

Background: Whether ribonucleotide and base excision repair (RER and BER) interfere during repair synthesis events is unknown.
Results: Complex mispairs containing ribonucleotides and oxidized bases are formed by polymerase β and processed by RER and BER enzymes.
Conclusion: Complex lesions modify the efficiency of DNA/RNA repair systems.
Significance: This work explains how complex mispairs can compromise BER and RER.
Keywords: 8-oxoguanine (8-oxoG), 8-oxoguanine glycosylase (OGG1), DNA damage, DNA polymerase, DNA repair, mutY homolog (MUTYH), ribonuclease
Abstract
The cellular pool of ribonucleotide triphosphates (rNTPs) is higher than that of deoxyribonucleotide triphosphates. To ensure genome stability, DNA polymerases must discriminate against rNTPs and incorporated ribonucleotides must be removed by ribonucleotide excision repair (RER). We investigated DNA polymerase β (POL β) capacity to incorporate ribonucleotides into trinucleotide repeated DNA sequences and the efficiency of base excision repair (BER) and RER enzymes (OGG1, MUTYH, and RNase H2) when presented with an incorrect sugar and an oxidized base. POL β incorporated rAMP and rCMP opposite 7,8-dihydro-8-oxoguanine (8-oxodG) and extended both mispairs. In addition, POL β was able to insert and elongate an oxidized rGMP when paired with dA. We show that RNase H2 always preserves the capacity to remove a single ribonucleotide when paired to an oxidized base or to incise an oxidized ribonucleotide in a DNA duplex. In contrast, BER activity is affected by the presence of a ribonucleotide opposite an 8-oxodG. In particular, MUTYH activity on 8-oxodG:rA mispairs is fully inhibited, although its binding capacity is retained. This results in the reduction of RNase H2 incision capability of this substrate. Thus complex mispairs formed by an oxidized base and a ribonucleotide can compromise BER and RER in repeated sequences.
Introduction
Reactive oxygen species, produced by numerous exogenous and endogenous factors, are a major source of DNA damage. Between 1000 and 7000 7,8-dihydro-8-oxoguanine (8-oxodG)3 lesions are generated in the DNA of a single cell each day (1). 8-OxodG can mispair with adenine during DNA replication to generate G:C to T:A transversion mutations (2). The precursors of DNA/RNA synthesis are also targets for damage during oxidative stress. DNA repair and nucleotide pool sanitation systems conserved from bacteria to mammalian cells counteract both types of damage (3). The cooperative action of the base excision repair (BER) DNA glycosylases OGG1 and MUTYH prevents 8-oxodG-induced mutagenesis. OGG1 removes 8-oxodG from 8-oxodG:dC pairs and MUTYH excises adenine mispaired with 8-oxodG (for reviews, see Refs. 4–6). The resulting abasic sites are repaired by the sequential action of apurinic/apyrimidinic (AP)-endonuclease-1 (APE1), DNA polymerase β (pol β), and DNA ligase. The oxidized purine nucleoside triphosphatase encoded by the MutT homolog MTH1 provides an additional level of protection by hydrolyzing 8-oxodGTP and 8-oxorGTP to the corresponding monophosphates to prevent their incorporation into DNA and RNA (7). Consistent with their protective role against mutation, inactivation of OGG1, MUTYH, or MTH1 is associated with a mutator phenotype (7–9).
BER factors can affect the length of trinucleotide repeat regions in several neurodegenerative diseases (10–12), suggesting that the intervention of DNA repair proteins might be deleterious. The relationship between oxidative stress and repeat expansion is not fully understood. Current models envisage strand interruptions introduced by BER enzymes, POL β-mediated insertion of a limited number of nucleotides, and “toxic oxidation cycles” leading to trinucleotide repeat expansion (13).
Recently genome instability due to erroneous incorporation of ribonucleotides by replicative DNA polymerases has been reported (for reviews, see Refs. 14 and 15). To prevent rNTP incorporation, the active sites of DNA polymerases operate selection via “steric gates” in which specific amino acid residues cause a steric clash with the C2′ oxygen of an incoming rNTP (16). This mechanism is not completely effective, however, and replicative DNA polymerases incorporate numerous ribonucleotides (17).
Ribonucleotide excision repair (RER) is the process that excises DNA ribonucleotides (18, 19). In RER, RNase H2 nicks duplex DNA 5′ to a misincorportated ribonucleotide. DNA repair is completed via strand displacement by proliferating cell nuclear antigen/replicative DNA polymerases, release of the ribonucleotide-containing DNA strand by ExoI and FEN1 and DNA ligation. In the absence of RNase H2, removal of DNA ribonucleotides may also occur via a DNA topoisomerase I-dependent mechanism (20).
Erroneous ribonucleotide incorporation might also occur during repair synthesis, as DNA polymerases involved in DNA repair (POL β, POL λ, POL μ) can all utilize rNMPs (21–24). We investigated the ability of pol β to incorporate rNTPs opposite canonical and/or oxidized guanines in repeated DNA sequences. We also examined incorporation of 8-oxorGTP. We demonstrate that complex mispairs, formed by an oxidized base and a ribonucleotide, can compromise BER in repeat sequences. In addition MUTYH binding at rA:8-oxodG mispair can adversely affect the efficiency of RNase H2.
Experimental Procedures
Materials
8-OxodGTP was purchased from TriLink BioTechnologies (San Diego, CA), 8-oxorGTP from Jena Biosciences GmbH (Jena, Germany), and dNTP and rNTP from Sigma. Synthetic oligonucleotides, HPLC purified, were purchased from Thermo (ThermoFisher Scientific, Ulm, Germany). Oligomers containing a single internal modification (8-oxodG or ribonucleotide base) were 5′ end labeled with 6-carboxyfluorescein (6-FAM) or Texas Red dye. Oligomers were further purified by PAGE (20% polyacrylamide/bisacrylamide (37.5:1 ratio) (Bio-Rad) in 7 m urea. Samples were generally annealed in a 1:1 concentration ratio. Duplexes homogeneity was always checked on a 10% nondenaturing PAGE.
Human recombinant BER proteins OGG1, APE1, and POL β were obtained from Trevigen Inc. (Gaithersburg, MD) and RNase H2 from New England Biolabs (UK). Human recombinant MUTYH was obtained by expression and purification as fusion maltose-binding protein as previously described (25). The active fraction concentration was evaluated as described by Turco et al. (26).
Primer Extension Experiments
Incorporation of dAMP/rAMP and dCMP/rCMP by POLβ were analyzed using a substrate obtained by annealing a 5′ end (6-FAM)-labeled primer (5′-CGA GTC ATC TAG CAT CCG TAC) to a template strand (5′-CTA CGA ATG AGT (CTG)5 CG*G TAC GGA TGC TAG ATG ACT CG) containing an 8-oxodG (G*) at the template position for the incoming nucleotide. Insertion of dAMP/rAMP and dCMP/rCMP were performed on substrates obtained by annealing 5′ end (6-FAM)-labeled primers (5′-CGA GTC ATC TAG CAT CCG TAC) and (5′-CGA GTC ATC TAG CAT CCG TA) to a template strand (5′-CTA CGA ATG AGT (CTG)6 TAC GGA TGC TAG ATG ACT CG) that contained, respectively, a T or a G at the templating position of the incoming nucleotide. Incorporation of 8-oxodGMP or 8-oxorGMP were analyzed by using a 5′ end (6-FAM)-labeled primer (5′-GCA ATG AGT AAG TCT ACG TAC TGC) annealed to a template (5′-CGA GTC ATC TAG CAT CCG TA (CAG)20 TAC GTA GAC TTA CTC ATT GC). In a standard reaction, primer/templates (10 nm) were preincubated with POL β (0.1 unit) at 4 °C for 1 min in the assay buffer (50 mm Tris-HCl, 10 mm MgCl2, 10 mm KCl, 1 mm DTT, 1% glycerol), then rNTP were added at increasing concentrations and the reaction allowed for 1 h at 37 °C. Elongation was performed by adding dNTP and the reaction allowed for an additional 30 min. Reactions were halted by addition of a loading buffer solution containing 95% formamide, 20 mm EDTA, 0.05% bromphenol blue and products denatured at 95 °C for 2 min. Reaction products were analyzed by electrophoresis in a denaturing 15% polyacrylamide gel at 500 V for 2–3 h at 40 °C. The insertion products were detected by fluorescence by using a Typhoon scanner (Typhoon 9200 Gel Imager, GE Healthcare, Uppsala, Sweden).
Incorporation Kinetics Analysis
The analysis of the band intensities was performed by ImageJ software (NIH Image software, rsb.info.nih.gov/ij/) and the percentage of incorporated dNMP was plotted as a function of the added dNTPs. Data were fitted by Kaleidagraph software (Kaleidagraph, Synergy Corp.). To calculate the kinetics parameters for nucleotide incorporation Vmax and Km, the values of the integrated gel band intensities were plotted as a function of the dNTP concentration and fitted to Equation 1,
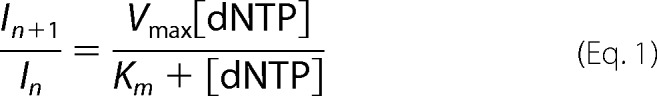 |
where n corresponds to the primer. All band intensities were normalized to the total band intensities to avoid effects due to loading differences. The selectivity index was calculated as Equation 2.
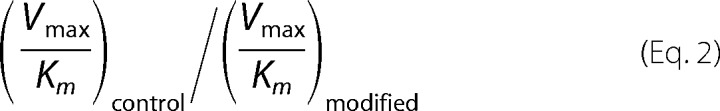 |
DNA Glycosylases and RNase H2 Assays
OGG1 substrates were obtained by annealing a 36-mer oligomer labeled at the 5′ end with Texas Red dye (5′ Texas red-CGA ATG AGT CTG* (CTG)5 GAT GAC TGC) with a 5′ end 6-FAM-labeled 36-mer oligomer (5′-(6-FAM)-GCA GTC ATC (CAG)5 rCAGACT CAT TCG) or (5′-(6-FAM)-GCA GTC ATC (CAG)6 ACT CAT TCG) to produce duplexes containing a single 8-oxodG:rC or 8-oxodG:dC mispair. MUTYH substrates were obtained by annealing the 5′ end Texas Red-labeled oligomer (5′-Texas Red-CGA ATG AGT CTG CTG CTG CTG CG*G CTG GAT GAC TGC) with the oligomers 6-FAM-GCA GTC ATC CAG CrAG (CAG)4 ACT CAT TCG) and 6-FAM-GCA GTC ATC CAG CrAG (CAG)4 ACT CAT TCG), to produce duplexes containing a single 8-oxodG:rA and a single 8-oxodG:dA mispair, respectively. In a standard OGG1 cleavage reaction, DNA substrate (10 nm) was incubated with the purified enzyme (0–20 nm active fraction) in 20 mm Tris-HCl, pH 8, 1 mm EDTA, 1 mm DTT, 0.1 mg/ml of BSA (Buffer A) at 37 °C for 30 min. Reactions were stopped by NaOH addition (100 mm final concentration) and heating at 90 °C for 2 min. After addition of gel loading solution (95% formamide, 10 mm EDTA, and 0.05% bromphenol blue) and heating at 95 °C for 2 min, reaction products were resolved on a 20% polyacrylamide gel made in 7 m urea, Tris borate buffer. Samples were run at 500 V for 2 h 30 min at 40 °C. Gel images were visualized by Typhoon scanner and data analysis was performed by using ImageJ and Kaleidagraph software as described before. MUTYH adenine removal capacity (both dA and rA) was analyzed by a similar procedure, but reaction buffer A was implemented with 80 mm NaCl (Buffer B). In addition, the incision at the abasic site, produced by the MUTYH reaction on DNA substrates, was performed by incubating the samples with APE1 enzyme (0.1 unit/reaction volume). For RNase H2 assays were allowed by incubating the DNA substrates labeled at the ribonucleotide strand (10 nm) with increasing concentrations of the purified enzyme at 37 °C for 1 h. Electrophoresis and data analysis were performed as described before. RNase H2 and MUTYH assays on 8-oxorG-dA containing substrate were performed on duplexes built by incorporation of the 8-oxorGTP triphosphate and extension of the terminal mispair by POL β as described before. Duplexes were EtOH precipitated and resuspended in the RNase H2 or MUTYH buffer (10 μl) and incision reactions were allowed at 37 °C for 1 h. Enzymes and dNTP concentrations are reported in the figure legends. Reaction products were resolved by PAGE and data analysis was performed as described before.
MUTYH Binding and RNase H2 Activity Inhibition
Electrophoretic mobility shift assay (EMSA) was used to evaluate MUTYH binding capacity to an 8-oxodG:rA substrate. DNA substrate (30 nm) was incubated with increasing MUTYH concentrations at 4 °C for 1 h. DNA-protein complexes were analyzed on nondenaturing 8% polyacrylamide gel in Tris borate buffer, run at 350 V for 3 h at 4 °C. The percentage of substrate bound to MUTYH was estimated by densitometric scan of the fluorescent bands (bound versus bound plus free DNA fraction). In competition experiments, 36-mer duplexes (30 nm), labeled at the 5′ end of the strand containing dA or rA opposite an 8-oxodG, were preincubated with MUTYH (10 nm) in 10 μl of buffer B, at 37 °C for time ranging from 0 up to 60 min. RNase H2 (1 unit) was added and MgCl2 concentration adjusted at 2 mm final concentration before 60 min incubation at 37 °C. Reactions were stopped by addition of loading buffer solution and heating at 95 °C for 2 min. No hydrolysis was detected in the control sample in these conditions.
Results
Ribonucleotide Incorporation and Extension by POL β at Repeated Sequences
We examined the capacity of POL β to incorporate ribonucleotides into a 50-mer template comprising six CAG repeats flanked by 20 and 12 nucleotides at the 3′ and 5′ sides, respectively. Primers that terminated at different positions in the first triplet of the sequence were used to direct the incorporation of ribo- and deoxyadenine or cytosine monophosphates (Fig. 1, A–C). To investigate ribonucleotide incorporation opposite 8-oxodG, a template thymine in the first triplet was substituted by 8-oxodG (G* in Fig. 1C). POL β strongly discriminates against ribonucleotides and insertion of rAMP occurred >3 orders of magnitude less efficiently than dAMP opposite template dT (selectivity index of 2273)(Fig. 1, A and D). A similar selectivity was also observed for dCMP over rCMP opposite template dG (selectivity index of 2941) (Fig. 1, B and D). These data confirm previously reported findings for POL β ribonucleotide incorporation into random DNA sequences (23). POLβ incorporated rAMP and rCMP opposite 8-oxodG at 1298- and 800-fold lower efficiency than dAMP and dCMP, respectively (Fig. 1, C and E). Finally the small difference measured in incorporating rCMP or rAMP opposite the oxidized purine (selectivity index of 1.5) indicates that POL β can equally use either ribonucleotides and is unable to discriminate well between them (Fig. 1, F and G). We then investigated whether POL β could extend a 3′ terminal rNMP:8-oxodG base pair. Addition of dNTPs resulted in extension from a terminal rAMP opposite either 8-oxodG or dT (Fig. 2, A and B, lanes 2). At least 50% of a terminal rAMP:8-oxodG or rAMP:dT was extended (lanes 3). Similarly, POL β efficiently extended rCMP:8-oxodG or dG pairs (compare lanes 2 and 3 in Fig. 2, C and D). No apparent difference was observed in the efficiency of extension of rCMP:dG and rCMP:8-oxodG pairs by POL β (Fig. 2, C and D, lanes 3). When dCTP was omitted from the dNTP mix, extension still occurred, albeit less efficiently. Multiple pause sites were identified along the DNA template in correspondence of the bases that followed the rC:dG mispairs (Fig. 2D, lane 4).
FIGURE 1.

Incorporation of rNTPs and dNTPs by human POL β. A–C, 20–21-mer primers (top strand), 5′ end labeled with 6-FAM, were annealed to different templates (50-mer, bottom strand) without (A and B) or with (C) the 8-oxodG base, indicated with G*. D, kinetic parameters for rAMP/dAMP and rCMP/dCMP incorporation opposite their canonical complementary base. E, kinetic parameters for rAMP/dAMP and rCMP/dCMP incorporation opposite a G* template. F, discrimination plot for incorporation of deoxynucleotide and ribonucleotide monophosphates opposite template T, C, and 8-oxo-dG bases. The ratios between Vmax and Km are reported on the y axis. The horizontal lines indicate the Vmax/Km for the insertion of a dNMP or rNMP. The vertical lines joining the two bars give a measure “at a glance” of the difference in incorporation efficiency between the two cases. G, discrimination plot for incorporation of dATP/dCTP and rATP/rCTP opposite template 8-oxo-dG bases. The ratios between Vmax and Km are reported on y axis.
FIGURE 2.

Elongation of mispairs from rAMP and rCMP incorporated opposite canonical or 8-oxodG bases. Incorporation of rAMP opposite template 8-oxo-dG (panel A, lane 2) or opposite template T (panel B, lane 2) and subsequent mispair extension (panels A and B, lanes 3). Primer/templates (10 nm) were incubated with POL β (0.1 unit) in the presence of rATP (400 μm final concentration) (lanes 2). Elongation of the mispair (lanes 3) was obtained by adding an equimolar mixture of dNTPs (10 μm final concentration) and incubating the samples at 37 °C for 1 h. Control DNA (lane 1) was incubated in the absence of POLβ or rATP/dNTP. Incorporation of rCMP (400 μm final concentration) opposite template 8-oxo-dG (panel C) or template G (panel D) and subsequent mispair extension (panels C and D) were studied using the same experimental conditions described above. Control DNA (lanes 1), rCMP insertion (lanes 2), elongation after addition of 10 μm dNTP (lanes 3), elongation of the rC:G mismatch by addition of dATP, dCTP, dTTP (10 μm) (panel D, lane 4) are shown.
These findings demonstrate that POL β-mediated ribonucleotide incorporation opposite a correct complementary base is less efficient than that of the corresponding deoxyribonucleotide. This effect is independent of the base to be inserted. They indicate further that the presence of a template 8-oxodG does not significantly affect the efficiency of ribonucleotide incorporation by POL β. Thus the presence of the 2′ oxygen does not change the dual nature of 8-oxodG coding and both rCMP and rAMP are incorporated opposite 8-oxodG (27). Although ribonucleotide incorporation is less efficient than that of dNMPs, Pol β is able to elongate a terminal rNMP:dN or rNMP:8-oxodG. In this regard, our results are in agreement with previous reports that POL β can extend ribonucleotide-containing primers (23) and insert multiple ribonucleotides when supplied with an excess of rCTP (21).
Processing of rC:8-OxodG Mispairs by OGG1 and RNase H2
The ability of BER and RER to process base pairs comprising a ribonucleotide and 8-oxodG was investigated (Fig. 3A). Fig. 3D shows the cleavage patterns of OGG1 DNA glycosylase on an 8-oxodG:dC or 8-oxodG:rC substrate. OGG1 digestion of a 36-bp duplex with the mispair located in the first of six CTG/GAC repeats generated the expected 11-mer cleavage product (indicated with an arrow in Fig. 3, B and D). Comparison of the incision efficiency indicated that the presence of a ribonucleotide opposite the oxidized purine (8-oxodG:rC) only slightly (3-fold) decreased 8-oxodG excision.
FIGURE 3.

OGG1 and RNase H2 incision of 8-oxo-dG:rC mispairs. A, scheme of the DNA/RNA repair enzymes involved in 8-oxo-dG and ribonucleotide base removal in a DNA duplex. B, scheme of the potential OGG1 incision site of a DNA duplex containing 8-oxo-dG:C or 8-oxo-dG:rC mispair. The 8-oxo-dG containing strand (36-mer) was 5′ end labeled with 6-FAM. C, scheme of the potential RNase H2 incision site of a DNA duplex containing 8-oxo-dG:rC or dG:dC mispairs. The rC containing strand (36-mer) was 5′ end labeled with 6-FAM. D, representative gel of the products of OGG1 incision assays. A 36-bp DNA duplex (10 nm), labeled at the 5′ end with 6-FAM, and containing single dG*:dC (lanes 1–6) or dG*:rC mispairs was reacted with increasing amounts of OGG1 enzyme (0–1 unit, lanes 1-6) for 1 h at 37 °C. OGG1 activity expressed as the amount of incision products/h was plotted against the OGG1 amount in the reaction volume (solid circle, dG*:dC; empty circle, dG*:rC). The mean of triplicate experiments and S.D. are reported in the panel on the right of the figure. E, representative gel of the product of an RNase H2 incision assay. 36-bp DNA duplex (10 nm), labeled with 6-FAM at the 5′ end of the strand containing the ribonucleotide, and containing single dG*:dC or dG*:rC mispairs (lanes 1–6) was reacted with increasing amounts of H2 enzyme (0–2 units, lanes 1-5) for 1 h at 37 °C. Data analysis was performed as described under “Experimental Procedures.” RNase H2 activity expressed as the amount of reaction products obtained following the reaction was plotted against the RNase H2 amount in the reaction volume (full circle, dG*:dC; empty circle, dG*:rC). The mean of triplicate experiments and S.D. are plotted in the panel on the right of the figure.
RNase H2 assays indicated that activity on a DNA duplex containing a single ribonucleotide was unaffected by the presence of the oxidized purine paired with rC (Fig. 3E). Representative gels showing the formation of the expected RNase H2 incision products (25-mer) on 36-bp duplexes containing either the dG:rC or 8-oxodG:rC mispairs (Fig. 3C) are presented in Fig. 3E. Data analysis indicates that both substrates are efficiently and equally cleaved by RNase H2 (Fig. 3E). We conclude that the presence of a ribonucleotide opposite an 8-oxodG lesion only moderately reduces OGG1 excision efficiency, whereas RNase H2 activity is not detectably affected by the presence of an oxidized purine opposite the DNA ribonucleotide.
Processing of rA:8-OxodG Mispairs by RNase H2 and MUTYH
The efficiency of RNase H2 incision of a 36-bp duplex containing an rA:8-oxodG mispair (Fig. 4, A and B) was unaffected by the presence of an 8-oxodG opposite the ribonucleotide. Incision kinetics were indistinguishable on substrates containing single rA:dT and rA:8-oxodG mispairs (Fig. 4, B and D). As expected no incision occurred on a DNA substrate containing paired dA:dT (Fig. 4D).
FIGURE 4.

MUTYH and RNase H2 enzymatic activity on 8-oxodG:rA mispairs. A, scheme of the RNA/DNA repair enzymes involved in 8-oxodG:rA removal in a DNA duplex. B and C, scheme of the potential incision site on a DNA duplex containing 8-oxodG:rA or T:rA mispair by MUTYH or RNase H2 enzyme. The rA containing strand (36-mer) was 5′ end labeled with 6-FAM dye. D, RNase H2 incision assay. The DNA duplex substrate was a 36-bp DNA (10 nm), labeled with 6-FAM at the 5′ end of the strand containing the rA or dA base. A single dG*:rA or dT:rA mispair was present at the 14-base position from the labeled 5′ end. The DNA substrate was reacted with increasing amount of RNase H2 enzyme (0–4 units) for 1 h at 37 °C. Products were separated by PAGE and data analysis was performed as described under “Experimental Procedures.” RNase H2 activity, expressed as the percentage of incision products, was plotted as a function of the RNase H2 amount in the reaction volume (full circle, rA:8-oxodG; empty circle, rA/dT; full diamond, dA/dT). E, MUTYH incision assay. DNA duplex substrate was a 36-bp DNA (10 nm), labeled at the 5′ end of the strand containing the rA or dA base. The substrates containing 8-oxodG:dA (lanes 1 and 2) and 8-oxodG-rA mispairs (lanes 3 and 4) were incubated with purified MUTYH (20 nm, active fraction concentration) as described under “Experimental Procedures.” Products were separated by PAGE and data analysis was performed as described before.
Processing of rNMP-containing base pairs by MUTYH was carried out using the purified human enzyme and the rA:8-oxodG- or dA:8-oxodG-containing duplex (Fig. 4, C and E). APE1 incubation did not result in cleavage of the rA-containing strand indicating that MUTYH had not removed the mispaired A (Fig. 4E). MUTYH was fully active on the analogous dA:8-oxodG-containing duplex (Fig. 4E).
Although unable to remove the mispaired A from the rA:8-oxodG-containing duplex, EMSA revealed that MUTYH efficiently bound both rA:8-oxodG and dA:8-oxoG substrates (Fig. 5, A and B, lanes 2–4 and 6–8). Non-productive MUTYH binding to the rA:8-oxodG duplex affected the incision by RNase H2. RNase H2 activity was assayed following a 10- or 30-min preincubation with MUTYH. Without preincubation, more than 90% of the substrate was incised to generate the expected 11-mer fragment (Fig. 5C, lane 2). In contrast, only 50% of the substrate was incised following 10 or 30 min preincubation with purified MUTYH (Fig. 5C, lane 3). This inhibition did not further increase following a prolonged MUTYH preincubation (Fig. 5C, lane 4).
FIGURE 5.

MUTYH binding and MUTYH/RNase H2 competition. A, sequence of the DNA duplexes used for band-shift experiments and incision assays. B, band-shift experiments were performed on 8-oxodG:dA/rA substrates (30 nm) and increasing MUTYH concentration (1–10 nm) for 1 h at 4 °C in a 10-μl reaction volume. Products were electrophoresed on 8% nondenaturing polyacrylamide gels. C, RNase H2 incision assay. DNA duplex substrate (10 nm) (lane 1) was reacted with RNase H2 enzyme (1 unit) for 1 h at 37 °C without (lane 2) or with a previous incubation with MUTYH (10 nm) for 10 (lane 3) and 30 (lane 4) min. Products were separated by denaturing PAGE and data analysis was performed as described before.
In summary, RNase H2 can efficiently remove a ribonucleotide paired to an oxidized guanine. However, catalytically ineffective MUTYH binding to this substrate interferes with RNase H2 processing.
POL β-mediated Incorporation of Oxidized Ribonucleotides and Processing by RNase H2 and MUTYH
Both dNTP and rNTP pools are susceptible to oxidation and might provide substrates for repair synthesis by POL β. We examined the ability of POL β to use oxidized ribonucleotides in primer extension reactions opposite correct or incorrect nucleotides (Fig. 6A). A 5′ end (6-FAM)-labeled primer was annealed to a normal template in which 20 CAG repeats were flanked by 20 bases long non-repetitive regions. POL β-mediated incorporation was analyzed in the presence of increasing amounts of 8-oxorGTP, 8-oxodGTP, and UTP. 8-OxorGMP incorporation opposite dA was 36-fold lower than that of 8-oxodGTP. This selectivity is of the same order as that of UTP incorporation (Fig. 6, B and C). In contrast we were unable to detect any incorporation of 8-oxorGTP opposite dC (Fig. 6, B and C) in either the repeated sequence or an unrelated random non-repetitive sequence (data not shown). We conclude that incorporation by POL β of an 8-oxo-rGTP derived from an oxidized pool will most likely give rise to 8-oxo-rG:dA mismatches, with formation of 8-oxo-rG:dC mispairs being strongly unfavored.
FIGURE 6.

Incorporation of 8-oxorGTP by POL β and incision of 8-oxorG:dA mispairs by RNase H2 and MUTYH. A, scheme of the primer/template DNA substrate used for measurements of POL β incorporation activity. B, kinetics parameters for incorporation of 8-oxorGMP, 8-oxodGMP, and UMP opposite template dA. Selectivity index was calculated to compare the incorporation efficiency of oxidized rNMP versus oxidized dNMP and oxidized rNMP versus UTP. C, discrimination plot showing the incorporation efficiency, described by the Vmax/Km ratio of 8-oxo-rGMP and 8-oxo-dGMP opposite dA or dC template. The meaning of the vertical lanes and horizontal bars have been described in the legend to Fig. 1. D, elongation of an 8-oxorG:dA mismatch by POL β and incision by RNase H2. Incorporation of 8-oxorGMP was obtained on primer/substrate (10 nm) reported in the panel A. 8-Oxo-rGMP (50 μm final concentration) was added to the samples and incorporation shown in panel D (lane 1). After a 30-min incubation at 37 °C, dNTP (10 μm) were added and reactions were allowed for a further 30 min (lane 2). Triplicate samples of the reaction loaded on lane 2 were prepared and two were EtOH precipitated. The same procedure was applied to an unmodified primer control (lane C). Recovered DNA without (panel D, lane 3) and with (lane 4) RNase H2 (2 units). E, RNase H2 incision on extended duplex noncontaining ribonucleotides. Only dCTP and dGTP were used in this case. dGMP incorporation (10 μm) (lane 1), dGTP plus dCTP incorporation (10 μm) (lane 2), and products obtained by RNase H2 reaction (lane 3) are shown. The same experimental conditions reported in panel D were used. F, MUTYH activity on a dA:8-oxorG containing substrate. A primer/template, with the same sequence reported in panel A, labeled with 6-FAM at the 5′ end of the dA strand was used. Following incorporation of 8-oxorGMP and mispair extension by dNTP addition, at the same concentrations used for RNase H2 experiments reported in panel E, samples were EtOH precipitated. Pellets were resuspended in the reaction buffer (buffer A plus 80 mm NaCl) and incubated for 1 h at 37 °C without (lane 2) or with (lane 3) MUTYH (10 nm). The reactions were parallel performed on a dA:8-oxodG substrate. Lane 4, control sample; lane 5, reaction products after MUTYH incubation. In lane 1 the reaction products obtained by using a substrate built by adding only dNTP are reported. M is a molecular weight marker.
The presence of an 8-oxorG:dA mispair does not significantly impede extension by POL β. In the presence of four dNTPs, POL β efficiently extended a terminal 8-oxorGMP (Fig. 6D, lane 1). This preferential incorporation of dA opposite 8-oxorG by POL β follows the same rule observed for incorporation of an oxidized dGMP (27).
To investigate whether RNase H2 can process an 8-oxorG:dA mispair we constructed DNA duplexes by primer extension from a terminal 8-oxorGMP:dA mispair at position 25 (numbered from the 5′ end of the labeled DNA strand) (Fig. 6A). Under our experimental conditions, almost all substrate was extended to generate products of different lengths, all of which contained a single 8-oxorG (Fig. 6D, lanes 2 and 3). RNase H2 digestion of these purified products generated fragments of the expected size (24-mer) (Fig. 6D, compare lanes 3 and 4) indicating that RNase H2 is fully active on an 8-oxorG:dA mispair. As expected no RNase H2-mediated incision was observed on DNA substrates built in a similar way and containing no ribonucleotides (Fig. 6E). We conclude that, notwithstanding oxidation of the ribonucleotide, RNase H2 is active on an 8-oxorG:dA mispair.
The same strategy was used to examine whether MUTYH can incise a substrate containing an 8-oxorG:dA mismatch (Fig. 6F). In this case an unlabeled primer was annealed to a 5′ end (6-FAM) template and DNA duplexes (100-mer) were obtained by POL β-mediated primer extension reactions. Following incubation with purified human MUTYH the expected 75-mer incision products derived from the dA-containing strand were observed (Fig. 6F, lanes 2 and 3). Similar levels of MUTYH-mediated cleavage (25%) were observed with an analogously built duplex containing an 8-oxodG:dA mispair indicating that MUTYH is fully active on an 8-oxorG:dA mismatch (Fig. 6F, lanes 4 and 5). No cleavage products were observed in control samples (lane 1).
Discussion
Our findings demonstrate that POL β is able to incorporate ribonucleotides during repair synthesis at repeated sequences. As in random sequences (22, 23), ribonucleotide insertion is always less favored than deoxynucleotide incorporation. Changing the coding base from G to 8-oxodG had only a minor effect on the efficiency of ribonucleotide incorporation. The slight preference of POL β for rCTP over rATP insertion opposite 8-oxodG mirrors a similar small discrimination between dCTP and dATP (27). The major determinant of dual coding by a template 8-oxodG is its ability to assume both syn and anti conformations (27–29). Our data indicate that this property is not detectably affected by the presence of a C2′ oxygen and ribonucleotide insertion opposite a template 8-oxodG appears to follow the same rules as deoxyribonucleotide incorporation.
POL β only incorporated 8-oxorGMP opposite a template dA and insertion was less favored than that of 8-oxodGMP. The structure of human POL β with an incoming 8-oxodGTP and template dA indicates that the pairing of 8-oxodGTP in syn- and anti-dA permits a planar arrangement of the base pair that is comfortably accommodated in the active site (30). The 8-oxodG:dA pairing is stabilized through Hoogsteen hydrogen bonding and the correct geometry in the active site is assured by a further hydrogen bond between the O8 oxygen of the incoming 8-oxodGTP with Asn-279 (Fig. 7, top). We postulate that the slightly disfavored 8-oxorGMP incorporation might be the consequence of destabilization of the planar arrangement of the base pair by the formation of an alternative hydrogen bond between Asn-279 and the C2′ oxygen (Fig. 7, bottom).
FIGURE 7.
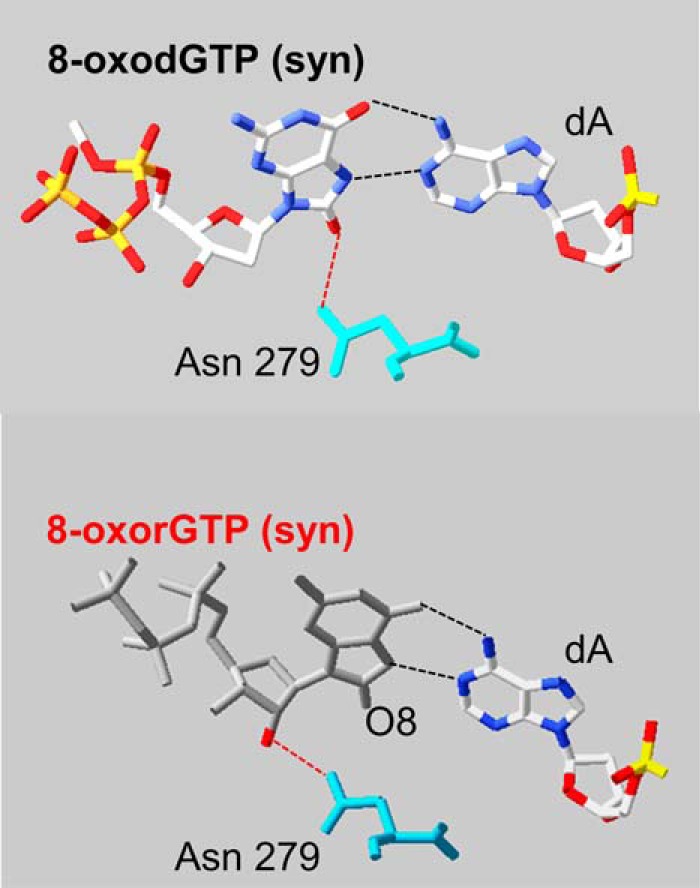
Comparison of 8-oxodGTP and 8-oxorGTP paired with a template adenine in the POL β active site. Comparison of the crystal structure of an incoming 8-oxodGTP (syn conformation, top) and 8-oxoGTP (bottom) paired with adenine (top figure was modified from Batra et al. (29)). The bottom figure was modeled from the crystal structure Protein Data Bank 3MBY by adding the hydroxyl group in C2′. The interaction with Asn-279 (turquoise) is shown. ArgusLab 4.0.1 software (M. Thompson, ArgusLab 4.0.1, Planaria Software LLC, Seattle, WA) and Swiss-PdbViewer (EXPASY) were used for Protein Data Bank manipulation.
Compared with 8-oxodGTP, utilization of 8-oxorGTP for insertion opposite a template dC was dramatically diminished, with this bias being independent of the template sequence context. It has been suggested that the steric repulsion between O8 of 8-oxodGTP in the anti-conformation, its sugar phosphate backbone, and the sugar (C2′) of the primer terminus induce a distortion in the active site of POL β that makes the incorporation of 8-oxodGMP opposite dC difficult (30). Our findings suggest that additional factors might come into play in the attempted formation of 8-oxorGTP:dC mispair. We conclude that, during repair synthesis events, POL β might incorporate ribonucleotides opposite normal bases as well as oxidized guanines. Based on a rCTP:dCTP pool imbalance close to 100 (17, 31), it has been calculated that POL β inserts one rNMP for every 81 dNMPs (23). In non-dividing cells, however, the rNTP/dNTP ratio may be more than 10-fold higher (32). In particular the large rGTP/dGTP ratio (2000) measured in growth-arrested cells suggests that POL β utilization of rGTP and its oxidized form, under conditions of oxidative stress, is likely. Information on the use of oxidized rNTPs or insertion of rNTPs opposite 8-oxodG is limited to Schizosaccharomyces pombe Pol4 polymerase (33). In contrast to our results with POL β, Pol4 showed better incorporation of 8-oxorGMP opposite dC than opposite dA template. This reflects structural differences of the active sites of POL β and Pol4 polymerases, with the last one being able to use rNTPs and dNTPs with similar efficiency (34). Thus although both DNA polymerases belong to the same POL X family, results obtained with Pol4 are not comparable with those obtained by other human repair polymerases.
We also showed that 8-oxodG:rC/rA and 8-oxorG:dA will be easily elongated during DNA repair resulting in the formation of complex mispairs at repeated sequences. Our data show that RNase H2 does not discriminate between canonical and oxidized ribonucleotides and can remove ribonucleotides also when these are paired with 8-oxodG. These results are consistent with the findings that RNase H2 interacts only with the DNA backbone, with a conserved tyrosine contacting the 2′ OH group (35). Similarly no interactions occur with the base opposite the ribonucleotide (35).
In contrast to RNase H2, OGG1 and MUTYH interact extensively with both the DNA 8-oxodG and the complementary strand (36, 37). In particular, OGG1 contacts with the opposed cytosine (36) and its activity is strongly inhibited by the presence of bases different from the canonical C or by an abasic site opposite 8-oxodG (38). Correct versus incorrect base pair (dA:dT versus dA:8-oxodG) discrimination by MUTYH is mostly due to the interaction of the carboxyl-terminal MUTYH domain (37). Our data confirm that a modification in 2′, whereas not affecting MUTYH binding capacity, totally abrogates the catalytic activity. This has been already reported for a fluorine substitution at the same sugar position (39). This MUTYH nonproductive interaction with the rA:8-oxodG substrate can potentially interfere with removal of rA by RNase H2.
Our data imply that formation of complex lesions, due to incorporation of rNTPs during DNA repair at repeated sequences, might modify the efficiency of repair systems (Fig. 8). The independent action of RNase H2 and OGG1 at 8-oxodG:rC mispairs would restore normal base pairing (Fig. 8, top). The removal of 8-oxorG or dA from 8-oxorG:dA mispair by, respectively, RNase H2 and MUTYH (Fig. 8, bottom), constitute a second level of protection against the mutagenic effects of oxidized ribonucleotides. Because in these cases BER and RER are active on two different strands, depending on the enzymes concentration and on the timing of repair events, production of double strand breaks might be also envisaged. A more complex scenario is present when BER and RER enzymes are acting on the same strand and compete for the same substrate (Fig. 8, middle). In this case MUTYH binding to the 8-oxodG:rA mispair in a catalytically inert fashion might interfere with RNase H2-mediated repair activity.
FIGURE 8.

BER and RER activity on complex mispairs containing oxidized bases and ribonucleotides. When POL β incorporates rCMP opposite 8-oxodG (dG*), RNase H2 is going to efficiently remove rC from the resulting 8-oxodG:rC mispair, whereas OGG1 repair of dG* is slightly reduced. Should 8-oxodG:rA arise after rAMP incorporation, RER will process the rA containing strand, whereas MUTYH-mediated BER will be inhibited. Possible interference on RER activity might occur by concurrent recognition of the lesion. In the likelihood of limiting MTH1 hydrolytic activity, 8-oxorGTP (rG*TP) might be used by POL β to produce rG*:dA mispairs. These substrates will be efficiently processed by MUTYH and RNase H2. Simultaneous BER and RER activities might lead to the formation of double strand breaks (DSB) or intermediate repair products of unknown reparability.
Our data have important implications on expansion/contraction events triggered by DNA repair at repeated sequences in non-dividing cells. The formation of complex mispairs containing mono-ribonucleotides by long-patch BER will eventually lead to new repair cycles mediated by RNase H2 and/or MUTYH intervention. These would create new strand interruptions, possibly leading to hairpin formation in opposite strands, with strand realignments causing expansion of the repeated sequences. Whether processing of complex mispairs, by compromising either BER and/or RER, affects the stability of trinucleotide repeat regions remains to be investigated.
Author Contributions
M. B. and F. M. conceived and designed the study and wrote the paper. A. M. provided technical assistance throughout the study. P. C. and A. M. performed the primer extension experiments with POL β. P. C. and F. M. performed the experiments and analyzed the results for OGG1, MUTYH, and RNase H2 activity, C. B. performed the PDB manipulation. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Peter Karran and Pietro Pichierri for data discussion.
This work was supported in part by Associazione Italiana Ricerca sul Cancro Investigator Grant 11755 (to M. B. and F. M.) and Ministry of Health Project “Malattie Rare” (to M. B. and F. M.). The authors declare that there are no competing financial interests in relationship to the work described.
- 8-oxodG
- 7,8-dihydro-8-oxoguanine
- rNTPs
- ribonucleotide triphosphates
- RER
- ribonucleotide excision repair
- POL β
- DNA polymerase β
- BER
- base excision repair
- AP
- apurinic/ apyrimidinic
- APE1
- apurinic/apyrimidinic-endonuclease-1.
References
- 1.Helbock H. J., Beckman K. B., Shigenaga M. K., Walter P. B., Woodall A. A., Yeo H. C., and Ames B. N. (1998) DNA oxidation matters: the HPLC-electrochemical detection assay of 8-oxo-deoxyguanosine and 8-oxo-guanine. Proc. Natl. Acad. Sci. U.S.A. 95, 288–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shibutani S., Takeshita M., and Grollman A. P. (1991) Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature 349, 431–434 [DOI] [PubMed] [Google Scholar]
- 3.Michaels M. L., and Miller J. H. (1992) The GO system protects organisms from the mutagenic effect of the spontaneous lesion 8-hydroxyguanine (7,8-dihydro-8-oxoguanine). J. Bacteriol. 174, 6321–6325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.David S. S., O'Shea V. L., and Kundu S. (2007) Base-excision repair of oxidative DNA damage. Nature 447, 941–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Loon B., Markkanen E., and Hübscher U. (2010) Oxygen as a friend and enemy: how to combat the mutational potential of 8-oxo-guanine. DNA Repair 9, 604–616 [DOI] [PubMed] [Google Scholar]
- 6.Mazzei F., Viel A., and Bignami M. (2013) Role of MUTYH in human cancer. Mutat. Res. 743, 33–43 [DOI] [PubMed] [Google Scholar]
- 7.Furuichi M., Yoshida M. C., Oda H., Tajiri T., Nakabeppu Y., Tsuzuki T., and Sekiguchi M. (1994) Genomic structure and chromosome location of the human mutT homologue gene MTH1 encoding 8-oxo-dGTPase for prevention of A:T to C:G transversion. Genomics 24, 485–490 [DOI] [PubMed] [Google Scholar]
- 8.Klungland A., Rosewell I., Hollenbach S., Larsen E., Daly G., Epe B., Seeberg E., Lindahl T., and Barnes D. E. (1999) Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc. Natl. Acad. Sci. U.S.A. 96, 13300–13305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruggieri V., Pin E., Russo M. T., Barone F., Degan P., Sanchez M., Quaia M., Minoprio A., Turco E., Mazzei F., Viel A., and Bignami M. (2013) Loss of MUTYH function in human cells leads to accumulation of oxidative damage and genetic instability. Oncogene 32, 4500–4508 [DOI] [PubMed] [Google Scholar]
- 10.Kovtun I. V., Liu Y., Bjoras M., Klungland A., Wilson S. H., and McMurray C. T. (2007) OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature 447, 447–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Y., Prasad R., Beard W. A., Hou E. W., Horton J. K., McMurray C. T., and Wilson S. H. (2009) Coordination between polymerase β and FEN1 can modulate CAG repeat expansion. J. Biol. Chem. 284, 28352–28366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goula A. V., Berquist B. R., Wilson D. M. 3rd, Wheeler V. C., Trottier Y., and Merienne K. (2009) Stoichiometry of base excision repair proteins correlates with increased somatic CAG instability in striatum over cerebellum in Huntington's disease transgenic mice. PLoS Genet. 5, e1000749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McMurray C. T. (2010) Mechanisms of trinucleotide repeat instability during human development. Nat. Rev. Genet. 11, 786–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clausen A. R., Zhang S., Burgers P. M., Lee M. Y., and Kunkel T. A. (2013) Ribonucleotide incorporation, proofreading and bypass by human DNA polymerase δ. DNA Repair 12, 121–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vaisman A., and Woodgate R. (2015) Redundancy in ribonucleotide excision repair: competition, compensation, and cooperation. DNA Repair 29, 74–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown J. A., and Suo Z. (2011) Unlocking the sugar “steric gate” of DNA polymerases. Biochemistry 50, 1135–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nick McElhinny S. A., Watts B. E., Kumar D., Watt D. L., Lundström E. B., Burgers P. M., Johansson E., Chabes A., and Kunkel T. A. (2010) Abundant ribonucleotide incorporation into DNA by yeast replicative polymerases. Proc. Natl. Acad. Sci. U.S.A. 107, 4949–4954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sparks J. L., Chon H., Cerritelli S. M., Kunkel T. A., Johansson E., Crouch R. J., and Burgers P. M. (2012) RNase H2-initiated ribonucleotide excision repair, Mol. Cell 47, 980–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reijns M. A., Rabe B., Rigby R. E., Mill P., Astell K. R., Lettice L. A., Boyle S., Leitch A., Keighren M., Kilanowski F., Devenney P. S., Sexton D., Grimes G., Holt I. J., Hill R. E., Taylor M. S., Lawson K. A., Dorin J. R., and Jackson A. P. (2012) Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell 149, 1008–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim N., Huang S. N., Williams J. S., Li Y. C., Clark A. B., Cho J. E., Kunkel T. A., Pommier Y., and Jinks-Robertson S. (2011) Mutagenic processing of ribonucleotides in DNA by yeast topoisomerase I. Science 332, 1561–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bergoglio V., Ferrari E., Hübscher U., Cazaux C., and Hoffmann J. S. (2003) DNA polymerase β can incorporate ribonucleotides during DNA synthesis of undamaged and CPD-damaged DNA. J. Mol. Biol. 331, 1017–1023 [DOI] [PubMed] [Google Scholar]
- 22.Nick McElhinny S. A., and Ramsden D. A. (2003) Polymerase μ is a DNA-directed DNA/RNA polymerase. Mol. Cell. Biol. 23, 2309–2315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cavanaugh N. A., Beard W. A., and Wilson S. H. (2010) DNA polymerase β ribonucleotide discrimination: insertion, misinsertion, extension, and coding. J. Biol. Chem. 285, 24457–24465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gosavi R. A., Moon A. F., Kunkel T. A., Pedersen L. C., and Bebenek K. (2012) The catalytic cycle for ribonucleotide incorporation by human DNA Pol λ. Nucleic Acids Res. 40, 7518–7527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.D'Agostino V. G., Minoprio A., Torreri P., Marinoni I., Bossa C., Petrucci T. C., Albertini A. M., Ranzani G. N., Bignami M., and Mazzei F. (2010) Functional analysis of MUTYH mutated proteins associated with familial adenomatous polyposis. DNA Repair 9, 700–707 [DOI] [PubMed] [Google Scholar]
- 26.Turco E., Ventura I., Minoprio A., Russo M. T., Torreri P., Degan P., Molatore S., Ranzani G. N., Bignami M., and Mazzei F. (2013) Understanding the role of the Q338H MUTYH variant in oxidative damage repair. Nucleic Acids Res. 41, 4093–4103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller H., Prasad R., Wilson S. H., Johnson F., and Grollman A. P. (2000) 8-OxodGTP incorporation by DNA polymerase β is modified by active site residue Asn 279. Biochemistry 39, 1029–1033 [DOI] [PubMed] [Google Scholar]
- 28.Beard W. A., Batra V. K., and Wilson S. H. (2010) DNA polymerase structure-based insight on the mutagenic properties of 8-oxoguanine. Mutat. Res. 703, 18–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Batra V. K., Shock D. D., Beard W. A., McKenna C. E., and Wilson S. H. (2012) Binary complex crystal structure of DNA polymerase β reveals multiple conformations of the templating 8-oxoguanine lesion. Proc. Natl. Acad. Sci. U.S.A. 109, 113–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Batra V. K., Beard W. A., Hou E. W., Pedersen L. C., Prasad R., and Wilson S. H. (2010) Mutagenic conformation of 8-oxo-7,8-dihydro-2′-dGTP in the confines of a DNA polymerase active site. Nat. Struct. Mol. Biol. 17, 889–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Traut T. W. (1994) Physiological concentrations of purines and pyrimidines. Mol. Cell Biochem. 140, 1–22 [DOI] [PubMed] [Google Scholar]
- 32.Ferraro P., Franzolin E., Pontarin G., Reichard P., and Bianchi V. (2010) Quantitation of cellular deoxynucleoside triphosphates. Nucleic Acids Res. 38, e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sastre-Moreno G., Sánchez A., Esteban V., and Blanco L. (2014) ATP insertion opposite 8-oxo-deoxyguanosine by Pol4 mediates error-free tolerance in Schizosaccharomyces pombe. Nucleic Acids Res. 42, 9821–9837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bebenek K., Garcia-Diaz M., Patishall S. R., and Kunkel T. A. (2005) Biochemical properties of Saccharomyces cerevisiae DNA polymerase IV. J. Biol. Chem. 280, 20051–20058 [DOI] [PubMed] [Google Scholar]
- 35.Rychlik M. P., Chon H., Cerritelli S. M., Klimek P., Crouch R. J., and Nowotny M. (2010) Crystal structures of RNase H2 in complex with nucleic acid reveal the mechanism of RNA-DNA junction recognition and cleavage. Mol. Cell 40, 658–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bruner S. D., Norman D. P., and Verdine G. (2000) Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature 403, 860–866 [DOI] [PubMed] [Google Scholar]
- 37.Fromme J. C., Banerjee A., Huang S. J., and Verdine G. L. (2004) Structural basis for removal of adenine mispaired with 8-oxoguanine by MutY adenine DNA glycosylase. Nature 427, 652–656 [DOI] [PubMed] [Google Scholar]
- 38.Bjorâs M., Luna L., Johnsen B., Hoff E., Haug T., Rognes T., and Seeberg E. (1997) Opposite base-dependent reactions of a human base excision repair enzyme on DNA containing 7,8-dihydro-8-oxoguanine and abasic sites. EMBO J. 16, 6314–6322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sharer O. D., Deng L., and Verdine G. L. (1995) Chemical approaches toward understanding base excision DNA repair. J. Am. Chem. Soc. 117, 10781–10782 [DOI] [PubMed] [Google Scholar]