

Background: Insulin regulates metabolism via the PI3K/Akt pathway.
Results: A kinome siRNA screen identified PFKFB3, a glycolysis regulator, as a modulator of insulin action. Manipulation of PFKFB3 activity or glycolysis affected insulin signaling.
Conclusion: Intracellular metabolism modulates important signal transduction pathways.
Significance: The novel link between glycolysis and growth factor signaling has important implications for the treatment of metabolic diseases.
Keywords: Akt PKB, glucose metabolism, glucose transporter type 4 (GLUT4), glycolysis, insulin, Warburg effect, PFKFB3
Abstract
The insulin/insulin-like growth factor (IGF)-1 signaling pathway (ISP) plays a fundamental role in long term health in a range of organisms. Protein kinases including Akt and ERK are intimately involved in the ISP. To identify other kinases that may participate in this pathway or intersect with it in a regulatory manner, we performed a whole kinome (779 kinases) siRNA screen for positive or negative regulators of the ISP, using GLUT4 translocation to the cell surface as an output for pathway activity. We identified PFKFB3, a positive regulator of glycolysis that is highly expressed in cancer cells and adipocytes, as a positive ISP regulator. Pharmacological inhibition of PFKFB3 suppressed insulin-stimulated glucose uptake, GLUT4 translocation, and Akt signaling in 3T3-L1 adipocytes. In contrast, overexpression of PFKFB3 in HEK293 cells potentiated insulin-dependent phosphorylation of Akt and Akt substrates. Furthermore, pharmacological modulation of glycolysis in 3T3-L1 adipocytes affected Akt phosphorylation. These data add to an emerging body of evidence that metabolism plays a central role in regulating numerous biological processes including the ISP. Our findings have important implications for diseases such as type 2 diabetes and cancer that are characterized by marked disruption of both metabolism and growth factor signaling.
Introduction
The insulin/insulin-like growth factor (IGF)-1 signaling pathway (ISP)10 regulates a number of cellular processes, particularly those relating to metabolism including glucose transport, glycogen synthesis, protein synthesis, lipid metabolism, and gene expression (1). One of these processes specific to fat and muscle cells is the translocation of the glucose transporter GLUT4 to the plasma membrane (PM) to facilitate glucose uptake. This process is defective in metabolic diseases such as insulin resistance and type 2 diabetes (T2D). Insulin triggers GLUT4 translocation via the PI3K/Akt pathway, resulting in phosphorylation of downstream substrates including the Akt substrate AS160. In addition, a number of other protein kinases have been implicated in insulin action including Cdk5 (2, 3), the G protein-coupled receptor kinase 2 (GRK2) (4, 5), Src family kinases (6, 7), members of the PKC family (8), and JNK (9). To understand the role of protein kinases and phosphorylation in insulin action we recently performed global analysis of insulin-regulated protein phosphorylation in 3T3-L1 adipocytes. This identified changes in >5,000 phosphorylation sites in response to insulin (10). Although this comprised substrates of Akt and other kinases known to play a role in insulin action, clearly there are many insulin-regulated substrates with no known kinase.
In this study, we set out to identify additional kinases that regulate insulin action. We chose insulin regulation of GLUT4 translocation to the PM as our end point because this is one of the most complex and important actions of insulin and it is postulated to represent one of the earliest contributors to the development of insulin resistance (11). We established a novel GLUT4 translocation assay in HeLa cells, facilitating high throughput analysis. We then performed a global kinase screen using this system in combination with siRNA knockdown of all kinases in the human genome. This resulted in identification of ∼300 kinases with a putative involvement in insulin action. Surprisingly there was an enrichment of kinases that play an important role in glucose metabolism. We next focused on one of these kinases, PFKFB3 (6-phosphofructo-2-kinase fructose-2,6-bisphosphatase 3). This enzyme regulates glycolysis through the production of fructose 2,6-bisphosphate (Fru-2,6-BP) (12) a potent allosteric activator of 6-phosphofructo-1-kinase (PFK-1), the rate-limiting step in glycolysis. Using a variety of pharmacological and genetic approaches we confirmed that PFKFB3 has an important role in insulin action. The mechanism for this effect involves a positive feedback regulation of glycolysis onto the Akt signaling pathway. These data have important implications for the Warburg effect in tumor cells.
Experimental Procedures
Reagents, Antibodies and Constructs
General chemicals were purchased from Sigma unless otherwise stated. FCS, DMEM, antibiotics, Glutamax, bicinchoninic acid reagent, and SuperSignal West Pico chemiluminescent substrate were obtained from Thermo Scientific. BSA was purchased from Bovostar. Inhibitors were purchased from Calbiochem (3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one, 3-PO) and Thermo Scientific (α-cyano-8-(1-phenylindol-3-yl)acrylate, UK-5099). GPR81 agonists were from Calbiochem (3-chloro-5-hydroxybenzoic acid) and Toronto Research Chemicals (3,5-dihydroxybenzoic acid). Antibodies were purchased from Covance Research Products (HA), Sigma (FLAG, α-tubulin), Roche Applied Science (GFP), Santa Cruz Biotechnology (14-3-3), Cell Signaling Technology (pSer473-Akt, pThr308-Akt, Akt, pThr642-AS160, pThr246-PRAS40, PRAS40, pSer235/236-S6, pThr389-S6K, pSer21/9-GSKα/β, and β-actin), Invitrogen (Alexa 488-conjugated antibody), Rockland Immunochemicals (IRDye 700- or 800-conjugated antibodies), GE Healthcare (HRP-conjugated antibodies), and Abgent (PFKFB3, N-terminal). The antibody against AS160 was previously described (13). Human PFKFB3 in pDEST47 was a gift from Charles Watt (Garvan Institute) and was originally derived from a Open Biosystems clone (catalog number MHS1010-9203644). PFKFB3 was subcloned into Gateway converted p3xFLAG-CMV-10 (Sigma) using Gateway cloning (Clontech).
Cell Culture and Transfection
3T3-L1 fibroblasts were obtained from Howard Green (14) and cultured and differentiated to adipocytes as previously described (15). HA-GLUT4 retrovirus was produced using Plat-E cells and used for infection of 3T3-L1 cells as previously described (15). HEK293 cells were transfected with FLAG-PFKFB3, PFKFB3 in pDEST47, or empty vector control p3xFLAG-CMV-10 or pEGFP-N1 (Clontech) using Lipofectamine 2000 (Thermo Scientific) according to the manufacturer's instructions 48 h prior to experiments. To establish the HA-GLUT4-HeLa cell line, HeLa cells were transfected with linearized HA-GLUT4 in pBABE-puro (15) using FuGENE HD (Thermo Scientific) according to the manufacturer's instructions. HeLa cell colonies stably expressing HA-GLUT4 were selected with 1 μg/ml of puromycin and screened for GLUT4 expression and insulin-stimulated GLUT4 translocation to the PM.
siRNA Screen and HA-GLUT4 Translocation Assay
Reverse transfection was performed using Dharmafect1 and siGenome SMARTpool and On Target-plus pool and single oligo siRNAs (Dharmacon) in HA-GLUT4 HeLa cells in 96-well plates. Cells were incubated for 72 h after transfection before the GLUT4 translocation assay. Cells were serum-starved for 2 h, followed by incubation in the presence or absence of 100 ng/ml of IGF-1 for 15 min, fixed, and immunostained for surface (and total in secondary screen) HA-GLUT4 as previously described (16). The assay was performed in duplicate 96-well plates per condition with controls on each plate amounting to a total of 10 plates in quadruplicates (duplicates of basal and IGF-1). Nuclei were stained with Hoechst nuclear stain. Fluorescence intensity was quantified using an IN Cell Analyzer 2000 (GE Healthcare). Images were collected at ×20 magnification and 9 fields/well.
Image and Data Analysis
Image analysis and processing was performed using IN Cell developer toolbox 1.8 to obtain fluorescence intensity of surface HA-GLUT4, normalized to cell number (Fig. 1A). Image processing involved object and intensity segmentation and post processing including binary sieving and erosion. Fluorescence intensity and area were determined for surface HA-GLUT4 staining and nuclei count for DAPI staining and the ratio of fluorescence intensity × area divided by nuclei was calculated. Z-scores were calculated for each plate independently. Mean ± S.D. (n = 2) of the duplicates were calculated for raw values (fluorescence intensity × area/nuclei count) and Z-scores. Z-score mean was used for filtering and selection for secondary screen.
FIGURE 1.

Establishment of siRNA screen in HA-GLUT4-HeLa cells. A, HA-GLUT4-HeLa cells were subject to IGF-1-stimulated HA-GLUT4 translocation assay and images taken at ×20 magnification using an IN Cell Analyzer detecting nuclei (1) or surface GLUT4 (3). Images were analyzed using IN Cell developer toolbox 1.8 software. Object segmentation and post-processing including binary sieving and erosion was applied to identify and count the nuclei (2) from the unprocessed image (1). To quantify surface GLUT4, intensity segmentation was applied to exclude background fluorescence and post-processing (binary sieving) was applied to measure the fluorescence intensity and the area (4). Surface GLUT4 (fluorescence intensity × area) was divided by number of nuclei for normalization. B, HA-GLUT4-HeLa cells were serum starved for 2 h before treatment with or without 200 nm insulin, 100 ng/ml of IGF-1, 100 ng/ml of PDGF, or 100 ng/ml of EGF for 15 min. GLUT4 translocation to the PM was determined by surface HA-GLUT4 staining normalized to total HA-GLUT4, n = 4, standard deviation is shown, *, p < 0.001; **, p < 0.0001 versus basal. C, immunofluorescence of HA-GLUT4 in HA-GLUT4-HeLa cells in the presence or absence of 100 ng/ml of IGF-1 for 15 min was performed. Scale bar is 5 μm. D, HA-GLUT4-HeLa cells and HA-GLUT4–3T3-L1 adipocytes were serum-starved for 2 h, followed by incubation with or without 100 nm PI3K inhibitor wortmannin, and stimulation with or without 200 nm insulin or 100 ng/ml of IGF-1 for 15 min. Cell lysates were immunoblotted with the indicated antibodies (loading control: β-actin). E and F, validation of the siRNA screen in HA-GLUT4-HeLa cells with siRNAs against components of the insulin signaling pathway, including insulin receptor (INSR), AS160, IRS1, Akt1, Akt2, Akt1 + 2, and non-targeting control (NTC). 72 h after siRNA transfection, cells were serum starved, stimulated with or without 100 ng/ml of IGF-1 for 15 min, followed by HA-GLUT4 translocation assay and image analysis. Mean ± S.D. of raw values of 2 replicates (E) and representative images taken by IN Cell Analyzer (F) are shown. Scale bar is 70 μm.
Lactate Determination
Lactate efflux into the extracellular medium was determined as described previously (17). Cells were lysed in PBS containing 2% SDS and the protein concentration was determined by bicinchoninic acid assay and media lactate levels were normalized to protein content. For intracellular lactate measurements cells were washed twice with ice-cold PBS and quenched in a freezing cold solution of methanol and H2O. Metabolites were extracted with an equal volume of ice-cold chloroform, the supernatant was evaporated at 45 °C and resuspended in H2O. Lactate was measured using a tetrazolium-coupled lactate assay (104) with modifications. Briefly, the sample was incubated with 2× reaction buffer (1.1 mm thiazolyl blue tetrazolium bromide, 0.45 mm phenazine methosulfate, 2.1 mm NAD+, 39 IU/ml of lactate dehydrogenase (Roche), 5.3 g/liter of glycine, 1.4% (v/v) Triton X-100). The reaction was incubated for 15 min in the dark at room temperature and absorbance was measured at 590 nm.
Extracellular Acidification Rate
Extracellular acidification rate was measured using the Seahorse Bioanalyser XF24 system (Seahorse Biosciences). Cells growing in XF24 plates were incubated in buffer-free DMEM for the duration of the experiment, with additions made by injection according to the manufacturer's instructions.
siRNA Transfection in 3T3-L1 Adipocytes
3T3-L1 adipocytes were transfected with scrambled and PFKFB3 siRNA, as previously described (18). A pool of 4 siRNAs was used for PFKFB3 (#1, caacgaaagtgttcaatgttt; #2, ccaagaagctgactcgctatt; #3 gttctacgctgcctactagtt; #4, cgaattgtatactacctgatt) and one scrambled siRNA (gacttaactcatccaacgatt).
Quantitative Real-time RT-PCR Assays
RNA extraction and real-time PCR analysis were performed as previously described (19). Primers and probes for the PFKFB3 mouse gene were selected according to the Universal Probe Library System (Roche Applied Science). The Cyclophilin gene was used as a control. The following primers were used for PFKFB3, cactgcgtgaacaggacaag and tggcgctctaattccatgat, and for Cyclophilin, ttcttcataaccacagtcaagacc and accttccgtaccacatccat.
Fructose-2,6-bisphosphate (Fru-2,6-BP) Assay
Fru-2,6-BP was measured in 3T3-L1 adipocytes using a fluorescent assay as previously described (20). Fru-2,6-BP levels were normalized to protein amount as determined by bicinchoninic acid assay.
Glucose Uptake
[3H]2-Deoxyglucose (PerkinElmer Life Sciences) uptake into 3T3-L1 adipocytes was determined as described previously (21).
Immunoblotting
Cells were incubated in serum-free DMEM containing 0.2% BSA for 2 h prior to stimulation with insulin or IGF-1. Cell lysates were subjected to SDS-PAGE analysis and immunoblotting with indicated antibodies as previously described (22).
Immunofluorescence Microscopy
HA-GLUT4-HeLa cells were incubated in serum-free medium for 2 h, followed by incubation with or without 100 ng/ml of IGF-1 for 15 min and subsequently fixed, permeabilized, and stained with HA antibody as previously described (23).
Pathway and Kinase Enrichment Analysis
A hypergeometric test was performed to identify over-representation of KEGG pathways in identified kinases using gene set collection C2 CP:KEGG from the molecular signature database (MSigDB) (24) and a universe of ∼45,000 genes. The false discovery rate q-value for the hypergeometric p value was obtained by correcting for multiple hypothesis testing using a Benjanmin and Hochberg method and q-value <0.05 was considered significant. Broad disease-related enriched pathways were excluded from further analysis. The kinase composition of each of the remaining significantly over-represented pathways was further characterized and the overlap between the kinases identified in the screen and the kinases in each pathway was calculated. Pathways with at least 2 overlapping kinases and >25% kinase enrichment were selected.
Statistical Analysis
Data are expressed as mean ± S.E. unless otherwise stated. p values were calculated by t test, one-way analysis of variance or two-way analysis of variance using GraphPad Prism.
Results
Establishment of siRNA Screen in HA-GLUT4-HeLa Cells
To identify novel kinases that play a role in insulin action we chose GLUT4 translocation as the end point and developed a GLUT4 translocation assay in HeLa cells stably expressing a GLUT4 reporter for a number of reasons: (a) GLUT4 translocation is a complex action of insulin involving several processes including signal transduction, metabolism, and regulated vesicle trafficking; (b) impaired GLUT4 translocation is one of the earliest defects in the development of insulin resistance and type 2 diabetes and hence is an important action of insulin; (c) HeLa cells provide an excellent system for high throughput siRNA screening due to their ease of transfection and knockdown efficiency; (d) cell lines that do not endogenously express GLUT4 including CHO (25, 26), HeLa (27), and HEK cells (28) have previously been used to study trafficking of transfected GLUT4; and (e) as shown below, HeLa cells recapitulate many of the characteristics of insulin-regulated GLUT4 trafficking in adipocytes. We developed HeLa cells stably expressing a GLUT4 reporter (HA-GLUT4) that facilitates measurement of surface GLUT4 in intact cells (16) (Fig. 1A). GLUT4 translocation to the PM was quantified in the presence of several growth factors. Among these, IGF-1 and EGF induced the most significant increase in PM GLUT4 (Fig. 1B). IGF-1 was selected for the siRNA screen because IGF-1 signaling, like insulin, requires IRS1 phosphorylation for PI3K activation. IGF-1 signals via the insulin and IGF-1 receptors and increases GLUT4 translocation and glucose uptake via pathways common to insulin (29, 30). The HA-GLUT4-HeLa cell system recapitulated many of the features of insulin-regulated GLUT4 trafficking in adipocytes. HA-GLUT4 immunostaining in HeLa cells exhibited basal and IGF-1-stimulated localization analogous to endogenous GLUT4 in 3T3-L1 adipocytes (31) (Fig. 1C). IGF-1 stimulation resulted in a robust increase in phosphorylation of components of the insulin signaling pathway in HA-GLUT4-HeLa cells similar to HA-GLUT4 expressing 3T3-L1 adipocytes (Fig. 1D). HA-GLUT4 translocation to the PM in HA-GLUT4-HeLa cells was measured as previously described (16) except an IN CELL Analyzer was used for quantitation (Fig. 1A). To validate the use of these cells for siRNA kinome screen, we transfected cells with siRNAs targeting a range of components of the ISP. Knockdown of the insulin receptor, IRS1, Akt1, Akt2, and Akt1 + 2 (Akt1/2) impaired IGF-1-stimulated GLUT4 translocation to the PM in HA-GLUT4-HeLa cells (Fig. 1, E and F), whereas knockdown of AS160, a negative regulator of GLUT4 translocation (13, 32, 33), increased GLUT4 translocation in unstimulated (basal) conditions (Fig. 1, E and F). These data confirmed that IGF-1-stimulated GLUT4 translocation in HA-GLUT4-HeLa cells shared many of the essential features of insulin-stimulated GLUT4 translocation in 3T3-L1 adipocytes and hence HA-GLUT4-HeLa cells provide an ideal system for high throughput analysis of insulin action.
siRNA Kinase Screen of GLUT4 Translocation to the PM
The siRNA screen targeted 779 human kinases, including protein, lipid, and metabolite kinases, as well as kinase regulatory proteins (Fig. 2). Z-scores were calculated for each siRNA treatment in both basal and IGF-1-stimulated conditions (Fig. 2B, supplemental Table S1). These were compared with several controls, including a non-targeting control (NTC, Z-score basal −0.54, stimulated 0.22) and siRNAs targeting Akt1 + 2 (Z-score stimulated −0.75), and AS160 (Z-score basal 0.9). Hits from the screen were defined as either positive or negative kinases. Positive kinases were those that impaired IGF-1-stimulated GLUT4 translocation when knocked down with a Z-score ≤−0.75. Negative kinases were those that stimulated PM GLUT4 levels under basal conditions upon knockdown with a Z-score basal ≥0.9 (Fig. 2, A and B). We identified 112 negative kinases and 189 positive kinases in the primary screen.
FIGURE 2.

High throughput siRNA screen identifies kinases involved in GLUT4 translocation. A, the screen workflow is shown. In the primary screen, HA-GLUT4-HeLa cells were transfected with siRNA pools targeting 779 kinases and HA-GLUT4 translocation to the PM in response to IGF-1 stimulation was determined as described under “Experimental Procedures.” B, results of the primary screen with siRNA pools targeting 779 kinases are shown. Z-score of stimulated PM GLUT4 was plotted against the Z-score of basal PM GLUT4 for each of the kinases, with 3 controls (NTC, AS160, and Akt1 + 2) indicated. Dashed lines indicate Z-score cut-off to identify hits (Z-score stimulated ≤−0.75 = positive kinases, Z-score basal ≥ 0.9 = negative kinases). Components of enriched KEGG pathways are indicated. C, secondary screen with 4 single siRNAs was performed on 64 candidate kinases (34 positive and 30 negative kinases) and the number of oligos that reproduced the result of the primary screen (greater effect or within 10% of primary screen result) are indicated. Validated kinases (≥2 oligos) are shown in bold.
We performed pathway over-representation analysis followed by kinase enrichment analysis for the 301 kinases identified as “hits” in the primary screen. This revealed an enrichment of kinases involved in signaling pathways known to play a role in GLUT4 translocation, thus validating the screen. These included signaling pathways for insulin, mechanistic target of rapamycin, and phosphatidylinositol (Fig. 3, supplemental Table S2). Interestingly, 10 of 32 ranked pathways identified in this analysis were involved in metabolism and the majority were directly involved in glucose metabolism: glycolysis/gluconeogenesis, pentose phosphate pathway, fructose and mannose metabolism, purine metabolism, and galactose and amino/nucleotide sugar metabolism. This implicates glucose metabolism as a potential regulator of GLUT4 translocation, in addition to being downstream of GLUT4-mediated glucose uptake.
FIGURE 3.
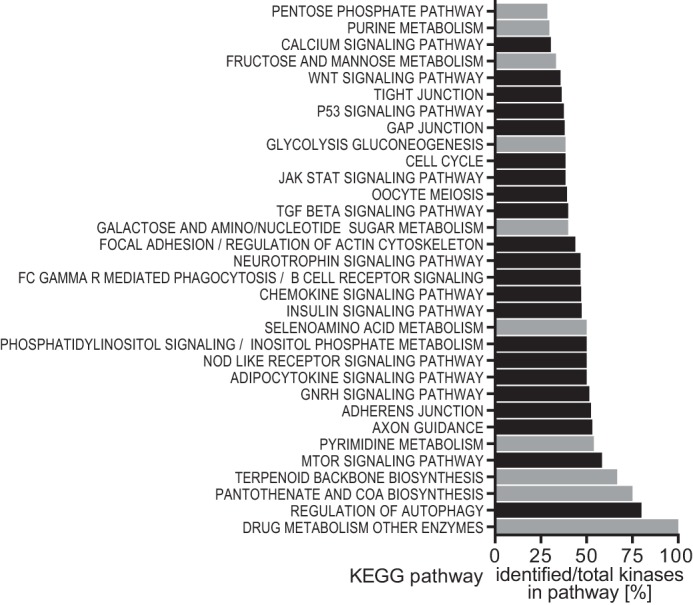
Pathway and kinase enrichment analysis reveals a role for metabolic pathways in GLUT4 translocation. Identified kinases from the primary screen were subjected to pathway enrichment analysis, followed by kinase enrichment analysis of significantly enriched pathways as described under “Experimental Procedures.” The percentage of identified kinases out of total kinases is shown for all significantly enriched pathways that showed more than 25% kinase enrichment. Metabolic pathways are shown in gray.
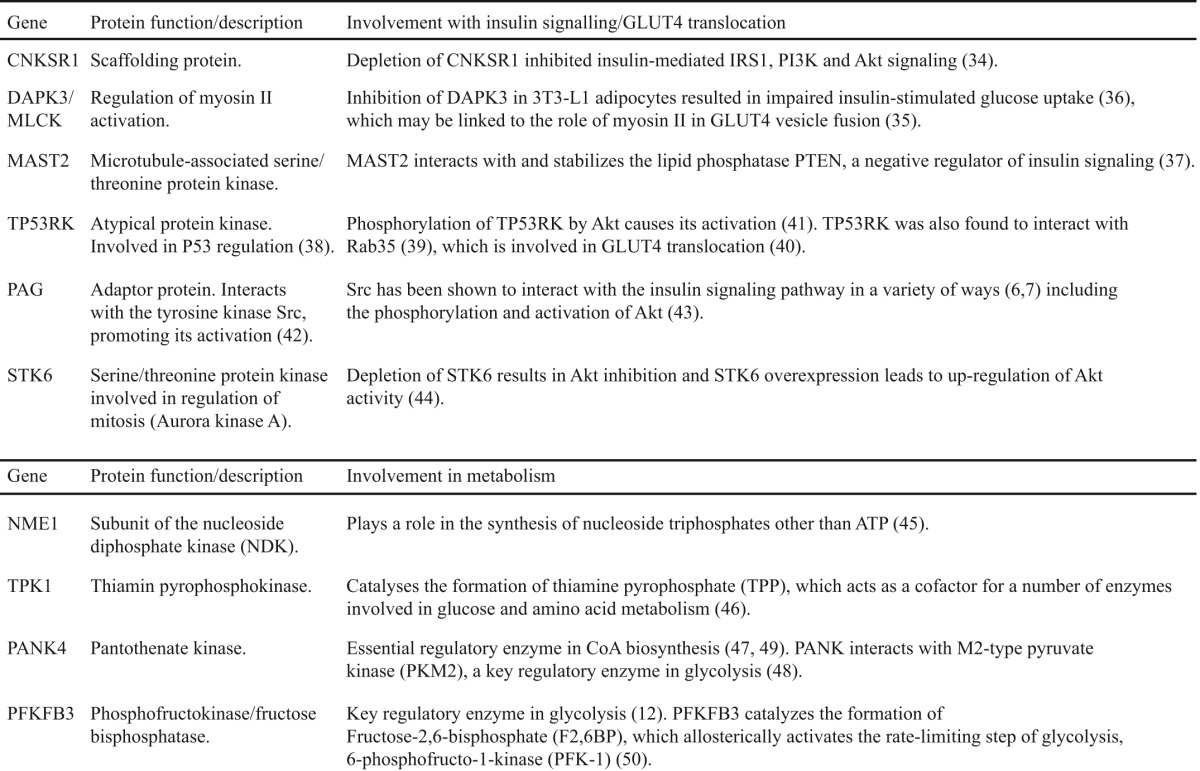
For the secondary screen we chose the top 30 negative kinases (basal Z-score ≥1.6) and the top 28 positive kinases (stimulated Z-score ≤−1.27) that were expressed in HeLa cells by gene expression. In addition, we included 6 lower ranked positive kinases (stimulated Z-score ≤−0.8) because they had also been identified in a separate screen for insulin action in adipocytes (10). In the secondary screen, we deconvoluted the siRNA pools used in the primary screen to test each of the four siRNAs separately (Fig. 2A). In addition to basal and IGF-1-stimulated GLUT4 translocation, we also determined total GLUT4 levels in the secondary screen and eliminated siRNA targets that modulated total GLUT4 levels. Hits were considered as validated if at least two oligonucleotides in the pool yielded the same phenotype as observed in the primary screen. As a result of the secondary screen, 10 negative and 23 positive kinases were validated (Fig. 2C). Raw values of the GLUT4 translocation to the PM and Z-scores from the primary screen of these 33 hits are shown in Fig. 4. These candidates include a number of kinases that have previously been implicated in insulin signaling: connector enhancer of kinase suppressor of Ras 1 (CNKSR1) (34), death-associated protein kinase 3 (DAPK3)/myosin light chain kinase (35, 36), microtubule-associated serine/threonine-protein kinase 2 (MAST2) (37), TP53 regulating kinase (TP53RK) (38–41), the Src adaptor protein phosphoprotein associated with glycosphingolipid microdomains (PAG) (6, 7, 42, 43), and STK6 (44) (Fig. 4 and Table 1). Among these candidates we also identified a number of metabolic regulatory kinases: NME1, a subunit of the nucleoside diphosphate kinase (45), thiamin pyrophosphokinase (TPK1) (46), pantothenate kinase (PANK4) (47–49), and PFKFB3 (12, 50) (Fig. 4 and Table 1).
FIGURE 4.

siRNA screen identifies 23 positive and 10 negative kinases. HA-GLUT4-HeLa cells were transfected with the indicated siRNAs and GLUT4 translocation to the PM was determined as described under “Experimental Procedures.” Raw values (fluorescence intensities/nuclei counts) (A) and Z-scores (B) of basal and IGF-1-stimulated GLUT4 translocation to the PM are shown. Mean ± S.D. of n = 2 for kinases and n = 10 for controls are shown. Controls (NTC, AS160, and Akt1 + 2) and PFKFB3 are indicated in bold and yellow. Dotted lines (in B) indicate Z-score cut-offs for the secondary screen, Z-score basal ≥1.6 for negative kinases (blue dotted line) and Z-score stimulated ≤ −0.8 for positive kinases (red dotted line). *, p < 0.0001; #, p < 0.01; §, p < 0.05 versus NTC.
TABLE 1.
Selected validated hits
PFKFB3 Inhibition in 3T3-L1 Adipocytes Decreases Insulin-stimulated GLUT4 Translocation to the PM, Reduces Lactate Efflux and Impairs Akt Signaling
In view of the enrichment of glucose metabolism pathways in the pathway analysis of the identified kinases in our screen, we selected PFKFB3, a key regulator of glycolysis, for further characterization (12). PFKFB3 catalyzes the formation of Fru-2,6-BP, which allosterically activates the rate-limiting step of glycolysis, PFK-1 (50). PFKFB3 is highly expressed in adipocytes and its expression increases during the differentiation process, accompanied by an increase in its product Fru-2,6-BP (51, 52). Disruption of PFKFB3 in mice causes decreased insulin signaling and exacerbates high fat diet-induced insulin resistance (52). Furthermore, PFKFB3 protein and its product Fru-2,6-BP are increased in several cancers and are suggested to contribute to the Warburg effect (12). Finally, our analysis of the insulin-regulated phosphoproteome in adipocytes revealed PFKFB3 as one of the most highly regulated insulin-dependent phosphoproteins (10).
In the present screen, PFKFB3 was identified as a positive regulatory kinase of GLUT4 translocation, as its knockdown impaired IGF-1-stimulated GLUT4 translocation (Figs. 1F, 2B, and 4, Z-score −1.54). To validate this finding in 3T3-L1 adipocytes, we used siRNA to knockdown PFKFB3 mRNA in 3T3-L1 adipocytes by 69% (Fig. 5A). PFKFB3 knockdown resulted in reduced extracellular lactate by 38%, consistent with reduced glycolytic flux, which is a key action of PFKFB3 activity (Fig. 5B). PFKFB3 knockdown also led to a reduction in insulin-stimulated 2-deoxyglucose uptake by 22% (Fig. 5C), confirming a role for PFKFB3 in insulin-stimulated GLUT4 translocation and validating the siRNA screen. To further characterize this kinase in 3T3-L1 adipocytes, we employed 3-PO, a competitive inhibitor of PFKFB3 that binds to the substrate binding site in PFKFB3 (53). 3-PO is specific for PFKFB3 as verified in cells with reduced PFKFB3 (53, 54), and it has been widely used to inhibit PFKFB3 in vitro and in vivo (53–59). In 3T3-L1 adipocytes, insulin significantly increased the levels of Fru-2,6-BP, the product of PFKFB3 activity, by 1.7-fold and incubation of cells with 3-PO blocked this increase, thus confirming that 3-PO inhibited PFKFB3 (Fig. 5D). Insulin markedly increased lactate efflux from 3T3-L1 adipocytes, measured by the extracellular acidification rate, and this effect was inhibited by 3-PO (Fig. 5E), consistent with the siRNA data. In addition, 3-PO inhibited insulin-stimulated glucose uptake (Fig. 5F) and GLUT4 translocation in 3T3-L1 adipocytes (Fig. 5G). The effect with 3-PO was greater than with PFKFB3 knockdown and this likely involves residual PFKFB3 activity due to incomplete knockdown. To explore the mechanism for this effect, we examined insulin signaling. 3-PO blunted insulin-mediated phosphorylation of Akt at Ser-473, whereas phosphorylation at the other key regulatory site Thr-308 was significantly increased with 3-PO (Fig. 5H). Although it is thought that Akt phosphorylation at Thr-308 and Ser-473 are representative of Akt activity, it was recently reported that Akt hyperphosphorylation can occur when Akt kinase activity is inhibited, due to a conformational change in Akt that shields the phosphosites from phosphatases (60, 61). We therefore assessed Akt activity by measuring phosphorylation of the Akt substrate GSK3 and the downstream substrate S6K. Despite the increase in Thr-308 phosphorylation, Akt kinase activity was inhibited with 3-PO as indicated by reduced insulin-dependent phosphorylation of GSK3 and S6K (Fig. 5H). These data suggest that PFKFB3 and/or glycolysis regulates insulin signaling possibly at the level of Akt.
FIGURE 5.
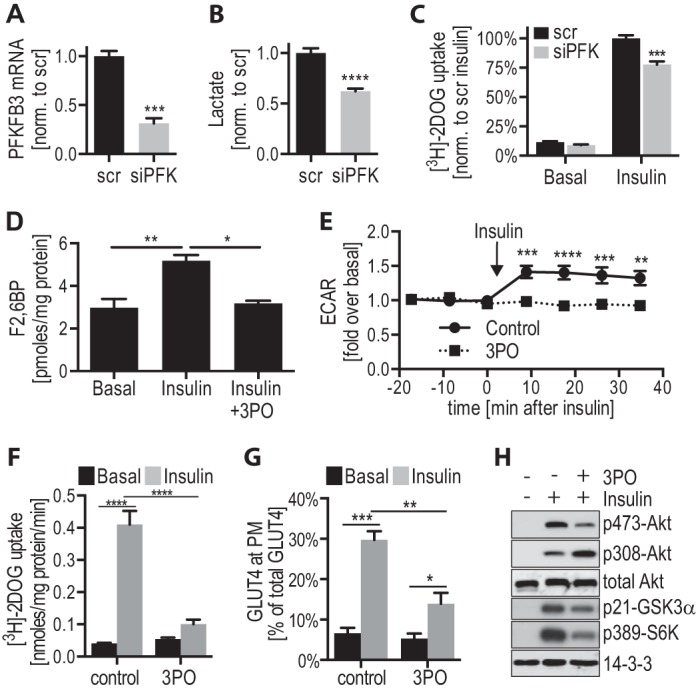
Inhibition of PFKFB3 activity inhibits insulin action in 3T3-L1 adipocytes. A–C, 3T3-L1 adipocytes were transfected with scrambled (scr) siRNA or PFKFB3 siRNA (siPFK) 96 h prior to experiments. A, cell medium that was incubated on the cells for 16 h was collected and lactate levels were measured and normalized to Scr control (n = 11). B, mRNA was isolated from Scr- and siPFK-transfected adipocytes, quantitative PCR was performed for PFKFB3 and normalized to cyclophilin control (n = 3). C, Scr- and siPFK-transfected 3T3-L1 adipocytes were incubated in serum-free medium, followed by insulin (100 nm) stimulation. Glucose uptake was measured using [3H]2-deoxyglucose ([3H]-2DOG) (n = 3). D–F, 3T3-L1 adipocytes were incubated in serum-free medium for 1.5 h, followed by incubation with 100 μm 3-PO or dimethyl sulfoxide control for 30 min, prior to stimulation with or without 100 nm insulin for 20 min (or indicated time). D, Fru-2,6-BP (F2,6BP) was measured in 3T3-L1 adipocytes ± 3-PO. Mean ± S.D. of n = 2 is shown. E, extracellular acidification rate (ECAR) was measured during insulin stimulation ±3-PO using a Seahorse Bioanalyser (n = 3). F, insulin-stimulated glucose uptake ± 3-PO was measured using [3H]-2DOG (n = 5). G, insulin-stimulated HA-GLUT4 translocation to the PM ± 3-PO was determined in HA-GLUT4 expressing 3T3-L1 adipocytes (n = 7). H, insulin-stimulated phosphorylation of components of the insulin signaling pathway ± 3-PO was determined by immunoblotting cell lysates with the indicated antibodies (loading control: 14-3-3) and representative immunoblots are shown (n = 3). *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001, error bars represent S.E. (except D).
PFKFB3 Overexpression Increases Insulin Signaling and Lactate Efflux
Because PFKFB3 disruption perturbed insulin action and signaling, we next sought to explore the effects of PFKFB3 overexpression on insulin signaling. Because 3T3-L1 adipocytes have high levels of PFKFB3 expression, we switched to HEK293 cells because they have lower PFKFB3 expression (data not shown). PFKFB3 overexpression in HEK293 cells resulted in a 1.7-fold increase in lactate efflux (Fig. 6A) and augmented insulin-stimulated phosphorylation of Akt on Ser-473 and Thr-308 by 1.5- and 2-fold, respectively (Fig. 6, B and C). Akt activity was increased as determined by the 1.5-fold increase in phosphorylation of the Akt substrate, AS160.
FIGURE 6.
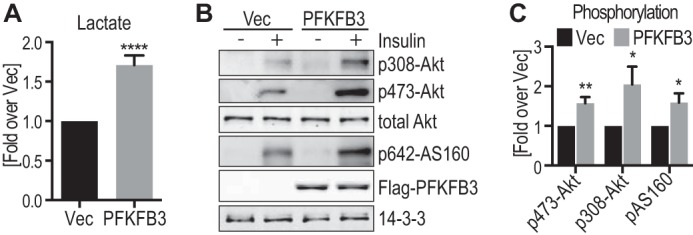
PFKFB3 overexpression results in increased lactate production and Akt activity in HEK293 cells. HEK293 cells were transfected with PFKFB3 or vector control (Vec) 48 h prior to serum starvation for 2 h, followed by stimulation with or without 100 nm insulin for 20 min. A, prior to starvation, cell medium that was incubated on the cells for 16 h was collected and lactate levels were measured and normalized to vector control between experiments (n = 14). B, cell lysates were immunoblotted with the indicated antibodies and representative immunoblots are shown. C, quantification of insulin-stimulated Akt and AS160 phosphorylation (data in B) is shown (phospho/total), normalized to insulin-stimulated cells transfected with vector control (n = 11–21). *, p < 0.05; **, p < 0.01; ****, p < 0.0001 versus Vec control, error bars represent S.E.
Exogenous Lactate Did Not Increase Insulin Signaling
Given that modulating PFKFB3 resulted in a change in lactate levels concomitant with a change in insulin signaling, we next explored whether lactate per se affected insulin signaling. Lactate has been shown to act as a signaling molecule in adipocytes by binding to the GPR81 receptor (62, 63). To test if this could explain the effects of PFKFB3 overexpression on Akt activity, we tested the effects of extracellular lactate or GPR81 agonists on insulin signaling. Neither exogenous lactate nor the GPR81 agonists, 3-chloro-5-hydroxybenzoic acid and 3,5–3,5-dihydroxybenzoic acid had any significant effect on insulin signaling in 3T3-L1 adipocytes (Fig. 7 and data not shown). These data indicate that autocrine effects of lactate were not responsible for the increase in insulin signaling.
FIGURE 7.
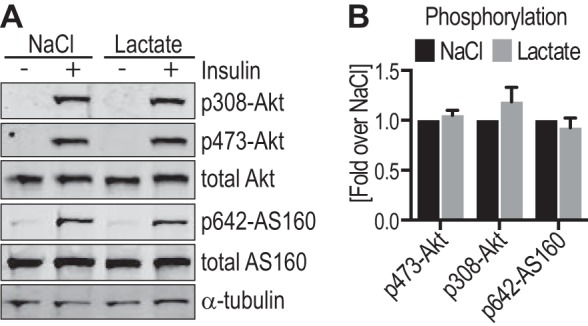
Extracellular lactate had no effect on insulin signaling in 3T3-L1 adipocytes. 3T3-L1 adipocytes were incubated either with 25 mm l-lactate (sodium salt) or 25 mm NaCl in serum-free medium, followed by stimulation with or without 10 nm insulin for 20 min. A, cell lysates were immunoblotted with the indicated antibodies (loading control: α-tubulin) and representative immunoblots are shown. B, quantification of insulin-stimulated Akt and AS160 phosphorylation (data in A) is shown (phospho/total), normalized to insulin-stimulated cells incubated with NaCl (n = 9–11). Error bars represent S.E.
Modulation of Aerobic Glycolysis Affects Insulin Signaling
We next hypothesized that glycolysis per se might potentiate Akt signaling. To test this we used two separate approaches to manipulate the distribution of glucose between glycolysis and mitochondria, thereby enhancing or reducing glycolysis. Initially we used UK-5099, a potent and specific inhibitor of the mitochondrial pyruvate carrier (MPC) (64, 65) in 3T3-L1 adipocytes. UK-5099 dose-dependently potentiated insulin-stimulated lactate efflux (Fig. 8A), suggesting that MPC inhibition redirected pyruvate metabolism toward lactate, thus enhancing glycolysis. Inhibition of MPC significantly increased insulin-stimulated phosphorylation of Akt at Thr-308 by 1.5-fold, whereas phosphorylation at Ser-473 was unchanged (Fig. 8, B and C). These data show that increasing glycolysis potentiated insulin signaling. We next used α-cyano-4-hydroxycinnamate, an inhibitor of the lactate transporter (MCT1) (66, 67), to determine whether increased intracellular lactate and the concomitant reduction in glycolytic flux (68) had an effect on insulin signaling. α-Cyano-4-hydroxycinnamate had a potent effect on MCT1 activity as indicated by a dose-dependent increase in intracellular lactate and by reduced extracellular lactate, which was undetectable at doses ≥2 mm α-cyano-4-hydroxycinnamate (Fig. 8D). Notably, MCT1 inhibition also impaired insulin signaling with similar dose-response characteristics to that observed for lactate efflux (Fig. 8, E and F). In summary, enhancing or inhibiting glycolysis resulted in respective changes in insulin signaling.
FIGURE 8.

Modulation of aerobic glycolysis affects insulin signaling in 3T3-L1 adipocytes. A-C, 3T3-L1 adipocytes were treated with the indicated doses of MPC inhibitor UK-5099 in serum-free medium for 2 h, prior to stimulation with or without 10 nm insulin for 20 min. A, lactate was measured in the medium that was collected after stimulation with insulin, normalized to protein levels in each experiment, and normalized to insulin-stimulated control cells between experiments (n = 3 (basal) to 5 (insulin)). B, cell lysates were immunoblotted with the indicated antibodies (loading control: α-tubulin) and representative immunoblots are shown. C, quantification of insulin-stimulated Akt phosphorylation (data in B) is shown (phospho/total), normalized to insulin-stimulated control cells (n = 4–5). D–F, 3T3-L1 adipocytes were treated with the indicated doses of MCT1 inhibitor α-cyano-4-hydroxycinnamate in serum-free medium for 2 h, prior to stimulation with or without 10 nm insulin for 20 min. D, extracellular and intracellular lactate was measured in the medium or cells, respectively, after stimulation with insulin, normalized to insulin-stimulated control cells between experiments (n = 3–4). #, indicates not detected. E, cell lysates were immunoblotted with the indicated antibodies (loading control: 14-3-3) and representative immunoblots are shown. F, quantification of insulin-stimulated Akt phosphorylation (data in E) is shown (phospho/total), normalized to insulin-stimulated control cells (n = 5). *, p < 0.05; **, p < 0.01; ***, p < 0.0001 versus insulin-stimulated control cells, error bars represent S.E.
Discussion
The insulin/IGF-1 pathway plays a pleiotropic role in long term health. It is intricately linked to a number of major diseases including cancer, diabetes, cardiovascular disease, and neurological disorders (69, 70). In the present study we sought to identify kinases that act as co-regulators of the insulin/IGF-1 pathway. We used GLUT4 trafficking to the PM as a measure of insulin/IGF-1 action because we have previously established a robust high throughput assay for PM GLUT4 (16) and this metabolic output is linked to a number of intracellular signal transduction pathways including Akt, AMP-activated protein kinase, and other stress kinase pathways (31, 71). Our screen identified 33 kinases that either positively or negatively impacted upon IGF-1 regulated GLUT4 trafficking (Figs. 2 and 4). Among these were kinases that intersect with pathways previously implicated in insulin/IGF-1 action including p53 (38), myosin light chain kinase (36), and the Ras scaffold protein CNKSR1 (34) (Table 1). Surprisingly, there was an enrichment of kinases that play a role in metabolism and that phosphorylate metabolites rather than proteins (Fig. 3). These included NME1, a subunit of nucleoside diphosphate kinase, TPK1, PANK4, and PFKFB3 (Fig. 4, Table 1). Although we have focused on PFKFB3 in the present study the role of these other three metabolic enzymes in insulin action is deserved of further research.
Metabolic switching between mitochondrial and glycolytic metabolism is a highly regulated process. One enzyme that has received much attention in terms of metabolic switching is PFKFB3. Despite being a bifunctional enzyme, possessing both phosphatase and kinase activity, its kinase activity is 700-fold more active than its bisphosphatase activity favoring production of Fru-2,6-BP (72, 73), a potent allosteric activator of PFK-1. PFKFB3 plays an important role in ensuring high glycolytic flux in a range of cell types including tumor cells (74), endothelial cells (75), and activated lymphocytes (T cells (76), dendritic cells (77), and M1 macrophages (78)). Inhibition of PFKFB3 activity using 3-PO or via genetic means has a range of physiological consequences including reduced tumorigenesis, reduced angiogenesis (54), and reduced activation of T cells (56). These and other findings have contributed to the notion first proposed by Otto Warburg (79, 80) that aerobic glycolysis favors specific biologic outcomes. Several reasons have been proposed to explain this association. Glycolysis, although less efficient in energy production compared with mitochondria, generates energy rapidly. Second, intermediates generated during glycolysis are used for synthesis of a range of macromolecules essential for rapidly proliferating cells (81). Third, glycolysis plays a key role in intracellular redox control and avoids potential deleterious consequences of mitochondrial reactive oxygen species production (82).
Here we provide another potential explanation for the Warburg effect, which is to potentiate signal transduction pathways that promote cell growth and survival. In this study we provide evidence that a range of pharmacologic and genetic perturbations that modulate glycolysis have parallel effects on Akt signaling (Figs. 5, 6, and 8). Although much of our study has focused on PFKFB3, it is unlikely that the feedback onto signaling is specific to this enzyme or its products as other manipulations that affected glycolysis independently of PFKFB3 also perturbed Akt signaling (Fig. 8). This unveils a novel cyclical relationship between metabolism and Akt signaling (Fig. 9) that has potential implications for diseases involving defects in the Akt/metabolism axes. For example, in tumor cells stabilization of the PI3K/Akt signaling pathway will ensure both rapid proliferation and inhibition of cell death, two of the major end points of this signaling nexus (83). It is not surprising that hyper-activation of Akt is frequently observed in human tumors and in some cancer types 70–100% of tumors show hyper-activated Akt (84), presumably due at least in part to the high rates of glycolysis in these cells. The reverse situation is insulin resistance, a pathological situation involving reduced Akt activity (85). It has long been assumed that reduced Akt activity is a driver of insulin resistance but another interpretation, based on the current study, is that reduced glucose metabolism, a prominent feature of insulin resistance, leads to reduced Akt activity. Consistent with reduced Akt activity being a consequence rather than a cause of insulin resistance, we and others have observed defects in glucose metabolism earlier than defects in Akt signaling in a range of models of insulin resistance (86, 87).
FIGURE 9.
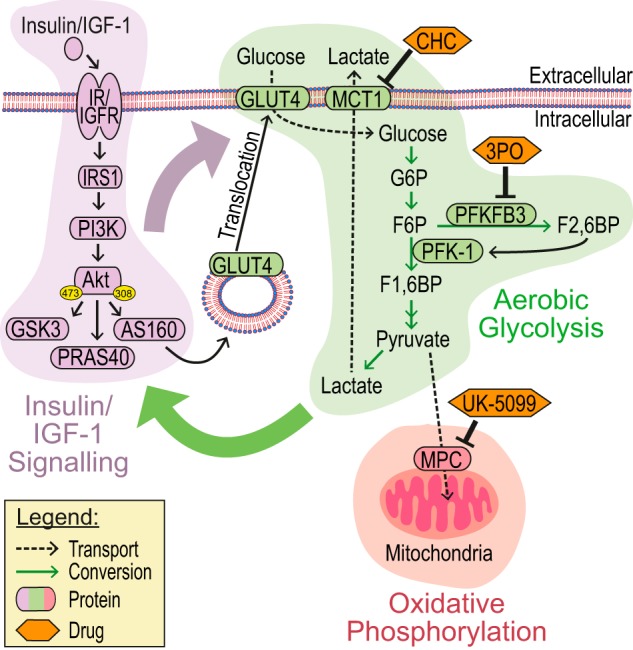
Model. The insulin/IGF-1 signaling pathway (purple) results in translocation of GLUT4 vesicles to the PM, facilitating increased glucose uptake. Glucose can either be converted to lactate (aerobic glycolysis, green) or undergo oxidative phosphorylation in the mitochondria (red). PFKFB3 generates Fru-2,6-BP (F2,6BP), a potent activator of PFK-1 and thus glycolysis. The insulin/IGF-1 signaling pathway positively regulates glucose uptake and glucose metabolism (purple arrow) and our data show that partitioning of glucose toward aerobic glycolysis potentiates insulin signaling (green arrow), whereas we predict that this effect will either not be seen or even inhibited when glucose undergoes mitochondrial oxidative phosphorylation. Drugs/inhibitors used in the study and their targets are indicated. G6P, glucose 6-phosphate; F6P, fructose 6-phosphate; F1,6BP, fructose 1,6-bisphosphate.
The current study raises several key questions, which are deserved of future study. First, why do adipocytes express such high levels of PFKFB3? Intriguingly, adipocytes display a Warburg type of metabolism as the majority of glucose taken up by adipocytes is converted to lactate (88, 89). We speculate this high rate of glycolysis might endow adipocytes with a highly sensitive insulin-regulated Akt network. Indeed, adipocytes are among the most insulin-sensitive cells in the body, displaying much greater insulin sensitivity than other bona fide insulin-sensitive organs such as muscle (90). The rationale for this hierarchical effect of insulin among different tissues is unclear and we speculate this may be related to the unusually high rate of PFKFB3-mediated glycolysis in the fat cell, which potentiates Akt signaling. Consistent with these observations, transgenic mice overexpressing PFKFB3 selectively in adipocytes, exhibit enhanced insulin sensitivity on high fat diet despite increased adiposity (91). Conversely, reduced PFKFB3 in PFKFB3+/− mice exacerbates diet-induced insulin resistance (52). This fits with the emerging role of the fat cell as an energy sensor and so future studies of this aspect of fat cell metabolism will likely yield new insights into insulin resistance and whole body metabolic homeostasis.
The second question is which metabolite(s) regulate Akt signaling? Lactate has a number of effects aside from its role in glycolysis including increased HIF1α activity (92) and activation of receptor tyrosine kinases AXL, TIE2, and VEGFR-2 (93). Lactate also binds to GPR81, a G protein-coupled receptor that is highly enriched in adipocytes (62). However, we were unable to observe a significant effect of external lactate (Fig. 7) or other GPR81 agonists on Akt signaling. We postulate that the effect that we have observed is not specific to PFKFB3 per se, but rather due to the up-regulation of glycolysis. One possibility is that the positive effect on Akt signaling is due to the partitioning of glucose toward lactate thereby preventing mitochondrial oxidation as well as its consequences on cellular redox (increased reactive oxygen species) and changes in metabolite levels (e.g. increased acetyl-CoA, the substrate for acetylation) (Fig. 9). Moreover, in an RNAi-mediated loss of function screen in Drosophila melanogaster cells for Akt-mediated proliferation and morphology, several glycolytic enzymes were identified including GLUT1, hexokinase, and PFK-1 as major regulators of Akt signaling (94). In addition to phosphorylation, many signaling proteins such as Akt are subject to regulation by a range of other post-translational modifications including acetylation, ubiquitination, nitrosylation, and O-GlcNAcylation (95–98). These kinds of modifications are subject to exquisite control by metabolism and are often regulated in a non-enzymatic manner and so this is also an area that requires further investigation.
The final question is which components within the PI3K/Akt pathway are the regulatory target(s) of this glycolytic feedback mechanism? Notably, despite a reduction in Akt kinase activity with PFKFB3 inhibition (Fig. 5), we observed insulin-dependent hyper-phosphorylation of Akt Thr-308. Hyper-phosphorylation of inactive Akt has been observed with direct Akt kinase inhibitors (60, 61) and oxidation (99). This phenotype is inconsistent with a block upstream in the ISP. For example, inhibition of PI3K completely ablates Akt phosphorylation. Therefore we predict that perturbations in glycolysis affect the ISP at the level of Akt itself rather than upstream of Akt.
In summary, our kinome screen has identified an important role for glucose metabolism in regulating the insulin/IGF-1 signaling pathway. PFKFB3, a key regulator of glycolysis, facilitates cross-talk between metabolism and signaling, generating a positive feedback loop (Fig. 9). Therefore, PFKFB3 expression/activity allows cells to perform high rates of glycolysis independently of their energy status. The product of PFKFB3, Fru-2,6-BP, overrides the inhibitory effect of ATP on PFK-1 and thereby drives glycolysis in the presence of high energy. PFKFB3 has been identified as an important target in cancer cells (100, 101). It is highly expressed in many cancers and efforts are underway to develop inhibitors and indeed 3-PO emerged from one such screen (53). Based on the current work we postulate that such drugs will not only inhibit glycolysis but also the signaling pathways required for cancer cell survival. The development of anti-cancer agents that selectively target the PI3K-Akt signaling pathway have had limited success, mainly due to adaptive changes in feedback inhibition pathways allowing reactivation of signaling (102). One of the intriguing implications of our data is that a major purpose of the “Warburg pathway,” aside from saving carbon backbones for biomass, might be to preserve the activity of signaling kinases that are essential for both cell proliferation and cell survival, two of the essential features of the Akt pathway. Thus, combination therapies involving metabolic/glycolytic inhibitors together with PI3K/Akt inhibitors may prove to be powerful antagonists of cancer cells at a broad level (103).
Author Contributions
S. T., P. S. K., D. E. J., J. R. J., and J. S. designed and coordinated the study. S. T. and P. S. K. performed the majority of the experiments. J. R. K., B. L. P., G. S., C. K., and J. S. performed experiments. R. C. performed the kinase enrichment analysis and D. J. F. provided intellectual input. J. L., J. P. S., E. T., and K. J. assisted in the kinome screen and data analysis. D. E. J. and J. S. wrote the manuscript. All authors reviewed and edited the manuscript and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Drs. Charles Watt and Howard Green for providing reagents. We thank Dr. Ellen van Dam and Kristen Thomas for technical assistance.
This work was supported in part by National Health and Medical Research Council (NHMRC) project Grants GNT1068469 (to J. S.) and GNT1086850 (to D. E. J.). The contents of the published material are solely the responsibility of the individual authors and do not reflect the views of NHMRC.

This article contains supplemental Tables S1 and S2.
- ISP
- insulin-like growth factor-1/insulin signalling pathway
- Fru-2,6-BP
- fructose 2,6-bisphosphate
- IGF
- insulin-like growth factor
- IRS1
- insulin receptor substrate 1
- KEGG
- Kyoto Encyclopedia of Genes and Genomes
- NTC
- non-targeting control
- PFKFB3
- 6-phosphofructo-2-kinase fructose-2,6-bisphosphatase 3
- PFK
- 1,6-phosphofructo-1-kinase
- UK-5099
- α-cyano-8-(1-phenylindol-3-yl)acrylate
- 3-PO
- 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one
- MPC
- mitochondrial pyruvate carrier.
References
- 1.Saltiel A. R., and Kahn C. R. (2001) Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414, 799–806 [DOI] [PubMed] [Google Scholar]
- 2.Lalioti V., Muruais G., Dinarina A., van Damme J., Vandekerckhove J., and Sandoval I. V. (2009) The atypical kinase Cdk5 is activated by insulin, regulates the association between GLUT4 and E-Syt1, and modulates glucose transport in 3T3-L1 adipocytes. Proc. Natl. Acad. Sci. U.S.A. 106, 4249–4253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Okada S., Yamada E., Saito T., Ohshima K., Hashimoto K., Yamada M., Uehara Y., Tsuchiya T., Shimizu H., Tatei K., Izumi T., Yamauchi K., Hisanaga S., Pessin J. E., and Mori M. (2008) CDK5-dependent phosphorylation of the Rho family GTPase TC10α regulates insulin-stimulated GLUT4 translocation. J. Biol. Chem. 283, 35455–35463 [DOI] [PubMed] [Google Scholar]
- 4.Garcia-Guerra L., Nieto-Vazquez I., Vila-Bedmar R., Jurado-Pueyo M., Zalba G., Díez J., Murga C., Fernández-Veledo S., Mayor F. Jr., and Lorenzo M. (2010) G protein-coupled receptor kinase 2 plays a relevant role in insulin resistance and obesity. Diabetes 59, 2407–2417 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5.Usui I., Imamura T., Satoh H., Huang J., Babendure J. L., Hupfeld C. J., and Olefsky J. M. (2004) GRK2 is an endogenous protein inhibitor of the insulin signaling pathway for glucose transport stimulation. EMBO J. 23, 2821–2829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bromann P. A., Korkaya H., and Courtneidge S. A. (2004) The interplay between Src family kinases and receptor tyrosine kinases. Oncogene 23, 7957–7968 [DOI] [PubMed] [Google Scholar]
- 7.Bülow M. H., Bülow T. R., Hoch M., Pankratz M. J., and Jünger M. A. (2014) Src tyrosine kinase signaling antagonizes nuclear localization of FOXO and inhibits its transcription factor activity. Sci. Rep. 4, 4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turban S., and Hajduch E. (2011) Protein kinase C isoforms: mediators of reactive lipid metabolites in the development of insulin resistance. FEBS Lett. 585, 269–274 [DOI] [PubMed] [Google Scholar]
- 9.Hirosumi J., Tuncman G., Chang L., Görgün C. Z., Uysal K. T., Maeda K., Karin M., and Hotamisligil G. S. (2002) A central role for JNK in obesity and insulin resistance. Nature 420, 333–336 [DOI] [PubMed] [Google Scholar]
- 10.Humphrey S. J., Yang G., Yang P., Fazakerley D. J., Stöckli J., Yang J. Y., and James D. E. (2013) Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab. 17, 1009–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rothman D. L., Shulman R. G., and Shulman G. I. (1992) 31P nuclear magnetic resonance measurements of muscle glucose-6-phosphate: evidence for reduced insulin-dependent muscle glucose transport or phosphorylation activity in non-insulin-dependent diabetes mellitus. J. Clin. Investig. 89, 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yalcin A., Telang S., Clem B., and Chesney J. (2009) Regulation of glucose metabolism by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases in cancer. Exp. Mol. Pathol. 86, 174–179 [DOI] [PubMed] [Google Scholar]
- 13.Larance M., Ramm G., Stöckli J., van Dam E. M., Winata S., Wasinger V., Simpson F., Graham M., Junutula J. R., Guilhaus M., and James D. E. (2005) Characterization of the role of the Rab GTPase-activating protein AS160 in insulin-regulated GLUT4 trafficking. J. Biol. Chem. 280, 37803–37813 [DOI] [PubMed] [Google Scholar]
- 14.Todaro G. J., and Green H. (1963) Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol. 17, 299–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shewan A. M., Marsh B. J., Melvin D. R., Martin S., Gould G. W., and James D. E. (2000) The cytosolic C-terminus of the glucose transporter GLUT4 contains an acidic cluster endosomal targeting motif distal to the dileucine signal. Biochem. J. 350, 99–107 [PMC free article] [PubMed] [Google Scholar]
- 16.Govers R., Coster A. C., and James D. E. (2004) Insulin increases cell surface GLUT4 levels by dose dependently discharging GLUT4 into a cell surface recycling pathway. Mol. Cell. Biol. 24, 6456–6466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prabhu A. V., Krycer J. R., and Brown A. J. (2013) Overexpression of a key regulator of lipid homeostasis, Scap, promotes respiration in prostate cancer cells. FEBS Lett. 587, 983–988 [DOI] [PubMed] [Google Scholar]
- 18.Tan S. X., Fisher-Wellman K. H., Fazakerley D. J., Ng Y., Pant H., Li J., Meoli C. C., Coster A. C., Stöckli J., and James D. E. (2015) Selective insulin resistance in adipocytes. J. Biol. Chem. 290, 11337–11348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stöckli J., Meoli C. C., Hoffman N. J., Fazakerley D. J., Pant H., Cleasby M. E., Ma X., Kleinert M., Brandon A. E., Lopez J. A., Cooney G. J., and James D. E. (2015) The RabGAP TBC1D1 plays a central role in exercise-regulated glucose metabolism in skeletal muscle. Diabetes 64, 1914–1922 [DOI] [PubMed] [Google Scholar]
- 20.Van Schaftingen E., Lederer B., Bartrons R., and Hers H. G. (1982) A kinetic study of pyrophosphate: fructose-6-phosphate phosphotransferase from potato tubers: application to a microassay of fructose 2,6-bisphosphate. Eur. J. Biochem. 129, 191–195 [DOI] [PubMed] [Google Scholar]
- 21.Tan S. X., Ng Y., and James D. E. (2010) Akt inhibitors reduce glucose uptake independently of their effects on Akt. Biochem. J. 432, 191–197 [DOI] [PubMed] [Google Scholar]
- 22.Tan S. X., Ng Y., Meoli C. C., Kumar A., Khoo P. S., Fazakerley D. J., Junutula J. R., Vali S., James D. E., and Stöckli J. (2012) Amplification and demultiplexing in insulin-regulated Akt protein kinase pathway in adipocytes. J. Biol. Chem. 287, 6128–6138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stöckli J., Davey J. R., Hohnen-Behrens C., Xu A., James D. E., and Ramm G. (2008) Regulation of glucose transporter 4 translocation by the Rab guanosine triphosphatase-activating protein AS160/TBC1D4: role of phosphorylation and membrane association. Mol. Endocrinol. 22, 2703–2715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., and Mesirov J. P. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bogan J. S., McKee A. E., and Lodish H. F. (2001) Insulin-responsive compartments containing GLUT4 in 3T3-L1 and CHO cells: regulation by amino acid concentrations. Mol. Cell. Biol. 21, 4785–4806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vijayakumar M. V., Ajay A. K., and Bhat M. K. (2010) Demonstration of a visual cell-based assay for screening glucose transporter 4 translocation modulators in real time. J. Biosci. 35, 525–531 [DOI] [PubMed] [Google Scholar]
- 27.Hernandez R., Teruel T., and Lorenzo M. (2001) Akt mediates insulin induction of glucose uptake and up-regulation of GLUT4 gene expression in brown adipocytes. FEBS Lett. 494, 225–231 [DOI] [PubMed] [Google Scholar]
- 28.Liu F., Dallas-Yang Q., Castriota G., Fischer P., Santini F., Ferrer M., Li J., Akiyama T. E., Berger J. P., Zhang B. B., and Jiang G. (2009) Development of a novel GLUT4 translocation assay for identifying potential novel therapeutic targets for insulin sensitization. Biochem. J. 418, 413–420 [DOI] [PubMed] [Google Scholar]
- 29.Weiland M., Bahr F., Höhne M., Schürmann A., Ziehm D., and Joost H. G. (1991) The signaling potential of the receptors for insulin and insulin-like growth factor I (IGF-I) in 3T3-L1 adipocytes: comparison of glucose transport activity, induction of oncogene c-fos, glucose transporter mRNA, and DNA-synthesis. J. Cell Physiol. 149, 428–435 [DOI] [PubMed] [Google Scholar]
- 30.Wilson C. M., Mitsumoto Y., Maher F., and Klip A. (1995) Regulation of cell surface GLUT1, GLUT3, and GLUT4 by insulin and IGF-I in L6 myotubes. FEBS Lett. 368, 19–22 [DOI] [PubMed] [Google Scholar]
- 31.Stöckli J., Fazakerley D. J., and James D. E. (2011) GLUT4 exocytosis. J. Cell Sci. 124, 4147–4159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brewer P. D., Romenskaia I., Kanow M. A., and Mastick C. C. (2011) Loss of AS160 Akt substrate causes Glut4 protein to accumulate in compartments that are primed for fusion in basal adipocytes. J. Biol. Chem. 286, 26287–26297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eguez L., Lee A., Chavez J. A., Miinea C. P., Kane S., Lienhard G. E., and McGraw T. E. (2005) Full intracellular retention of GLUT4 requires AS160 Rab GTPase activating protein. Cell Metab. 2, 263–272 [DOI] [PubMed] [Google Scholar]
- 34.Lim J., Zhou M., Veenstra T. D., and Morrison D. K. (2010) The CNK1 scaffold binds cytohesins and promotes insulin pathway signaling. Genes Dev. 24, 1496–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steimle P. A., Fulcher F. K., and Patel Y. M. (2005) A novel role for myosin II in insulin-stimulated glucose uptake in 3T3-L1 adipocytes. Biochem. Biophys. Res. Commun. 331, 1560–1565 [DOI] [PubMed] [Google Scholar]
- 36.Fulcher F. K., Smith B. T., Russ M., and Patel Y. M. (2008) Dual role for myosin II in GLUT4-mediated glucose uptake in 3T3-L1 adipocytes. Exp. Cell Res. 314, 3264–3274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Valiente M., Andrés-Pons A., Gomar B., Torres J., Gil A., Tapparel C., Antonarakis S. E., and Pulido R. (2005) Binding of PTEN to specific PDZ domains contributes to PTEN protein stability and phosphorylation by microtubule-associated serine/threonine kinases. J. Biol. Chem. 280, 28936–28943 [DOI] [PubMed] [Google Scholar]
- 38.Abe Y., Matsumoto S., Wei S., Nezu K., Miyoshi A., Kito K., Ueda N., Shigemoto K., Hitsumoto Y., Nikawa J., and Enomoto Y. (2001) Cloning and characterization of a p53-related protein kinase expressed in interleukin-2-activated cytotoxic T-cells, epithelial tumor cell lines, and the testes. J. Biol. Chem. 276, 44003–44011 [DOI] [PubMed] [Google Scholar]
- 39.Abe Y., Takeuchi T., Imai Y., Murase R., Kamei Y., Fujibuchi T., Matsumoto S., Ueda N., Ogasawara M., Shigemoto K., and Kito K. (2006) A small Ras-like protein Ray/Rab1c modulates the p53-regulating activity of PRPK. Biochem. Biophys. Res. Commun. 344, 377–385 [DOI] [PubMed] [Google Scholar]
- 40.Davey J. R., Humphrey S. J., Junutula J. R., Mishra A. K., Lambright D. G., James D. E., and Stöckli J. (2012) TBC1D13 is a RAB35 specific GAP that plays an important role in GLUT4 trafficking in adipocytes. Traffic 13, 1429–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Facchin S., Ruzzene M., Peggion C., Sartori G., Carignani G., Marin O., Brustolon F., Lopreiato R., and Pinna L. A. (2007) Phosphorylation and activation of the atypical kinase p53-related protein kinase (PRPK) by Akt/PKB. Cell Mol. Life Sci. 64, 2680–2689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brdicka T., Pavlistová D., Leo A., Bruyns E., Korínek V., Angelisová P., Scherer J., Shevchenko A., Hilgert I., Cerný J., Drbal K., Kuramitsu Y., Kornacker B., Horejsí V., and Schraven B. (2000) Phosphoprotein associated with glycosphingolipid-enriched microdomains (PAG), a novel ubiquitously expressed transmembrane adaptor protein, binds the protein tyrosine kinase csk and is involved in regulation of T cell activation. J. Exp. Med. 191, 1591–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lodeiro M., Theodoropoulou M., Pardo M., Casanueva F. F., and Camiña J. P. (2009) c-Src regulates Akt signaling in response to ghrelin via β-arrestin signaling-independent and -dependent mechanisms. PLoS ONE 4, e4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yao J. E., Yan M., Guan Z., Pan C. B., Xia L. P., Li C. X., Wang L. H., Long Z. J., Zhao Y., Li M. W., Zheng F. M., Xu J., Lin D. J., and Liu Q. (2009) Aurora-A down-regulates IκBα via Akt activation and interacts with insulin-like growth factor-1 induced phosphatidylinositol 3-kinase pathway for cancer cell survival. Mol. Cancer 8, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mehta A., and Orchard S. (2009) Nucleoside diphosphate kinase (NDPK, NM23, AWD): recent regulatory advances in endocytosis, metastasis, psoriasis, insulin release, fetal erythroid lineage and heart failure; translational medicine exemplified. Mol. Cell. Biochem. 329, 3–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nosaka K., Onozuka M., Kakazu N., Hibi S., Nishimura H., Nishino H., and Abe T. (2001) Isolation and characterization of a human thiamine pyrophosphokinase cDNA. Biochim. Biophys. Acta 1517, 293–297 [DOI] [PubMed] [Google Scholar]
- 47.Jackowski S., and Rock C. O. (1981) Regulation of coenzyme A biosynthesis. J. Bacteriol. 148, 926–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li Y., Chang Y., Zhang L., Feng Q., Liu Z., Zhang Y., Zuo J., Meng Y., and Fang F. (2005) High glucose upregulates pantothenate kinase 4 (PanK4) and thus affects M2-type pyruvate kinase (Pkm2). Mol. Cell. Biochem. 277, 117–125 [DOI] [PubMed] [Google Scholar]
- 49.Rock C. O., Calder R. B., Karim M. A., and Jackowski S. (2000) Pantothenate kinase regulation of the intracellular concentration of coenzyme A. J. Biol. Chem. 275, 1377–1383 [DOI] [PubMed] [Google Scholar]
- 50.Hue L., and Rider M. H. (1987) Role of fructose 2,6-bisphosphate in the control of glycolysis in mammalian tissues. Biochem. J. 245, 313–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Atsumi T., Nishio T., Niwa H., Takeuchi J., Bando H., Shimizu C., Yoshioka N., Bucala R., and Koike T. (2005) Expression of inducible 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase/PFKFB3 isoforms in adipocytes and their potential role in glycolytic regulation. Diabetes 54, 3349–3357 [DOI] [PubMed] [Google Scholar]
- 52.Huo Y., Guo X., Li H., Wang H., Zhang W., Wang Y., Zhou H., Gao Z., Telang S., Chesney J., Chen Y. E., Ye J., Chapkin R. S., and Wu C. (2010) Disruption of inducible 6-phosphofructo-2-kinase ameliorates diet-induced adiposity but exacerbates systemic insulin resistance and adipose tissue inflammatory response. J. Biol. Chem. 285, 3713–3721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Clem B., Telang S., Clem A., Yalcin A., Meier J., Simmons A., Rasku M. A., Arumugam S., Dean W. L., Eaton J., Lane A., Trent J. O., and Chesney J. (2008) Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol. Cancer Ther. 7, 110–120 [DOI] [PubMed] [Google Scholar]
- 54.Schoors S., De Bock K., Cantelmo A. R., Georgiadou M., Ghesquière B., Cauwenberghs S., Kuchnio A., Wong B. W., Quaegebeur A., Goveia J., Bifari F., Wang X., Blanco R., Tembuyser B., Cornelissen I., Bouché A., Vinckier S., Diaz-Moralli S., Gerhardt H., Telang S., Cascante M., Chesney J., Dewerchin M., and Carmeliet P. (2014) Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell Metab. 19, 37–48 [DOI] [PubMed] [Google Scholar]
- 55.Klarer A. C., O'Neal J., Imbert-Fernandez Y., Clem A., Ellis S. R., Clark J., Clem B., Chesney J., and Telang S. (2014) Inhibition of 6-phosphofructo-2-kinase (PFKFB3) induces autophagy as a survival mechanism. Cancer Metab. 2, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Telang S., Clem B. F., Klarer A. C., Clem A. L., Trent J. O., Bucala R., and Chesney J. (2012) Small molecule inhibition of 6-phosphofructo-2-kinase suppresses t cell activation. J. Transl. Med. 10, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xintaropoulou C., Ward C., Wise A., Marston H., Turnbull A., and Langdon S. P. (2015) A comparative analysis of inhibitors of the glycolysis pathway in breast and ovarian cancer cell line models. Oncotarget 6, 25677–25695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xie N., Tan Z., Banerjee S., Cui H., Ge J., Liu R. M., Bernard K., Thannickal V. J., and Liu G. (2015) Glycolytic reprogramming mediates myofibroblast differentiation and promotes lung fibrosis. Am. J. Respir. Crit. Care Med. 10.1164/rccm.201504-0780OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu Y., An X., Guo X., Habtetsion T. G., Wang Y., Xu X., Kandala S., Li Q., Li H., Zhang C., Caldwell R. B., Fulton D. J., Su Y., Hoda M. N., Zhou G., Wu C., and Huo Y. (2014) Endothelial PFKFB3 plays a critical role in angiogenesis. Arterioscler. Thromb. Vasc. Biol. 34, 1231–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chan T. O., Pascal J. M., Armen R. S., and Rodeck U. (2012) Autoregulation of kinase dephosphorylation by ATP binding in AGC protein kinases. Cell Cycle 11, 475–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin K., Lin J., Wu W. I., Ballard J., Lee B. B., Gloor S. L., Vigers G. P., Morales T. H., Friedman L. S., Skelton N., and Brandhuber B. J. (2012) An ATP-site on-off switch that restricts phosphatase accessibility of Akt. Sci. Signal. 5, ra37. [DOI] [PubMed] [Google Scholar]
- 62.Ahmed K., Tunaru S., Tang C., Müller M., Gille A., Sassmann A., Hanson J., and Offermanns S. (2010) An autocrine lactate loop mediates insulin-dependent inhibition of lipolysis through GPR81. Cell Metab. 11, 311–319 [DOI] [PubMed] [Google Scholar]
- 63.Cai T. Q., Ren N., Jin L., Cheng K., Kash S., Chen R., Wright S. D., Taggart A. K., and Waters M. G. (2008) Role of GPR81 in lactate-mediated reduction of adipose lipolysis. Biochem. Biophys. Res. Commun. 377, 987–991 [DOI] [PubMed] [Google Scholar]
- 64.Halestrap A. P. (1975) The mitochondrial pyruvate carrier. Kinetics and specificity for substrates and inhibitors. Biochem. J. 148, 85–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Halestrap A. P. (1976) The mechanism of the inhibition of the mitochondrial pyruvate transportater by α-cyanocinnamate derivatives. Biochem. J. 156, 181–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Manning Fox J. E., Meredith D., and Halestrap A. P. (2000) Characterisation of human monocarboxylate transporter 4 substantiates its role in lactic acid efflux from skeletal muscle. J. Physiol. 529, 285–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sonveaux P., Végran F., Schroeder T., Wergin M. C., Verrax J., Rabbani Z. N., De Saedeleer C. J., Kennedy K. M., Diepart C., Jordan B. F., Kelley M. J., Gallez B., Wahl M. L., Feron O., and Dewhirst M. W. (2008) Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 118, 3930–3942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Doherty J. R., Yang C., Scott K. E., Cameron M. D., Fallahi M., Li W., Hall M. A., Amelio A. L., Mishra J. K., Li F., Tortosa M., Genau H. M., Rounbehler R. J., Lu Y., Dang C. V., Kumar K. G., Butler A. A., Bannister T. D., Hooper A. T., Unsal-Kacmaz K., Roush W. R., and Cleveland J. L. (2014) Blocking lactate export by inhibiting the Myc target MCT1 disables glycolysis and glutathione synthesis. Cancer Res. 74, 908–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hers I., Vincent E. E., and Tavaré J. M. (2011) Akt signalling in health and disease. Cell Signal. 23, 1515–1527 [DOI] [PubMed] [Google Scholar]
- 70.Spielman L. J., Little J. P., and Klegeris A. (2014) Inflammation and insulin/IGF-1 resistance as the possible link between obesity and neurodegeneration. J. Neuroimmunol. 273, 8–21 [DOI] [PubMed] [Google Scholar]
- 71.Richter E. A., and Hargreaves M. (2013) Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol. Rev. 93, 993–1017 [DOI] [PubMed] [Google Scholar]
- 72.Chesney J., Mitchell R., Benigni F., Bacher M., Spiegel L., Al-Abed Y., Han J. H., Metz C., and Bucala R. (1999) An inducible gene product for 6-phosphofructo-2-kinase with an AU-rich instability element: role in tumor cell glycolysis and the Warburg effect. Proc. Natl. Acad. Sci. U.S.A. 96, 3047–3052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sakakibara R., Kato M., Okamura N., Nakagawa T., Komada Y., Tominaga N., Shimojo M., and Fukasawa M. (1997) Characterization of a human placental fructose-6-phosphate, 2-kinase/fructose-2,6-bisphosphatase. J. Biochem. 122, 122–128 [DOI] [PubMed] [Google Scholar]
- 74.Cordero-Espinoza L., and Hagen T. (2013) Increased concentrations of fructose 2,6-bisphosphate contribute to the Warburg effect in phosphatase and tensin homolog (PTEN)-deficient cells. J. Biol. Chem. 288, 36020–36028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.De Bock K., Georgiadou M., Schoors S., Kuchnio A., Wong B. W., Cantelmo A. R., Quaegebeur A., Ghesquière B., Cauwenberghs S., Eelen G., Phng L. K., Betz I., Tembuyser B., Brepoels K., Welti J., Geudens I., Segura I., Cruys B., Bifari F., Decimo I., Blanco R., Wyns S., Vangindertael J., Rocha S., Collins R. T., Munck S., Daelemans D., Imamura H., Devlieger R., Rider M., Van Veldhoven P. P., Schuit F., Bartrons R., Hofkens J., Fraisl P., Telang S., Deberardinis R. J., Schoonjans L., Vinckier S., Chesney J., Gerhardt H., Dewerchin M., and Carmeliet P. (2013) Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 154, 651–663 [DOI] [PubMed] [Google Scholar]
- 76.Maciver N. J., Jacobs S. R., Wieman H. L., Wofford J. A., Coloff J. L., and Rathmell J. C. (2008) Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. J. Leukocyte Biol. 84, 949–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Krawczyk C. M., Holowka T., Sun J., Blagih J., Amiel E., DeBerardinis R. J., Cross J. R., Jung E., Thompson C. B., Jones R. G., and Pearce E. J. (2010) Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115, 4742–4749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Haschemi A., Kosma P., Gille L., Evans C. R., Burant C. F., Starkl P., Knapp B., Haas R., Schmid J. A., Jandl C., Amir S., Lubec G., Park J., Esterbauer H., Bilban M., Brizuela L., Pospisilik J. A., Otterbein L. E., and Wagner O. (2012) The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab. 15, 813–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Warburg O., Wind F., and Negelein E. (1927) The metabolism of tumors in the body. J. Gen. Physiol. 8, 519–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Warburg O. (1956) On the origin of cancer cells. Science 123, 309–314 [DOI] [PubMed] [Google Scholar]
- 81.Vander Heiden M. G., Cantley L. C., and Thompson C. B. (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hamanaka R. B., and Chandel N. S. (2010) Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 35, 505–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Robey R. B., and Hay N. (2009) Is Akt the “Warburg kinase”?-Akt-energy metabolism interactions and oncogenesis. Semin. Cancer Biol. 19, 25–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bellacosa A., Kumar C. C., Di Cristofano A., and Testa J. R. (2005) Activation of AKT kinases in cancer: implications for therapeutic targeting. Adv. Cancer Res. 94, 29–86 [DOI] [PubMed] [Google Scholar]
- 85.Krook A., Roth R. A., Jiang X. J., Zierath J. R., and Wallberg-Henriksson H. (1998) Insulin-stimulated Akt kinase activity is reduced in skeletal muscle from NIDDM subjects. Diabetes 47, 1281–1286 [DOI] [PubMed] [Google Scholar]
- 86.Hoehn K. L., Hohnen-Behrens C., Cederberg A., Wu L. E., Turner N., Yuasa T., Ebina Y., and James D. E. (2008) IRS1-independent defects define major nodes of insulin resistance. Cell Metab. 7, 421–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hoy A. J., Brandon A. E., Turner N., Watt M. J., Bruce C. R., Cooney G. J., and Kraegen E. W. (2009) Lipid and insulin infusion-induced skeletal muscle insulin resistance is likely due to metabolic feedback and not changes in IRS-1, Akt, or AS160 phosphorylation. Am. J. Physiol. Endocrinol. Metab. 297, E67–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hodson L., Humphreys S. M., Karpe F., and Frayn K. N. (2013) Metabolic signatures of human adipose tissue hypoxia in obesity. Diabetes 62, 1417–1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sabater D., Arriarán S., Romero Mdel M., Agnelli S., Remesar X., Fernández-López J. A., and Alemany M. (2014) Cultured 3T3L1 adipocytes dispose of excess medium glucose as lactate under abundant oxygen availability. Sci. Rep. 4, 3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Challiss R. A., Espinal J., and Newsholme E. A. (1983) Insulin sensitivity of rates of glycolysis and glycogen synthesis in soleus, stripped soleus, epitrochlearis, and hemi-diaphragm muscles isolated from sedentary rats. Biosci. Rep. 3, 675–679 [DOI] [PubMed] [Google Scholar]
- 91.Huo Y., Guo X., Li H., Xu H., Halim V., Zhang W., Wang H., Fan Y. Y., Ong K. T., Woo S. L., Chapkin R. S., Mashek D. G., Chen Y., Dong H., Lu F., Wei L., and Wu C. (2012) Targeted overexpression of inducible 6-phosphofructo-2-kinase in adipose tissue increases fat deposition but protects against diet-induced insulin resistance and inflammatory responses. J. Biol. Chem. 287, 21492–21500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.De Saedeleer C. J., Copetti T., Porporato P. E., Verrax J., Feron O., and Sonveaux P. (2012) Lactate activates HIF-1 in oxidative but not in Warburg-phenotype human tumor cells. PLoS ONE 7, e46571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ruan G. X., and Kazlauskas A. (2013) Lactate engages receptor tyrosine kinases Axl, Tie2, and vascular endothelial growth factor receptor 2 to activate phosphoinositide 3-kinase/Akt and promote angiogenesis. J. Biol. Chem. 288, 21161–21172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wheeler D. B., Bailey S. N., Guertin D. A., Carpenter A. E., Higgins C. O., and Sabatini D. M. (2004) RNAi living-cell microarrays for loss-of-function screens in Drosophila melanogaster cells. Nat. Methods 1, 127–132 [DOI] [PubMed] [Google Scholar]
- 95.Lu X. M., Tompkins R. G., and Fischman A. J. (2013) Nitric oxide activates intradomain disulfide bond formation in the kinase loop of Akt1/PKBα after burn injury. Int. J. Mol. Med. 31, 740–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Park S. Y., Ryu J., and Lee W. (2005) O-GlcNAc modification on IRS-1 and Akt2 by PUGNAc inhibits their phosphorylation and induces insulin resistance in rat primary adipocytes. Exp. Mol. Med. 37, 220–229 [DOI] [PubMed] [Google Scholar]
- 97.Sundaresan N. R., Pillai V. B., Wolfgeher D., Samant S., Vasudevan P., Parekh V., Raghuraman H., Cunningham J. M., Gupta M., and Gupta M. P. (2011) The deacetylase SIRT1 promotes membrane localization and activation of Akt and PDK1 during tumorigenesis and cardiac hypertrophy. Sci. Signal. 4, ra46. [DOI] [PubMed] [Google Scholar]
- 98.Yang W. L., Wang J., Chan C. H., Lee S. W., Campos A. D., Lamothe B., Hur L., Grabiner B. C., Lin X., Darnay B. G., and Lin H. K. (2009) The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science 325, 1134–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wani R., Qian J., Yin L., Bechtold E., King S. B., Poole L. B., Paek E., Tsang A. W., and Furdui C. M. (2011) Isoform-specific regulation of Akt by PDGF-induced reactive oxygen species. Proc. Natl. Acad. Sci. U.S.A. 108, 10550–10555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Clem B. F., O'Neal J., Tapolsky G., Clem A. L., Imbert-Fernandez Y., Kerr D. A. 2nd, Klarer A. C., Redman R., Miller D. M., Trent J. O., Telang S., and Chesney J. (2013) Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol. Cancer Ther. 12, 1461–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Seo M., Kim J. D., Neau D., Sehgal I., and Lee Y. H. (2011) Structure-based development of small molecule PFKFB3 inhibitors: a framework for potential cancer therapeutic agents targeting the Warburg effect. PLoS ONE 6, e24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Seton-Rogers S. (2014) Anticancer drugs: a clearer pathway view. Nat. Rev. Drug Discov. 13, 176–177 [DOI] [PubMed] [Google Scholar]
- 103.Zhao Y., Butler E. B., and Tan M. (2013) Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 4, e532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Owen O. E. (March 3, 1981) U.S Patent 4254222
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.