

Background: Mutations in the Ca2+ sensing protein calmodulin (CaM) cause lethal cardiac arrhythmias.
Results: CaM mutations impair the activation and termination of store overload-induced Ca2+ release via the cardiac ryanodine receptor (RyR2).
Conclusion: CaM mutations alter RyR2-CaM interaction, thereby affecting RyR2-mediated Ca2+ release.
Significance: Aberrant regulation of RyR2 store Ca2+ sensing is a potential component of calmodulin-mediated cardiac arrhythmias.
Keywords: calcium intracellular release, calmodulin (CaM), excitation-contraction coupling (E-C coupling), protein-protein interaction, receptor regulation, ryanodine receptor, arrhythmia
Abstract
The intracellular Ca2+ sensor calmodulin (CaM) regulates the cardiac Ca2+ release channel/ryanodine receptor 2 (RyR2), and mutations in CaM cause arrhythmias such as catecholaminergic polymorphic ventricular tachycardia (CPVT) and long QT syndrome. Here, we investigated the effect of CaM mutations causing CPVT (N53I), long QT syndrome (D95V and D129G), or both (CaM N97S) on RyR2-mediated Ca2+ release. All mutations increased Ca2+ release and rendered RyR2 more susceptible to store overload-induced Ca2+ release (SOICR) by lowering the threshold of store Ca2+ content at which SOICR occurred and the threshold at which SOICR terminated. To obtain mechanistic insights, we investigated the Ca2+ binding of the N- and C-terminal domains (N- and C-domain) of CaM in the presence of a peptide corresponding to the CaM-binding domain of RyR2. The N53I mutation decreased the affinity of Ca2+ binding to the N-domain of CaM, relative to CaM WT, but did not affect the C-domain. Conversely, mutations N97S, D95V, and D129G had little or no effect on Ca2+ binding to the N-domain but markedly decreased the affinity of the C-domain for Ca2+. These results suggest that mutations D95V, N97S, and D129G alter the interaction between CaM and the CaMBD and thus RyR2 regulation. Because the N53I mutation minimally affected Ca2+ binding to the C-domain, it must cause aberrant regulation via a different mechanism. These results support aberrant RyR2 regulation as the disease mechanism for CPVT associated with CaM mutations and shows that CaM mutations not associated with CPVT can also affect RyR2. A model for the CaM-RyR2 interaction, where the Ca2+-saturated C-domain is constitutively bound to RyR2 and the N-domain senses increases in Ca2+ concentration, is proposed.
Introduction
During cardiac excitation, Ca2+ entry into the cytoplasm of cardiomyocytes through sarcolemmal voltage-gated Ca2+ channels (CaV1.2) activates RyR22 channels in the SR, giving rise to the so-called Ca2+-induced Ca2+ release (1–3). Ca2+ released from the SR eventually leads to increases in cytosolic free Ca2+ ([Ca2+]cyt) throughout the cardiomyocyte, where binding of Ca2+ to myofilaments results in contraction (4). RyR2 channels are, however, not only sensitive to [Ca2+]cyt but also the SR luminal free Ca2+ ([Ca2+]SR), and both calcium concentrations modulate the activation and termination of Ca2+ release (1, 5–9).
In the SR membrane, RyR2s arrange as homotetrameric channels extending into the cytosol, where the interaction with numerous proteins and ligands regulate the Ca2+ release activity of the channel (4, 10). Among these RyR2 modulators, CaM is a cytosolic inhibitor of Ca2+ release both at diastolic and systolic [Ca2+]cyt and may also serve additional regulatory purposes (11–14). CaM is a ubiquitous Ca2+ sensing protein in vertebrates and confers its sensing of intracellular Ca2+ signals onto a multitude of protein targets, including ion channels and pumps responsible for excitation and contraction in cardiomyocytes (15). Although a cytosolic protein, CaM does affect the response of the RyR2 channel to [Ca2+]SR, most likely as part of an extensive allosteric regulation of RyR2 (14).
CaM consists of two domains (C- and N-domains, respectively) with a flexible linker between them that enables independent and correlated functions. Each domain is comprised of two structurally integrated EF hand motifs that each binds one Ca2+ ion (Fig. 1). Although highly homologous with extensive sequence identity, the two domains of CaM display distinct Ca2+ binding properties and both independent and correlated interactions with protein targets. CaM binds to RyR2 with a stoichiometry of four per channel, primarily via the CaM binding domain (CaMBD) (Arg3581–Pro3607, human RyR2) (15–18). In the Ca2+ saturated form, the CaM C-domain appears to bind RyR2 around Trp3587 in the CaMBD, and the CaM N-domain appears to bind RyR2 in the vicinity of Phe3603, although the CaM N-domain interaction is more promiscuous, especially at low free Ca2+ concentrations ([Ca2+]free) (18–20). Even in the absence of Ca2+, CaM can bind to the RyR2 CaMBD, most likely via the C-domain (18, 19). Also, an engineered CaM mutant (CaM E31A/E67A/E104A/E140A, CaM1234) defective in Ca2+ binding is a competitive inhibitor of the native regulation of RyR2 by the CaM WT (21). Hence, CaM binding to RyR2 and Ca2+ binding to CaM are each critical determinants of RyR2 regulation by CaM.
FIGURE 1.
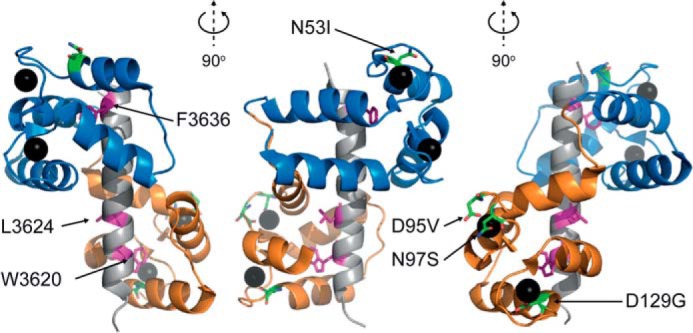
Representative structure of CaM. Ca2+-saturated CaM is shown binding a 27-residue peptide corresponding to part of the CaMBD in RyR1(Lys3614–Pro3640) (PDB ID: 2BCX), differing in one residue from the CaMBD in RyR2 (RyR1 Thr3639). Protein and peptide secondary structures are shown as cartoon representation, the N-domain of CaM (Ala1–Thr79) is indicated in blue, and the C-domain (Asp80–Lys148) is in orange, and the peptide is in gray. Sites of CaM mutations (N53I, N97S, D95V, and D129G) are highlighted as green stick representations (nonmutated residues), and Ca2+ ions are shown as black spheres. The RyR1 CaMBD residues (Trp3620, Leu3624, and Phe3636) corresponding to RyR2 CaMBD residues Trp3587, Leu3591, and Phe3603 are highlighted as magenta stick representations. Numbering is according to human RyR2 and mature CaM without initiating Met residue.
Three genes (CALM1–3) in the human genome encode the exact same CaM protein, and we previously found that two separate mutations in the CALM1 gene (N53I and N97S, mature CaM numbering) each lead to dominantly inherited CPVT (22). Subsequently, Crotti et al. (23) identified a CaM mutation in CALM2 (D95V) and two in CALM1 (D129G and F141L) each dominantly causing long QT syndrome (LQTS). More recently, Makita et al. (24) found another five mis-sense mutations in CALM2, three in individuals with severe LQTS (N97I, D133H, and, interestingly, N97S) and two in individuals showing features of both LQTS and CPVT (D131E and Q135P). In addition, a CaM F89L mutation was identified in a family with idiopathic ventricular fibrillation (25).
Both CPVT and LQTS lead to perturbations of excitation-contraction cycles in cardiomyocytes, which in turn can lead to ventricular fibrillation and sudden cardiac death. CPVT is characterized by syncope or sudden cardiac death following exercise or acute emotion, whereas LQTS also affects the resting heart and with increasing effect upon adrenergic stimulation (25, 26). Despite the universal function of CaM, all mutations identified so far confer arrhythmia phenotypes with few or no other symptoms observed in carriers (1, 22–24, 27).
The RyR2 channel accounts for the bulk of Ca2+ introduced into the cardiomyocyte cytosol during contraction, and mutations in RyR2 and auxiliary proteins cause CPVT (28–30). Thus, the aberrant regulation of RyR2 by CaM is a likely convergence point for the CaM N53I and N97S disease mechanisms causing CPVT (1, 15). This is also consistent with the observation that these CaM mutations increase spontaneous Ca2+ release in permeabilized cardiomyocytes (15, 31). Conversely, LQTS is mainly associated with dysfunction of sarcolemmal voltage-gated Na+, Ca2+, or K+ channels in control of the action potential. Notably the activities of a majority of these channels are regulated by CaM, for example, CaM mutations N97S, D95V, F141L and D129G all confer reduced Ca2+-dependent inhibition of the cardiac L-type voltage-gated Ca2+ channel (CaV1.2) (15, 26, 32).
In this study, we investigated the impact of CaM mutations linked to CPVT (N53I), LQTS (D95V and D129G), or both (N97S) on the CaM-dependent regulation of RyR2 channels. To this end, we monitored the endoplasmic reticulum (ER) Ca2+ dynamics in RyR2-expressing HEK293 cells transfected with CaM WT or mutants. The RyR2-mediated Ca2+ release in this system is triggered by increasing the Ca2+ load in the ER, which mimics the store overload-induced Ca2+ release (SOICR) model for CPVT (14, 33). Furthermore, we investigated domain-specific interactions of CaM with Ca2+ in the presence of a peptide corresponding to the CaMBD of RyR2. Our results demonstrate that both CPVT and LQTS-associated CaM mutations alter RyR2-mediated Ca2+ release.
Experimental Procedures
Plasmid Constructs
Plasmid constructs based on the pMAL vector (New England Biolabs) for recombinant expression and purification of native, full-length CaM were prepared as described previously (1, 22). For expression of CaM variants in HEK293 cells, CaM coding sequences from the pMAL vectors were PCR-amplified and the products ligated into pcDNA3.1 vectors (Invitrogen). Chemically competent Escherichia coli DH5a cells (in-house stock) were transformed with pcDNA3.1 vectors, and overnight cultures were used to prepare purified plasmid preparations using the Qiagen plasmid maxi kit. pcDNA3.1 vectors were eluted in double-distilled water, and Sanger sequencing verified the sequence of CaM encoding inserts in all plasmids.
Model Fitting and Statistical Analysis
All fitting of data to mathematical models and statistical analyses was done using GraphPad Prism 6 for Mac (version 6.0f). Models and statistical method details are described below.
Endoplasmic Reticulum Luminal Ca2+ Imaging of HEK293 Cells Expressing RyR2 during Store Overload-induced Ca2+ Release
Stable expression of murine RyR2 in HEK293 cells co-transfected with plasmids encoding CaM, and the D1ER Ca2+ probe was done as previously described (14). Briefly, D1ER FRET signals reflecting ER luminal [Ca2+]free were monitored for individual cells in an epifluorescent microscope setting with perfusion (6, 14) (Fig. 2A). Each FRET signal trace was used to measure the Ca2+ release properties of the RyR2 channels relative to the ER Ca2+ content: the activation and termination thresholds (Fig. 2A) and their difference, the fractional Ca2+ release. The ER Ca2+ store capacities were calculated as the difference between maximum and minimum FRET signal (Fmax − Fmin). Experiments were also done with HEK293 cells expressing a RyR2 mutant with the CaMBD deleted (RyR2 ΔCaMBD, murine RyR2 ΔLys3583–Phe3603). Measured parameters were compared using one-way analysis of variance with Holm-Sidak multiple comparison test for all possible combinations and with p < 0.01 (against the CaM WT values) chosen as a conservative measure of significance.
FIGURE 2.

Store overload-induced Ca2+ release from RyR2-expressing cells transfected with CaM WT and CPVT-linked CaM mutations. A–C, the FRET signal from the ER luminal D1ER [Ca2+]free indicator oscillates as Ca2+ is released through RyR2. Rectangles and labels indicate the concentrations of Ca2+, tetracaine (RyR2 channel inhibitor), and caffeine (activator) in the perfusion solution. Stepwise increase in Ca2+ concentration elicited RyR2 SOICR oscillations, tetracaine blocked Ca2+ release filling ER to maximum [Ca2+]free, and finally caffeine opened RyR2 channels depleting ER Ca2+. Tetracaine and caffeine were used to establish maximum and minimum ER [Ca2+]free as measured using D1ER (Fmax and Fmin), respectively. The activation and termination thresholds were calculated relative to Fmax and Fmin. Example traces for transfection with CaM WT (A), N53I (B), and N97S (C) are shown. D–G, the activation threshold (D), termination threshold (E), fractional release (F), and store capacity (G) averaged from multiple traces are shown as bar graphs. The error bars show S.D., and asterisks indicate significant changes compared with CaM WT (p < 0.01).
Protein Expression and Purification
CaM was expressed from the pMAL (CaM N53I and N97S) or the pET15b (D95V and D129G) vector in E. coli Rosetta B cells (EMD Chemicals) or E. coli BL21 (DE3) cells (Novagen), respectively, and purified as previously described (1, 22, 23). The identity, purity, and integrity of each protein preparation were confirmed by SDS-PAGE and MALDI-TOF mass spectrometry of trypsin-digested proteins.
Peptides Corresponding to the CaM Binding Domain of RyR2
A peptide corresponding to the CaMBD of RyR2 (RyR2(Arg3581–Leu3611), human RyR2 3581RSKKAVWHKLLSKQRKRAVVACFRMAPLYNL3611) was purchased from Peptide 2.0 Inc. (Chantilly, VA) at >98% purity.
Titration Buffers and Verification of Free Ca2+ Concentrations
Titration experiments were performed by mixing different volumes of pH- and Ca2+-buffered solution (50 mm HEPES, 100 mm KCl, 0.5 mm EGTA, and 2 mm nitrilotriacetic acid at pH 7.2 (25 °C)) with the same buffer spiked to 3, 7, or 22 mm CaCl2 to reach precalculated [Ca2+]free levels (5). In practice, ×1.5 concentrated buffer stocks were prepared and proteins, peptide, Ca2+ probes (final concentration, 0.75 μm), and reducing agent (tris(2-carboxyethyl)phosphine; final concentration, 16.5 μm) were added to the double distilled water used for diluting concentrated buffers. The calculated buffer ionic strength, which affects Ca2+ binding to CaM, was stable at 0.15 m. For all titration experiments, the [Ca2+]free was followed by including the Ca2+ probe Fura-2 or Fura-6F in solutions (Invitrogen) and indirectly via measuring of Ca2+ binding to CaM WT (1). Based on these measurements, a 15% error for the [Ca2+]free was included throughout data sets and fitting procedures.
Titrations of CaM/ RyR2(Arg3581–Leu3611) and Free CaM with Ca2+
15 μm CaM in the presence of 16.5 μm RyR2(Arg3581–Leu3611) was titrated with Ca2+ as previously described (1, 5). Briefly, intrinsic protein fluorescence from the N- and C-domain of CaM were measured as partial Phe and Tyr emission spectra, respectively (Table 1), using a spectrofluorometer (HORIBA Jobin Yvon, FluoroMax®-4P) (1, 34, 35). In addition, the Trp fluorescence from the RyR2(Arg3581–Leu3611) peptide was also measured (Table 1). Titrations of free CaM WT, D95V, and D129G with Ca2+ were done by mixing discontinuous titration points in an automated liquid handler (Hamilton, Microlab STARlet) and serially transferring these to a 2-mm cuvette for emission spectra recording. Measurements were done in triplicate with 10 μm CaM. Each of the fractional saturations (Y) for the N- and C-domains of CaM were fitted to the raw fluorescence intensity (FI) signals from the partial Phe and Tyr spectra at 280 and 320 nm, respectively, according to the following,
where the constants b and a indicate the initial FI and the span in FI from low to high [Ca2+]free, respectively. Y is the fractional saturation of the monitored CaM domain binding to two Ca2+ as described by a two-site Adair model (36, 37),
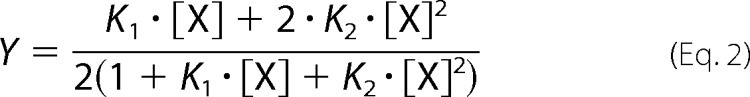 |
where K1 is the sum of the microscopic equilibrium constants, and K2 is the equilibrium constant for the particular domain binding to two Ca2+. The apparent dissociation constants for either domain in the free CaM or in the presence of RyR2(Arg3581–Leu3611) (appKDfree and appKDbound, respectively) were calculated as the reciprocal square root of K2. When CaM binds to RyR2(Arg3581–Leu3611), the peptide Trp fluorescence shows a peak shift from ∼350 to 340 nm, and furthermore the FI increases markedly upon Ca2+ binding to CaM (22). The raw FI for Trp fluorescence at 340 nm was also fitted to the model described above. Titration curves were normalized using the fitted a and b parameters for figure plotting purposes only. Statistical significances of differences in K2 were evaluated via nonoverlapping 95% confidence intervals or one-way analysis of variance with Dunnett's post hoc test against values measured for the CaM WT and p < 0.05 considered significant. Furthermore, the K2 values were also used to calculate the mutation-induced change in Gibb's free energy of Ca2+ binding to the domains of CaM (ΔΔGofree and ΔΔGobound respectively) according to the following,
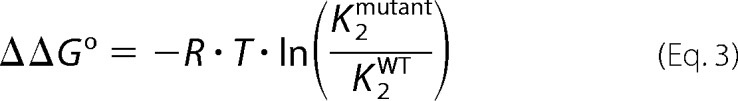 |
using standard conditions of 1 m and 298.15 K (25 °C).
TABLE 1.
Fluorescence spectrophotometry measurements
Acquisition settings for measuring Trp, Phe, and Tyr fluorescence spectra are shown. Integration time was 0.2 s for all recordings. Exc and Em are excitations and emission wavelengths, respectively.
| Spectra | Exc | Em | Bandwidths | Spectra averaged | Increments |
|---|---|---|---|---|---|
| nm | nm | nm | nm | ||
| Phe | 250 | 264–284 | 7/7 | 3 | 2 |
| Tyr | 270 | 310–330 | 3/3 | 2 | 3 |
| Trp | 295 | 336–370 | 3/3 | 2 | 2 |
Results
Arrhythmogenic CaM Mutations Decrease the Activation and Termination Thresholds for Spontaneous Ca2+ Release
Spontaneous Ca2+ release can occur in cardiomyocytes during increased Ca2+ load in the SR lumen. This spontaneous SOICR is arrhythmogenic, because it may lead to delayed afterdepolarizations and triggered activity, a hallmark of CPVT (25, 38). To determine whether CaM mutations affect this arrhythmogenic SOICR, we transfected RyR2-expressing HEK293 cells with CaM and monitored the ER Ca2+ dynamics using a FRET-based ER luminal Ca2+ probe D1ER (14). Perfusion of these transfected cells with increasing extracellular Ca2+ concentrations induced SOICR in the form of ER Ca2+ oscillations (Fig. 2A), as reported previously (14, 39). The oscillating D1ER FRET signal was then used to calculate the ER Ca2+ level required for activating SOICR, the activation threshold, and the ER Ca2+ depletion required for terminating SOICR, the termination threshold (Fig. 2A and “Experimental Procedures”). Perfusion with the RyR2 inhibitor tetracaine and then the agonist caffeine established maximum (Fmax) and minimum (Fmin) ER Ca2+ content. The ER capacity for storing Ca2+, the store capacity, was calculated as Fmax − Fmin.
In the SOICR experiments, the CPVT-linked CaM mutation, N53I, lowered the SOICR activation threshold (5%) relative to CaM WT, and so did the CPVT- and LQTS-linked mutation CaM N97S (4%) (Fig. 2, B–D, and Table 2). Similarly, the two LQTS-linked CaM mutations, D95V and D129G, also lowered the SOICR activation threshold by 6 and 4% (Fig. 3, B–D, and Table 2), respectively. The effects on the activation thresholds are modest, however, in cardiomyocytes SR Ca2+ release increases with increasing SR luminal Ca2+ concentrations in a steep and nonlinear fashion (40). Hence, even modest effects of CaM mutations on the response of RyR2 to SR luminal Ca2+ may have major impacts on SR Ca2+ release.
TABLE 2.
Quantified change in Ca2+ release properties for RyR2 channels expressed in HEK293 cells transfected with CaM
Numbers in parentheses indicate the percentages of statistically significant change in the measured properties for RyR2 Ca2+ release relative to RyR2 expressing cells transfected with CaM WT (p < 0.01, one-way analysis of variance). Transfections of RyR2 ΔCaMBD expressing cells with CaM variants did not change the Ca2+ release properties (Fig. 4, A–E). For comparison to RyR2 WT expressing cells, an average for all RyR2 ΔCaMBD experiments was calculated (Average RyR2 ΔCaMBD).
| CaM variant | Activation | Termination | Fractional release |
|---|---|---|---|
| % | % | % | |
| WT | 91 | 56 | 35 |
| N53I | 86 (−5) | 44 (−21) | 42 (20) |
| N97S | 87 (−4) | 39 (−30) | 48 (38) |
| D95V | 85 (−6) | 38 (−33) | 48 (38) |
| D129G | 87 (−4) | 40 (−28) | 47 (34) |
| (Average RyR2 ΔCaMBD) | 92 | 34 (−38) | 58 (65) |
FIGURE 3.

Store overload-induced Ca2+ release from RyR2-expressing cells transfected with CaM WT and LQTS-linked CaM mutations. A–C, example traces of the D1ER FRET signal for cells transfected with CaM WT (A, same as Fig. 2A), D95V (B), and D129G (C). D–G, the activation threshold (D), termination threshold (E), fractional release (F), and store capacity (G) averaged from multiple traces are shown as bar graphs. Error bars show S.D., and asterisks indicate significant changes compared with CaM WT (p < 0.01).
More strikingly, all of these CaM mutations markedly affected the ER Ca2+ level at which SOICR terminated. The CaM mutation N53I decreased the termination threshold by 21% relative to CaM WT (Fig. 2, B and E), and, likewise, the CaM mutations N97S, D95V and D129G also decreased the termination threshold by 30, 33, and 28% (Figs. 2, C and E, and 3, B, C, and E), respectively.
The amount of Ca2+ released from the ER during SOICR was calculated by subtracting the termination threshold from the activation threshold. As shown in Figs. 2 (B–E) and 3 (B–E), the disease-causing CaM mutations decreased the termination thresholds proportionally more than the activation thresholds and hence increased ER fractional Ca2+ release (Figs. 2F and 3F). CaM N53I increased the ER fractional Ca2+ release by 20% relative to the CaM WT, and CaM D95V, D129G, and N97S increased this release by 38, 38, and 34%, respectively (Table 2). Interestingly, the effect of CaM-N53I, located in the N-domain of CaM, on the ER fractional release (20%) was significantly less than that of the three CaM mutations located in the C-domain (average 33%), (p < 0.001 for each multiple comparison).
Notably, none of these CaM mutations affected the SOICR activation or termination thresholds in HEK293 cells expressing a RyR2 mutant with a deletion of the CaMBD (Fig. 4, A–D). This observation demonstrates that the CaMBD in RyR2, and not secondary effects of CaM overexpression (e.g. altered Ca2+ buffering), mediate the observed effects (Fig. 4, C–E, all individual data sets not shown). Furthermore, the deletion of the CaMBD in RyR2 increased the ER fractional Ca2+ release by 65% relative to that of WT RyR2 (Table 2), whereas the maximum increase in the ER fractional Ca2+ release caused by the CaM mutations was less than 40% (Figs. 2F and 3F and Table 2). It is noteworthy that this comparison clearly shows that the arrhythmogenic CaM mutations did not suppress CaM-mediated inhibition of Ca2+ release to the same extent as ablating the interaction between CaM and RyR2 by deleting the CaMBD in RyR2. In other words, the arrhythmogenic CaMs did interact with and inhibit RyR2, only not to the same extent as the CaM WT.
FIGURE 4.

Store overload-induced Ca2+ release from RyR2 ΔCaMBD-expressing cells transfected with CaM WT or D129G. A and B, example traces of the D1ER FRET signal for cells transfected with CaM WT (A) and D129G (B). Transfections with all CaM variants showed no significant changes to the properties of RyR2 ΔCaMBD Ca2+ release when comparing to CaM WT transfection. C–E, the activation threshold (C), termination threshold (D), and store capacity (E) averaged for multiple traces are shown as bar graphs. Because there were no significant changes to activation or termination thresholds, the bar graph for fractional Ca2+ release was omitted but averaged 58% (Table 2). The error bars show S.D.
Ca2+ homeostasis in HEK293 cells differs from that in cardiomyocytes. The [Ca2+]cyt in HEK293 cells before RyR2 Ca2+ release (60 nm) is similar to that of diastolic cardiomyocytes (100 nm), but ER Ca2+ release is unlikely to increase [Ca2+]cyt to the same peak levels as within the cardiomyocyte dyadic clefts during systole (>100 μm) (41, 42). High Ca2+ concentrations mitigate the effects of mutations in the CaM C-domain (see below), and therefore their effect on RyR2 Ca2+ release termination may be less at peak systole [Ca2+]cyt in cardiomyocytes than in the HEK293 cells. However, the CaM mutations will on average lead to a suppressed inhibition of RyR2 by CaM and consequently increase the fractional Ca2+ release by RyR2 in vivo (32). No significant differences in the HEK293 cell ER Ca2+ store capacities were observed (Figs. 2–4), and Ca2+ release in cardiomyocytes terminates at ∼60% of diastole [Ca2+]SR, similar to the RyR2 termination thresholds in HEK293 cells (Table 2), which supports similar ER and SR luminal [Ca2+]free (7, 8).
Taken together, each of the CaM mutations N53I, N97S, D95V, and D129G conferred a slightly increased propensity for SOICR and reduced the capability for terminating SOICR. The difference in termination threshold and ER fractional Ca2+ release between CaM N53I and the other CaM mutations may support a mechanistically distinct effect of the CaM N53I mutation.
Arrhythmogenic Calmodulin Mutations Affect Binding of Ca2+ to Calmodulin in the Presence of the RyR2 CaM Binding Domain
Regulation of RyR2 by CaM critically depends on (a) CaM binding to the CaMBD in RyR2 and (b) Ca2+ binding to CaM (43, 44). Furthermore, this regulation of RyR2 by CaM is dependent on the characteristics of binding of Ca2+ to each of the CaM domains (11, 14, 16, 21). Hence, we investigated whether the mutations would affect Ca2+ binding to CaM in the presence of a RyR2(Arg3581–Leu3611) peptide corresponding to part of the CaMBD in RyR2. Ca2+ binding was investigated by monitoring changes in CaM protein fluorescence (Phe for the N-domain and Tyr for the C-domain) as Ca2+ was titrated to the CaM-peptide complex (34). Although available from a previous study, Ca2+ binding to CaM in the absence of RyR2(Arg3581–Leu3611) was measured for D95V and D129G as reported for CaM N53I and N97S to ensure the reliability of comparisons made here (1, 23). Average [Ca2+]cyt varies between ∼0.1 and 1 μm in cardiomyocytes during each heartbeat, but there is a large spatial heterogeneity of systolic [Ca2+]cyt. For example systolic [Ca2+]cyt can exceed 100 μΜ in the dyadic cleft in vicinity of RyR2 channels (41, 45). Thus, a wide range of Ca2+ concentrations (1 nm to 2 mΜ) was used in our Ca2+ titration experiments with RyR2(Arg3581–Leu3611).
The presence of the RyR2(Arg3581–Leu3611) increased the apparent Ca2+ affinity of the CaM N-domain and CaM C-domain ∼20- and ∼80-fold, respectively (Fig. 5A and Table 3). These increases clearly demonstrate the thermodynamic coupling between the Ca2+-CaM and the CaM-RyR2(Arg3581–Leu3611) binding events (Table 3) (46–48). Importantly, the presence of RyR2(Arg3581–Leu3611) increased the binding affinities of N-domain and C-domain for Ca2+ differentially, such that the Ca2+-CaM interaction for both domains occurred in the range 0.001–6 μm [Ca2+]free. In the absence of peptide, both CaM domains bind Ca2+ within the range of 0.1–100 μm [Ca2+]free (Fig. 5A). Ca2+ binding to the N-domain of CaM in the presence of RyR2(Arg3581–Leu3611) occurred in the range 0.1–6 μm [Ca2+]free with an appKDbound of 0.8 μm. This is ∼8-fold higher than the 0.1 μm [Ca2+]cyt at diastole in cardiomyocytes (Fig. 5A and Table 3) and within [Ca2+]cyt range of the diastole to systole cycling. On the other hand, Ca2+ binding to the C-domain in the presence of RyR2(Arg3581–Leu3611) showed a much higher binding affinity with an appKDbound of 0.03 μm, i.e. 3-fold lower than the diastolic [Ca2+]cyt. This suggests that the C-domain will be nearly Ca2+-saturated (>90%) at 0.1 μm [Ca2+]free. In other words, these results imply that the C-domain of CaM in complex with RyR2 is inherently Ca2+-loaded at diastolic levels of [Ca2+]cyt. Conversely, the N-domain of CaM in the same protein is poised for sensing increases in [Ca2+]cyt. Hence, the N-domain but not the C-domain will switch between apo and Ca2+-loaded states during diastole to systole cycles and alter its interactions with RyR2.
FIGURE 5.

Domain-specific titration curves for Ca2+ binding to CaM in the presence of RyR2(Arg3581–Leu3611). A–E, CaM/RyR2(Arg3581–Leu3611) (black) Ca2+ titration points for CaM N-domain (circles) and C-domain (squares) were fitted with a two-site Adair model (dashed and solid, respectively), and normalized Y is plotted as a function of [Ca2+]free. The dotted vertical line indicates the cardiomyocyte diastolic equivalent 0.1 μm [Ca2+]free. Also in A, Ca2+ titration of free CaM WT is shown in gray (1), illustrating the thermodynamic coupling of CaM binding Ca2+ and RyR2(Arg3581–Leu3611). In B–E, gray lines represents the Adair fits for CaM WT/RyR2(Arg3581–Leu3611) binding Ca2+ to illustrate the effects of CaM mutations. F shows the quantified mutation effect (ΔΔGobound) on Ca2+ binding to CaM in the presence of RyR2(Arg3581–Leu3611). Error bars indicate S.D., and asterisks indicate statistical significant differences (p < 0.05).
TABLE 3.
Measured thermodynamic parameters for Ca2+ binding to CaM
Apparent Ca2+ dissociation constants for RyR2(Arg3581–Leu3611) bound and free CaM (appKDbound and appKDfree, respectively) were measured using CaM intrinsic protein Phe (N-domain) and Tyr (C-domain) fluorescence. Mutation-imposed changes in Gibb's free energy of Ca2+ binding to CaM (ΔΔGobound and ΔΔGofree, respectively) were calculated relative to each CaM WT value. appKDfree and ΔΔGofree values for free CaM WT, N53I, and N97S are from a previous study (1). Dissociations constants and changes in Gibb's free energy are given in μm and kJ/mol, respectively. Bold font highlights significant changes versus CaM WT values (p < 0.05, one-way analysis of variance).
| appKDbound | appKDfree | ΔGobound | ΔΔGofree | |
|---|---|---|---|---|
| N-domain | ||||
| WT | 0.78 | 16 | ||
| N53I | 1.21 | 19 | 2.1 | 1.1 |
| N97S | 0.71 | 16 | −0.5 | 0.1 |
| D95V | 0.48 | 5.2 | −2.5 | −6.4 |
| D129G | 0.22 | 13 | −6.4 | −1.9 |
| C-domain | ||||
| WT | 0.03 | 2.5 | ||
| N53I | 0.03 | 2.3 | −0.3 | −0.3 |
| N97S | 0.15 | 10.4 | 8.4 | 7.2 |
| D95V | 0.14 | 31 | 8.3 | 13.2 |
| D129G | 4.0 | 84 | 25.3 | 18.2 |
An additional observation was made during the titrations of the CaM-peptide complex with Ca2+. During the titrations, we also monitored the change in RyR2(Arg3581–Leu3611) Trp fluorescence (data not shown), which coincided with the change in Tyr fluorescence from the CaM C-domain, but not with the change in Phe fluorescence from the CaM N-domain. This strongly supports that the C-domain of CaM WT binds around RyR2 Trp3587 and its Ca2+ binding induces a structural shift of the C-domain position on RyR2(Arg3581–Leu3611) as is also hinted by previous studies (18, 19, 22).
The CPVT- and/or LQTS-linked CaM mutations differentially perturbed this domain-specific interaction of Ca2+ with CaM. The CPVT-linked CaM-N53I mutation slightly decreased (appKDbound 1.2 μm) the Ca2+ affinity of the N-domain in the presence of RyR2(Arg3581–Leu3611) and showed no measurable effect on Ca2+ binding to the C-domain (Fig. 5B and Table 3). Conversely, the CaM-N97S mutation linked to both CPVT- and LQTS, had no measurable effect on Ca2+ binding to the N-domain, but markedly decreased (appKDbound 0.15 μm) the Ca2+ affinity of the C-domain (Fig. 5C and Table 3).
The LQTS-linked CaM mutations, D95V and D129G, both increased (appKDbound 0.48 and 0.22 μm) the Ca2+ affinity of the N-domain in the presence of RyR2(Arg3581–Leu3611) and both also markedly decreased the Ca2+ affinity of the C-domain (appKDbound 0.14 and 4 μm) (Fig. 4, D and E, and Table 3). The effect of the D129G mutation was so marked that the Ca2+ affinity of the C-domain was weaker than that of the N-domain. This suggests that the N-domain of CaM D129G may bind the CaMBD in RyR2 at a lower Ca2+ concentration than the C-domain. Our data support the notion that mutations in the CaM C-domain (D95V, N97S, and D129G) all markedly perturb the interaction between Ca2+ and CaM bound to the CaMBD in RyR2. Because CaM will bind to the CaMBD even in the absence of Ca2+, the reduction in C-domain Ca2+ affinity for the mutants may result in binding to the CaMBD in RyR2 in a partially saturated or even Ca2+-unbound form (18). The small increase in the Ca2+ affinity of the N-domain in the presence of RyR2(Arg3581–Leu3611), as conferred by the LQTS-linked CaM mutations D95V and D129G, suggests that the N-domain CaM mutations will still respond to an increase in [Ca2+]cyt from diastole to systole. In contrast to the marked perturbations of Ca2+ binding by the CaM C-domain mutations, the CPVT-linked CaM N53I mutation showed only a small decrease in the CaM N-domain affinity for Ca2+ in the presence of RyR2(Arg3581–Leu3611), yet a substantial effect on the interaction with RyR2 is expected because there is a substantial effect on SOICR.
Discussion
The domain-specific affinities of CaM for Ca2+ in the presence of the RyR2(Arg3581–Leu3611) offer novel insight into the distinct roles of the two domains of CaM in regulating the Ca2+ release from RyR2 (Fig. 6, A–C). Our results support an interaction between CaM and the CaMBD in RyR2 where the Ca2+-saturated C-domain of CaM is constitutively tethered around RyR2 Trp3587 even at the low diastole level of 0.1 μm Ca2+free (Fig. 6B) and thus not releasing Ca2+ during cardiac contraction cycles (Fig. 6, B and C). This implies a critical role for Ca2+ binding to the C-domain in inhibiting Ca2+ release from RyR2. In line with this view, Tian et al. (14) showed that a CaM double mutant (D93A/D129A) with loss of C-domain Ca2+ binding is defective in regulating the termination of RyR2 SOICR. Both the work of Tian et al. and this study also demonstrate that the interaction between the Ca2+-unbound CaM C-domain with the CaMBD in RyR2 inhibits Ca2+ release (Fig. 6A), but not to nearly the same extent as the Ca2+-saturated C-domain. Thus, the novel observation in this study is that the CaM C-domain under physiologically relevant conditions binds to the RyR2 CaMBD in a Ca2+-saturated form.
FIGURE 6.

Schematic model for the proposed Ca2+-dependent interaction between domains of CaM and the CaMBD in RyR2. RyR2 CaMBD is shown as an α-helical representation of RyR2(Arg3581–Leu3611) with Trp3587, Leu3591, and Phe3603 highlighted as magenta sticks. The apoCaM C-domain (orange) binds in the vicinity of Trp3587 and likely involves Leu3591 (A) (16), and this interaction changes conformation to fully encompass W3587 upon Ca2+ binding to CaM (B). Both the apoCaM and Ca2CaM interactions with the CaMBD (A and B) inhibit RyR2 Ca2+ release, with the latter interaction being a markedly stronger inhibitor. The N-domain of CaM (blue) interacts with another region of RyR2, indicated by the black α-helix, in the Ca2+-unbound form (A and B) and clamps down on Phe3603 upon Ca2+ binding (C). appKDbound for the CaM WT C-domain and N-domain, respectively, are indicated above equilibrium symbols, and black circles indicate Ca2+ bound to domains of CaM. The physiologically relevant interactions between CaM and the RyR2 CaMBD, in our model, are those shown in B and C. In our model, mutations conferring a reduced affinity of the CaM C-domain for binding to Ca2+, shift the physiological state of the C-domain at diastole (B) and systole (C) toward the pathological apoCaM interaction (A).
The pivotal role of the tethered CaM C-domain for inhibiting RyR2 Ca2+ release is consistent with studies showing that >70% of RyR2 channels in intact cardiomyocytes have CaM bound and that dissociation of CaM leads to excessive Ca2+ release through RyR2 channels (13, 43, 49, 50). In addition, several studies show that single mutations in the binding site for the CaM C-domain in the CaMBD of RyR2 (W3587A or L3591D) decrease the affinity for CaM to RyR2 at 0.4 and 1 μm [Ca2+]free, and that this decrease also leads to impaired inhibition of RyR2-mediated Ca2+ release (14, 16, 44, 51).
With the region of the RyR2 CaMBD around Trp3587 occupied by the C-domain of CaM, the binding site for the N-domain would be adjacent and likely covering RyR2 Phe3603, similar to the structure of CaM binding to the CaMBD in RyR1 (Fig. 1) (20). In contrast to the C-domain, our results suggest that the N-domain is Ca2+-unbound at the diastole level of [Ca2+]free but binds Ca2+ upon increases in [Ca2+]cyt above the diastolic level. Hence, the N-domain appears to cycle between the apo and Ca2+ saturated states during a heartbeat, thereby alternating between low and high affinity for binding to the RyR2 CaMBD. Accordingly, our data suggest that the CaM N-domain is a sensor of [Ca2+]cyt for RyR2 channels, albeit with a so far unknown function. Conversely, the CaM C-domain may not be a Ca2+ sensor because of its Ca2+-saturated state at physiological [Ca2+]cyt, when bound to the RyR2 CaMBD. Noteworthy, several Ca2+ binding proteins that interact with RyR2, as well as the endogenous RyR2 sensor of [Ca2+]cyt, all contribute to the response of the RyR2 macro complex to [Ca2+]cyt in vivo (52, 53). We further propose that the Ca2+-unbound N-domain interacts with regions of RyR2 outside the canonical CaMBD and then binds in the vicinity of Phe3603 upon cardiomyocyte excitation (Fig. 6, B and C) (18, 20). This proposed interaction would be consistent with the CaMBD in RyR2 not being the only RyR2 region reported to intact with CaM (11, 18, 54, 55). Moreover, at low [Ca2+]free, the N-domain may interact with a region in the skeletal muscle SR Ca2+ release channel, ryanodine receptor (RyR1) that is noncontiguous with the RyR1 CaMBD (18–20, 56). On the other hand, cryo-electron microscopy models of RyR2 channels indicate that CaM does not change position on RyR2 when comparing samples prepared in low and high Ca2+ (55, 57). However, because activation of the RyR2 channel involves substantial movements in the channel structure, it cannot be ruled out that the N-domain may bind different RyR2 regions in the open and closed channel, respectively, without CaM markedly shifting position.
In summary, we propose that the interaction between CaM and RyR2 involves constitutive tethering of the Ca2+-saturated CaM C-domain to the CaMBD in RyR2, which serves to inhibit Ca2+ release by stabilizing the RyR2 channel closed state throughout Ca2+ release (14, 21, 43, 58). In our model, N-domain binding to RyR2 is a Ca2+ sensing step that responds to increased [Ca2+]cyt upon cardiomyocyte excitation. However, determining the exact function of Ca2+ binding to CaM N-domain in regulating RyR2 Ca2+ release will require further investigation.
Ca2+ binding to the CaM N-domain does not appear to affect the SOICR activation and termination thresholds of RyR2 Ca2+ release, because no effect was detected in experiments using a CaM mutant with dramatically decreased N-domain Ca2+ affinity (CaM D20A/D56A) (14). Also, in the current study only minor effects of the CaM N53I mutation on the binding of Ca2+ to the N-domain in the presence of RyR2(Arg3581–Leu3611) were observed (see below). The binding of Ca2+ to the N-domain of CaM, when CaM is tethered to RyR2, may instead be part of inhibiting Ca2+-induced Ca2+ release during the high systolic [Ca2+]cyt. Alternatively, Ca2+ binding to the N-domain could be part of the triggering mechanism for the activation of RyR2 channel Ca2+ release during Ca2+-induced Ca2+ release. Both of these functions would be in combination with endogenous cytosolic and SR luminal Ca2+ sensors in RyR2 (52, 59). An inhibiting or facilitating function of CaM on Ca2+-induced Ca2+ release would not be detected in the SOICR experiments performed in this study (6, 14).
Our results support the notion that arrhythmogenic CaM mutations adversely affect the native interaction between CaM and RyR2. We show that the C-domain mutations (D95V, N97S, and D129G) perturb the interactions between CaM, Ca2+ and the CaMBD in RyR2. Notably, this perturbation occurs within physiologic relevant Ca2+ concentrations. We suggest that at diastole [Ca2+]cyt, CaM with either of these C-domain mutations will bind to the RyR2 CaMBD with the CaM C-domain in a partially saturated or Ca2+-unbound form as opposed to the native Ca2+-saturated state (transition from Figs. 6B to 6A). This in turn leads to an insufficient inhibition of the RyR2 Ca2+ release during SOICR in intact cells, similar to the aberrant regulation of RyR2 by CaM1234 and CaM D93A/D129G mutants (14, 21). In vivo CaM D95V, N97S, and D129G are likely to still tether to RyR2 because each mutated CaM retains a significant affinity for intact RyR2 channels, despite reduced affinity for Ca2+ (31).
Interestingly, the N53I mutation in the N-domain of CaM adversely affected the regulation of RyR2-mediated Ca2+ release during SOICR, without any changes to the affinity of the CaM N53I C-domain for binding Ca2+ in the presence of RyR2(Arg3581–Leu3611). This strongly suggests that the integrity of the CaM N-domain is important in inhibiting RyR2 Ca2+ release but not dependent on Ca2+ binding to the N-domain, as discussed above (14). In strong support of this hypothesis is that CaM D20A/D56A, with markedly decreased N-domain Ca2+ affinity, does not affect activation or termination thresholds for RyR2-mediated SOICR, whereas CaM N53I does. A likely explanation is that the N53I mutation affects an interaction of the Ca2+-unbound N-domain of CaM with a region of RyR2 that is outside the CaMBD. The significantly different impact of the CaM N53I mutation on RyR2 Ca2+ release, compared with the C-domain mutations, also supports this notion. Furthermore, we previously found divergent effects of the N53I and N97S mutations on protein properties and the binding of Ca2+ to CaM, which supports different molecular disease mechanisms for the two mutations (1).
Overexpression of CaM in HEK293 cells could theoretically lead to buffering of [Ca2+]cyt, which in turn may affect RyR2 Ca2+ release through mechanisms independent of the interaction between CaM and RyR2, and lead to differences between the CaM mutations caused by differences in Ca2+ buffering capacities. This was, however, not the case in the SOICR experiments as judged from three observations: (a) CaM N53I had marked effects on RyR2-mediated Ca2+ release, although it displayed Ca2+ binding properties highly similar to those of CaM WT both in the RyR2-bound and free form (Fig. 2, A and B, and Table 3); (b) CaM WT and CaM D129G had no effect on Ca2+ release from the RyR2 ΔCaMBD mutant, although they exhibited widely different Ca2+ binding properties (Fig. 4, A and B) (1, 14); and (c) because the Ca2+ affinity (appKDfree) of the CaM C-domain not bound to a protein target is ∼2.5 μm, the buffering capacity of free CaM is highly limited at the ∼60 nm [Ca2+]cyt in HEK293 cells.
The combined results of this study show that both CPVT- and LQTS-linked CaM mutations can lead to excessive Ca2+ release from RyR2 channels, primarily from insufficient termination of RyR2-mediated Ca2+ release. Also, the same CaM mutations lower the activation thresholds for SOICR, which would increase the propensity for SOICR in vivo during conditions with increased SR Ca2+ load. Excessive Ca2+ release and leaky RyR2 channels are a well documented hallmark of CPVT-linked RyR2 mutations. It follows that dysregulation of RyR2 likely underlies the disease mechanisms of CaM N53I and N97S mutation-associated CPVT (38). Taken together, we propose that the regulation of RyR2 Ca2+ release is highly sensitive to CaM and that aberrant regulation of RyR2 may be a common component of both CPVT and LQTS arrhythmias caused by CaM mutations.
Author Contributions
M. T. S., S. R. W. C., and M. T. O. designed the research; M. T. S., X. T., Y. L., and R. W. performed research; M. T. S., X. T., Y. L., S. R. W. C., and M. T. O. analyzed data; and M. T. S., W. J. C., S. R. W. C., and M. T. O. wrote the paper.
Acknowledgment
We thank laboratory coordinator Zuzana V. Hansen (CamAgon Aps, Aarhus, Denmark) for excellent technical assistance.
This study was supported by research grants from the Obelske Family Foundation, the Novo Nordic Foundation, and the Lundbeck Foundation (to M. T. O.); by a postdoctoral fellowship from the Danish Council for Independent Research (Technology and Production Sciences) (to M. T. S.); by institutional support from Vanderbilt University (to W. J. C.); and by research grants from the Canadian Institutes of Health Research, the Heart and Stroke Foundation of Alberta, Northwest Territories, and Nunavut, the Canada Foundation for Innovation, and the Heart and Stroke Foundation/Libin Professorship in Cardiovascular Research (to S. R. W. C.). The authors declare that they have no conflicts of interest with the contents of this article.
- RyR2
- ryanodine receptor 2
- RyR1
- ryanodine receptor 1
- CaM
- calmodulin
- CPVT
- catecholaminergic polymorphic ventricular tachycardia
- LQTS
- long QT syndrome
- SOICR
- store overload-induced Ca2+ release
- N-domain
- N-terminal domain
- C-domain
- C-terminal domain
- CaMBD
- CaM-binding domain
- SR
- sarcoplasmic reticulum
- ER
- endoplasmic reticulum
- FI
- fluorescence intensity.
References
- 1.Søndergaard M. T., Sorensen A. B., Skov L. L., Kjaer-Sorensen K., Bauer M. C., Nyegaard M., Linse S., Oxvig C., and Overgaard M. T. (2015) Calmodulin mutations causing catecholaminergic polymorphic ventricular tachycardia confer opposing functional and biophysical molecular changes. FEBS J. 282, 803–816 [DOI] [PubMed] [Google Scholar]
- 2.Bers D. M. (2002) Cardiac excitation-contraction coupling. Nature 415, 198–205 [DOI] [PubMed] [Google Scholar]
- 3.Berridge M. J. (2014) Cell signalling biology: module 7: cellular processes. Biochem. J. 10.1042/csb0001007 [DOI] [Google Scholar]
- 4.Berridge M. J. (2014) Cell signalling biology: module 3: ion channels. Biochem. J. 10.1042/csb0001003 [DOI] [Google Scholar]
- 5.Dweck D., Reyes-Alfonso A. Jr., and Potter J. D. (2005) Expanding the range of free calcium regulation in biological solutions. Anal. Biochem. 347, 303–315 [DOI] [PubMed] [Google Scholar]
- 6.Jiang D., Wang R., Xiao B., Kong H., Hunt D. J., Choi P., Zhang L., and Chen S. R. (2005) Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ. Res. 97, 1173–1181 [DOI] [PubMed] [Google Scholar]
- 7.Zima A. V., Picht E., Bers D. M., and Blatter L. A. (2008) Termination of cardiac Ca2+ sparks: role of intra-SR [Ca2+], release flux, and intra-SR Ca2+ diffusion. Circ. Res. 103, e105–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Györke S., and Terentyev D. (2008) Modulation of ryanodine receptor by luminal calcium and accessory proteins in health and cardiac disease. Cardiovasc Res. 77, 245–255 [DOI] [PubMed] [Google Scholar]
- 9.Chen W., Wang R., Chen B., Zhong X., Kong H., Bai Y., Zhou Q., Xie C., Zhang J., Guo A., Tian X., Jones P. P., O'Mara M. L., Liu Y., Mi T., Zhang L., Bolstad J., Semeniuk L., Cheng H., Zhang J., Chen J., Tieleman D. P., Gillis A. M., Duff H. J., Fill M., Song L.-S., and Chen S. R. (2014) The ryanodine receptor store-sensing gate controls Ca2+ waves and Ca2+-triggered arrhythmias. Nat. Med. 20, 184–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Petegem F. (2012) Ryanodine receptors: structure and function. J. Biol. Chem. 287, 31624–31632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balshaw D. M., Xu L., Yamaguchi N., Pasek D. A., and Meissner G. (2001) Calmodulin binding and inhibition of cardiac muscle calcium release channel (ryanodine receptor). J. Biol. Chem. 276, 20144–20153 [DOI] [PubMed] [Google Scholar]
- 12.Xu L., and Meissner G. (2004) Mechanism of calmodulin inhibition of cardiac sarcoplasmic reticulum Ca2+ release channel (ryanodine receptor). Biophys. J. 86, 797–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu X., Yano M., Uchinoumi H., Hino A., Suetomi T., Ono M., Tateishi H., Oda T., Okuda S., Doi M., Kobayashi S., Yamamoto T., Ikeda Y., Ikemoto N., and Matsuzaki M. (2010) Defective calmodulin binding to the cardiac ryanodine receptor plays a key role in CPVT-associated channel dysfunction. Biochem. Biophys. Res. Commun. 394, 660–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tian X., Tang Y., Liu Y., Wang R., and Chen S. R. (2013) Calmodulin modulates the termination threshold for cardiac ryanodine receptor-mediated Ca2+ release. Biochem. J. 455, 367–375 [DOI] [PubMed] [Google Scholar]
- 15.Sorensen A. B., Søndergaard M. T., and Overgaard M. T. (2013) Calmodulin in a heartbeat. FEBS J. 280, 5511–5532 [DOI] [PubMed] [Google Scholar]
- 16.Yamaguchi N., Xu L., Pasek D. A., Evans K. E., and Meissner G. (2003) Molecular basis of calmodulin binding to cardiac muscle Ca2+ release channel (ryanodine receptor). J. Biol. Chem. 278, 23480–23486 [DOI] [PubMed] [Google Scholar]
- 17.Kimlicka L., and Van Petegem F. (2011) The structural biology of ryanodine receptors. Sci. China Life Sci. 54, 712–724 [DOI] [PubMed] [Google Scholar]
- 18.Lau K., Chan M. M., and Van Petegem F. (2014) Lobe-specific calmodulin binding to different ryanodine receptor isoforms. Biochemistry 53, 932–946 [DOI] [PubMed] [Google Scholar]
- 19.Xiong L.-W., Newman R. A., Rodney G. G., Thomas O., Zhang J.-Z., Persechini A., Shea M. A., and Hamilton S. L. (2002) Lobe-dependent regulation of ryanodine receptor type 1 by calmodulin. J. Biol. Chem. 277, 40862–40870 [DOI] [PubMed] [Google Scholar]
- 20.Maximciuc A. A., Putkey J. A., Shamoo Y., and Mackenzie K. R. (2006) Complex of calmodulin with a ryanodine receptor target reveals a novel, flexible binding mode. Structure 14, 1547–1556 [DOI] [PubMed] [Google Scholar]
- 21.Fruen B. R., Black D. J., Bloomquist R. A., Bardy J. M., Johnson J. D., Louis C. F., and Balog E. M. (2003) Regulation of the RYR1 and RYR2 Ca2+ release channel isoforms by Ca2+-insensitive mutants of calmodulin. Biochemistry 42, 2740–2747 [DOI] [PubMed] [Google Scholar]
- 22.Nyegaard M., Overgaard M. T., Søndergaard M. T., Vranas M., Behr E. R., Hildebrandt L. L., Lund J., Hedley P. L., Camm A. J., Wettrell G., Fosdal I., Christiansen M., and Børglum A. D. (2012) Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am. J. Hum. Genet. 91, 703–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crotti L., Johnson C. N., Graf E., De Ferrari G. M., Cuneo B. F., Ovadia M., Papagiannis J., Feldkamp M. D., Rathi S. G., Kunic J. D., Pedrazzini M., Wieland T., Lichtner P., Beckmann B.-M., Clark T., Shaffer C., Benson D. W., Kääb S., Meitinger T., Strom T. M., Chazin W. J., Schwartz P. J., and George A. L. Jr. (2013) Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation 127, 1009–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Makita N., Yagihara N., Crotti L., Johnson C. N., Beckmann B. M., Roh M. S., Shigemizu D., Lichtner P., Ishikawa T., Aiba T., Homfray T., Behr E. R., Klug D., Denjoy I., Mastantuono E., Theisen D., Tsunoda T., Satake W., Toda T., Nakagawa H., Tsuji Y., Tsuchiya T., Yamamoto H., Miyamoto Y., Endo N., Kimura A., Ozaki K., Motomura H., Suda K., Tanaka T., Schwartz P. J., Meitinger T., Kääb S., Guicheney P., Shimizu W., Bhuiyan Z. A., Watanabe H., Chazin W. J., and George A. L. Jr. (2014) Novel calmodulin mutations associated with congenital arrhythmia susceptibility. Circ. Cardiovasc. Genet. 7, 466–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Venetucci L., Denegri M., Napolitano C., and Priori S. G. (2012) Inherited calcium channelopathies in the pathophysiology of arrhythmias. Nat. Rev. Cardiol. 9, 561–575 [DOI] [PubMed] [Google Scholar]
- 26.Amin A. S., Pinto Y. M., and Wilde A. A. (2013) Long QT syndrome: beyond the causal mutation. J. Physiol. 591, 4125–4139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marsman R. F., Barc J., Beekman L., Alders M., Dooijes D., van den Wijngaard A., Ratbi I., Sefiani A., Bhuiyan Z. A., Wilde A. A., and Bezzina C. R. (2014) A mutation in CALM1 encoding calmodulin in familial idiopathic ventricular fibrillation in childhood and adolescence. J. Am. Coll. Cardiol. 63, 259–266 [DOI] [PubMed] [Google Scholar]
- 28.Laitinen P. J., Brown K. M., Piippo K., Swan H., Devaney J. M., Brahmbhatt B., Donarum E. A., Marino M., Tiso N., Viitasalo M., Toivonen L., Stephan D. A., and Kontula K. (2001) Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation 103, 485–490 [DOI] [PubMed] [Google Scholar]
- 29.Lahat H., Pras E., and Eldar M. (2003) RYR2 and CASQ2 mutations in patients suffering from catecholaminergic polymorphic ventricular tachycardia. Circulation 107, e29. [DOI] [PubMed] [Google Scholar]
- 30.Roux-Buisson N., Cacheux M., Fourest-Lieuvin A., Fauconnier J., Brocard J., Denjoy I., Durand P., Guicheney P., Kyndt F., Leenhardt A., Le Marec H., Lucet V., Mabo P., Probst V., Monnier N., Ray P. F., Santoni E., Trémeaux P., Lacampagne A., Fauré J., Lunardi J., and Marty I. (2012) Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum. Mol. Genet. 21, 2759–2767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hwang H.-S., Nitu F. R., Yang Y., Walweel K., Pereira L., Johnson C. N., Faggioni M., Chazin W. J., Laver D., George A. L. Jr., Cornea R. L., Bers D. M., and Knollmann B. C. (2014) Divergent regulation of ryanodine receptor 2 calcium release channels by arrhythmogenic human calmodulin missense mutants. Circ. Res. 114, 1114–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Limpitikul W. B., Dick I. E., Joshi-Mukherjee R., Overgaard M. T., George A. L. Jr., and Yue D. T. (2014) Calmodulin mutations associated with long QT syndrome prevent inactivation of cardiac L-type Ca2+ currents and promote proarrhythmic behavior in ventricular myocytes. J. Mol. Cell. Cardiol. 74, 115–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang D., Xiao B., Yang D., Wang R., Choi P., Zhang L., Cheng H., and Chen S. R. (2004) RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR). Proc. Natl. Acad. Sci. U.S.A. 101, 13062–13067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.VanScyoc W. S., Sorensen B. R., Rusinova E., Laws W. R., Ross J. B., and Shea M. A. (2002) Calcium binding to calmodulin mutants monitored by domain-specific intrinsic phenylalanine and tyrosine fluorescence. Biophys. J. 83, 2767–2780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang J., Zhou Y., Zou J., Chen Y., Patel P., Yang J. J., and Balog E. M. (2010) Site-specific modification of calmodulin Ca2+ affinity tunes the skeletal muscle ryanodine receptor activation profile. Biochem. J. 432, 89–99 [DOI] [PubMed] [Google Scholar]
- 36.Adair G. (1925) The hemoglobin system. J. Biol. Chem. 63, 529–545 [Google Scholar]
- 37.Shea M. A., Verhoeven A. S., and Pedigo S. (1996) Calcium-induced interactions of calmodulin domains revealed by quantitative thrombin footprinting of Arg37 and Arg106. Biochemistry 35, 2943–2957 [DOI] [PubMed] [Google Scholar]
- 38.Priori S. G., and Chen S. R. (2011) Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ. Res. 108, 871–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones P. P., Jiang D., Bolstad J., Hunt D. J., Zhang L., Demaurex N., and Chen S. R. (2008) Endoplasmic reticulum Ca2+ measurements reveal that the cardiac ryanodine receptor mutations linked to cardiac arrhythmia and sudden death alter the threshold for store-overload-induced Ca2+ release. Biochem. J. 412, 171–178 [DOI] [PubMed] [Google Scholar]
- 40.Xie L.-H., Sato D., Garfinkel A., Qu Z., and Weiss J. N. (2008) Intracellular Ca alternans: coordinated regulation by sarcoplasmic reticulum release, uptake, and leak. Biophys. J. 95, 3100–3110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bers D. M. (2000) Calcium fluxes involved in control of cardiac myocyte contraction. Circ. Res. 87, 275–281 [DOI] [PubMed] [Google Scholar]
- 42.Breitwieser G. E., and Gama L. (2001) Calcium-sensing receptor activation induces intracellular calcium oscillations. Am. J. Physiol. Cell Physiol. 280, C1412–C1421 [DOI] [PubMed] [Google Scholar]
- 43.Ono M., Yano M., Hino A., Suetomi T., Xu X., Susa T., Uchinoumi H., Tateishi H., Oda T., Okuda S., Doi M., Kobayashi S., Yamamoto T., Koseki N., Kyushiki H., Ikemoto N., and Matsuzaki M. (2010) Dissociation of calmodulin from cardiac ryanodine receptor causes aberrant Ca2+ release in heart failure. Cardiovasc. Res. 87, 609–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arnáiz-Cot J. J., Damon B. J., Zhang X.-H., Cleemann L., Yamaguchi N., Meissner G., and Morad M. (2013) Cardiac calcium signalling pathologies associated with defective calmodulin regulation of type 2 ryanodine receptor. J. Physiol. 591, 4287–4299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berridge M. J., Bootman M. D., and Roderick H. L. (2003) Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4, 517–529 [DOI] [PubMed] [Google Scholar]
- 46.Peersen O. B., Madsen T. S., and Falke J. J. (1997) Intermolecular tuning of calmodulin by target peptides and proteins: differential effects on Ca2+ binding and implications for kinase activation. Protein Sci. 6, 794–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bayley P. M., Findlay W. A., and Martin S. R. (1996) Target recognition by calmodulin: dissecting the kinetics and affinity of interaction using short peptide sequences. Protein Sci. 5, 1215–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Evans T. I., Hell J. W., and Shea M. A. (2011) Thermodynamic linkage between calmodulin domains binding calcium and contiguous sites in the C-terminal tail of CaV1.2. Biophys. Chem. 159, 172–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guo T., Fruen B. R., Nitu F. R., Nguyen T. D., Yang Y., Cornea R. L., and Bers D. M. (2011) FRET detection of calmodulin binding to the cardiac RyR2 calcium release channel. Biophys. J. 101, 2170–2177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang Y., Guo T., Oda T., Chakraborty A., Chen L., Uchinoumi H., Knowlton A. A., Fruen B. R., Cornea R. L., Meissner G., and Bers D. M. (2014) Cardiac myocyte Z-line calmodulin is mainly RyR2-bound, and reduction is arrhythmogenic and occurs in heart failure. Circ. Res. 114, 295–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamaguchi N., Takahashi N., Xu L., Smithies O., and Meissner G. (2007) Early cardiac hypertrophy in mice with impaired calmodulin regulation of cardiac muscle Ca release channel. J. Clin. Invest. 117, 1344–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li P., and Chen S. R. (2001) Molecular basis of Ca2+ activation of the mouse cardiac Ca2+ release channel (ryanodine receptor). J. Gen. Physiol. 118, 33–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Van Petegem F. (2015) Ryanodine receptors: allosteric ion channel giants. J. Mol. Biol. 427, 31–53 [DOI] [PubMed] [Google Scholar]
- 54.Chen S. R., and MacLennan D. H. (1994) Identification of calmodulin-, Ca2+-, and ruthenium red-binding domains in the Ca2+ release channel (ryanodine receptor) of rabbit skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. 269, 22698–22704 [PubMed] [Google Scholar]
- 55.Huang X., Liu Y., Wang R., Zhong X., Liu Y., Koop A., Chen S. R., Wagenknecht T., and Liu Z. (2013) Two potential calmodulin-binding sequences in the ryanodine receptor contribute to a mobile, intra-subunit calmodulin-binding domain. J. Cell Sci. 126, 4527–4535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang H., Zhang J.-Z., Danila C. I., and Hamilton S. L. (2003) A noncontiguous, intersubunit binding site for calmodulin on the skeletal muscle Ca2+ release channel. J. Biol. Chem. 278, 8348–8355 [DOI] [PubMed] [Google Scholar]
- 57.Huang X., Fruen B., Farrington D. T., Wagenknecht T., and Liu Z. (2012) Calmodulin-binding locations on the skeletal and cardiac ryanodine receptors. J. Biol. Chem. 287, 30328–30335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oda T., Yang Y., Nitu F. R., Svensson B., Lu X., Fruen B. R., Cornea R. L., and Bers D. M. (2013) In cardiomyocytes, binding of unzipping peptide activates ryanodine receptor 2 and reciprocally inhibits calmodulin binding. Circ. Res. 112, 487–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang J., Chen B., Zhong X., Mi T., Guo A., Zhou Q., Tan Z., Wu G., Chen A. W., Fill M., Song L.-S., and Chen S. R. (2014) The cardiac ryanodine receptor luminal Ca2+ sensor governs Ca2+ waves, ventricular tachyarrhythmias and cardiac hypertrophy in calsequestrin-null mice. Biochem. J. 461, 99–106 [DOI] [PMC free article] [PubMed] [Google Scholar]