

Background: TREM2 is associated with several neurodegenerative diseases.
Results: ApoE bound to TREM2 and increased phagocytosis of apoptotic neurons by microglia. Alzheimer disease (AD) risk-associated TREM2-R47H mutant had a reduced binding to apoE.
Conclusion: ApoE is a novel ligand for TREM2. Interaction between apoE and TREM2 likely regulates phagocytosis of apoE-bound apoptotic neurons.
Significance: Interaction between two AD risk-associated proteins modulates microglial function.
Keywords: Alzheimer disease, apolipoprotein, apolipoprotein E (apoE), microglia, neuroinflammation, TREM2
Abstract
Several heterozygous missense mutations in the triggering receptor expressed on myeloid cells 2 (TREM2) have recently been linked to risk for a number of neurological disorders including Alzheimer disease (AD), Parkinson disease, and frontotemporal dementia. These discoveries have re-ignited interest in the role of neuroinflammation in the pathogenesis of neurodegenerative diseases. TREM2 is highly expressed in microglia, the resident immune cells of the central nervous system. Along with its adaptor protein, DAP12, TREM2 regulates inflammatory cytokine release and phagocytosis of apoptotic neurons. Here, we report apolipoprotein E (apoE) as a novel ligand for TREM2. Using a biochemical assay, we demonstrated high-affinity binding of apoE to human TREM2. The functional significance of this binding was highlighted by increased phagocytosis of apoE-bound apoptotic N2a cells by primary microglia in a manner that depends on TREM2 expression. Moreover, when the AD-associated TREM2-R47H mutant was used in biochemical assays, apoE binding was vastly reduced. Our data demonstrate that apoE-TREM2 interaction in microglia plays critical roles in modulating phagocytosis of apoE-bound apoptotic neurons and establish a critical link between two proteins whose genes are strongly linked to the risk for AD.
Introduction
Alzheimer disease (AD)2 is the most common form of dementia in the elderly with the overwhelming majority of cases being classified as late onset (LOAD; >65 years). A pathological hallmark of AD is the accumulation of toxic amyloid-β (Aβ) in the central nervous system (CNS). Although AD patients do not normally develop clinical symptoms until later in life, key disease-initiating events likely begin decades before deficits in cognitive functions are observed (1). During homeostasis, amyloid precursor protein is cleaved by secretases yielding Aβ peptides that are continuously removed from the brain parenchyma (2). However, mounting evidence indicates that under pathogenic conditions, an imbalance between Aβ production and clearance leads to Aβ accumulation and subsequent formation of toxic Aβ aggregates including oligomers and amyloid plaques (3).
Recently, heterozygous rare mutations in the triggering receptor expressed on myeloid cells 2 (TREM2) have been linked to increased risk of AD, Parkinson disease, frontotemporal dementia, amyotrophic lateral sclerosis, and essential tremor (4–19). Importantly, in regard to AD, two research groups independently discovered that a TREM2 SNP, rs75932628-T encoding the R47H variant, conferred a significantly increased risk of LOAD with odds ratios of 5.05 (14) and 2.92 (15), which are comparable with that of a well established AD risk gene APOE4 (20). Following this seminal finding, a number of other studies have confirmed the association of TREM2-R47H with LOAD (7, 11, 16–19).
TREM2 is a type I transmembrane protein and a member of the immunoglobulin (Ig) receptor superfamily. It contains an ectodomain, a transmembrane domain, and a short cytoplasmic tail. Signal transduction is mediated through its adaptor protein, DNAX-activating protein of 12 kDa (DAP12) (21), which associates with TREM2 via electrostatic interaction within the transmembrane domains. The cytoplasmic domain of DAP12 contains a single immunoreceptor tyrosine-based activation motif. TREM2-mediated signaling occurs through phosphorylation of tyrosine residues within the immunoreceptor tyrosine-based activation motif of DAP12 by Src kinases (22). This, in turn, recruits Syk via Src homology domain 2 and subsequent activation of downstream targets. Although the exact signaling mechanisms are unknown, studies utilizing TREM2-deficint mice and cells have shown that TREM2 is able to modulate key aspects of cellular homeostasis by suppressing inflammatory cytokine production (23–27) and facilitating phagocytosis of apoptotic cells (24, 28, 29). Within the periphery, TREM2 is found on the surface of osteoclasts, immature dendritic cells, and macrophages. In the CNS, TREM2 is primarily expressed in microglia, the resident immune cells of the CNS (30, 31).
The apolipoprotein E (APOE) gene is the strongest genetic risk factor for LOAD (32, 33). Apolipoproteins are a class of proteins that bind to and transport cholesterol and other lipids. Notably, apoE is a major apolipoprotein in the CNS expressed primarily by astrocytes, vascular cells, and to a lesser extent in microglia and in stressed neurons (34). Three isoforms, apoE2, apoE3, and apoE4, are encoded by APOE ϵ2, ϵ3, and ϵ4 alleles, respectively. APOE4/4 individuals are 10–30 times more likely to develop AD than APOE3/3 individuals. Conversely, the APOE2 allele is protective against LOAD (35). Several pathways have been proposed to explain the risk associated with APOE4 (33, 36). Importantly, apoE has been shown to modulate Aβ clearance and aggregation in cellular and mouse models (33, 37–40).
It has previously been reported that TREM2 is capable of binding microbial and damage-associated molecular signatures found on bacteria (41, 42), lipids exposed during axonal injury (23, 43), and nucleic acid released from dying cells (29). Here, we report apoE as a novel TREM2 ligand. Using a biochemical approach, we verified high-affinity binding of apoE to human TREM2, with a dissociation constant (Kd) in the low nanomolar range. The biological importance of this association was demonstrated using a microglial phagocytosis assay where increased uptake of apoE-coated apoptotic neurons was observed in a TREM2-mediated manner. Importantly, apoE exhibited significantly reduced binding affinity to the AD-associated TREM2-R47H variant. Our data suggest a novel pathway in which two AD risk genes are able to interact and modulate microglial function that may contribute to the initiation and propagation of the inflammatory process in AD.
Experimental Procedures
Materials
Recombinant hIgG-Fc and TREM1-Fc chimeric proteins and mouse anti-Fc antibody were purchased from R&D Systems. Recombinant apoE and apoA1 proteins were from Fitzgerald Industries. Goat anti-apoE biotin antibody was purchased from Meridian Life Science. Mouse anti-β-actin antibody was from Sigma.
Dot Blot
Recombinant or purified proteins diluted in PBS were spotted onto a nitrocellulose membrane using a dot blot manifold apparatus (GE Healthcare). Membrane strips were blocked with 1% Block Ace (Bio-Rad) in PBS, and incubated with the indicated proteins overnight at 4 °C. Bound proteins were detected with biotin-conjugated primary and IRDye®-conjugated streptavidin. Blots were imaged and quantified using Odyssey Infrared Imaging System (LI-COR Biosciences). For determination of binding affinity, the integrated infrared signal (K Count) of each dot was analyzed using Prism (GraphPad).
Solid Phase Binding Assay
A 96-well plate was coated with 40 nm recombinant apoE in PBS overnight at 4 °C. After washing and blocking with 4% BSA in PBS for 1 h at 37 °C, 12.5 or 25 nm recombinant TREM2-Fc or TREM2-R47H-Fc diluted in PBS containing 0.5% BSA were added and incubated for 20 min at 37 °C. After washing, the bound Fc proteins were detected with biotinylated anti-Fc antibody for 1 h at 37 °C. Plates were washed and then incubated with avidin-HRP for 30 min at 37 °C, washed again, and developed with TMB substrate solution (Sigma), and read at 650 nm.
Generation of Trem2 Knock-out (Trem2−/−) Mice
All animal procedures were approved by the Mayo Clinic Institutional Animal Care and Use Committee (IACUC) and were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. TREM2 knock-out mice (Trem2-KO on C57BL/6N background) and their control C57BL/6N mice were obtained from the University of California Davis Knock-out Mouse Project (KOMP) repository. These Trem2-KO mice were originally generated by Velocigene as a “definitive null” and have a LacZ reporter cassette that replaces the entire coding region of the Trem2 locus and is identical to the line recently reported (44).
Mouse Primary Microglial Culture
Mice at postnatal days 1 to ∼3 were used to prepare mixed glial cultures according to a previously published protocol (45). Briefly, mixed glial cells were plated onto polylysine-coated culture flasks in DMEM containing 10% fetal bovine serum (FBS), and medium was changed the next day to a medium containing 25 ng/ml of GM-CSF and 10% FBS. Primary microglia cells were harvested by shaking after 10–12 days in culture and once a week thereafter (up to three times total).
Purification of ApoE from Culture Medium
HEK293 cells were stably transfected with human apoE3 cDNA using FuGENE 6 transfection reagent (Roche) and Zeocin (300 μg/ml) as a selection reagent. Immortalized mouse astrocytes derived from apoE-targeted replacement mice expressing human apoE2, E3, and E4 were cultured as described previously (46). Culture medium was conditioned with serum-free medium for 36–48 h. Conditioned medium was concentrated using a Amicon centrifugal filter unit (Millipore), and run through a HiTrap heparin column on an AKTA FPLC system (GE Healthcare). Heparin-bound apoE was eluted with NaCl gradient from 0 to 1 m in Tris buffer. Peak fractions containing pure apoE were concentrated, and quantified by measuring band intensity of silver-stained SDS-PAGE gel against BSA standards or by apoE ELISA (47).
Phagocytosis of Apoptotic N2a
Phagocytosis of apoptotic N2a by primary microglia was carried out as described by Hsieh et al. (28). Briefly, apoptosis of CM-DiI (Life Technologies) labeled mouse neuroblastoma N2a cells was induced with 0.5 μm staurosporine for 16 h. Apoptotic N2a cells were washed with PBS twice, and fed to primary microglia at a ratio of 1:3 microglia to N2a with or without 10 nm HEK-produced apoE. After incubating the cells for 2 h at 37 °C, cells were harvested, and microglia were labeled with anti-CD11b-APC antibody (eBioscience). For flow cytometry analysis, a total of 50,000 cells were counted with a FACS Accuri (BD Biosciences), and analyzed using CFlow software.
ApoE Binding to Apoptotic N2a
Control or apoptotic N2a were incubated with 10 nm HEK-produced apoE in serum-free medium for 1 h at 4 °C. After washing with Dulbecco's PBS twice, cells were lysed in TBS containing 1% Triton X-100 and 1× protease inhibitor mixture (Roche). Cell lysates were then analyzed by Western blotting.
Purification of TREM2-Fc and TREM2-R47H-Fc
The extracellular region (amino acids 1–174) of TREM2 and TREM2-R47H mutant were cloned into pFUSE-hIgG-Fc (Invivogen) with EcoRI and XhoI. HEK293 cells were transfected with TREM2-Fc and TREM2-R47H Fc along with a vector control using Lipofectamine® 2000 (Life Technologies). Medium was changed to serum-free DMEM 24 h after transfection and conditioned for 36 h. Fc chimeric proteins were purified with protein A-agarose beads (Pierce), eluted with acidic glycine buffer, and neutralized with Tris-HCl. Purified proteins were quantified with a BCA protein assay kit (Pierce) and the purity was determined on silver-stained SDS-PAGE gels.
Results
ApoE Specifically Binds to TREM2 with High Affinity
To examine if apoE binds to TREM2, a recombinant chimeric protein of human TREM2 extracellular domain fused with the Fc region of human IgG (TREM2-Fc) was used in dot blot binding assays. Recombinant human IgG Fc (hIgG-Fc) and another human TREM family member, TREM1 extracellular domain fused with Fc (TREM1-Fc), were used as controls. Nitrocellulose membrane strips were spotted with 200 ng each of the chimeric proteins and incubated with recombinant or immortalized astrocyte-secreted apoE2, E3, or E4 (10 nm) at 4 °C overnight. Membrane-bound apoE was then detected with biotinylated anti-apoE antibody. Significantly, we found that all three isoforms of apoE bound to TREM2-Fc, but not to the Fc region alone or TREM1-Fc (Fig. 1A). The amount of apoE bound to TREM2 was then quantified by normalizing the signal intensities of the TREM2-apoE blot and the signal intensities of the loading control blot in which apoE was spotted onto a membrane and detected by an anti-apoE antibody. We found that the binding of different isoforms of apoE to TREM2-Fc was comparable among each other, and that the lipidation status of apoE did not affect its binding to TREM2-Fc (Fig. 1B). Furthermore, because the ϵ3 allele represents ∼78% of the worldwide APOE genetic frequency (48), we quantitatively measured the affinity of the apoE3-TREM2 interaction. Membrane strips spotted with hIgG-Fc, TREM1-Fc, and TREM2-Fc were incubated with increasing concentrations (0 to 80 nm) of recombinant apoE3. Using these data, we constructed a saturation binding curve and extrapolated an apoE3-TREM2-Fc Kd of 6.7 nm (Fig. 1C).
FIGURE 1.
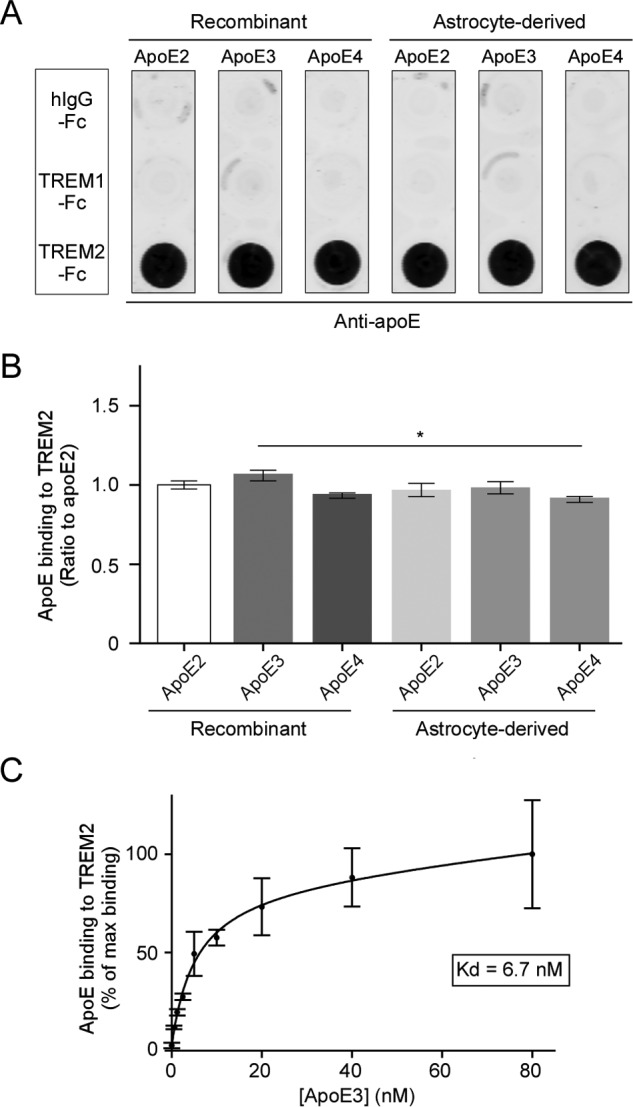
ApoE specifically binds to TREM2 with high affinity. A, representative dot blot of apoE binding to TREM2. A nitrocellulose membrane was spotted with 200 ng each of recombinant human IgG Fc region (hIgG-Fc), recombinant human TREM1, or TREM2 extracellular region conjugated to Fc (TREM1-Fc and TREM2-Fc). Membrane strips were then incubated with 10 nm recombinant or immortalized astrocyte-derived apoE2, -E3, or -E4. Bound apoE was detected using biotinylated anti-apoE antibody. B, quantification of three independent dot blots. Data are plotted as mean ± S.E. (n = 3, one-way analysis of variance with Tukey post hoc analysis, *, p < 0.05). C, saturation binding curve and dissociation constant (Kd) of apoE3 binding to TREM2-Fc. Membrane strips spotted with hIgG-Fc, TREM1-Fc, or TREM2-Fc were incubated with increasing concentrations of recombinant apoE3, and bound apoE was detected using appropriate antibodies. The curve fit and Kd were derived using GraphPad Prism nonlinear fit for one site total binding. Data are plotted as mean ± S.E. (n = 3).
ApoA1 and ApoB Also Bind to TREM2
To evaluate if other apolipoproteins, in addition to apoE, are able to bind to TREM2, we tested apolipoprotein family members apoA1 and apoB in a dot blot assay. Membrane strips were incubated with hIgG-Fc, TREM1-Fc, or TREM2-Fc (80 nm), and the Fc-chimeric proteins bound to mIgG, apoA1, or apoB were detected with biotinylated anti-Fc antibody. Our dot blot results demonstrated that both apoA1 and apoB bound to TREM2-Fc, but not to hIgG-Fc or TREM1-Fc (Fig. 2A). Classically, endogenous apoE binds to the low-density lipoprotein receptor-related protein 1 (LRP1), a major neuronal receptor for apoE (49). Therefore, we used other LRP1 ligands, tissue-type plasminogen activator (tPA), and receptor-associated protein (RAP), to investigate whether TREM2 was able to interact with other LRP1 ligands. Membrane strips were spotted with apoE3, tPA, or RAP and incubated with hIgG-Fc, TREM1-Fc, or TREM2-Fc, and detected with biotinylated anti-Fc antibody. Dot blot analysis revealed that TREM2 binds to apoE, but not to tPA or RAP (Fig. 2B). Because apoE and apoA1 are two of the apolipoprotein subtypes found in the CNS, and both constitute the protein component of high-density lipoprotein (HDL), we next examined whether apoE and apoA1 compete with one another for TREM2 binding. Membrane strips spotted with hIgG-Fc, TREM1-Fc, or TREM2-Fc were incubated with a constant apoE3 concentration (10 nm) and increasing concentrations of apoA1, and the amount of apoE bound to TREM2-Fc was quantified. Strikingly, the presence of apoA1 dose-dependently reduced apoE binding to TREM2-Fc, suggesting that these two apolipoproteins share a similar binding mechanism to TREM2 (Fig. 2C).
FIGURE 2.

ApoA1 and apoB also bind to TREM2. A, a representative dot blot of TREM2 binding to apoA1 and apoB. Membrane strips spotted with 200 ng each of normal mouse IgG (mIgG), apoA1, or apoB were incubated with 80 nm hIgG-Fc, TREM1-Fc, or TREM2-Fc. Bound Fc protein was detected with biotinylated anti-Fc antibody. B, a representative dot blot showing binding specificity of TREM2 to apoE and not to two other LRP1 ligands, tPA or RAP. Membrane strips spotted with 200 ng each of mIgG, apoE3, tPA, or RAP were incubated with 80 nm hIgG-Fc, TREM1-Fc, or TREM2-Fc, and bound Fc protein was detected with biotinylated anti-Fc antibody. C, apoA1 competes with apoE for binding to TREM2. Membrane strips spotted with hIgG-Fc, TREM1-Fc, or TREM2-Fc were incubated with 10 nm apoE3 along with increasing concentrations of apoA1, and bound apoE was detected and quantified using biotinylated anti-apoE antibody. Data are plotted as mean ± S.E. (n = 3).
ApoE Binds to Apoptotic Neurons and Increases Phagocytosis by Microglia
Next, we investigated the functional role of the apoE/TREM2 interaction. ApoE has been shown to co-localize with amyloid plaques, and neuronal expression of apoE has been reported following injury (34). For experiments using mammalian cells, to reduce the possibility of endotoxin effects in Escherichia coli-derived recombinant apoE, we switched to using apoE expressed in HEK293 cells stably transfected with apoE plasmid. ApoE in the conditioned medium was purified by a heparin column run on FPLC. To evaluate if apoE acts as an opsonizing agent for apoptotic neurons, we incubated apoE with apoptotic neuronal N2a cells in which apoptosis was induced using 0.5 μm staurosporine for 16 h. Western blot analysis of the cell lysates revealed that apoE bound strongly to apoptotic N2a cells, and to a much lesser extent to control (viable) N2a cells (Fig. 3A). Next, we assessed the effect of exogenous apoE on the phagocytic capacity of primary mouse microglia. Microglia from WT mice were incubated with CM-DiI-labeled apoptotic N2a cells with or without 10 nm HEK-derived apoE3. Microglia were harvested after 2 h of incubation at 37 °C, and then labeled with anti-CD11b APC-conjugated antibody. The CD11b+/CM-DiI+ population, which corresponds to microglia that have phagocytosed apoptotic N2a cells, were analyzed by flow cytometry. Importantly, we found that co-incubation of apoE3 significantly increased phagocytosis of apoptotic N2a cells (Fig. 3B). To determine whether this increase was facilitated by TREM2, we also incubated apoptotic N2a cells with primary microglia from Trem2-KO mice under the same experimental conditions. Significantly, we found that Trem2-KO microglia exhibited reduced phagocytic activity compared with WT controls (Fig. 3C) suggesting the apoE promotes phagocytosis of apoptotic neurons through a TREM2-mediated pathway.
FIGURE 3.
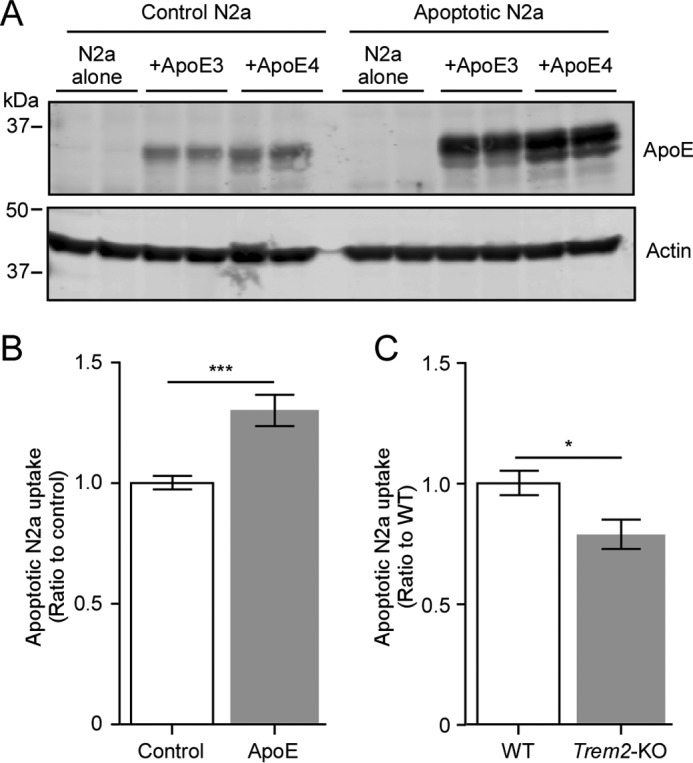
ApoE binds to apoptotic neurons and increases TREM2-mediated phagocytosis by microglia. A, Western blot analysis of neuronal N2a cells incubated with apoE3 or apoE4. Control or staurosporin-induced apoptotic N2a cells were incubated with 10 nm apoE for 1 h. After washing with PBS, cells were lysed and analyzed for cell-bound apoE. Actin was used as a loading control. B, phagocytic uptake of apoptotic N2a cells by WT mouse primary microglia. CM-DiI-labeled apoptotic N2a cells were added to mouse primary microglia with or without 10 nm apoE for 2 h. Microglia were labeled with CD11b-APC antibody, and analyzed by flow cytometry. Percent of CM-DiI+/CD11b+ cells were quantified using CFlow. Data are plotted as mean ± S.E. (n = 4, Student's t test, ***, p < 0.001). C, phagocytosis assay of apoptotic N2a cells by WT and Trem2-KO mouse primary microglia. WT or Trem2-KO microglia were incubated with 10 nm apoE3 and CM-DiI-labeled apoptotic N2a for 2 h. Percent of CM-DiI+/CD11b+ cells were quantified using CFlow. Data are plotted as mean ± S.E. (n = 3, Student's t test; *, p < 0.05).
AD-associated R47H Mutation in TREM2 Impairs ApoE Binding
The majority of disease-associated TREM2 mutations are clustered within the extracellular putative ligand-binding region. Arginine at amino acid 47 substituted with histidine (R47H) is the mutation most strongly associated with AD (14). To examine if the TREM2-R47H mutation affects apoE binding, the extracellular domain of TREM2 or TREM2-R47H mutant was fused with hIgG-Fc and purified from conditioned medium of HEK cells transfected with the appropriate plasmid (Fig. 4A). Silver staining of SDS-PAGE gels of the purified proteins showed highly purified TREM2-Fc fusion proteins at the expected size of ∼60 kDa (Fig. 4B). Western blot analysis of the purified proteins showed that both TREM2 and TREM2-R47H mutant Fc fusion proteins were recognized by anti-TREM2 and anti-Fc antibodies (Fig. 4, C and D). To evaluate if the TREM2-R47H mutation affects apoE binding, purified proteins were spotted onto nitrocellulose membranes that were subsequently incubated with all three apoE isoforms. The bound apoE was detected using anti-apoE antibody (Fig. 5A). A control membrane strip was also incubated with anti-Fc antibody to confirm equal loading of the recombinant proteins. Significantly, quantification of dot blot assays revealed that the TREM2-R47H mutant exhibited reduced binding to apoE2 (Fig. 5B), apoE3 (Fig. 5C), and apoE4 (Fig. 5D) compared with native TREM2. To further confirm our results, we performed a solid phase binding assay. In agreement with dot blot data, the TREM2-R47H mutant protein exhibited significantly reduced binding to immobilized apoE2 (Fig. 5E), apoE3 (Fig. 5F), and apoE4 (Fig. 5G).
FIGURE 4.

Expression and purification of soluble TREM2-Fc. A, schematic representations of human full-length TREM2, and TREM2-Fc and TREM2-R47H-Fc chimeric proteins. SP, signal peptide; TM, transmembrane domain. The cDNA encoding the extracellular domain of TREM2 or TREM2-R47H mutant (amino acids 1–174) was transfected into HEK293 cells and TREM2 protein in the conditioned medium was purified by protein A-agarose beads. The purified TREM2-Fc and TREM2-R47H-Fc fusion proteins were analyzed by silver-stained SDS-PAGE (B) and Western blotting using anti-TREM2 (C) and anti-Fc (D) antibodies.
FIGURE 5.

The AD-associated R47H mutation in TREM2 impairs apoE binding. A, a representative dot blot of apoE binding to TREM2. Membrane strips spotted with 200 ng each of hIgG-Fc, TREM2-Fc, or TREM2-R47H-Fc were incubated with 10 nm apoE, and bound apoE was detected using biotinylated anti-apoE antibody. Membrane strips spotted with the same proteins were also incubated with anti-Fc antibody to ensure equal protein loading. B–D, quantification of dot blot of TREM2 and mutant TREM2-R47H binding to apoE2 (B), apoE3 (C), and apoE4 (D). Data are plotted as mean ± S.E. (n = 3, Student's t test, **, p < 0.01; ***, p < 0.001). E-G, solid phase binding analysis of TREM2 and TREM2-R47H mutant to apoE2 (E), apoE3 (F), and apoE4 (G). A 96-well plate coated with apoE was incubated with 12.5 and 25 nm TREM2 or mutant TREM2-R47H-Fc. The bound TREM2-Fc proteins were detected using biotinylated anti-Fc antibody. Uncoated wells incubated with TREM2-Fc proteins were used as background controls. Data are plotted as mean ± S.E. (n = 3, two-way analysis of variance with Sidak post hoc correction, *, p < 0.05; **, p < 0.01; ***, p < 0.001).
Discussion
In this study, we provide strong evidence that apoE binds with high affinity to the myeloid cell-surface receptor TREM2. Furthermore, we show that phagocytosis is increased when apoptotic neurons are coated with apoE in a TREM2-mediated mechanism. In the context of neurodegenerative disease, our most intriguing finding is that the AD-associated TREM2-R47H mutation abolished physical interaction of apoE and TREM2.
Within the CNS, TREM2 and its signaling adaptor, DAP12, are preferentially expressed in microglia (30, 31). Genetic depletion studies of TREM2 or DAP12 in primary mouse microglia or the microglial BV2 cell line have demonstrated that pro-inflammatory cytokine levels (including IL-1β, TNFα, and IL-6) are significantly increased following co-incubation with bacterial lipopolysaccharide (27), apoptotic neurons (24), or Aβ (50). Overexpression of microglial TREM2, however, suppressed this excessive cytokine production under the same stimulatory conditions (27). Furthermore, TREM2 deficiency has been shown to result in reduced phagocytic activity of apoptotic neurons (24, 28, 29), Aβ (50, 51), and E. coli (51) by microglia. A recent report elegantly demonstrated a direct role for TREM2 in facilitating phagocytosis both in vitro and in vivo. Kawabori et al. (29) showed that TREM2 knockdown in cultured mouse primary microglia attenuated their capacity to phagocytose oxygen/glucose-deprived neurons. Similarly, in a murine experimental stroke model, Trem2-KO mice showed fewer myeloid cells within the CNS that stained positive for phagocytosed intracellular lipids when compared with WT controls. In this model, less infarcted brain resorption in Trem2-KO mice also correlated with increased severity of neurological deficits (29).
Collectively, these findings establish critical roles for TREM2 in regulating the neuroinflammatory environment and in mediating phagocytosis of microbial and damage-associated self-derived motifs. Therefore, during experimental conditions that mimic microbial invasion or neural tissue damage, microglial TREM2 is able to bind bacterial fragments (41, 42), anionic, zwitterionic, and myelin-associated lipids (23, 43), and nucleic acid released from dead cells (29).
Our current study identifies apoE as a high-affinity, AD-associated TREM2 ligand. Using a biochemical approach, we show that all three isoforms of apoE specifically bind to human TREM2. Furthermore, we quantitatively measured the affinity of the apoE/TREM2 interaction and determined that the Kd was in the low nanomolar range. In the CNS, apoE is secreted primarily by astrocytes as an apolipoprotein and then matures to a lipoprotein particle. Here, we demonstrate that different isoforms of lipidated apoE also show similar binding to TREM2.
Previous studies have reported that TREM2-depleted microglia exhibit a reduced capacity to phagocytose apoptotic neurons; here we expand on those findings. In the current study, exogenous apoE preferentially bound to apoptotic neuronal N2a cells, and the presence of apoE promoted phagocytosis of apoptotic neurons by WT primary mouse microglia. Importantly, we found that increased phagocytosis of apoE-coated apoptotic neurons is mediated by TREM2. Because apoE has previously been shown to facilitate receptor-mediated uptake of other endogenous ligands (e.g. soluble Aβ) (33, 37), TREM2 may function under physiological or pathophysiological conditions to facilitate phagocytosis of apoE-coated cells or protein aggregates, which can include apoE-coated Aβ aggregates in AD brains. Moreover, apoE expression in the CNS is up-regulated in response to excitotoxic injury (52, 53). Therefore, it is plausible that stress-induced apoE may promote phagocytosis of apoptotic cells by TREM2-expressing microglia.
Recently, the missense TREM2-R47H mutation has been identified and validated as a risk variant for LOAD (6, 7, 11, 15–19). Although numerous studies have investigated the genetic link between TREM2 variants and neurodegenerative diseases, only two other studies have empirically tested the functional outcome of disease-associated mutations (23, 51). In our study, we provide evidence that the AD-associated TREM2-R47H mutation exhibits reduced ability to bind its endogenous ligand, apoE. Using two different biochemical methods, we showed that all three isoforms of apoE were able to bind native TREM2. Conversely, when the TREM2-R47H mutant was used in our assays, all three apoE isoforms exhibited significantly attenuated binding.
In a study by Wang et al. (23) the authors concluded that TREM2 is a sensor that detects lipids that are exposed during conditions of brain injury or insult. However, the TREM2-R47H mutation significantly reduced the ability of TREM2 to bind damage-associated lipids when compared with non-mutant TREM2 (23). Consistent with results from this study, we conclude that TREM2-R47H is also unable to bind its AD-associated ligand, apoE.
Although the immunomodulatory function of TREM2 is an increasingly focused area of investigation, the lack of specified endogenous ligands has hindered attempts to understand which molecular mechanisms may be affected by disease-associated TREM2 mutation. Further studies are needed to investigate the extent of functional outcomes that may be impacted when apoE is no longer able to engage TREM2 due to mutation. Does TREM2-R47H solely impart loss-of-phagocytic function, or does TREM2-R47H ectopically engage other epitopes and assume novel roles via aberrant signaling or by sequestration of critical ligands?
Most importantly, the implication of our finding that two AD risk genes are mechanistically linked is exemplified in a recent publication that evaluated disease risk in families with high numbers of LOAD cases (19). Significantly, in one family, 11 of 16 members with dementia had APOE4 and TREM2-R47H genotypes. Genetic, behavioral, and histological assessments led this team to conclude that microglial TREM2 is important in the clearance of aggregation-prone proteins and that this function is compromised in R47H carriers (19). Our data suggest a novel pathway in which the two strongest genetic links to LOAD are able to physically interact to modulate microglial function in AD and other neurodegenerative diseases.
Author Contributions
Y. A., C. L., M. M. P., J. D. F., and G. B. conceived and coordinated the study and wrote the paper. C. L. and X. L. generated stably transfected HEK cells. X. C. designed and constructed vectors for expression of TREM2-Fc. C. V., H. Z., R. R., S. S. K., H. X., S. Y., and P. D. contributed to the conception of the study and the interpretation of data. All authors examined the results and approved the final version of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants R01AG027924, R01AG035355, R01AG046205, P01AG30128, P01NS074969, and P50AG016574 (to G. B.), and grants from the Alzheimer's Association and Cure Alzheimer's Fund (to G. B.). We declare no competing financial interests.
- AD
- Alzheimer disease
- LOAD
- late onset Alzheimer disease
- TREM2
- triggering receptor expressed on myeloid cells 2
- Aβ
- amyloid beta
- apoE
- apolipoprotein E
- APC
- allophycocyanin
- tPA
- tissue-type plasminogen activator
- RAP
- receptor-associated protein.
References
- 1.Jack C. R. Jr., Knopman D. S., Jagust W. J., Petersen R. C., Weiner M. W., Aisen P. S., Shaw L. M., Vemuri P., Wiste H. J., Weigand S. D., Lesnick T. G., Pankratz V. S., Donohue M. C., and Trojanowski J. Q. (2013) Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 12, 207–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kanekiyo T., Liu C. C., Shinohara M., Li J., and Bu G. (2012) LRP1 in brain vascular smooth muscle cells mediates local clearance of Alzheimer's amyloid-β. J. Neurosci. 32, 16458–16465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marques F., Sousa J. C., Sousa N., and Palha J. A. (2013) Blood-brain-barriers in aging and in Alzheimer's disease. Mol. Neurodegener. 8, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paloneva J., Manninen T., Christman G., Hovanes K., Mandelin J., Adolfsson R., Bianchin M., Bird T., Miranda R., Salmaggi A., Tranebjaerg L., Konttinen Y., and Peltonen L. (2002) Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am. J. Hum. Genet. 71, 656–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kondo T., Takahashi K., Kohara N., Takahashi Y., Hayashi S., Takahashi H., Matsuo H., Yamazaki M., Inoue K., Miyamoto K., Yamamura T. (2002) Heterogeneity of presenile dementia with bone cysts (Nasu-Hakola disease): three genetic forms. Neurology 59, 1105–1107 [DOI] [PubMed] [Google Scholar]
- 6.Guerreiro R. J., Lohmann E., Brás J. M., Gibbs J. R., Rohrer J. D., Gurunlian N., Dursun B., Bilgic B., Hanagasi H., Gurvit H., Emre M., Singleton A., and Hardy J. (2013) Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia-like syndrome without bone involvement. JAMA Neurol. 70, 78–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giraldo M., Lopera F., Siniard A. L., Corneveaux J. J., Schrauwen I., Carvajal J., Muñoz C., Ramirez-Restrepo M., Gaiteri C., Myers A. J., Caselli R. J., Kosik K. S., Reiman E. M., and Huentelman M. J. (2013) Variants in triggering receptor expressed on myeloid cells 2 are associated with both behavioral variant frontotemporal lobar degeneration and Alzheimer's disease. Neurobiol. Aging 34, 2077.e2011–2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rayaprolu S., Mullen B., Baker M., Lynch T., Finger E., Seeley W. W., Hatanpaa K. J., Lomen-Hoerth C., Kertesz A., Bigio E. H., Lippa C., Josephs K. A., Knopman D. S., White C. L. 3rd, Caselli R., Mackenzie I. R., Miller B. L., Boczarska-Jedynak M., Opala G., Krygowska-Wajs A., Barcikowska M., Younkin S. G., Petersen R. C., Ertekin-Taner N., Uitti R. J., Meschia J. F., Boylan K. B., Boeve B. F., Graff-Radford N. R., Wszolek Z. K., Dickson D. W., Rademakers R., and Ross O. A. (2013) TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson's disease. Mol. Neurodegener. 8, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Le Ber I., De Septenville A., Guerreiro R., Bras J., Camuzat A., Caroppo P., Lattante S., Couarch P., Kabashi E., Bouya-Ahmed K., Dubois B., and Brice A. (2014) Homozygous TREM2 mutation in a family with atypical frontotemporal dementia. Neurobiol. Aging 35, 2419.e23–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borroni B., Ferrari F., Galimberti D., Nacmias B., Barone C., Bagnoli S., Fenoglio C., Piaceri I., Archetti S., Bonvicini C., Gennarelli M., Turla M., Scarpini E., Sorbi S., and Padovani A. (2014) Heterozygous TREM2 mutations in frontotemporal dementia. Neurobiol. Aging 35, 934.e7–10 [DOI] [PubMed] [Google Scholar]
- 11.Cuyvers E., Bettens K., Philtjens S., Van Langenhove T., Gijselinck I., van der Zee J., Engelborghs S., Vandenbulcke M., Van Dongen J., Geerts N., Maes G., Mattheijssens M., Peeters K., Cras P., Vandenberghe R., De Deyn P. P., Van Broeckhoven C., Cruts M., Sleegers K., and BELNEU Consortium (2014) Investigating the role of rare heterozygous TREM2 variants in Alzheimer's disease and frontotemporal dementia. Neurobiol. Aging 35, 726.e11–9 [DOI] [PubMed] [Google Scholar]
- 12.Cady J., Koval E. D., Benitez B. A., Zaidman C., Jockel-Balsarotti J., Allred P., Baloh R. H., Ravits J., Simpson E., Appel S. H., Pestronk A., Goate A. M., Miller T. M., Cruchaga C., and Harms M. B. (2014) TREM2 variant p.R47H as a risk factor for sporadic amyotrophic lateral sclerosis. JAMA Neurol. 71, 449–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ortega-Cubero S., Lorenzo-Betancor O., Lorenzo E., Agúndez J. A., Jiménez-Jiménez F. J., Ross O. A., Wurster I., Mielke C., Lin J.-J., Coria F., et al. (2015) TREM2 R47H variant and risk of essential tremor: a cross-sectional international multicenter study. Parkinsonism Relat. Disord. 21, 306–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guerreiro R., Wojtas A., Bras J., Carrasquillo M., Rogaeva E., Majounie E., Cruchaga C., Sassi C., Kauwe J. S., Younkin S., et al. (2013) TREM2 variants in Alzheimer's disease. N. Engl. J. Med. 368, 117–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jonsson T., Stefansson H., Steinberg S., Jonsdottir I., Jonsson P. V., Snaedal J., Bjornsson S., Huttenlocher J., Levey A. I., Lah J. J., et al. (2013) Variant of TREM2 associated with the risk of Alzheimer's disease. N. Engl. J. Med. 368, 107–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benitez B. A., Cooper B., Pastor P., Jin S. C., Lorenzo E., Cervantes S., and Cruchaga C. (2013) TREM2 is associated with the risk of Alzheimer's disease in Spanish population. Neurobiol. Aging 34, 1711.e15–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin S. C., Carrasquillo M. M., Benitez B. A., Skorupa T., Carrell D., Patel D., Lincoln S., Krishnan S., Kachadoorian M., Reitz C., et al. (2015) TREM2 is associated with increased risk for Alzheimer's disease in African Americans. Mol. Neurodegener. 10, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosenthal S. L., Bamne M. N., Wang X., Berman S., Snitz B. E., Klunk W. E., Sweet R. A., Demirci F. Y., Lopez O. L., and Kamboh M. I. (2015) More evidence for association of a rare TREM2 mutation (R47H) with Alzheimer's disease risk. Neurobiol. Aging 36, 2443 e2421–2446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Korvatska O., Leverenz J. B., Jayadev S., McMillan P., Kurtz I., Guo X., Rumbaugh M., Matsushita M., Girirajan S., Dorschner M. O., et al. (2015) R47H variant of TREM2 associated with Alzheimer disease in a large late-onset family: clinical, genetic, and neuropathological study. JAMA Neurol. 72, 920–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neumann H., and Daly M. J. (2013) Variant TREM2 as risk factor for Alzheimer's disease. N. Engl. J. Med. 368, 182–184 [DOI] [PubMed] [Google Scholar]
- 21.Bouchon A., Hernández-Munain C., Cella M., and Colonna M. (2001) A DAP12-mediated pathway regulates expression of CC chemokine receptor 7 and maturation of human dendritic cells. J. Exp. Med. 194, 1111–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sessa G., Podini P., Mariani M., Meroni A., Spreafico R., Sinigaglia F., Colonna M., Panina P., and Meldolesi J. (2004) Distribution and signaling of TREM2/DAP12, the receptor system mutated in human polycystic lipomembraneous osteodysplasia with sclerosing leukoencephalopathy dementia. Eur. J. Neurosci. 20, 2617–2628 [DOI] [PubMed] [Google Scholar]
- 23.Wang Y., Cella M., Mallinson K., Ulrich J. D., Young K. L., Robinette M. L., Gilfillan S., Krishnan G. M., Sudhakar S., Zinselmeyer B. H., Holtzman D. M., Cirrito J. R., and Colonna M. (2015) TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell 160, 1061–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takahashi K., Rochford C. D., and Neumann H. (2005) Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 201, 647–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ito H., and Hamerman J. A. (2012) TREM-2, triggering receptor expressed on myeloid cell-2, negatively regulates TLR responses in dendritic cells. Eur. J. Immunol. 42, 176–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turnbull I. R., Gilfillan S., Cella M., Aoshi T., Miller M., Piccio L., Hernandez M., and Colonna M. (2006) Cutting edge: TREM-2 attenuates macrophage activation. J. Immunol. 177, 3520–3524 [DOI] [PubMed] [Google Scholar]
- 27.Zhong L., Chen X.-F., Zhang Z.-L., Wang Z., Shi X.-Z., Xu K., Zhang Y.-W., Xu H., and Bu G. (2015) DAP12 stabilizes the C-terminal fragment of the triggering receptor expressed on myeloid cells-2 (TREM2) and protects against LPS-induced pro-inflammatory response. J. Biol. Chem. 290, 15866–15877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsieh C. L., Koike M., Spusta S. C., Niemi E. C., Yenari M., Nakamura M. C., and Seaman W. E. (2009) A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J. Neurochem. 109, 1144–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawabori M., Kacimi R., Kauppinen T., Calosing C., Kim J. Y., Hsieh C. L., Nakamura M. C., and Yenari M. A. (2015) Triggering receptor expressed on myeloid cells 2 (TREM2) deficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J. Neurosci. 35, 3384–3396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hickman S. E., Kingery N. D., Ohsumi T. K., Borowsky M. L., Wang L.-C., Means T. K., and El Khoury J. (2013) The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 16, 1896–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hickman S. E., and El Khoury J. (2014) TREM2 and the neuroimmunology of Alzheimer's disease. Biochem. Pharmacol. 88, 495–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Strittmatter W. J., Saunders A. M., Schmechel D., Pericak-Vance M., Enghild J., Salvesen G. S., and Roses A. D. (1993) Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 90, 1977–1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu C.-C., Kanekiyo T., Xu H., and Bu G. (2013) Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol 9, 106–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu Q., Bernardo A., Walker D., Kanegawa T., Mahley R. W., and Huang Y. (2006) Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J. Neurosci. 26, 4985–4994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Corder E. H., Saunders A. M., Strittmatter W. J., Schmechel D. E., Gaskell P. C., Small G. W., Roses A. D., Haines J. L., and Pericak-Vance M. A. (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261, 921–923 [DOI] [PubMed] [Google Scholar]
- 36.Bu G. (2009) Apolipoprotein E and its receptors in Alzheimer's disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 10, 333–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kanekiyo T., Zhang J., Liu Q., Liu C.-C., Zhang L., and Bu G. (2011) Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid-β uptake. J. Neurosci. 31, 1644–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tai L. M., Mehra S., Shete V., Estus S., Rebeck G. W., Bu G., and LaDu M. J. (2014) Soluble apoE/Aβ complex: mechanism and therapeutic target for APOE4-induced AD risk. Mol. Neurodegener. 9, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Verghese P. B., Castellano J. M., Garai K., Wang Y., Jiang H., Shah A., Bu G., Frieden C., and Holtzman D. M. (2013) ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc. Natl. Acad. Sci. U.S.A. 110, E1807–1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kanekiyo T., Xu H., and Bu G. (2014) ApoE and Aβ in Alzheimer's disease: accidental encounters or partners? Neuron 81, 740–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Daws M. R., Sullam P. M., Niemi E. C., Chen T. T., Tchao N. K., and Seaman W. E. (2003) Pattern recognition by TREM-2: binding of anionic ligands. J. Immunol. 171, 594–599 [DOI] [PubMed] [Google Scholar]
- 42.N'Diaye E.-N., Branda C. S., Branda S. S., Nevarez L., Colonna M., Lowell C., Hamerman J. A., and Seaman W. E. (2009) TREM-2 (triggering receptor expressed on myeloid cells 2) is a phagocytic receptor for bacteria. J. Cell Biol. 184, 215–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poliani P. L., Wang Y., Fontana E., Robinette M. L., Yamanishi Y., Gilfillan S., and Colonna M. (2015) TREM2 sustains microglial expansion during aging and response to demyelination. J. Clin. Invest. 125, 2161–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jay T. R., Miller C. M., Cheng P. J., Graham L. C., Bemiller S., Broihier M. L., Xu G., Margevicius D., Karlo J. C., Sousa G. L., Cotleur A. C., Butovsky O., Bekris L., Staugaitis S. M., Leverenz J. B., Pimplikar S. W., Landreth G. E., Howell G. R., Ransohoff R. M., and Lamb B. T. (2015) TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer's disease mouse models. J. Exp. Med. 212, 287–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu W., Zheng H., Shao X., Wang W., Yao Q., and Li Z. (2010) Excitotoxicity of TNFα derived from KA activated microglia on hippocampal neurons in vitro and in vivo. J. Neurochem. 114, 386–396 [DOI] [PubMed] [Google Scholar]
- 46.Morikawa M., Fryer J. D., Sullivan P. M., Christopher E. A., Wahrle S. E., DeMattos R. B., O'Dell M. A., Fagan A. M., Lashuel H. A., Walz T., Asai K., and Holtzman D. M. (2005) Production and characterization of astrocyte-derived human apolipoprotein E isoforms from immortalized astrocytes and their interactions with amyloid-β. Neurobiol. Dis. 19, 66–76 [DOI] [PubMed] [Google Scholar]
- 47.Casey C. S., Atagi Y., Yamazaki Y., Shinohara M., Tachibana M., Fu Y., Bu G., and Kanekiyo T. (2015) Apolipoprotein E inhibits cerebrovascular pericyte mobility through a RhoA protein-mediated pathway. J. Biol. Chem. 290, 14208–14217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Farrer L. A., Cupples L. A., Haines J. L., Hyman B., Kukull W. A., Mayeux R., Myers R. H., Pericak-Vance M. A., Risch N., and van Duijn C. M. (1997) Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 278, 1349–1356 [PubMed] [Google Scholar]
- 49.Liu Q., Zerbinatti C. V., Zhang J., Hoe H. S., Wang B., Cole S. L., Herz J., Muglia L., and Bu G. (2007) Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron 56, 66–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiang T., Tan L., Zhu X.-C., Zhang Q.-Q., Cao L., Tan M.-S., Gu L.-Z., Wang H.-F., Ding Z.-Z., and Zhang Y.-D. (2014) Upregulation of TREM2 ameliorates neuropathology and rescues spatial cognitive impairment in a transgenic mouse model of Alzheimer's disease. Neuropsychopharmacology 39, 2949–2962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kleinberger G., Yamanishi Y., Suárez-Calvet M., Czirr E., Lohmann E., Cuyvers E., Struyfs H., Pettkus N., Wenninger-Weinzierl A., Mazaheri F., Tahirovic S., Lleó A., Alcolea D., Fortea J., Willem M., Lammich S., Molinuevo J. L., Sánchez-Valle R., Antonell A., Ramirez A., Heneka M. T., Sleegers K., van der Zee J., Martin J. J., Engelborghs S., Demirtas-Tatlidede A., Zetterberg H., Van Broeckhoven C., Gurvit H., Wyss-Coray T., Hardy J., Colonna M., and Haass C. (2014) TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med. 6, 243ra86. [DOI] [PubMed] [Google Scholar]
- 52.Petegnief V., Saura J., de Gregorio-Rocasolano N., and Paul S. M. (2001) Neuronal injury-induced expression and release of apolipoprotein E in mixed neuron/glia co-cultures: nuclear factor κB inhibitors reduce basal and lesion-induced secretion of apolipoprotein E. Neuroscience 104, 223–234 [DOI] [PubMed] [Google Scholar]
- 53.Page K. J., Hollister R. D., and Hyman B. T. (1998) Dissociation of apolipoprotein and apolipoprotein receptor response to lesion in the rat brain: an in situ hybridization study. Neuroscience 85, 1161–1171 [DOI] [PubMed] [Google Scholar]