

Background: The α6β4 integrin associates with syndecans and, via an unknown mechanism, with receptor tyrosine kinases.
Results: Syndecans-1 and -4 capture HER2 and EGFR, along with α3β1 integrin, via docking sites in their ectodomains.
Conclusion: Syndecans organize integrins and receptor tyrosine kinases into signaling complexes that stimulate epithelial invasion.
Significance: Novel peptides (synstatins) are defined that block kinase capture and are potential cancer therapeutics.
Keywords: epidermal growth factor receptor (EGFR), epithelial cell, extracellular matrix, integrin, invasion, peptide interaction, protein complex, syndecan, wound healing
Abstract
The α6β4 integrin is known to associate with receptor tyrosine kinases when engaged in epithelial wound healing and in carcinoma invasion and survival. Prior work has shown that HER2 associates with α6β4 integrin and syndecan-1 (Sdc1), in which Sdc1 engages the cytoplasmic domain of the β4 integrin subunit allowing HER2-dependent motility and carcinoma cell survival. In contrast, EGFR associates with Sdc4 and the α6β4 integrin, and EGFR-dependent motility depends on cytoplasmic engagement of β4 integrin with Sdc4. However, how HER2 and EGFR assimilate into a complex with the syndecans and integrin, and why kinase capture is syndecan-specific has remained unknown. In the present study, we demonstrate that HER2 is captured via a site, comprised of amino acids 210–240, in the extracellular domain of human Sdc1, and EGFR is captured via an extracellular site comprised of amino acids 87–131 in human Sdc4. Binding assays using purified recombinant proteins demonstrate that the interaction between the EGFR family members and the syndecans is direct. The α3β1 integrin, which is responsible for the motility of the cells, is captured at these sites as well. Peptides based on the interaction motifs in Sdc1 and Sdc4, called synstatins (SSTN210–240 and SSTN87–131) competitively displace the receptor tyrosine kinase and α3β1 integrin from the syndecan with an IC50 of 100–300 nm. The syndecans remain anchored to the α6β4 integrin via its cytoplasmic domain, but the activation of cell motility is disrupted. These novel SSTN peptides are potential therapeutics for carcinomas that depend on these HER2- and EGFR-coupled mechanisms for their invasion and survival.
Introduction
The α6β4 integrin is typically found in hemidesmosomes in resting epithelial cells where its extracellular domain engages laminin 332 (LN332, also known as laminin 5)2 in the basement membrane, and its cytoplasmic domain engages scaffolding proteins, e.g. plectin, BP180, BP230, that link the β4 subunit to the keratin filament cytoskeleton inside the cell (1). For this purpose, the β4 subunit is equipped with a cytoplasmic domain over 1,000 amino acids long (2, 3) comprised of two pairs of FNIII repeats spaced by a connecting segment, and a distal tail. Binding sites within the connecting segment and 3rd FN III repeat provide docking sites for plectin, a central feature of the hemidesmosomes (4, 5).
When growth factor receptors are activated during epithelial wounding or carcinogenesis, several serines in the connecting segment and C-terminal tail of the β4 subunit are targeted by PKC and other kinases that causes the dissociation of the integrin from plectin and breakdown of the hemidesosome (6–8). Following this phosphorylation, the free integrin associates with several kinases, including EGFR, HER2, or c-Met in the activated cells (9–13). This association appears critical for cell motility during morphogenesis, wound healing, or tumor invasion, as well as for sustaining survival signaling in tumor cells (14–22). Although it does not appear to do so directly, the kinase associated with the integrin causes tyrosine phosphorylation of the β4 cytoplasmic domain, thereby converting it into a signaling scaffold (9, 10, 23). This phosphorylated scaffold provides docking sites for Shc, IRS-1/2, and other scaffolding proteins, leading ultimately to activation of PI-3K, Erk1/2, Src, Rho family GTPases, and other signaling cascades (14, 17, 23–28).
The importance of this mechanism has been shown in transgenic mice expressing a β4 mutant in which the distal half of the β4 cytoplasmic domain bearing this “signaling domain” has been deleted (β41355T) (9, 29, 30). This construct leads to reduced tumorigenesis in the MMTV-Neu breast cancer model (9), expansion of prostate cancer progenitors in the PB-Tag prostate cancer model (30), as well as reduced angiogenesis in multiple tumor types (29). Analysis of these models shows that downstream signaling is severely diminished by this integrin mutant.
We have shown recently that another feature of the integrin cytoplasmic signaling domain is its ability to engage syndecans, a family of cell surface heparan-sulfate proteoglycans that serve as matrix receptors (2, 3). This discovery implicated syndecans as central organizers of the α6β4 integrin/growth factor receptor complexes. All four syndecan family members have been shown by yeast-two hybrid analysis to engage the cytoplasmic domain of the β4 integrin (3). Recent work (2) has shown that syndecan-1 (Sdc1) engages arginine 1733 (Arg-1733) near the distal tip of the β4 integrin cytoplasmic domain. When the syndecan and integrin are engaged, HER2 is captured by the Sdc1-coupled complex and undergoes autophosphorylation when the complex is clustered to sites of matrix engagement. Mutation of Arg-1733 prevents capture of the integrin by Sdc1, blocks HER2-dependent migration of the cells and prevents the growth and survival of HER2+ SKBr3 carcinoma cells.
Syndecan-4 (Sdc4) is implicated in a similar mechanism (2). It also binds the distal tip of the β4 integrin cytoplasmic domain, requiring glutamate 1729 (Glu-1729) in the integrin. This binding appears necessary for EGFR to activate β4 signaling. Mutation of the Sdc4 binding site prevents EGFR capture by the α6β4 integrin and blocks EGF stimulated motility of HaCat keratinocytes and MCF10A breast epithelial cells. Thus, association of the syndecans with the integrin appears necessary for the functional coupling of the integrin and receptor tyrosine kinases. But how this occurs is not clear.
In this study, we examine how syndecan docking with the integrin leads to capture of HER2 or EGFR, and question why the capture is syndecan-specific. Our findings build on a paradigm first established several years ago when Sdc1 was shown to organize a different receptor complex, namely, the insulin-like growth factor-1 receptor (IGF-1R) associated with the αvβ3 or αvβ5 integrin (31–34). In this example, the association is mediated by a 28 amino acid site in the Sdc1 extracellular domain, which binds to both the integrin and IGF-1R and captures them into a functional complex. When captured and clustered by the syndecan to sites of matrix anchorage, IGF-1R undergoes autoactivation and initiates signaling downstream that activates the integrin. Lastly, a peptide based on the docking site in Sdc1 called “synstatin” (or SSTN), which we now call SSTNIGF-1R to denote its specificity for the IGF-1R kinase, blocks assembly of the receptor complex and disrupts IGF-1R activation.
With this paradigm in mind, we questioned whether the specificity for HER2 or EGFR docking with the α6β4 integrin might also reside in the extracellular domains of Sdc1 and Sdc4, respectively. Whereas the syndecans have highly homologous transmembrane domains and have exactly conserved regions within their short cytoplasmic domains, their extracellular domains are highly divergent (35). We find that sites in the extracellular domains of Sdc1 and Sdc4 are responsible for capturing HER2 and EGFR, respectively, along with the α3β1 integrin. This ternary receptor complex docks with the α6β4 integrin via its cytoplasmic link with the syndecan, leading to β4 phosphorylation and downstream signaling (2, 3). Peptides based on these sites (SSTN210–240, with numbering based on the amino acid sequence of human Sdc1, and SSTN87–131 based on human Sdc4) disrupt HER2 and EGFR-dependent motility, respectively. Last, we show by expressing recombinant HER2 and EGFR ectodomains that the binding of the syndecans to these receptor tyrosine kinases is direct. This defines a novel site in EGFR and HER2 that is necessary for their participation in adhesion signaling and cell survival, and defines potential therapeutic peptides to target this mechanism.
Experimental Procedures
Antibodies
Antibodies used were: mouse mAb 3E1 to the human β4 integrin extracellular domain (hybridoma facility, Memorial Sloan Kettering, New York); rabbit polyclonal antibody Ab1922 to the β4 cytoplasmic domain and P1B5 to the α3β1 integrin (Millipore, Billerica, MA); anti-penta-histidine antibody (Qiagen, Valencia, CA); mouse mAb F94–8G3 (kindly provided by Dr. Guido David, University of Leuven, Belgium) and B-A38 (Accurate Chemical & Scientific, Westbury, NY) against human Sdc4 and Sdc1, respectively. Mouse mAb anti-EGFR (528) and anti-EGFR extracellular domain (20E12), rabbit polyclonal anti-HER2/Neu (c-18) and mouse mAb anti-Her-2/Neu extracellular (c-3) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), were used against human EGFR and HER2. Goat anti-mouse IgG (H+L) secondary was from Jackson Immuno Research (West Grove, PA).
Reagents
Dulbecco's modified Eagle's medium (DMEM), Schneider's Drosophila Medium (Revised) (1×) and Express Five® SFM (1×) were from Invitrogen (Life Technologies, Grand Island, NY); glutathione-conjugated Sepharose beads were from GEHealthcare; GammaBind G-Sepharose beads were from Amersham Biosciences; Nickel Agarose (for purification of His-tagged proteins) was from Gold Biotechnology (St. Louis, MO); heparinase II and chondroitin ABC lyase were from IBEX Pharmaceuticals, Inc. (Montreal, Canada) and Sigma, respectively. Human recombinant epidermal growth factor, thrombin and oleoyl-l-α-lysophosphatidic acid sodium salts (LPA) were from Sigma. Human Sdc1 and Sdc4 extracellular-domain peptides were synthesized by LifeTein, LLC (Hillsborough, NJ).
Cell Culture
Human HaCat keratinocytes were cultured as described previously (3). MCF10A human breast epithelial cells were cultured in DMEM F12 50/50, 15 mm Hepes, 5% horse serum, 10 μg/ml insulin, 0.5 μg/ml hydrocortisone, and 0.02 μg/ml EGF (2).
Fusion Protein Expression and Purification
Native or mutagenized human GST-syndecan-1 extracellular domain (GST-S1ED) and mouse GST-syndecan-4 extracellular domain (GST-S4ED) inserted into pGEX4T vector in BamHI (thrombin cleavage site)/Xhol site were expressed in E. coli and purified on glutathione-Sepharose beads following cell lysis in 150 mm NaCl, 20 mm sodium phosphate (pH 7.4), 1% Triton X-100 and 0.5% sodium deoxycholate.
Human HER2 extracellular domain, consisting of amino acids 1–654 and human EGFR extracellular domain, consisting of amino acids 1–646, were expressed as 6×His-tagged recombinant proteins in S2 insect cells. The cDNAs were inserted into vector pMT containing puromycin-selection marker, with the insect signal peptide (sequence MKLCILLAVVAFVGLSLG at the N terminus and a 6×His tag at C terminus, then transfected into S2 cells using transfection kit from Mirus (Madison, WI). The secreted proteins from conditioned medium were purified on nickel agarose.
Wound Healing Assay
HaCat or MCF10A cells were grown to confluence in 48 well-plates, and then were starved by serum deprivation for 8 h followed by scraping a wound in the monolayer with a 200 μl pipette tip. Cells were cultured an additional 15 h in DMEM (serum-free) in the presence or absence of GST-fusion proteins or peptide inhibitors and containing either LPA (3 μm), mAb 3E1 (10 μg/ml), and goat anti-mouse IgG (30 μg/ml) to cluster α6β4 integrin and mimic matrix-stimulated chemokinesis, or EGF (10 ng/ml) to stimulate EGF chemokinesis as described previously (2). Note that short sequences of S4ED appear to become masked by GST in the fusion proteins. Thus, all GST-S4ED fusion proteins used in Fig. 4 were pre-treated with thrombin (2 units/mg) for 4 h at 4 °C to remove the GST prior to use. Wounds were imaged using a PlanApo 20 (0.75 numerical aperture) objective and a Photometrics CoolSnap ES camera on a Nikon Eclipse TE2000U microscope. Wound closure was quantified by using Metamorph®.
FIGURE 4.
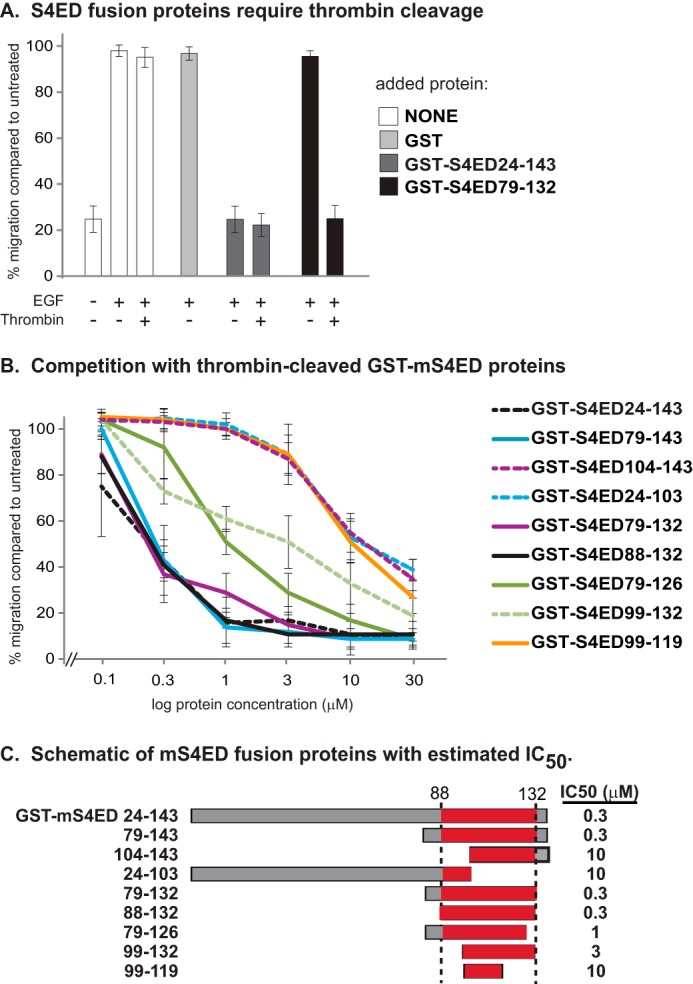
Testing truncation mutants of mouse Sdc4 ectodomain for inhibitory motif. A, wound healing assay is conducted with HaCat cells in the presence or absence of 10 ng/ml EGF, thrombin (2 units/mg) and added mouse GST-S4ED fusion protein (10 μm). Note that neither thrombin nor 10 μm GST has an effect on migration. However, GST-S4ED79–132 fusion protein, in which GST is close to the active site, requires the removal of GST for activity. B, concentration range of thrombin-cleaved GST-S4ED fusion protein or truncation mutants are tested for inhibition of MCF10A cell migration in response to 10 ng/ml EGF in a wound healing assay. C, schematic showing fusion proteins with the putative binding site (amino acids 88–132) highlighted and estimated IC50.
Immunoprecipitation and Bead Capture Assays
Immunoprecipitations were carried out as described previously (3). For bead capture assays, lysates of cells (lysed in 150 mm NaCl, 20 mm sodium phosphate (pH 7.4), 1% Triton X-100, and 0.5% sodium deoxycholate) or 6× His-tagged HER2 or EGFR extracellular domains (2 μm) were incubated at 4 °C overnight with 100 μl of glutathione-Sepharose 4B beads and GST-S4ED or GST-S1ED in the presence or absence of competing peptides. Samples were analyzed under reducing conditions on a 10% Laemmli electrophoresis gel, transferred to Immobilon P, and stained with primary antibody followed by alkaline phosphatase-conjugated secondary antibody. Immune-reactive bands were visualized using ECF reagent (GE Healthcare) and scanned using a Typhoon Trio Variable Mode Imager (GE-Healthcare).
Results
Our prior work has shown that epithelial cell motility in response to laminin 332 (matrix chemokinesis) or EGF (EGF chemokinesis) depends on coupling of α6β4 integrin to HER2 and EGFR, respectively (2). Also required is engagement of the cytoplasmic domain of the β4 integrin subunit by syndecans. Sdc1 engagement is necessary for HER2-dependent signaling, whereas Sdc4 engagement is necessary for signaling that depends on EGFR (2).
To test the hypothesis that syndecan specificity in these two mechanisms may reflect a novel ability of HER2 or EGFR to engage “organizer” sites in the extracellular domains of specific syndecans, we tested whether or not recombinant Sdc1 or Sdc4 extracellular domains expressed as GST fusion proteins (e.g. GST-S1ED or GST-S4ED) could act as competitors of HER2- or EGFR-stimulated cell migration. MCF10A cells were stimulated to close a scratch wound by clustering the α6β4 integrin with integrin-specific antibodies in the presence of LPA (Fig. 1, A and B), a method shown previously to mimic laminin-induced chemokinesis (matrix-chemokinesis) that depends on HER2 and Sdc1 (2). S1ED nearly completely blocks wound closure when added at 1 μm concentrations, with an IC50 observed at 0.3–1 μm (Figs. 1 and 2). The fusion protein has no effect on cell adhesion, which we have shown previously depends on the α3β1 integrin engaging LN332 deposited by the cells (2). In contrast to S1ED, GST-S4ED has no effect on this mechanism when added at 30 μm, the maximum concentration tested.
FIGURE 1.
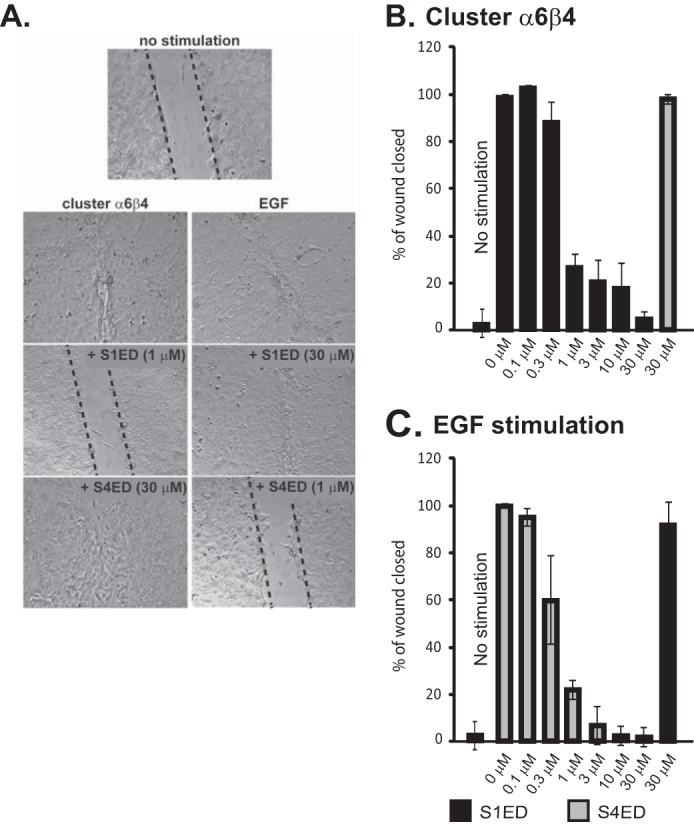
Inhibition of HER2- and EGFR-dependent migration of MCF10A breast epithelial cells by recombinant Sdc1 and Sdc4 extracellular domains. A, MCF10A cells migrate for 15 h following serum-starvation to close scratch wounds in response to artificial clustering of α6β4 integrin with antibodies in the presence of LPA (cluster α6β4), a mechanism that requires HER2, or in response to stimulation of EGFR with 20 ng/ml EGF. GST-S1ED or GST-S4ED are added to the culture medium throughout the assay at the concentrations shown. B, quantification of wound closure following stimulation with β4 integrin clustering antibodies + LPA in the presence of a concentration range of GST-S1ED protein, or 30 μm GST-S4ED. C, quantification of wound closure following stimulation with 20 ng/ml EGF in the presence of a concentration range of GST-S4ED protein, or 30 μm GST-S1ED.
FIGURE 2.
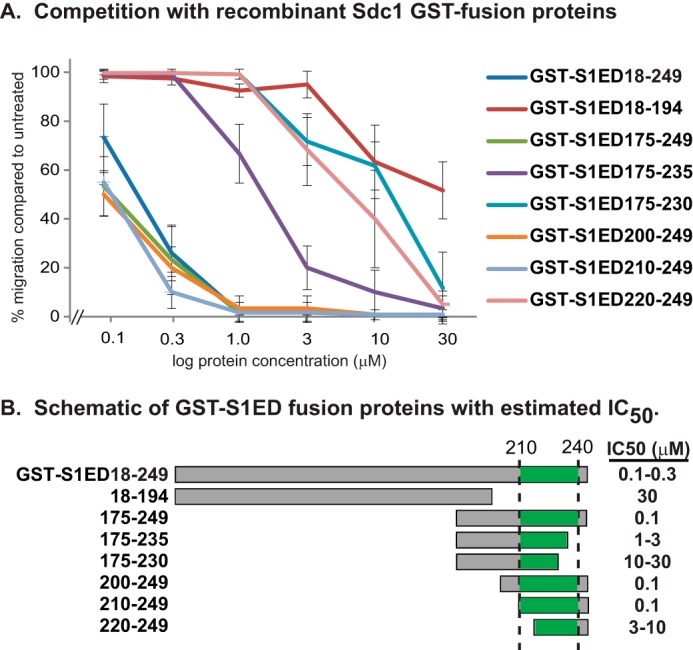
Testing truncation mutants of Sdc1 ectodomain for inhibitory motif. A, concentration range of GST-S1ED fusion protein or truncation mutants are tested for inhibition of MCF10A migration in response to α6β4 integrin clustering in wound healing assay. B, schematic showing fusion proteins with the putative binding site (amino acids 210–240) highlighted and estimated IC50.
In contrast to its lack of effect on matrix-chemokinesis involving HER2, S4ED used at 1 μm does block migration stimulated by 20 ng/ml EGF (EGF chemokinesis) (Fig. 1, A and C), a process shown previously to depend on Sdc4, α6β4, and α3β1 integrins and EGFR (2). S4ED displays an IC50 of 0.3 μm, similar to the inhibitory activity of S1ED during matrix-chemokinesis. S1ED, although active against the HER2 mechanism, fails to disrupt EGF-chemokinesis when used at 30 μm (Fig. 1, A and C).
The ability of GST-S1ED to block cell motility implies that an active site highly specific for the HER2 mechanism exists within the Sdc1 extracellular domain. To identify this site, we expressed recombinant proteins representing overlapping thirds of the Sdc1 extracellular domain and discovered that the juxtamembrane third (GST-S1ED175–249) retains the competitive ability (Fig. 2A). GST-S1ED18–194, which lacks most of this region (note that amino acids 1–17 are the signal peptide), has reduced inhibitory activity, displaying an IC50 of ∼30 μm. Note that S1ED18–194 also contains the distal organizer site (amino acids 93–120 in human Sdc1) described previously that captures the αvβ3 or αvβ5 integrin together with IGF-1R (see Fig. 3A) (31, 32). However, SSTNIGF-1R, the 28 amino acid peptide representing the distal capture site, displays no competitive activity when used at 30 μm in the matrix chemokinesis assay (data not shown).
FIGURE 3.
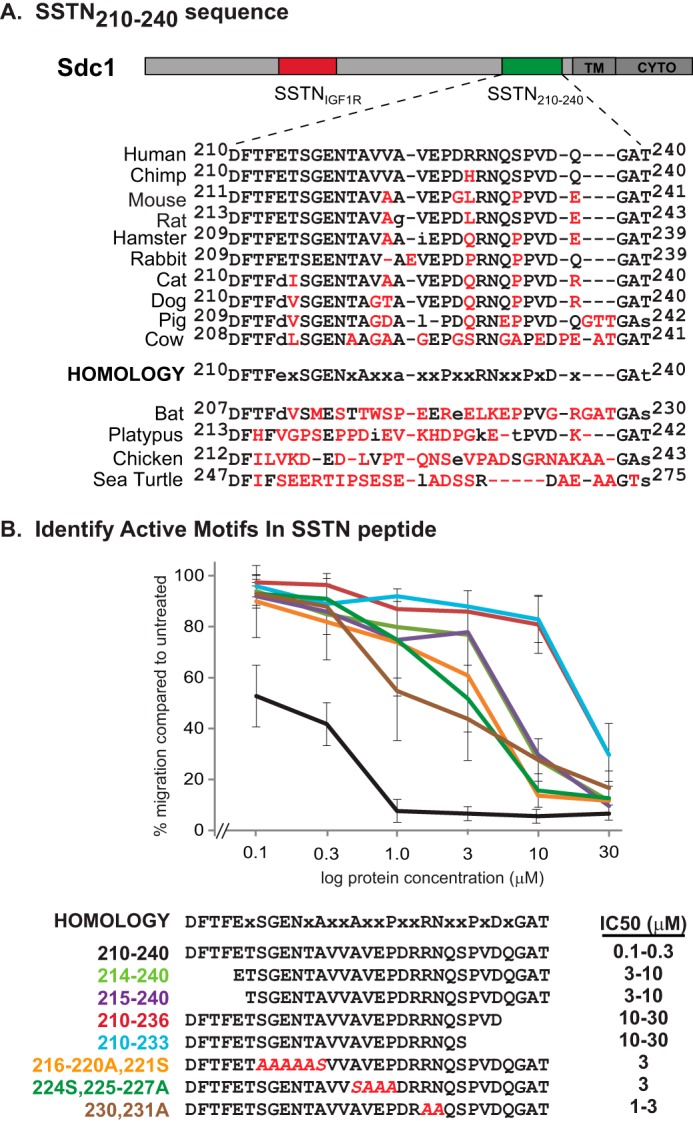
Identification of active motifs in SSTN210–240 peptide. A, schematic of human Sdc1 ectodomain depicting location of amino acids 210–240 relative to previously described active site (amino acids 93–120) and transmembrane (TM) and cytoplasmic domain (CYTO). Sequence conservation is shown over a range of mammalian and other vertebrate species, with nonconserved amino acids shown in red. A putative homology sequence conserved in higher mammals is shown. B, peptides bearing truncations or alanine mutations of the putative mammalian homology sequence are tested in MCF10A cells migrating to close scratch wounds in response to clustering α6β4 integrin. The estimated IC50 is shown.
We then tested other fusion proteins bearing truncations within the putative active region to map the functional site (Fig. 2, A and B). All proteins that retain all of the amino acids between 210 and 240 within their sequence (e.g. 175–249, 200–249, 210–240) are highly effective, displaying IC50's of 0.1–0.3 μm in the wound healing assay. Proteins that retain only a portion of the 210–240 sequence (e.g. 175–235, 175–230, 220–240) display competitive ability as well, but it is reduced. These findings suggest that the active site that regulates HER2 signaling resides between amino acids 210–240 in human Sdc1.
Scanning for homology within this region of human Sdc1 across mammalian species reveals a number of conserved amino acids, e.g. DFTFeXSGENXAXXaXXPXXRNXXPXDXGat (Fig. 3A). However, although conserved in primates and rodents, the sequence is not consistently maintained across all mammals, and shows significant deterioration across lower vertebrates. To evaluate which of these conserved amino acids are critical for activity, we turned to synthetic peptides as competitors in the wound healing assay (Fig. 3B). The 31 amino acid peptide consisting of amino acids 210–240 is highly competitive, displaying an IC50 of 100–300 nm. We call this inhibitory peptide synstatin210–240 (SSTN210–240) reflecting its derivation from a syndecan, its inhibitory activity, and its sequence numbering within the human Sdc1 extracellular domain. Truncation of the peptide at either end causes a 30–100-fold loss of activity, indicating the importance of the N-terminal DFTF motif and the C-terminal QGAt/s motifs. Further truncation from the C terminus, removing the conserved PVD motif has no further effect on activity, suggesting that the conserved PVD is not involved in the HER2 mechanism. An alanine screen of internal conserved amino acids demonstrates that the SGENXA, aXXP and RN motifs also contribute to the activity, as their mutation reduces activity 10–30-fold.
Next, we turned our attention to the active site in Sdc4. A juxtamembrane site consisting of amino acids 56–109 (numbering not including the signal sequence, but 79–132 from the start of translation) in mouse Sdc4 has been identified previously, initially by McFall and Rapraeger as a site that supported the attachment of fibroblasts and endothelial cells (e.g. a “cell binding domain”) (36, 37), and then more extensively by Whiteford and Couchman as a ligand for β1 integrin-mediated attachment of fibroblasts and leukocytes (38). To determine whether or not the presence of this site is responsible for the inhibitory activity of GST-S4ED protein in the EGF-induced wound healing assay, we tested mouse GST fusion proteins containing all or parts of this sequence as potential inhibitors. We soon discovered that whereas the full-length mouse S4ED acts as an inhibitor when expressed as a fusion protein with GST (GST-mS4ED24–143), smaller lengths of the syndecan (e.g. GST-mS4ED79–132) appear to be inhibited, presumably by steric hindrance (Fig. 4A). Thus, all of the constructs were pretreated with thrombin to remove the GST. Note that neither GST added alone, nor thrombin added alone, affects the assay. We find that GST-mS4ED79–132 is inhibitory, displaying an IC50 of 300 nm, as is GST-S4ED88–132, suggesting that amino acids 79–87 are not part of the active site (Fig. 4, B and C). Further truncations of this sequence from either end cause reduced activity but do not completely abolish activity, suggesting that multiple interaction sites likely exist within this sequence.
We next tested synthetic peptides based on a homologous site (amino acids 87–131) in the human sequence (Fig. 5). Scanning for sequence homology reveals several groups of conserved amino acids across mammalian species (87NXIPeXXXXGXXvPtXXKXlXXNEViPKXXXPXeXXXDXSNKVSM) (Fig. 5A). As noted previously (38), several of these, namely, NxIP (87–90), NEV (109–111), and SNXvXM (126–131), are highly conserved across vertebrates, including zebrafish, whereas others are not, similar to the level of sequence conservation noted for the active site in Sdc1. Using either synthetic human peptides or mouse GST-fusion proteins to screen these motifs for inhibitory activity (Fig. 5B), we find that the human peptide consisting of amino acids 87–131 retains full inhibitory activity, displaying an IC50 of 0.1–0.3 μm and qualifying it as a synstatin (SSTN87–131). Alanine substitution of Ile89 within its NXIP motif, an amino acid shown to be critical for other activities (38), reduces activity 10-fold. Truncation of the C-terminal SNKVSM motif in the mouse GST fusion protein (GST-mS4ED79–125) shows only a 3-fold reduction in activity (Fig. 4, A and B). However, we find in addition that alanine substitution of the vPt (30–100-fold), KXl(10–30-fold), NEV (3-fold), and IPKX (10–30-fold) motifs reduce activity as well, suggesting that multiple interactions within this site are necessary for full activity.
FIGURE 5.
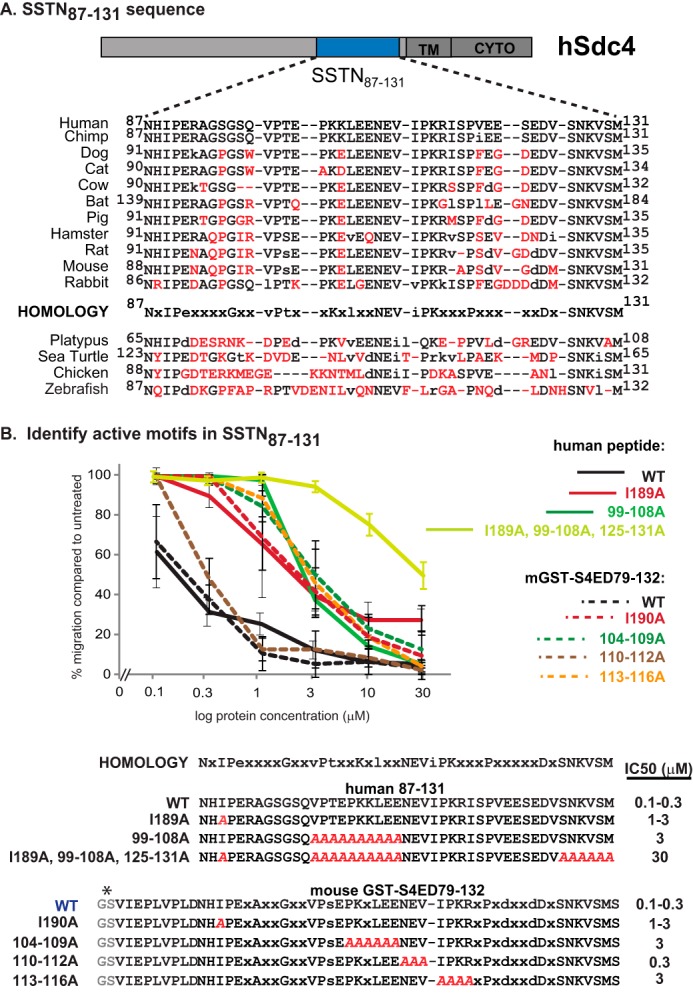
Identification of active motifs in human SSTN87–131 peptide. A, schematic of human Sdc4 ectodomain depicting location of amino acids 87–131 (equivalent to mouse 88–132) relative to transmembrane (TM) and cytoplasmic domains (CYTO) is shown. Sequence conservation is shown over a range of mammalian and other vertebrate species, with nonconserved amino acids shown in red. A putative homology sequence conserved in higher mammals is shown. B, human peptides or thrombin-cleaved mouse GST-fusion proteins bearing truncations or alanine mutations of the putative mammalian homology sequence are tested in MCF10A cells migrating to close scratch wounds in response to EGF stimulation. The estimated IC50 is shown. *, GS is from thrombin-cleavage site in fusion protein.
Our prior work has shown HER2 and EGFR-dependent epithelial cell migration relies on the α3β1 integrin and capture of α6β4 integrin by either Sdc1 or Sdc4, respectively (2, 3). We have shown that the syndecans capture the α6β4 integrin via syndecan-specific sites in the β4 cytoplasmic domain. We now hypothesize that HER2 and EGFR are incorporated into these receptor complexes via their association with the juxtamembrane sites in the Sdc1 (HER2) or Sdc4 (EGFR) defined by the SSTN peptides. To test this, individual receptors were immunoprecipitated from either HaCat or MCF10A cells and probed for co-immunoprecipitation of either HER2 or EGFR (Fig. 6A). HER2 is observed to co-precipitate with Sdc1, α6β4, and α3β1 integrin. It also precipitates with EGFR as the classical heterodimer known to exist on epithelial cells (39). However, HER2 is not observed to precipitate with Sdc4 in either cell type. In contrast, EGFR is observed to co-precipitate with Sdc4, α6β4, and α3β1 integrins, as well as in the HER2-EGFR heterodimer. But little or no EGFR is observed to co-precipitate with Sdc1. These are difficult immunoprecipitations as they are all conducted in the same experiment rather than being optimized for each receptor.
FIGURE 6.

Analysis of receptors associated with Sdc1 and Sdc4 in HaCat keratinocytes and MCF10A breast epithelial cells. A, receptors shown previously (2) to comprise the functional complexes responsible for matrix chemokinesis (HER2, Sdc1, α3β1 integrin, α6β4 integrin) or EGF-chemokinesis (EGFR, Sdc4, α3β1 integrin, α6β4 integrin) of HaCat and MCAF10A cells are imunoprecipitated, then probed for the presence of either HER2 or EGFR. (*, Note splicing of blot for the purpose of re-arranging the sample order for MCF10A cells). B, α6β4 integrin is immunoprecipitated from MCF10A cell lysates in the presence or absence of SSTN210–240 or SSTN87–131, then probed for Sdc1, Sdc4, α3β1 integrin, HER2, EGFR, and α6β4 integrin. Each of the receptors is also immunoprecipitated using receptor-specific antibodies and stained to provide a receptor marker. C, quantification of α3β1 integrin co-precipitation with α6β4 integrin in the presence or absence of SSTN peptides (*, p < 0.01).
Next, to specifically investigate the role of the syndecan extracellular capture sites in coupling receptors to the α6β4 integrin, we immunoprecipitated the β4 integrin in the presence or absence of SSTN peptides and probed for co-precipitation of each of the other receptors. Confirming the finding in Fig. 6A, we find that Sdc1, Sdc4, α3β1 integrin, HER2, and EGFR co-precipitate with the α6β4 integrin (Fig. 6B). SSTN210–240, representing the putative capture site in Sdc1, displaces all of HER2 and approximately half of the α3β1 integrin (Fig. 6, B and C). The peptide does not displace Sdc1, which is linked to the integrin via a cytoplasmic interaction (2), nor does it displace EGFR. These findings suggest that HER2 and α3β1 integrin are linked to Sdc1 via its juxtamembrane binding site, which in turn links these receptors to α6β4 integrin. In contrast, SSTN87–131, representing the active site in Sdc4, displaces all of EGFR and also displaces approximately half of the α3β1 integrin (Fig. 6, B and C), but fails to displace Sdc4 or HER2 (Fig. 6B), suggesting that EGFR and α3β1 integrin are linked to Sdc4, which in turn links them to α6β4. Competition with SSTN210–240 and SSTN87–131 together displaces nearly all of α3β1 integrin from the β4 subunit (Fig. 6C), suggesting that the two portions of α3β1 integrin displaced by either peptide alone are distinct and are associated with either Sdc1 or Sdc4.
To test this model using a different approach, we used the GST-S1ED or GST-S4ED fusion proteins to see if they can directly capture either HER2, EGFR, α3β1 integrin, or α6β4 integrin from cell lysates. S1ED captures HER2 and α3β1 integrin, but not if the capture site at amino acids 210–240 is deleted from the fusion protein (GST-S1EDΔ210–240) (Fig. 7A). As predicted, the syndecan extracellular domain does not capture α6β4 integrin, which instead is captured via a cytoplasmic interaction with the syndecan. Capture of HER2 and α3β1 integrin are significantly reduced by competition with SSTN210–240. SSTN214–240 shows a significantly reduced ability to compete for HER2 capture, suggesting that the asp-phe-thr-phe213 motif that is lacking in this peptide is involved in the interaction with HER2. In contrast, SSTN210–236 is largely ineffective at competing for capture of the α3β1 integrin, implicating the C-terminal gln-gly-ala-thr240 motif in integrin capture.
FIGURE 7.

Identification of receptors captured by the binding motifs in the extracellular domains of Sdc1 and Sdc4. A, MCF10A cell lysates are incubated overnight with 2 μm GST-S1EDΔ210–240 or GST-S1ED (bait) in the presence of 30 μm SSTN210–240, SSTN214–240, or SSTN210–236 (competitors), then the fusion proteins are captured on glutathione beads and subjected to Western blot analysis to probe for capture of HER2, EGFR, α6β4 integrin, or α3β1 integrin. The relative capture of HER2 versus α3β1 integrin in the presence of the peptides is quantified in the bar graph as a percentage of capture in the absence of peptide. (*, p < 0.01). B, MCF10A cell lysates are incubated overnight with 2 μm GST-S4EDΔ87–131 or GST-S4ED (bait) in the presence of 30 μm SSTN87–131, or SSTN89A,99–108A,125–131A (SSTN-triple alanine mutant (TAM), then the fusion proteins are captured on glutathione beads and subjected to Western blot analysis to probe for capture of HER2, EGFR, α6β4 integrin, or α3β1 integrin. The relative capture of EGFR versus α3β1 integrin in the presence of the peptides is quantified in the bar graph as a percentage of capture in the absence of peptide (*, p < 0.01).
In contrast to S1ED, S4ED captures EGFR and α3β1 integrin, but fails to capture HER2 or the α6β4 integrin (Fig. 7B). Capture of both receptors is abolished if amino acids 87–131 are deleted (GST-S4EDΔ87–131). Capture of EGFR and α3β1 integrin are competed by SSTN87–131. No single mutation within this sequence abolishes capture of EGFR or α3β1 integrin (data not shown), suggesting that their capture utilizes multiple interaction sites within the sequence. However, SSTN competition is minimized if several conserved sites are all converted to alanine (e.g. a triple alanine mutant (TAM) consisting of 89A, 99–108A, and 125–131A) (Fig. 7B).
These findings strongly suggest that HER2 and EGFR interact directly with Sdc1 and Sdc4, respectively. To test this using yet another approach, we elected to conduct capture assays using purified recombinant proteins (Fig. 8). The extracellular domains of HER2 (HER2ecto) and EGFR (EGFRecto) were expressed as 6×His-tagged proteins in insect cells, purified, and incubated overnight with recombinant GST-S1ED or GST-S4ED attached to glutathione beads. HER2ecto is captured by S1ED and this is effectively blocked by SSTN210–240 (Fig. 8A). EGFR is not captured by Sdc1. In contrast, EGFRecto, and not HER2ecto, is captured by S4ED and this capture is effectively blocked by SSTN87–131 (Fig. 8B).
FIGURE 8.
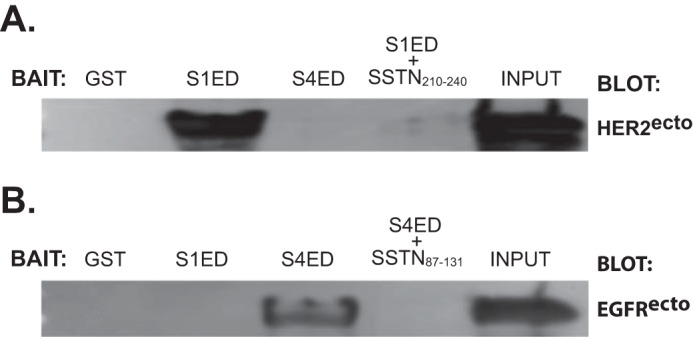
The extracellular domains of HER2 and EGFR interact directly with Sdc1 and Sdc4, respectively. A, recombinant HER2ecto expressed and purified from S2 insect cell conditioned medium is incubated overnight with recombinant GST, GST-S1ED with or without SSTN210–240 or with GST-S4ED, then the GST fusion proteins are captured on glutathione beads and probed for binding of HER2ecto. An equivalent amount of HER2ecto used in the capture assay is shown as input. B, recombinant EGFRecto expressed in insect cells is incubated overnight with recombinant GST, GST-S4ED with or without SSTN87–131 or with GST-S1ED, then the GST fusion proteins are captured on glutathione beads and probed for binding of EGFRecto. An equivalent amount of EGFRecto used in the capture assay is shown as input.
Discussion
These findings describe two novel mechanisms via which syndecans act as organizers at sites of cell-matrix adhesion to capture receptor tyrosine kinases and couple them to integrins to stimulate cell motility. This work extends a prior study in which Sdc1 and Sdc4 were shown to capture the α6β4 integrin via cytoplasmic interactions with syndecan-specific sites at the extreme C terminus of the β4 subunit (2, 3) (see model in Fig. 9). Mutation of Arg-1733 in the integrin abolishes capture by Sdc1 and generates a dominant-negative integrin mutant that prevents HER2-mediated cell motility that depends on the α3β1 integrin (Fig. 9). The coupling of α6β4 integrin to Sdc1 also appears necessary for HER2+ breast carcinoma cell survival (2).
FIGURE 9.

Model of SSTN peptides and receptor interactions. A, SSTN210–240 and SSTN87–131 are shown, along with highlighted homology sequences and potential binding sites for α3β1 integrin, HER2 or EGFR. Solid line denotes binding site, whereas dashed line denotes site of possible interaction. B, model showing association of HER2, α3β1 integrin and α6β4 integrin with Sdc1, and their displacement by SSTN210–240. The β4 subunit is phosphorylated in its “signaling domain” by Fyn kinase when bound to Sdc1 and HER2 is activated by clustering of the receptor complex (3). The engagement of α6β4 integrin with Sdc1 is prevented by mutation of Arg-1733 in the β4 cytoplasmic domain, which acts as a dominant negative mutant to blocks the HER2-dependent mechanism, as shown by Wang et al. (2). The model also depicts the association of EGFR, α3β1 integrin, and α6β4 integrin with Sdc4, and their displacement by SSTN87–131. The engagement of α6β4 integrin with Sdc4 is prevented by mutation of E1729A in the β4 cytoplasmic domain, which acts as a dominant negative mutant to block the EGF-stimulated mechanism (2). The model also depicts the heterodimerization of HER2 and EGFR, which is not affected by SSTN peptides. EGFR homodimerization, or heterodimerization with other family members is not shown.
Similarly, mutation of Glu-1729 in the β4 integrin cytoplasmic domain disrupts capture by Sdc4 and generates a dominant negative integrin mutant that blocks EGFR-stimulated cell motility involving the α3β1 integrin (2) (Fig. 9). Our present findings appear to explain the syndecan-specificity of these two mechanisms. HER2, but not EGFR, is captured along with the α3β1 integrin via an extracellular binding site (amino acids 210–240) in Sdc1 and in this manner is coupled by the cytoplasmic domain of Sdc1 to the α6β4 integrin. A peptide representing this juxtamembrane site, SSTN210–240, competitively disrupts binding of HER2 and α3β1 integrin to Sdc1 and blocks cell motility stimulated when LN332 is engaged by the syndecan or the α6β4 integrin (matrix-chemokinesis) (Fig. 9). In an analogous fashion, EGFR, but not HER2, is captured along with the α3β1 integrin via an extracellular site in Sdc4, which in turn couples them via its cytoplasmic domain to the α6β4 integrin (Fig. 9). SSTN87–131, which mimics the extracellular site in Sdc4, disrupts the capture of EGFR and α3β1 integrin by Sdc4 and blocks EGF stimulated cell motility (Fig. 9). We have no evidence that these inhibitory peptides have any effect on the formation of HER2-EGFR heterodimers, EGFR-EGFR homodimers, or dimerization of HER2 or EGFR with other EGFR family members.
Amino acid conservation within the sequences from both Sdc1 and Sdc4 deteriorates significantly across vertebrate species. Nonetheless, several sets of conserved amino acids in the Sdc1 sequence can be discerned in higher mammals and appear to be necessary for full activity, as deletion or alanine mutation of these discrete sets within S1ED fusion proteins or peptides impair their ability to competitively block cell migration in wound healing assays. Deletion of asp-phe-thr-phe213 at the N terminus of SSTN210–240 reduces activity by 30-fold and analysis of peptides lacking this sequence shows a reduced ability to compete for HER2 binding, suggesting that this motif interacts with HER2 (Fig. 9A). Similarly, deletion of gln-gly-ala-thr240 at the C terminus reduces activity by 100-fold, and this deletion reduces the ability of peptides to compete with capture of the α3β1 integrin, suggesting that this is a binding site for the α3β1 integrin. But the overall interaction is likely more complex, as mutation of conserved amino acids elsewhere in the peptide affect its overall competitive ability as well. An exception is the pro-val-asp236 sequence, which is highly conserved, but can be deleted without further loss of activity; this hints that these amino acids may have other functions and that this juxtamembrane region may be a multifunctional site. Indeed, other work in the Rapraeger laboratory3 indicates that this same region serves to capture the α4β1 integrin (VLA-4) and VEGFR2 on myeloma cells and vascular endothelial cells. This capture requires the region between amino acids 210–236, with the C-terminal gln-gly-ala-thr240 shown to be important in the HER2/α3β1 mechanism not required; in contrast, the pro-val-asp236 motif that is not necessary for capture of HER2 or α3β1 integrin is required for the VLA-4/VEGFR2 mechanism. Prior work has also implicated the ala-val-val-ala-val225 motif within this sequence as an inhibitory rather than stimulatory sequence during the invasion of type I collagen gels by human ARH-77 myeloma cells, although the receptors involved in this mechanism were not identified (40).
The binding site in Sdc4 also appears to be a multifunctional site. It corresponds to a so-called “cell binding domain” originally identified by using the Sdc4 extracellular domain as an artificial substratum for the attachment of NIH 3T3, dermal, and Swiss3T3 fibroblasts, as well as aortic endothelial cells by McFall and Rapraeger, although its binding partners on the cell were not identified (36, 37). This motif was characterized more extensively by Whiteford and Couchman, who demonstrated that it functions as an artificial cell binding domain for the β1-integrin-mediated adhesion of fibroblasts and leukocytes but not for epithelial cells (38), contrasting starkly with the epithelial role described here. Its binding partners again remained obscure, but seem unlikely to involve EGFR, α3β1, or α6β4 integrin given the cell types involved.
This site contains multiple sets of amino acids conserved across vertebrate species to zebrafish, namely, asn-x-iso-pro90, asn-asp-val111, and ser-asn-x-val-x-met131. Their deletion or mutation to alanine in S4ED or peptides used as competitors shows that asn-x-iso-pro90 and ser-asn-x-val-x-met131 have a role in this mechanism, but the conserved asn-asp-val111 motif does not. Isoleucine 89 within the N-terminal motif is clearly important for its activity, mimicking the importance of this amino acid in zebrafish and fibroblasts. However, other conserved sites, found in higher mammals but lacking in lower mammals and zebrafish are necessary for full activation of the Sdc4-coupled EGFR and α3β1 integrin, including val-pro-thr-x-x-lys-x-leu106 and iso-pro-lys114. In fact, even mS4ED99–119 retains significant competitive ability despite the fact that the only amino acids within this sequence that are conserved in zebrafish, asn-asp-val, are not required for activity. Clearly, a full understanding of the capture mechanism used here will require in-depth structural work beyond the scope of the present studies.
These findings show clearly that the interaction between Sdc1 and HER2, and between Sdc4 and EGFR are direct. This is an entirely novel interaction for these well-described receptor tyrosine kinases. It is also intriguing that the syndecan extracellular domains capture only their respective kinase from cell lysates, despite the ability of HER2 and EGFR to form heterodimers and the presence of such heterodimers on these cells. This was observed in prior studies in which Sdc1 and Sdc4 were observed to regulate cell migration stimulated by these kinases and in which only HER2 acted in concert with Sdc1 and only EGFR acted with Sdc4 (2). This raises the intriguing possibility that heterodimers comprised of EGFR family members are not capable of associating with these syndecans, or even that the kinases associate with the syndecan as a monomer.
It is interesting to note that both extracellular motifs are juxtamembrane in their respective syndecan. The sequence in human Sdc1, amino acids 210–240, is located eleven amino acids from the plasma membrane whereas the site in Sdc4, amino acids 87–131, is 14 amino acids from its transmembrane domain. Thus, the interaction sites on the receptor tyrosine kinases and α3β1 integrin may also be juxtamembrane sites. This stretch of amino acids in the two syndecans is near to or contains the sites for attachment of chondroitin sulfate glycosaminoglycan chains (e.g. EXSG207 and EXSG217 in Sdc1); although Sdc1 is commonly referred to as a heparan sulfate proteoglycan, it was actually the first proteoglycan shown to be a hybrid, that is, to display both heparan sulfate and chondroitin sulfate chains (41). The importance of chondroitin sulfate at this site remains largely unknown, although one could speculate given the current findings that its attachment serves to inhibit the interaction with HER2 and integrin, and that its enzymatic removal could facilitate the interactions and promote cell motility.
In summary, the assimilation of receptor tyrosine kinases into signaling complexes with the α6β4 integrin have been shown by a variety of workers to be critical for carcinogenesis (9–14, 18, 20, 23, 25, 29). At least in the case of HER2 and EGFR, this assimilation appears to depend on syndecans. The synstatin peptides described here, which prevent capture of the kinase and its stimulation of integrin-mediate motility and cell survival, are potential therapeutics for the treatment of these diseases.
Author Contributions
A. C. R. conceived and coordinated the study, designed the experiments, and wrote the paper. H. W. performed and analyzed the experiments. H. J. generated the protein expression constructs used in these studies. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank the University of Wisconsin Carbone Cancer Center for the use of its shared services, supported by the National Cancer Institute (P30 CA014520). We are especially thankful for reagents from Filippo Giancotti, Memorial Sloan-Kettering Cancer Center, that have made this work possible.
This work was supported by National Institutes of Health Grants R01-CA109010 and R01-CA163662 (to A. C. R.). The authors declare that they have no conflicts of interest with the contents of this article
O. Jung, V. Trapp-Stamborski, A. Purushothaman, R. D. Sanderson, and A. C. Rapraeger, manuscript submitted.
- LN
- laminin
- Sdc
- syndecan
- LPA
- oleoyl-l-α-lysophosphatidic acid.
References
- 1.Nievers M. G., Schaapveld R. Q., and Sonnenberg A. (1999) Biology and function of hemidesmosomes. Matrix Biol. 18, 5–17 [DOI] [PubMed] [Google Scholar]
- 2.Wang H., Jin H., Beauvais D. M., and Rapraeger A. C. (2014) Cytoplasmic domain interactions of syndecan-1 and syndecan-4 with α6β4 integrin mediate human epidermal growth factor receptor (HER1 and HER2)-dependent motility and survival. J. Biol. Chem. 289, 30318–30332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang H., Leavitt L., Ramaswamy R., and Rapraeger A. C. (2010) Interaction of syndecan and α6β4 integrin cytoplasmic domains: Regulation of ErbB2-mediated integrin activation. J. Biol. Chem. 285, 13569–13579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Pereda J. M., Lillo M. P., and Sonnenberg A. (2009) Structural basis of the interaction between integrin α6β4 and plectin at the hemidesmosomes. EMBO J. 28, 1180–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koster J., van Wilpe S., Kuikman I., Litjens S. H., and Sonnenberg A. (2004) Role of binding of plectin to the integrin β4 subunit in the assembly of hemidesmosomes. Mol. Biol. Cell 15, 1211–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frijns E., Kuikman I., Litjens S., Raspe M., Jalink K., Ports M., Wilhelmsen K., and Sonnenberg A. (2012) Phosphorylation of threonine 1736 in the C-terminal tail of integrin β4 contributes to hemidesmosome disassembly. Mol. Biol. Cell 23, 1475–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilhelmsen K., Litjens S. H., Kuikman I., Margadant C., van Rheenen J., and Sonnenberg A. (2007) Serine phosphorylation of the integrin β4 subunit is necessary for epidermal growth factor receptor induced hemidesmosome disruption. Mol. Biol. Cell 18, 3512–3522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rabinovitz I., Tsomo L., and Mercurio A. M. (2004) Protein kinase C-α phosphorylation of specific serines in the connecting segment of the β4 integrin regulates the dynamics of type II hemidesmosomes. Mol. Cell Biol. 24, 4351–4360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo W., Pylayeva Y., Pepe A., Yoshioka T., Muller W. J., Inghirami G., and Giancotti F. G. (2006) β4 integrin amplifies ErbB2 signaling to promote mammary tumorigenesis. Cell 126, 489–502 [DOI] [PubMed] [Google Scholar]
- 10.Mariotti A., Kedeshian P. A., Dans M., Curatola A. M., Gagnoux-Palacios L., and Giancotti F. G. (2001) EGF-R signaling through Fyn kinase disrupts the function of integrin α6β4 at hemidesmosomes: role in epithelial cell migration and carcinoma invasion. J. Cell Biol. 155, 447–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bertotti A., Comoglio P. M., and Trusolino L. (2005) β4 integrin is a transforming molecule that unleashes Met tyrosine kinase tumorigenesis. Cancer Res. 65, 10674–10679 [DOI] [PubMed] [Google Scholar]
- 12.Trusolino L., Bertotti A., and Comoglio P. M. (2001) A signaling adapter function for α6β4 integrin in the control of HGF-dependent invasive growth. Cell 107, 643–654 [DOI] [PubMed] [Google Scholar]
- 13.Falcioni R., Antonini A., Nisticò P., Di Stefano S., Crescenzi M., Natali P. G., and Sacchi A. (1997) α6β4 and α6β1 integrins associate with ErbB-2 in human carcinoma cell lines. Exp. Cell Res. 236, 76–85 [DOI] [PubMed] [Google Scholar]
- 14.Bertotti A., Comoglio P. M., and Trusolino L. (2006) β4 integrin activates a Shp2-Src signaling pathway that sustains HGF-induced anchorage-independent growth. J. Cell Biol. 175, 993–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilhelmsen K., Litjens S. H., and Sonnenberg A. (2006) Multiple functions of the integrin α6β4 in epidermal homeostasis and tumorigenesis. Mol. Cell Biol. 26, 2877–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Russell A. J., Fincher E. F., Millman L., Smith R., Vela V., Waterman E. A., Dey C. N., Guide S., Weaver V. M., and Marinkovich M. P. (2003) α6β4 integrin regulates keratinocyte chemotaxis through differential GTPase activation and antagonism of α3β1 integrin. J. Cell Sci. 116, 3543–3556 [DOI] [PubMed] [Google Scholar]
- 17.Sehgal B. U., DeBiase P. J., Matzno S., Chew T. L., Claiborne J. N., Hopkinson S. B., Russell A., Marinkovich M. P., and Jones J. C. (2006) Integrin β4 regulates migratory behavior of keratinocytes by determining laminin-332 organization. J. Biol. Chem. 281, 35487–35498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldfinger L. E., Hopkinson S. B., deHart G. W., Collawn S., Couchman J. R., and Jones J. C. (1999) The α3 laminin subunit, α6β4 and α3β1 integrin coordinately regulate wound healing in cultured epithelial cells and in the skin. J. Cell Sci. 112, 2615–2629 [DOI] [PubMed] [Google Scholar]
- 19.Folgiero V., Avetrani P., Bon G., Di Carlo S. E., Fabi A., Nisticò C., Vici P., Melucci E., Buglioni S., Perracchio L., Sperduti I., Rosanò L., Sacchi A., Mottolese M., and Falcioni R. (2008) Induction of ErbB-3 expression by α6β4 integrin contributes to tamoxifen resistance in ERβ1-negative breast carcinomas. PLoS ONE 3, e1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Folgiero V., Bachelder R. E., Bon G., Sacchi A., Falcioni R., and Mercurio A. M. (2007) The α6β4 integrin can regulate ErbB-3 expression: implications for α6β4 signaling and function. Cancer Res. 67, 1645–1652 [DOI] [PubMed] [Google Scholar]
- 21.Gambaletta D., Marchetti A., Benedetti L., Mercurio A. M., Sacchi A., and Falcioni R. (2000) Cooperative signaling between α(6)β(4) integrin and ErbB-2 receptor is required to promote phosphatidylinositol 3-kinase-dependent invasion. J. Biol. Chem. 275, 10604–10610 [DOI] [PubMed] [Google Scholar]
- 22.Nisticò P., Di Modugno F., Spada S., and Bissell M. J. (2014) β1 and β4 integrins: from breast development to clinical practice. Breast Cancer Res. 16, 459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dans M., Gagnoux-Palacios L., Blaikie P., Klein S., Mariotti A., and Giancotti F. G. (2001) Tyrosine phosphorylation of the β4 integrin cytoplasmic domain mediates Shc signaling to extracellular signal-regulated kinase and antagonizes formation of hemidesmosomes. J. Biol. Chem. 276, 1494–1502 [DOI] [PubMed] [Google Scholar]
- 24.Dutta U., and Shaw L. M. (2008) A key tyrosine (Y1494) in the β4 integrin regulates multiple signaling pathways important for tumor development and progression. Cancer Res. 68, 8779–8787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Merdek K. D., Yang X., Taglienti C. A., Shaw L. M., and Mercurio A. M. (2007) Intrinsic signaling functions of the β4 integrin intracellular domain. J. Biol. Chem. 282, 30322–30330 [DOI] [PubMed] [Google Scholar]
- 26.Shaw L. M. (2001) Identification of insulin receptor substrate 1 (IRS-1) and IRS-2 as signaling intermediates in the α6β4 integrin-dependent activation of phosphoinositide 3-OH kinase and promotion of invasion. Mol. Cell Biol. 21, 5082–5093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shaw L. M., Rabinovitz I., Wang H. H., Toker A., and Mercurio A. M. (1997) Activation of phosphoinositide 3-OH kinase by the α6β4 integrin promotes carcinoma invasion. Cell 91, 949–960 [DOI] [PubMed] [Google Scholar]
- 28.Yang X., Dutta U., and Shaw L. M. (2010) SHP2 mediates the localized activation of Fyn downstream of the α6β4 integrin to promote carcinoma invasion. Mol. Cell Biol. 30, 5306–5317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nikolopoulos S. N., Blaikie P., Yoshioka T., Guo W., and Giancotti F. G. (2004) Integrin β4 signaling promotes tumor angiogenesis. Cancer Cell 6, 471–483 [DOI] [PubMed] [Google Scholar]
- 30.Yoshioka T., Otero J., Chen Y., Kim Y. M., Koutcher J. A., Satagopan J., Reuter V., Carver B., de Stanchina E., Enomoto K., Greenberg N. M., Scardino P. T., Scher H. I., Sawyers C. L., and Giancotti F. G. (2013) β4 Integrin signaling induces expansion of prostate tumor progenitors. J. Clin. Invest. 123, 682–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beauvais D. M., Ell B. J., McWhorter A. R., and Rapraeger A. C. (2009) Syndecan-1 regulates αvβ3 and αvβ5 integrin activation during angiogenesis and is blocked by synstatin, a novel peptide inhibitor. J. Exp. Med. 206, 691–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beauvais D. M., and Rapraeger A. C. (2010) Syndecan-1 couples the insulin-like growth factor-1 receptor to inside-out integrin activation. J. Cell Sci. 123, 3796–3807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rapraeger A. C. (2013) Synstatin: a selective inhibitor of the syndecan-1-coupled IGF1R-αvβ3 integrin complex in tumorigenesis and angiogenesis. FEBS J 280, 2207–2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rapraeger A. C., Ell B. J., Roy M., Li X., Morrison O. R., Thomas G. M., and Beauvais D. M. (2013) Vascular endothelial-cadherin stimulates syndecan-1-coupled insulin-like growth factor-1 receptor and cross-talk between αVβ3 integrin and vascular endothelial growth factor receptor 2 at the onset of endothelial cell dissemination during angiogenesis. FEBS J 280, 2194–2206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bernfield M., Götte M., Park P. W., Reizes O., Fitzgerald M. L., Lincecum J., and Zako M. (1999) Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 68, 729–777 [DOI] [PubMed] [Google Scholar]
- 36.McFall A. J., and Rapraeger A. C. (1997) Identification of an adhesion site within the syndecan-4 extracellular protein domain. J. Biol. Chem. 272, 12901–12904 [DOI] [PubMed] [Google Scholar]
- 37.McFall A. J., and Rapraeger A. C. (1998) Characterization of the high affinity cell-binding domain in the cell surface proteoglycan syndecan-4. J. Biol. Chem. 273, 28270–28276 [DOI] [PubMed] [Google Scholar]
- 38.Whiteford J. R., and Couchman J. R. (2006) A conserved NXIP motif is required for cell adhesion properties of the syndecan-4 ectodomain. J. Biol. Chem. 281, 32156–32163 [DOI] [PubMed] [Google Scholar]
- 39.Hynes N. E., and MacDonald G. (2009) ErbB receptors and signaling pathways in cancer. Curr. Opin. Cell Biol. 21, 177–184 [DOI] [PubMed] [Google Scholar]
- 40.Langford J. K., Yang Y., Kieber-Emmons T., and Sanderson R. D. (2005) Identification of an invasion regulatory domain within the core protein of syndecan-1. J. Biol. Chem. 280, 3467–3473 [DOI] [PubMed] [Google Scholar]
- 41.Rapraeger A., Jalkanen M., Endo E., Koda J., and Bernfield M. (1985) The cell surface proteoglycan from mouse mammary epithelial cells bears chondroitin sulfate and heparan sulfate glycosaminoglycans. J. Biol. Chem. 260, 11046–11052 [PubMed] [Google Scholar]