

Background: Interferon-induced transmembrane (IFITM) proteins limit a broad range of RNA viruses.
Results: Tyrosine phosphorylation of IFITM2 and IFITM3, and S-palmitoylation of the IFITM proteins, are crucial for anti-hepatitis C virus (HCV) activity.
Conclusion: IFITM2 and IFITM3 are able to limit HCV infection by targeting the late entry stages of the virus.
Significance: IFITM proteins inhibit HCV at early and late stages of entry.
Keywords: cellular immune response, hepatitis C virus (HCV), interferon, protein palmitoylation, protein phosphorylation, interferon-induced transmembrane (IFITM) proteins
Abstract
The interferon-induced transmembrane (IFITM) family of proteins have recently been identified as important host effector molecules of the type I interferon response against viruses. IFITM1 has been identified as a potent antiviral effector against hepatitis C virus (HCV), whereas the related family members IFITM2 and IFITM3 have been described to have antiviral effects against a broad range of RNA viruses. Here, we demonstrate that IFITM2 and IFITM3 play an integral role in the interferon response against HCV and act at the level of late entry stages of HCV infection. We have established that in hepatocytes, IFITM2 and IFITM3 localize to the late and early endosomes, respectively, as well as the lysosome. Furthermore, we have demonstrated that S-palmitoylation of all three IFITM proteins is essential for anti-HCV activity, whereas the conserved tyrosine residue in the N-terminal domain of IFITM2 and IFITM3 plays a significant role in protein localization. However, this tyrosine was found to be dispensable for anti-HCV activity, with mutation of the tyrosine resulting in an IFITM1-like phenotype with the retention of anti-HCV activity and co-localization of IFITM2 and IFITM3 with CD81. In conclusion, we propose that the IFITM proteins act in a coordinated manner to restrict HCV infection by targeting the endocytosed HCV virion for lysosomal degradation and demonstrate that the actions of the IFITM proteins are indeed virus and cell-type specific.
Introduction
Hepatitis C virus (HCV)2 is a major public health problem, with over 185 million people infected worldwide (1, 2). Approximately 80% of infected individuals develop a chronic life long infection with a proportion progressing to significant liver disease such as fibrosis, cirrhosis, and in some cases hepatocellular carcinoma (3). The newly approved direct acting antivirals have improved sustained virological response rates (upwards of 95%), however, interferon-α (IFN-α) therapy is still required in combination to reduce the development of viral resistance (4). IFN, whether endogenous or exogenous, induces the expression of hundreds of interferon-stimulated genes (ISGs), the primary host effectors to mediate an antiviral state against viral infection. Only a handful of ISGs have been characterized in limiting HCV replication, and although the spectrum of ISGs capable of controlling HCV is emerging many more anti-HCV ISGs remain to be discovered and characterized.
Five interferon-induced transmembrane (IFITM) proteins have been identified in humans to date: IFITM1, IFITM2, IFITM3, IFITM5, and IFITM10. IFITM1, IFITM2, and IFITM3 are inducible by both type I and II IFN (5). IFITM5 is not IFN-inducible and little is known about the function of IFITM10 (6, 7). IFITM1, IFITM2, and IFITM3 have recently been identified as antiviral mediators conferring resistance against a broad range of viruses including influenza A virus (8, 9), West Nile virus (WNV) (8, 10), Dengue virus (11, 12), vesicular stomatis virus (VSV) (13), human immunodeficiency virus (HIV) (14), SARS coronavirus and Marbug virus (15). The IFITM proteins are the first known ISGs to target the late entry step of viral entry by preventing viral-cell fusion, although exact mechanisms still remain unclear. They primarily inhibit RNA viruses that require low pH-dependent entry into target cells (16, 17). The localization of IFITM3 to the late endosome and lysosome explains the unique antiviral actions of these ISGs, whereas IFITM1 has been found to localize to the plasma membrane and the early endosome. IFITM1 has recently been demonstrated to significantly restrict HCV entry into hepatocytes by disrupting the sequential interactions between the virus and the essential host co-receptors, in particular CD81 (18). Raychoudhri et al. (19, 20) have also reported the anti-HCV actions of IFITM1, whereas the expression of IFITM1 has been shown to be regulated by HCV through the up-regulation of mir-130a. Several genomic screens have identified IFITM2 and IFITM3 as possible anti-HCV effectors at the level of HCV RNA replication and translation, however, more comprehensive studies are required to characterize the relative anti-HCV activity and the mode of action (21).
HCV entry into the hepatocyte is coordinated through sequential interactions with the essential co-receptors: SR-BI, CD81, Claudin-1 (CLDN1), and Occludin (OCLN). The entry pathway is not completely understood, although it is postulated that HCV binds LDL receptors and heparin sulfate glycosaminoglycans leading to high-affinity interactions with SR-BI and CD81 on the hepatic surface (22). The interaction of HCV bound CD81 with the tight junction molecule CLDN1 initiates clathrin-mediated endocytosis of the HCV virion resulting in its traffic along actin stress fibers to Rab5a-containing early endosomes (22, 23). Fusion and acidification of the endosome results in the release of the viral genome to the cytoplasm where it is directly translated. Because HCV entry into the hepatocyte is low pH-dependent and utilizes the endocytic pathway, it is plausible that IFITM2 and IFITM3 may contribute to the anti-HCV response of IFN in a mechanism similar to that observed for other viruses.
To this end, we investigated the role of IFITM2 and IFITM3 in the context of the full HCV life cycle, including entry, translation, replication, and egress. We present evidence that IFITM2 and IFITM3 display anti-HCV activity that may complement the anti-HCV activity of IFITM1 (18) by inhibiting the late stages of HCV entry, possibly in a coordinated manner by trapping the virion in the endosomal pathway and targeting it for degradation at the lysosome. Furthermore, we demonstrate that post-translational modifications, in particular S-palmitoylation and tyrosine phosphorylation to contribute to the cellular localization and the anti-HCV activities of the IFITM proteins.
Experimental Procedures
Plasmid DNA and Transfections
Human IFITM genes were PCR-amplified from cDNA synthesized from Huh-7 cells stimulated with IFN-α (1000 units/ml) for 16 h using primers and cloned, in-frame, into BamHI and XhoI sites of pLenti6/V5-D-TOPO (Invitrogen). A FLAG tag was attached to the N terminus of each IFITM. Transfection of all plasmids was performed using FuGENE6 (Roche Applied Science) according to the manufacturer's recommendations. Mutant versions of each protein were constructed into pLenti6/V5-D-TOPO utilizing a QuikChange II XL Site-directed Mutagenesis system (Stratagene, La Jolla, CA). Plasmids pLenti6-mCherry-Rab5a, pRC-CMV-Viperin, and pRL-HL have been previously described (24–26).
Establishment of Cell Lines and Culture Conditions
The human hepatoma cell lines Huh-7 and the HCV genomic replicon line NNeoC-5B(RG) were maintained as previously described (27). Huh-7 cells stably expressing the IFITM proteins and an empty vector control were generated and designated Huh-7+IFITM1, Huh-7+IFITM2, Huh-7+IFITM3, and Huh-7+Vector. Lentiviral particles were prepared by co-transfecting 293T cells with equivalent amounts of the packaging vectors psPAX2 (Addgene 12260), pRSV-Rev (Addgene 12253), and pMD2.G (Addgene 12259), along with the IFITM and empty plasmid DNAs. Supernatant containing virus was collected 48 and 72 h post-transfection, filtered (0.45 μm), and applied to Huh-7 cells at a 1:5 ratio with normal culture medium. Polyclonal cell populations were selected with 3 μg/ml of blasticidin.
Virus Generation and Infection
Cell culture-propagated HCV particles (HCVcc, Jc1) were generated as described elsewhere (28) and used experimentally at m.o.i. 0.03. Pseudoviruses encoding luciferase were generated by co-transfection of 293T cells with the packaging plasmid pNL43-LucRE and the VSV-G envelope expression plasmid pMD2.G (Addgene plasmid 12259) for generation of VSVpp, the expression plasmid pE1E2-GT1b (212) for generation of HCVpp, the expression plasmid pMLV for generation of MLVpp, or empty plasmid pcDNA3 for generation of Env-pp. Supernatants were harvested at 48 h post-transfection and filtered (0.45 μm). Virus-containing cell culture supernatants were incubated with target cells, seeded at 8 × 103 cells/cm2 the day before (unless otherwise specified), overnight before washing with PBS, and returned to culture. Pseudoparticle infections were performed in the presence of 10 μg/ml of Polybrene. At 72 h post-infection luciferase activity was measured using a Luciferase Assay System (Promega) and a GloMax 96 microplate luminometer (Promega). Specific HCVpp, MLVpp, and VSVpp infectivity levels were determined by subtraction of the luciferase signals associated with the use of non-enveloped pseudoparticles (Env-pp).
Huh-7+IFITM stables were electroporated as described (3) and plated into 10-cm dishes for 24 h. To determine the amount of intracellular infectious virus, the cells were harvested via trypsinization, resuspended in complete medium, washed twice with 1× PBS, and lysed via 4 freeze/thaw cycles at −80 °C. Lysates were then clarified by centrifugation at 2300 × g for 5 min prior to inoculation on to naive Huh-7 cells. Extracellular medium was collected at the same time. Amounts of intracellular and extracellular infectious virus were determined by focus forming assay (3).
Immunoblotting
Western blotting was performed as described elsewhere (29) and used the following antibodies: mouse anti-FLAG (Sigma) diluted at 1/1000 and mouse anti-phosphotyrosine (Millipore) diluted at 1/1000. Mouse anti-human β-actin (Sigma) was used to control loading of protein at 1:10,000. Appropriate secondary antibodies labeled with horseradish peroxidase (Cell Signaling) were used, and bound protein was detected by chemiluminescence using SuperSignal West Femto (Pierce).
Immunoprecipitation
Immunoprecipitation of FLAG-tagged proteins was carried out as described (29), with samples harvested in the presence of phosphatase inhibitor (Calbiochem).
Immunofluorescence Microscopy
Cells were grown on 0.2% gelatin-coated coverslips overnight, transfected where applicable, and fixed the following day using acetone/methanol (1:1) for 5 min on ice for standard fluorescence microscopy or with 4% parafomaldehyde for 10 min on ice followed by a 10-min incubation in 0.1% Triton X-100 in PBS for confocal microscopy, prior to incubation with primary antibodies at room temperature for 1 h. Cells were washed in PBS and incubated with secondary antibodies for 1 h at 4 °C before being mounted with ProLong Gold reagent (Invitrogen). Images were acquired with a Bio-Rad Radiance 2100 Confocal or a Nikon TiE inverted microscope. Mouse monoclonal anti-FLAG and rabbit polyclonal anti-FLAG were obtained from Sigma. Rabbit monoclonal antibodies against Rab5a, Rab7, and Lamp1 were obtained from Cell Signaling. Mouse monoclonal anti-CD81 was obtained from BD Pharmingen, rabbit polyclonal anti-mCherry was obtained from BioVision, and human anti-HCV serum was generated as previously described (3).
HCV RNA Quantitation
Extraction of total cellular RNA, first-strand cDNA synthesis, and real-time RT-PCR was performed as described elsewhere (26).
Fluorescence Energy Resonance Transfer (FRET) Analysis
FRET by acceptor photobleaching was carried out essentially as described previously (30).
Proximity Ligation Assay (PLA)
Cells were cultured on 0.2% (w/v) gelatin-coated coverslips in 24-well culture plates prior to fixation with 4% paraformaldehyde. Proximity ligation assay (PLA) was conducted using the Duolink® In situ kit (Olink® Biosciences) as per the manufacturer's instructions. Positive interactions visualized using a Nikon Eclipse TiE fluorescence inverted microscope and images were captured using NIS Elements software.
Statistics
Results are expressed as mean ± S.E. Student's t test was used for statistical analysis. p < 0.05 was considered to be significant. All statistical analysis was performed using Prism 6 (GraphPad Software).
Results
IFITM1, IFITM2, and IFITM3 Limit HCV Infection in Vitro
Several functional genomics screens have identified IFITM1, IFITM2, and IFITM3 as potential anti-HCV effector molecules (21, 31, 32), with a recent study by Wilkins et al. (18) characterizing the anti-HCV nature of IFITM1. Examination of the IFITM family of proteins (Fig. 1A) revealed that all three IFITM proteins share high amino acid homology, containing two hydrophobic membrane-associated domains (M1 and M2) separated by a conserved intracellular loop (CIL) but differing at their N- and C-terminal domains. In contrast to IFITM1, IFITM2 and IFITM3 contain 20 and 21 amino acid extensions at the N-terminal domain, respectively, whereas IFITM1 contains a 13-amino acid extension at the C terminus (5, 33). It has also been shown that the promoter regions of all three IFITM genes contain ISRE and GAS elements (5). Thus we hypothesized that based on the high similarity in protein sequence and the recent findings in the literature, IFITM2 and IFITM3 may also have a significant antiviral activity against HCV. Initially we investigated the ability of Huh-7 cells and primary human hepatocytes to express IFITM1 in the presence of IFN-α/λ. As expected IFN-α and -λ induced expression of IFITM1 mRNA and protein in a concentration- and time-dependent manner (data not shown). Due to the high level of protein conservation between IFITM2 and IFITM3, we were unable to design specific RT-PCR primers or obtain specific antibodies for either protein and thus we were unable to extend these observations to endogenous IFITM2 or IFITM3. To determine the role of endogenous IFITM1 following IFN-α stimulation, we generated a polyclonal Huh-7 cell line stably expressing shRNA specifically targeting IFITM1 mRNA (Fig. 1B, i). These cells were either pretreated with 50 IU/ml of IFN-α for 24 h followed by HCV Jc1 infection or infected with Jc1 prior to stimulation with IFN-α (Fig. 1B, ii). Interestingly the anti-HCV activity of IFN-α is attenuated in IFITM1 shRNA cells compared with controls only in the pre-IFN-α treatment setting. These results suggest that IFITM1 plays an important, but not exclusive, role in the antiviral effects of IFN-α against the early stages of HCV infection in vitro.
FIGURE 1.

IFITM2 and IFITM3 inhibit HCV infection. A, schematic representation of human IFITM1, IFITM2, and IFITM3. IFITM1 differs from IFITM2 and IFITM3 with a 21-amino acid truncation at the N terminus and a 13-amino acid extension at the C terminus. B, i, Huh7+shControl and Huh-7+shIFITM1 cells were stimulated with 100 IU/ml of IFN-α for 16 h. Total RNA was harvested for RT-quantitative PCR for IFITM1 mRNA levels (data represented as a mean ± S.E. with a significance of ****, p < 0.0001 calculated using a Student's t test). ii, Huh7+shControl and Huh-7+shIFITM1 cells were either pretreated for 24 h before Jc1 infection (m.o.i. = 0.03) or post-treated 24 h after Jc1 infection for 16 h with 50 IU/ml of IFN-α. Total RNA was harvested for RT-quantitative PCR for HCV RNA levels (data represented as a mean ± S.E. with a significance of *, p = 0.035, calculated using a Student's t test). C, Huh-7+IFITM and Huh-7+vector control cells were either stained with a mouse monolconal anti-FLAG antibody, followed by an Alexa 555-conjugated anti-mouse IgG (i) or cellular lysate harvested to detect specific IFITM protein expression (ii). D, Huh-7+IFITM and Huh-7+vector control cells were infected with HCV Jc1 (m.o.i. 0.03). Total RNA was harvested at the indicated time points for RT-quantitative PCR for HCV RNA levels (data represented as a mean ± S.E. with a significance of *, p < 0.05; **, p < 0.005 calculated using a Student's t test). E, Huh-7+IFITM and Huh-7+vector control cells were infected with HCV Jc1 (m.o.i. 0.03) and immunofluorescence analysis conducted 72 h later using mouse monoclonal anti-FLAG and human anti-HCV serum antibodies, followed by an Alexa 555-conjugated anti-mouse IgG and an Alexa 488-conjugated anti-human IgG, respectively.
To characterize the relative anti-HCV roles of IFITM1, IFITM2, and IFITM3 and their cellular localization, we generated polyclonal Huh-7 cells stably expressing each of the IFITM proteins (Huh-7+IFITM) with a N-terminal FLAG tag to facilitate detection. Protein expression in these stable cell lines was confirmed by immunofluorescence and immunoblot analysis (Fig. 1C). In Huh-7 cells, IFITM1 localized predominantly to the cell surface with some intracellular localization, whereas IFITM2 and IFITM3 localized to specific intracellular compartments within the cytoplasm. HCV infection (Jc1) of the stable Huh-7+IFITM cell lines and analysis of HCV RNA 24 h thereafter demonstrated that IFITM1, IFITM2, and IFITM3 were able to significantly decrease HCV RNA levels by 77, 61, and 57%, respectively (Fig. 1D), compared with vector control. Similar results were obtained upon extending HCV infection to 48 and 72 h. Interestingly, Huh-7+IFITM cells infected with Jc1 for 72 h depicted a “viral exclusion” phenotype, where cells expressing the IFITM proteins do not appear to be infected with HCV, whereas neighboring cells lacking IFITM expression were infected (Fig. 1E). These results corroborate the known anti-HCV activity of IFITM1, and for the first time demonstrate the anti-HCV nature of IFITM2 and IFITM3.
IFITM Proteins Inhibit HCV Infection at an Early Stage of Infection
Recent studies have identified IFITM1 to be able to limit HCV infection at the level of HCV entry through an interaction with the essential entry co-receptor CD81 (18). To determine whether this mechanism was also true for IFITM2 and IFITM3, we compared the ability of HCV (E1/E2) psuedoparticles (HCVpp) to enter Huh-7+IFITM cells in comparison to vector control cells (Fig. 2A). As expected, we observed a significant reduction in the entry of HCVpp into cells expressing IFITM1 compared with control. Similar reductions in HCVpp entry were observed in cells expressing IFITM2 and IFITM3, demonstrating that the anti-HCV activity observed for these proteins is also at the level of HCV entry. Based on these results and even though IFITM2 and IFITM3 show a predominantly intracellular localization, we next examined the localization of IFITM2 and IFITM3 in relationship to all HCV entry factors (CD81, SR-BI, CLDN1, and OCLN). We confirmed the co-localization (Fig. 2B) of IFITM1 with CD81 and extended this to show a physical interaction between IFITM1 and CD81 via FRET (Fig. 2C) and PLA (data not shown). In contrast, IFITM2 localized solely to the cytoplasm and hence did not co-localize with CD81, however, IFITM3, whereas predominantly cytoplasmic, did partially co-localize with CD81 in some instances (Fig. 2B). None of the other HCV entry receptors tested (SR-BI, CLDN1, and OCLN) were found to interact with any of the IFITM proteins as determined by FRET and PLA (data not shown).
FIGURE 2.

IFITM1, IFITM2, and IFITM3 limit HCV entry into Huh-7 cells. A, Huh-7+IFITM and Huh-7+vector control cells were transduced with pseudoparticles containing envelopes of HCV, MLV, and VSV. Viral entry was determined by luciferase activity 72 h post-transduction (data represented as a mean ± S.E. with a significance of *, p < 0.02; ***, p < 0.003; ****, p < 0.0001 calculated using a Student's t test). B, Huh-7+IFITM cells were stained with rabbit polyclonal anti-FLAG and mouse anti-CD81 antibodies, followed by an Alexa 488-conjugated anti-rabbit IgG and an Alexa 555-conjugated anti-mouse IgG. C, Huh-7+IFITM1 cells were stained with a rabbit polyclonal anti-FLAG and mouse anti-CD81, followed by an Alexa 555-conjugated anti-rabbit IgG and a CY5 goat anti-mouse IgG, respectively. Cells were analyzed on a Zeiss Axioplan microscope using FRET (Carl Zeizz, Oberkochen, Germany). DIF, difference in fluorescence.
We next investigated the role of the IFITM proteins on the rest of the HCV life cycle. Transient expression of each of the IFITM proteins did not significantly effect HCV RNA replication in genomic HCV replicon cell lines (Fig. 3A, i). This is in contrast to the anti-HCV activity of the well characterized ISG viperin that served as a positive control (30, 34). Interestingly, the viral exclusion phenotype observed using the HCVcc system was no longer evident upon expression of the IFITM proteins in these cells (Fig. 3A, ii), with co-expression of IFITM proteins and cells harboring HCV replication suggesting that the IFITM proteins do not impact upon HCV RNA replication. Furthermore, the IFITM proteins had no effect on HCV IRES promoter activity, upon transfection of the Huh-7+IFITM cells with a construct containing the HCV IRES driving a luciferase reporter gene (Fig. 3B). Finally we examined HCV egress using an extracellular:intracellular infectivity assay, where Huh-7+IFITM and vector control cells were electroporated with HCV Jc1 RNA to bypass entry and after 24 h the extracellular and intracellular supernatant was applied onto naive Huh-7 cells prior to measurement of HCV infectivity by a focus-forming assay. IFITM1, IFITM2, and IFITM3 did not limit HCV egress following analysis of both raw HCV focus-forming units (FFU) (Fig. 3C, i) and the ratio of extracellular FFU:intracellular FFU (Fig. 3C, ii). Collectively this data demonstrates that all three IFITM proteins block de novo HCV infection with no detectable effect on other stages of the HCV life cycle, indicating that IFITM1, IFITM2, and IFITM3 primarily act on the early stages of HCV infection.
FIGURE 3.

IFITM1, IFITM2, and IFITM3 have no effect on HCV RNA replication, translation, or egress. A, i, HCV genomic replicon cells were transduced with each of the IFITM proteins, viperin, and the vector control for 48 h and total RNA harvested for RT-quantitative PCR for HCV RNA levels (data represented as a mean ± S.E.). ii, HCV genomic replicon cells were transduced with IFITM proteins and the vector control and immunofluorescence analysis conducted 48 h later using mouse monoclonal anti-FLAG and human anti-HCV serum antibodies, followed by an Alexa 555-conjugated anti-mouse IgG and an Alexa 488-conjugated anti-human IgG respectively. B, Huh-7+IFITM and Huh-7+vector control cells were transiently transfected with IRES-luc for 48 h. 24 h post-transfection vector control cells were treated with 100 ng/ml of IL-29 for 24 h. Luciferase activity determined 48 h post-transfection (data represented as a mean ± S.E.). C, Huh-7+IFITM and Huh-7+vector control cells were electroporated with Jc1 and extracellular and intracellular supernatant was applied to naive Huh-7 cells 24 h post-electroporation. At 72 h post-infection focus-forming units were enumerated using human anti-HCV serum antigens. i, extracellular FFU and intracellular FFU titers expressed as FFU/ml. ii, FFU/ml is expressed as a ratio of extracellular FFU to intracellular FFU (data represented as a mean ± S.E.).
IFITM2 and IFITM3 Co-localize with Early and Late Endosomes and Lysosomes in Hepatocytes
The cellular localization of IFITM2 and IFITM3, particularly IFITM3, has been examined in a number of different cell lines (A549, HEK293, and HeLa) in relationship to their antiviral activity against other viruses (9, 15, 16, 35). These studies have identified that IFITM2 and IFITM3 partially co-localize with late endosomes and lysosomes within the majority of cells. We sought to investigate this localization within Huh-7 cells, and probed the Huh-7+IFITM cells with specific antibodies targeting early endosomes (Rab5a), late endosomes (Rab7), and lysosomes (Lamp1). Co-localization immunofluorescence analysis found that in Huh-7 cells, IFITM2 and IFITM3 partially co-localized with late and early endosomes, respectively (Fig. 4, A and B). Interestingly, both IFITM2 and IFITM3 were found to co-localize with lysosomes, whereas partial co-localization between intracellular IFITM1 and lysosomes was also observed in some cells (Fig. 4C). Although these observations were not solely unexpected, it is interesting to note the difference in localization of the IFITM proteins between cell types. In the context of this study, the endosomal localization of IFITM2 and IFITM3 is most noteworthy, as HCV entry requires Rab5a-positive endosomes, whereas the formation of the HCV replication complex requires both Rab5a and Rab7 (36).
FIGURE 4.

IFITM2 and IFITM3 partially co-localize with early and late endosomes, whereas all three IFITM proteins co-localize with lysosomes. A, Huh-7+IFITM2 and Huh-7+IFITM3 cells were transfected with mCherry-Rab5a expression plasmid. 24 h later cells were stained with mouse monoclonal anti-FLAG and rabbit polyclonal anti-mCherry antibodies, followed by an Alexa 488-conjugated anti-mouse IgG and an Alexa 555-conjugated anti-rabbit IgG (20 μm scale bar). B, Huh-7+IFITM2 and Huh-7+IFITM3 cells were stained with mouse monoclonal anti-FLAG and rabbit monoclonal anti-Rab7 antibodies, followed by Alexa 488-conjugated anti-mouse IgG and Alexa 555-conjugated anti-rabbit IgG (20 μm scale bar). C, Huh-7+IFITM cells were stained with mouse monoclonal anti-FLAG and rabbit monoclonal anti-Lamp1 antibodies, followed by an Alexa 488-conjugated anti-mouse IgG and an Alexa 555-conjugated anti-rabbit IgG (20 μm scale bar).
The Conserved N-terminal Tyrosine Residue Common to IFITM2 and IFITM3 Crucial for Cellular Localization but Does Not Affect Anti-HCV Activity
Tyrosine residue 20 (Tyr-20) in the N-terminal domain of IFITM3 has recently been identified to be important for both cellular localization and antiviral activity, particularly against influenza A virus, Dengue virus, and VSV (16, 37). Furthermore, recent studies have demonstrated Tyr-20 to be part of an endocytic signal (YEML) targeting IFITM3 to the late endosome (38, 39). We examined the protein sequences between the three IFITM proteins and discovered that this N-terminal tyrosine residue is conserved in IFITM2 (Tyr-19) but is not present in IFITM1 due to the 21-amino acid N-terminal truncation (Fig. 5A). Based on this observation, we decided to investigate the importance of this conserved N-terminal tyrosine residue on the anti-HCV activity of IFITM2 and IFITM3. We generated tyrosine to alanine mutants for IFITM2 and IFITM3, Y19A and Y20A, respectively, and created polyclonal constitutively expressing cell lines as previously described. First we sought to confirm the loss in phosphorylation upon mutating the tyrosine residue, where IFITM2, IFITM3, and the tyrosine mutant cells were treated with a phosphatase inhibitor mixture prior to harvesting for immunoprecipitation. The precipitated samples were probed by immunoblot with antibodies specifically targeting phosphorylated tyrosine (Tyr(P)). A complete loss in phosphorylation was observed for Y19A and Y20A compared with wild-type IFITM2 and IFITM3, respectively (Fig. 5B). We extended our observations to the cellular localization of IFITM2:Y19A and IFITM3:Y20A compared with wild-type, as mutation of Tyr-20 has previously been reported to change IFITM3 localization from the endosome to the plasma membrane and abrogate antiviral activity. Immunofluorescence analysis confirmed this redistribution, with the localization of Y20A within Huh-7 cells resembling that of wild-type IFITM1 on the cell surface (Fig. 5C). Y19A also displayed a change in localization although not as striking as that of Y20A, where the majority of protein localization was redistributed to the plasma membrane with some perinuclear localization still present (Fig. 5C). We next sought to determine whether this change in localization would alter the anti-HCV activity of IFITM2 and IFITM3. HCV infection of cells stably expressing either wild-type or tyrosine mutant IFITM2 and IFITM3 for 24 h revealed no loss in anti-HCV activity in cells expressing the tyrosine mutants compared with vector control (Fig. 5D, i). The viral exclusion phenotype was retained in cells expressing the tyrosine mutants, where Jc1 infection was only observed in neighboring cells lacking IFITM2:Y19A and IFITM3:Y20A expression (Fig. 5D, ii). Furthermore, it can be noted that the Y19A and Y20A mutants exhibited enhanced anti-HCV properties compared with wild-type. These results were unexpected based on previous studies where the mutation of the tyrosine resulted in loss of antiviral activity for IFITM3. Interestingly, the enhanced anti-HCV activity of both Y19A and Y20A resemble the level of anti-HCV activity observed in Fig. 1C for IFITM1. To investigate whether the loss of a single tyrosine residue reverted IFITM2 and IFITM3 to an IFITM1-like phenotype, we examined the localization of Y19A and Y20A in the context of CD81. Significant co-localization was observed between the IFITM2 and IFITM3 tyrosine mutants and CD81 (Fig. 6), mirroring the localization of IFITM1. These results demonstrate for the first time that the conserved N-terminal tyrosine residue in IFITM2 and IFITM3 is important for cellular localization but not for the anti-HCV properties of these proteins.
FIGURE 5.

Conserved N-terminal tyrosine residue is important for IFITM2 and IFITM3 cellular localization but not anti-HCV activity. A, schematic representation of IFITM2 and IFITM3 and the tyrosine mutant derivatives. B, Huh-7+IFITM2, Huh-7+IFITM3, Huh-7+IFITM2:Y19A, and Huh-7+IFITM3:Y20A cells treated with a phosphatase inhibitor mixture before cellular lysates were harvested. IFITM proteins were precipitates with anti-FLAG antibodies and then tested in Western blotting with anti-tyrosine phosphorylation antibodies. C, IFITM2, IFITM3, and tyrosine mutant overexpression cells were stained with mouse monoclonal anti-FLAG, followed by an Alexa 555-conjugated anti-mouse IgG. D, i, Huh-7+IFITM2, Huh-7+IFITM3, Huh-7+IFITM2:Y19A, Huh-7+IFITM3:Y20A, and Huh-7+vector control cells were infected with HCV Jc1 (m.o.i. 0.03). Total RNA was harvested at the indicated time points for RT-quantitative PCR for HCV RNA levels (data are represented as mean ± S.E. with a significance of p = 0.004 calculated using a Student's t test); ii, Huh-7+IFITM2:Y19A and Huh-7+IFITM3:Y20A were infected with HCV Jc1 (m.o.i. 0.03) and immunofluorescence analysis was conducted 72 h later using mouse monoclonal anti-FLAG and human anti-HCV serum antibodies, followed by an Alexa 555-conjugated anti-mouse IgG and an Alexa 488-conjugated anti-human IgG, respectively. IP, immunoprecipitation.
FIGURE 6.
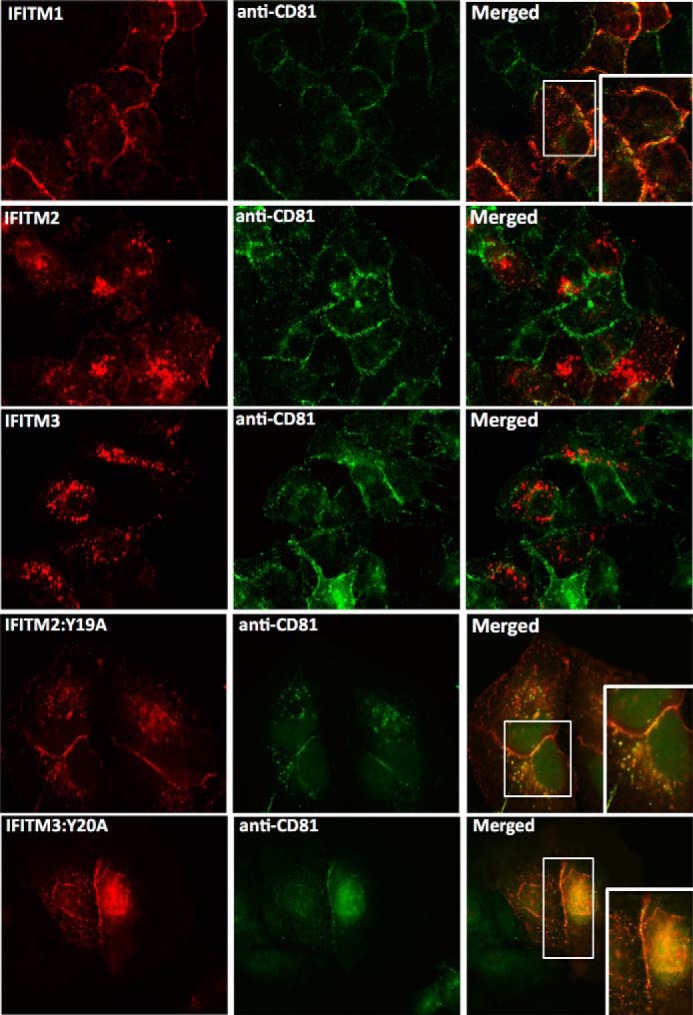
IFITM2:Y19A and IFITM3:Y20A mutants co-localize with CD81 on the cell surface. Huh-7+IFITM cells and Huh-7+IFITM2:Y19A and Huh-7+IFITM3:Y20A cells were stained with rabbit polyclonal anti-FLAG and mouse anti-CD81 antibodies, followed by an Alexa 555-conjugated anti-rabbit IgG and an Alexa 488-conjugated anti-mouse IgG.
Palmitoylation of the IFITM Proteins Is Important for Anti-HCV Activity
S-palmitoylation is a post-translational modification resulting in the addition of a palmitoyl group to cytosolic cysteine residues and in many instances is essential for protein stability, localization, and association with lipid rafts (40). Yount et al. (41) identified for the first time that the IFITM proteins undergo S-palmitoylation at three specific cysteine residues: two consecutive cysteine residues found in the first membrane-associated domain (M1), whereas the other cysteine residue is in the CIL. Additional studies have identified S-palmitoylation of the IFITM proteins, particularly the Cys-72 residue of IFITM3 to be vital for antiviral activity against influenza A virus and Dengue virus (16, 41, 42). Interestingly, it has been noted that S-palmitoylation of the IFITM proteins is not required for their anti-HIV activity (43). To examine whether S-palmitoylation of the IFITM proteins was important for anti-HCV activity, double (M1, C50A/C51A; M2, C70A/C71A; M3, C71A/C72A) and single (M1, C84A; M2, C104A; M3, C105A) cysteine to alanine mutants were generated for each of the IFITM proteins (Fig. 7A). Transient expression of wild-type and palmitoylation mutants prior to infection with HCV (Jc1) for 24 h revealed the single cysteine residue in the CIL of each IFITM proteins to be important for anti-HCV activity, as a complete loss of anti-HCV activity was observed for the CIL mutants compared with vector control (Fig. 7B). IFITM3, however, also requires the two cysteine residues found in the first membrane-associated domain (M1) for its anti-HCV activity. Previous studies have reported conflicting roles of S-palmitoylation on IFITM localization within the cell. Yount et al. (41) demonstrated that S-palmitoylation played a role in the clustering of IFITM3 near the ER, whereas John et al. (16) showed the converse, where the removal of the cysteine residues resulted in significant clustering within the cell. To determine whether a similar change in localization occurred in Huh-7 cells, the cellular localization of the palmitoylation mutants was examined in relationship to previously identified subcellular markers (Fig. 8). We demonstrate for the first time that mutation of the cysteine residue within the CIL resulted in each of the IFITM proteins predominantly localizing to the lysosome. This redistribution was also observed for M1 palmitoylation mutants C50A/C51A and C71A/C72A in IFITM1 and IFITM3, respectively. Interestingly, the C70A/C71A mutant of IFITM2 retained partial co-localization at the late endosome and lysosome similar to wild-type. The loss of localization of the CIL mutants, and IFITM3, C71A/C72A, validates the complete abrogation of anti-HCV activity observed for these mutants in Fig. 7B and suggest possible degradation of these mutants at the lysosome. This indicates the importance of this cysteine residue and hence conceivably S-palmitoylation for the anti-HCV activity of these proteins. It is interesting, however, to note, that the C50A/C51A and C70A/C71A mutations in IFITM1 and IFITM2, respectively, retain partial wild-type localization and this provides an explanation for the anti-HCV activity observed for these mutants. Nevertheless, the partial retention of subcellular localization for these mutants indicates a possibility that these proteins are also able to associate with either the virus or a new host protein to limit HCV infection, highlighting the versatility of these proteins to retain anti-HCV activity.
FIGURE 7.

IFITM1, IFITM2, and IFITM3 require palmitoylation for anti-HCV activity. A, schematic representation of IFITM1, IFITM2, and IFITM3 and the palmitoylation mutant derivatives. B, Huh-7 cells were transfected with wild-type IFITM proteins, the palmitoylation mutants, and vector control (empty) for 24 h prior to infection with HCV Jc1 (m.o.i. 0.003). Total RNA was harvested 24 h post-infection for RT-quantitative PCR for HCV RNA levels (data represented as a mean ± S.E. with a significance of **, p < 0.001; ***, p = 0.003; ****, p < 0.0001 calculated using a Student's t test).
FIGURE 8.
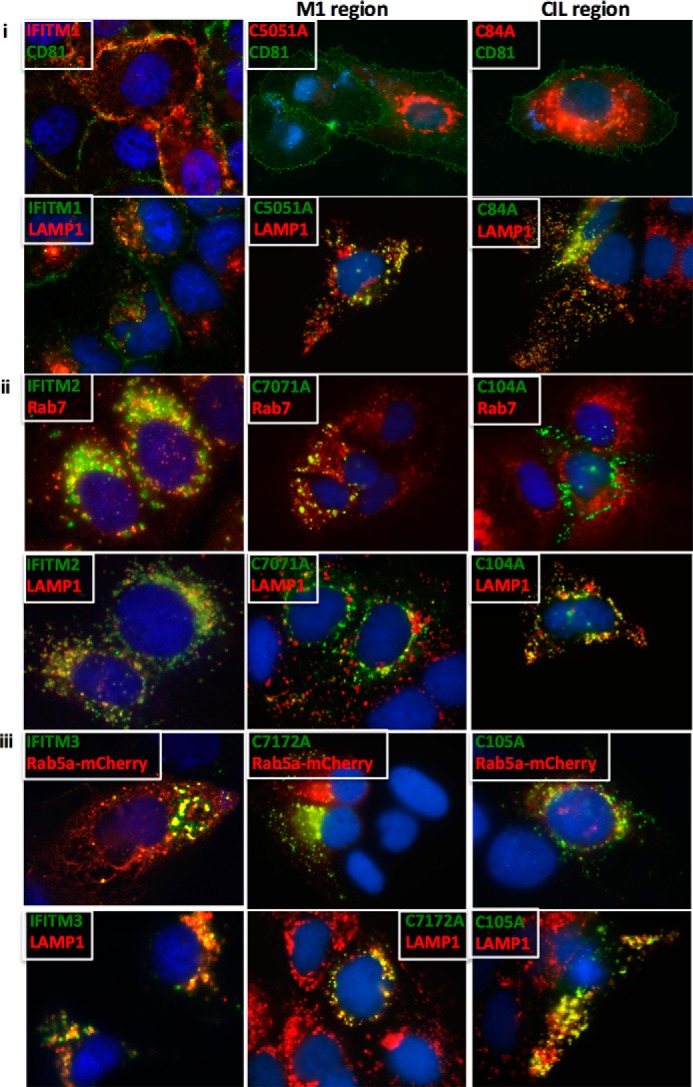
Cysteine mutations targeting the CIL region of the IFITM proteins re-localize to the lysosome. i, Huh-7 cells transiently transfected for 24 h with either wild-type IFITM1 or the respective IFITM1 palmitoylation mutants were stained with mouse monoclonal anti-FLAG and either mouse anti-CD81 or rabbit monoclonal anti-Lamp1 antibodies, followed by an Alexa 488-conjugated anti-mouse IgG and an Alexa 555-conjugated anti-rabbit IgG. ii, Huh-7 cells transiently transfected or 24 h with either wild-type IFITM2 or the respective IFITM2 palmitoylation mutants were stained with mouse monoclonal anti-FLAG and either rabbit monoclonal anti-Rab7 or rabbit monoclonal anti-Lamp1 antibodies, followed by an Alexa 488-conjugated anti-mouse IgG and an Alexa 555-conjugated anti-rabbit IgG. iii, Huh-7 cells transiently transfected for 24 h with either wild-type IFITM1 or the respective IFITM1 palmitoylation mutants, as well as a mCherry-Rab5a expression plasmid, were stained with mouse monoclonal anti-FLAG and either rabbit polyclonal anti-mCherry or rabbit monoclonal anti-Lamp1 antibodies, followed by an Alexa 488-conjugated anti-mouse IgG and an Alexa 555-conjugated anti-rabbit IgG.
Discussion
The antiviral actions of the type I interferon response is key in the control of viral infection and the foundation behind interferon therapy for chronic hepatitis C virus infection that is still used in conjunction with the new wave of direct acting antivirals. IFN induces the expression of hundreds of antiviral ISGs; however, the subset of ISGs essential to mediate the IFN response against HCV and many other viruses is yet to be elucidated.
Previous studies using genome-wide siRNA screens have identified the importance of IFITM1, IFITM2, and IFITM3 in the antiviral response against HCV, whereas recent studies have identified a specific role for IFITM1 against HCV entry (18). Here we confirm and extend these studies further by first corroborating the ability of IFITM1 to interact with CD81 to limit HCV entry, whereas also establishing IFITM2 and IFITM3 as anti-HCV ISGs that target the late-entry stages of viral infection. IFITM2 and IFITM3 have no discernable effect on the post-entry stages of HCV infection such as RNA replication, translation, or viral egress but were able to limit HCV entry. Although the observed restriction of HCV entry was not as significant as that observed for IFITM1 (using a HCVpp assay), the localization of IFITM2 and IFITM3 at both early and late endosomes as well as lysosomes indicates that these proteins are following the established paradigm of acting at the late entry stages of HCV entry. Our data suggests that the IFITM proteins may be acting in a sequential and combined manner to limit HCV entry by directly targeting HCV-host receptor interactions as well as the processes of uncoating and release of the HCV genome into the cytoplasm (Fig. 9).
FIGURE 9.

Model of the role of IFITM1, −2, and -3 on the HCV life cycle. IFITM1 interacts with CD81 on the cells surface to limit HCV entry into the hepatocyte. HCV virions that bypass this inhibition enter the cell via clathrin-mediated endocytosis and enter Rab5a- and IFITM3-positive early endosomes. Alterations to endosomal membrane function traps the HCV virion within the early endosome, which is targeted for endosomal-lysosmal degradation via interactions between IFITM proteins, found in opposing endosomal and lysosomal membranes.
Endosomal-lysosomal degradation is a sequential process requiring endosomal maturation starting at the early endosome, moving to the late endosome and culminating in the fusion of the late endosome with the lysosome, resulting in the degradation of trapped particles (44). We hypothesize that although IFITM1 limits HCV infection by disrupting HCV co-receptor assembly as demonstrated by Wilkins et al. (18), IFITM2 and IFITM3 limit HCV infection by preventing viral-endosomal fusion thereby “trapping” the endocytosed virion within the endocytic pathway targeting it for lysosomal degradation. The ability of the IFITM proteins to alter membrane fluidity and curvature to prevent viral hemifusion provides an explanation for this hypothesis (45). Furthermore, IFITM3-mediated enrichment of endolysosomal membranes has also been shown by several independent studies, and whereas it is possible that IFITM3-induced cholesterol accumulation contributes to changes in endosomal membrane function within hepatocytes, it is not through an interaction with VAPA, as was shown by Amini-Bavil-Olyaee et al. (35) (data not shown). Li et al. (46) proposed that IFITM-induced alterations to endosomal membrane function promoted fusion with opposing membranes expressing IFITM proteins. Thus, we postulate that HCV is trapped in the early endosome after clathrin-mediated endocytosis due to IFITM3-mediated alterations at the early endosomal membrane preventing fusion and subsequently targeting the virion for degradation via the late endosome and lysosome through interactions between the different IFITM proteins found at these organelles.
Post-translational modifications of the IFITM proteins have been identified to play a crucial role in the localization and function of these proteins. Our data indicates for the first time that the conserved cysteine residue found in the CIL is important for cellular localization and anti-HCV activity of all three IFITM proteins. In addition, Cys-71 and Cys-72 found within the first membrane-associated domain of IFITM3 are also important for anti-HCV activity. S-palmitoylation of Cys-72 has been shown to be essential for the ability of IFITM3 to restrict influenza A virus and Dengue virus (16, 41). The addition of palmitoyl groups is associated with protein stability, localization, and the association of proteins with lipid rafts, thus explaining the importance of S-palmitoylation for the antiviral activity of the IFITM proteins. Interestingly, we also noted that mutation of the M1 cysteine residues in IFITM1 and IFITM2 retained anti-HCV activity despite partial retention of wild-type localization, perhaps suggesting a novel association with either a viral or host factor. We also confirmed the importance of the highly conserved YXXF motif and the phosphorylation of this tyrosine residue in the IFITM2 and IFITM3 N-terminal domain for the localization of these proteins at endosomal compartments, but unexpectedly found that the N-terminal domain tyrosine mutants enhanced anti-HCV activity compared with wild-type. The IFITM2 and IFITM3 tyrosine mutants co-localize with CD81 on the hepatic cell surface thus mirroring the localization and potentially the anti-HCV activity of IFITM1. For the first time we demonstrate a unique feature of the IFITM proteins against HCV infection, where a single mutation in IFITM1, IFITM2, and IFITM3 can still maintain significant anti-HCV activity despite significantly altered localization. This suggests that whereas the IFITM proteins probably act broadly at the site of viral-cell fusion, the exact mechanisms of antiviral activity of these proteins is both virus and cell-type specific.
In this study we demonstrate that the IFITM proteins may act sequentially to limit HCV infection, and support the theory that the IFITM proteins, in particular IFITM2 and IFITM3 exhibit antiviral activity by altering virus-cell fusion. Further questions remain to define the mechanism of action of these proteins, in particular the site of this potential disruption and whether any cofactors are involved. Insights into the cellular functions of the IFITM proteins could lead to strategies to specifically induce IFITM protein expression or mimic the function of these proteins to complement not only existing anti-HCV therapies but to also target other viral infections.
Author Contributions
M. R. B. and S. K. N. designed the study and wrote the paper. S. K. N. and E. M. M. produced all IFITM constructs and performed all antiviral and immunofluorescence assays. N. S. E. produced confocal images and provided Rab5a-mCherry constructs. R. A. W. and A. E. performed HCVpp experiments. K. J. H. and A. R. L. provided technical advice and critical evaluation of the manuscript. All authors analyzed the results and approved the final version of the manuscript.
This work was supported in part by National Health & Medical Research Council of Australia Grant 1053206. The authors declare that they have no conflicts of interest with the contents of this article.
- HCV
- hepatitis C virus
- IFITM
- interferon-induced transmembrane protein
- ISG
- interferon stimulated gene
- VSV
- vesicular stomatis virus
- CLDN1
- Claudin-1
- OCLN
- occludin
- PLA
- proximity ligation assay
- FFU
- focus-forming units
- m.o.i.
- multiplicity of infection
- CIL
- conserved intracellular loop
- IRES
- internal ribosome entry site.
References
- 1.Thomas D. L. (2013) Global control of hepatitis C: where challenge meets opportunity. Nat. Med. 19, 850–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCartney E. M., Helbig K. J., Narayana S. K., Eyre N. S., Aloia A. L., and Beard M. R. (2013) Signal transducer and activator of transcription 3 is a proviral host factor for hepatitis C virus. Hepatology 58, 1558–1568 [DOI] [PubMed] [Google Scholar]
- 3.Zhong J., Gastaminza P., Cheng G., Kapadia S., Kato T., Burton D. R., Wieland S. F., Uprichard S. L., Wakita T., and Chisari F. V. (2005) Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U.S.A. 102, 9294–9299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aloia A. L., Locarnini S., and Beard M. R. (2012) Antiviral resistance and direct-acting antiviral agents for HCV. Antivir. Ther. 17, 1147–1162 [DOI] [PubMed] [Google Scholar]
- 5.Lewin A. R., Reid L. E., McMahon M., Stark G. R., and Kerr I. M. (1991) Molecular analysis of a human interferon-inducible gene family. Eur. J. Biochem. 199, 417–423 [DOI] [PubMed] [Google Scholar]
- 6.Moffatt P., Gaumond M. H., Salois P., Sellin K., Bessette M. C., Godin E., de Oliveira P. T., Atkins G. J., Nanci A., and Thomas G. (2008) Bril: a novel bone-specific modulator of mineralization. J. Bone Miner. Res. 23, 1497–1508 [DOI] [PubMed] [Google Scholar]
- 7.Hickford D., Frankenberg S., Shaw G., and Renfree M. B. (2012) Evolution of vertebrate interferon inducible transmembrane proteins. BMC Genomics 13, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brass A. L., Huang I. C., Benita Y., John S. P., Krishnan M. N., Feeley E. M., Ryan B. J., Weyer J. L., van der Weyden L., Fikrig E., Adams D. J., Xavier R. J., Farzan M., and Elledge S. J. (2009) The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and Dengue virus. Cell 139, 1243–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feeley E. M., Sims J. S., John S. P., Chin C. R., Pertel T., Chen L. M., Gaiha G. D., Ryan B. J., Donis R. O., Elledge S. J., and Brass A. L. (2011) IFITM3 inhibits influenza A virus infection by preventing cytosolic entry. PLoS Pathog. 7, e1002337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang D., Weidner J. M., Qing M., Pan X. B., Guo H., Xu C., Zhang X., Birk A., Chang J., Shi P. Y., Block T. M., and Guo J. T. (2010) Identification of five interferon-induced cellular proteins that inhibit West Nile virus and Dengue virus infections. J. Virol. 84, 8332–8341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang D., Guo H., Xu C., Chang J., Gu B., Wang L., Block T. M., and Guo J. T. (2008) Identification of three interferon-inducible cellular enzymes that inhibit the replication of hepatitis C virus. J. Virol. 82, 1665–1678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan Y. K., Huang I. C., and Farzan M. (2012) IFITM proteins restrict antibody-dependent enhancement of Dengue virus infection. PloS One 7, e34508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weidner J. M., Jiang D., Pan X. B., Chang J., Block T. M., and Guo J. T. (2010) Interferon-induced cell membrane proteins, IFITM3 and tetherin, inhibit vesicular stomatitis virus infection via distinct mechanisms. J. Virol. 84, 12646–12657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu J., Pan Q., Rong L., He W., Liu S. L., and Liang C. (2011) The IFITM proteins inhibit HIV-1 infection. J. Virol. 85, 2126–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang I. C., Bailey C. C., Weyer J. L., Radoshitzky S. R., Becker M. M., Chiang J. J., Brass A. L., Ahmed A. A., Chi X., Dong L., Longobardi L. E., Boltz D., Kuhn J. H., Elledge S. J., Bavari S., Denison M. R., Choe H., and Farzan M. (2011) Distinct patterns of IFITM-mediated restriction of filoviruses, SARS coronavirus, and influenza A virus. PLoS Pathog. 7, e1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.John S. P., Chin C. R., Perreira J. M., Feeley E. M., Aker A. M., Savidis G., Smith S. E., Elia A. E., Everitt A. R., Vora M., Pertel T., Elledge S. J., Kellam P., and Brass A. L. (2013) The CD225 domain of IFITM3 is required for both IFITM protein association and inhibition of influenza A virus and Dengue virus replication. J. Virol. 87, 7837–7852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bailey C. C., Zhong G., Huang I. C., and Farzan M. (2014) IFITM-family proteins: the cell's first line of antiviral defense. Annu. Rev. Virol. 1, 261–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilkins C., Woodward J., Lau D. T., Barnes A., Joyce M., McFarlane N., McKeating J. A., Tyrrell D. L., and Gale M. Jr. (2013) IFITM1 is a tight junction protein that inhibits hepatitis C virus entry. Hepatology 57, 461–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raychoudhuri A., Shrivastava S., Steele R., Kim H., Ray R., and Ray R. B. (2011) ISG56 and IFITM1 proteins inhibit hepatitis C virus replication. J. Virol. 85, 12881–12889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhanja Chowdhury J., Shrivastava S., Steele R., Di Bisceglie A. M., Ray R., and Ray R. B. (2012) Hepatitis C virus infection modulates expression of interferon stimulatory gene IFITM1 by upregulating miR-130A. J. Virol. 86, 10221–10225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Q., Zhang Y. Y., Chiu S., Hu Z., Lan K. H., Cha H., Sodroski C., Zhang F., Hsu C. S., Thomas E., and Liang T. J. (2014) Integrative functional genomics of hepatitis C virus infection identifies host dependencies in complete viral replication cycle. PLoS Pathog. 10, e1004163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dubuisson J., and Cosset F. L. (2014) Virology and cell biology of the hepatitis C virus life cycle: an update. J. Hepatol. 61, S3-S13 [DOI] [PubMed] [Google Scholar]
- 23.Blanchard E., Belouzard S., Goueslain L., Wakita T., Dubuisson J., Wychowski C., and Rouillé Y. (2006) Hepatitis C virus entry depends on clathrin-mediated endocytosis. J. Virol. 80, 6964–6972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eyre N. S., Fiches G. N., Aloia A. L., Helbig K. J., McCartney E. M., McErlean C. S., Li K., Aggarwal A., Turville S. G., and Beard M. R. (2014) Dynamic imaging of the hepatitis C virus NS5A protein during a productive infection. J. Virol. 88, 3636–3652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ikeda M., Yi M., Li K., and Lemon S. M. (2002) Selectable subgenomic and genome-length dicistronic RNAs derived from an infectious molecular clone of the HCV-N strain of hepatitis C virus replicate efficiently in cultured Huh7 cells. J. Virol. 76, 2997–3006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Helbig K. J., Lau D. T., Semendric L., Harley H. A., and Beard M. R. (2005) Analysis of ISG expression in chronic hepatitis C identifies viperin as a potential antiviral effector. Hepatology 42, 702–710 [DOI] [PubMed] [Google Scholar]
- 27.Helbig K. J., Ruszkiewicz A., Semendric L., Harley H. A., McColl S. R., and Beard M. R. (2004) Expression of the CXCR3 ligand I-TAC by hepatocytes in chronic hepatitis C and its correlation with hepatic inflammation. Hepatology 39, 1220–1229 [DOI] [PubMed] [Google Scholar]
- 28.Wakita T., Pietschmann T., Kato T., Date T., Miyamoto M., Zhao Z., Murthy K., Habermann A., Kräusslich H. G., Mizokami M., Bartenschlager R., and Liang T. J. (2005) Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11, 791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eyre N. S., Drummer H. E., and Beard M. R. (2010) The SR-BI partner PDZK1 facilitates hepatitis C virus entry. PLoS Pathog. 6, e1001130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Helbig K. J., Eyre N. S., Yip E., Narayana S., Li K., Fiches G., McCartney E. M., Jangra R. K., Lemon S. M., and Beard M. R. (2011) The antiviral protein viperin inhibits hepatitis C virus replication via interaction with nonstructural protein 5A. Hepatology 54, 1506–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schoggins J. W., Wilson S. J., Panis M., Murphy M. Y., Jones C. T., Bieniasz P., and Rice C. M. (2011) A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao H., Lin W., Kumthip K., Cheng D., Fusco D. N., Hofmann O., Jilg N., Tai A. W., Goto K., Zhang L., Hide W., Jang J. Y., Peng L. F., and Chung R. T. (2012) A functional genomic screen reveals novel host genes that mediate interferon-α's effects against hepatitis C virus. J. Hepatol. 56, 326–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siegrist F., Ebeling M., and Certa U. (2011) The small interferon-induced transmembrane genes and proteins. J. Interferon Cytokine Res. 1, 183–197 [DOI] [PubMed] [Google Scholar]
- 34.Wang S., Wu X., Pan T., Song W., Wang Y., Zhang F., and Yuan Z. (2012) Viperin inhibits hepatitis C virus replication by interfering with binding of NS5A to host protein hVAP-33. J. Gen. Virol. 93, 83–92 [DOI] [PubMed] [Google Scholar]
- 35.Amini-Bavil-Olyaee S., Choi Y. J., Lee J. H., Shi M., Huang I. C., Farzan M., and Jung J. U. (2013) The antiviral effector IFITM3 disrupts intracellular cholesterol homeostasis to block viral entry. Cell Host Microbe 13, 452–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Manna D., Aligo J., Xu C., Park W. S., Koc H., Heo W. D., and Konan K. V. (2010) Endocytic Rab proteins are required for hepatitis C virus replication complex formation. Virology 398, 21–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jia R., Pan Q., Ding S., Rong L., Liu S. L., Geng Y., Qiao W., and Liang C. (2012) The N-terminal region of IFITM3 modulates its antiviral activity by regulating IFITM3 cellular localization. J. Virol. 86, 13697–13707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jia R., Xu F., Qian J., Yao Y., Miao C., Zheng Y. M., Liu S. L., Guo F., Geng Y., Qiao W., and Liang C. (2014) Identification of an endocytic signal essential for the antiviral action of IFITM3. Cell. Microbiol. 16, 1080–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chesarino N. M., McMichael T. M., Hach J. C., and Yount J. S. (2014) Phosphorylation of the antiviral protein IFITM3 dually regulates its endocytosis and ubiquitination. J. Biol. Chem. 289, 11986–11992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blaskovic S., Blanc M., and van der Goot F. G. (2013) What does S-palmitoylation do to membrane proteins? FEBS J. 280, 2766–2774 [DOI] [PubMed] [Google Scholar]
- 41.Yount J. S., Moltedo B., Yang Y. Y., Charron G., Moran T. M., López C. B., and Hang H. C. (2010) Palmitoylome profiling reveals S-palmitoylation-dependent antiviral activity of IFITM3. Nat. Chem. Biol. 6, 610–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yount J. S., Karssemeijer R. A., and Hang H. C. (2012) S-palmitoylation and ubiquitination differentially regulate interferon-induced transmembrane protein 3 (IFITM3)-mediated resistance to influenza virus. J. Biol. Chem. 287, 19631–19641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chutiwitoonchai N., Hiyoshi M., Hiyoshi-Yoshidomi Y., Hashimoto M., Tokunaga K., and Suzu S. (2013) Characteristics of IFITM, the newly identified IFN-inducible anti-HIV-1 family proteins. Microbes Infect. 15, 280–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schulze H., Kolter T., and Sandhoff K. (2009) Principles of lysosomal membrane degradation: cellular topology and biochemistry of lysosomal lipid degradation. Biochim. Biophys. Acta 1793, 674–683 [DOI] [PubMed] [Google Scholar]
- 45.Desai T. M., Marin M., Chin C. R., Savidis G., Brass A. L., and Melikyan G. B. (2014) IFITM3 restricts influenza A virus entry by blocking the formation of fusion pores following virus-endosome hemifusion. PLoS Pathog. 10, e1004048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li K., Markosyan R. M., Zheng Y. M., Golfetto O., Bungart B., Li M., Ding S., He Y., Liang C., Lee J. C., Gratton E., Cohen F. S., and Liu S. L. (2013) IFITM proteins restrict viral membrane hemifusion. PLoS Pathog. 9, e1003124. [DOI] [PMC free article] [PubMed] [Google Scholar]