

Background: Surface proteins fine-tune insulin secretion from pancreatic β-cells.
Results: Kirrel2 is regulated by multiple post-translational modifications, and its expression influences basal insulin secretion.
Conclusion: Kirrel2 expression is tightly regulated by phosphorylation and suppresses basal insulin secretion.
Significance: Genetic and biochemical characterization of Kirrel2 in pancreatic β-cells reveals insight into regulation of protein stability and insulin secretion.
Keywords: adherens junction, insulin secretion, membrane protein, phosphorylation, receptor protein serine/threonine kinase, kinase, phosphorylation
Abstract
Direct interactions among pancreatic β-cells via cell surface proteins inhibit basal and enhance stimulated insulin secretion. Here, we functionally and biochemically characterized Kirrel2, an immunoglobulin superfamily protein with β-cell-specific expression in the pancreas. Our results show that Kirrel2 is a phosphorylated glycoprotein that co-localizes and interacts with the adherens junction proteins E-cadherin and β-catenin in MIN6 cells. We further demonstrate that the phosphosites Tyr595–596 are functionally relevant for the regulation of Kirrel2 stability and localization. Analysis of the extracellular and intracellular domains of Kirrel2 revealed that it is cleaved and shed from MIN6 cells and that the remaining membrane spanning cytoplasmic domain is processed by γ-secretase complex. Kirrel2 knockdown with RNA interference in MIN6 cells and ablation of Kirrel2 from mice with genetic deletion resulted in increased basal insulin secretion from β-cells, with no immediate influence on stimulated insulin secretion, total insulin content, or whole body glucose metabolism. Our results show that in pancreatic β-cells Kirrel2 localizes to adherens junctions, is regulated by multiple post-translational events, including glycosylation, extracellular cleavage, and phosphorylation, and engages in the regulation of basal insulin secretion.
Introduction
Pancreatic β-cells sense fluctuations in metabolic demand, particularly minute changes in circulating glucose levels, and uniquely respond by secreting adequate amounts of insulin to regulate glucose homeostasis and metabolism (1). Loss of β-cell mass or impairment of normal insulin secretion results in diabetes, the most common metabolic disease in man that is characterized by hyperglycemia over a prolonged period (1). Hence, uncovering molecular mechanisms underlying normal β-cell function as well as those that lead to its dysfunction is crucial for understanding the pathology of diabetes and developing novel approaches for its treatment.
During embryonic development, scattered β-cells aggregate with other β-cells and endocrine cells into small clusters that subsequently mature through a series of morphogenetic events to form endocrine micro-organs of the pancreas termed islets of Langerhans (2, 3). In pancreatic islets, several surface proteins mediate direct communication among β-cells and their neighbors (4). These integral membrane proteins selectively interact to adopt one or more of the following functions: establishment of adhesive links among adjacent cells (5, 6); assistance in the formation of characteristic islet architecture (3, 7, 8); contribution to functional polarity of the β-cells (7, 9); formation of channels that facilitate direct exchange of cytosolic components (10); and propagation of intracellular signals in response to receptor-ligand interactions (11). These interactions at the cell surface collectively assist β-cells to optimize their insulin gene expression, insulin content, as well as basal and stimulated insulin secretion (12–19). Hence, physical separation of β-cells from their native contacts in pancreatic islets results in impairment of their insulin secretory responses (20, 21).
Kin of irregular chiasm-like 2 (Kirrel2, also known as nephrin-like 3/NEPH3) is an immunoglobulin superfamily protein with β-cell-specific expression in human and mouse pancreas (22). It forms a family of three closely related proteins together with Kirrel3 (nephrin-like 1/NEPH1) and Kirrel3 (nephrin-like 2/NEPH2). The extracellular region of Kirrel family proteins shows strong structural homology to nephrin, a molecule with a central role in formation and function of the glomerular slit diaphragm of the mammalian kidney (23–27). Kirrel2 was previously shown to form homotypic interactions with itself, heterotypic interactions with nephrin and NEPH1, and to induce cell adhesion through these interactions when overexpressed in mouse L fibroblasts, which do not exhibit cell adhesion activity due to a lack of adherens junction proteins (28). Apart from the important roles of nephrin/Kirrel proteins in specialized cell-cell contacts and formation of the size-selective filter barrier in the kidney, nephrin has also been localized to the surface of insulin granules and the plasma membrane of the pancreatic β-cells, where it was proposed to function as an active component of insulin vesicle machinery that may affect vesicle-actin interactions and mobilization of insulin granules to the plasma membrane (29). Autoantibodies against Kirrel2 have been detected in patients with type 1 diabetes (30), raising the possibility that shedding of this membrane protein within the pancreatic islet, uptake by resident or circulating macrophages or dendritic cells, and antigen presentation in lymph nodes draining the pancreas are some of the mechanisms contributing to autoimmunity (31).
Because of the β-cell-restricted expression, the structural homology of Kirrel family proteins and nephrin (32–34), and the possible involvement in the pathogenesis of type 1 diabetes, we aimed to characterize the role of Kirrel2 in pancreatic β-cell function, cell adhesion, as well as insulin secretion using biochemical, cell biological, and genetic approaches.
Experimental Procedures
Eukaryotic Expression Constructs
Mammalian expression plasmids for Kirrel2-V5 and Fyn-V5 were constructed by amplification of cDNA fragments coding for full-length mouse Kirrel2 (NM_172898, 5′-tcttggggtacacacactcg-3′ and 5′-cacatgagtctggagacgct-3′) and Fyn (NM_008054, 5′-gggagaggaccatgtgaatg-3′ and 5′-caggttttcaccgggctgat-3′) by PCR from murine (C57Bl/6, male, 12 weeks) pancreatic islet cDNA and ligation of these fragments into pCDNA3.1/V5-His vector (Life Technologies, Inc.). An HA tag sequence was inserted between Pro21 and His22 of Kirrel2-V5 to obtain the Kirrel2-HA-V5 expression vector. Kirrel2-GFP was constructed by directional cloning of full-length mouse Kirrel2 cDNA into pEGFP-N1 vector (Clontech) with PCR primers containing HindIII and SacII cut sites (5′-ctagggaagctttggggtacacacactcg-3′ and 5′-caacaaccgcggcacatgagtctggagac-3′). Plasmids encoding Kirrel2-V5 substitution mutants Y595F/Y596F, Y595D/Y596D, Y595A/Y596A, Y631F/Y632F, Y653F, Y595F/Y596F/Y631F/Y632F/Y653F, and the kinase-dead Fyn K299M mutant were prepared using QuikChange site-directed mutagenesis kit (Agilent Technologies) with primers that were designed by the manufacturer's software. Full-length human IGF1R cDNA from the human ORFeome library was subcloned into pcDNA3.1 and used as control construct for immunoprecipitation experiments. All plasmids were validated by DNA sequencing.
Cell Culture and Transfection
MIN6 cells were cultured in DMEM (Gibco) supplemented with 25 mm glucose, 16% FBS (PAA), penicillin/streptomycin (Gibco), and 55 μm β-mercaptoethanol. For shedding experiments, the medium was exchanged to Opti-MEM (Gibco) supplemented with penicillin/streptomycin (Gibco). Transfections were performed with Lipofectamine 2000 reagent (Life Technologies, Inc.) following the manufacturer's instructions.
Cell Treatments
Pervanadate was prepared by incubation of activated orthovanadate with 0.18% H2O2 at the dark for 15 min and used in culture medium at 100 μm for 15 min prior to cell lysis unless stated otherwise. Src kinase inhibitors PP2 and SU6656 (Sigma) were used at 10 and 5 μm, respectively, for 30 min. For N-linked glycan removal, lysates of MIN6 cells were treated with the indicated units of N-glycanase enzyme (Prozyme) following the manufacturer's instructions for 1 h at 37 °C, and the reactions were stopped by addition of Laemmli buffer with DTT. For protein half-life assessment, cycloheximide (Sigma) was used at 50 μg/ml in culture medium, and cells were either directly lysed (t = 0) or incubated for the indicated time periods before cell lysis.
Immunoblotting and Immunoprecipitation
Cells were washed twice with ice-cold PBS and lysed with Triton X-100 lysis buffer (1% Triton X-100, 20 mm HEPES, pH 7.9, 0.3 m NaCl, 0.2 mm EDTA, 1.5 mm MgCl2, 100 μm pervanadate, protease, and phosphatase inhibitor mixtures (Roche Applied Science)) for 30 min on ice. Cell debris was removed by centrifugation (14,000 × g, 15 min, 4 °C). Equal amounts of total protein were dissolved with SDS-PAGE and transferred to nitrocellulose membrane to be immunoblotted with indicated antibodies. For immunoprecipitation, equal amounts of total protein were incubated with anti-HA or anti-V5-agarose affinity gels (Sigma) for 4 h at 4 °C by rotation. Beads were washed with lysis buffer; bound proteins were eluted with 2× reducing Laemmli buffer and dissolved with SDS-PAGE.
Surface Protein Isolation
As instructed in the Pierce® cell surface protein isolation kit (Thermo Scientific), cells were washed twice with ice-cold PBS followed by incubation with 0.25 mg/ml Sulfo-NHS-SS-Biotin or PBS on a rocking platform for 30 min at 4 °C. After quenching of the reaction, cells were washed twice with ice-cold TBS and lysed with Triton X-100 cell lysis buffer for 30 min on ice. After removal of cell debris, a fraction of the clarified supernatant was spared as input material. An equal amount of total protein was incubated with the NeutrAvidin-agarose slurry for 2 h at 4 °C in a closed column. The unbound proteins in the flow-through, representing the intracellular fraction, were collected by centrifugation. Any remaining unbound proteins were removed by extensive washing with the lysis buffer. The captured surface proteins were eluted with 2× Laemmli buffer with DTT.
Subcellular Fractionation
Cells were incubated in fractionation buffer (250 mm sucrose, 20 mm HEPES, pH 7.4, 10 mm KCl, 1.5 mm MgCl2, 1 mm EDTA, 1 mm EGTA, 1 mm DTT, protease inhibitor mixture) for 30 min and were homogenized by passing through a 27-gauge needle 15 times. A small portion of the homogenate was spared as total lysate and was mixed with 3× the volume of lysis buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 10% glycerol, protease inhibitor mixture (Roche Applied Science)). The rest of the cell suspension was centrifuged for 10 min at 10,000 × g. The cleared supernatant was centrifuged for 1 h at 100,000 × g and procured as the cytosolic fraction. The pellet was washed once with 3 ml of fractionation buffer and centrifuged for 1 h at 100,000 × g. The supernatant was removed, and the membrane-enriched pellet was dissolved in lysis buffer. Equal amounts of protein from each fraction were analyzed by Western blotting.
Reagents, Antibodies and siRNAs
Rabbit polyclonal anti-β-catenin (Sigma), mouse monoclonal anti-CPE (BD Biosciences), rabbit monoclonal anti-E-cadherin (Cell Signaling), rabbit polyclonal anti-EGFR (Cell Signaling), rabbit polyclonal anti-Fyn (Cell Signaling), mouse monoclonal anti-γ-tubulin (Sigma), rabbit polyclonal GAPDH (Cell Signaling), rabbit polyclonal anti-GFP (Invitrogen), mouse monoclonal anti-HA (Covance), rabbit polyclonal anti-glucagon (Millipore), guinea pig polyclonal anti-insulin (Linco), goat polyclonal anti-Kirrel2 (R & D Systems), mouse monoclonal anti-phosphotyrosine (clone 4G10®, Millipore), and mouse monoclonal anti-V5 (Sigma) antibodies were obtained commercially and used at dilutions recommended by the manufacturer. Secondary antibodies linked to horseradish peroxidase (Calbiochem) and to Alexa-Fluor® (Life Technologies, Inc.) were used at 1:10,000 and 1:500 dilutions, respectively. siRNAs against Kirrel2 (SASI_Mm01_00162935 and SASI_Mm01_ 00162937) and Fyn (SASI_Mm02_00309539 and SASI_Mm02_00309540) were purchased from Sigma.
Immunohistochemistry
MIN6 cells were fixed with 4% paraformaldehyde in PBS for 15 min at room temperature and washed with PBS. Cells were blocked and permeabilized in blocking buffer (3% of serum of the secondary antibody species, 1% BSA in PBS) with 0.1% Triton X-100 for 30 min and incubated overnight at 4 °C with the primary antibody and for 1 h at room temperature with the secondary antibody both diluted in the blocking buffer. Excessive fixing solution and unbound primary and secondary antibodies were removed by repetitive PBS washes. For nuclei and filamentous actin detection, Hoechst 33342 stain (Life Technologies, Inc.) and Alexa Fluor® 568 phalloidin (Life Technologies, Inc.) were added to the secondary antibody solutions in dilutions recommended by manufacturer. Cells were mounted with CC/MountTM tissue mounting medium (Sigma) and visualized with Zeiss LSM 510 or LSM 780 confocal microscope.
Generation of Kirrel2 KO Mice
An embryonic stem (ES) cell line JM8A1.N3 was obtained from the KOMP repository, in which the trapping cassette “En2SA-lacZ-pA” and a downstream loxP site were inserted in introns 2 and 4, respectively, in the Kirrel2 gene. ES cells were injected into C57Bl/6N blastocysts at the Rockefeller University Gene Targeting Center. Kirrel2 null mice were generated by crossing targeted mice with Deleter Cre mice, thereby generating alleles in which the neo-cassette and exons 3 and 4 were deleted, resulting in early truncation of the endogenous transcript.
Pancreatic Islet Isolation
The bile duct was clamped at the ampulla of Vater, and the pancreas was perfused through the common duct with 0.2 mg/ml Liberase (Roche Applied Science) in serum-free RPMI 1640 medium (Gibco). The perfused pancreas was removed and digested for 18 min in 1 ml of Liberase (0.2 mg/ml) in serum-free RPMI 1640 medium at 37 °C. Digestion was blocked by addition of 20 ml of RPMI 1640 medium with 10% FBS, and the digested tissue was disassociated from the exocrine tissue with vigorous shaking. With repeated mild centrifugation and resuspension steps, the exocrine pancreas and collagenase were partially removed from the contents of the tube. Final pellet was resuspended in 10 ml of Histopaque 1077 (Sigma) covered with 10 ml of serum-free RPMI 1640 medium creating a sharp interface. Following centrifugation at 2200 rpm for 20 min, the islets were collected from Histopaque medium interface. Intact islets were picked under a light microscope and cultured in RPMI 1640 medium with 11 mm glucose, 10% FBS, and penicillin/streptomycin.
Insulin Secretion Assay
MIN6 cells or isolated islets were preincubated for 1 h at 37 °C in KRBH/BSA buffer (Krebs-Ringer bicarbonate HEPES buffer; 129 mm NaCl, 5 mm NaHCO3, 4.8 mm KCl, 1.2 mm KH2PO4, 1.2 mm MgSO4, 10 mm HEPES, and 1 mm CaCl2 at pH 7.4, 0.1% bovine serum albumin) containing 2 or 5 mm glucose, respectively. First, they were incubated for 1 h in KRBH/BSA with low glucose, and later they were incubated for 1 h in secretion buffer containing the indicated concentrations of glucose or other secretagogues. Insulin content of collected supernatants, acid/ethanol islet extracts, and cell lysates were determined by insulin ELISA (Crystal Chem Inc.).
Metabolic Measurements and Experimental Animals
Blood glucose was measured using a Contour glucometer (Bayer). For IPGTT, mice fasted overnight (13 h) were injected with 2 g/kg (chow diet) or 1.5 g/kg (HFD) d-glucose solution. After weaning (3 weeks), mice were housed in a pathogen-free animal facility and were maintained on a 12-h light/12-h dark cycle with free access to either standard rodent chow or HFD containing 60% kJ of fat. All animals were on the C57BL/6N background. Plasma insulin and glucagon (Phoenix Pharmaceuticals) concentrations were determined by ELISA.
Statistical Analysis
All values are described as mean ± S.D. Statistical significance was determined by Student's t test or analysis of variance with Bonferroni post hoc test, rejecting the null hypothesis at p = 0.05.
MS Sample Preparation
Tryptic digestion of immunopurified Kirrel2 protein was performed as described in part previously (35). In brief, digestion was performed in a 100 mm ammonium bicarbonate, 0.1% Rapigest-containing buffer. Cysteine bridges were reduced by addition of 5 mm tris(2-carboxyethyl)phosphine (Pierce), and free cysteine was carbamidomethylated by 10 mm iodoacetamide (Pierce). An enzyme to protein ratio of 1:50 (Trypsin, Promega) was used to digest proteins at 37 °C for 12 h. Peptide mixtures were desalted by Ultra Microtip Columns (The Nest Group). Samples were dried in a vacuum concentrator and stored at −20 °C until further use.
MS Analysis
Kirrel2 samples were analyzed with an LTQ-Orbitrap XL mass spectrometer (Thermo Scientific). Peptides were loaded on a 10-cm reversed phase HPLC column (75 μm diameter) packed with C18 material (Magic C18 AQ 3 μm; Michrom Bioresources). Peptides were separated using a linear gradient of 5–30% Buffer B (2% H2O, 0.1% formic acid in acetonitrile) in Buffer A (2% acetonitrile, 0.1% formic acid in H2O) for 60 min at a flow rate of 300 nl/min. Samples were injected in duplicates. The first duplicate was recorded in data-dependent mode, and the other duplicate was recorded in data-dependent mode with the addition of a preferred mass list for the peptide “DPTNGYYR” in unphosphorylated ([M + 2H]2+, 493.222) and singly phosphorylated form ([M + 2H]2+, 533.218). Peptide ion mass to charge range of 350–1600 (400–1600 for injections run with preferred mass list) was monitored with one high resolution (60,000) MS1 scan followed by five MS2 fragmentation scans (TOP5) on the five most intense ions in collision-induced dissociation mode. Singly charged ions were excluded from MS2 fragmentation. Essential full MS settings were as follows: automatic gain control = 106; maximum ion time = 500 ms; resolution = 60,000 full width at half-maximum. MS2 settings were as follows: AGC = 30,000; maximum ion time = 10 ms; minimum signal threshold = 250; dynamic exclusion time = 30 s; isolation width = 2 Da; normalized collision energy = 32; activation Q = 0.25.
MS Database Search and Data Analysis
MS data were searched against the mouse UniProt Database (May 2014) (36), including common protein contaminants using the MaxQuant search engine (version 1.305) (37). MS data analysis included search for variable modifications of N termini (acetylation), oxidized methionines, and phosphorylation of serine, threonine, and tyrosine residues. Carbamidomethylation of cysteines was included as static modification. A protein and peptide false discovery rate of 1% was determined by target-decoy-based search (reverse database search). Peptides with a score of <60, a posterior error probability of >0.05, and a δ score of <5 were removed from the data set. For phosphopeptides, additional filters of “score difference” of >10 and “localization score” of >0.75 were applied. Common contaminants, proteins only identified by a modified site, and proteins identified by only one unique peptide were removed. Furthermore, proteins with a low abundance score (<1000 intensity-based absolute quantification score) were removed. For identified protein groups, the major protein identification was used. Identified Kirrel2 peptides and their positions in the Kirrel2 amino acid sequence were visualized using Protter (38).
Results
Kirrel2 Is Glycosylated and Localizes to Cell Contacts
To study the localization of Kirrel2, we constructed different tagged vectors expressing mouse Kirrel2 (Fig. 1A). A specific antibody against the extracellular domain of Kirrel2 recognized the endogenous and overexpressed Kirrel2 protein with an apparent molecular mass of 95 kDa in cell lysates of the pancreatic β-cell line MIN6 (Fig. 1, B and C). To determine whether Kirrel2 is N-linked glycosylated, we used N-glycanase, an enzyme that releases intact N-linked oligosaccharides from glycoproteins. We applied an enzyme gradient to lysates of MIN6 cells and analyzed its effect on electrophoretic mobility of Kirrel2-V5. A shift of the apparent molecular mass of Kirrel2 from ≈95 to ≈80 kDa was observed with a multistep pattern of glycan removal, demonstrating that Kirrel2 is an N-linked glycoprotein (Fig. 1D).
FIGURE 1.

Kirrel2 is predominantly expressed at the cell junctions and co-localizes with adherens junction proteins. A, schematic representation of wild type N- and C-terminally tagged Kirrel2 proteins. B, Western blot of MIN6 cells transfected with control or Kirrel2 targeting siRNAs using an antibody against the N-terminal domain of Kirrel2. γ-Tub, γ-tubulin. C, immunoblots (IB) of MIN6 cells transfected with control (pcDNA) or Kirrel2-V5 expression vectors using anti-Kirrel2 antibody. D, lysates of MIN6 cells transfected with control (−) or Kirel2-V5-expressing (+) plasmids were treated with indicated units of N-glycanase enzyme and immunoblotted with anti-V5 antibodies. E–H, MIN6 cells expressing Kirrel2-V5 were immunolabeled with indicated antibodies or phalloidin (F-actin). I, MIN6 cells were transfected with Kirrel2-V5 or Kirrel2-GFP vectors. After 16 h, the two cell populations were trypsinized, mixed, and cultured on chambered microscopy slides for 16 h prior to immunolabeling with anti-V5 antibodies. Images were obtained by confocal microscopy. Bar, 10 μm.
To understand the functions of Kirrel2 in pancreatic β-cells, we next examined its cellular localization, in particular with respect to co-localization with insulin granules and plasma membrane distribution. MIN6 cells transiently expressing Kirrel2-V5 were immunolabeled with anti-V5 and anti-insulin antibodies. Kirrel2 staining marked distinct vesicles and did not co-localize with insulin granules, indicating that it is not a constituent of the insulin vesicle machinery (Fig. 1E). Kirrel2 predominantly localized to the plasma membrane as dots or “puncta” along the sites of the cell contacts (Fig. 1, F–I). To test whether any other characterized adherens junction proteins localize to these structures, we immunolabeled MIN6 cells with antibodies against epithelial cadherin (E-cadherin) (Fig. 1F), β-catenin (Fig. 1G), and fluorescent phalloidin that labels filamentous actin (F-actin) (Fig. 1H). We observed that E-cadherin and β-catenin staining at the cell contacts largely co-localized with Kirrel2, although F-actin staining was rather continuous along the cell edges. To assess whether Kirrel2 proteins of two opposing cells co-localize within the same puncta, we transiently transfected two separate pools of MIN6 cells that were either expressing Kirrel2-V5 or Kirrel2-GFP. The two cell populations were trypsinized, mixed at a 1:1 ratio, seeded on microscopy slides, and prepared for analysis by immunofluorescent confocal microscopy. We observed that at contact sites of neighboring Kirrel2-V5- and Kirrel2-GFP-expressing cells, each signal obtained by Kirrel2-V5 staining was directly adjoined by a Kirrel2-GFP signal from the opposing cell (Fig. 1I) indicating adhesive trans-interactions between Kirrel2 molecules of neighboring cells. In line with this observation, the Kirrel2 signal at intracellular contacts of a transfected and a non-transfected cell was significantly less compared with the signal observed at contacts with other transfected cells. This suggests that significantly lower endogenous levels of Kirrel2 in non-transfected cells are insufficient to efficiently stabilize similar amounts of overexpressed Kirrel2 at the cell-to-cell contacts (Fig. 1, E–I). Additionally, no Kirrel2 signal was observed at the contact-free cell membranes (Fig. 1, E–I).
Fyn Interacts with and Phosphorylates Kirrel2
Cytoplasmic domains of cell surface proteins are frequent targets of intracellular protein kinases, and phosphorylated residues are often involved in assembly of cell-cell adhesions and cell adhesion-initiated signaling pathways. Likewise, phosphorylation of several cytoplasmic tyrosine residues of nephrin and NEPH1 has been associated with activation of signaling cascades, actin polymerization, and protein endocytosis (39–42). To determine whether Kirrel2 is a target of tyrosine phosphorylation, MIN6 cells expressing Kirrel2-V5 were treated with the tyrosine phosphatase inhibitor pervanadate prior to cell lysis. We then immunoprecipitated Kirrel2-V5 with anti-V5-agarose affinity gel and immunoblotted with anti-V5 or anti-phosphotyrosine antibodies. As shown in Fig. 2A, pervanadate treatment induced marked tyrosine phosphorylation of Kirrel2. A corresponding upward mobility shift of the anti-V5 tag antibody signal was also evident in pervanadate-treated samples.
FIGURE 2.

Kirrel2 is tyrosine-phosphorylated by Src family kinase Fyn. A, MIN6 cells were transfected with control (−) or Kirrel2-V5 (+) vectors and treated with PBS (−) or pervanadate (+) prior to cell lysis. Lysates were incubated with anti-V5 agarose affinity gel, and precipitated protein complexes were immunoblotted (IB) with anti-phosphotyrosine (4G10) and anti-V5 antibodies. B, transfected MIN6 cells were treated with 10 μm DMSO, 10 μm PP2, or 5 μm SU6656 for 20 min, followed by 10 min of pervanadate treatment prior to lysis. Following purification with anti-V5-agarose affinity gel, immunoprecipitates (IP) were immunoblotted with anti-phosphotyrosine (4G10) and anti-V5 antibodies. C, MIN6 cells expressing Kirrel2-HA, wild type (WT), or kinase-dead (KD) Fyn were treated with PBS (−) or pervanadate (+) prior to cell lysis. After immunoprecipitation with anti-HA-agarose affinity gel, immunoprecipitates were blotted with 4G10, anti-HA, or anti-V5 antibodies. Immunoblotting of total cell lysates with anti-V5 antibody demonstrates Fyn-V5 expression. D, MIN6 cells were co-transfected with control (−) or Kirrel2-V5 (+) vectors and control or anti-Fyn siRNAs. Prior to lysis, cells were treated with PBS or pervanadate. Lysates were purified with anti-V5-agarose affinity gel, and immune complexes were blotted with 4G10 or anti-V5 antibodies. 50 μg of total protein was immunoblotted with 4G10 antibody to evaluate changes in total tyrosine phosphorylation in A, B, and D. F, Protter images of identified peptides and phosphopeptides of the intracellular (left) and extracellular (right) domains were obtained from three independent Kirrel2-HA-V5 immunopurifications. Identified peptides (blue) and modified residues (green) are highlighted. Arrow indicates a potential putative cleavage site, identified through a semi-tryptic peptide FLVEAFPAPEVEGGQGPGLISVLH, with a correct tryptic cleavage at the N terminus and a non-tryptic cleavage at the C terminus (H) of the extracellular domain. Dashed lines show trypsin cleavage sites. PM, plasma membrane. E, conservation in the cytoplasmic tail of Kirrel2 among mammals.
Previous studies have demonstrated that Fyn, a member of Src family tyrosine kinases (SFKs), tyrosine phosphorylates cytoplasmic domain of nephrin and NEPH1 in podocytes (39, 43, 44). Various SFKs are also expressed in pancreatic β-cells and exert a tonic inhibitory role in Ca2+-dependent insulin secretion (45). These observations prompted us to investigate the effect of SFK-specific inhibitors on tyrosine phosphorylation of Kirrel2 in pancreatic β-cells. For this purpose, we treated MIN6 cells with two structurally unrelated SFK inhibitors, PP2 and SU6656. Pervanadate was added to culture medium 10 min prior to lysis, and Kirrel2-V5 was immunoprecipitated with anti-V5-agarose affinity gel. As shown in Fig. 2B, tyrosine phosphorylation of Kirrel2 was strongly inhibited by both PP2 and SU6656. These data demonstrate that Kirrel2, at least in part, is phosphorylated by SFKs in MIN6 cells.
Next, we investigated whether Fyn directly interacts with and phosphorylates Kirrel2. For this purpose, we co-transfected Kirrel2-HA with either a wild type (Fyn-V5WT) or a kinase-dead (Fyn-V5KD) form of V5-tagged Fyn in MIN6 cells. Co-expression of Fyn-V5WT was sufficient to trigger tyrosine phosphorylation of Kirrel2-HA even without the pervanadate treatment (Fig. 2C). Phosphorylation was drastically increased when these cells were treated with pervanadate. Increased tyrosine phosphorylation correlated with a significant upward mobility shift in HA tag antibody signal. On the contrary, expression of Fyn-V5KD neither enhanced Kirrel2 phosphorylation nor changed the electrophoretic mobility of Kirrel2. Additionally, Fyn-V5WT co-immunoprecipitated with Kirrel2-HA, indicating a direct interaction between the two proteins. The amount of co-immunoprecipitated Fyn-V5WT significantly increased when the cells were treated with pervanadate. In contrast, Fyn-V5KD co-immunoprecipitated with Kirrel2 only after pervanadate treatment. Neither form of Fyn-V5 was immunoprecipitated with anti-HA-agarose affinity gel alone. To further corroborate that Kirrel2 is phosphorylated by Fyn, we co-transfected MIN6 cells with Kirrel2-V5 and either anti-Fyn or control siRNAs. A significant reduction in total amount of Fyn as well as a corresponding decrease in Kirrel2-V5 phosphorylation was observed (Fig. 2D). These findings demonstrate that Fyn mediates tyrosine phosphorylation of Kirrel2 in pancreatic β-cells.
Kirrel2 Is Phosphorylated at Residues Tyr595,596, Tyr631/632, and Tyr653
Cytoplasmic domain of full-length mouse Kirrel2 protein contains eight tyrosine residues, seven of which are conserved in humans and other mammals (Fig. 2E). We employed mass spectrometry (MS) to identify potentially phosphorylated tyrosine residues in Kirrel2. MIN6 cells co-expressing Kirrel2-HA and Fyn-V5WT were treated with pervanadate prior to lysis, and Kirrel2-HA was immunoprecipitated with anti-HA-agarose affinity gel prior to digestion with trypsin and MS analysis. In total, 154 proteins were consistently identified in three independent experiments. As expected, in all three studies the Kirrel2 protein was identified as the most abundant protein providing 39.8, 22.4, and 24.5% of the total protein mass in the samples (based on the intensity-based absolute quantification score (iBAQ score) (46). The peptide coverage included 42 unique peptides with five phosphorylation sites in the cytoplasmic domain (Fig. 2F). The peptide “DPTNGYYR596” was identified as a double-phosphorylated peptide at amino acids Tyr595,596 (underlined). Another tyrosine phosphorylation was detected on peptide (R)AGYLTTPHPR(A), where underline indicates Tyr653. A serine phosphorylation was identified on peptide “GPEEETGSSEDR, where underline indicates Ser563. Additionally, another single serine phosphorylation on (R)IPGSSEGSSR(G) peptide was detected, where underlines indicate Ser548,549. The phosphorylation was restricted to either Ser548 or Ser549; however, due to site ambiguity, the exact position of the serine phosphorylation could not be determined from the spectrum. None of the identified phosphosites are currently listed in the UniProt or the PhosphoSitePlus database.
To confirm phosphorylation of the mapped tyrosine residues and to study their functional relevance, we introduced phenylalanine substitutions into Kirrel2-V5 at positions Tyr595–596 and Tyr653. Both mutations resulted in a reduction of the tyrosine phosphorylation signal of Kirrel2 in immunoblot analysis (Fig. 3A). Total tyrosine phosphorylation of triple mutant Kirrel2-V5Y595F/Y596F/Y653F was markedly reduced compared with the wild type Kirrel2; however, a considerable amount of residual signal indicated phosphorylation of additional tyrosine residues (data not shown). By introducing a series of phenylalanine substitutions for tyrosine residues located within the peptides that were not covered by the MS data, we discovered that mutations at the evolutionarily conserved Tyr631 and neighboring Tyr632 also resulted in a significant decrease in total tyrosine phosphorylation of Kirrel2 (Fig. 3A). The total intensity of the phosphotyrosine signal was clearly reduced for the quintuple mutant Kirrel2Y595F/Y596F/Y631F/Y632F/Y653F, but not completely abolished, indicating phosphorylation of other additional residues that need further investigation. These findings complement MS data showing phosphorylation of Tyr595,596 and Tyr653 and demonstrate phosphorylation at residues Tyr631 and/or Tyr632.
FIGURE 3.

Mutant Kirrel2Y595F/Y596F protein displays enhanced stability. A, MIN6 cells expressing wild type or mutant Kirrel2-V5 were treated with pervanadate. V5-tagged proteins were immunoprecipitated (IP) with anti-V5-agarose affinity gels and immunoblotted (IB) with 4G10 and anti-V5 antibodies. B, MIN6 cells were transfected with IGF1R-V5, control (pcDNA), wild type, or mutant Kirrel2 vectors. Duplicate transfections were performed using independent maximum preparations. Total protein lysates were immunoblotted with indicated antibodies. γ-Tub, γ-tubulin. C, MIN6 cells, co-transfected with GFP and wild type or mutant Kirrel2-V5 expression vectors, were treated with cycloheximide for the indicated time. Cell extracts were immunoblotted with V5 tag and GFP antibodies. D, quantification of Kirrel-V5 signals from two independent experiments by densitometry. Two-way analysis of variance with Bonferroni post hoc test was performed. **, p < 0.01; ***, p < 0.001.
Phosphodeficient Kirrel2 Mutants Exhibit Altered Stability and Cell Surface Localization
When MIN6 cells were transfected with equal amounts of Kirrel2 expression plasmids, protein levels of Kirrel2Y595F/Y596F or Kirrel2Y595F/Y596F/Y631F/Y632F/Y653F were detected at higher levels than Kirrel2WT, Kirrel2Y631F/Y632F and Kirrel2Y653 (Fig. 3B). Recurrent observations with independent plasmid preparations prompted us to investigate potential differences in turnover rates of mutant Kirrel2 proteins. To assess protein half-lives, we blocked novel protein biosynthesis with cycloheximide in MIN6 cells co-expressing GFP and Kirrel2-V5 (Fig. 3C). Total protein extracts were immunoblotted with anti-V5 and anti-GFP antibodies. The levels of co-transfected GFP were used as transfection and loading controls. Quantification of the remaining Kirrel2-V5 signal by densitometric scanning of the immunohybridization signals revealed that Kirrel2Y595F/Y596F mutants exhibit a ≈30% prolonged half-life (Fig. 3D).
Next, we assessed whether differences in stabilities of phospho-mutant Kirrel2 proteins would correlate with differences in their subcellular localization. For this purpose, we performed co-stainings in MIN6 cells expressing wild type or mutant Kirrel2-V5 using anti-V5 antibodies and phalloidin. In MIN6 cells expressing wild type Kirrel2, the F-actin and Kirrel2 signal was linearly distributed along the cell contacts (Fig. 4A). A similar pattern of Kirrel2 and F-actin staining was observed for Kirrel2-V5Y631F/Y632F- and Kirrel2Y653F-expressing cells (Fig. 4, C and D). However, the signal observed in most of the contact sites of Kirrel2-V5Y595F/Y596F- and Kirrel2Y595F/Y596F/Y631F/Y632F/Y653F-expressing cells appeared less distinct and demarcated (Fig. 4, B and E). Frequently, the signal was distributed to three-dimensional reticular structures instead of a narrow straight line. The F-actin staining was also disturbed in these contacts. These reticular structures largely co-localized with both E-cadherin and β-catenin (Fig. 5, A and B).
FIGURE 4.

Expression of Kirrel2Y595F/Y596F results in formation of reticular cell junctions. MIN6 cells were transfected with plasmids encoding for wild type or indicated mutants of Kirrel2-V5. Kirrel2 was immunolabeled with anti-V5 antibody (green), F-actin with phalloidin 568 (red), and nuclei with Hoechst stain (blue). Bar, 10 μm
FIGURE 5.
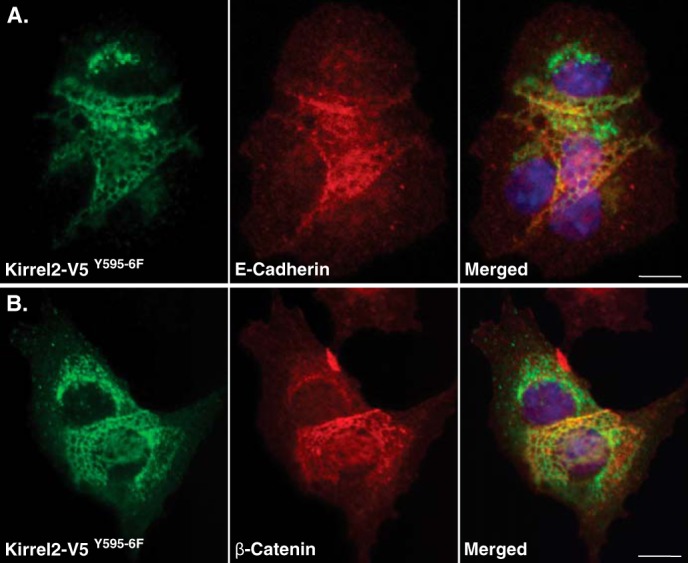
Reticular junctions of Kirrel2Y595F/Y596F co-localizes with E-cadherin and β-catenin. MIN6 cells expressing Kirrel2-V5 or Kirrel2Y595F/Y596F-V5 were immunolabeled with anti-V5 (green), anti-E-cadherin (A), or β-catenin (B) antibodies (red). Nuclei were labeled with Hoechst stain (blue). Bar, 10 μm
Having shown that mutant Kirrel2Y595F/Y596F is more stable and modifies the structure of cell contacts, we asked whether the mutation results in accumulation of Kirrel2 at the plasma membrane. For this purpose, we first quantitatively assessed endogenous levels of Kirrel2 located at the plasma membrane of MIN6 cells and compared it with other well characterized integral membrane proteins using a surface protein isolation technique (47). We labeled surface proteins containing accessible lysine residues with a cell-impermeable cleavable biotinylation reagent, Sulfo-NHS-SS-Biotin, followed by affinity purification with streptavidin-coated beads. We then resolved the collected fractions, including the total protein lysate (Fig. 6, Input), unbound proteins in the flow-through, and captured surface proteins (Eluate) by SDS-PAGE, and immunoblotted with antibodies against Kirrel2, E-cadherin, epidermal growth factor receptor (EGFR), and CPE (Fig. 6A). E-cadherin, a β-cell adhesion molecule (3), EGFR, an intensely trafficking cell surface receptor (48), and CPE, an insulin granule membrane resident protein (49), were used as representative β-cell transmembrane proteins with distinct cell surface exposure. As expected, we observed that most of the E-cadherin, a small fraction of the total EGFR, and no CPE was found in the eluate enriched for plasma membrane proteins (Fig. 6A). Similar to E-cadherin, Kirrel2 was mainly localized at the plasma membrane. This observation indicates that endogenous Kirrel2 resides mostly on the MIN6 cell surface in standard culture conditions. We next assessed potential differences between surface to intracellular protein fractions of mutant and wild type Kirrel2 molecules. For this purpose, we once again isolated surface proteins of MIN6 cells expressing wild type or mutant Kirrel2-V5. Immunoblotting analysis of the collected fractions with V5 antibodies revealed that ratios of surface to intracellular levels were significantly higher for Kirrel2-V5Y595F/Y596F and Kirrel2-V5Y595F/Y596/Y631F/Y632F/Y653F (Fig. 6B). In contrast, levels of E-cadherin in the plasma membrane remained unchanged. These results show that Tyr595–596 are involved in regulation of Kirrel2 localization at the plasma membrane.
FIGURE 6.

Kirrel2Y595F/Y596F stabilization at plasma membrane is independent of its dimerization. A, intact MIN6 cells treated with or without Sulfo-NHS-SS-Biotin were processed according to the kit instructions. Collected fractions, including input, flow-through, and eluate, were analyzed by immunoblotting (IB) with antibodies against Kirrel2, E-cadherin (E-cad), EGFR, and CPE. Ratios of biotinylated (surface) to unbiotinylated (intracellular) proteins based on densitometry of the immunohybridization signals are shown in the right panel. B, MIN6 cells, transfected with V5-tagged wild type or mutant Kirrel2 expression plasmids, were treated as described in A. Collected fractions were analyzed by immunoblotting with anti-V5 (upper panel) and with anti-E-cadherin antibodies (lower panel). Ratios of biotinylated (surface) to unbiotinylated (intracellular) proteins are shown in right panels. *, p < 0.05 at Student's t test. C and D, MIN6 cells were transfected with Kirrel2-V5 or Kirrel2-GFP expression plasmids together with their respective controls. Protein lysates were resolved with reducing (+DTT) or non-reducing (−DTT) SDS-PAGE and blotted with anti-V5 or -GFP antibodies. E, Western blot of MIN6 cell lysates expressing the indicated Kirrel2-V5 proteins, resolved with reducing (+DTT) and non-reducing (−DTT) SDS-PAGE following immunoblotting with anti-V5 antibodies. F, lysates of MIN6 cells co-expressing wild type Kirrel2-HA and mutant Kirrel2-V5 were incubated with anti-HA affinity gel; immunoprecipitates were immunoblotted with anti-V5 and anti-HA antibody. A total of 50 μg of protein was immunoblotted with anti-V5 antibodies as input control. G, anti-V5 affinity gel immunoprecipitates of MIN6 cell lysates transiently expressing V5 tagged wild type or mutant Kirrel2 immunoblotted with anti-E-cadherin, -β-catenin, and -V5 antibodies.
Kirrel2 Forms Homotypic Dimers That Are Not Influenced by Tyrosine Phosphorylation
Previous findings indicate that the cytoplasmic fragment of Kirrel2 can homo- and heterodimerize with the related proteins nephrin and NEPH1 in podocytes (28). Homotypic and heterotypic interactions of NEPH1 are crucial for glomerular slit diaphragm function (24, 33, 50). We asked whether Kirrel2 could form homotypic multimers in MIN6 cells, and whether phosphorylation of Kirrel2 affects this process. For this purpose, we performed Western blotting in the presence and absence of the reducing agent DTT. When SDS-PAGE is performed under non-reducing conditions, covalent interactions among proteins can be preserved and identified with immunoblotting due to a size shift they introduce to the inspected proteins. In Western blots of MIN6 lysates transiently expressing Kirrel2-V5 or Kirrel2-GFP, both anti-V5 and anti-GFP antibodies interacted with additional bands that correspond to twice the molecular weight of the Kirrel2-V5 or Kirrel2-GFP monomers, indicating Kirrel2 forms homotypic dimers in MIN6 (Fig. 6, C and D).
Immunohistochemistry results suggested that Kirrel2 localization at the cell surface depends on trans-homotypic interactions of Kirrel2 molecules from neighboring cells. In case of another adhesion molecule C-cadherin, formation of trans-protein dimers requires prior cis-homodimerization (51). Therefore, we next assessed whether the increased amount of Kirrel2Y596F/Y596F levels at the cell surface is mediated by an increase in Kirrel2 dimers. Lysates of MIN6 cells expressing wild type or mutant Kirrel2-V5 were resolved with reducing and non-reducing SDS-PAGE followed by immunoblotting with anti-V5 antibodies (Fig. 6E). Densitometric scanning of the immunohybridization signals revealed that dimer to monomer ratio of Kirrel2Y596F/Y596F was not changed compared with wild type Kirrel2. Next, we tested whether interactions between wild type and mutant Kirrel2 molecules are altered by performing co-immunoprecipitation with lysates from MIN6 cells co-expressing Kirrel2-HA and respective phospho-mutants (Fig. 6F). Kirrel2-HA efficiently precipitated both wild type and mutant Kirrel2-V5 molecules, but not V5-tagged IGF1R, another type I transmembrane protein of similar molecular weight. We concluded that the phosphorylation status of cytoplasmic tyrosine residues does not influence Kirrel2 dimerization.
We have shown that both wild type and phospho-mutant Kirrel2 proteins co-localize with the adherens junction molecules E-cadherin and β-catenin. To investigate whether Kirrel2 forms stable protein-protein interactions with E-cadherin and β-catenin, we expressed V5-tagged wild type or phospho-mutant Kirrel2 in MIN6 cells and performed immunoprecipitation with anti-V5 agarose affinity gel. Both E-cadherin and β-catenin were detected in the immune complexes, indicating that these proteins interact with Kirrel2 (Fig. 6G).
Kirrel2 Is Constitutively Shed from MIN6 Cells
In Western blotting experiments using anti-V5 antibodies, we noticed another specific band with a lower molecular mass (∼28 kDa) in whole cell lysates of Kirrel2-V5-expressing MIN6 cells (Fig. 7A). The membrane spanning cytoplasmic domain of Kirrel2-V5, including the protein tag and linker peptides, has a molecular mass around 24 kDa. Therefore, we hypothesized that the extracellular Kirrrel2 domain might be cleaved and possibly shed, with the lower band representing the residual C-terminal membrane spanning fragment of Kirrel2 (Fig. 2F). Interestingly, when SDS-PAGE was performed in non-reducing conditions (−DTT), another band with a molecular mass of ≈55 kDa appeared (Fig. 7A). It has previously been shown that the cytosolic domain of Kirrel2 can co-immunoprecipitate full-length protein, indicating that Kirrel2 molecules dimerize through their cytoplasmic domains (28). The 55-kDa band observed in non-reducing conditions therefore might represent dimers of the cleaved C-terminal fragment. In subcellular fractionation experiments, the 28-kDa C-terminal fragment was exclusively detected in membrane-enriched fractions and was not found in the cytosolic fractions (Fig. 7B). To confirm extracellular cleavage of Kirrel2 and to assess whether its ectodomain is shed, we analyzed supernatants from MIN6 cells by Western blotting with an antibody against the N-terminal fragment of Kirrel2. MIN6 cells expressing Kirrel2-V5 or control plasmids were cultured for 24 h in reduced serum medium. Culture media were collected, concentrated, and analyzed together with whole cell lysates by Western blotting (Fig. 7C). In the supernatants, Kirrel2 antibodies interacted with two bands with molecular masses of ≈50 and ≈65 kDa. These signals were not detected in cell lysates. These results indicate that the Kirrel2 ectodomain is constitutively shed from MIN6 cells, conceivably with two cleavage events. Next, we investigated the mechanisms through which the cytoplasmic Kirrel2 fragment is processed following the extracellular cleavage. Previous studies show that various type I integral membrane proteins such as E-cadherin (52), N-cadherin (53), and ephrinB2 (54) that undergo ectodomain shedding are subject to subsequent intramembrane cleavage by γ-secretase complex. To assess whether the C-terminal fragment of Kirrel2 is processed by γ-secretase, we repressed its functions with the potent γ-secretase inhibitor DAPT. Incubation of MIN6 cells with DAPT in three independent studies resulted in stabilization of the C-terminal fragment in the cell lysate compared with controls (p = 0.02), thereby confirming Kirrel2 as a substrate of the γ-secretase complex (Fig. 7D).
FIGURE 7.

Kirrel2 is cleaved and shed from MIN6 cells. A, lysates of MIN6 cells transfected with Kirrel2-V5 (+) or with control (−) plasmids were resolved with reducing (+DTT) or non-reducing (−DTT) SDS-PAGE and immunoblotted (IB) with anti-V5 antibody. B, total lysates and cytoplasmic and membrane-enriched fragments of MIN6 cells transfected with control (−) or Kirrel2-V5 (+) vectors were immunoblotted with anti-V5 tag, E-cadherin, and GAPDH antibodies. C, lysates and concentrated supernatants of MIN6 cells transfected with control (−) or Kirrel2-V5 (+) vectors were immunoblotted with antibodies against the extracellular domain of Kirrel2. D, MIN6 cells, transfected with control (−) or Kirrel2-V5 (+) vectors, were treated with DMSO or γ-secretase inhibitor DAPT (10 μm) for 16 h. Lysates were immunoblotted with anti-V5 antibodies. E, lysates and concentrated supernatants of MIN6 cells, expressing wild type or mutant Kirrel2-V5, were immunoblotted with anti-Kirrel2 antibody.
Regulated proteolysis is one of the most efficient mechanisms that cells exploit to control protein function to rapidly respond to alterations in internal and environmental cues (55). Therefore, we next assessed whether enhanced stability of Kirrel2Y595F/Y596F is due to decreased ectodomain shedding. To test this, we analyzed supernatants of MIN6 cells expressing wild type and phospho-mutant Kirrel2 molecules by immunoblotting. The ratio of shed to full-length Kirrel2 remained similar for both wild type and mutant Kirrel2 proteins, indicating that regulation of Kirrel2 stability by phosphorylation is independent of ectodomain cleavage (Fig. 7E).
Kirrel2 Alters Basal Insulin Secretion In Vitro and In Vivo
To assess functional relevance of Kirrel2 in pancreatic β-cells, we modulated Kirrel2 levels and measured insulin content and secretion in MIN6 cells. Kirrel2 knockdown with RNAi resulted in a small but significant increase in basal insulin secretion, whereas transient overexpression of Kirrel2-V5 suppressed basal insulin secretion from MIN6 cells (Fig. 8, A and B). Neither knockdown nor overexpression of Kirrel2 altered glucose-induced insulin secretion and total insulin content of MIN6 (Fig. 8, A and B).
FIGURE 8.

Kirrel2 regulates basal insulin secretion from pancreatic β-cells. Effect of RNAi mediated silencing (A) and overexpression (B) of Kirrel2 on basal and glucose-stimulated insulin secretion from MIN6 cells (n = 6 for A and B). *, p < 0.05 at Student's t test. IB, immunoblot. C, quantitative PCR with two different primer pairs that amplify fragments of Kirrel2 genomic locus upstream (primer pair 1) and downstream (primer pair 2) of the trapping cassette insertion site (n = 2). D, immunofluorescent images of islets from Kirrel2+/+, Kirrel2−/+, and Kirrel2−/− mice with anti-insulin (green) and anti-glucagon (red) antibodies. Nuclei (blue) are labeled with Hoechst stain. Bar, 50 μm. In vitro insulin secretion from E and total insulin content of F size-matched wild type and knock-out islets (n >4). **, p < 0.01 at Student's t test. G and H, circulating fasting insulin and glucagon in Kirrel2+/+, Kirrel2−/+, and Kirrel2−/− mice fed chow diet (CD) (n >5). I, total pancreatic insulin content normalized to pancreatic weight of 12-week-old, chow-fed males (n = 6). J, IPGTT with overnight-fasted 12-week-old male mice on chow diet (n >8). K and L, weight and circulating fasted glucose concentrations of Kirrel2 null, heterozygous, and littermate wild type male mice, kept on a high-fed diet (HFD), starting at 3 weeks of age (n >5). M and N, circulating fasting insulin and glucagon levels of Kirrel2+/+, Kirrel2−/+, and Kirrel2−/− mice fed a HFD (n >5). O, IPGTT with overnight-fasted, 12-week-old male mice that were fed a HFD for 8 weeks (n >5).
To investigate the physiological functions of Kirrel2, we generated mice with a null mutation in the Kirrel2 gene by targeted insertion of a trapping cassette within intron 2, resulting in premature termination of the endogenous transcript. Loss of Kirrel2 expression was confirmed by quantitative PCR (Fig. 8C). Expression of Kirrel1 and -3 was similar in Kirrel2 KO and control mice, demonstrating that there was no compensation (data not shown). Surprisingly, we found nephrin levels 50% reduced in Kirrel2 KO mice, most likely due to interference of the targeting construct with the bidirectional promoter/transcriptional unit regulating the expression of nephrin and Kirrel2 (56). Kirrel2−/− mice were born at expected Mendelian frequencies of normal size, fertile, and appeared healthy up to 12 months of age. To determine whether Kirrel2 is required for normal islet morphogenesis, we performed immunohistochemistry analysis with anti-insulin (β-cells) and glucagon (α-cells) antibodies on wild type and Kirrel2 null mice on pancreatic sections (Fig. 8D). In adult murine islets, structural organization of the endocrine cells creates a β-cell core and a discontinuous non-β-cell (mainly α-cells) mantle architecture (57). No apparent abnormalities were noted in the distribution of α- and β-cells of Kirrel2-deficient islets compared with littermate controls, indicating that Kirrel2 is not required for the formation and maintenance of the mantle-core arrangement in rodent islets. To assess other possible morphological changes in Kirrel2 null islets, we further investigated whether adherens junctions are altered. Confocal imaging of immunolabeled pancreatic sections of Kirrel2 null mice revealed that E-cadherin and β-catenin (data not shown) are expressed and distributed similarly in knock-out and control animals.
To study the functional significance of Kirrel2 in β-cells, we isolated pancreatic islets from Kirrel2 null and wild type littermates and performed basal and glucose-stimulated insulin secretion assays. Consistent with observations made with MIN6 cells, islets from mice with genetic deletion of Kirrel2 exhibited increased basal insulin secretion (Fig. 8E). However, no changes in insulin secretion were measured in isolated islets in response to high glucose concentrations (16 mm), glucagon-like peptide 1 (GLP-1, 10 nm), and KCl (30 mm). Total insulin content of size-matched islets used for static insulin secretion was similar (Fig. 8F). Kirrel2−/− mice were normoglycemic and had similar fasting plasma insulin and glucagon levels (Fig. 8, G and H). Total pancreatic insulin content (Fig. 8I) and glucose tolerance (Fig. 8J) were also comparable in mutant and wild type mice.
Finally, to assess the long term consequences of Kirrel2 ablation on pancreatic β-cell function and glucose whole body homeostasis during metabolic stress, we fed Kirrel2−/− and wild type littermate controls a HFD and monitored the starting at time of their weaning. No differences in body weight, fasting plasma glucose, insulin, and glucagon levels as well as in IPGTT were measured in Kirrel2 null and control mice (Fig. 8, K–O). These results demonstrate that Kirrel2 is dispensable for glycemic control in physiological and metabolic stress conditions in mice.
Discussion
In glomeruli of the kidney, nephrin and Kirrel family molecules localize to the slit diaphragms, which are highly specialized cell junctions of the podocytes (57). Despite the morphological and functional differences among the two cell types, multiple protein components of the slit diaphragms are also expressed in pancreatic β-cells (58). Although the functional importance of these proteins in the glomerular filtration barrier was established more than a decade ago, they have only recently been investigated in detail in pancreatic in β-cells. Nephrin, for instance, is expressed on the surface of insulin granules and the plasma membrane, its cellular localization is highly dynamic, and it facilitates glucose-stimulated insulin release (29). Our results show that Kirrel2 is unlikely to be a constituent of insulin granules because we found it predominantly at cell-to-cell contacts that co-localize with the adherens junction proteins E-cadherin and β-catenin as well as Kirrel2 molecules from neighboring cells. Additionally, we have demonstrated that both E-cadherin and β-catenin co-precipitate with Kirrel2, indicating that they might be members of the same protein supercomplex that is formed at the cell junctions.
In podocytes, both nephrin and NEPH1 involvement in the dynamic organization of podocyte morphology and function are regulated by phosphorylation of several cytoplasmic residues (57). More recently, tyrosine phosphorylation of nephrin was shown to be altered by glucose (59). In this study, we have demonstrated that Kirrel2 is also regulated by post-translational modifications. Our results revealed phosphorylation at two serine and several tyrosine residues in the cytoplasmic tail of Kirrel2. Serine phosphorylations have not been reported in any Kirrel family member or nephrin, and its function is currently unknown. In contrast, tyrosine phosphorylation in NEPH1 and nephrin family is well studied with established links to signal transduction pathways and protein-protein interactions. Two of the identified phosphosites Tyr595 and Tyr596 in Kirrel2 are located in a highly conserved stretch of nine amino acids (DPTNGYYXV) that is shared by all three Kirrel family members. Phosphorylation of both tyrosines in this stretch is required for NEPH1 interactions with an Src homology 2 domain-containing adaptor molecule, growth factor receptor-bound protein (Grb2) in podocytes (40). Additionally, phosphorylation of the first tyrosine residue in the conserved peptide is critical for the interaction of Kirrel family members with podocin, another essential member of the glomerular slit diaphragm (60). In podocytes, podocin is crucial for membrane docking of nephrin and nephrin signaling (61) as well as facilitating NEPH1-nephrin interactions (33). Unlike Kirrel family proteins and nephrin, podocin is not expressed in pancreatic islets (58) raising the question whether a functionally similar protein can substitute for it in pancreatic β-cells.
The significance of tyrosine phosphorylation at Tyr595 and Tyr596 in the regulation of Kirrel2 function was demonstrated by the drastic changes in the morphology of cell junctions elicited by the phospho-mutant Kirrel2Y595F/Y596F. This effect was not mediated by increased protein expression of Kirrel2Y595F/Y596F because wild type Kirrel2 does not induce such perturbations even when expressed at higher levels than the phospho-mutant. The main effects observed upon disruption of Tyr595 and Tyr596 phosphorylation in Kirrel2 were an increase in protein stability and cell surface localization. These results suggest a mechanism of Kirrel2 turnover that is functionally coupled to its signaling, most likely by Kirrel2 internalization and subsequent degradation. Indeed, tyrosine phosphorylation of nephrin for instance has been shown to increase its raft-mediated endocytosis thereby creating a negative feedback loop in the nephrin signaling pathway (41). It is important to point out that although mutations at Tyr595 and Tyr596 significantly alter the amount of Kirrel2 that localizes to the cell junctions, they are not sufficient to localize Kirrel2 at contact free membranes. These observations indicate that phosphorylation alone does not regulate Kirrel2 localization at the cell surface. It is possible that cis- and trans-dimerization as well as interactions with other molecules are also required for stabilization of Kirrel2 at the cell contacts similar to the model that was proposed for nephrin sorting in podocytes (41). Our results indicate that the signals transduced from other phosphorylated tyrosine residues on Kirrel2 are not involved in the regulation of protein stability or localization. However, it would be interesting to investigate whether phosphorylation at these residues alters Kirrel2 signaling or its interactions with other molecules.
Fyn kinase is a tyrosine-specific phosphotransferase and a member of the Src family and has been shown to phosphorylate key targets involved in a variety of different signaling pathways, including integrin-mediated signaling, growth factor and cytokine receptor signaling, ion channel and cell adhesion (62). The role of Fyn in pancreatic β-cells is currently unknown; however, the related kinase YES has been shown to be required for granule mobilization/replenishment, and F-actin remodeling in sustained insulin secretion (63) and Fyn-related kinase FRK influences β-cell number during embryogenesis and neonatal life (64). In this study, we have shown that Fyn is also responsible for Kirrel2 phosphorylation in pancreatic β-cells. We did not study the contribution of other members of the Src family kinases to Kirrel2 phosphorylation because Fyn knockdown alone was sufficient to significantly reduce tyrosine phosphorylation of Kirrel2. Our data suggest that tyrosine phosphorylation regulates the stability of Kirrel2 in pancreatic β-cells, which might be a prerequisite for destabilizing cell-cell contacts in response to changes in islet growth or regulated exocytosis of insulin granules.
Our analysis of MIN6 cell supernatants revealed that Kirrel2 is extracellularly cleaved giving rise to two different N-terminally shed fragments. Ectodomain shedding of cell adhesion molecules can regulate their adhesive properties or signaling functions and be induced during natural processes such as protein turnover, cell division, differentiation, and morphological alterations linked to development as well as stress and tissue injury (65). For example, proteinuria that develops during preeclampsia is induced by nephrin shedding from podocytes as a result of endothelin-1 release from endothelial glomerular cells (66). Similarly, the amount of NEPH2 ectodomain that is shed from human podocytes is significantly higher in the urine of patients with membranous glomerulonephritis (67). Interestingly, NEPH2 shedding from podocytes produces two extracellular cleavage products of the same molecular size as the shed fragments of Kirrel2 that were detected in the MIN6 supernatants (67). Considering the fact that the two proteins display highly homologous extracellular domain architecture, it is possible that the molecular mechanisms that are involved in their shedding are also similar. Additionally, we show that following its ectodomain shedding the membrane spanning cytoplasmic domain of Kirrel2 is processed by the γ-secretase complex. There are various examples that show shed extracellular domains or cleaved intracellular domains of γ-secretase substrates are not always simple intermediates destined for degradation but can continue to interact with signaling partners and promote direct or indirect intracellular signaling (68). Therefore, functional characterization of Kirrel2 cleavage products as well as external and internal stimuli that influence Kirrel2 shedding could provide additional insight into the cellular and physiological functions of Kirrel2.
We found that basal insulin secretion was modestly increased in MIN6 cells with knockdown of Kirrel2 and in primary islets of Kirrel2 null mice compared with control littermates. The reduced expression of nephrin in Kirrel2 KO mice is unlikely to contribute to the increased basal insulin secretion in Kirrel2 KO mice because nephrin null mice exhibit reduced glucose-stimulated insulin release in MIN6 cells and human islets (29). To corroborate these findings, we silenced nephrin expression by RNAi in Min6 cells and found no changes in insulin secretion at low (3 mm) but increased insulin release at high glucose concentration (data not shown). Interestingly, an insulin tolerance test did not reveal altered insulin sensitivity (data not shown), and we did not measure increased plasma insulin or decreased blood glucose levels under fasting or randomly fed conditions in Kirrel2 null mice. The reason for this discrepancy is currently unknown but is most likely due to the many hypoglycemic counter-regulatory responses known to regulate glucose homeostasis in intact animals, including the autonomic nervous system, metabolites, and gut hormones.
It is well established that in vitro close to normal secretory responses of intact islets are significantly impaired upon islet dispersion resulting in blunted stimulated and elevated basal insulin secretion (20). Furthermore, it is widely accepted that individual β-cells require direct cell contact to optimize their inherently heterogeneous basal and stimulated insulin secretion (69, 70). Here, we have shown that decreased expression levels of Kirrel2 in MIN6 cells and genetic ablation of Kirrel2 from pancreatic islets of mice result in elevated basal insulin secretion. Interestingly, this phenotype has also been noted in MIN6 cells with lower expression levels of E-cadherin (71) and in knock-out mice of β-cell gap junction molecule connexin36 (14). Our results may indicate that Kirrel2 is part of an extracellular cell adhesion scaffold that is required for normal basal insulin secretion. However, genetic deletion of Kirrel2 does not result in significant changes in glucose-stimulated insulin secretion, clearly indicating that Kirrel2 is dispensable for normal glucose homeostasis in mice.
Author Contributions
B. Y. designed, performed, analyzed, and interpreted all biochemical, cell biological, and physiological experiments and drafted the article. M. S. designed, analyzed, and interpreted data and edited and revised the manuscript. T. B. and B. W. performed and interpreted the phospho-proteomic analysis. All authors reviewed the results, contributed to the writing, and approved the final version of the manuscript.
Acknowledgments
We thank Regina Kubsch and Hasan Kabakci for technical help; Dr. Chingwen Yang for ES cell microinjections; Dr. Eckhard Lammert for providing us MIN6 cells; Dr. Mathieu Latreille and Dr. Daria Esterházy for their contributions to the designing of various experiments; and Dr. Bengt-Frederik Belgardt, Dr. Faraz Quazzi, and Dr. Dominik Waluk for critical reading of the manuscript.
The authors declare that they have no conflicts of interest with the contents of this article.
- Kirrel
- Kin of IRRE-like protein
- CPE
- carboxypeptidase E
- E-cadherin
- epithelial cadherin
- EGFR
- epidermal growth factor receptor
- HFD
- high fat diet
- IGF1R
- insulin-like growth factor 1 receptor
- IPGTT
- intraperitoneal glucose tolerance test
- NEPH
- nephrin-like protein
- SFK
- Src family kinase.
References
- 1.Bell G. I., and Polonsky K. S. (2001) Diabetes mellitus and genetically programmed defects in beta-cell function. Nature 414, 788–791 [DOI] [PubMed] [Google Scholar]
- 2.Jeon J., Correa-Medina M., Ricordi C., Edlund H., and Diez J. A. (2009) Endocrine cell clustering during human pancreas development. J. Histochem. Cytochem. 57, 811–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dahl U., Sjødin A., and Semb H. (1996) Cadherins regulate aggregation of pancreatic beta-cells in vivo. Development 122, 2895–2902 [DOI] [PubMed] [Google Scholar]
- 4.Meda P. (2013) Protein-mediated interactions of pancreatic islet cells. Scientifica 2013, 621249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rogers G. J., Hodgkin M. N., and Squires P. E. (2007) E-cadherin and cell adhesion: a role in architecture and function in the pancreatic islet. Cell. Physiol. Biochem. 20, 987–994 [DOI] [PubMed] [Google Scholar]
- 6.Rouiller D. G., Cirulli V., and Halban P. A. (1991) Uvomorulin mediates calcium-dependent aggregation of islet cells, whereas calcium-independent cell adhesion molecules distinguish between islet cell types. Dev. Biol. 148, 233–242 [DOI] [PubMed] [Google Scholar]
- 7.Esni F., Täljedal I. B., Perl A. K., Cremer H., Christofori G., and Semb H. (1999) Neural cell adhesion molecule (N-CAM) is required for cell type segregation and normal ultrastructure in pancreatic islets. J. Cell Biol. 144, 325–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cirulli V., Baetens D., Rutishauser U., Halban P. A., Orci L., and Rouiller D. G. (1994) Expression of neural cell adhesion molecule (N-CAM) in rat islets and its role in islet cell type segregation. J. Cell Sci. 107, 1429–1436 [DOI] [PubMed] [Google Scholar]
- 9.Steed E., Balda M. S., and Matter K. (2010) Dynamics and functions of tight junctions. Trends Cell Biol. 20, 142–149 [DOI] [PubMed] [Google Scholar]
- 10.Serre-Beinier V., Le Gurun S., Belluardo N., Trovato-Salinaro A., Charollais A., Haefliger J. A., Condorelli D. F., and Meda P. (2000) Cx36 preferentially connects beta-cells within pancreatic islets. Diabetes 49, 727–734 [DOI] [PubMed] [Google Scholar]
- 11.Konstantinova I., Nikolova G., Ohara-Imaizumi M., Meda P., Kucera T., Zarbalis K., Wurst W., Nagamatsu S., and Lammert E. (2007) EphA-Ephrin-A-mediated beta cell communication regulates insulin secretion from pancreatic islets. Cell 129, 359–370 [DOI] [PubMed] [Google Scholar]
- 12.Wellershaus K., Degen J., Deuchars J., Theis M., Charollais A., Caille D., Gauthier B., Janssen-Bienhold U., Sonntag S., Herrera P., Meda P., and Willecke K. (2008) A new conditional mouse mutant reveals specific expression and functions of connexin36 in neurons and pancreatic beta-cells. Exp. Cell Res. 314, 997–1012 [DOI] [PubMed] [Google Scholar]
- 13.Speier S., Gjinovci A., Charollais A., Meda P., and Rupnik M. (2007) Cx36-mediated coupling reduces beta-cell heterogeneity, confines the stimulating glucose concentration range, and affects insulin release kinetics. Diabetes 56, 1078–1086 [DOI] [PubMed] [Google Scholar]
- 14.Ravier M. A., Güldenagel M., Charollais A., Gjinovci A., Caille D., Söhl G., Wollheim C. B., Willecke K., Henquin J. C., and Meda P. (2005) Loss of connexin36 channels alters beta-cell coupling, islet synchronization of glucose-induced Ca2+ and insulin oscillations, and basal insulin release. Diabetes 54, 1798–1807 [DOI] [PubMed] [Google Scholar]
- 15.Olofsson C. S., Håkansson J., Salehi A., Bengtsson M., Galvanovskis J., Partridge C., SörhedeWinzell M., Xian X., Eliasson L., Lundquist I., Semb H., and Rorsman P. (2009) Impaired insulin exocytosis in neural cell adhesion molecule−/− mice due to defective reorganization of the submembrane F-actin network. Endocrinology 150, 3067–3075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vozzi C., Ullrich S., Charollais A., Philippe J., Orci L., and Meda P. (1995) Adequate connexin-mediated coupling is required for proper insulin production. J. Cell Biol. 131, 1561–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Charollais A., Gjinovci A., Huarte J., Bauquis J., Nadal A., Martín F., Andreu E., Sánchez-Andrés J. V., Calabrese A., Bosco D., Soria B., Wollheim C. B., Herrera P. L., and Meda P. (2000) Junctional communication of pancreatic beta cells contributes to the control of insulin secretion and glucose tolerance. J. Clin. Invest. 106, 235–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hauge-Evans A. C., Squires P. E., Persaud S. J., and Jones P. M. (1999) Pancreatic beta-cell-to-beta-cell interactions are required for integrated responses to nutrient stimuli: enhanced Ca2+ and insulin secretory responses of MIN6 pseudoislets. Diabetes 48, 1402–1408 [DOI] [PubMed] [Google Scholar]
- 19.Hodson D. J., Mitchell R. K., Bellomo E. A., Sun G., Vinet L., Meda P., Li D., Li W. H., Bugliani M., Marchetti P., Bosco D., Piemonti L., Johnson P., Hughes S. J., and Rutter G. A. (2013) Lipotoxicity disrupts incretin-regulated human beta cell connectivity. J. Clin. Invest. 123, 4182–4194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Halban P. A., Wollheim C. B., Blondel B., Meda P., Niesor E. N., and Mintz D. H. (1982) The possible importance of contact between pancreatic islet cells for the control of insulin release. Endocrinology 111, 86–94 [DOI] [PubMed] [Google Scholar]
- 21.Jaques F., Jousset H., Tomas A., Prost A. L., Wollheim C. B., Irminger J. C., Demaurex N., and Halban P. A. (2008) Dual effect of cell-cell contact disruption on cytosolic calcium and insulin secretion. Endocrinology 149, 2494–2505 [DOI] [PubMed] [Google Scholar]
- 22.Sun C., Kilburn D., Lukashin A., Crowell T., Gardner H., Brundiers R., Diefenbach B., and Carulli J. P. (2003) Kirrel2, a novel immunoglobulin superfamily gene expressed primarily in beta cells of the pancreatic islets. Genomics 82, 130–142 [DOI] [PubMed] [Google Scholar]
- 23.Ruotsalainen V., Ljungberg P., Wartiovaara J., Lenkkeri U., Kestilä M., Jalanko H., Holmberg C., and Tryggvason K. (1999) nephrin is specifically located at the slit diaphragm of glomerular podocytes. Proc. Natl. Acad. Sci. U.S.A. 96, 7962–7967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu G., Kaw B., Kurfis J., Rahmanuddin S., Kanwar Y. S., and Chugh S. S. (2003) Neph1 and nephrin interaction in the slit diaphragm is an important determinant of glomerular permeability. J. Clin. Invest. 112, 209–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kestilä M., Lenkkeri U., Männikkö M., Lamerdin J., McCready P., Putaala H., Ruotsalainen V., Morita T., Nissinen M., Herva R., Kashtan C. E., Peltonen L., Holmberg C., Olsen A., and Tryggvason K. (1998) Positionally cloned gene for a novel glomerular protein–nephrin–is mutated in congenital nephrotic syndrome. Mol. Cell 1, 575–582 [DOI] [PubMed] [Google Scholar]
- 26.Putaala H., Soininen R., Kilpeläinen P., Wartiovaara J., and Tryggvason K. (2001) The murine nephrin gene is specifically expressed in kidney, brain and pancreas: inactivation of the gene leads to massive proteinuria and neonatal death. Hum. Mol. Genet. 10, 1–8 [DOI] [PubMed] [Google Scholar]
- 27.Rantanen M., Palmén T., Pätäri A., Ahola H., Lehtonen S., Aström E., Floss T., Vauti F., Wurst W., Ruiz P., Kerjaschki D., and Holthöfer H. (2002) nephrin TRAP mice lack slit diaphragms and show fibrotic glomeruli and cystic tubular lesions. J. Am. Soc. Nephrol. 13, 1586–1594 [DOI] [PubMed] [Google Scholar]
- 28.Heikkilä E., Ristola M., Havana M., Jones N., Holthöfer H., and Lehtonen S. (2011) Trans-interaction of nephrin and Neph1/Neph3 induces cell adhesion that associates with decreased tyrosine phosphorylation of nephrin. Biochem. J. 435, 619–628 [DOI] [PubMed] [Google Scholar]
- 29.Fornoni A., Jeon J., Varona Santos J., Cobianchi L., Jauregui A., Inverardi L., Mandic S. A., Bark C., Johnson K., McNamara G., Pileggi A., Molano R. D., Reiser J., Tryggvason K., Kerjaschki D., et al. (2010) nephrin is expressed on the surface of insulin vesicles and facilitates glucose-stimulated insulin release. Diabetes 59, 190–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rinta-Valkama J., Aaltonen P., Lassila M., Palmén T., Tossavainen P., Knip M., and Holthöfer H. (2007) Densin and filtrin in the pancreas and in the kidney, targets for humoral autoimmunity in patients with type 1 diabetes. Diabetes Metab. Res. Rev. 23, 119–126 [DOI] [PubMed] [Google Scholar]
- 31.Mathis D., Vence L., and Benoist C. (2001) beta-Cell death during progression to diabetes. Nature 414, 792–798 [DOI] [PubMed] [Google Scholar]
- 32.Donoviel D. B., Freed D. D., Vogel H., Potter D. G., Hawkins E., Barrish J. P., Mathur B. N., Turner C. A., Geske R., Montgomery C. A., Starbuck M., Brandt M., Gupta A., Ramirez-Solis R., Zambrowicz B. P., and Powell D. R. (2001) Proteinuria and perinatal lethality in mice lacking NEPH1, a novel protein with homology to NEPHRIN. Mol. Cell. Biol. 21, 4829–4836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barletta G. M., Kovari I. A., Verma R. K., Kerjaschki D., and Holzman L. B. (2003) nephrin and Neph1 co-localize at the podocyte foot process intercellular junction and form cis hetero-oligomers. J. Biol. Chem. 278, 19266–19271 [DOI] [PubMed] [Google Scholar]
- 34.Gerke P., Huber T. B., Sellin L., Benzing T., and Walz G. (2003) Homodimerization and heterodimerization of the glomerular podocyte proteins nephrin and NEPH1. J. Am. Soc. Nephrol. 14, 918–926 [DOI] [PubMed] [Google Scholar]
- 35.Bock T., Moest H., Omasits U., Dolski S., Lundberg E., Frei A., Hofmann A., Bausch-Fluck D., Jacobs A., Krayenbuehl N., Uhlen M., Aebersold R., Frei K., and Wollscheid B. (2012) Proteomic analysis reveals drug accessible cell surface N-glycoproteins of primary and established glioblastoma cell lines. J. Proteome Res. 11, 4885–4893 [DOI] [PubMed] [Google Scholar]
- 36.UniProt Consortium (2014) Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res. 42, D191–D198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 38.Omasits U., Ahrens C. H., Müller S., and Wollscheid B. (2014) Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics 30, 884–886 [DOI] [PubMed] [Google Scholar]
- 39.Verma R., Wharram B., Kovari I., Kunkel R., Nihalani D., Wary K. K., Wiggins R. C., Killen P., and Holzman L. B. (2003) Fyn binds to and phosphorylates the kidney slit diaphragm component nephrin. J. Biol. Chem. 278, 20716–20723 [DOI] [PubMed] [Google Scholar]
- 40.Garg P., Verma R., Nihalani D., Johnstone D. B., and Holzman L. B. (2007) Neph1 cooperates with nephrin to transduce a signal that induces actin polymerization. Mol. Cell. Biol. 27, 8698–8712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qin X. S., Tsukaguchi H., Shono A., Yamamoto A., Kurihara H., and Doi T. (2009) Phosphorylation of nephrin triggers its internalization by raft-mediated endocytosis. J. Am. Soc. Nephrol. 20, 2534–2545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones N., Blasutig I. M., Eremina V., Ruston J. M., Bladt F., Li H., Huang H., Larose L., Li S. S., Takano T., Quaggin S. E., and Pawson T. (2006) Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature 440, 818–823 [DOI] [PubMed] [Google Scholar]
- 43.Li H., Lemay S., Aoudjit L., Kawachi H., and Takano T. (2004) SRC-family kinase Fyn phosphorylates the cytoplasmic domain of nephrin and modulates its interaction with podocin. J. Am. Soc. Nephrol. 15, 3006–3015 [DOI] [PubMed] [Google Scholar]
- 44.Harita Y., Kurihara H., Kosako H., Tezuka T., Sekine T., Igarashi T., and Hattori S. (2008) Neph1, a component of the kidney slit diaphragm, is tyrosine-phosphorylated by the Src family tyrosine kinase and modulates intracellular signaling by binding to Grb2. J. Biol. Chem. 283, 9177–9186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheng H., Straub S. G., and Sharp G. W. (2007) Inhibitory role of Src family tyrosine kinases on Ca2+-dependent insulin release. Am. J. Physiol. Endocrinol. Metab. 292, E845–E852 [DOI] [PubMed] [Google Scholar]
- 46.Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., and Selbach M. (2011) Global quantification of mammalian gene expression control. Nature 473, 337–342 [DOI] [PubMed] [Google Scholar]
- 47.Esterházy D., Akpinar P., and Stoffel M. (2012) Tmem27 dimerization, deglycosylation, plasma membrane depletion, and the extracellular Phe-Phe motif are negative regulators of cleavage by Bace2. Biol. Chem. 393, 473–484 [DOI] [PubMed] [Google Scholar]
- 48.Sorkin A., and Goh L. K. (2008) Endocytosis and intracellular trafficking of ErbBs. Exp. Cell Res. 314, 3093–3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dhanvantari S., Arnaoutova I., Snell C. R., Steinbach P. J., Hammond K., Caputo G. A., London E., and Loh Y. P. (2002) Carboxypeptidase E, a prohormone sorting receptor, is anchored to secretory granules via a C-terminal transmembrane insertion. Biochemistry 41, 52–60 [DOI] [PubMed] [Google Scholar]
- 50.Aaltonen P., and Holthöfer H. (2007) The nephrin-based slit diaphragm: new insight into the signalling platform identifies targets for therapy. Nephrol. Dial. Transplant. 22, 3408–3410 [DOI] [PubMed] [Google Scholar]
- 51.Brieher W. M., Yap A. S., and Gumbiner B. M. (1996) Lateral dimerization is required for the homophilic binding activity of C-cadherin. J. Cell Biol. 135, 487–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marambaud P., Shioi J., Serban G., Georgakopoulos A., Sarner S., Nagy V., Baki L., Wen P., Efthimiopoulos S., Shao Z., Wisniewski T., and Robakis N. K. (2002) A presenilin-1/γ-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J. 21, 1948–1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marambaud P., Wen P. H., Dutt A., Shioi J., Takashima A., Siman R., and Robakis N. K. (2003) A CBP binding transcriptional repressor produced by the PS1/ϵ-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell 114, 635–645 [DOI] [PubMed] [Google Scholar]
- 54.Georgakopoulos A., Litterst C., Ghersi E., Baki L., Xu C., Serban G., and Robakis N. K. (2006) Metalloproteinase/presenilin1 processing of ephrinB regulates EphB-induced Src phosphorylation and signaling. EMBO J. 25, 1242–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lal M., and Caplan M. (2011) Regulated intramembrane proteolysis: signaling pathways and biological functions. Physiology 26, 34–44 [DOI] [PubMed] [Google Scholar]
- 56.Ristola M., and Lehtonen S. (2014) Functions of the podocyte proteins nephrin and Neph3 and the transcriptional regulation of their genes. Clin. Sci. 126, 315–328 [DOI] [PubMed] [Google Scholar]
- 57.Brissova M., Fowler M. J., Nicholson W. E., Chu A., Hirshberg B., Harlan D. M., and Powers A. C. (2005) Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy. J. Histochem. Cytochem. 53, 1087–1097 [DOI] [PubMed] [Google Scholar]
- 58.Rinta-Valkama J., Palmén T., Lassila M., and Holthöfer H. (2007) Podocyte-associated proteins FAT, α-actinin-4 and filtrin are expressed in Langerhans islets of the pancreas. Mol. Cell. Biochem. 294, 117–125 [DOI] [PubMed] [Google Scholar]
- 59.Kapodistria K., Tsilibary E. P., Politis P., Moustardas P., Charonis A., and Kitsiou P. (2015) nephrin, a transmembrane protein, is involved in pancreatic beta-cell survival signaling. Mol. Cell. Endocrinol. 400, 112–128 [DOI] [PubMed] [Google Scholar]
- 60.Sellin L., Huber T. B., Gerke P., Quack I., Pavenstädt H., and Walz G. (2003) NEPH1 defines a novel family of podocin interacting proteins. FASEB J. 17, 115–117 [DOI] [PubMed] [Google Scholar]
- 61.Huber T. B., Kottgen M., Schilling B., Walz G., and Benzing T. (2001) Interaction with podocin facilitates nephrin signaling. J. Biol. Chem. 276, 41543–41546 [DOI] [PubMed] [Google Scholar]
- 62.Parsons S. J., and Parsons J. T. (2004) Src family kinases, key regulators of signal transduction. Oncogene 23, 7906–7909 [DOI] [PubMed] [Google Scholar]
- 63.Yoder S. M., Dineen S. L., Wang Z., and Thurmond D. C. (2014) YES, a Src family kinase, is a proximal glucose-specific activator of cell division cycle control protein 42 (Cdc42) in pancreatic islet beta cells. J. Biol. Chem. 289, 11476–11487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Akerblom B., Annerén C., and Welsh M. (2007) A role of FRK in regulation of embryonal pancreatic beta cell formation. Mol. Cell. Endocrinol. 270, 73–78 [DOI] [PubMed] [Google Scholar]
- 65.Stützer I., Esterházy D., and Stoffel M. (2012) The pancreatic beta cell surface proteome. Diabetologia 55, 1877–1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Collino F., Bussolati B., Gerbaudo E., Marozio L., Pelissetto S., Benedetto C., and Camussi G. (2008) Preeclamptic sera induce nephrin shedding from podocytes through endothelin-1 release by endothelial glomerular cells. Am. J. Physiol. Renal Physiol. 294, F1185–F1194 [DOI] [PubMed] [Google Scholar]
- 67.Gerke P., Sellin L., Kretz O., Petraschka D., Zentgraf H., Benzing T., and Walz G. (2005) NEPH2 is located at the glomerular slit diaphragm, interacts with nephrin and is cleaved from podocytes by metalloproteinases. J. Am. Soc. Nephrol. 16, 1693–1702 [DOI] [PubMed] [Google Scholar]
- 68.Kopan R., and Ilagan M. X. (2004) γ-Secretase: proteasome of the membrane? Nat. Rev. Mol. Cell Biol. 5, 499–504 [DOI] [PubMed] [Google Scholar]
- 69.Pipeleers D. G. (1992) Heterogeneity in pancreatic beta-cell population. Diabetes 41, 777–781 [DOI] [PubMed] [Google Scholar]
- 70.Salomon D., and Meda P. (1986) Heterogeneity and contact-dependent regulation of hormone secretion by individual B cells. Exp. Cell Res. 162, 507–520 [DOI] [PubMed] [Google Scholar]
- 71.Calabrese A., Caton D., and Meda P. (2004) Differentiating the effects of Cx36 and E-cadherin for proper insulin secretion of MIN6 cells. Exp. Cell Res. 294, 379–391 [DOI] [PubMed] [Google Scholar]