

Background: The C452F mutation in the mitochondrial fission protein Drp1 leads to heart failure through an unknown mechanism.
Results: C452F impairs Drp1 disassembly, leading to impaired mitophagy, failed bioenergetics, and inflammation.
Conclusion: Drp1-mediated mitochondrial fission is essential for normal cardiac function.
Significance: Mutations in mitochondrial quality control proteins are a likely cause of human cardiomyopathy.
Keywords: cardiomyopathy, heart failure, mitochondria, mitochondrial metabolism, mitophagy
Abstract
We have reported previously that a missense mutation in the mitochondrial fission gene Dynamin-related protein 1 (Drp1) underlies the Python mouse model of monogenic dilated cardiomyopathy. The aim of this study was to investigate the consequences of the C452F mutation on Drp1 protein function and to define the cellular sequelae leading to heart failure in the Python monogenic dilated cardiomyopathy model. We found that the C452F mutation increased Drp1 GTPase activity. The mutation also conferred resistance to oligomer disassembly by guanine nucleotides and high ionic strength solutions. In a mouse embryonic fibroblast model, Drp1 C452F cells exhibited abnormal mitochondrial morphology and defective mitophagy. Mitochondria in C452F mouse embryonic fibroblasts were depolarized and had reduced calcium uptake with impaired ATP production by oxidative phosphorylation. In the Python heart, we found a corresponding progressive decline in oxidative phosphorylation with age and activation of sterile inflammation. As a corollary, enhancing autophagy by exposure to a prolonged low-protein diet improved cardiac function in Python mice. In conclusion, failure of Drp1 disassembly impairs mitophagy, leading to a downstream cascade of mitochondrial depolarization, aberrant calcium handling, impaired ATP synthesis, and activation of sterile myocardial inflammation, resulting in heart failure.
Introduction
Dilated cardiomyopathy (DCM)6 is a heart failure syndrome characterized by progressive cardiomyocyte loss, contractile dysfunction, and sudden death. Its etiology is diverse, including ischemia, inflammation, and genetic disease (1). Rare monogenic forms of DCM are mechanistically tractable models, yielding insights into disease pathogenesis. For example, energy deficiency is a common, albeit typically late, feature of DCM (2). Understanding the cellular regulation of mitochondria and control of energetics is of fundamental importance for developing novel therapies.
We have reported previously that the C452F mutation in the gene encoding the mitochondrial fission protein Dynamin-related protein 1 (Drp1, also known as Dnm1l) causes DCM in mice (3). Mitochondrial morphology and function are regulated dynamically by conserved fission and fusion proteins. Drp1, a member of the dynamin superfamily of large GTPases, is the principal mitochondrial fission protein in eukaryotic cells. After activation, Drp1 translocates from a cytosolic pool to the outer mitochondrial membrane, where it oligomerizes, hydrolyzes GTP, and constricts to divide mitochondria (4).
Mitochondrial remodeling has multiple roles, including quality control and clearance of mitochondria by mitophagy, segregation of mitochondrial DNA and proteins, and apoptosis (5). As a corollary, Drp1 contributes to Parkin-induced mitophagy in MEFs (6). Changes in mitochondrial morphology reflect active cellular control over mitochondrial function. For example, mitochondrial elongation under conditions of nutrient deprivation protects against mitophagic degradation and preserves ATP synthesis (7, 8) Failure of cellular control over mitochondria triggers inflammation by Toll-like receptors or the inflammasome, leading to heart failure (9, 10).
A single human mutation in DRP1 has been identified (A395D), lying close to the C452F mutation in the middle domain of the protein (11). DRP1 A395D is lethal in infancy, causing abnormal development, lactic acidosis, and encephalopathy. Biochemically, the mutant A395D protein has been found to be defective for self-assembly and GTP hydrolysis (12).
Here we show that the C452F mutation in Drp1 dysregulates protein disassembly. In a MEF model, this results in abnormal mitochondrial morphology, mitophagy, mitochondrial membrane potential, and calcium signaling. In the hearts of Drp1 C452F (Python) mice, there is a corresponding progressive decline in oxidative phosphorylation and activation of sterile inflammation. Prolonged exposure of Python mice to a low-protein diet augments intrinsic cellular macroautophagy and improves cardiac function. We conclude that, in the heart, failure of Drp1-mediated mitochondrial regulation leads to impairment of mitophagy, bioenergetic failure, and inflammation, ultimately resulting in DCM.
Experimental Procedures
Animal Studies
Drp1 C452F mice (Python) have been described previously (3). Procedures involving live animals were performed in line with United Kingdom Home Office guidelines (project license 30/2977). Unless stated otherwise, mice were backcrossed onto a C57BL/6 genetic background for at least five generations. Where indicated, mice were fed an isocaloric 6% low-protein diet (Teklad TD.90016 diet, Harlan Teklad) from 4 weeks of age onwards.
Mouse Embryonic Fibroblasts
Drp1 C452F MEFs were derived from embryonic day 13.5 embryos. Cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and immortalized using SV40 large T antigen. Transient transfections were performed using Lipofectamine 2000 (Invitrogen) according to the protocol of the manufacturer. Where indicated, MEFs were transfected and imaged by confocal microscopy as described previously (13). Drp1−/− MEFs (14) were originally a gift from Prof. Mihara to Prof. Scorrano. For starvation experiments, MEFs were washed four times and then incubated in Hanks' balanced salt solution supplemented with 10 mm HEPES (pH 7.4) at 37 °C for the indicated time.
Mitochondrially Targeted, Photoactivatable GFP (mito-PAGFP) Imaging
MEFs were cotransfected as above with mito-PAGFP (Addgene) and dsRed. After focal activation of mitochondria, whole cells were imaged by confocal microscopy at 5, 10, and 30 min, and the fluorescence intensity of the mito-PAGFP signal in the photoactivated region was plotted against time. Values of the percentage of mito-PAGFP fluorescence decrease in the region of activation were plotted against time for each experimental group as described previously (15).
DNA Constructs for Protein Expression
For protein expression, Drp1 isoform 1 (GenBank accession no. AB006965) was amplified by PCR with Pfu Turbo DNA polymerase (Stratagene) as an NdeI/XhoI fragment, with a tobacco etch virus protease site (ENLYFQS) preceding the XhoI restriction site. The resulting DNA fragment was then ligated into the bacterial expression vector pET29b (EMD Millipore, Billerica, MA), which contains a C-terminal His6 tag. The C452F and A401D mutations were introduced into pET29b-Drp1-His6 using the QuikChange method (Stratagene). All constructs were verified by DNA sequencing (Retrogen).
Expression and Purification of His6-tagged Drp1, Drp1 C452F, and Drp1 A401D
Drp1 activity was evaluated by recombinant expression of the human enzyme isoform 1, which shares 94.5% sequence identity with mice. The human mutation A395D is equivalent to mouse A401D. For expression of His6-tagged Drp1, C452F-His6, and A401D-His6 fusion proteins, plasmids were transformed into Escherichia coli BL21 (DE3). Cells were grown at 37 °C in Super Broth with kanamycin (30 μg/ml) to an A600 of ∼1.5 with shaking at 250 rpm. The temperature was lowered to 14 °C, and, after 30 min, protein expression was induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside for 12–16 h. Cells were harvested by centrifugation using a Sorvall JLA-8.1000 rotor at 5000 rpm for 10 min at 4 °C and resuspended in column buffer A (20 mm HEPES (pH 7.4), 400 mm NaCl, 5 mm MgCl2, and 40 mm imidazole) containing protease inhibitors (Complete, EDTA-free protease inhibitor mixture, Roche Applied Science). Cells were lysed by five passes through Emulsiflex C-3 (Avestin) operating at ∼15,000 p.s.i. DNase was added to a final concentration of 1 μg/ml, and lysates were clarified by centrifugation using a Sorvall SS34 rotor at 15,000 rpm for 30 min at 4 °C. His6-tagged fusion proteins were isolated from the resulting supernatant by affinity chromatography using Ni2+-Sepharose high-performance beads (GE Healthcare). Bound fusion proteins were washed with 200–300 ml of column buffer A, 100 ml of column buffer B (20 mm HEPES (pH 7.4), 400 mm NaCl, 5 mm MgCl2, 40 mm imidazole, 1 mm ATP, and 10 mm KCl), 100 ml of column buffer C (20 mm HEPES (pH 7.4), 40 mm imidazole, 400 mm NaCl, 0.5% (w/v) CHAPS), and 100 ml of column buffer A. Bound fusion proteins were then eluted with column buffer D (20 mm HEPES (pH 7.4), 400 mm NaCl, 5 mm MgCl2, and 500 mm imidazole). Fractions containing His6-tagged Drp1 were pooled and dialyzed overnight at 4 °C into buffer containing 20 mm HEPES (pH 7.4), 5 mm MgCl2, and 400 mm NaCl with Fisherbrand regenerated cellulose dialysis tubing with a 6000–8000 molecular weight cutoff (Thermo Fisher Scientific). Proteins were concentrated by centrifugation in Vivaspin 20 ultrafiltration devices with a 50,000 molecular weight cutoff (GE Healthcare). Drp1 concentration was quantified by UV absorbance at 280 nm using an extinction coefficient of 36380 m−1 cm−1 after incubation for 1 h in 6 m guanidine hydrochloride at 65 °C. Purified protein was stored at 4 °C until use and used within 36 h of cell lysis or frozen in 1-ml aliquots using a dry ice and ethanol bath. Frozen aliquots were stored at −80 °C and showed no loss of activity compared with fresh protein when used within 12 h of thawing.
GTPase Activity Assay
A continuous GTPase assay was used in which the rate of GTP hydrolysis was determined through coupling to a GTP regeneration system as described previously (12). GTPase activity was assayed in 150 μl of GTPase reaction buffer (25 mm HEPES, 25 mm PIPES, 7.5 mm KCl, 5 mm MgCl2, 1 mm phosphoenolpyruvate, 20 units/ml pyruvate kinase/lactate dehydrogenase, and 600 μm NADH (pH 7.0)), which was placed into the wells of a 96-well flat, clear-bottom plate. Depletion of NADH over time was measured for 40 min at 25 °C at indicated GTP, enzyme, and NaCl concentrations by using a Molecular Devices Flexstation 3 multi-detection reader with integrated fluid transfer. GTPase assays were started by the addition of GTP to the desired concentration. Substrate kinetics at each indicated NaCl concentration were measured but did not achieve Vmax at higher NaCl concentrations. For this reason, we report kobs at 1 mm GTP. For the determination of salt dependence, the final NaCl concentration varied between 50 and 500 mm as indicated, whereas enzyme concentrations were held at 2.5 μm. Enzyme activity was also measured in the presence of ATP as a substrate, but no measurable activity was observed.
Drp1 Light-scattering Assay
To assess the relative assembly and disassembly of Drp1 and mutations, the known NaCl dependence on Drp1 assembly/disassembly was measured by monitoring the light-scattering signal at 450 nm during the course of the continuous GTPase assay by robotic addition of 100 μl of reaction buffer with 0, 150, or 525 mm NaCl concentration to give a final [NaCl] of 90, 150, or 300 mm, respectively. Added buffers contained 1 mm GTP and the coupled reagents as described above. The linear range of these data, typically over an 8-min window, were fit to a straight line, with the resulting slope normalized for total protein and reported as the ΔA450 min−1 mg of enzyme−1.
Western Blotting
Standard protocols were utilized. Briefly, tissue lysates were obtained following homogenization in radioimmune precipitation assay buffer (50 mm Tris-HCl, 150 mm NaCl, 2 mm EDTA, 1% Nonidet P-40, 0.1% SDS, and 1% sodium deoxycholate) containing protease inhibitors, separated by SDS/PAGE, and transferred onto a PVDF membrane. The antibodies used were anti-LC3 (Cell Signaling Technology), anti-β-tubulin (Sigma-Aldrich), anti-Opa1 (Cell Signaling Technology), and anti-Mfn1 and anti-Mfn2 (Abcam).
Cloning
Drp1-YFP was mutated to Drp1-C452F YFP by a PCR-based method, and the mutation was confirmed by Sanger sequencing.
RNA Preparation and Quantitative PCR
RNA was extracted from harvested hearts using TRIzol reagent, followed by RNA cleanup using the RNeasy kit (Qiagen) according to the instructions of the manufacturer. 2 μg of RNA was used to generate cDNA using a high-capacity cDNA synthesis kit (Applied Biosystems). IL-6, IFIT-1, TNFα, and IFNγ primer/probe sets were obtained from Applied Biosystems. mRNA expression was determined by quantitative real-time PCR using TaqMan Fast Universal PCR Master Mix (Applied Biosystems) on a 7900 fast real-time PCR system machine (Applied Biosystems). Data were analyzed according to the ΔCt method, with hypoxanthine-guanine phosphoribosyltransferase serving as the reference housekeeping gene.
Mitochondrial:Nuclear DNA Ratio
DNA was extracted from powdered ventricular myocardium using the DNeasy blood and tissue kit (Qiagen) according to the instructions of the manufacturer. The ratio of mitochondrial ND1 to nuclear NDUFV1 was calculated by quantitative PCR using the following primers/probe sets: Mt-NDP1 forward, TCGACCTGACAGAAGGAGAATCA; MT-ND1 reverse, GGGCCGGCTGCGTAT; MT-ND1 probe, AATTAGTATCAGGGTTTAACG; Nuc-Ndufv1 forward, GAGCAGGACTTCTCCTTCACATC; Nuc-Ndufv1 reverse, CCCGTCTCAGGGCACCTT; and Nuc-Ndufv1 probe, TTTCCTACTCTGTCCAGGCT.
Mitochondrial Membrane Potential
MEFs were cultured on coverslips in growth medium until 80% confluent. The cells were then incubated in a final concentration of 2 μm JC-1 (Invitrogen) for 30 min at 37 °C, 5% CO2. Images were captured using a Zeiss LSM 5.10 confocal microscope. Fluorescence was quantified using ImageJ 1.42q software.
ATP Levels
Cells were harvested and washed twice in 1 ml of incubation medium (140 mm NaCl, 5 mm KCl, 1 mm MgCl2, 1.2 mm CaCl2, 6 mm glucose, 20 mm sodium-HEPES (pH7.4), and 0.04% phenol red). After the final wash, cells were resuspended in 70 μl of permeabilizing solution (30 μm digitonin, 140 mm potassium glutamate, 1 mm EGTA, 1 mm MgCl2, 6 mm glucose, 20 mm potassium-PIPES (pH 7.0), and 0.04% phenol red) and incubated for 10 min at 30 °C. Each cell line was analyzed in triplicate, with volumes of 20 μl aliquoted into three wells of a white 96-well plate. 180 μl of the standard reaction solution (1 μm DTT, 0.5 mm d-luciferin, and 12.5 μm firefly luciferase (Invitrogen)) was added to the cells in suspension. Luciferase activity was measured using the Berthold Mithras LB940 system with Mikrowin 2000 software. A standard curve of known ATP concentrations against luminescence was used to deduce ATP concentrations.
Mitochondrion-LC3 Colocalization
Cells were seeded on a 96-well plate. The next day, the medium was changed, and either bafilomycin (10 nm) or dimethyl sulfoxide vehicle was added for incubation overnight. Three replicates of each treatment were performed. Following fixation in 4% paraformaldehyde, cells were incubated with a primary mouse monoclonal antibody to pyruvate dehydrogenase (1:200, MSP03, Cambridge Bioscience) and a primary rabbit monoclonal antibody to LC3 (1:1000, PM036, Caltag-Medsystems Ltd.) for 1 h at room temperature. Cells were then washed three times with PBS before incubation with rabbit Alexa Fluor 488-conjugated secondary antibody (1:500, Invitrogen) and mouse Alexa Fluor 546-conjugated secondary antibody (1:500, Invitrogen) for 40 min at room temperature. Cells were again washed three times with PBS for 5 min before DAPI staining, at 0.5 μg/ml diluted with PBS, for 5 min. DAPI was replaced with PBS before foci were detected using an IN Cell Analyzer 1000 automated epifluorescence microscope (GE Healthcare). 8–12 images (200 cells) were obtained per well. The immunofluorescence signal was quantified using IN Cell Analyzer workstation software (version 3.5). Mitophagy was measured by colocalization of mitochondrial and LC3 signals as the number of colocalizing LC3-positive puncta.
Calcium Studies
Cells were perfused with 2.5 mm Ca2+-containing buffer, followed by Ca2+-free buffer containing 1 mm EGTA for the duration indicated by the clear horizontal bar. For the CPA/oligomycin experiment, cells were initially perfused with 2.5 mm Ca2+-containing buffer and then switched to Ca2+-free buffer containing 1 mm EGTA for the duration marked by the clear horizontal bar. 10 μm CPA was applied for the duration shown by the horizontal black bar. Mitochondrial Ca2+ stores were emptied by applying 10 μm carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) to collapse the mitochondrial membrane potential in the presence of 2.5 μg/ml oligomycin. CPA was still present in the perfusate when FCCP and oligomycin was applied to prevent reuptake of Ca2+ into the endoplasmic reticulum. A similar protocol was used to measure cytosolic Ca2+ following the application of 0.1 mm ATP to deplete intracellular Ca2+ stores. Mitochondrial Ca2+ concentration was measured following transfection of a mitochondria-targeted aequorin using Lipofectamine 2000 (Invitrogen). Wild-type and Python MEFs were washed twice in Krebs-Ringer modified buffer (125 mm NaCl, 5 mm KCl, 1 mm Na3PO4, 1 mm MgSO4, 5.5 mm glucose, and 20 mm HEPES (pH 7.4)) containing 1 mm CaCl2 and then incubated in Krebs-Ringer modified buffer containing 1 mm CaCl2 and 5 μm coelenterazine (Promega) for 30 min on ice to reconstitute the aequorin protein. Following a final centrifugation, cells were resuspended in 90 μl of Krebs-Ringer modified buffer containing 1 mm CaCl2 and 5 μm coelenterazine. The cells were then transferred to a white 96-well plate. Luminescence was measured every 0.5 s using the Berthold Mithras LB940 system with Mikrowin 2000 software. After 30 s, tert-butylhydroquinone was added to the cells to a final concentration of 10 mm. Subsequent conversion to Ca2+ concentration was achieved using a standard curve of known Ca2+ concentrations against luminescence.
Transthoracic Echocardiography
Mouse echocardiography was performed as described previously (16). Briefly, mice were anesthetized lightly using 1–1.5% isoflurane and imaged with a 22- to 55-MHz linear array transducer using the Vevo 2100 ultrasound system (Visualsonics). M mode tracings of short-axis images were obtained via the left parasternal window and used to measure the left ventricular end-diastolic chamber dimension and end-systolic chamber dimensions and the anterior and posterior wall thickness on the basis of measurements averaged from at least three cardiac cycles.
Transverse Aortic Constriction
Male C57Bl/6J mice (body weight, ∼25 g) underwent transverse aortic constriction or sham surgery. Briefly, mice were anesthetized with isoflurane and intubated and underwent trans-sternal thoracotomy. The transverse aorta was constricted with a 7-0 polypropylene monofilament suture (Ethicon) tied against a 27-gauge needle. In sham-operated mice, the aortic arch was dissected, but no suture was tied. Mice were given subcutaneous buprenorphine 0.8 mg/kg) for pain relief and overnight heat therapy to minimize surgery-induced hypothermia. All mice underwent echocardiography at 4–6 weeks to confirm left ventricular dysfunction prior to tissue harvest.
Tissue Harvesting
All mice were weighed and euthanized by cervical dislocation. The heart was excised, washed briefly with ice-cold PBS, blotted, and weighed before snap-freezing in liquid nitrogen and storage at −80 °C.
Oxidative Phosphorylation (OXPHOS)
Heart tissue was excised from Python and wild-type mice at various ages and placed directly in Mir05 solution (0.5 mm EGTA, 3 mm MgCl2·6H2O, 60 mm potassium-lactibionate, 20 mm taurine, 10 mm KH2PO4, 20 mm HEPES, 110 mm sucrose, and 1 mg/ml BSA) on ice to preserve mitochondrial function. The tissue was then teased apart under the microscope into very fine segments and placed in 50 g/ml saponin on ice for 30 min to lyse cells and expose the mitochondria. The saponin was then removed by three subsequent washes in Mir05 and placed into the chamber of the OROBOROS Oxygraph-2K. Although O2 flux was measured constantly, 10 mm pyruvate, 2 mm malate, and 10 mm glutamate were added to show total respiration. Complexes V and II were assessed with the addition of 2.5 mm ADP and 10 mm succinate, respectively. FCCP was added gradually from 0.5–1.25 μm to completely uncouple O2 consumption and ATP production, indicating the maximum capacity of OXPHOS. The complex I inhibitor rotenone was added at 0.5 μm. Finally, the addition of 2.5 μm antimycin A, a complex III inhibitor, completely terminated the process of OXPHOS, and any remaining O2 consumption at this stage occurred by non-mitochondrial respiration.
Transmission Electron Microscopy
Tissue for electron microscopy was processed as described previously (17). To count autophagosomes by EM, only structures that were double-membraned, spatially distinct (i.e. separated from other organelles such as mitochondria, suggesting mature autophagosomes), and had cytosolic content were included in the analysis. Where indicated, MEFs were incubated with bafilomycin A (500 nm, Sigma).
Statistics
All data are expressed as mean ± S.E. Statistical analysis was performed using unpaired Student's t test or analysis of variance followed by Bonferroni correction where appropriate. Statistical analysis was carried out using GraphPad Prism version 6. Differences were considered statistically significant when p < 0.05.
Results
The C452F Mutation Impairs Disassembly of Drp1 Oligomers
The Drp1 C452F mutation underlying the Python DCM model lies in close proximity to the reported human A395D mutation, labeled here as (murine position) A401D (Fig. 1A). We suspected similar mechanisms of protein dysfunction, namely interference with Drp1 self-association and GTP hydrolysis. We measured GTP hydrolysis by Drp1 C452F compared with wild-type Drp1 and the assembly-deficient mutation Drp1 A401D. Unexpectedly, we found that the Drp1 C452F mutation stimulates GTP hydrolysis (Fig. 1B). Because both Drp1 assembly and activity are known to be salt-dependent (where high [NaCl] favors disassembly), we compared the activity of the C452F mutant protein with the wild type under varying salt conditions. We found that GTP hydrolysis by Drp1 C452F is less sensitive to [NaCl] than the wild type, with supranormal GTP hydrolysis at all concentrations (Fig. 1C).
FIGURE 1.

A, structure of the Drp1 protein. The C452F mutation identified in the Python mouse model of DCM lies in close proximity to the reported human A395D (equivalent to mouse A401D) mutation in the middle domain. VD, variable domain; GED, GTPase effector domain. B, the Drp1 C452F mutation stimulates GTP hydrolysis compared with wild-type Drp1 and the assembly-deficient mutation Drp1 A401D. C, the C452F mutation impairs the NaCl dependence of GTP hydrolysis observed with wild type Drp1. D, assembly was measured via absorbance at 450 nm in reactions containing 20 μm Drp1 WT, 10 μm Drp1 A401D, or 10 μm Drp1 C452F. Reactions were started by the addition of 1 mm GTP. After 20 min of reaction time, buffers that altered the salt concentration of the reaction were added to measure the overall effect on assembly at final [NaCl] of 90 mm, 150 mm, and 300 mm. Representative data at [NaCl]final = 300 mm. E, light-scattering at varying [NaCl], showing a persistently high signal from the C452F mutant compared with the WT and A401D, consistent with failure of disassembly. F, to visualize the distribution of Drp1 C452F, Drp1−/− MEFs were transfected with a YFP-tagged Drp1 WT or Drp1 C45F construct. Drp1 C452F is seen in cytosolic clumps, consistent with increased assembly, in contrast to the diffuse cytosolic pattern of Drp1 WT.
We hypothesized that, given the linkage between Drp1 hydrolytic activity and assembly state, the Drp1 C452F mutant may also exhibit an increase in higher-ordered assembly. Protein assembly was measured by light scattering at 450 nm, and Drp1 was indeed shown to be more assembled by this measure compared with WT enzyme or the A401D mutant (2.5 μm protein, data not shown). To test whether this observation was caused by an inability of the mutant to properly disassemble because of a stabilization of the higher-ordered oligomeric state, we sought to observe Drp1 disassembly in real time. In reactions containing 10 μm protein (wild-type or mutant) and 150 mm NaCl, assembly was allowed to progress for 20 min after starting the reactions with 1 mm GTP. After 20 min of reaction time, buffers that altered the salt concentration of the reaction were added to measure the overall effect on assembly. The C452F mutant shows a significant increase in light scattering compared with the wild type (Fig. 1D), consistent with increased assembly. We found that, with increasing [NaCl] promoting disassembly, there is a persistently high light-scattering signal from the C452F mutant compared with the WT and A401D (Fig. 1E). Overall, these data show that the C452F mutation impairs protein disassembly after oligomerization with futile GTP hydrolysis.
To assess the effect of the mutation on the distribution of cellular Drp1, we transfected Drp1−/− MEFs with the Drp1 WT construct or the C452F mutant tagged with YFP. In keeping with the biochemical evidence of a failure of disassembly, Drp1 C452F-YFP was aggregated and clumped within cells rather than being distributed throughout the cytosol, as seen in the Drp1 WT-YFP cells (Fig. 1F).
Mitochondria in Drp1 C452F MEFs Are Morphologically and Functionally Abnormal
Assessment of mitochondrial fission-fusion in the heart is challenging, possibly because of relative low event frequency or constraints on mitochondrial morphology imposed by the “sardine tin” architecture of the cardiomyocyte (11, 18, 19). Accordingly, we generated an immortalized Drp1 C452F MEF cell line to characterize the effect of the mutation on mitochondrial morphology and function.
Drp1−/− MEFs are known to exhibit abnormalities of mitochondrial morphology, with highly interconnected networks of mitochondria (11, 14). As anticipated, we found that the mitochondrial morphology in Drp1 C452F MEFs was also grossly abnormal, with a large increase in the percentage of cells displaying an elongated, interconnected mitochondrial network (Fig. 2A). This suggests that the C452F mutation leads to a reduction in mitochondrial fission, with unopposed fusion driving an interconnected, tubular phenotype.
FIGURE 2.

A, representative confocal microscopy images of Drp1 C452F and Drp1 WT MEFs after transfection with mitochondrial red fluorescent protein to show mitochondria. C452F leads to an elongated, tubular, and interconnected mitochondrial morphological phenotype. Cell population analysis of mitochondrial morphology confirms a significant shift to an elongated mitochondrial morphology (50 cells/experiment; n = 3; ***, p < 0.001). B, Western blots of Drp1 C452F and WT lysates for key mitochondrial fusion proteins. Levels of Mfn1 and Mfn2 were unchanged. Levels of Opa1 were increased mildly (three independent experiments). C, Drp1 C452F and WT MEFs were cotransfected with mito-DsRed and mito-PAGFP for imaging the functional connectivity of the mitochondrial network. After activation of GFP in randomly selected regions of interest (circled), the spread of the GFP signal through the highly interconnected mitochondrial network of Drp1 C452F MEFs was evident by 10 min. This was associated with a drop in GFP fluorescence intensity in the original region of interest. In contrast, in Drp1 WT MEFs, the reduction in GFP signal in the region of interest was significantly less, indicating reduced connectivity (n = >10 cells/group, with 1–2 regions of interest/cell; *, p < 0.05). D, autophagosome LC3 and mitochondrial pyruvate dehydrogenase were stained by immunofluorescence and colocalization assessed on an IN Cell Analyzer 1000 automated epifluorescence microscope. Drp1 C452F and Drp1−/− MEFs displayed mitochondrial-autophagosome colocalization after inhibition of autophagosome degradation by bafilomycin (Baf), ***, p < 0.001. E, quantitative PCR analysis of mitochondrial MT-ND1 DNA normalized to nuclear NDUFV1 DNA in MEFs exposed to nutrient deprivation (culture in Hanks' balanced salt solution) for the time shown (n = 3; *, p < 0.05; ns, not significant). In WT but not C452F MEFs, nutrient deprivation results in a drop in mitochondrial DNA consistent with mitochondrial degradation by mitophagy. This decrease is not seen in the Drp1 C452F cells. F, after 4 h of nutrient deprivation, C452F MEFs form fewer mature, spatially separated autophagosomes, as counted by EM (20 cells/group, n = 3). G, large numbers of smaller autophagosomes are present in close apposition to mitochondria in Drp1 C452F cells, possibly consistent with stalled ingestion of mitochondria within autophagosomes (arrows). MV, multiple-membraned vesicles; G, Golgi body; Mi, mitochondrion; N, nucleus. Scale bars = 500 nm. H, representative EM image of MEFs after combined nutrient deprivation and 500 nm bafilomycin A1 treatment for 4 h, showing normal activation of generalized macroautophagy after supramaximal stimulation, with diffuse autophagosome formation in the cytosol. Scale bars = 2 μm. I, ATP concentration measured by firefly luciferase reporter luminescence. **, p < 0.01. J, mitochondrial membrane potential measured by JC1 fluorescence in Drp1 WT compared with C452F MEFs. ***, p < 0.001.
To exclude a significant contribution from altered levels of mitochondrial fusion proteins in the elongated mitochondrial phenotype, we assessed the levels of Mfn1, Mfn2, and Opa1 by Western blot analysis. Levels of Mfn1 and Mfn2 were unchanged in C452F cells, and levels of Opa1 increased, although we did not consider this change sufficient to explain the dramatic phenotype observed (Fig. 2B).
To further study the connectivity of the mitochondrial network, we transfected Drp1 C452F MEFs with a mito-PAGFP construct as described previously (15). After targeted photoactivation of a region of interest, redistribution of the photoactivated GFP protein can be tracked over time as it disperses within the mitochondrial network. The rate of decrease of the GFP signal within the photoactivated region of interest, as it disperses, is a marker of mitochondrial connectivity. We found that the mito-PAGFP signal in Drp1 C452F MEFs dispersed significantly faster than Drp1 WT MEFs, confirming a highly fused and functionally interconnected mitochondrial network (Fig. 2C).
Drp1 is required for mitophagy (8). By quantifying the colocalization of mitochondrial pyruvate dehydrogenase with GFP tagged to the mature autophagosomal marker LC3, we assessed mitophagy at baseline and after exposure to bafilomycin (an inhibitor of the vacuolar-type proton ATPase), which inhibits degradation of the late autophagosome. We found a reduction in mitochondrion-autophagosome colocalization in both Drp1 C452F and Drp1−/− MEFs compared with the wild type, suggesting impaired mitophagy (Fig. 2D).
In Drp1−/− MEFs, there is reduced autophagic clearance of mitochondria under starvation conditions, suggestive of impaired mitophagy (8). We assessed whether the mitochondria in Drp1 C452F MEFs were resistant to autophagic clearance. After induction of generalized autophagy through nutrient deprivation, Drp1 WT MEFs showed a significant reduction in the mitochondrial:nuclear DNA ratio after 10 h, consistent with mitochondrial degradation by mitophagy, which was not seen in Drp1 C452F cells (Fig. 2E).
By electron microscopy, starvation in Drp1 WT MEFs triggered the formation of numerous complex autophagosome structures and vacuoles with multiple membranes (Fig. 2G, i and ii). In Drp1 C452F MEFs, there were fewer mature autophagosomes in the cytosol (Fig. 2G), but we observed numerous small autophagosomes in close apposition with mitochondria (Fig. 2G, iii and iv). We hypothesize that these may represent the attempted ingestion of mitochondria into early autophagosomes. Supramaximal stimulation of generalized autophagy by combining starvation with bafilomycin A1 incubation, which blocks autophagosome-lysosome fusion, stimulated the normal formation of multiple large autophagosomes, suggesting no primary defect with autophagy in Drp1 C452F MEFs (Fig. 2H).
Overall, these data are consistent with a specific defect in the clearance of mitochondria by the autophagosomal degradation pathway. We hypothesized that inefficient clearance of mitochondria results in the accumulation of inefficient, damaged mitochondria, consistent with the increase in mitochondrial DNA identified previously in the failing Python heart (3). Indeed, Drp1 C452F mitochondria were depolarized, as measured by JC1 fluorescence, and produced lower levels of ATP (Fig. 2, I and J).
The Python Heart Shows an Age-related Decline in OXPHOS Efficiency
OXPHOS is central to ATP synthesis and is essential for normal cardiomyocyte function. Because dysfunction of mitochondrial fission-fusion proteins has been linked to reduced OXPHOS capacity, we hypothesized that energy deficiency resulting from impaired OXPHOS contributes to DCM in Python (20, 21).
The efficiency of OXPHOS enzyme complexes was measured in Python cardiac tissue samples in a respirometer with specific substrates and inhibitors (Fig. 3A). Between 3 and 5 months of age, we observed a progressive and profound decrease in the overall level of mitochondrial respiration in the Python heart, not seen in the wild-type heart (Fig. 3B). This severe reduction in OXPHOS is likely to be initiated by decreased activity of ATP synthase at 4 months of age, followed by a reduction in complex I and II activity at 5 months of age, and then complex III activity at 6 months. These changes immediately precede the onset of overt heart failure, which occurs at approximately 6 months of age (3).
FIGURE 3.
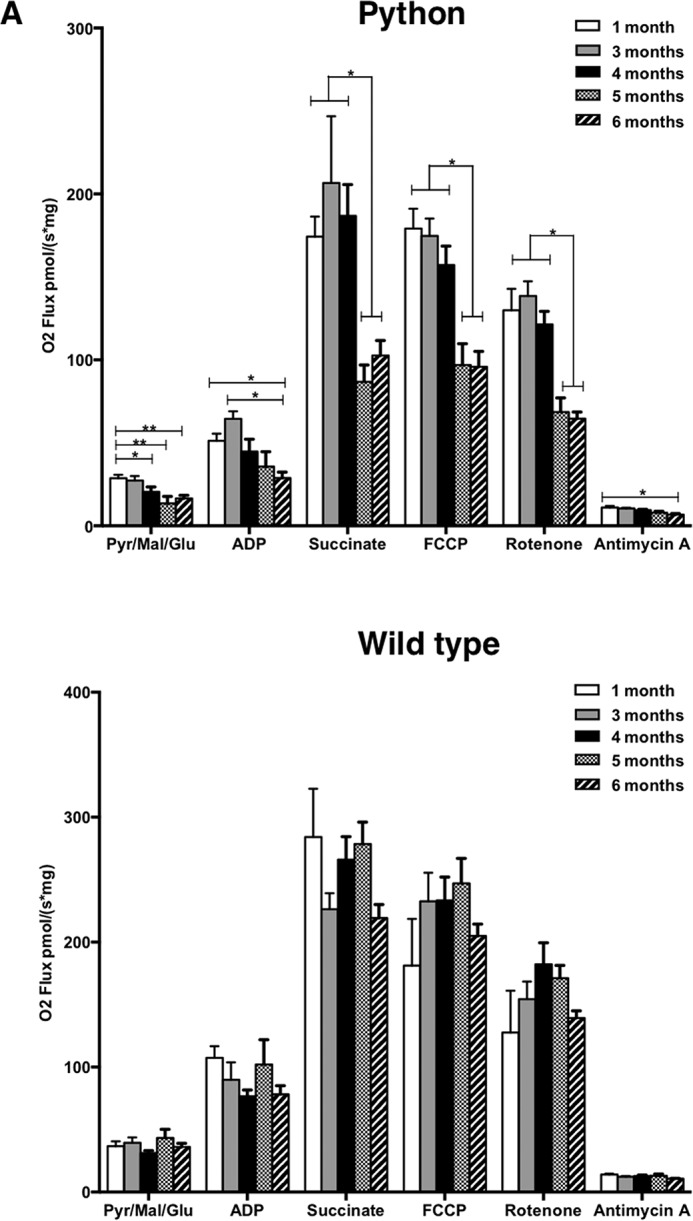
A, mitochondria were analyzed from the hearts of Python C57BL/6J mice after exposure to different conditions. Pyruvate, glutamate, and malate (Pyr/Mal/Glu) were added to initiate respiration. ADP and succinate are substrates of ATP synthase and complex II, respectively. FCCP uncouples the OXPHOS system, representing the maximum capacity of the electron transport chain. Rotenone and antimycin A are inhibitors of complexes I and III, respectively. General respiration begins to decline at 4 months of age. ATP synthase activity gradually declines from 3 months of age until onset of heart failure. Complexes I and II both show no alteration of OXPHOS activity until 5 months of age, when a dramatic reduction becomes evident for both complexes and remains the same until the onset of overt heart failure at 6 months. The maximum capacity of the electron transport chain also follows a dramatic decline from 5 months. Inhibition with antimycin A displays a gradual reduction from 3 months of age (*, p < 0.05, **, p < 0.01). B, mitochondrial OXPHOS activity in wild-type hearts. No significant decreases in O2 flux were observed between any of the time points measured.
Drp1 C452F Alters Mitochondrial and Cytosolic Calcium Transients
Mitochondrial calcium uptake is intrinsically linked with the regulation of oxidative metabolism (13, 22–25). The mitochondrial membrane potential is the major driving force for calcium uptake across the inner mitochondrial membrane, which, in turn, activates calcium-dependent enzymes of the Krebs cycle (26). MEFs are well established as a cellular model to probe the interaction of mitochondrial fission-fusion dynamics and calcium signaling (27). Knowing that the mitochondrial membrane potential and OXPHOS were impaired in C452F cells and Python hearts, we characterized both mitochondrial and cytosolic calcium transients in Drp1 C452F MEFs.
First, we exposed cells to cyclopiazonic acid (CPA), an inhibitor of the endoplasmic reticulum (ER) Ca2+ ATPase. Following store depletion, we added FCCP together with oligomycin (to prevent ATP consumption by the F1F0-ATP synthase functioning in reverse mode). In WT MEFs, this led to a rise of [Ca2+]i because of release of Ca2+ from mitochondria. This response was attenuated significantly in Drp1 C452F cells (Fig. 4, A and B), suggesting impaired mitochondrial uptake of Ca2+ released from the ER after CPA treatment.
FIGURE 4.

A, representative traces showing [Ca2+]i in WT (top panel) and C452F (bottom panel) MEFs following emptying of ER stores with the reversible ER Ca2+ ATPase inhibitor CPA, followed by emptying of mitochondrial Ca2+ stores using FCCP in the presence of oligomycin (Oligo). r.u., relative units. B, results (mean ± S.E.) for the peak response to CPA and the peak response to FCCP and oligomycin in WT and C452F MEFs (n = 10/group). Ab units, absorbance units. *, p < 0.05; ns, not significant. C, representative traces and area under the curve (AUC) of mitochondrial Ca2+ uptake in MEFs (measured by mitochondrially targeted aequorin) after ATP-evoked ER Ca2+ release (n = 2/group). *, p < 0.05. D, representative traces of [Ca2+]i during ATP-evoked Ca2+ release from ER stores, followed by capacitative calcium entry in WT and C452F MEFs. 100 μm ATP was applied for the duration of the black bar to evoke Ca2+ release from the ER prior to reintroducing Ca2+ (2.5 mm) in the perfusate to determine the capacitative calcium entry. E, results (mean ± S.E.) for resting Ca2+-containing baseline, the peak response to ATP, the integral of the response (AUC), and the t½ in WT and C452F MEFs (n = 12 recordings for WT and 11 for C452F). ***, p < 0.01.
To directly measure the mitochondrial calcium transient, we transfected Drp1 WT and Drp1 C452F MEFs with a targeted mitochondrial aequorin. The mitochondrial transient after addition of ATP confirmed a significant reduction in mitochondrial calcium uptake in the mutant cells (Fig. 4C).
Next we assessed inositol 1,4,5-trisphosphate-mediated release of [Ca2+]i from intracellular stores by application of ATP in the absence of extracellular Ca2+ (Fig. 4, D and E). Basal [Ca2+]i was raised significantly in the C452F MEFs in both the absence or presence of extracellular calcium. After application of 0.1 mm ATP, the amplitude and duration of the C452F calcium transient was significantly greater than that of the WT. There was no effect on the magnitude of the subsequent capacitative Ca2+ transient, which was similar to that of WT cells (data not shown). In summary, these data show that Drp1 C452F is associated with significantly reduced mitochondrial calcium uptake, a critical regulator of energy supply and demand matching in the heart (28).
Cardiac Drp1 Dysfunction Leads to Inflammation
Failure of cellular control over mitochondria leads to the activation of innate immune signaling (29–31). In the heart, free mitochondrial DNA because of loss of DNase II has been shown to activate TLR9, resulting in heart failure in mice (9). We investigated whether the failure of mitophagy observed in our cellular model was associated with activation of inflammation in the Python mouse heart. We measured the expression of pro-inflammatory genes encoding IL-6, TNFα, and IFNγ in the hearts of young animals (at 50 days of age) prior to the development of cardiomyopathy (Fig. 5A) and old animals (150 days of age) with incipient heart failure (Fig. 5B). In both young and old mouse hearts, there was significant activation of the inflammatory cascade (Fig. 5B). This pattern of immune activation was not seen in a control model of heart failure induced in age-matched mice by transverse aortic constriction (TAC, Fig. 5B).
FIGURE 5.

A, quantitative PCR of inflammatory cytokines in young (age, 50 days) Python mice prior to the development of left ventricular dysfunction (n = 10/group). B, inflammatory cytokines in old (age, 150 days) Python mice, which display severe left ventricular dysfunction, compared with age-matched littermate WT mice and age-matched mice with left ventricular failure induced by transverse aortic constriction (TAC) mice (n = 9–11/group). *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant.
Induction of Autophagy by Low-protein Diet Rescues Cardiac Failure
In light of defective mitochondrial recycling by mitophagy, we sought to assess the therapeutic potential of activating generalized cellular macroautophagy in the Python model (32). Activation of autophagy by low protein diet (LPD) has been shown previously to be beneficial in a model of congenital muscular dystrophy caused by abnormal autophagy, leading to improved myofiber survival (33).
We commenced a cohort of mice on an isocaloric 6% LPD from 4 weeks of age. The LPD led to activation of the macroautophagy pathway, as demonstrated by an increased ratio of LC3-I:LC3-II (Fig. 6A). At 150 days of age, mice underwent echocardiography and were sacrificed. Compared with Python littermates on a control chow diet, LPD animals were significantly lighter and had a reduced heart weight to tibial length ratio (Fig. 6, B and C). Echocardiography at 150 days of age showed substantial improvements in the left ventricular end-diastolic dimension and ejection fraction, although this was not completely restored to normal (Fig. 6, C–F).
FIGURE 6.

A, Western blot analysis for LC3 in Python cardiac samples demonstrating activation of macroautophagy by an isocaloric LPD. B and C, overall body weight and ratio of heart weight to tibia length was significantly lower in the Python group on LPD (n = >5/group) compared with Python mice on a normal chow diet. ***, p < 0.01 Python (Py)/LPD versus Py/chow. D, representative M mode echocardiography images performed at an age of 150 days, showing improved cardiac function. E, LPD decreased left ventricular end-diastolic dimension (LVEDD). ***, p < 0.01 Py/LPD versus Py/chow. F, improved ejection fraction (EF). ***, p < 0.01 Py/LPD versus Py/chow. G, the overall mitochondrial:nuclear DNA ratio was unchanged by LPD, suggesting that intact mitochondrial DNA is not the primary driver of cardiomyopathy.
The improvement in cardiac function with LPD was not associated with reduced expression of the inflammatory cytokines IL-6, TNFα, or IFNγ (data not shown) or a significant change in the mitochondrial:nuclear DNA ratio (Fig. 6G), suggesting that intact mitochondrial DNA is not the primary driver of cardiomyopathy. These data show that activation of macroautophagy by LPD leads to significant improvement in cardiac function in Python mice, albeit without an evident decrease in mitochondrial copy number.
Discussion
We present the first model manifesting stabilization of Drp1 oligomers. Our data suggest that dysfunctional Drp1 C452F has multiple downstream cellular consequences driving DCM, including inefficient mitophagy, impaired mitochondrial calcium handling, deficient ATP biosynthesis, and inflammation. In this setting, activation of macroautophagy by exposure to a low-protein diet partially protects against DCM. Our results provide support for the critical role of Drp1-mediated mitochondrial regulation and quality control in cardiac metabolism.
Drp1 C452F protein function is abnormal, with supranormal GTPase function but a failure of disassembly after oligomerization. Although this is a novel finding for Drp1, there is a precedent for the similar influence of middle-domain mutations in the closely related human dynamin 2 gene, where disassembly mutants have preserved GTPase function but are functionally deficient, leading to congenital myopathy (34, 35).
Three recent studies have employed an inducible knockdown approach to study the role of cardiac Drp1 (36–38). All showed that loss of Drp1 leads to progressive heart failure but with important differences in cellular pathogenesis. Ikeda et al. (36) have found that inducible down-regulation of Drp1 leads to reduced mitophagy with accumulation of damaged mitochondria. Kageyama et al. (38) have shown increased levels of the mitophagy markers p62 and ubiquitin, consistent with accelerated early mitophagy, but impaired delivery of mitochondria to lysosomes. Finally, Song et al. (37) have reported abundant formation of mitophagosomes and decreased mitochondrial content, proposing a model of retained fission, but inappropriate mitophagic degradation and generalized mitochondrial loss. A central feature of the studies by Ikeda et al. (36) and Song et al. (37) was necrotic cell death by activation of the mitochondrial permeability transition pore.
Collectively, this approach has established a critical role for cardiac Drp1 in mitophagy/autophagy (39). However, in contrast to the inducible knockout approach, our model of a Drp1 missense mutation is functionally hypomorphic with substantially retained protein activity. Impaired Drp1 depolymerization may lead to futile GTP hydrolysis and Drp1 sequestration, resulting in functional Drp1 haploinsufficiency. This may explain the cardiac specificity of the Python phenotype. The retained Drp1 activity is likely sufficient for other organs with limited mitochondrial remodeling, but the accelerated requirements of the heart become limiting over time, with an increasing burden of mitochondrial dysfunction manifesting as DCM. Our data should be viewed in this context of residual cellular Drp1 activity. Unlike the inducible Drp1-KO, where heart failure develops in ∼4–6 weeks, Python undergoes a chronic accumulation of defective, depolarized mitochondria, which permits viability. The chronicity and long-term viability of our model makes it likely to be of greater relevance to human disease.
We propose that accumulation of damaged mitochondria occurs through the relative failure of mitophagy. Although difficult to demonstrate directly in the heart, under conditions of acute nutrient deprivation or bafilomycin exposure, we observed reduced colocalization of the mature autophagosome marker LC3 and mitochondrial pyruvate dehydrogenase; visualization of numerous immature, fused mitochondrial autophagosomes; and reduced clearance of mitochondrial DNA after nutrient deprivation. Our previous work has shown the accumulation of mitochondrial DNA in the aging Python heart, also consistent with a failure of completed mitophagy (3).
In the Python heart, we found evidence of activated innate immune signaling, consistent with recent work demonstrating that a failure of cellular control of mitochondria can result in the activation of the inflammasome, Toll-like receptors, stimulator of interferon genes (STING) and mitochondrial antiviral signaling protein (MAVS) (31). Activation of cardiac inflammatory nodes, specifically TLR9, influences mitochondrial calcium by inhibition of SERCA2, reducing mitochondrial calcium uptake and ATP synthesis (40). This has been interpreted as a stress protection response in cardiomyocytes but might also be detrimental when activated long-term, leading to functional ATP deficiency. Our work in MEFs confirms reduced mitochondrial calcium uptake.
Drp1 C452F leads to profound ATP deficiency in MEFs and the Python heart, supporting a wider body of work linking Drp1 with respiratory failure in both cell models and in the heart (36, 38, 41). In the Python heart, two models can be invoked to explain energetic failure: first, a direct effect of reduced mitochondrial calcium uptake (occurring by inflammation-mediated deactivation of SERCA2), leading to reduced stimulation of OXPHOS, or, alternatively, through accumulation of a pool of damaged, inefficient mitochondria. Therapeutic intervention on the cardiac Drp1 pathway has so far focused on inhibition of fission in acute ischemia-reperfusion, but low-level chronic activation to promote mitophagy and high ATP synthesis might be of benefit in the failing heart (42, 43).
Although increasing autophagy normalized ventricular function, there was a less prominent effect on inflammation and mitochondrial copy number. This likely reflects the presence of multiple parallel drivers of inflammation downstream of failed mitophagy, not all of which are resolved by intervention on the macroautophagy pathway.
In this study, we analyzed the downstream consequences of a Drp1 mutation underlying heart failure in mice. Drp1 has a non-redundant role in cardiomyocyte mitophagy and, likely, mitochondrial quality control in the heart. Although, as inferred from our model, this is evident at baseline, it is likely that the role of Drp1 is also manifest during diverse cardiac stresses (36). We describe a model of mitophagic failure with activation of inflammation and aberrant calcium handling downstream. Importantly, our model does not make use of an inducible knockout approach but demonstrates that dilated cardiomyopathy can follow a single pathogenic missense mutation in Drp1. As exemplified by Drp1 C452F in mice, key proteins in the fission-fusion-mitophagy machinery are good candidates for familial DCM in humans.
Author Contributions
T. J. C. coordinated the study. T. J. C., V. L., M. K., A. S., N. W. K., L. B., G. M. C., A. R. H., G. C., C. L., V. S., A. M., D. F., and N. T. H. performed and analyzed experiments. M. B. carried out transverse aortic constriction surgery. A. Y., J. P., L. S., C. P., J. B., R. B. H., A. S., H. W., T. N. D., and H. A. contributed to the study design and analysis of results. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Cara Marie Manlandro for technical expertise in subcloning and protein purification protocols for recombinant Drp1.
The authors declare that they have no conflicts of interest with the contents of this article.
- DCM
- dilated cardiomyopathy
- mito-PAGFP
- mitochondrially targeted photoactivatable GFP
- OXPHOS
- oxidative phosphorylation
- CPA
- cyclopiazonic acid
- ER
- endoplasmic reticulum
- LPD
- low-protein diet.
References
- 1.Cahill T. J., Ashrafian H., and Watkins H. (2013) Genetic cardiomyopathies causing heart failure. Circ. Res. 113, 660–675 [DOI] [PubMed] [Google Scholar]
- 2.Neubauer S. (2007) The failing heart: an engine out of fuel. N. Engl. J. Med. 356, 1140–1151 [DOI] [PubMed] [Google Scholar]
- 3.Ashrafian H., Docherty L., Leo V., Towlson C., Neilan M., Steeples V., Lygate C. A., Hough T., Townsend S., Williams D., Wells S., Norris D., Glyn-Jones S., Land J., Barbaric I., Lalanne Z., Denny P., Szumska D., Bhattacharya S., Griffin J. L., Hargreaves I., Fernandez-Fuentes N., Cheeseman M., Watkins H., and Dear T. N. (2010) A mutation in the mitochondrial fission gene Dnm1l leads to cardiomyopathy. PLoS Genet. 6, e1001000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Youle R. J., and van der Bliek A. M. (2012) Mitochondrial fission, fusion, and stress. Science 337, 1062–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Otera H., Ishihara N., and Mihara K. (2013) New insights into the function and regulation of mitochondrial fission. Biochim. Biophys. Acta 1833, 1256–1268 [DOI] [PubMed] [Google Scholar]
- 6.Tanaka A., Cleland M. M., Xu S., Narendra D. P., Suen D.-F., Karbowski M., and Youle R. J. (2010) Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 191, 1367–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liesa M., and Shirihai O. S. (2013) Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 17, 491–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gomes L. C., Di Benedetto G., and Scorrano L. (2011) During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 13, 589–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oka T., Hikoso S., Yamaguchi O., Taneike M., Takeda T., Tamai T., Oyabu J., Murakawa T., Nakayama H., Nishida K., Akira S., Yamamoto A., Komuro I., and Otsu K. (2012) Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485, 251–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou R., Yazdi A. S., Menu P., and Tschopp J. (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 [DOI] [PubMed] [Google Scholar]
- 11.Waterham H. R., Koster J., van Roermund C. W., Mooyer P. A., Wanders R. J., and Leonard J. V. (2007) A lethal defect of mitochondrial and peroxisomal fission. N. Engl. J. Med. 356, 1736–1741 [DOI] [PubMed] [Google Scholar]
- 12.Chang C.-R., Manlandro C. M., Arnoult D., Stadler J., Posey A. E., Hill R. B., and Blackstone C. (2010) A lethal de novo mutation in the middle domain of the dynamin-related GTPase Drp1 impairs higher order assembly and mitochondrial division. J. Biol. Chem. 285, 32494–32503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cereghetti G. M., Stangherlin A., Martins de Brito O., Chang C. R., Blackstone C., Bernardi P., and Scorrano L. (2008) Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. U.S.A. 105, 15803–15808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishihara N., Nomura M., Jofuku A., Kato H., Suzuki S. O., Masuda K., Otera H., Nakanishi Y., Nonaka I., Goto Y., Taguchi N., Morinaga H., Maeda M., Takayanagi R., Yokota S., and Mihara K. (2009) Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 11, 958–966 [DOI] [PubMed] [Google Scholar]
- 15.Karbowski M., Arnoult D., Chen H., Chan D. C., Smith C. L., and Youle R. J. (2004) Quantitation of mitochondrial dynamics by photolabeling of individual organelles shows that mitochondrial fusion is blocked during the Bax activation phase of apoptosis. J. Cell Biol. 164, 493–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ashrafian H., Czibik G., Bellahcene M., Aksentijević D., Smith A. C., Mitchell S. J., Dodd M. S., Kirwan J., Byrne J. J., Ludwig C., Isackson H., Yavari A., Støttrup N. B., Contractor H., Cahill T. J., Sahgal N., Ball D. R., Birkler R. I., Hargreaves I., Tennant D. A., Land J., Lygate C. A., Johannsen M., Kharbanda R. K., Neubauer S., Redwood C., de Cabo R., Ahmet I., Talan M., Günther U. L., Robinson A. J., Viant M. R., Pollard P. J., Tyler D. J., and Watkins H. (2012) Fumarate Is cardioprotective via activation of the Nrf2 antioxidant pathway. Cell Metab. 15, 361–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ling Y. M., Shaw M. H., Ayala C., Coppens I., Taylor G. A., Ferguson D. J., and Yap G. S. (2006) Vacuolar and plasma membrane stripping and autophagic elimination of Toxoplasma gondii in primed effector macrophages. J. Exp. Med. 203, 2063–2071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beraud N., Pelloux S., Usson Y., Kuznetsov A. V., Ronot X., Tourneur Y., and Saks V. (2009) Mitochondrial dynamics in heart cells: very low amplitude high frequency fluctuations in adult cardiomyocytes and flow motion in non beating Hl-1 cells. J. Bioenerg. Biomembr. 41, 195–214 [DOI] [PubMed] [Google Scholar]
- 19.Dorn G. W., 2nd (2013) Mitochondrial dynamics in heart disease. Biochim. Biophys. Acta 1833, 233–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zanna C., Ghelli A., Porcelli A. M., Karbowski M., Youle R. J., Schimpf S., Wissinger B., Pinti M., Cossarizza A., Vidoni S., Valentino M. L., Rugolo M., and Carelli V. (2008) OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain 131, 352–367 [DOI] [PubMed] [Google Scholar]
- 21.Twig G., Elorza A., Molina A. J., Mohamed H., Wikstrom J. D., Walzer G., Stiles L., Haigh S. E., Katz S., Las G., Alroy J., Wu M., Py B. F., Yuan J., Deeney J. T., Corkey B. E., and Shirihai O. S. (2008) Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saotome M., Safiulina D., Szabadkai G., Das S., Fransson A., Aspenstrom P., Rizzuto R., and Hajnóczky G. (2008) Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc. Natl. Acad. Sci. U.S.A. 105, 20728–20733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szabadkai G., Simoni A. M., Chami M., Wieckowski M. R., Youle R. J., and Rizzuto R. (2004) Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol. Cell 16, 59–68 [DOI] [PubMed] [Google Scholar]
- 24.Hom J., Yu T., Yoon Y., Porter G., and Sheu S.-S. (2010) Regulation of mitochondrial fission by intracellular Ca2+ in rat ventricular myocytes. Biochim. Biophys. Acta 1797, 913–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Redpath C. J., Bou Khalil M., Drozdzal G., Radisic M., and McBride H. M. (2013) Mitochondrial hyperfusion during oxidative stress is coupled to a dysregulation in calcium handling within a C2C12 cell model. PLoS ONE 8, e69165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murgia M., Giorgi C., Pinton P., and Rizzuto R. (2009) Controlling metabolism and cell death: at the heart of mitochondrial calcium signalling. J. Mol. Cell Cardiol. 46, 781–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Brito O. M., and Scorrano L. (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610 [DOI] [PubMed] [Google Scholar]
- 28.Carley A. N., Taegtmeyer H., and Lewandowski E. D. (2014) Matrix revisited: mechanisms linking energy substrate metabolism to the function of the heart. Circ. Res. 114, 717–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Green D. R., Galluzzi L., and Kroemer G. (2011) Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 333, 1109–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Q., Raoof M., Chen Y., Sumi Y., Sursal T., Junger W., Brohi K., Itagaki K., and Hauser C. J. (2010) Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cloonan S. M., and Choi A. M. (2013) Mitochondria: sensors and mediators of innate immune receptor signaling. Curr. Opin. Microbiol. 16, 327–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kanamori H., Takemura G., Maruyama R., Goto K., Tsujimoto A., Ogino A., Li L., Kawamura I., Takeyama T., Kawaguchi T., Nagashima K., Fujiwara T., Fujiwara H., Seishima M., and Minatoguchi S. (2009) Functional significance and morphological characterization of starvation-induced autophagy in the adult heart. Am. J. Pathol. 174, 1705–1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grumati P., Coletto L., Sabatelli P., Cescon M., Angelin A., Bertaggia E., Blaauw B., Urciuolo A., Tiepolo T., Merlini L., Maraldi N. M., Bernardi P., Sandri M., and Bonaldo P. (2010) Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat. Med. 16, 1313–1320 [DOI] [PubMed] [Google Scholar]
- 34.Wang L., Barylko B., Byers C., Ross J. A., Jameson D. M., and Albanesi J. P. (2010) Dynamin 2 mutants linked to centronuclear myopathies form abnormally stable polymers. J. Biol. Chem. 285, 22753–22757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.James N. G., Digman M. A., Ross J. A., Barylko B., Wang L., Li J., Chen Y., Mueller J. D., Gratton E., Albanesi J. P., and Jameson D. M. (2014) A mutation associated with centronuclear myopathy enhances the size and stability of dynamin 2 complexes in cells. Biochim. Biophys. Acta 1840, 315–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ikeda Y., Shirakabe A., Maejima Y., Zhai P., Sciarretta S., Toli J., Nomura M., Mihara K., Egashira K., Ohishi M., Abdellatif M., and Sadoshima J. (2015) Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ. Res. 116, 264–278 [DOI] [PubMed] [Google Scholar]
- 37.Song M., Mihara K., Chen Y., Scorrano L., and Dorn G. W. 2nd. (2015) Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse Hearts and cultured fibroblasts. Cell Metab. 21, 273–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kageyama Y., Hoshijima M., Seo K., Bedja D., Sysa-Shah P., Andrabi S. A., Chen W., Höke A., Dawson V. L., Dawson T. M., Gabrielson K., Kass D. A., Iijima M., and Sesaki H. (2014) Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J. 33, 2798–2813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dorn G. W., 2nd (2015) Gone Fission … : diverse consequences of cardiac Drp1 deficiency. Circ. Res. 116, 225–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shintani Y., Drexler H. C., Kioka H., Terracciano C. M., Coppen S. R., Imamura H., Akao M., Nakai J., Wheeler A. P., Higo S., Nakayama H., Takashima S., Yashiro K., and Suzuki K. (2014) Toll-like receptor 9 protects non-immune cells from stress by modulating mitochondrial ATP synthesis through the inhibition of SERCA2. EMBO Rep. 15, 438–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parone P. A., Da Cruz S., Tondera D., Mattenberger Y., James D. I., Maechler P., Barja F., and Martinou J.-C. (2008) Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS ONE 3, e3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ong S.-B., Subrayan S., Lim S. Y., Yellon D. M., Davidson S. M., and Hausenloy D. J. (2010) Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 121, 2012–2022 [DOI] [PubMed] [Google Scholar]
- 43.Disatnik M. H., Ferreira J. C. B., Campos J. C., Gomes K. S., Dourado P. M. M., Qi X., and Mochly-Rosen D. (2013) Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long-term cardiac dysfunction. J. Am. Heart Assoc. 2, e000461. [DOI] [PMC free article] [PubMed] [Google Scholar]