] [Google Scholar]
- 57.Lapchak P. A. (2002) Hemorrhagic transformation following ischemic stroke: significance, causes, and relationship to therapy and treatment. Curr. Neurol. Neurosci. Rep. 2, 38–43 [DOI] [PubMed] [Google Scholar]
- 58.Bennett W. F., Paoni N. F., Keyt B. A., Botstein D., Jones A. J., Presta L., Wurm F. M., and Zoller M. J. (1991) High-resolution analysis of functional determinants on human tissue-type plasminogen-activator. J. Biol. Chem. 266, 5191–5201 [PubMed] [Google Scholar]