

Background: Mutations in the triggering receptor on myeloid cells 2 (TREM2) are associated with several neurodegenerative diseases, including Alzheimer disease, but the relevant TREM2 ligand is unknown.
Results: TREM2 binds to apolipoprotein E (ApoE).
Conclusion: TREM2 is a cellular receptor for ApoE.
Significance: Identification of ApoE as a TREM2 ligand helps explain the role of TREM2 role in neurodegenerative disorders.
Keywords: Alzheimer disease, apolipoprotein E (ApoE), genetic polymorphism, high-density lipoprotein (HDL), myeloid cell, neurodegenerative disease, Nasu-Hakola disease, PLOSL, TREM2
Abstract
The triggering receptor expressed on myeloid cells 2 (TREM2) is an Ig-like V-type receptor expressed by populations of myeloid cells in the central nervous system and periphery. Loss-of-function mutations in TREM2 cause a progressive, fatal neurodegenerative disorder called Nasu-Hakola disease. In addition, a TREM2 R47H coding variant was recently identified as a risk factor for late-onset Alzheimer disease. TREM2 binds various polyanionic molecules but no specific protein ligands have been identified. Here we show that TREM2 specifically binds apolipoprotein E, a well established participant in Alzheimer disease. TREM2-Ig fusions efficiently precipitate ApoE from cerebrospinal fluid and serum. TREM2 also binds recombinant ApoE in solution and immobilized ApoE as detected by ELISA. Furthermore, the Alzheimer disease-associated R47H mutation, and other artificial mutations introduced in the same location, markedly reduced the affinity of TREM2 for ApoE. These findings reveal a link between two Alzheimer disease risk factors and may provide important clues to the pathogenesis of Nasu-Hakola disease and other neurodegenerative disorders.
Introduction
Triggering receptor expressed on myeloid cells 2 (TREM2)2 is a type I transmembrane protein with a single, extracellular, Ig-like V-type domain (1, 2). The cytoplasmic tail of TREM2 is short and contains no signaling motifs. Instead, TREM2 forms a signaling complex with TYROBP, a small immunoreceptor tyrosine-based activation motif-containing transmembrane protein. Regulation of TREM2 signaling is complex. TREM2 is cleaved by multiple proteases, including the γ-secretase complex, yielding a soluble ectodomain fragment and a transmembrane fragment that remains associated with TYROBP (3, 4).
Interest in TREM2 stems from its role in several neurodegenerative disorders. TREM2 was first implicated in a disease called polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy, also known as Nasu-Hakola disease (5, 6). Nasu-Hakola disease is a rare, autosomal recessive disorder characterized by the appearance of fatty cysts in the metaphyses of long bones during early adulthood followed by progressive, and ultimately fatal dementia. Homozygous loss-of-function mutations in either TREM2 or TYROBP cause the Nasu-Hakola phenotype underscoring the importance to TREM2 signaling to homeostasis (7, 8). Although less extensively characterized, TREM2 polymorphisms have been subsequently implicated in frontotemporal dementia-like syndromes, Parkinson disease, and amyotrophic lateral sclerosis (9–14).
In 2013, two genome-wide association studies identified the TREM2 SNP rs75932628 as a risk factor for Alzheimer disease (15, 16). The significance of this association was subsequently confirmed in several additional studies and meta-analyses (9, 17–19). rs75932628 causes an R47H missense mutation in the TREM2 ectodomain. Although the impact of this mutation is not completely understood, it appears to result in aberrant glycosylation and trafficking of the protein and impairs its ability to recognize plastic-immobilized lipids (20, 21).
The biological role of TREM2 in neurodegenerative disease remains uncertain, in part due to the conflicting results of mouse studies. Even the question of in vivo localization is contentious. Some investigations have localized TREM2 to cytoplasmic puncta of neurons (15, 22), but most have shown that TREM2 is expressed by myeloid cells of the CNS (22–25). The TREM2+ myeloid cells are usually reported as microglia, but at least one recent study has shown that these cells have a CD46hiLy6C+P2RY12− phenotype typical of infiltrating peripheral macrophages rather than microglia (24).
Clinical studies show a clear protective effect for TREM2 given the variety of neurodegenerative conditions that arise when TREM2 is mutated. Mouse studies, however, are less consistent. In some mouse experiments, TREM2 deficiency is protective against pathology or has minimal effect on disease progression (24, 26). In others TREM2 deficiency results in exacerbation of Alzheimer pathology, or pathology associated with demyelination or ischemic damage (21, 27, 28).
Although TREM2 is known to bind various polyanionic molecules including bacterial antigens, dextran sulfate, nucleic acids, and immobilized phospholipids, the relevance of these findings to neurodegenerative disorders is unclear (21, 28–30). Here we report that TREM2 binds to apolipoprotein E. The identification of ApoE as a TREM2 ligand is interesting due to the association of ApoE genotype with Alzheimer disease. We show that TREM2 recognizes ApoE in several contexts including in cerebrospinal fluid, as a soluble protein, and when ApoE is immobilized to plastic. Identification of ApoE as a TREM2 ligand should open various avenues of investigation into the pathogenesis of several neurodegenerative disorders.
Experimental Procedures
Ig Fusion Constructs
TREM2- and CD4-Ig fusion constructs were generated by cloning the protein ectodomains (without signal peptides) into a human IgG1 fusion construct encoding the signal peptide from CD5. The TIM1-Ig fusion construct was a generous gift from Dr. Hyeryun Choe. HEK293T cells maintained in DMEM with 10% FBS were transfected with Ig fusion plasmids via the calcium phosphate method. 6–8 h post-transfection, cells were washed with PBS and the medium was changed to a serum-free formulation (Freestyle, Life Technologies). Alternatively, 30-ml cultures of Expi293 cells in suspension were transfected by the Expifectamine reagent (Life Technologies). 48–120 h post-transfection, the medium was collected, clarified by centrifugation at 4000 × g for 30 min, and vacuum filtered through a 0.45-μm pore-size, surfactant-free cellulose acetate membrane (Nalgene). Clarified supernatant was passed through a protein A affinity column (GE Healthcare) by means of a peristaltic pump. Columns were washed first with PBS containing 0.1% Tween 20 and then with PBS. Fusion constructs were eluted with a pH 2.8, amine-based buffer (IgG Elution Buffer, Thermo Scientific) directly into a 1/10th volume of 1 m Tris, pH 9.0. The neutralized elution buffer was replaced with PBS by repeated centrifugal filtration through a 3-kDa cutoff filter (Centricon Plus-70, Millipore). Yield was quantified via the Pierce 660 nm Protein Assay Reagent (Life Technologies). Product size and purity were verified by SDS-PAGE and Coomassie staining.
Lipid Arrays
Membranes pre-spotted with various synthetic phospho- and sphingolipids (Membrane Lipid Strips, Echelon Biosciences) were probed with TREM2-Ig or a human IgG1 isotype control antibody following the manufacturer's protocol. Briefly, membranes were hydrated and blocked in a blocking solution of PBS with 3% BSA and 0.1% Tween 20 followed by staining with primary immunoreagents in the same. Following a PBS-T wash, membranes were incubated with a goat anti-human HRP-conjugated secondary antibody (Life Technologies) in blocking buffer and imaged via chemiluminescence (SuperSignal West Femto ECL, Life Technologies) with an ImageQuant LAS4000 Mini ECL reader (GE Healthcare).
Flow Cytometry
Jurkat cells were suspended in growth medium (HEPES-buffered RPMI 1640 with 10% FBS and supplemental penicillin/streptomycin) at a density of 2.5 × 105 cells/ml. To induce apoptosis, an equal volume of growth medium containing 2 μm actinomycin D1 (Life Technologies) was added. 12 h later, cells were resuspended in binding buffer (140 mm NaCl, 2.5 mm CaCl2, 1% BSA, 10 mm HEPES, pH 7.4) and stained with the following primary reagents: buffer only, TREM2-Ig, TIM1-Ig, and human IgG1. After 30 min, cells were washed twice with PBS and stained with the following secondary reagents: Alexa 488-Annexin V (for previously unstained cells, Life Technologies), or Alexa 488-goat anti-human IgG (Life Technologies). After 30 min, cells were washed twice with PBS, resuspended in 1 μg/ml of propidium iodide (Life Technologies) in PBS. Stained cells were analyzed with a BD Accuri C6 flow cytometer (BD Biosciences).
Immunoprecipitation from Cerebrospinal Fluid and Serum
2 μg of Ig-fusion construct or human IgG1 isotype control antibody was pre-bound to 25 μl of protein G magnetic bead slurry (Protein G DynaBeads, Life Technologies). The beads were incubated with 300 μl of cynomolgus macaque cerebrospinal fluid (CSF) or serum (Bioreclamation). Beads were washed 3 times with 500 μl of PBS, resuspended in 100 μl of PBS, and transferred to fresh tubes. PBS was removed and bound Ig fusion eluted with IgG Elution Buffer (Thermo Scientific), which was neutralized with the addition of 1/10th volume of 1 m Tris, pH 9.0.
Immunoprecipitation of Soluble Apolipoproteins
Ig fusion-bound beads were prepared as described above, then blocked in a solution of 5% vegetable peptone (Sigma) in PBS for 20 min with rotation to prevent nonspecific binding of apoplipoproteins to beads. Recombinant apoplipoproteins ApoE and ApoA-I produced in Escherichia coli, and ApoA-II purified from human plasma (Fitzgerald) were bound to beads at a concentration of 1 μg/ml in the presence of peptone block for 15 min followed by 4 washes with PBS and eluted as described above.
SDS-PAGE and Western Blot
Protein samples were prepared in 1× Laemmli sample buffer, denatured at 75 °C for 10 min, and run on 4–20% gradient polyacrylamide Tris glycine gels (Life Technologies). Silver staining was performed using the SilverXpress Kit (Life Technologies). The primary antisera were used for Western blots were: goat polyclonal anti-ApoE, 1:2000 (Merk Millipore), goat polyclonal anti-ApoA-I, 1:1000 (Merk Millipore), and rabbit anti-ApoA-II, 1:1000 (Meridian Life Sciences), and mouse IgG1 anti-TREM2 ectodomain clone B-3, 1:200 (Santa Cruz Biotechnology).
Co-immunoprecipitation of Cell-expressed ApoE and TREM2
HEK293T cells cultured in DMEM with 10% FBS were co-transfected with the indicated plasmids by the calcium phosphate method. 6 h post-transfection, cells were washed with PBS and growth medium was replaced with DMEM, 1% BSA to remove bovine apolipoproteins. 24 h post-transfection, cells were lysed by scraping into 0.2 ml/well of a 1% solution of Triton X-100 in PBS with a protease inhibitor mixture (Roche Applied Science). Lysis was completed by rotation at 4 °C for 30 min followed by clarification of the lysate by centrifugation. Supernatants were incubated with M2 anti-FLAG-conjugated magnetic Sepharose beads (Sigma) for 10 min at room temperature with rotation. Following four washes with PBS-T, samples were eluted with IgG Elution Buffer (Thermo Scientific).
ELISA
Apolipoproteins for Fig. 5B were purchased from Fitzgerald Industries International (ApoE and ApoA-I were produced in E. coli). Mamallian cell culture origin ApoJ was purchased from R&D Systems. Highly purified, human plasma-derived ApoE, ApoA-I, and ApoA-II (Fig. 5, C and D) were obtained from Fitzgerald Industries International. To produce control and ApoE e2, e3, and e4 supernatants, 293T cells in 10-cm dishes were transfected with vector pQCXIP or pQCXIP containing the appropriate cDNAs. 24 h post-transfection, 293T growth medium was exchanged with protein-free Freestyle growth medium. 72 h post-transfection, supernatants were clarified via centrifugation and 0.2-μm filtration.
FIGURE 5.

The TREM2 R47H polymorphism reduces the affinity of TREM2 for ApoE. A, immunoprecipitation (IP) of macaque CSF was performed as in the legend to Fig. 1A with the addition of a TREM2-Ig bait containing the R47H mutation. CSF input (leftmost lane) and precipitates were separated by SDS-PAGE and blotted for the indicated apolipoproteins. The R47H mutation decreases, but does not abolish, the precipitation of ApoE from CSF. This experiment is representative of two. B, wild-type TREM2-Ig, mutant TREM2-Ig constructs, and CD4-Ig were used as primary immunoreagents in ELISAs against recombinant ApoE and ApoA-I (E. coli produced), ApoA-II (from human plasma), and ApoJ (of NS-20 NS0 mouse cell origin). TREM2-Ig (wild-type) bound strongly to ApoE. The R47H mutation severely impaired binding of TREM2-Ig as did similar mutations. Error bars show 95% confidence intervals for triplicate wells. C, the effects R47H and related experimental mutations on TREM2/ApoE interaction were confirmed with ApoE purified from human plasma. In this, and following experiments, the concentration of ApoE used to coat the plate was varied rather than the concentration of Ig fusion. Error bars show 95% confidence intervals of triplicate wells of one of two experiments. D, the specificity of TREM2-Ig for ApoE was confirmed via ELISA with plates coated with apoproteins E, A-I, and A-II all purified from human plasma. The dashed gray line indicates the mean level of CD4-Ig binding to the highest concentration of ApoE on the plate (mean signal from 3 wells). Error bars show 95% confidence intervals for triplicate wells from a representative experiment. E, either ApoE antiserum (AS) or control goat serum (CS) were titered onto immobilized purified plasma-derived ApoE or a BSA control ligand. ApoE antiserum, but not the control serum, blocked TREM2-Ig binding in a concentration-dependent manner. Error bars show upper (AS) or lower (CS) 95% confidence intervals for the TREM2 binding signal. BSA error bars are omitted for clarity. The serum dilution series begins at a 1:50 dilution (corresponding to a relative dilution factor of 1 on the graph). The dotted gray line shows the TREM2/ApoE binding signal in the absence of serum (mean value from 6 wells).
96-Well, half-area Costar Assay Plates (Corning Incorporated) were coated with 25 μl of apolipoproteins or apolipoprotein-containing supernatants diluted 1:1 with PBS-T for 1 h at 37 °C. Wells were washed with PBS and blocked with 5% milk for 1–2 h at room temperature. The primary reagent consisted of 25 μl/well of Ig-fusion construct diluted in 5% milk. Primary incubation was carried out for 1 h at room temperature. Wells were washed with PBS-T prior to 30 min of incubation with a 1:3000 dilution of HRP-conjugated goat anti-human IgG (Life Technologies). Plates were washed and assayed with a 15–60 min room temperature incubation with TMB substrate (1-Step Ultra TMB Elisa Substrate Solution, ThermoFisher) depending on the assay. Reactions were quenched with TMB Stop Solution (Southern Biotech) and optical density measurements were taken at 450 nm.
The ApoE-antiserum blocking ELISA in Fig. 5E was performed with the following modifications. Wells were coated with 25 μl of a 120 nm solution of ApoE in PBS-T. Dilution series of both 0.2 μm filtered goat serum or a polyclonal goat antiserum directed against ApoE (Merck Millipore AB947), ranging from 1:50 to 1:3200, were prepared in 5% milk. After blocking the plate, 75 μl of these diluted sera were added to wells. Following a 1-h incubation, the plate was rinsed three times in 5% milk. Incubation with TREM2-Ig was carried out for only 15, rather than 60 min.
Results
A TREM2-Ig Fusion Construct Binds Lipid in Planar Arrays, but Not Cell Membranes
We initially hypothesized that TREM2 was a cellular receptor for phosphatidylserine (PS) based on reports of its ability to bind apoptotic cells and promote clearance of apoptotic debris (31, 32). To test this hypothesis, we created a TREM2-Ig fusion construct consisting of the ectodomain of human TREM2 fused to the Fc region of human IgG1 and used it to probe commercially prepared arrays of lipids. On these arrays, TREM2-Ig labeled spots of PS, as well as spots of phosphatidic acid and cardiolipin (Fig. 1A). However, TREM2-Ig failed to stain PS on the membranes of apoptotic cells under conditions in which two known PS-binding proteins (Annexin V and TIM-1) bound (Fig. 1B) (33). The failure of TREM2 to bind apoptotic cellular membranes prompted us to question the physiologic relevance of its PS binding in the context of lipid arrays, prompting us to continue searching for a ligand.
FIGURE 1.

TREM2-Ig recognizes PS and other lipids on lipid arrays, but not on the surface of apoptotic cells. A, the TREM2-Ig fusion was used to probe an array spotted with various lipids as indicated in the legend on the right. Dark spots show lipids recognized by TREM2-Ig as detected by chemiluminescence. As shown by the left membrane, TREM2-Ig bound strongly to spots containing phosphatidic acid (PA), PS, and cardiolipin (CL). A human IgG1 isotype control exhibited no appreciable lipid binding. These results were reproducible with at least two different lots of arrays. PA and PS binding were also observed on sphingolipid strips (that do not include CL, not shown). B, Jurkat cells were treated for 12 h with actinomycin D1 (actD1) to induce apoptosis and expose PS on the cell surface. Cells were then stained with PS-binding reagents ANXAV and TIM1-Ig, with TREM2-Ig, or with a human IgG1 isotype control antibody. The membrane-impermeant vital stain propidium iodide (PI) was used to discriminate between dead cells (PI+) and apoptotic or viable cells (PI−). The vertical axis of each plot shows the relative intensity of ANXAV/Ig fusion staining and the horizontal axis shows the intensity of PI staining. Known PS-binding proteins ANXAV and TIM-1 labeled apoptotic cells (ANXAV+, PI−) as indicated in the bottom left plot. TREM2-Ig, however, failed to label apoptotic cells, indicating that it does not bind PS in the context of intact cellular membranes.
TREM2 Precipitates Apolipoproteins from CSF
Because TREM2 is primarily implicated in neurodegenerative disorders, we next searched for a ligand within CSF. We performed immunoprecipitation of cynomolgus macaque cerebrospinal fluid with the TREM2-Ig construct and found that TREM2-Ig, but not a human IgG1 isotype control, precipitated two proteins. SDS-PAGE and silver staining of the precipitate revealed a doublet band of ∼36 kDa and a single band that ran beneath the IgG1 light chain at ∼22 kDa (Fig. 2A). A second gel was stained with Coomassie in parallel (not shown) and the corresponding bands were excised and analyzed by mass spectrometry. Mass spectrometry identified the 36-kDa doublet as apolipoprotein E (ApoE) and the 22-kDa band as apolipoprotein A-I (indicated by bold in Table 1).
FIGURE 2.

TREM2-Ig precipitates ApoE from cerebrospinal fluid. A, TREM2-Ig, a human IgG1 isotype control antibody (hIgG), or unbound protein G beads were used as bait for immunoprecipitation of cynomolgus macaque CSF. Input (CSF), the TREM2-Ig regent by itself, and the precipitated products were separated by reducing SDS-PAGE and visualized by silver staining. TREM2-Ig, but neither hIgG nor beads alone, precipitated a doublet band of ∼36 kDa (white arrowhead), and a single band of ∼22 kDa (black arrowhead). Dashed lines indicate bands corresponding to TREM2-Ig, the 50-kDa IgG heavy chain (HC), and the 25-kDa IgG light chain (LC). B, immunoprecipitation (IP) of macaque CSF was repeated and analyzed by Western blot. The 36-kDa doublet was confirmed as ApoE and the 22-kDa single band as ApoA-I. ApoA-II, although not visible with silver staining, was detected by Western blot in both the CSF and TREM2-Ig precipitate lanes. The band indicated by the white arrowhead is the result of secondary antibody cross-reaction with the hIgG1 light chain. IB, immunoblot.
TABLE 1.
Proteins from the two bands identified in Fig. 1, or a control band derived from the hIgG1 lane were identified by mass spectrometry
Shown are the numbers of peptide spectra from each protein (excluding keratins and other contaminants). Peptides from human IgG and TREM2 were presumably derived from the TREM2-Ig fusion used for immunoprecipitation.
| Protein | Number of spectra |
||
|---|---|---|---|
| 36-kDa band | 22-kDa band | Control band | |
| ApoE | 44 | 0 | 0 |
| ApoA-I | 0 | 18 | 0 |
| IgG heavy chain | 15 | 5 | 0 |
| TREM2 | 4 | 1 | 0 |
| ApoD | 2 | 1 | 0 |
| GAPDH | 0 | 0 | 2 |
The mass spectrometry result was confirmed by Western blot with antisera to ApoE and ApoA-I (Fig. 2B). Although no band was visible on the silver-stained gel at the predicted molecular weight, Western blot also demonstrated the precipitation of small amounts of ApoA-II (Fig. 2B).
TREM2-Ig Precipitates Apolipoproteins from Serum
To determine whether the ability of TREM2 to precipitate apolipoproteins was somehow limited to CSF, we also performed immunoprecipitation of cynomolgus macaque serum. By SDS-PAGE and silver staining, we visualized the same 22-kDa band (ApoA-I) that previously precipitated from CSF (Fig. 3A). Although no bands corresponding to ApoE and ApoA-II were visible on the silver-stained gel, their presence was confirmed by Western blot (Fig. 3B).
FIGURE 3.
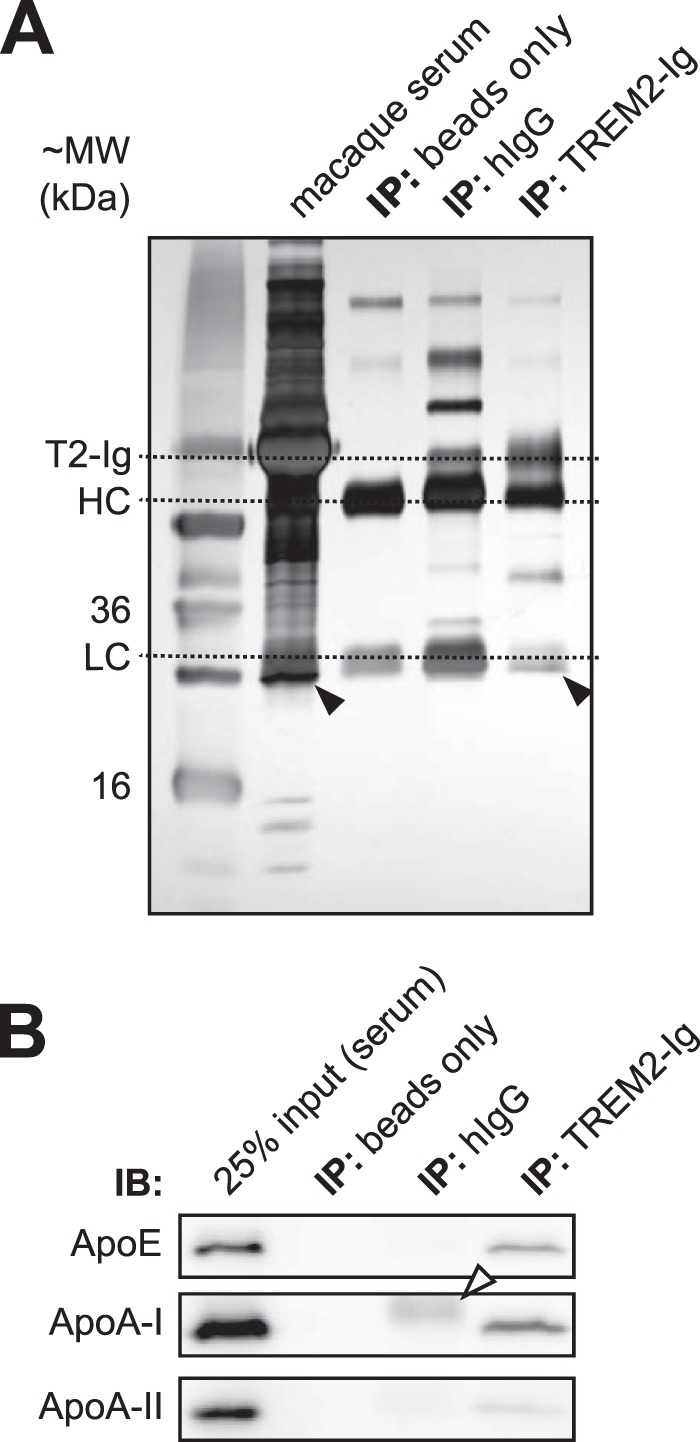
TREM2-Ig precipitates ApoE from serum. A, immunoprecipitation (IP) of cynomolgus macaque serum was performed in an experiment analogous to that shown in Fig. 1. The precipitated proteins are shown next to input (serum) following reducing SDS-PAGE and silver staining. ApoE was not visible on the silver-stained gel but ApoA-I was visible beneath the IgG light chain in both serum and the TREM2-Ig IP lane. B, Western blot confirmed the presence of ApoE and ApoA-I in the TREM2-Ig precipitate. The white arrowhead indicates the hIgG1 light chain band. IB, immunoblot.
TREM2 Binds ApoE Specifically
TREM2-Ig precipitated apolipoproteins E, A-I, and, to a lesser extent, A-II from CSF and serum. These apolipoproteins co-exist on the same lipoprotein particles, however, so these experiments could not distinguish which apolipoprotein, if any, TREM2 bound. Indeed, TREM2 may have been binding some other component of the apolipoprotein particle altogether that was not visible on the silver-stained gel. We therefore tested the ability of TREM2-Ig to precipitate various purified or recombinant apolipoproteins. As shown in Fig. 4A, TREM2-Ig, but neither CD4-Ig nor a human IgG1 isotype control, bound recombinant ApoE. Similarly, ApoE co-immunoprecipitated with a C terminally FLAG-tagged TREM2 when both proteins were co-expressed in HEK293T cells (Fig. 4B). We also measured the affinity of TREM2-Ig for immobilized ApoE of various genotypes. Supernatants of HEK293T cells expressing ApoE2, ApoE3, and ApoE4 were immobilized to plastic and used as ligands in direct ELISA (Fig. 4C). Although TREM2-Ig showed an apparent preference for ApoE4, the same preference was observed when the ELISA was repeated with a polyclonal anti-ApoE antiserum, suggesting that ApoE4 more efficiently coats the plate. Therefore, the TREM2/ApoE interaction may not be heavily influenced by the ApoE genotype.
FIGURE 4.

TREM2-Ig binds ApoE specifically and irrespective of genotype. A, TREM2-Ig or control beads were used to immunoprecipitate (IP) solutions of the indicated purified apolipoproteins. Shown here are the results of SDS-PAGE and Western blots for each of the indicated apolipoproteins. The leftmost lane in each blot contains a sample of the input as a positive control. These results are representative of three independent experiments. B, ApoE co-immunoprecipitates (IP) with C terminally FLAG-tagged TREM2 when both constructs are co-transfected into HEK293T cells. Cells were co-transfected with three different plasmid mixtures as indicated at the top of the figure. The top two blots (Input) show immunostaining of the clarified lysates and the bottom two blots (IP FLAG) show staining of the corresponding immunoprecipitates. The left lane demonstrates the specificity of the ApoE antibody. The middle lane shows that FLAG-tagged TREM2 bound to ApoE, whereas the right lane includes an untagged, rather than FLAG-tagged, TREM2 as a negative control. The faint ApoE band in the rightmost lane of the IP FLAG blot shows the level of background binding of ApoE to the Sepharose support of the anti-FLAG beads. Recovery of ApoE from cell lysates was generally inefficient compared with other experiments so the (IP FLAG) ApoE blot was exposed 5 times longer than the corresponding (Input) ApoE blot. These results are representative of three independent experiments. C, ApoE2, E3, and E4 were expressed in HEK293T cells. Levels of ApoE in culture supernatants were approximately equivalent as shown by SDS-PAGE and Coomassie staining (left). A dilution series of these supernatants were used as immobilized ligands for ELISA. The left graph shows the binding of TREM2-Ig to each of the ApoE variants, whereas the right graph shows the binding of a polyclonal control serum. Error bars show 95% confidence intervals for triplicate wells. Similar results were obtained with a second preparation of ApoE (not shown).
The R47H Mutation Reduces ApoE Binding Affinity
The TREM2 R47H variant allele is linked to an increased risk of late-onset Alzheimer disease (15, 16). Therefore, we tested the effects of this polymorphism on the TREM2/ApoE interaction. A TREM2 R47H-Ig fusion construct still precipitated ApoE and ApoA-I from cerebrospinal fluid but with reduced affinity compared with wild type TREM2-Ig (Fig. 5A). We also tested the ability of TREM2-Ig to bind various apolipoproteins in the context of an ELISA. Consistent with its ability to precipitate recombinant ApoE, TREM2-Ig bound ApoE strongly in the ELISA but exhibited weak affinity toward ApoA-I and ApoA-II. In contrast, TREM2-Ig showed no affinity for ApoJ. Strikingly, the R47H mutation (and similar mutations R47A, R47E, and R46A) effectively abolished binding of TREM2-Ig to plastic-bound apolipoproteins (Fig. 5B). To exclude the possibility that TREM2-Ig was binding a contaminant from the bacterial expression system rather than ApoE, we preformed another ELISA using highly purified, human plasma-derived ApoE as a ligand. As shown in Fig. 5C, wild-type TREM2-Ig, but not the panel of mutants, also bound plasma-derived ApoE with high affinity. In Fig. 5D, we tested the affinity of TREM2-Ig for plasma-derived ApoA-I and A-II in addition to ApoE. Consistent, with the previous experiments, TREM2-Ig showed specific affinity for ApoE. Last, to prove that TREM2-Ig was not binding a plasma-derived contaminant that co-purified with ApoE, for example, lipid material, we demonstrated that pre-blocking the immobilized ApoE with an polyclonal ApoE antibody could effectively abolish TREM2-Ig/ApoE interaction (Fig. 5E).
Discussion
Previous investigations have proposed several different ligands for TREM2. The best characterized include polysaccharides, nucleic acids, and planar arrays of phospholipids, essentially repetitive arrays of charge (21, 28–30). Here we provide the first report of a specific TREM2 protein ligand. TREM2 recognizes ApoE in multiple contexts: in CSF and serum (Figs. 2 and 3), as a soluble protein (Fig. 4), and as an immobilized ELISA ligand (Fig. 5). The identification of ApoE as a TREM2 ligand is especially interesting due to the well established role of the ApoE genotype as a determinant of Alzheimer disease risk (34, 35).
For the assays shown here, the isoforms of commercially sourced human plasma-derived ApoE were unknown. Human TREM2-Ig also bound ApoE in cynomolgus macaque bodily fluids. At the amino acid level, the mature human and cynomolgus proteins are 96% identical. We do not know whether cynomolgus macaques have ApoE isoforms that correspond to human ApoE2 or E3 but the reference cynomolgus ApoE sequence has arginine residues at positions 112 and 158, similar to human ApoE4. In our hands, however, the ApoE genotype did not appear to have a major impact on its affinity for TREM2, at least when ApoE was used as a ligand in ELISA. In a more physiologically relevant context, however, it is possible that the ApoE isoform might impact binding affinity.
Although TREM2 binds ApoE, it bears little resemblance to other apolipoprotein receptors. LDL receptor proteins have large, repetitive ectodomains and their cytoplasmic tails contain NPXY motifs that modulate internalization and signaling (36). In contrast, TREM2 has a single, comparatively small, Ig family ectodomain, and the TREM2 cytoplasmic tail has no known signaling or internalization motifs.
These structural differences imply differences in function. Typical LDL receptors extend far from the cell surface to facilitate capture of lipoprotein particles. Similarly, because they act to internalize these particles, LDL receptor cytosolic domains contain endocytosis motifs. TREM2 exhibits neither of these features, so we speculate that TREM2 acts primarily as a modulator of phagocytic activity and not as an lipoprotein particle receptor.
This model is consistent with our data that show that TREM2 recognizes ApoE not only in its lipidated state but also as a soluble protein, and in its plastic-bound conformation. In vivo, ApoE is present not only as a free protein, but as a constituent of lipoprotein particles, and as an integral component of the senile plaques of Alzheimer disease (35, 37). These different forms probably result in differences in binding and signaling through TREM2. Free ApoE, for example, lacks avidity and therefore the ability to cluster TREM2. Thus free ApoE would not be expected to result in TREM2 signaling or phagocyte activation. Plaque-associated ApoE, however, presents multiple opportunities for TREM2 binding across the plaque surface and the potential for TREM2 clustering and signaling.
A physiologic role for TREM2 as an ApoE sensor in Alzheimer disease makes sense for two reasons: first, ApoE accumulates in senile plaques, and second, TREM2+ cells accumulate around these plaques in vivo (24, 37). We hypothesize that phagocytes in the CNS recognize senile plaques, in part, via a TREM2/ApoE interaction. TREM2/TYROBP signaling activates the phagocytes, prevents their apoptosis, and enhances their ability to clear Aβ (21). Failure of Aβ clearance, however, may lead to increased neuroinflammation from activated cells (24). This model may explain the differing results from mouse studies that have shown both protective and detrimental effects of TREM2 knock-out.
In Nasu-Hakola disease, TREM2 and TYROBP mutations are associated with fatty cysts in the bone marrow and, more rarely, in other peripheral organs in which Aβ is unlikely to be involved. Similarly, the degenerative CNS lesions of Nasu-Hakola disease, frontotemporal dementia, and other TREM2-associated neurodegenerative disorders arise independently of Aβ deposition. How might a TREM2/ApoE interaction contribute to the pathogenesis of these other diseases?
One thread potentially common to the pathogenesis of these diseases is the involvement of lipid. Nasu et al. (5) speculated based on the fatty degenerative changes in the bone marrow and the primarily white matter-associated lesions of the CNS that Nasu-Hakola disease was the result of a failure of lipid metabolism. Similarly, in mouse models of cerebral ischemic damage, multiple sclerosis, and chemically induced demyelination, Trem2 promotes clearance of myelin and other cellular debris following injury (27, 28, 38).
A few investigations have also shown a role for TREM2 in recognizing or promoting clearance of apoptotic bodies or cellular debris (31, 32, 38). Consistent with the results of previous investigations, we found that TREM2 shows some affinity for PS, a lipid exposed on the membranes of apoptotic cells (21, 30). The inability of TREM2 to bind apoptotic cell membranes (Fig. 1B), however, suggests that TREM2 is not primarily a PS receptor. Intriguingly, though, ApoE may provide the link between TREM2 and recognition of apoptotic debris. ApoE associates with apoptotic bodies and promotes their clearance by macrophages (39). Thus, TREM2 may contribute to apoptotic debris clearance not by PS recognition but via ApoE binding. Similarly, ApoE may play a role in binding and targeting other damaged lipids, such as myelin, for degradation.
We propose, as illustrated in Fig. 6, that TREM2 plays a somewhat different role in Alzheimer disease than in other neurodegenerative diseases, based on the context in which it binds ApoE. The TREM2 R47H mutation associated with Alzheimer disease reduces the ability of TREM2+ phagocytes to bind ApoE within senile plaques, thereby decreasing the clearance of Aβ from the brain. Accumulation of toxic Aβ eventually leads to the disease phenotype. Similarly, ApoE may mark deposits of extracellular lipid for degradation. In the absence of TREM2/TYROBP signaling, as in Nasu-Hakola disease, phagocytic clearance of lipid debris is impaired resulting in the accumulation of fatty cysts in the periphery, and CNS lesions in the lipid-rich white matter of the brain.
FIGURE 6.

A model for TREM2/ApoE interaction in neurodegenerative disease. We speculate that, under normal conditions, TREM2 recognizes ApoE in at least two contexts: in association with Aβ and in association with lipid debris. TREM2 promotes clearance of these injurious substances by activating phagocytes and possibly modulating the inflammatory response. Hypomorphic mutations lead to mild impairment of ApoE recognition that may ultimately pre-dispose to Aβ accumulation and Alzheimer disease late in life. Complete loss of TREM2 function impairs phagocytic clearance of lipid debris (such as myelin) resulting in the early onset leukoencephalopathy and fatty, cystic degenerative changes in peripheral organs that constitute Nasu-Hakola disease.
In summary, we have identified apolipoprotein E as a TREM2 ligand. TREM2 recognizes ApoE in various contexts, including both lipidated and non-lipidated forms. The precise biological function of TREM2 remains to be determined but our data suggest a role in recognition of senile plaques and extracellular lipid via interaction with ApoE.
Author Contributions
C. C. B. designed experiments, performed experiments, interpreted data, and wrote the manuscript. L. B. D. performed experiments and analyzed data. M. F. designed experiments, interpreted data, and approved the final version of the manuscript.
Acknowledgment
We thank the Scripps Florida Mass Spectrometry and Proteomics core facility for their assistance with mass spectrometry and data interpretation.
Note Added in Proof
The wrong scatter plot was used to show hIgG1 staining of Jurkat cells not treated with actinomycin D1 in Fig. 1 in the version of this article that was published on September 15, 2015 as a Paper in Press. This error has now been corrected. This correction does not affect the interpretation of the results or the conclusions.
This work was supported by startup funds provided by the Scripps Research Institute Florida. The authors declare that they have no conflicts of interest with the contents of this article.
- TREM2
- triggering receptor expressed on myeloid cells 2
- TYROBP
- TYRO protein kinase-binding protein
- CD4
- cluster of differentiation 4
- TIM1
- T cell immunoglobulin and mucin domain 1
- CSF
- cerebrospinal fluid
- PS
- phosphatidylserine.
References
- 1.Bouchon A., Dietrich J., and Colonna M. (2000) Cutting edge: inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J. Immunol. 164, 4991–4995 [DOI] [PubMed] [Google Scholar]
- 2.Daws M. R., Lanier L. L., Seaman W. E., and Ryan J. C. (2001) Cloning and characterization of a novel mouse myeloid DAP12-associated receptor family. Eur. J. Immunol. 31, 783–791 [DOI] [PubMed] [Google Scholar]
- 3.Wunderlich P., Glebov K., Kemmerling N., Tien N. T., Neumann H., and Walter J. (2013) Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) protein by ectodomain shedding and γ-secretase-dependent intramembranous cleavage. J. Biol. Chem. 288, 33027–33036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhong L., Chen X.-F., Zhang Z.-L., Wang Z., Shi X.-Z., Xu K., Zhang Y.-W., Xu H., and Bu G. (2015) DAP12 Stabilizes the C-terminal fragment of the triggering receptor expressed on myeloid cells-2 (TREM2) and protects against LPS-induced pro-inflammatory response. J. Biol. Chem. 290, 15866–15877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nasu T., Tsukahara Y., and Terayama K. (1973) A lipid metabolic disease-“membranous lipodystrophy”-an autopsy case demonstrating numerous peculiar membrane-structures composed of compound lipid in bone and bone marrow and various adipose tissues. Acta Pathol. Jpn. 23, 539–558 [DOI] [PubMed] [Google Scholar]
- 6.Hakola H. P. (1972) Neuropsychiatric and genetic aspects of a new hereditary disease characterized by progressive dementia and lipomembranous polycystic osteodysplasia. Acta Psychiatr. Scand. Suppl. 232, 1–173 [PubMed] [Google Scholar]
- 7.Paloneva J., Kestilä M., Wu J., Salminen A., Böhling T., Ruotsalainen V., Hakola P., Bakker A. B., Phillips J. H., Pekkarinen P., Lanier L. L., Timonen T., and Peltonen L. (2000) Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat. Genet. 25, 357–361 [DOI] [PubMed] [Google Scholar]
- 8.Paloneva J., Manninen T., Christman G., Hovanes K., Mandelin J., Adolfsson R., Bianchin M., Bird T., Miranda R., Salmaggi A., Tranebjaerg L., Konttinen Y., and Peltonen L. (2002) Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am. J. Hum. Genet. 71, 656–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cuyvers E., Bettens K., Philtjens S., Van Langenhove T., Gijselinck I., van der Zee J., Engelborghs S., Vandenbulcke M., Van Dongen J., Geerts N., Maes G., Mattheijssens M., Peeters K., Cras P., Vandenberghe R., De Deyn P. P., Van Broeckhoven C., Cruts M., Sleegers K., and BELNEU Consortium (2014) Investigating the role of rare heterozygous TREM2 variants in Alzheimer's disease and frontotemporal dementia. Neurobiol. Aging 35, 726.e11–19 [DOI] [PubMed] [Google Scholar]
- 10.Borroni B., Ferrari F., Galimberti D., Nacmias B., Barone C., Bagnoli S., Fenoglio C., Piaceri I., Archetti S., Bonvicini C., Gennarelli M., Turla M., Scarpini E., Sorbi S., and Padovani A. (2014) Heterozygous TREM2 mutations in frontotemporal dementia. Neurobiol. Aging 35, 934.e7–10 [DOI] [PubMed] [Google Scholar]
- 11.Chouery E., Delague V., Bergougnoux A., Koussa S., Serre J.-L., and Mégarbané A. (2008) Mutations in TREM2 lead to pure early-onset dementia without bone cysts. Hum. Mutat. 29, E194–204 [DOI] [PubMed] [Google Scholar]
- 12.Guerreiro R. J., Lohmann E., Brás J. M., Gibbs J. R., Rohrer J. D., Gurunlian N., Dursun B., Bilgic B., Hanagasi H., Gurvit H., Emre M., Singleton A., and Hardy J. (2013) Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia-like syndrome without bone involvement. JAMA Neurol. 70, 78–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rayaprolu S., Mullen B., Baker M., Lynch T., Finger E., Seeley W. W., Hatanpaa K. J., Lomen-Hoerth C., Kertesz A., Bigio E. H., Lippa C., Josephs K. A., Knopman D. S., White C. L. 3rd, Caselli R., Mackenzie I. R., Miller B. L., Boczarska-Jedynak M., Opala G., Krygowska-Wajs A., Barcikowska M., Younkin S. G., Petersen R. C., Ertekin-Taner N., Uitti R. J., Meschia J. F., Boylan K. B., Boeve B. F., Graff-Radford N. R., Wszolek Z. K., Dickson D. W., Rademakers R., and Ross O. A. (2013) TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson's disease. Mol. Neurodegener. 8, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cady J., Koval E. D., Benitez B. A., Zaidman C., Jockel-Balsarotti J., Allred P., Baloh R. H., Ravits J., Simpson E., Appel S. H., Pestronk A., Goate A. M., Miller T. M., Cruchaga C., and Harms M. B. (2014) TREM2 variant p.R47H as a risk factor for sporadic amyotrophic lateral sclerosis. JAMA Neurol. 71, 449–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guerreiro R., Wojtas A., Bras J., Carrasquillo M., Rogaeva E., Majounie E., Cruchaga C., Sassi C., Kauwe J. S., Younkin S., Hazrati L., Collinge J., Pocock J., Lashley T., Williams J., Lambert J. C., Amouyel P., Goate A., Rademakers R., Morgan K., Powell J., St George-Hyslop P., Singleton A., Hardy J., and Alzheimer Genetic Analysis Group (2013) TREM2 variants in Alzheimer's disease. N. Engl. J. Med. 368, 117–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jonsson T., Stefansson H., Steinberg S., Jonsdottir I., Jonsson P. V., Snaedal J., Bjornsson S., Huttenlocher J., Levey A. I., Lah J. J., Rujescu D., Hampel H., Giegling I., Andreassen O. A., Engedal K., Ulstein I., Djurovic S., Ibrahim-Verbaas C., Hofman A., Ikram M. A., van Duijn C. M., Thorsteinsdottir U., Kong A., and Stefansson K. (2013) Variant of TREM2 associated with the risk of Alzheimer's disease. N. Engl. J. Med. 368, 107–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin S. C., Benitez B. A., Karch C. M., Cooper B., Skorupa T., Carrell D., Norton J. B., Hsu S., Harari O., Cai Y., Bertelsen S., Goate A. M., and Cruchaga C. (2014) Coding variants in TREM2 increase risk for Alzheimer's disease. Hum. Mol. Genet. 23, 5838–5846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosenthal S. L., Bamne M. N., Wang X., Berman S., Snitz B. E., Klunk W. E., Sweet R. A., Demirci F. Y., Lopez O. L., and Kamboh M. I. (2015) More evidence for association of a rare TREM2 mutation (R47H) with Alzheimer's disease risk. Neurobiol. Aging 36, 2443.e21–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lill C. M., Rengmark A., Pihlstrøm L., Fogh I., Shatunov A., Sleiman P. M., Wang L.-S., Liu T., Lassen C. F., Meissner E., Alexopoulos P., Calvo A., Chio A., Dizdar N., Faltraco F., Forsgren L., Kirchheiner J., Kurz A., Larsen J. P., Liebsch M., Linder J., Morrison K. E., Nissbrandt H., Otto M., Pahnke J., Partch A., Restagno G., Rujescu D., Schnack C., Shaw C. E., Shaw P. J., Tumani H., Tysnes O.-B., Valladares O., Silani V., van den Berg L. H., van Rheenen W., Veldink J. H., Lindenberger U., Steinhagen-Thiessen E., SLAGEN Consortium, Teipel S., Perneczky R., Hakonarson H., Hampel H., von Arnim C. A. F., Olsen J. H., Van Deerlin V. M., Al-Chalabi A., Toft M., Ritz B., and Bertram L. (April 30, 2015) The role of TREM2 R47H as a risk factor for Alzheimer's disease, frontotemporal lobar degeneration, amyotrophic lateral sclerosis, and Parkinson's disease. Alzheimers Dement. 10.1016/j.jalz.2014.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park J.-S., Ji I. J., An H. J., Kang M.-J., Kang S.-W., Kim D.-H., and Yoon S.-Y. (2015) Disease-associated mutations of TREM2 alter the processing of N-linked oligosaccharides in the Golgi apparatus. Traffic 16, 510–518 [DOI] [PubMed] [Google Scholar]
- 21.Wang Y., Cella M., Mallinson K., Ulrich J. D., Young K. L., Robinette M. L., Gilfillan S., Krishnan G. M., Sudhakar S., Zinselmeyer B. H., Holtzman D. M., Cirrito J. R., and Colonna M. (2015) TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell 160, 1061–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sessa G., Podini P., Mariani M., Meroni A., Spreafico R., Sinigaglia F., Colonna M., Panina P., and Meldolesi J. (2004) Distribution and signaling of TREM2/DAP12, the receptor system mutated in human polycystic lipomembraneous osteodysplasia with sclerosing leukoencephalopathy dementia. Eur. J. Neurosci. 20, 2617–2628 [DOI] [PubMed] [Google Scholar]
- 23.Jiang T., Tan L., Zhu X.-C., Zhang Q.-Q., Cao L., Tan M.-S., Gu L.-Z., Wang H.-F., Ding Z.-Z., Zhang Y.-D., and Yu J.-T. (2014) Upregulation of TREM2 ameliorates neuropathology and rescues spatial cognitive impairment in a transgenic mouse model of Alzheimer's disease. Neuropsychopharmacology 39, 2949–2962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jay T. R., Miller C. M., Cheng P. J., Graham L. C., Bemiller S., Broihier M. L., Xu G., Margevicius D., Karlo J. C., Sousa G. L., Cotleur A. C., Butovsky O., Bekris L., Staugaitis S. M., Leverenz J. B., Pimplikar S. W., Landreth G. E., Howell G. R., Ransohoff R. M., and Lamb B. T. (2015) TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer's disease mouse models. J. Exp. Med. 212, 287–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Savage J. C., Jay T., Goduni E., Quigley C., Mariani M. M., Malm T., Ransohoff R. M., Lamb B. T., and Landreth G. E. (2015) Nuclear receptors license phagocytosis by trem2+ myeloid cells in mouse models of Alzheimer's disease. J. Neurosci. 35, 6532–6543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ulrich J. D., Finn M. B., Wang Y., Shen A., Mahan T. E., Jiang H., Stewart F. R., Piccio L., Colonna M., and Holtzman D. M. (2014) Altered microglial response to Aβ plaques in APPPS1–21 mice heterozygous for TREM2. Mol. Neurodegener. 9, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poliani P. L., Wang Y., Fontana E., Robinette M. L., Yamanishi Y., Gilfillan S., and Colonna M. (2015) TREM2 sustains microglial expansion during aging and response to demyelination. J. Clin. Invest. 125, 2161–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawabori M., Kacimi R., Kauppinen T., Calosing C., Kim J. Y., Hsieh C. L., Nakamura M. C., and Yenari M. A. (2015) Triggering receptor expressed on myeloid cells 2 (TREM2) deficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J. Neurosci. 35, 3384–3396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Daws M. R., Sullam P. M., Niemi E. C., Chen T. T., Tchao N. K., and Seaman W. E. (2003) Pattern recognition by TREM-2: binding of anionic ligands. J. Immunol. 171, 594–599 [DOI] [PubMed] [Google Scholar]
- 30.Cannon J. P., O'Driscoll M., and Litman G. W. (2012) Specific lipid recognition is a general feature of CD300 and TREM molecules. Immunogenetics 64, 39–47 [DOI] [PubMed] [Google Scholar]
- 31.Takahashi K., Rochford C. D., and Neumann H. (2005) Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 201, 647–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsieh C. L., Koike M., Spusta S. C., Niemi E. C., Yenari M., Nakamura M. C., and Seaman W. E. (2009) A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J. Neurochem. 109, 1144–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kobayashi N., Karisola P., Peña-Cruz V., Dorfman D. M., Jinushi M., Umetsu S. E., Butte M. J., Nagumo H., Chernova I., Zhu B., Sharpe A. H., Ito S., Dranoff G., Kaplan G. G., Casasnovas J. M., Umetsu D. T., Dekruyff R. H., and Freeman G. J. (2007) TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity 27, 927–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corder E. H., Saunders A. M., Strittmatter W. J., Schmechel D. E., Gaskell P. C., Small G. W., Roses A. D., Haines J. L., and Pericak-Vance M. A. (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261, 921–923 [DOI] [PubMed] [Google Scholar]
- 35.Strittmatter W. J., Saunders A. M., Schmechel D., Pericak-Vance M., Enghild J., Salvesen G. S., and Roses A. D. (1993) Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 90, 1977–1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lane-Donovan C., Philips G. T., and Herz J. (2014) More than cholesterol transporters: lipoprotein receptors in CNS function and neurodegeneration. Neuron 83, 771–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Namba Y., Tomonaga M., Kawasaki H., Otomo E., and Ikeda K. (1991) Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer's disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 541, 163–166 [DOI] [PubMed] [Google Scholar]
- 38.Takahashi K., Prinz M., Stagi M., Chechneva O., and Neumann H. (2007) TREM2-transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. PLoS Med. 4, e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grainger D. J., Reckless J., and McKilligin E. (2004) Apolipoprotein E modulates clearance of apoptotic bodies in vitro and in vivo, resulting in a systemic proinflammatory state in apolipoprotein E-deficient mice. J. Immunol. 173, 6366–6375 [DOI] [PubMed] [Google Scholar]