

Background: Angiogenesis is a pathological component of chronic inflammatory diseases including inflammatory bowel disease (IBD).
Results: An angiogenic molecule VEGF-A is produced by the corticotropin-releasing hormone (CRH) family of peptides through the cAMP/CREB pathway.
Conclusion: The CRH family acts as angiogenic stimulators in intestinal epithelial cells.
Significance: Angiogenic regulation by the cAMP/CREB pathway constitutes novel strategies to treat IBD.
Keywords: angiogenesis, cAMP response element-binding protein (CREB), colitis, inflammatory bowel disease (IBD), vascular endothelial growth factor (VEGF), corticotropin-releasing hormone, urocortin
Abstract
Colonic epithelium is the first line of defense against various pathological offenses in the gut. Previous studies have shown that the peptides of the corticotropin-releasing hormone (CRH) family modulate vascular endothelial growth factor (VEGF)-A production in other cells. Here we sought to investigate whether CRH and urocortin (Ucn) 3 regulate VEGF-A secretion in colonocytes through CRH receptors and to elucidate the underlying mechanism of action. CRH and Ucn 3 significantly increased the expression levels of VEGF-A mRNA and protein through CRH receptor 1 and 2, respectively, in human colonic epithelial cells and primary mouse intestinal epithelial cells. Underlying mechanisms involve activation of adenylyl cyclase with subsequent increase of intracellular cAMP level and increased DNA binding activity of transcription factor CREB on VEGF-A promoter region. Finally, genetic deficiency of CREB decreased intestinal inflammation and VEGF-A expression in a dextran sodium sulfate-induced colitis model. These results show that activation of CRH receptors by CRH ligands stimulates VEGF-A expression in intestinal epithelial cells through the cAMP/CREB pathway. Since VEGF-A boosts inflammatory responses through angiogenesis, these data suggest that CREB may be a key effector of CRH and Ucn 3-dependent inflammatory angiogenesis.
Introduction
Vascular endothelial growth factor (VEGF)2 is the most potent inducer of angiogenesis. The VEGF family is composed of at least five members: VEGF-A, placental growth factor, VEGF-B, VEGF-C, and VEGF-D. Despite various implication of each VEGF family member, VEGF-A, original termed VEGF, is considered to be primarily effective in vascular endothelium, although VEGF-A stimulation in macrophages or kidney epithelial cells has previously been suggested. Because of the requirement of vascularization in maintaining tissue homeostasis, VEGF is an essential factor not only in a physiological conditions, but also in pathological environments such as neoplasia and chronic inflammatory disorders (reviewed in Ref. 1). Since VEGF is a potent inducer of angiogenesis, which is required for tumor growth and metastasis, the important role of VEGF has been intensively investigated with a promising perspective to develop effective anti-cancer approaches. Meanwhile, a growing body of evidence suggests that VEGF can be implicated in regulating the pathogenesis and prognosis of inflammatory diseases such as inflammatory bowel disease (IBD). For instance, the expression levels of VEGF-A are increased in the colonic mucosa, serum, and plasma of IBD patients compared with the healthy controls (2, 3). In line with this observation, experimental mouse colitis is ameliorated by inhibition of VEGF expression and/or activity with neutralizing antibodies or thalidomide treatment (4, 5). Even though emerging evidence indicates an important role of VEGF in regulating IBD, the molecular mechanism by which VEGF regulates colonic inflammation is still under investigation.
Several mechanisms regulating vegf gene expression have been postulated (6). Among them, an oxygen-sensing signaling pathway regulates vegf gene expression by determining the stability of the key transcription factor hypoxia-inducible factor-1 (HIF-1), by increasing the kinase activity of c-Src or by increasing VEGF mRNA stability (7–9). VEGF levels are also regulated by inflammatory cytokines, growth factors, and proteases like matrix metalloproteinase (MMP)-9 and cathepsins (1, 6, 10, 11). Moreover, the cAMP and cAMP response element-binding protein (CREB) cascade is implicated in neuronal VEGF induction (12). In addition, activation of adenylyl cyclase by phorbol esters and forskolin increases VEGF mRNA expression (13). Accordingly, VEGF mRNA expression is induced by protein kinase A activation (14).
The corticotropin-releasing hormone (CRH) family of peptides including CRH, urocortin (Ucn) 1, Ucn 2 (stresscopin-related peptide), and Ucn 3 (stresscopin) is hypothalamic peptides, which modulate the synthesis and release of the adrenocorticotropic hormone from the pituitary, leading to the release of corticosteroid from the adrenal gland (15–19). Upon binding to CRH receptors (CRHR1 and CRHR2), these peptides activate Gαs protein and the adenylyl cyclase/cAMP signaling pathway (20). An important function of the CRH family of peptides includes coordinating stress-related responses, regulating gut motility, and modulating intestinal inflammation (21–24).
Studies over the last few years have elucidated a pivotal role of the CRH family of peptides in the regulation of angiogenesis. For instance, CRHR2-deficient mice become hypervascularized postnatally, the expression level of CRHR2 is diminished in tumor tissues along with increased microvessels, and the viral expression of Ucn 2 inhibits tumor vascularization (25–28). Our recent study also suggests that activation of CRHR1 increases, while activation of CRHR2 decreases colitis-associated angiogenesis (29).
Furthermore, the CRH family of peptides regulates VEGF expression levels in various settings. In rat vascular smooth muscle cells (SMCs), activation of CRHR2 by Ucn 2 treatment reduces VEGF expression levels (25). In contrast, CRH increases VEGF secretion in human mast cells and VEGF mRNA levels in human extravillous trophoblasts (30, 31). Our study also demonstrated that VEGF levels were decreased in the colon of CRHR1-deficient mice with colitis but increased in the colon of CRHR2-deficient mice (29). Thus, the CRH family of peptides regulates angiogenesis through the production of VEGF. Previous studies, however, did not address important questions regarding whether the CRH family of peptides could regulate VEGF expression in colonic epithelial cells, representing key players in intestinal inflammation and angiogenesis. In the present study, we sought to define a role of the CRH family of peptides in VEGF induction in colonic epithelial cells and to elucidate the underlying molecular mechanisms.
Experimental Procedures
Cell Culture
Human colonic epithelial cells (NCM460) were cultivated in M3D medium (INCELL Corp., San Antonio, TX) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (Invitrogen, Carlsbad, CA) and 10 units/ml penicillin and 100 μg/ml streptomycin (Invitrogen) at 37 °C in air supplemented with 5% CO2 as previously described (32). Primary mouse intestinal epithelial cells (mIECs) from mouse small intestine were isolated as previously described (33, 34). After isolation, cells spread out from organoids were then cultivated in medium containing equal volumes of DMEM and Ham's 12 medium (Lonza, Basel, Switzerland) with 10% FBS and antibiotics in 5% CO2 at 37 °C. The medium was replaced every other day. Human colonic smooth muscle cells (hCSMC) and human aortic smooth muscle cells (hASMC) were purchased from ScienCell Research Laboratory (Carlsbad, CA) and cultivated in smooth muscle cell medium which consists of basal medium, 2% FBS, 1% smooth muscle cell growth supplement, and 1% penicillin/streptomycin solution using poly-L-lysine coated plates.
Reagents
Antalarmin, astressin 2B, and 8-bromoadenosine 3′:5′-cyclic monophosphate (cAMP) sodium were purchased from Sigma. Human/rat (h/r) CRH, hUcn 3, and mouse Ucn 3 were purchased from Bachem (Torrance, CA). SQ 22536 (9-(tetrahydro-2′-furyl) adenine) was purchased from EMD-Calbiochem (Gibbstown, NJ).
Luciferase Reporter Assay
VEGF-A promoter constructs are a kind gift from Dr. Mukhopadhyay (Mayo Clinic, Rochester, MN). NCM460 cells were plated in six-well plates (1.2 × 106 cells/well) and transfected with the VEGF-A promoter constructs and β-galactosidase expression plasmid (HSP70-β-gal) as an internal control using SuperFect transfect reagent (Qiagen, Valencia, CA) according to the manufacturer's instruction. One day after transfection, relative luciferase activity was determined by normalization with β-galactosidase activity as described previously (35). Assays were performed in triplicate, and a single representative experiment is shown.
CRHR1 and CRHR2 Expression Vector
Constructs containing a full-length cDNA of human CRHR1 or CRHR2 were purchased from University of Missouri-Rolla cDNA Rescource Center (Rolla, MO) and transfected to NCM460 cells using the SuperFect transfection reagent (Qiagen). The clones expressing CRHR1 or CRHR2 were selected in the presence of G418 (0.8 mg/ml).
Chromatin Immunoprecipitation (ChIP) Assay
ChIP was performed according to a manufacturer's protocol (ChIP-IT kit, Active Motif, Carlsbad, CA) with modification. Briefly, NCM460 cells were cultured in three 15-cm cell culture plates (∼4 × 107 cells), and treated with 100 nm CRH or Ucn 3. At the indicated time points, cells were fixed in 37% formaldehyde solution, lysed, and homogenized to release chromatin. Chromatin was then sonicated to an average of 500–1000 bp DNA fragments and immunoprecipitated with anti-CREB antibody, anti-RNA Pol II (positive control) antibody, or IgG (negative control) conjugated with protein A-agarose beads. Precipitated DNA was extracted and amplified by PCR using the following primers: forward, 5-GCGTGTCTCT GGACAGAGTTTCCGG-3, and reverse, 5-CAG GCGAGCCTCAGCCCTTCCACACGC-3. With these primers, the −263 to −97 bp VEGF promoter sequence from the transcription start site was amplified.
Animal Models
CRHR1 heterozygote mice (Crhr1tm1Klee) were obtained from The Jackson Laboratory (Bar Harbor, ME). CRHR1−/− mice and their wild type littermates (M&F, 8–12 weeks) were derived from heterozygous breeding. CRHR2−/− mice were a gift from Dr. W. Vale (Salk Institute, La Jolla, CA) and had been backcrossed onto a B6 background (>N10). CRHR2−/− mice and their wild type littermates (M&F, 8–12 weeks) were derived from heterozygous breeding. CREB−/− mice were purchased from The Jackson Laboratory. CREB−/− mice and their wild type littermates (M&F, 8–12 weeks) were derived from heterozygous breeding.
To induce colitis, mice were fed with 3% dextran sodium sulfate (DSS, MP Biomedicals, Irvine, CA) dissolved in regular tap water for 14 days. Mice were weighed and monitored for clinical colitis symptoms every day. For histological evaluation, mice were fed with 3% DSS for 7 days and then euthanized. The entire colon was excised, fixed, paraffin-embedded, and stained with hematoxylin & eosin (H&E) as we previously described (36). Experimental animal protocols were approved by the Institutional Animal Care and Use Committee of the University of California at Los Angeles.
Enzyme-linked Immunosorbent Assay (ELISA)
To measure the expression levels of VEGF-A or cAMP, NCM460 cells or the cells expressing CRHR1 or CRHR2 were stimulated with CRH or Ucn 3 at the indicated concentrations for 6 h. Supernatant was collected and applied to VEGF ELISA kit (R&D, Minneapolis, MN) or cAMP EIA kit (Biomedical Technologies Inc., Stoughton, MA), respectively. Additionally, supernatant from colon tissues of CREB+/+ and CREB−/− mice with DSS-induced colitis was collected and applied to mouse VEGF-A ELISA kit (R&D, Minneapolis, MN). Each experiment was carried out in triplicate.
Measurement of Transcription Factor Activation
After treatment with CRH (100 nm) or Ucn 3 (100 nm), nuclear extracts from NCM460 cells were prepared by using a commercial kit (Nuclear Extract Kit, Active Motif). TransAM transcription factor ELISA for NF-κB and pCREB was performed according to the manufacturer's recommendations. The levels of NF-κB and pCREB were measured in triplicate by a spectrophotometer at A450. Each sample was run in triplicate. Protein concentration in each sample was determined by using a standard BCA protein assay.
Immunofluorescence Staining
Segments of the transverse colon were embedded in OCT and frozen immediately. 5-micron sections were cut and then processed for immunofluorescence staining using a goat anti-rat CRHR1 antibody (1:200 dilution, Cat.: SC-1757, Santa Cruz Biotechnology, Santa Cruz, CA) or a goat-anti-human CRHR2 antibody (1:100 dilution, Cat.: SC-20550, Santa Cruz Biotechnology).
Immunohistochemistry
Segments of the transverse colon were embedded in OCT and frozen immediately. 5-micron sections were cut and then processed for peroxidase immunohistochemistry using a p-CREB/CREB antibody (1:50, overnight at 4 °C, Cell Signaling Technology, Danvers, MA) or its negative control rabbit IgG (BD Pharmingen, San Jose, CA), and then a biotinylated anti-rabbit antibody (1:200, 30 min, at room temperature, BD Pharmingen) as previously described (37). Hematoxylin solution (Vector Laboratories, Burlingame, CA) was used for counterstaining.
Immunoblotting Analysis
Equal amounts of protein from cell lysates were subjected to SDS-PAGE analysis and immunoblotting using anti-p-CREB antibody (1:1000, overnight at 4 °C, Cell Signaling Technology) and anti-CREB antibody (1:1000, overnight at room temperature, Cell Signaling Technology) as described previously (38).
Quantitative Real-time PCR (qPCR)
Total RNA from the mouse colon was isolated using RNeasy Plus Mini Kit (Qiagen), and an equal amount of RNA (2 μg) was transcribed into cDNA using a High Capacity Reverse Transcription kit (Applied Biosystems, Foster city, CA). Subsequently, qPCR was performed with FAMTM dye-labeled TaqMan® MGB (minor groove binding) probes using a 7500 Fast Real-Time PCR instrument (Applied Biosystems). The primer pairs and probes of hCRH (Cat.: Hs0 1921237_s1), Ucn 2 (Cat.: Hs00264218_s1), Ucn 3 (Cat.: Hs00846499_s1), CRHR1 (Cat.: Hs00366363_m1), CRHR2 (Cat.: Hs002 66401_m1), VEGF-A (Cat.: Hs00900055_m1), or GAPDH (Cat.: 4333764F) for the internal control were purchased from Applied Biosystems. The level of expression was calculated based upon the PCR cycle number (CT) at which the exponential growth in fluorescence from the probe passed a certain threshold value (CT). Relative gene expression was determined by differences in the CT values of the target genes after normalization to the RNA input level, using the CT value of GAPDH. Relative quantification was represented by standard 2−ΔCT calculations. ΔCT = (CT-target gene − CT-GAPDH). Each reaction was performed in triplicate.
Statistical Analysis
All results, except otherwise stated, are expressed as the mean ± S.D. Difference in survival was shown by Kaplan-Meier plot and the log-rank test was used to compare significant survival difference. Group data were compared by two-way ANOVA followed by the multiple-comparison Bonferroni t test or one-way ANOVA followed by a Newman-Keuls post-hoc test to assess differences between groups. The nonparametric Mann-Whitney U test was used to compare histological difference. Otherwise, paired and 2-tailed Student's t tests were used to compare results from the experiments. A p value of less than 0.05 was considered statistically significant.
Results
CRH and Ucn 3 Increase VEGF-A Production in Colonic Epithelial Cells
Previous studies indicated that the CRH family regulates VEGF-A secretion in human mast cells and rat vascular smooth muscle cells (25, 30). In colonic epithelial cells, however, there has been no report regarding the effect of the CRH system on VEGF-A expression. Thus, we first investigated whether CRH regulates VEGF-A expression in human colonic epithelial cells (NCM460) which endogenously express CRHR1 and CRHR2 (Fig. 1, A and B). When the cells were stimulated with CRH (100 nm) or Ucn 3 (100 nm) for 6 h, VEGF-A protein secretion was increased, compared with vehicle-treated cells (Fig. 1C). Although colonic epithelial cells expressed VEGF-A endogenously, stimulation by the CRH family increased VEGF-A expression even further (Vehicle, Fig. 1C). In addition, CRH- or Ucn 3-mediated VEGF-A production was completely inhibited in the presence of the CRHR1 inhibitor, antalarmin (10 nm) and the CRHR2 inhibitor, astressin 2B (10 nm), respectively (Fig. 1D). Since CRH and Ucn 3 exert their actions through binding to their preferential receptors CRHR1 and CRHR2, respectively, we further investigated the involvement of CRHRs in VEGF-A production. To test this, we isolated mouse intestinal epithelial cells (mIECs) as previously described (33, 34) and investigated the effect of CRH and Ucn 3 on VEGF-A production. In mIECs from CRHR1+/+ and CRHR2+/+ mice, treatment with CRH (100 nm) and Ucn 3 (100 nm) for 24 h increased VEGF-A production (Fig. 1E) as CRH and Ucn 3 were able to induce VEGF-A in NCM460 cells (Fig. 1, C and D). In mIECs from CRHR1−/− and CRHR2−/− mice, however, CRH- and Ucn 3-mediated VEGF-A protein production was greatly diminished (Fig. 1E) suggesting that VEGF-A production is mediated by CRHRs. In contrast, neither CRH nor Ucn 3 altered VEGF-A expression in other cells including human colonic smooth muscle cells (hCSMC) and human aortic smooth muscle cells (hASMC) (Fig. 1F), although a previous study showed Ucn 2-mediated VEGF-A expression in rat vascular smooth muscle cells (25).
FIGURE 1.
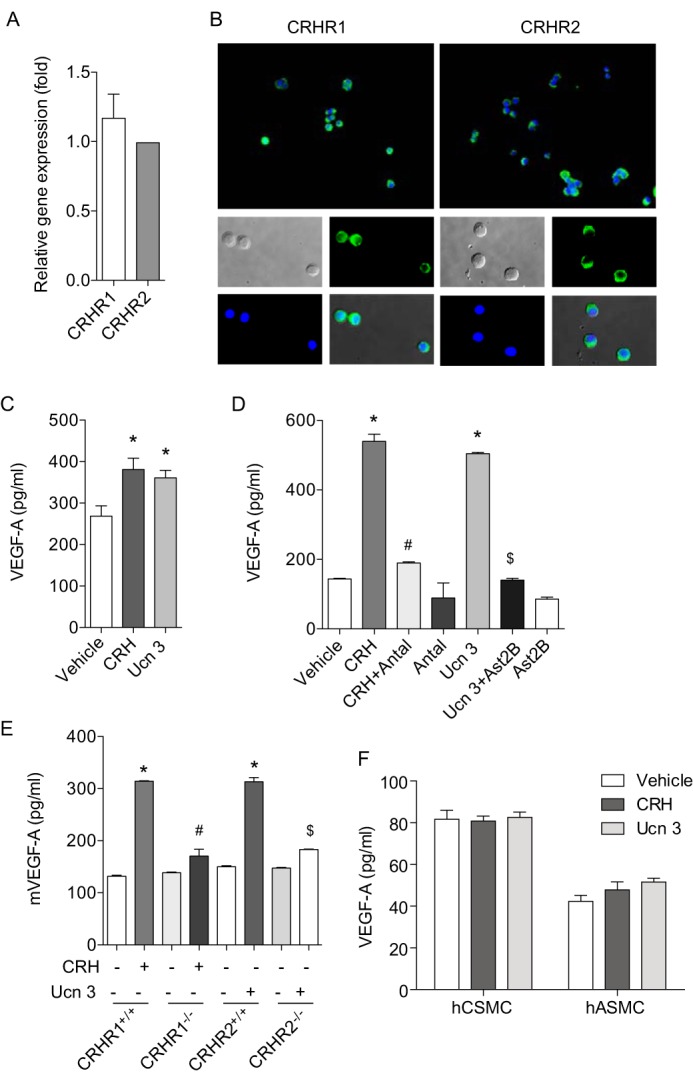
CRH and Ucn 3 increase the expression level of VEGF-A in the intestinal epithelium through CRHR1 and CRHR2, respectively. A, endogenous expression levels of CRHR1 and CRHR2 in NCM460 cells were determined by qPCR. The complementary DNA from human smooth muscle cells was used as a calibrator. B, expression levels of CRHR1 and CRHR2 were shown by immunofluorescence staining. C–F, ELISA was performed to measure VEGF-A levels in NCM460 cells (C and D; #, p < 0.05 versus CRH, $, p < 0.05 versus Ucn 3), mIECs (E; #, p < 0.05 versus CRH in CRHR1+/+ cells, $, p < 0.05 versus Ucn 3 in CRHR2+/+ cells), hCSMC and hASMC (F).
To further enhance CRH-mediated responses, we stably over-expressed either of the two CRH receptors in NCM460 cells. Since NCM460 cells express all the CRH receptor ligands, receptors are activated through an autocrine loop (Fig. 2A). Enhanced CRHR1 or CRHR2 expression was confirmed in cells transfected with CRHR1 or CRHR2 expressing constructs, compared with cells transfected with empty vector (EV) (Fig. 2B). As expected, CRHR1- or CRHR2-transfected cells exhibited substantially enhanced VEGF-A mRNA (Fig. 2C) and protein (Fig. 2D) levels compared with control cells in the absence of the ligand stimulation. Stimulation of the cells with CRH or Ucn 3 further enhanced VEGF-A production in this setting (Fig. 2E). Collectively, these results show that the CRH family of peptides is able to induce VEGF-A expression in colonic epithelial cells.
FIGURE 2.
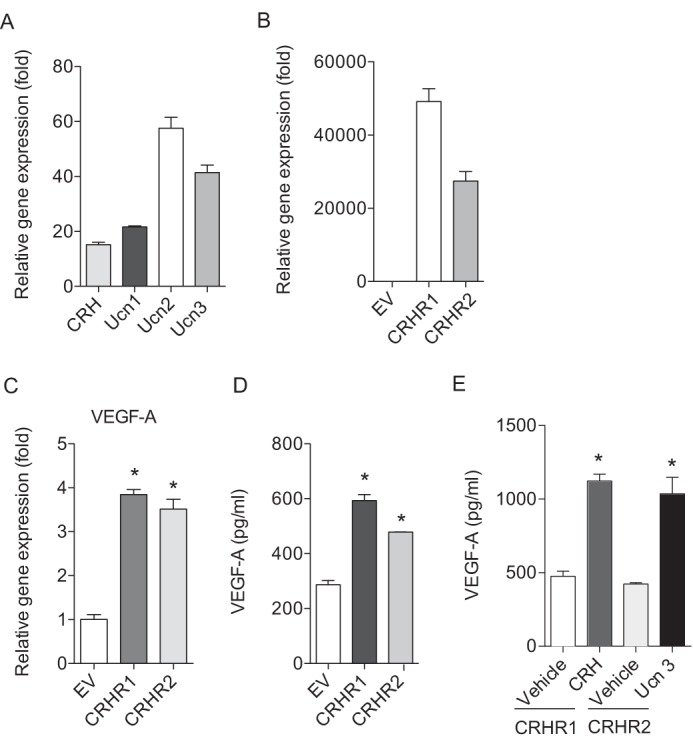
Overexpression of CRHR1 or CRHR2 further enhances VEGF-A production. A, expression levels of CRH and Ucn 1–3 were determined by qPCR. The complementary DNA from human smooth muscle cells was used as a calibrator. B, expression levels of CRHR1 and CRHR2 in NCM460 cells overexpressing CRHR1 or CRHR2 was determined by qPCR. The complementary DNA from NCM460 cells overexpressing empty vector (EV) was used as a calibrator. C, VEGF-A mRNA expression level was determined by qPCR. D and E, ELISA was performed to VEGF-A levels in NCM460 cells overexpressing CRHR1 or CRHR2. Data are mean ± S.D. *, p < 0.05 versus vehicle.
The CRH Family Induces VEGF-A Production through cAMP-dependent Signaling
To investigate a putative mechanism by which the CRH family modulates VEGF-A production, we evaluated the involvement of various signaling molecules in the CRHR-dependent downstream pathways (39). As a first step, we examined whether the adenylyl cyclase is involved in this regulation since the peptides of the CRH family are reported to activate adenylyl cyclase and subsequently increase cAMP levels in rat pituitary (40). To investigate whether the CRH family is also able to increase cAMP levels in colonic epithelial cells, we measured intracellular cAMP levels. Stimulation of colonic epithelial cells with CRH (10 nm) or Ucn 3 (10 nm) for 3 min substantially increased cAMP levels (Fig. 3A). Moreover, stimulation of the cells expressing CRHR1 with CRH (10 nm) or the cells expressing CRHR2 with Ucn 3 (10 nm) for 3 min also markedly increased cAMP levels (Fig. 3, B and C). We next tested whether exogenous addition of cAMP can enhance CRH-mediated VEGF-A production. Incubation of cells with 8-bromo-cAMP, a cell-permeable cAMP analog, increased VEGF-A production in the cells expressing EV (Fig. 3D) or CRHR1 or CRHR2 (Fig. 3E). In addition, inhibition of adenylyl cyclase activity by pretreatment (30 min) with the adenylyl cyclase inhibitor SQ22536 (8 mm) completely abolished CRH (100 nm) or Ucn 3 (100 nm)-induced VEGF-A expression both in NCM460 cells and in cells overexpressing CRHR1 or CRHR2 (Fig. 3, F–H). These results suggest that the CRH family of peptides regulates VEGF-A production by the activation of adenylyl cyclase and subsequent augmentation of cAMP.
FIGURE 3.

VEGF-A production by CRH and Ucn 3 is cAMP-dependent. cAMP EIA was monitored in NCM460 cells expressing an empty vector (EV) (A) or CRHR1 (B) or CRHR2 (C). VEGF-A ELISA was performed in NCM460 cells (D) or NCM460 cells expressing CRHR1 or CRHR2 (E) after adding 8-bromo-cAMP exogenously. In the presence of adenylyl cyclase inhibitor SQ22536, VEGF-A ELISA was performed in NCM460 cells (F) or NCM460 cells expressing CRHR1 (G) or CRHR2 (H). Data are mean ± S.D. *, p < 0.05 versus vehicle; #, p < 0.05 versus CRH; $, p < 0.05 versus Ucn 3.
CRH and Ucn 3 Increase VEGF Production via a HIF-1α-independent Pathway
It is generally recognized that VEGF expression is primarily regulated by the transcription factor HIF-1α. Thus, we tested whether HIF-1α is also involved in CRH-induced VEGF expression at the transcriptional level. To test the involvement of this pathway, a series of VEGF promoter constructs were transiently transfected into NCM460 cells. These constructs have various 5′ deletion sequences from the human vegf gene upstream of the luciferase reporter gene (shown schematically in Fig. 4A). Among those, the 2.6 kb promoter construct contains a well-known transcriptional regulator of VEGF, HIF-1α binding site at −965, whereas the 0.35 kb construct did not. CRH (10 nm) or Ucn 3 (10 nm) stimulation increased the reporter activity of the 2.6 kb promoter construct (−2361 to +298) by 10- or 8-fold, respectively, compared with vehicle treatment (Fig. 4B). The reporter activity of the 0.35 kb promoter construct (−196 to +157) was also increased by CRH (10 nm) or Ucn 3 (10 nm) stimulation by 15- or 20-fold, respectively, suggesting that HIF-1α may not be involved in CRH-mediated VEGF-A expression (Fig. 4B). We further examined whether there is an alteration of HIF-1α activity induced by CRH or Ucn 3 stimulation (10 nm) using a transcription factor ELISA system. Similar to the result in Fig. 4B, neither CRH nor Ucn 3 affected HIF-1α activation (Fig. 4, C and D). These results suggest that CRH-mediated VEGF-A production is independent of HIF-1α.
FIGURE 4.
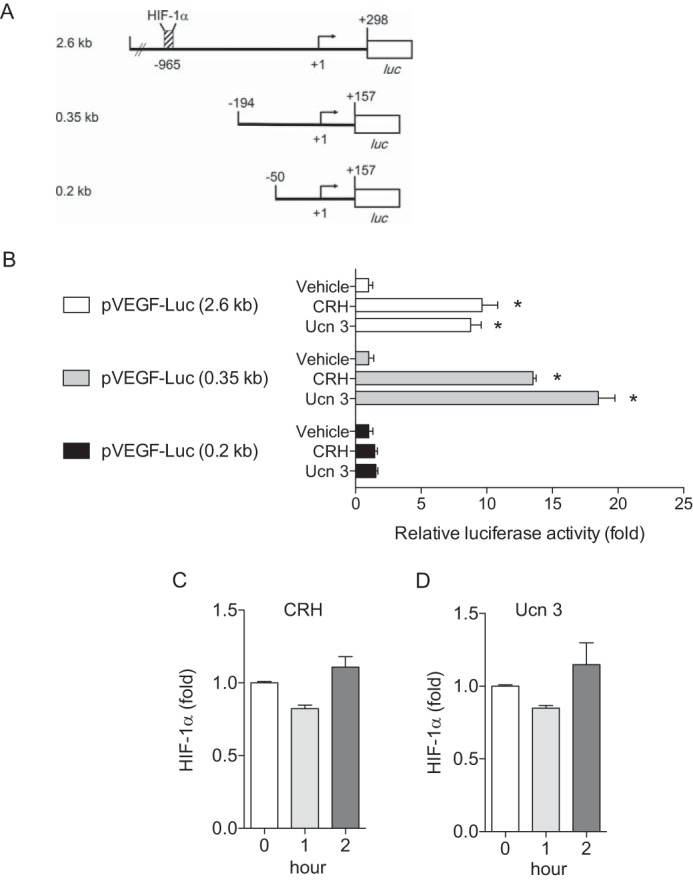
VEGF-A production by CRH and Ucn 3 is HIF-1α-independent. A, schematic illustration of VEGF promoter deletion constructs is presented. B, luciferase reporter assay is performed in NCM460 cells transfected with a series of VEGF promoter constructs. *, p < 0.05 versus vehicle. C, HIF-1α ELISA is performed in NCM460 cells after treatment with CRH (C) or Ucn 3 (D). Data are mean ± S.D.
Regulation of VEGF Promoter Activity by the CRH family Is Mediated by CREB
To further dissect out transcriptional regulation involved in CRH-regulated VEGF expression, the role of putative transcription factors which bind to VEGF promoter region was investigated. As shown in Fig. 4B, the cells transfected with the shortest, 0.2 kb promoter construct (−50 to +157) were not responsive to CRH or Ucn 3 stimulation (100 nm) suggesting that the sequence from −194 to −50 in the VEGF promoter is critical for the transcriptional regulation of CRH- and Ucn 3-mediated VEGF expression. The 144 bp of the VEGF promoter region (−194 to −51) that conferred CRH-mediated transcriptional activity contains putative binding sequences of several transcription factors including NF-κB (p65 and p50) and CREB. However, stimulation of the cells with CRH or Ucn 3 did not significantly increase the activities of NF-κB (p65 and p50) when measured by transcription factor ELISA in which activated transcription factor binds to the consensus-binding site on the oligo in the kit (data not shown). Since CREB activation is known to be essential for VEGF expression in various settings (12, 41, 42), we further tested whether the CRH family of peptides regulates CREB activation and subsequent VEGF-A expression. The DNA binding activity of the activated form of CREB (pCREB) was increased by CRH (100 nm) or Ucn 3 (100 nm) stimulation (Fig. 5A). Moreover, results from ChIP assays indicated that CRH (100 nm) or Ucn 3 (100 nm) stimulation induced the recruitment of CREB onto a putative CREB-binding site of VEGF promoter region in a time-dependent manner (Fig. 5B). In agreement with the above results, the level of phosphorylated CREB at Ser-133 was increased by CRH (100 nm) or Ucn 3 (100 nm) stimulation (Fig. 5C).
FIGURE 5.
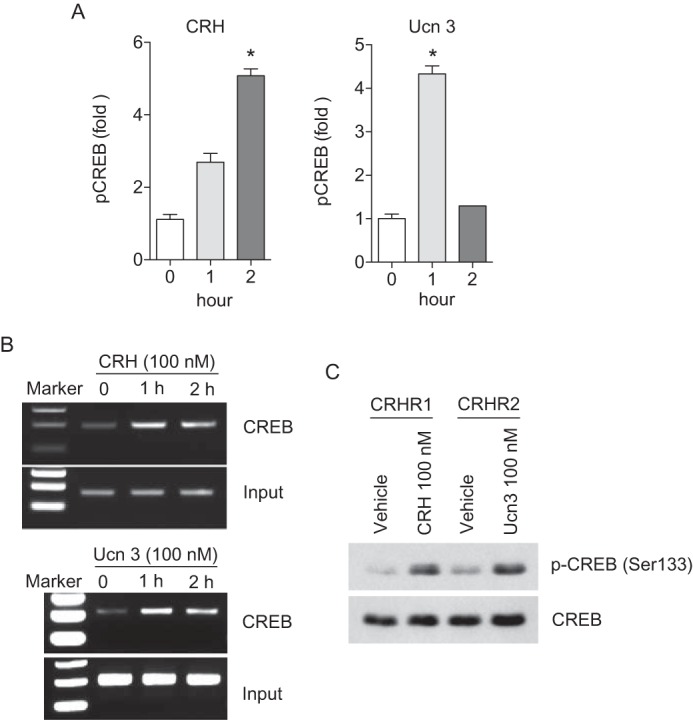
CRH and Ucn 3 increase VEGF-A production through the activation of CREB. A, transcription factor ELISA for pCREB is performed in CRH or Ucn 3-treated NCM460 cells. Data are mean ± S.D. *, p < 0.05 versus 0 h. B, ChIP assay is performed in CRH or Ucn 3-treated NCM460 cells. C, phosphorylation of CREB is examined in CRH or Ucn 3-treated NCM460 cells using Western blot analysis.
Furthermore, immunohistochemistry of pCREB was performed on the intestinal sections from CRHR1+/+ and CRHR1−/− or CRHR2+/+ and CRHR2−/− mice. As a result, the expression level of pCREB was decreased in the intestinal sections of CRHR1−/− and CRHR2−/− mice compared with the intestinal sections of WT mice (Fig. 6). Therefore, these results suggest that engagement of CRHRs with their ligands induces VEGF production via the CREB-dependent pathway.
FIGURE 6.
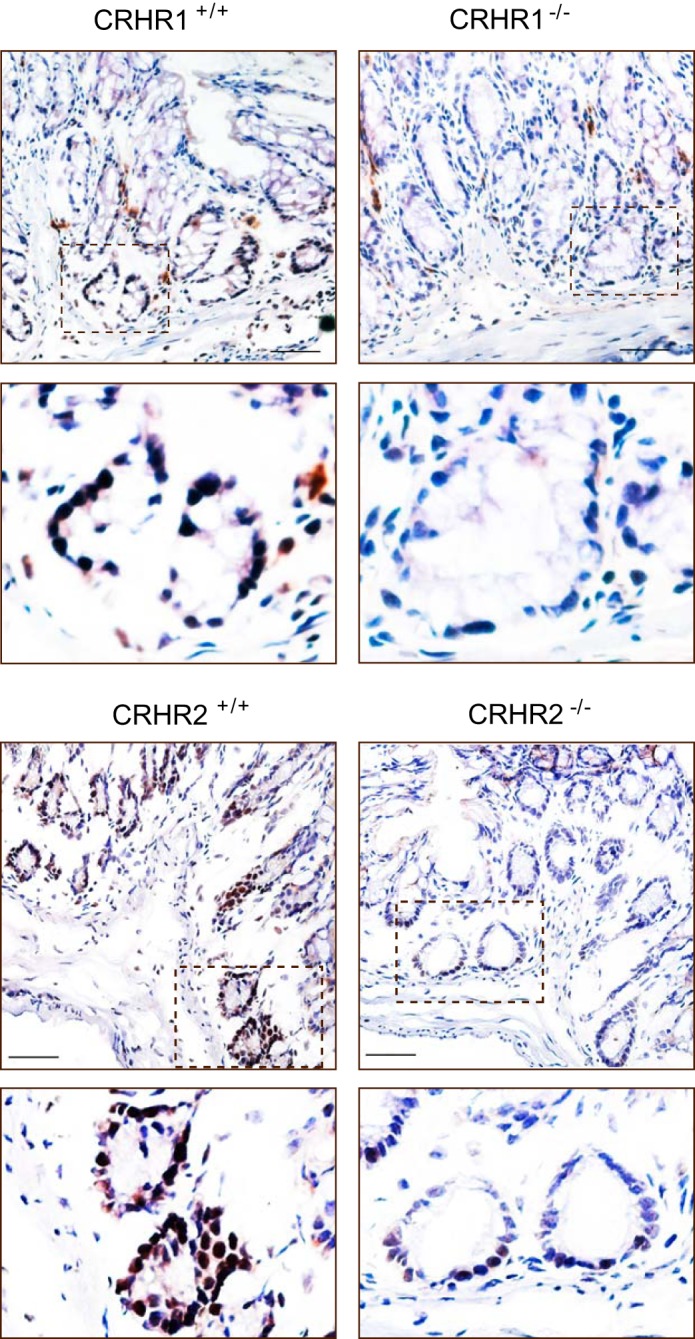
CRH and Ucn 3 regulate CREB phosphorylation. Immunohistochemistry results show CREB phosphorylation in the intestinal tissues of CRHR1+/+, CRHR1−/−, CRHR2+/+, and CRHR2−/− mice. Scale bar, 100 μm.
Genetic Deficiency of CREB Decreases Intestinal Inflammation
The above results showed that CRH-induced VEGF expression was regulated by the transcription factor CREB in colonic epithelial cells. Intriguingly, it has been previously suggested that increased expression level of VEGF could promote intestinal inflammation in animal and clinical studies (2–5). In light of this contention, we hypothesized that CREB−/− mice would express less VEGF than CREB+/+ mice and thereby exhibit less severe inflammatory response than the wild type control. To test this, we induced experimental colitis in CREB+/+ and CREB−/− mice by feeding 3% DSS in drinking water for 14 days and the inflammatory response was evaluated. Mortality and weight loss was reduced in CREB−/− mice compared with their littermate control CREB+/+ mice (Fig. 7, A and B). Clinical symptoms of colitis including rectal bleeding and diarrhea were also ameliorated in CREB−/− mice compared with CREB+/+ mice (data not shown). Additionally, shortening of colon length was dampened in CREB−/− mice compared with CREB+/+ mice, indicative of less severe colitis in CREB−/− mice than CREB+/+ mice (Fig. 7C). Colon tissues from CREB+/+ mice showed severe inflammatory lesions throughout the mucosa including loss of crypts, increased inflammatory cell infiltration and submucosal edema, whereas colon tissues from CREB−/− mice displayed less severe tissue damage than CREB+/+ mice and had partially intact epithelium (Fig. 7D). Since CREB plays a key role in CRH and Ucn 3-mediated VEGF expression, we performed mouse VEGF-A ELISA with colon tissues from CREB+/+ and CREB−/− mice and found that VEGF-A levels were significantly decreased in CREB−/− mice (Fig. 7E). Taken together, these results indicate that CREB deficiency suppresses inflammatory responses in the intestine.
FIGURE 7.

DSS-induced colitis is reduced in CREB−/− mice compared with littermate control CREB+/+ mice. A, difference in the survival is shown by the Kaplan-Meier plot. The log-rank test indicates significant survival differences between CREB+/+ and CREB−/− mice (n = 8–9 mice per genotype, p < 0.05). B, body weight data are mean ± S.E. (n = 8–9 mice per genotype; *, p < 0.05). C, difference in colon length is shown (n = 7 mice per genotype; *, p < 0.05). D, representative images of H&E-stained intestinal sections. Scale bar, 100 μm. E, mouse VEGF-A ELISA is performed in CREB+/+ and CREB−/− mice. Data are mean ± S.E. (n = 4–5 mice per genotype; *, p < 0.05).
Discussion
In our previous studies, we reported that the activation of CRHR1 by its preferential agonist, CRH promotes intestinal inflammation, whereas Ucn 3-mediated activation of CRHR2 inhibits the response (29). Most importantly, it was shown that CRH induces tube formation of intestinal endothelial cells in vitro, increases aortic vessel outgrowth ex vivo and enhances colitis-associated angiogenesis in vivo. Conversely, Ucn 3 decreases these activities. Additionally, we showed that the inflamed intestine in CRHR1−/− mice had decreased microvascular density and VEGF-A levels, whereas the intestine of CRHR2−/− mice had increased angiogenesis and VEGF-A expression. In the study presented herein, we expand these findings by demonstrating that the CRH family of peptides through activation of either CRHR1 or CRHR2 up-regulates VEGF-A expression in the intestinal epithelial cells. Underlying mechanisms of the VEGF-A production include the activation of adenylyl cyclase and increased cAMP induction along with transcriptional regulation of CREB. Thus, these data indicate that CREB deficiency confers a protective effect in intestinal inflammation through decreasing VEGF-A levels.
Reports from other groups, however, showed that the CRH family of peptides inhibits VEGF expression. For instance, in rat smooth muscle cells, Ucn 1 inhibited VEGF release and consequently suppressed angiogenesis (25). Moreover, either CRH or Ucn 3 decreased VEGF mRNA expression levels in cultured early placental extravillous trophoblasts and this effect was counteracted by the CRHR2 antagonist antisauvagine-30 (31). Additionally, Ucn 1 and Ucn 2 suppressed VEGF expression in hepatocellular carcinoma and small cell lung carcinoma cells via CRHR2 (43, 44).
The fact that both CRH and Ucn 3 trigger VEGF-A expression in the intestinal epithelial cells through CRHR1 and CRHR2, respectively was a somewhat unexpected characteristic of these peptides. One possibility that needs to be considered on the basis of this new information is that the activities of the CRH family of peptides depend on the types of cells and tissues. In this study, we measured the levels of VEGF expression in human colonic epithelial cells as well as primary mouse intestinal epithelial cells. This provides relevant tools to investigate the influence of the CRH family of peptides in terms of VEGF expression in the intestinal mucosa. Another possibility is that Ucn 3 may trigger production of an anti-angiogenic VEGF-A165b isoform instead of a pro-angiogenic VEGF-A165 isoform. VEGF-A exists in multiple isoforms of variable exon content. VEGF-A165b isoforms are generated by exon 8 distal splice site selection and constitute more than or close to half of the total VEGF-A expressed in many tissues including colonic epithelium (45). Unlike VEGF-A, VEGF-A165b does not stimulate endothelial cells migration and proliferation (46). In addition, VEGF-A165b reduces microvascular permeability and inhibits angiogenesis (46, 47). From our previous results, global deletion of CRHR2 gene increased angiogenesis during intestinal inflammation implying an anti-angiogenic effect of CRHR2. In the present study, however, Ucn 3 increases VEGF-A production, which is regarded as a pro-angiogenic factor. One explanation that might explain this contradiction is that Ucn 3 increases VEGF-A165b instead of VEGF-A, a distinction that is unable to be detected by most of VEGF-A ELISA kits. Our preliminary results showed that Ucn 3, but not CRH, increases VEGF-A165b production suggesting that there may be a differential regulation between CRH and Ucn 3 on the expression of the anti-angiogenic VEGF-A165b isoforms. Moreover, SP1 transcription factor activity is increased by only Ucn 3, suggesting that SP1 may play a key role in anti-angiogenic VEGF production (data not shown). This warrants a future study to elucidate a role of the CRH family in production of different VEGF isoforms.
The expression of VEGF is tightly regulated by hypoxia, cytokines, or growth factors (48). In response to hypoxia, HIF-1α is known to regulate VEGF transcription (7). However, recent studies indicate that HIF-1α can control VEGF in hypoxia-independent settings as well (10, 49). The 2.6-kb promoter construct used in figure 4 contains the HIF-1α binding site while the 0.35-kb promoter construct lacks this site. Yet, we found that VEGF reporter activity upon stimulation by CRH or Ucn 3 was similar in the cells transfected with either of the constructs, suggesting that HIF-1α is not involved in the CRH peptide-mediated VEGF regulation. In addition, HIF-1α ELISA showed no difference in protein levels in response to CRH or Ucn 3 stimulation. Moreover, the von Hippel-Lindau tumor suppressor gene (VHL)-dependent repression of VEGF promoter activity has been reported, and it is known that the sequence from bp −194 to −50 of the VEGF promoter confers most of the VHL response (50). The 0.35-kb promoter construct used in Fig. 4 contains the HIF-1α binding site while the 0.2-kb promoter construct lacks this site. Thus, we would expect increased VEGF promoter activity when the construct is missing the repressor VHL sequence. However, we found that the promoter activity was significantly reduced in the 0.2-kb promoter construct compared with the 0.35-kb promoter construct suggesting that the VHL-mediated repression of VEGF is not involved in this setting.
In a previous study, CRH signaling can be mimicked by the activation of cAMP/PKA/CREB signaling pathway in synovial tissue inflammation (51). On this basis, one might speculate that CRH-dependent CREB activation may regulate VEGF-A production. Indeed, activation of CRH signaling increased CREB phosphorylation and CREB binding activity on the VEGF promoter region. Furthermore, genetic deficiency of CREB decreased intestinal inflammation suggesting that reduction of VEGF levels because of loss of CREB may contribute to amelioration of colitis symptoms.
Finally, the findings reported here showed that activation of the either CRHR1 or CRHR2 signaling by CRH and Ucn 3, respectively increases VEGF-A in intestinal epithelial cells suggesting that these peptides could be a main player in inflammatory angiogenesis. Moreover, since CRH and Ucn 3 regulates VEGF-A production through CREB transcriptional activity, strategies to inactivate CREB could have therapeutic potential in devastating inflammatory diseases in the gut.
Author Contributions
S. H. R. performed experiments and wrote the paper. E. M. and Y. L. performed experiments. Y. T. and C. P. helped data interpretation. E. I. designed the study, performed experiments and wrote the paper. All authors analyzed the results and approved the final version of the manuscript.
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2014R1A1A2057432, to E. I.) and by the National Institutes of Health (R01DK101671, to C. P.). The authors declare that they have no conflicts of interest with the contents of this article.
- VEGF
- vascular endothelial growth factor
- CREB
- cAMP response element-binding protein
- CRH
- corticotropin releasing hormone
- CRHR
- CRH receptor
- DSS
- dextran sodium sulfate
- IBD
- inflammatory bowel disease
- Ucn
- urocortin.
References
- 1.Ferrara N., and Davis-Smyth T. (1997) The biology of vascular endothelial growth factor. Endocr. Rev. 18, 4–25 [DOI] [PubMed] [Google Scholar]
- 2.Scaldaferri F., Vetrano S., Sans M., Arena V., Straface G., Stigliano E., Repici A., Sturm A., Malesci A., Panes J., Yla-Herttuala S., Fiocchi C., and Danese S. (2009) VEGF-A links angiogenesis and inflammation in inflammatory bowel disease pathogenesis. Gastroenterology 136, 585–595 [DOI] [PubMed] [Google Scholar]
- 3.Tsiolakidou G., Koutroubakis I. E., Tzardi M., and Kouroumalis E. A. (2008) Increased expression of VEGF and CD146 in patients with inflammatory bowel disease. Dig. Liver Dis. 40, 673–679 [DOI] [PubMed] [Google Scholar]
- 4.Tolstanova G., Khomenko T., Deng X., Chen L., Tarnawski A., Ahluwalia A., Szabo S., and Sandor Z. (2009) Neutralizing anti-vascular endothelial growth factor (VEGF) antibody reduces severity of experimental ulcerative colitis in rats: direct evidence for the pathogenic role of VEGF. J. Pharmacol. Exp. Ther. 328, 749–757 [DOI] [PubMed] [Google Scholar]
- 5.Carvalho A. T., Souza H., Carneiro A. J., Castelo-Branco M., Madi K., Schanaider A., Silv F., Pereira Junior F. A., Pereira M. G., Tortori C., Dines I., Carvalho J., Rocha E., and Elia C. (2007) Therapeutic and prophylactic thalidomide in TNBS-induced colitis: synergistic effects on TNF-alpha, IL-12 and VEGF production. World J. Gastroenterol 13, 2166–2173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferrara N., Gerber H. P., and LeCouter J. (2003) The biology of VEGF and its receptors. Nat. Med. 9, 669–676 [DOI] [PubMed] [Google Scholar]
- 7.Forsythe J. A., Jiang B. H., Iyer N. V., Agani F., Leung S. W., Koos R. D., and Semenza G. L. (1996) Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 16, 4604–4613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levy A. P., Levy N. S., and Goldberg M. A. (1996) Post-transcriptional regulation of vascular endothelial growth factor by hypoxia. J. Biol. Chem. 271, 2746–2753 [DOI] [PubMed] [Google Scholar]
- 9.Ikeda E., Achen M. G., Breier G., and Risau W. (1995) Hypoxia-induced transcriptional activation and increased mRNA stability of vascular endothelial growth factor in C6 glioma cells. J. Biol. Chem. 270, 19761–19766 [DOI] [PubMed] [Google Scholar]
- 10.Im E., Venkatakrishnan A., and Kazlauskas A. (2005) Cathepsin B regulates the intrinsic angiogenic threshold of endothelial cells. Mol. Biol. Cell 16, 3488–3500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bergers G., Brekken R., McMahon G., Vu T. H., Itoh T., Tamaki K., Tanzawa K., Thorpe P., Itohara S., Werb Z., and Hanahan D. (2000) Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2, 737–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee J. S., Jang D. J., Lee N., Ko H. G., Kim H., Kim Y. S., Kim B., Son J., Kim S. H., Chung H., Lee M. Y., Kim W. R., Sun W., Zhuo M., Abel T., Kaang B. K., and Son H. (2009) Induction of neuronal vascular endothelial growth factor expression by cAMP in the dentate gyrus of the hippocampus is required for antidepressant-like behaviors. J. Neurosci. 29, 8493–8505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garrido C., Saule S., and Gospodarowicz D. (1993) Transcriptional regulation of vascular endothelial growth factor gene expression in ovarian bovine granulosa cells. Growth Factors 8, 109–117 [DOI] [PubMed] [Google Scholar]
- 14.Claffey K. P., Wilkison W. O., and Spiegelman B. M. (1992) Vascular endothelial growth factor. Regulation by cell differentiation and activated second messenger pathways. J. Biol. Chem. 267, 16317–16322 [PubMed] [Google Scholar]
- 15.Lewis K., Li C., Perrin M. H., Blount A., Kunitake K., Donaldson C., Vaughan J., Reyes T. M., Gulyas J., Fischer W., Bilezikjian L., Rivier J., Sawchenko P. E., and Vale W. W. (2001) Identification of urocortin III, an additional member of the corticotropin-releasing factor (CRF) family with high affinity for the CRF2 receptor. Proc. Natl. Acad. Sci. U.S.A. 98, 7570–7575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reyes T. M., Lewis K., Perrin M. H., Kunitake K. S., Vaughan J., Arias C. A., Hogenesch J. B., Gulyas J., Rivier J., Vale W. W., and Sawchenko P. E. (2001) Urocortin II: a member of the corticotropin-releasing factor (CRF) neuropeptide family that is selectively bound by type 2 CRF receptors. Proc. Natl. Acad. Sci. U.S.A. 98, 2843–2838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsu S. Y., and Hsueh A. J. (2001) Human stresscopin and stresscopin-related peptide are selective ligands for the type 2 corticotropin-releasing hormone receptor. Nat. Med. 7, 605–611 [DOI] [PubMed] [Google Scholar]
- 18.Im E. (2014) Corticotropin-releasing Hormone and Its Biological Diversity toward Angiogenesis. Intest Res. 12, 96–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vale W., Spiess J., Rivier C., and Rivier J. (1981) Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science 213, 1394–1397 [DOI] [PubMed] [Google Scholar]
- 20.Hillhouse E. W., and Grammatopoulos D. K. (2006) The molecular mechanisms underlying the regulation of the biological activity of corticotropin-releasing hormone receptors: implications for physiology and pathophysiology. Endocr. Rev. 27, 260–286 [DOI] [PubMed] [Google Scholar]
- 21.Taché Y., and Bonaz B. (2007) Corticotropin-releasing factor receptors and stress-related alterations of gut motor function. J. Clin. Invest. 117, 33–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anton P. M., Gay J., Mykoniatis A., Pan A., O'Brien M., Brown D., Karalis K., and Pothoulakis C. (2004) Corticotropin-releasing hormone (CRH) requirement in Clostridium difficile toxin A-mediated intestinal inflammation. Proc. Natl. Acad. Sci. U.S.A. 101, 8503–8508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kokkotou E., Torres D., Moss A. C., O'Brien M., Grigoriadis D. E., Karalis K., and Pothoulakis C. (2006) Corticotropin-releasing hormone receptor 2-deficient mice have reduced intestinal inflammatory responses. J. Immunol. 177, 3355–3361 [DOI] [PubMed] [Google Scholar]
- 24.Million M., Taché Y., and Anton P. (1999) Susceptibility of Lewis and Fischer rats to stress-induced worsening of TNB-colitis: protective role of brain CRF. Am. J. Physiol. 276, G1027–1036 [DOI] [PubMed] [Google Scholar]
- 25.Bale T. L., Giordano F. J., Hickey R. P., Huang Y., Nath A. K., Peterson K. L., Vale W. W., and Lee K. F. (2002) Corticotropin-releasing factor receptor 2 is a tonic suppressor of vascularization. Proc. Natl. Acad. Sci. U.S.A. 99, 7734–7739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hao Z., Huang Y., Cleman J., Jovin I. S., Vale W. W., Bale T. L., and Giordano F. J. (2008) Urocortin2 inhibits tumor growth via effects on vascularization and cell proliferation. Proc. Natl. Acad. Sci. U.S.A. 105, 3939–3944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tezval H., Jurk S., Atschekzei F., Becker J. U., Jahn O., Serth J., and Kuczyk M. A. (2009) Urocortin and corticotropin-releasing factor receptor 2 in human renal cell carcinoma: disruption of an endogenous inhibitor of angiogenesis and proliferation. World J. Urol. 27, 825–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tezval H., Jurk S., Atschekzei F., Serth J., Kuczyk M. A., and Merseburger A. S. (2009) The involvement of altered corticotropin releasing factor receptor 2 expression in prostate cancer due to alteration of anti-angiogenic signaling pathways. Prostate 69, 443–448 [DOI] [PubMed] [Google Scholar]
- 29.Im E., Rhee S. H., Park Y. S., Fiocchi C., Taché Y., and Pothoulakis C. (2010) Corticotropin-releasing hormone family of peptides regulates intestinal angiogenesis. Gastroenterology 138, 2457–2467, 2467, e2451–2455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao J., Papadopoulou N., Kempuraj D., Boucher W. S., Sugimoto K., Cetrulo C. L., and Theoharides T. C. (2005) Human mast cells express corticotropin-releasing hormone (CRH) receptors and CRH leads to selective secretion of vascular endothelial growth factor. J. Immunol. 174, 7665–7675 [DOI] [PubMed] [Google Scholar]
- 31.Wakahashi S., Nakabayashi K., Maruo N., Yata A., Ohara N., and Maruo T. (2008) Effects of corticotropin-releasing hormone and stresscopin on vascular endothelial growth factor mRNA expression in cultured early human extravillous trophoblasts. Endocrine 33, 144–151 [DOI] [PubMed] [Google Scholar]
- 32.Rhee S. H., Keates A. C., Moyer M. P., and Pothoulakis C. (2004) MEK is a key modulator for TLR5-induced interleukin-8 and MIP3α gene expression in non-transformed human colonic epithelial cells. J. Biol. Chem. 279, 25179–25188 [DOI] [PubMed] [Google Scholar]
- 33.Choi Y. J., Im E., Chung H. K., Pothoulakis C., and Rhee S. H. (2010) TRIF mediates Toll-like receptor 5-induced signaling in intestinal epithelial cells. J. Biol. Chem. 285, 37570–37578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choi Y. J., Jung J., Chung H. K., Im E., and Rhee S. H. (2013) PTEN regulates TLR5-induced intestinal inflammation by controlling Mal/TIRAP recruitment. FASEB J. 27, 243–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rhee S. H., and Hwang D. (2000) Murine TOLL-like receptor 4 confers lipopolysaccharide responsiveness as determined by activation of NF κB and expression of the inducible cyclooxygenase. J. Biol. Chem. 275, 34035–34040 [DOI] [PubMed] [Google Scholar]
- 36.Rhee S. H., Im E., Riegler M., Kokkotou E., O'brien M., and Pothoulakis C. (2005) Pathophysiological role of Toll-like receptor 5 engagement by bacterial flagellin in colonic inflammation. Proc. Natl. Acad. Sci. U.S.A. 102, 13610–13615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rhee S. H., Im E., and Pothoulakis C. (2008) Toll-like receptor 5 engagement modulates tumor development and growth in a mouse xenograft model of human colon cancer. Gastroenterology 135, 518–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Im E., and Kazlauskas A. (2006) Regulating angiogenesis at the level of PtdIns-4,5-P2. EMBO J. 25, 2075–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grammatopoulos D. K. (2012) Insights into mechanisms of corticotropin-releasing hormone receptor signal transduction. Br. J. Pharmacol 166, 85–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Labrie F., Veilleux R., Lefevre G., Coy D. H., Sueiras-Diaz J., and Schally A. V. (1982) Corticotropin-releasing factor stimulates accumulation of adenosine 3′, 5′-monophosphate in rat pituitary corticotrophs. Science 216, 1007–1008 [DOI] [PubMed] [Google Scholar]
- 41.Impey S., McCorkle S. R., Cha-Molstad H., Dwyer J. M., Yochum G. S., Boss J. M., McWeeney S., Dunn J. J., Mandel G., and Goodman R. H. (2004) Defining the CREB regulon: a genome-wide analysis of transcription factor regulatory regions. Cell 119, 1041–1054 [DOI] [PubMed] [Google Scholar]
- 42.Jeon S. H., Chae B. C., Kim H. A., Seo G. Y., Seo D. W., Chun G. T., Yie S. W., Eom S. H., and Kim P. H. (2007) The PKA/CREB pathway is closely involved in VEGF expression in mouse macrophages. Mol. Cells 23, 23–29 [PubMed] [Google Scholar]
- 43.Wang J., Xu Y., Xu Y., Zhu H., Zhang R., Zhang G., and Li S. (2008) Urocortin's inhibition of tumor growth and angiogenesis in hepatocellular carcinoma via corticotrophin-releasing factor receptor 2. Cancer Invest 26, 359–368 [DOI] [PubMed] [Google Scholar]
- 44.Wang J., Jin L., Chen J., and Li S. (2010) Activation of corticotropin-releasing factor receptor 2 inhibits the growth of human small cell lung carcinoma cells. Cancer Invest 28, 146–155 [DOI] [PubMed] [Google Scholar]
- 45.Harper S. J., and Bates D. O. (2008) VEGF-A splicing: the key to anti-angiogenic therapeutics? Nat. Rev. Cancer 8, 880–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bates D. O., Cui T. G., Doughty J. M., Winkler M., Sugiono M., Shields J. D., Peat D., Gillatt D., and Harper S. J. (2002) VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 62, 4123–4131 [PubMed] [Google Scholar]
- 47.Woolard J., Wang W. Y., Bevan H. S., Qiu Y., Morbidelli L., Pritchard-Jones R. O., Cui T. G., Sugiono M., Waine E., Perrin R., Foster R., Digby-Bell J., Shields J. D., Whittles C. E., Mushens R. E., Gillatt D. A., Ziche M., Harper S. J., and Bates D. O. (2004) VEGF165b, an inhibitory vascular endothelial growth factor splice variant: mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Res. 64, 7822–7835 [DOI] [PubMed] [Google Scholar]
- 48.Takahashi A., Kureishi Y., Yang J., Luo Z., Guo K., Mukhopadhyay D., Ivashchenko Y., Branellec D., and Walsh K. (2002) Myogenic Akt signaling regulates blood vessel recruitment during myofiber growth. Mol. Cell. Biol. 22, 4803–4814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Isaacs J. S., Jung Y. J., Mimnaugh E. G., Martinez A., Cuttitta F., and Neckers L. M. (2002) Hsp90 regulates a von Hippel Lindau-independent hypoxia-inducible factor-1α-degradative pathway. J. Biol. Chem. 277, 29936–29944 [DOI] [PubMed] [Google Scholar]
- 50.Mukhopadhyay D., Knebelmann B., Cohen H. T., Ananth S., and Sukhatme V. P. (1997) The von Hippel-Lindau tumor suppressor gene product interacts with Sp1 to repress vascular endothelial growth factor promoter activity. Mol. Cell. Biol. 17, 5629–5639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McEvoy A. N., Bresnihan B., Fitzgerald O., and Murphy E. P. (2002) Corticotropin-releasing hormone signaling in synovial tissue vascular endothelium is mediated through the cAMP/CREB pathway. Ann. N.Y. Acad. Sci. 966, 119–130 [DOI] [PubMed] [Google Scholar]